
Dynamics of dissociative electron attachment to aliphatic thiols
Sukanta
Das
 and
Vaibhav S.
Prabhudesai
*
and
Vaibhav S.
Prabhudesai
*
Tata Institute of Fundamental Research, Colaba, Mumbai 400005, India. E-mail: vaibhav@tifr.res.in
First published on 4th January 2024
Abstract
Dissociative electron attachment (DEA) shows functional group-dependent site selectivity in H− ion channels. In this context, thiol functional groups have yet to be studied in great detail, although they carry importance in radiation damage studies where low-energy secondary electrons are known to induce damage through the DEA process. In this context, we report detailed measurements of absolute cross-sections and momentum images of various anion fragments formed in the DEA process in simple aliphatic thiols. We also compare the observed dynamics with that reported earlier in hydrogen sulphide, the precursor molecule for this functional group, and with that in aliphatic alcohols. Our findings show substantial resemblance in the underlying dynamics in these compounds and point to a possible generalisation of these features in the DEA to thiols. In addition, we identify various pathways that contribute to the S− and SH− channels.
1. Introduction
Low-energy electron collisions with organic molecules have attracted much attention lately after the discovery of their direct1 and indirect2 role in the single- and double-strand breaks in DNA that may lead to radiation damage to living cells. Apart from the low-energy electrons formed in the energy range of 0–20 eV due to high-energy radiation interaction with organic matter, the radicals formed during such interactions also cause considerable damage to the living cells.3 In this context, many efforts have been made to understand the interaction of low-energy electrons with organic molecules. The functional group-dependent site-selectivity observed in dissociative electron attachment (DEA) to organic molecules has unravelled new facets of the low-energy electron molecule interaction.4 The molecules containing a hydroxyl group are particularly interesting and have been extensively studied in the past.5–8 Thiol is one of the important molecular functional groups performing various tasks inside biological systems. The molecules that have a thiol group primarily reside in the cell and play a vital role in different cellular functions such as glycolysis, mitochondrial energy production, and stimulation of glucose metabolism.9 Thiols are one of the main protective mechanisms against oxidative stress.10 They also play a role in enzymatic reactions, detoxification, and antioxidant protection in the body.11 However, DEA to thiol functional group-containing compounds has not been explored extensively, apart from one report on ethanethiol.12On the other hand, a comparison of DEA to water and hydrogen sulphide (H2S) provides an interesting case to understand the intricate molecular dynamics followed by the negative ion resonances containing the OH and SH groups. For example, the two molecules show distinct resonances dissociating into the H− channel in DEA. These resonances are at 6.5, 8.5 and 12 eV in water13 and around 5.2, 7.5 and 9.6 eV in H2S.14 Both these molecules belong to the C2v symmetry group. As O and S atoms belong to the same group in the periodic table, these molecules are expected to show similar structures and dynamics. On the other hand, the O atom is more electronegative than the S atom, making it a stronger hydrogen bond participant. The negative ion resonances corresponding to the three peaks observed in the H− channel in DEA are 2B1, 2A1 and 2B2 states, which are core excited resonances. However, there are distinct differences in their dynamics as observed in the momentum imaging.14,15 The H− channel at the first resonance in water shows substantial internal excitation of the OH fragment, whereas in H2S, the SH fragment shows very little internal excitation. The angular distribution of the H− channel from the second resonance in water shows significant internal motion before dissociation, making it deviate from the expected angular distribution from the axial recoil motion.16,17 In contrast, in H2S, the angular distribution at the second resonance is in accordance with the axial recoil motion.14 More interestingly, the OH− ions are reported to be observed experimentally by Fedor et al.18 as the direct dissociation product from all three resonances, while theoretical calculations do not find this channel as a direct product in these resonances and attribute it to nonadiabatic effects.19 On the other hand, in DEA to H2S, SH− ions are reported at 2.4 eV.14,20 Interestingly, both simple alcohols21,22 and thiols12 have shown the production of OH− and SH− ions, respectively. Moreover, the systematic investigations on simple organic alcohols have shown the presence of an H− channel at around 6.5 eV that arises from the hydroxyl group and shows strong functional group dependence.4 However, at 6.5 eV resonance, using the momentum imaging technique, it was found that the corresponding angular distribution of the H− channel is substantially different from that reported for H− from water at 6.5 eV despite a similar resonance state playing a role in the reaction. The difference in the angular distribution has been explained as due to the torsional modes of vibrations active at room temperature.6,8 These vibrational excitations have a substantial influence on the angular distribution. In light of these observations, it would be interesting to explore the similar functional group dependence of the DEA process in thiol groups.
Ibănescu and Allan have reported CH3CH2S−, CH3CHS−, SH−, and S− fragments from DEA to ethanethiol.12 CH3CHS− shows two peaks at 0.61 eV and 1.66 eV, CH3CH2S− shows a peak at 1.83 eV, and all these peaks are assigned as shape resonance. While SH−peaks at 8.7 eV and S− at 8.1 eV, they are inferred to arise from core excited Feshbach resonances.
Here, we report the detailed investigation of the DEA process in simple thiols, namely, ethanethiol and 1-butanethiol. Also, taking the cue from the H2S, we expect the thiol group-containing compounds to follow the axial recoil motion, throwing more light on the underlying DEA dynamics. In this work, we have obtained the resonance position of different fragments from ethanethiol and 1-butanethiol, calculated their kinetic energy (KE) and angular distribution from the momentum images taken using the velocity slice imaging (VSI) technique and compared them with those of the H2S and ethanol, especially at the 6.5 eV channel of ethanol. We have also found an energetic S−/SH− channel. This channel is important in atmospheric chemistry, particularly in its sulphur budget from organic mercaptans.23
