
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA povarov-type reaction to access tetrahydroquinolines from N-benzylhydroxylamines and alkenes in HFIP†
Valentyn
Pozhydaiev
a,
Daniella
Al-Othman
a,
Joseph
Moran
 *abc and
David
Lebœuf
*a
*abc and
David
Lebœuf
*a
aInstitut de Science et d’Ingénierie Supramoléculaires (ISIS), CNRS UMR 7006, Université de Strasbourg, 67000 Strasbourg, France. E-mail: moran@unistra.fr; dleboeuf@unistra.fr
bDepartment of Chemistry and Biomolecular Sciences, University of Ottawa, Ottawa, Ontario K1N 6N5, Canada
cInstitut Universitaire de France (IUF), 75005 Paris, France
First published on 2nd September 2024
Abstract
Here, we report the synthesis of tetrahydroquinolines between newly developed N-benzylhydroxylamine reagents and alkenes using HFIP as a solvent. This transformation is notably applicable to highly electronically deactivated styrenes and aliphatic alkenes, expanding the range of tetrahydroquinolines attainable.
Tetrahydroquinolines have a pivotal role across diverse industrial sectors as building blocks for the synthesis of pharmaceuticals, agrochemicals, and materials (Scheme 1A).1,2 Currently, the Povarov reaction and the partial reduction of quinolines are among the most popular approaches to synthesise tetrahydroquinolines (Scheme 1B).1–3 Nevertheless, both have their own limitations. In the case of the Povarov reaction, the transformation is mainly limited to alkenes bearing electron-donating groups (EDGs),4 while the reduction of quinolines requires the pre-installation of the desired functionalities through multi-step synthesis. More recently, new elegant strategies for the preparation of tetrahydroquinolines have been reported that rely on the intramolecular C–H amination of arenes via nitrogen-centered radicals formed from electrophilic aminating agents,5,6 as exemplified by the groups of Marsden, Morandi and Murphy (Scheme 1C).7–9 Other relevant variants to access tetrahydroquinolines were also reported by the groups of Falck and Bower via rhodium catalysis and Brønsted acid-promoted reactions.10,11 However, overall, the scaffolds attainable are limited, and engineered substrates are often required to access more complex molecules.