2. Experimental setup
The experimental setup is described in detail elsewhere,24 and here we provide its brief description. Magnetically collimated pulsed electron beam produced from a home-built thermionic emission-based electron gun is made to cross the molecular beam at a right angle. The gaseous molecular beam is produced using a capillary array connected to the glass bulb that contains the pure sample of the target molecule in liquid form. The bulb is evacuated before introducing the liquid and pumped using a dry pump until the sample volume is reduced to 1/3rd. The pulsed electron beam (width 100 ns and repeat rate of 10 kHz) is collected by a Faraday cup mounted coaxially with the electron gun but on the opposite side of the interaction volume. The electron current is measured using an electrometer (Kethley 6512). The molecular beam is introduced in the interaction region along the axis of the VSI spectrometer. The VSI spectrometer comprises an interaction region flanked by two electrodes, namely, a pusher and puller, with the latter having a molybdenum wire mesh of 64% transmission spot-welded across its central aperture of diameter 50 mm. The pusher–puller distance is kept at 20 mm. The puller electrode is followed by a four-element electrostatic lens assembly and a short flight tube of 25 mm in length. The two-dimensional position-sensitive detector comprises a pair of micro-channel plates (MCP) in the chevron configuration, and a phosphor screen with a P47 scintillator is mounted co-axially with the flight tube on its other end. The ions generated in the interaction region are pushed into the direction of the detector using a square pulsed voltage (−100 V amplitude and 1-microsecond duration) applied to the pusher electrode with the puller electrode kept at zero potential. The extraction pulse is synchronised with the electron pulse using a delay generator, maintaining a delay of 100 ns. The signal due to ions is obtained for the time-of-flight measurement using a coupling circuit connected to the MCP back. The ion–yield curves so obtained are put on the absolute scale using the relative flow technique,25 where the DEA to oxygen at 6.5 eV is used as a standard gas to obtain the absolute cross-section of the process.26 After identifying the ion, the position information of a particular ion hit is obtained by recording the light signal obtained from the phosphor screen using a CCD camera. The MCP detector is synchronously pulsed (pulse width of 80 ns) using a high-voltage switch to get a time slice of the Newton sphere of a particular ion species. This pulsing is synchronised with the electron pulse with an appropriate delay to coincide with the centre of the time-of-flight peak of that particular ion species. The ion hit distribution obtained using a CCD camera is added frame-by-frame to obtain their final distribution. As for the effusive molecular target, the background gas contributes substantially to the detected signal, especially when the projectile electron beam is not focused in the interaction region, and this background signal produces substantial distortion in the image. The imaging condition is optimised to minimise this distortion, and we have subtracted the background signal from the crossed-beam signal with appropriate normalisation with the electron current to avoid any additional artefacts.273. Results and discussion
The DEA measurements on ethanethiol and 1-butanethiol show H−as the most dominant channel apart from the S−/SH− and (M − 1)− ions (M being the mass of the parent molecule). The VSI spectrometer could not resolve the S− and SH− ions due to poor mass resolution. Similarly, the (M − 1)− ions may also have a contribution from (M − 2)−. However, the VSI spectrometer used in these measurements could not distinguish between the two. The absolute cross-section curves for various ions as a function of electron energy starting from 1 eV are shown in Fig. 1. The H−channel from both molecules shows three peaks around 5 eV, 7 eV, and 8 eV. It is also the most dominant channel in DEA in case of both molecules. The first two peak positions are close to those obtained from H2S, the precursor molecule of the SH functional group. As can be seen from Fig. 1, H− is the most dominant channel with a maximum cross-section of 4 × 10−19 cm2 at 5 eV for ethanethiol and 8.7 × 10−20 cm2 at 7.8 eV for 1-butanethiol. The absolute cross-sections of other H− peaks are given in Table 1. All the cross-section values have an uncertainty of about 15%, with the maximum contribution from the reference gas cross-section value.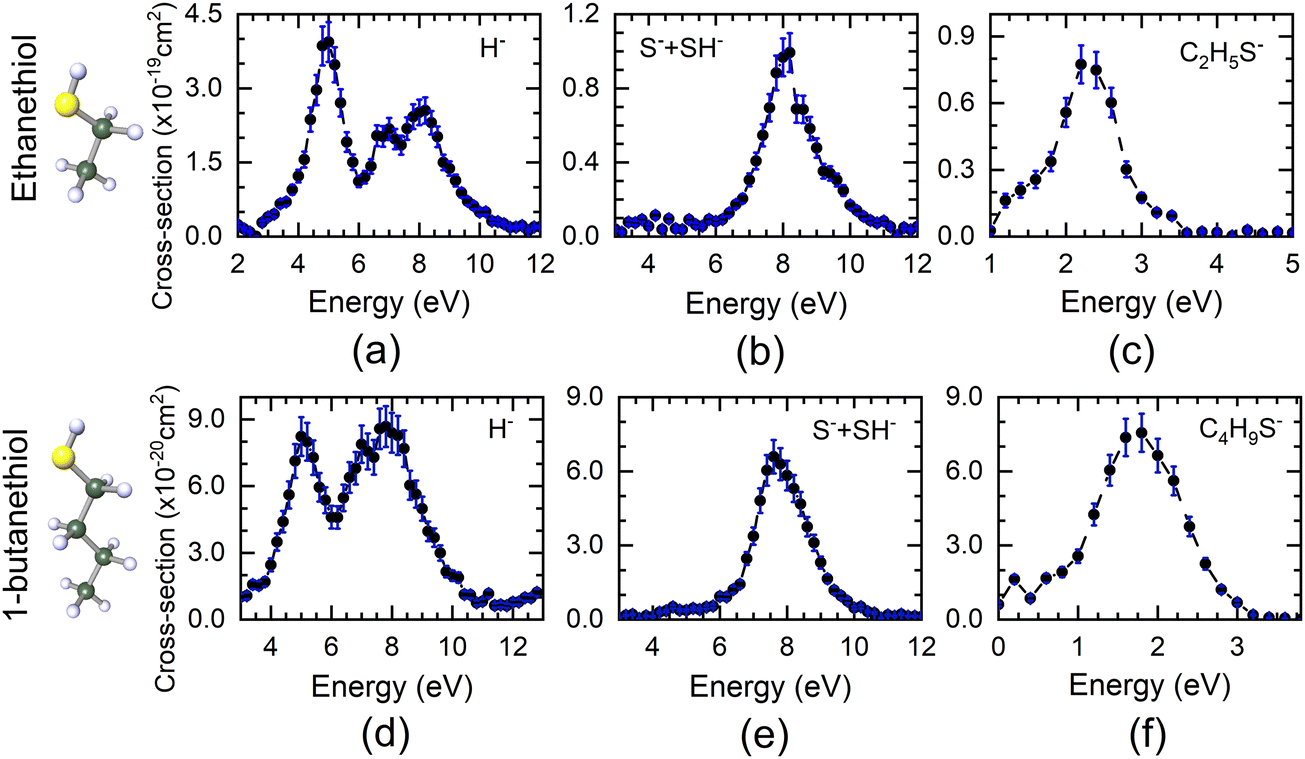 | ||
| Fig. 1 Ion–yield curve obtained for the DEA process and put on the absolute cross-section scale for the (a) H−, (b) (S− + SH−), and (c) (C2H5S− + C2H4S−) channels from ethanethiol and (d) H−, (e) (S− + SH−), and (f) (C4H9S− + C4H8S−) channels from 1-butanethiol as a function of electron energies. Structures of the ethanethiol and 1-butanethiol are shown, where the yellow, green and white spheres represent S, C and H atoms, respectively. | ||
| Ion species | Ethanethiol | 1-Butanethiol | ||
|---|---|---|---|---|
| Electron energy (eV) | Absolute cross-section (× 10−19 cm2) | Electron energy (eV) | Absolute cross-section (× 10−20 cm2) | |
| H− | 5 | 3.39 ± 0.40 | 5 | 8.23 ± 0.87 |
| 7 | 2.17 ± 0.22 | 7 | 7.88 ± 0.83 | |
| 8.2 | 2.55 ± 0.26 | 7.8 | 8.67 ± 0.90 | |
| S− + SH− | 8.2 | 0.96 ± 0.10 | 7.6 | 6.59 ± 0.67 |
| (M − 1)− | 2.2 | 0.77 ± 0.08 | 1.8 | 7.56 ± 0.77 |
Unlike H2S, neither of the thiol molecules shows any resonance in the S−/SH− channels at low electron energy. These channels peak around 8.2 eV for ethanethiol and 7.6 eV for 1-butanethiol, with the absolute cross-section of 9.6 × 10−20 cm2 and 6.59 × 10−20 cm2, respectively (Table 1). These values are lower by a factor of about 20 as compared to H2S.28 The (M − 1)− ion signal peaks around 2.2 eV and 1.8 eV for ethanethiol and 1-butanethiol with the absolute cross-section of 7.7 × 10−20 cm2 and 7.59 × 10−20 cm2, respectively (Table 1).
We have obtained the momentum images of all these channels at various electron energies using the VSI technique. We deduced the KE and angular distributions for these channels from the offline data analysis of these momentum images. Below, we provide details of these distributions for each channel and describe the inferred molecular dynamics behind these dissociation channels.