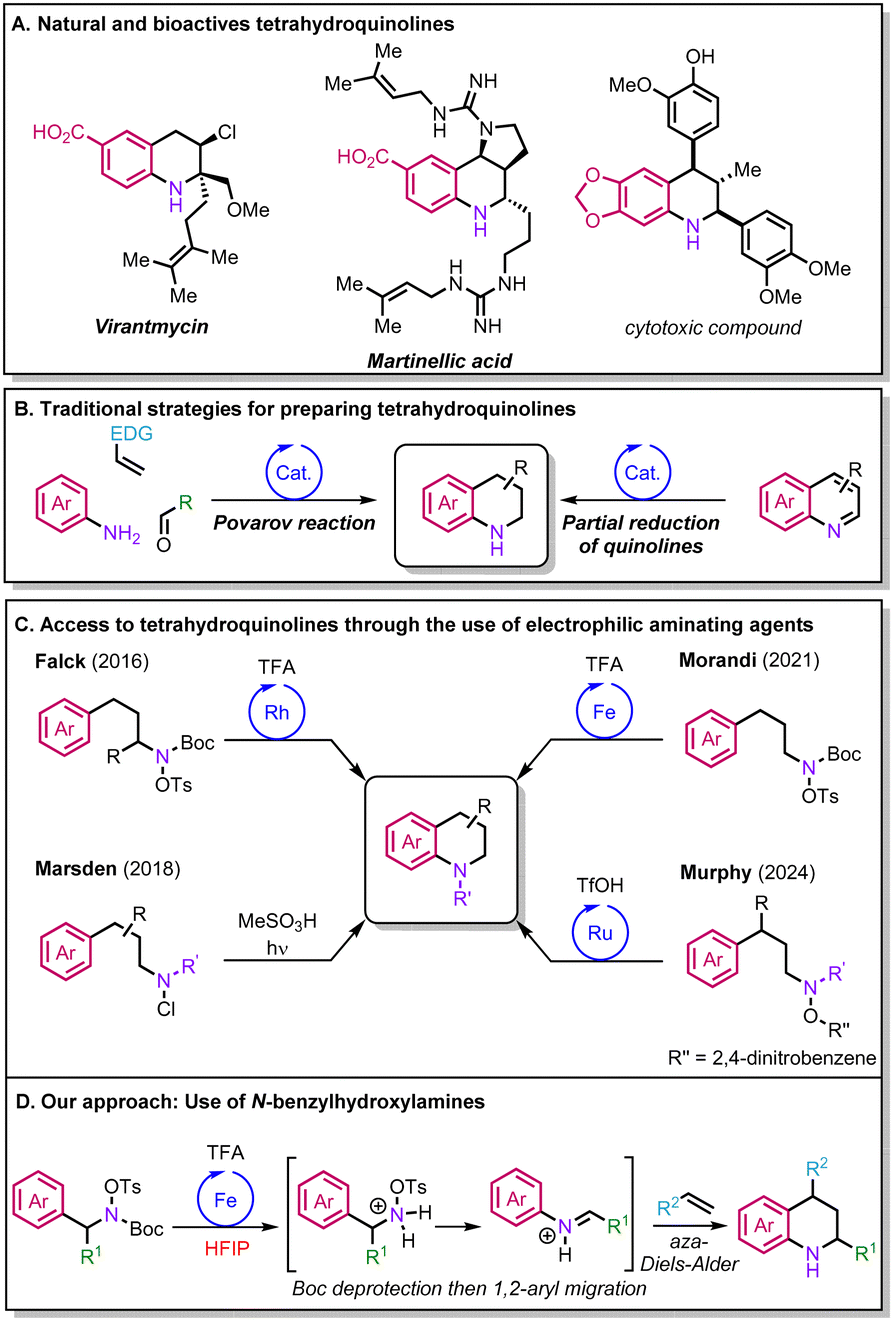 | ||
| Scheme 1 Importance of tetrahydroquinolines and synthetic approaches to access them. | ||
In this context, hydroxylammonium salts serve as promising sources for the incorporation of nitrogen in feedstock compounds.12,13 These reagents have attracted the attention of researchers owing to their chemical properties and versatility. Hydroxylammonium salts are prepared from relatively inexpensive and commercially available starting material which makes their synthesis straightforward. These reagents grant access to a variety of free unprotected amines; their nitrogen–oxygen bond can be homolytically cleaved to generate a nitrogen-centered radical that adds to C–C double bonds. Their use has been remarkably exploited by the group of Morandi in several iron(II)-catalysed alkene aminofunctionalisations.14–16 Our group has recently described efficient methods for the 1,2-aminoarylation and 1,2-diamination of highly electronically deactivated styrenes, affording unprotected amines in a one-pot sequence while displaying broad functional group compatibility.17,18 During our investigations, we noted that the reactions only occurred when hexafluoroisopropanol (HFIP) was used as a solvent,19–21 which we attributed to its ability to enhance the reactivity of the various reactive intermediates.
To exploit these reagents in intermolecular processes, we aimed to develop a new set of readily available and bench-stable hydroxylamine reagents that could react with alkenes to provide complex tetrahydroquinolines. By relying on the unique properties of HFIP, we hypothesised that we might unlock the reactivity of highly electronically deactivated styrenes to complement the scope of the Povarov reaction. Our design plan relied on the use N-benzylhydroxylamine derivatives that could be easily obtained by a Mitsunobu reaction (Scheme 1D). Following a Boc deprotection under acidic conditions, a 1,2-aryl migration could occur to give an N-aryliminium; this intermediate could then engage in an aza-Diels–Alder reaction to yield the corresponding tetrahydroquinoline. Here, we disclose our research efforts in the development of this transformation.