3.1 H− ions
This channel shows three peaks at 5, 7 and 8 eV. The momentum images obtained for this channel at various electron energies for both molecules are shown in Fig. 2.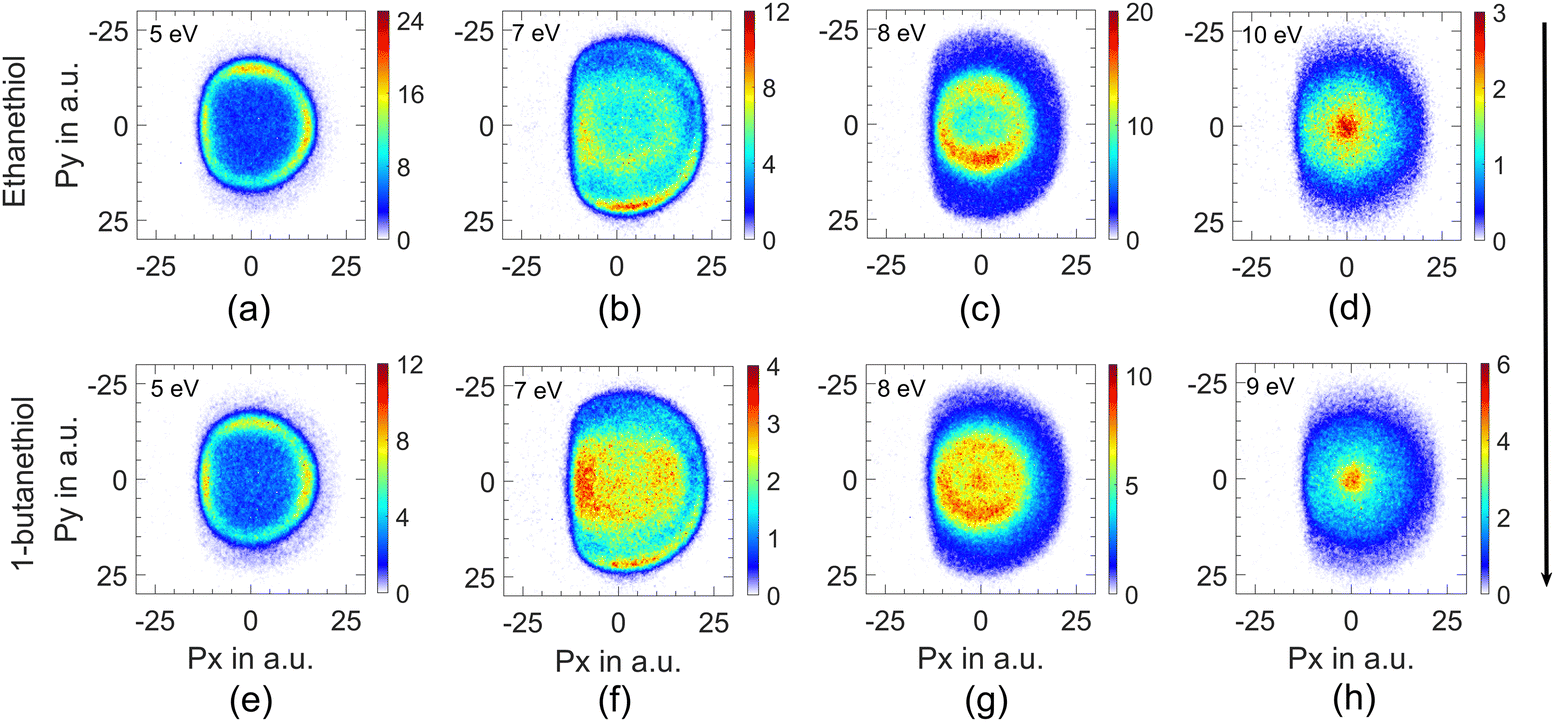 | ||
| Fig. 2 Momentum images for the H− ions at (a) 5 eV, (b) 7 eV, (c) 8 eV and (d) 10 eV from ethanethiol and (e) 5 eV, (f) 7 eV, (g) 8 eV and (h) 9 eV electron energy from 1-butanethiol. The direction of the electron beam is from top to bottom. | ||
Due to the presence of an electron-beam-collimating transverse magnetic field, the ion images are deflected away from the axis of the spectrometer. This deviation is maximum for the lightest H− ions. It causes the distortion of the half of the image (the left half in this case) that results from the ion trajectories passing closer to the electrodes’ aperture edges.29 As the DEA process has azimuthal symmetry about the electron beam, the momentum image is expected to show the left-right symmetry. For the KE and angular distribution estimation, we have used only the right half of the image, measured close to the detector axis. We have used this part of the image to determine the centre of the distribution and carried out further analysis. The KE distributions obtained for this channel from both the molecules at various electron energies are shown in Fig. 3.
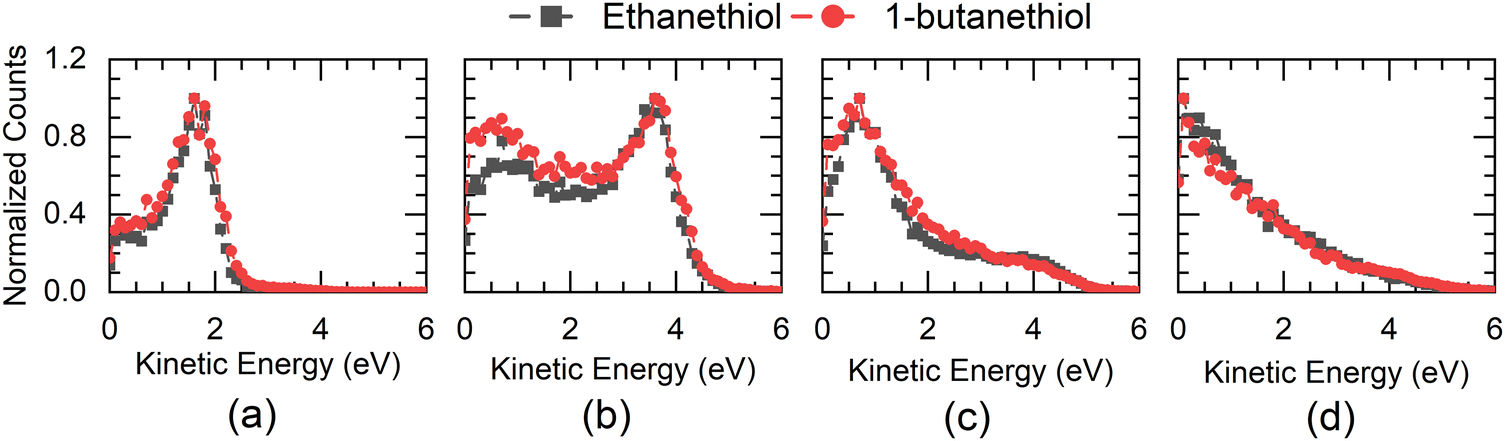 | ||
| Fig. 3 KE distribution for the H− ions from ethanethiol (–■–) and 1-butanethiol (–●–) at (a) 5 eV, (b) 7 eV, and (c) 8 eV electron energies. (d) shows the KE distribution obtained for ethanethiol at 10 eV and 1-butanethiol at 9 eV electron energy. | ||
As mentioned earlier, the first two peaks in the ion–yield curve, at 5 eV and 7 eV, are consistent with those observed in the H2S, the precursor molecule of the thiol (–SH) functional group. Based on the earlier work on the functional group-dependent site-selective fragmentation, we conclude that these two peaks arise exclusively from the S–H site of the molecules.