In our initial investigations, we evaluated the reaction between hydroxylamine 2a and an excess of p-nitrostyrene 1a (2 equiv.) in the presence of a catalytic amount of iron(II) sulfate heptahydrate and trifluoroacetic acid (TFA) using HFIP (0.1 M) as a solvent. The role of TFA is to promote the in situ deprotection of the Boc group. Attempts to prepare the corresponding hydroxylammonium salt [MsO–NH2Bn][OTf] from hydroxylamine 2a and triflic acid led to rapid decomposition of the product. Under the reaction conditions mentioned above, the target product 3 was isolated in 50% isolated yield (Table 1, entry 1). Other iron(II) salts were tested but did not improve the yields (Table 1, entries 2 and 3). As observed in our previous studies, the reaction only took place in HFIP (Table 1, entries 4–7). In the absence of either TFA or FeSO4·7H2O, a significant drop in yield was observed (Table 1, entries 8 and 9). In the same vein, decreasing the amount of styrene, operating at higher concentration or higher temperature proved detrimental for the reactivity (Table 1, entries 10–12). Using an excess of 2a also led to a decrease in efficacy (Table 1, entry 13). Replacing the mesyl group by a tosyl one on 2a did not affect the reactivity, delivering 3 in 52% yield (Table 1, entry 14). Some of the mass balance of the reaction was found to be diverted to the formation of side product 4 (10% yield), resulting from the reaction with the isobutene produced in situ during Boc group deprotection of hydroxylamine 2a.
|
|
||
|---|---|---|
| Entry | Variation from standard conditionsa | Yieldb |
| a Standard reaction conditions: 1a (0.4 mmol), 2 (0.2 mmol), FeSO4·7H2O (10 mol%) and TFA (0.4 mmol) in HFIP (0.1 M), rt, 1 h (in a sealed tube). b NMR yield using triethylsilane as an external standard (isolated yield in parentheses). c Product 4 obtained in 10% yield. n.r. = no reaction. | ||
| 1 | None | 52% (50%) |
| 2 | Fe(OTf)2 instead of FeSO4·7H2O | 41% (38%) |
| 3 | Fe(OAc)2 instead of FeSO4·7H2O | 44% (41%) |
| 4 | MeNO2 instead of HFIP | n.r. |
| 5 | TFE instead of HFIP | n.r. |
| 6 | 1,2-DCE instead of HFIP | n.r. |
| 7 | DCM instead of HFIP | n.r. |
| 8 | Without TFA | 32% |
| 9 | Without FeSO4·7H2O | 17% |
| 10 | 1 equiv. 1a | 24% |
| 11 | 0.2 M | 19% |
| 12 | 40 °C | 19% |
| 13 | 1 equiv. 1a, 3 equiv. 2a | 36% |
| 14 | TsO instead of MsO | 55% (52%)c |
| 16 | TsO instead of MsO and argon bubbling | 63% (60%) |
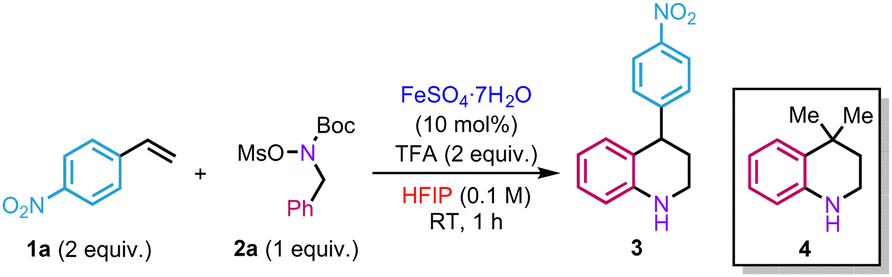
Thus, two different alkenes in the reaction medium compete to react with the hydroxylammonium salt. A similar side-product was observed by the group of Falck during their studies on amination of benzyl alcohols.22 Although we failed to completely suppress its formation, bubbling argon in the reaction mixture slightly improved the yield (60%) (Table 1, entry 15).