The threshold for obtaining this ion in a two-body break-up from the parent molecule can be determined using the heat of formation for various components. Taking the heat of formation (ΔHf) for C2H5SH as −46 kJ mol−1,30 C2H5S as 104 kJ mol−1,31 and H as 218 kJ mol−1 and taking the electron affinity (E.A.) of H as 73 kJ mol−1, we get 295 kJ mol−1 or 2.97 eV as the minimum electron energy required to obtain this ion in DEA to ethanethiol. For 1-butanethiol with the ΔHf of 1-C4H9SH as −87.9 kJ mol−1 and that for 1-C4H9S as 54.4 kJ mol−1,32 we get the threshold energy for the H− channel as 2.98 eV. Hence, the excess energy in the system would be 2.03 eV (2.02 eV) for ethanethiol (1-butanethiol) for the 5 eV electron. As H− is the lightest fragment, in the axial recoil motion, most of the excess energy will show up as its KE. The KE distribution peaks around 1.75 eV for this channel at 5 eV electron energy for both molecules, indicating fast dissociation. Although for 5 eV electron energy, the maximum expected KE of H− is 2 eV, the KE distribution shows a spread up to 2.5 eV. We attribute it to the electron beam's poor energy resolution (about 0.8 eV). For the 7 eV resonance as well, the KE distribution of H− has a peak between 3.5 and 3.75 eV, which is consistent with the fast two-body break-up scenario.
The angular distribution of H− at 5 eV has two distinct features (Fig. 4(a)). The angular distribution peaks around 90°–100°, dips close to 130° and then again rises close to 180°. The 100° peak matches fairly well with the angular distribution obtained for H− from H2S at 5 eV14 with one significant difference. The H− angular distribution for H2S becomes extremely small in the forward and backward direction with respect to the incoming electron beam, which is not the case with either of the thiol molecules. However, in this context, it resembles the angular distribution obtained for the first resonance in this channel in ethanol.8 In H2S, similar to water, the 5.5 eV resonance is understood to be the core excited resonance, with a significant contribution coming from the excitation of the lone pair of electrons from the sulphur atom to the 6a1 orbital with the incoming electron getting captured in it. This makes the resonant state a 2B1 state, and the H− angular distribution resembles that of water with zero intensity in the forward and backward directions,33i.e., no electron capture along the symmetry plane of the H2S leads to this channel. However, the presence of the DEA signal in the forward and backward direction in alcohols8 has been identified as the contribution of the torsionally excited molecules. The torsional modes break the planner symmetry of the alcohols (C–H–O plane) and make the capture along the symmetry plane possible, which leads to an enhanced signal in the forward–backward direction with respect to the electron beam.6 Similarly, we attribute the signal in the forward and backward direction from alkyl thiols to the torsionally excited molecules. The major difference between the angular distribution from ethanol and ethanethiol is that the contribution in the backward direction in the latter is far lower than that in the former. Moreover, the signal in the forward direction shows the reverse trend. This points to the possibility of differences in the dynamics of the torsionally excited molecules in both species on electron attachment. Interestingly, the spread in the KE distribution in both ethanol and ethanethiol is comparable, unlike in water and H2S, showing similarity in the overall dynamics followed by the anions formed by electron attachment to the ground state molecules. Another point to note is that the angular distribution observed in 1-butanethiol is similar to that observed for ethanethiol. The increase observed in the signal in the backward direction from methanol6 to ethanol8 is not observed in the ethanethiol and 1-butanethiol, although the number of torsional degrees of freedom increases, and their frequency decreases.
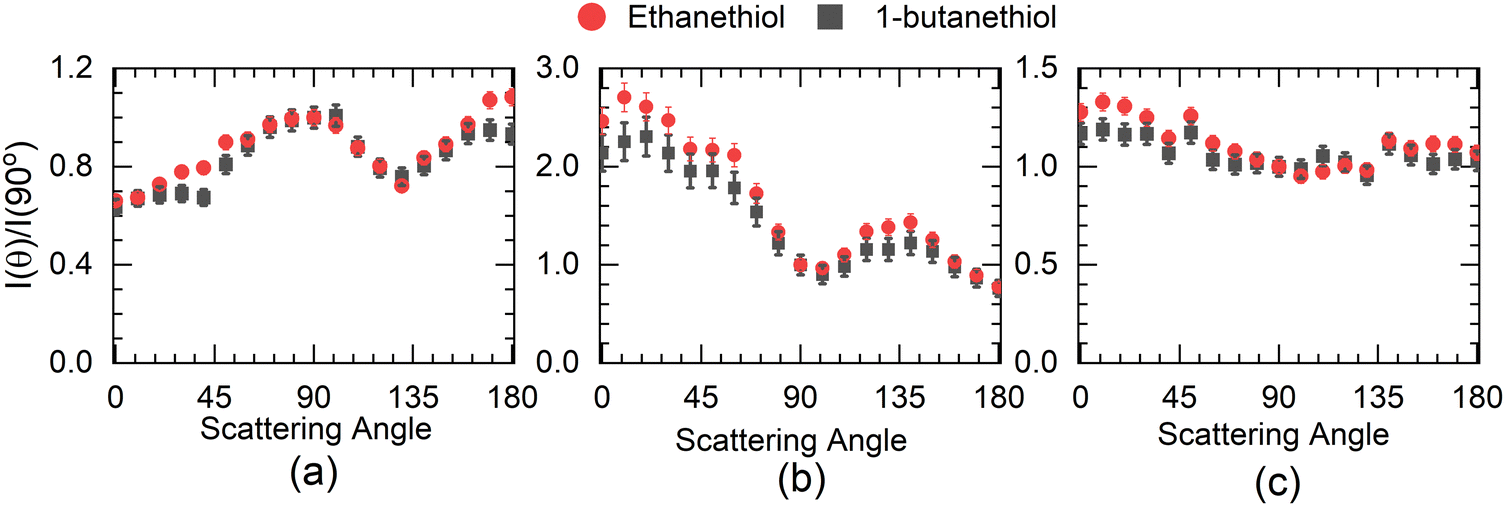 | ||
| Fig. 4 Angular distribution of H− ions from ethanethiol (–●–) and 1-butanethiol (–■–) at (a) 5 eV (KE range 1.1 to 2 eV), (b) 7 eV (KE range 3 to 4.2 eV) and (c) 8 eV (KE range 0.3 to 1.3 eV) peaks in the ion–yield curve. | ||
Compared with the first resonance, the second resonance in ethanethiol shows a substantially different angular distribution compared to ethanol. The angular distribution obtained at 7 eV electron energy for the H− channel with the KE of H− in the range of 3 to 4.2 eV (20 to 24 a.u. momentum) (Fig. 4(b)) resembles that obtained for the second resonance in this channel in H2S. In water, this resonance shows substantial internal dynamics.16,17 Similar internal dynamics also result in the second resonance in ethanol, which is reflected in a more smeared angular distribution with reduced anisotropy.8 However, H2S shows very little internal dynamics, resulting in the angular distribution following the expected pattern from the axial recoil approximation.14 Comparing the angular distribution for the second resonance in ethanethiol with ethanol and hydrogen sulphide, we conclude that this resonance also follows the axil recoil motion in the dissociation process. For 1-butanethiol, this resonance shows similar dynamics to that of ethanethiol.
The momentum image at the third peak at around 8 eV shows an annular distribution in the momentum range of 5 to 10 a.u. (Fig. 2(c)) along with a non-negligible signal at lower energies (momentum <5 a.u.) (Fig. 2(c)) for ethanethiol. This low-energy feature clearly appears as a blob in the case of 1-butanethiol (Fig. 2(g)). The 8 eV peak is broad in the ion–yield curve, and its contribution at lower electron energies can be seen in the momentum image at 7 eV as the inner ring (Fig. 2(b), region between 5 and 10 a.u. momentum). The images taken at higher electron energies (10 eV for ethanethiol and 9 eV for 1-butanethiol) show a clearly enhanced signal close to zero KE. This indicates the presence of another resonance in this electron energy range. On comparison with the momentum images obtained for methanol34 and ethanol8 around these electron energies, we identify this feature arising from the C–H site in the molecule. This site breakage results from at least a three-body break-up mechanism, leaving very little KE with the fragment. It is also known that the resonance peak in the H− channel from alkanes shifts to the lower electron energies.35 This is consistent with the observation that this low-KE feature appears prominently in 1-butanethiol compared to ethanethiol in the momentum image at 8 eV.