We then began to explore the scope of the reaction by using electronically varied hydroxylamines 2d–2l in reaction with p-nitrostyrene 1a (Scheme 2). The transformation tolerates the presence of various electron-donating and moderate electron-withdrawing groups at the para-position, including ether, thioether, aryl, and halide, to afford the corresponding tetrahydroquinolines 5–8 in 46–68% yields. On the other hand, in the case of the more electron-deficient nitro-containing hydroxylamine 2h, aziridine 9′ was obtained, as a major product (33% yield) with only traces of tetrahydroquinoline 9 (7% yield). The formation of product 9′ seems to indicate that the migration of the aryl does not occur in the presence of a strong electron-withdrawing group. Therefore, the reaction between the styrene and hydroxylammonium takes place to form, instead, the aziridine as observed in our previous studies.17,18
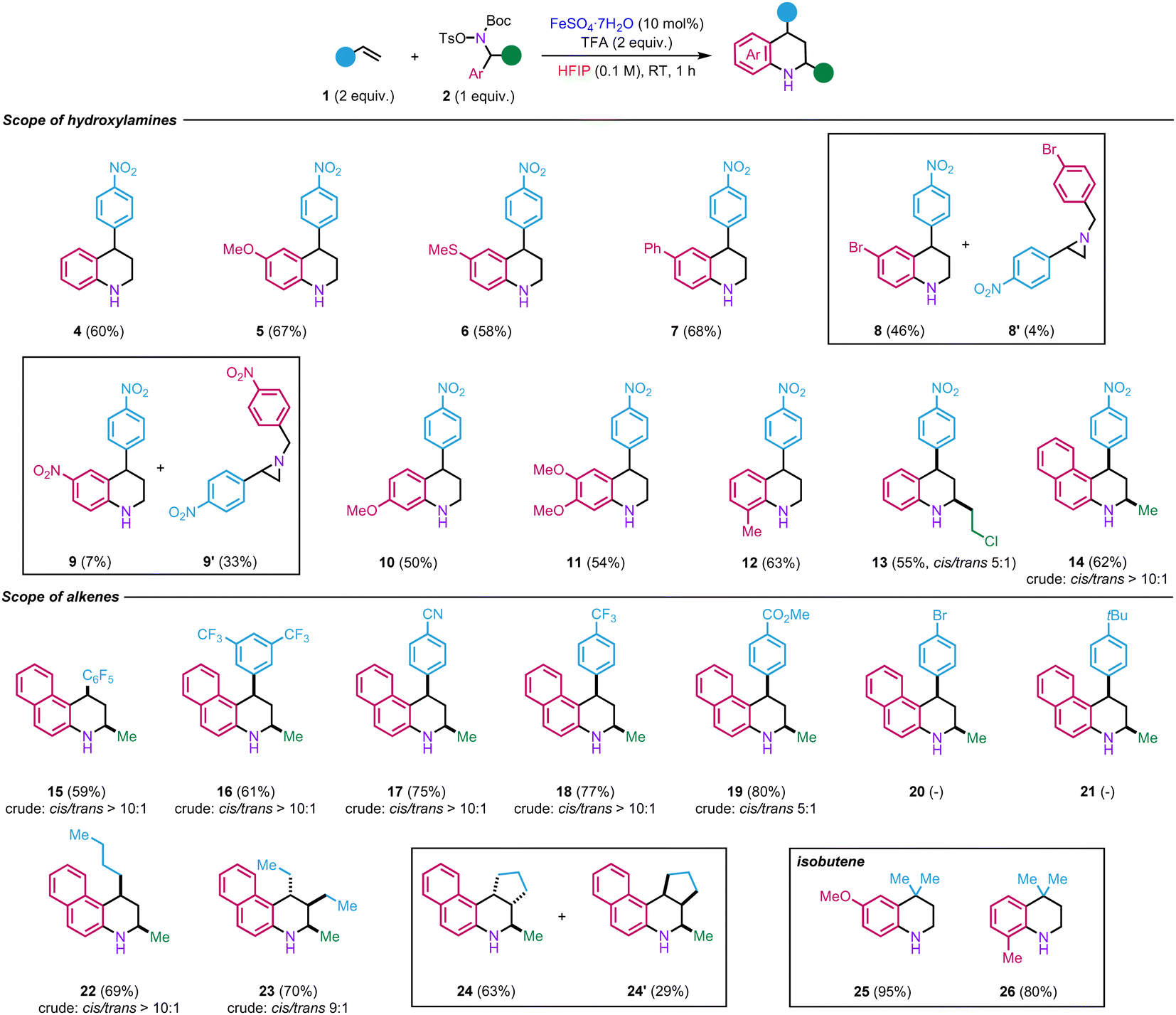 | ||
| Scheme 2 Scope of the transformation. | ||
The reaction is also compatible with the presence of electron-rich substituents at the ortho- and meta-position (10–12, 50–63%). The reaction is not limited to primary benzyl alcohols but could also be extended to secondary ones such as 13 (55%). Tetrahydrobenzo[f]quinoline scaffolds such as 14 (62%) are also accessible using this methodology from naphthyl hydroxylamine 2l. In these different examples, the reaction did not produce observe notable by-products, suggesting that some of the moderate yields result from the partial decomposition of the hydroxylamines during the reaction.
Regarding the reactivity of various alkenes, 2l was used as a model hydroxylamine. The functional group tolerance of this method was studied towards styrenes incorporating strong electron-withdrawing groups as they show limited reactivity in the existing Povarov reaction. To our delight, electron-deficient styrene derivatives afforded products 15–19 in high yields (59–80% yields). The