The higher KE feature (peaking between 5 and 10 a.u. momentum) shows an almost isotropic distribution from 1-butanethiol, whereas ethanethiol shows slightly higher intensity in the forward direction compared to the backward direction with respect to the electron beam. Its angular distribution at 8 eV electron energy (in the KE range 0.45–0.9 eV (0.35–0.8 eV) for ethanethiol (1-butanethiol)) is shown in Fig. 4(c). The KE and the angular distribution suggest that the source of this channel is a two-body dissociation. However, for 8 eV electron energy, the excess energy for the two-body breakage path with both the fragments in the ground state is almost 5 eV. The observed KE ranges from 0 to 2 eV (Fig. 3(c)) and indicates that the excess energy with the molecular fragment (C2H5S) is in the range of 3 to 5 eV. However, this fragment has an absorption band with the band origin at around 2.9 eV. The corresponding excited state is identified as the B2A′′ state.36 This implies that the observed H− channel arises from the dissociation path.
| e + C2H5SH → (C2H5SH−)* → C2H5S(B2A′′) + H− | (1) |
3.2 S− and SH− ions
The experimental setup used in these measurements cannot separate the ions from the S− and SH− channels. The combined ion yield curve for these channels shows a single peak at 8.2 eV (7.6 eV) for ethanethiol (1-butanethiol) (Fig. 1(b) and (e)). For the VSI condition used for the measurements, the time-of-flight signal for the two ions was expected to appear within the width of the time slice (80 ns). Hence, in the momentum images, we cannot distinguish between the two ions. According to Ibănescu and Allan, both ions have equal intensity and peak at 8.1 eV with an additional peak at 8.7 eV in the SH− channel.12 To separate the SH−/S− ions, we used another VSI spectrometer with a longer flight tube of length 10 cm and operated it with voltage conditions required to get the best possible mass resolution. We calibrated the electron energy in this spectrometer by performing DEA measurements on SO2.36 We also compared the ion yield curves for various ions arising from thiols with those obtained earlier. S−/SO2 are used for the mass comparison, as shown in Fig. 5. The modified spectrometer clearly shows both the S− and SH− ions (Fig. 5(e)). Their relative strengths as a function of electron energy show that the 8 eV peak observed in the combined-ion yield curve is dominated by the S− ions along the rising edge of the peak. At the peak, the two ions have almost equal contributions, and on the trailing edge, the signal is dominated by the SH− ions. We obtained the momentum images of these ions at various electron energies across the 8.2 eV peak for ethanethiol. These momentum images are shown in Fig. 5, along with the observed mass spectrometric identification of the ion species.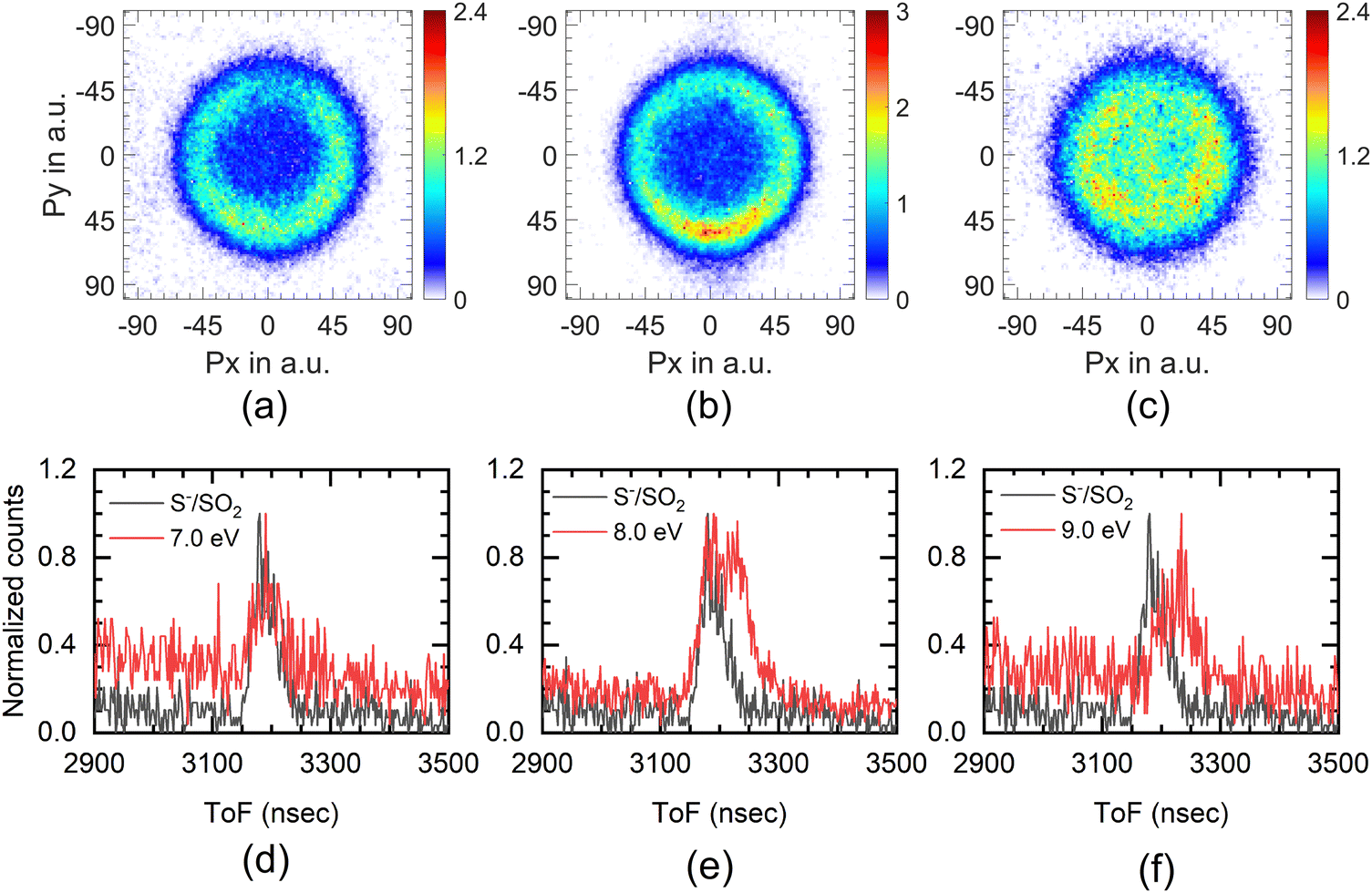 | ||
| Fig. 5 Momentum image of S−+SH− at (a) 7 eV, (b) 8 eV and (c) 9 eV electron energy from ethanethiol. The direction of the electron beam is from top to bottom. (d) to (f) are the mass spectra obtained for the corresponding electron energies from ethanethiol (red curve) in this mass range using another VSI spectrometer with relatively better mass resolution. In these spectra, the mass peak of the S−/SO2 (black curve) obtained at 4 eV electron energy is also shown for the mass reference. | ||
For 7 eV electron energy, the momentum image shows an annular pattern (spread in the momentum range of 30 to 70 a.u.) with an angular anisotropy, which appears to be on top of an isotropic distribution. The anisotropy increases at 8 eV. However, the distribution becomes slightly narrower and more intense. At 9 eV, the inner part of the image (<30 a.u. of momentum) gets filled, showing the production with low KE. We obtained the KE distribution from these images, and the results are shown in Fig. 6(a).
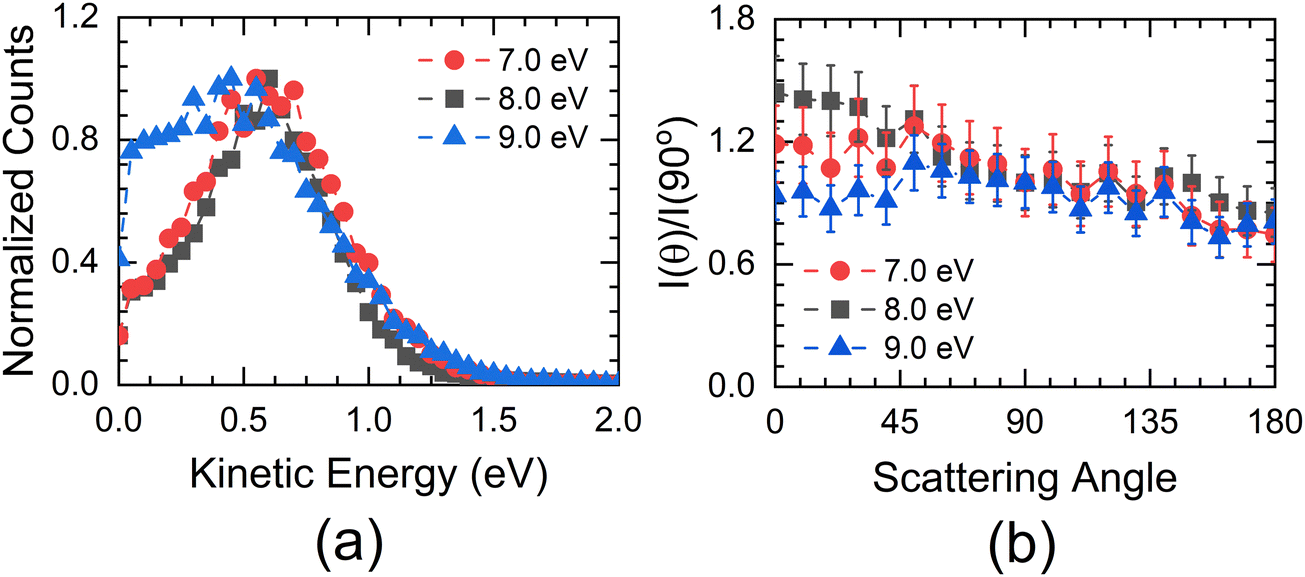 | ||
| Fig. 6 (a) KE distribution of S−+SH− ions obtained from the momentum images taken at 7 eV, 8 eV and 9 eV for ethanethiol. (b) Corresponding angular distribution of these ions taken in the KE range 0.4 eV to 0.85 eV. | ||
The KE distribution peaks between 0.5 and 0.75 eV for both 7 and 8 eV electron energies. As the ion signal at 7 eV is dominated by the S− ions, these ions are observed with some KE as well as anisotropy. Typically, there are three possibilities for getting these ions in DEA. The first one is from the concerted three-body dissociation, where two bonds are simultaneously broken.
| e + C2H5SH → (C2H5SH−)* → C2H5 + S− + H | (2) |
The thermodynamic threshold for this channel would be 4.77 eV (ΔHf (C2H5) = 119 kJ mol−1, ΔHf (S) = 277 kJ mol−1, E.A. (S) = 2.07 eV).30 In this case, one of the fragments, the H atom, is likely to take away most of the excess energy in the system as its KE. Interestingly, this channel is observed in H2S at this electron energy.13 Understandably, the S− channel there shows very low KE release as the other fragments are H atoms. Hence, this channel may contribute to the low KE part of the image.
The other mode would be a two-body break-up after the rearrangement of the H atom from the SH site. This channel would be slow due to the H migration in the molecule.
| e + C2H5SH → (C2H5SH−)* → C2H6 + S− | (3) |
The thermodynamic threshold for this channel is 0.47 eV (ΔHf (C2H6) = −84 kJ mol−1, ΔHf (S) = 277 kJ mol−1, E.A. (S) = 2.07 eV).30 Based on the amount of excess energy available in the system, this channel may contribute to the annular pattern. However, it requires substantial internal rearrangement; it is unlikely to show the anisotropy as seen in the momentum image.
The third channel of the formation of S− ions via a sequential dissociation, i.e., the formation of SH− ions followed by their further dissociation into S− ions.
| e + C2H5SH → (C2H5SH−)* → C2H5 + (SH−)* | (4a) |
| (SH−)* → S− + H | (4b) |
This is possible if the C2H5 radical is formed in the electronic ground state and SH− ions are formed in their dissociating part of an excited state. Based on the heat of formation of fragments (ΔHf (SH) = 139 kJ mol−1) and electron affinity of SH (E.A.(SH) = 2.3 eV), we estimate the dissociation energy of the SH− ion to be 3.92 eV. This implies that the minimum electron energy required to obtain the S− ions by this channel is 4.77 eV. However, the first step (4a) would have at most 2.23 eV as the KE for 7 eV electron energy. This implies that the maximum KE of the (SH−)* would be 1.04 eV. In this case, the S− ions would have very little KE in the centre of mass frame of the SH− ions. The corresponding linear momentum of SH− ion would be passed on to the S− ion formed in its centre of mass frame. This makes the maximum KE of the S− ion in this channel to be 1.07 eV. This is consistent with the observed KE distribution. Moreover, the angular distribution of the S− ions would be that of the SH− ions, explaining the observed anisotropy. In the absence of the SH− signal at 7 eV, we attribute the observed S− signal to this channel. It is worth noting that in H2S as well, only S− and no SH− were observed at this electron energy.20
At 8 eV, we also see the SH− ions with almost the same intensity as that of S− ions. However, we do not see a substantial change in the KE distribution, whereas the anisotropy increases compared to the 7 eV image.
The SH− channel in ethanethiol can result from either a two-body or a three-body break-up. The possible reaction paths are
| e + C2H5SH → (C2H5SH−)* → C2H5 + SH− | (5) |
| e + C2H5SH → (C2H5SH−)* → CH3 + CH2 + SH− | (6) |
The threshold for the path (5) is 0.85 eV. Similarly, for path (6) (ΔHf (CH3) = 145 kJ mol−1, ΔHf (CH2) = 386 kJ mol−1), we have obtained the threshold as 5.13 eV. There can be other three-body break-up paths where one of the fragments in the concerted break-up can be an H atom. However, in such cases, most of the excess energy will be carried by the H atom, the lightest fragment. As the KE distribution peaks at about 0.75 eV with a well-defined relatively narrow spread, we rule out any such concerted break-up mechanism resulting in this fragment at this electron energy. Moreover, from path (6), the KE distribution of the SH− fragment would also be spread from zero KE. Also, it may not show the anisotropy obtained in the momentum image. Hence, we conclude that this concerted break-up mechanism is contributing very little to the signal. At 8 eV electron energy, in the path (5), the excess energy available to the system is 7.15 eV. However, the C2H5 radical can also be electronically excited. Two electronic excited states of this radical are identified in the UV absorption spectra at 5.03 and 6.05 eV.37,38 This would provide the following additional dissociation paths for the resonance state:
| e + C2H5SH → (C2H5SH−)* → C2H5(3s) + SH− | (7) |
| e + C2H5SH → (C2H5SH−)* → C2H5(3p) + SH− | (8) |
The momentum image obtained at 8 eV for the 1-butanethiol also shows an annular pattern similar to that for ethanethiol (Fig. 7). Similar excited states are also observed in secondary butyl radical.37 We assume that the primary butyl radical would also have similar states and attribute this channel in 1-butanethiol to such electronically excited butyl radical similar to ethanethiol. 1-Butanethiol also shows comparatively lower anisotropy, indicating the role of a higher number of degrees of freedom in delaying the initial dissociation process.