cis configuration for the major products was ascertained by NOESY analyses (see ESI†). On the other hand, in the case of styrene bearing a moderate electron-withdrawing group (Br, 20) or electron-donating group (tBu, 21), oligomerisation of the styrene was observed. The versatility of the method was also tested with electron-rich aliphatic alkenes. For instance, product 22 was obtained from 1-hexene in 70% yield. In the case of trans-3-hexene, product 23 was obtained in 70% yield as major diastereoisomer. Its structure was evidenced by NOESY analyses (see ESI†). The fact that the stereochemistry of the starting material was retained in the product implies that the reaction might involve a concerted mechanism. We next examined cyclic alkenes. Satisfyingly, cyclopentene was well-tolerated in the reaction, yielding two diastereoisomers 24 and 24′ in a combined yield of 92%. Finally, different hydroxylamines were tested with isobutene generated in situ. First, para-substituted methoxy hydroxylamine afforded product 25 in a nearly quantitative yield (95%). Second, ortho-methyl substituted hydroxylamine led to the formation of 26 in 80% yield. However, no product was observed with naphthyl hydroxylamine 2l. In that case, it seems that the hydrolysis of the postulated iminium intermediate is faster than the reaction with isobutene since only 2-naphthylamine was recovered from the reaction.
Regarding this transformation, various observations make us lean towards a radical cation crossover mechanism: (1) in the presence of TEMPO, the reaction is completely inhibited, which strongly suggests the involvement of radical species; (2) as mentioned above, in the absence of an alkene partner, naphthyl hydroxylamine 2l led to 2-naphthylamine, which is consistent with the 1,2-aryl migration proposed; (3) the fact that the stereochemistry of the alkene is retained in the product implies that the reaction likely involves a concerted mechanism.