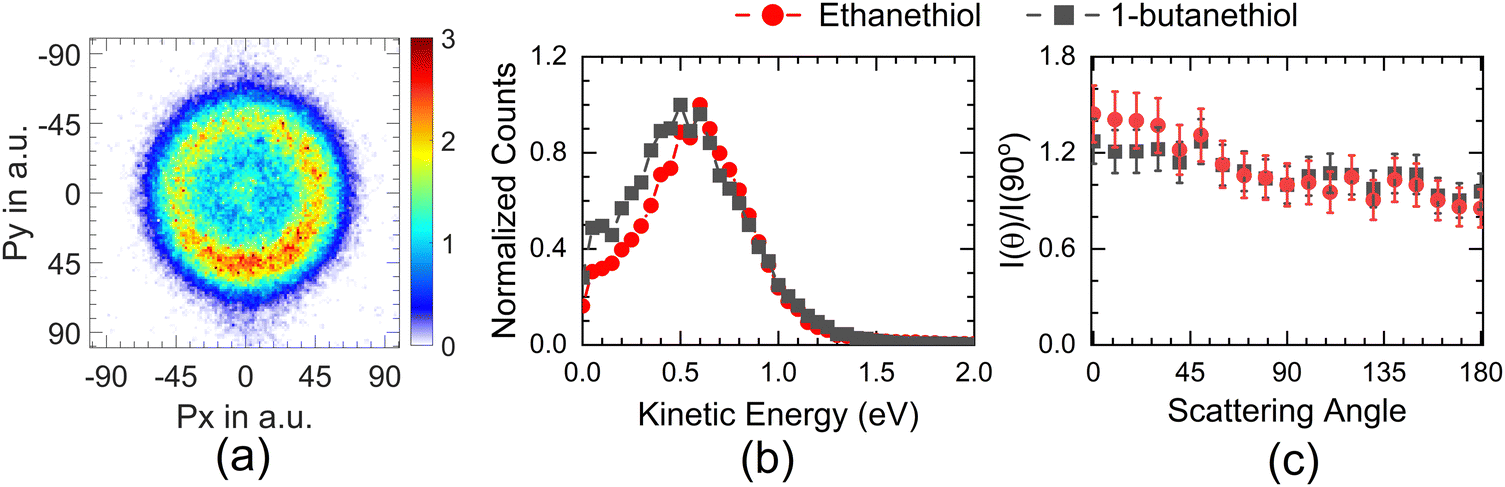 | ||
Fig. 7 (a) Momentum image obtained for the S− + SH− channel from 1-butanethiol at 8 eV. Corresponding (b) KE and (c) angular distribution of the S−+SH− channel from 1-butanethiol (–■–) along with those for ethanethiol ( ). ). | ||
At 9 eV, the ion signal is dominated by the SH− ions (Fig. 5(f)), and the corresponding momentum image shows an increase in the low KE ion signal (Fig. 5(c) and 6(a)). This indicates that the contribution from the path (8) starts showing up with significant intensity. It also shows that this particular path also results in internal excitation of the C2H5 radical.
In ethanol, the OH− formation peaks around 9 eV, and it shows the H atom scrambling from the alkyl site.8 Interestingly, unlike S− from ethanethiol, no O− has been observed with high KE from DEA.8,22 These findings indicate differences in the properties of the underlying resonant states in alcohols and thiols.
3.3 (M − 1)− ions
The (M − 1)− channel shows a peak at 2.2 eV in ethanethiol and 1.8 eV in 1-butanethiol, and the peak cross section of this channel is 7.70 × 10−20 and 7.56 × 10−20 cm2, in ethanethiol and 1-butanethiol, respectively. In ethanethiol, the (M − 2)− ions are also observed at lower energies.12 However, their signal is found to be extremely low at these energies. Hence, although we cannot distinguish between the two ions due to the limited mass resolution of our mass spectrometer, we attribute the observed signal at the peak to the (M − 1)− ions. This channel is attributed to the single particle shape resonance based on its appearance energy. We obtained the momentum image of this channel. The image shows a momentum distribution that peaks around zero but shows a slight spread along the electron beam. This corresponds to the KE of up to 50 meV. Considering the threshold of formation of this channel as 1.74 eV,12 the maximum KE expected in this channel will be 10 meV. We note that although the threshold energy for this channel is 1.74 eV, we attribute the substantial signal in this mass range below this energy to the (M − 2)− ions, which we could not mass resolve.4. Conclusion
We have observed the thiol functional group-dependent site selectivity in DEA. The dynamics of the H− channel from –SH resembles that of H2S, the precursor molecule for this functional group. This channel shows the effect of the molecule's torsional motion. However, unlike in aliphatic alcohols, aliphatic thiols do not show substantial variation in the torsion-related signal with the length of the alkyl chain. Moreover, the molecular fragment formed in this channel has very little energy in its internal degrees of freedom. In contrast with H2S, alkyl thiols show an additional peak in this channel with anisotropic angular distribution. In this dissociation process, the alkanethio radical is formed in its electronic excited state. The H− channel originating from the alkyl site shows the signature of at least a three-body dissociation consistent with the other aliphatic compounds. The single peak observed in the (S− + SH−) ions has a contribution from at least two resonances. The S− formation results from a sequential dissociation of the electronically excited SH− ion. The underlying resonant state in alkyl thiols has distinct properties in comparison with aliphatic alcohols. Such relatively faster sulphur radical anion may trigger further chemical reactions, making it interesting for the atmospheric sulphur budget with organic mercaptans.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the financial support from the Department of Atomic Energy, India, under project identification no. RTI4002.References
- B. Boudaïffa, P. Cloutier, D. Hunting, M. A. Huels and L. Sanche, Resonant Formation of DNA Strand Breaks by Low-Energy (3 to 20 eV) Electrons, Science, 2000, 287, 1658–1660 CrossRef PubMed.
- E. Alizadeh, A. G. Sanz, G. García and L. Sanche, Radiation Damage to DNA: The Indirect Effect of Low-Energy Electrons, J. Phys. Chem. Lett., 2013, 4, 820–825 CrossRef CAS PubMed.
- C. von Sonntag, Free-Radical-Induced DNA Damage and Its Repair, Springer-Verlag, Berlin, Heidelberg, Germany, 2006 Search PubMed.
- V. S. Prabhudesai, A. H. Kelkar, D. Nandi and E. Krishnakumar, Functional Group Dependent Site Specific Fragmentation of Molecules by Low Energy Electrons, Phys. Rev. Lett., 2005, 95, 143202 CrossRef PubMed.
- V. S. Prabhudesai, D. Nandi, A. H. Kelkar and E. Krishnakumar, Functional group dependent dissociative electron attachment to simple organic molecules, J. Chem. Phys., 2008, 128, 154309 CrossRef PubMed.
- D. S. Slaughter, D. J. Haxton, H. Adaniya, T. Weber, T. N. Rescigno, C. W. McCurdy and A. Belkacem, Ion-momentum imaging of resonant dissociative-electron-attachment dynamics in methanol, Phys. Rev. A: At., Mol., Opt. Phys., 2013, 87, 052711 CrossRef.
- B. C. Ibănescu, O. May, A. Monney and M. Allan, Electron-induced chemistry of alcohols, Phys. Chem. Chem. Phys., 2007, 9, 3163–3173 RSC.
- S. Das, S. Swain and V. S. Prabhudesai, Dynamics of the dissociative electron attachment to Ethanol, Arxiv, 2308.06662.
- N. Haugaard, Reflections on the Role of the Thiol Group in Biology, Ann. N. Y. Acad. Sci., 2000, 899, 148–158 CrossRef CAS PubMed.
- G. Pizzino, N. Irrera, M. Cucinotta, G. Pallio, F. Mannino, V. Arcoraci, F. Squadrito, D. Altavilla and A. Bitto, Oxidative Stress: Harms and Benefits for Human Health, Oxid. Med. Cell. Longevity, 2017, 2017, 1–13 Search PubMed.
- A. Kükürt, V. Gelen, Ö. Faruk Başer, H. Ahmet Deveci and M. Karapehlivan, Accenting Lipid Peroxidation, IntechOpen, 2021 Search PubMed.
- B. C. Ibănescu and M. Allan, Selectivity in bond cleavage in amines and thiols by dissociative electron attachment, J. Phys. Conf. Ser., 2009, 194, 012030 CrossRef.
- P. Rawat, V. S. Prabhudesai, G. Aravind, M. A. Rahman and E. Krishnakumar, Absolute cross sections for dissociative electron attachment to H2O and D2O, J. Phys. B: At., Mol. Opt. Phys., 2007, 40, 4625–4636 CrossRef CAS.
- N. B. Ram and E. Krishnakumar, Dissociative electron attachment to H2S probed by ion momentum imaging, Phys. Chem. Chem. Phys., 2011, 13, 13621 RSC.
- N. B. Ram, V. S. Prabhudesai and E. Krishnakumar, Resonances in dissociative electron attachment to water, J. Phys. B: At., Mol. Opt. Phys., 2009, 42, 225203 CrossRef.
- H. Adaniya, B. Rudek, T. Osipov, D. J. Haxton, T. Weber, T. N. Rescigno, C. W. McCurdy and A. Belkacem, Imaging the Molecular Dynamics of Dissociative Electron Attachment to Water, Phys. Rev. Lett., 2009, 103, 233201 CrossRef CAS PubMed.
- N. B. Ram, V. S. Prabhudesai and E. Krishnakumar, Comment on “Imaging the Molecular Dynamics of Dissociative Electron Attachment to Water”, Phys. Rev. Lett., 2011, 106, 049301 CrossRef PubMed.
- J. Fedor, P. Cicman, B. Coupier, S. Feil, M. Winkler, K. Głuch, J. Husarik, D. Jaksch, B. Farizon, N. J. Mason, P. Scheier and T. D. Märk, Fragmentation of transient water anions following low-energy electron capture by H2O/D2O, J. Phys. B: At., Mol. Opt. Phys., 2006, 39, 3935–3944 CrossRef CAS.
- D. J. Haxton, C. W. McCurdy and T. N. Rescigno, Dissociative electron attachment to the H2O molecule I. Complex-valued potential-energy surfaces for the 2B1, 2A1, and 2B2 metastable states of the water anion, Phys. Rev. A, 2007, 75, 012710 CrossRef.
- R. Abouaf and D. Teillet-Billy, Low energy electron collisions in H2S and H2Se: Structure in dissociative attachment cross-sections, Int. J. Mass Spectrom., 2008, 277, 79–83 CrossRef CAS.
- A. Kühn, H.-P. Fenzlaff and E. Illenberger, Formation and dissociation of negative ion resonances in methanol and allylalcohol, J. Chem. Phys., 1988, 88, 7453–7458 CrossRef.
- D. Chakraborty, D. S. Slaughter and S. Ptasinska, Dynamics of resonant low-energy electron attachment to ethanol-producing hydroxide anions, Phys. Rev. A, 2023, 108, 052806 CrossRef CAS.
- E. K. Berner and R. A. Berner, Global Environment, Princeton University Press, 2012 Search PubMed.
- K. Gope, V. Tadsare, V. S. Prabhudesai, N. J. Mason and E. Krishnakumar, Negative ion resonances in carbon monoxide, Eur. Phys. J. D, 2016, 70, 134 CrossRef.
- E. Krishnakumar and K. Nagesha, Dissociative attachment studies by negative-ion time-of-flight mass spectrometry, Rapid Commun. Mass Spectrom., 1995, 9, 336–343 CrossRef CAS.
- D. Rapp and D. D. Briglia, Total Cross Sections for Ionization and Attachment in Gases by Electron Impact. II. Negative-Ion Formation, J. Chem. Phys., 1965, 43, 1480–1489 CrossRef CAS.
- S. Das, S. Swain, K. Gope, V. Tadsare and V. S. Prabhudesai, Effect of Background Signal on Momentum Imaging, ArXiv, 2306.16708.
- F. Fiquet-Fayard, J. P. Ziesel, R. Azria, M. Tronc and J. Chiari, Formation of HS− and DS− by Dissociative Attachment in H2S, HDS, and D2S, J. Chem. Phys., 1972, 56, 2540–2548 CrossRef CAS.
- A. Kumar, S. Swain, J. Upadhyay, Y. Upalekar, R. Arya and V. S. Prabhudesai, Predissociation dynamics of negative-ion resonances of H2 near 12 and 14.5 eV using the velocity slice imaging technique, Phys. Rev. A, 2023, 107, 062803 CrossRef CAS.
- NIST Chemistry Webbook, https://webbook.nist.gov/Chemistry/, (accessed 1 November 2023).
- J. L. Franklin and H. E. Lumpkin, Some C–S, H–S and S–S Bond Strengths by the Electron Impact Method, J. Am. Chem. Soc., 1952, 74, 1023–1026 CrossRef CAS.
- D. H. Fine and J. B. Westmo, Heats of formation of some alkylthio radicals, Can. J. Chem., 1970, 48, 395 CrossRef CAS.
- D. J. Haxton, C. W. McCurdy and T. N. Rescigno, Angular dependence of dissociative electron attachment to polyatomic molecules: Application to the 2B1 metastable state of the H2O and H2S anions, Phys. Rev. A: At., Mol., Opt. Phys., 2006, 73, 062724 CrossRef.
- V. S. Prabhudesai, N. B. Ram, G. Aravind, P. Rawat and E. Krishnakumar, Probing site selective fragmentation of molecules containing hydroxyl group using Velocity Slice Imaging, J. Phys. Conf. Ser., 2007, 80, 012016 CrossRef.
- L. V. Trepka Von and H. Neuert, Über die Entstehung von negativen Ionen aus einigen Kohlenwasserstoffen und Alkoholen durch Elektronenstoß, Z. Natuforschg., 1963, 18a, 1295 Search PubMed.
- W. C. Hung, M. Shen, C. Yu and Y.-P. Lee, Vibronic analysis of the
![[B with combining tilde]](https://www.rsc.org/images/entities/char_0042_0303.gif) 2A′–
2A′–![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) 2A′′ laser-induced fluorescence of jet-cooled C2H5S, J. Chem. Phys., 1996, 105, 5722–5730 CrossRef CAS.
2A′′ laser-induced fluorescence of jet-cooled C2H5S, J. Chem. Phys., 1996, 105, 5722–5730 CrossRef CAS. - H. R. Wendt and H. E. Hunziker, The UV spectra of primary, secondary, and tertiary alkyl radicals, J. Chem. Phys., 1984, 81, 717–723 CrossRef CAS.
- J. Munk, P. Pagsberg, E. Ratajczak and A. Sillesen, Spectrokinetic studies of ethyl and ethylperoxy radicals, J. Phys. Chem., 1986, 90, 2752–2757 CrossRef CAS.
| This journal is © the Owner Societies 2024 |