We thus propose the following mechanism (Scheme 3): Initially, the Boc group is deprotected in the presence of TFA, generating the corresponding ammonium B. Then, a classical homolytic cleavage of the N–O bond occurs to provide aminium radical cation C. At this point, the reaction can diverge depending on the substitution pattern of the hydroxylamine. In the presence of an electron-withdrawing group (EWG), the 1,2-aryl migration is disfavoured and the aminium radical cation can directly add across the double bond (D) to finally provide aziridine E.23 On the other hand, in the presence of an EDG, a rare but not unprecedented radical 1,2-aryl migration can occur to provide α-aminomethyl radical G.24 From there, G can regenerate C by single electron transfer to B, a mechanism consistent with a precedent report by the group of Phipps.25 Lastly, iminium H would engage in a classical aza-Diels–Alder to deliver tetrahydroquinoline I. Regarding the positive effect of HFIP on the reactivity, it might be explained by its ability to strongly donate H-bonds, thereby increasing the electrophilicity of various intermediates such as C or H to facilitate the key steps of the process, namely the 1,2-aryl migration and aza-Diels–Alder.
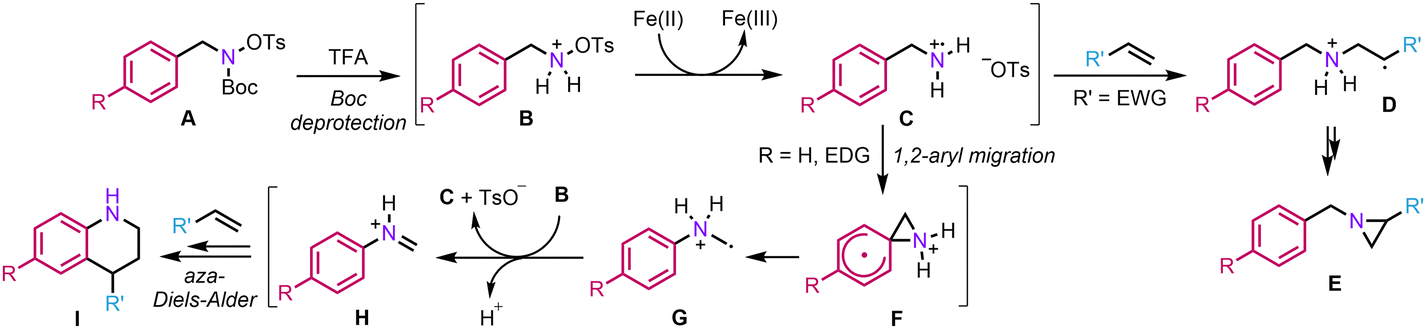 | ||
| Scheme 3 Plausible mechanistic pathway. | ||
In conclusion, through exploring the reactivity of new hydroxylamine reagents, we have demonstrated their efficacy in generating tetrahydroquinolines with different substitution patterns. In contrast to the classical Povarov reaction, our methodology accommodates electron-deficient and aliphatic alkenes, thereby expanding the chemical space of available tetrahydroquinoline scaffolds. The ability of these reagents to react with isobutene without the need to directly handle this hazardous compound also represents a significant advantage. Ongoing investigations are focusing on deciphering the mechanism of this transformation.
This work was supported by the Interdisciplinary Thematic Institute ITI-CSC via the IdEx Unistra (ANR-10-IDEX-0002) within the program Investissement d’Avenir. V. P. and D. O. thank the CSC Graduate School funded by the French National Research Agency (CSC-IGS ANR-17-EURE-0016). within the program Investissement d’Avenir. D. L. and J. M. thank the CNRS. D. L. thank Dr Sami Lakhdar for useful discussions.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- V. Sridharan, P. A. Suryavanshi and J. C. Menéndez, Chem. Rev., 2011, 111, 7157–7259 CrossRef CAS PubMed.
- I. Muthukrishnan, V. Sridharan and J. C. Menéndez, Chem. Rev., 2019, 119, 5057–5191 CrossRef CAS PubMed.
- W. Ferreira de Paiva, Y. de Freitas Rego, Â. de Fátima and S. A. Fernandes, Synthesis, 2022, 3162–3179 CAS.
- For a rare example of Povarov reaction with an electron-deficient alkene, see: J. A. Leitch, A. L. Fuentes de Arriba, J. Tan, O. Hoff, M. C. Martínez and D. J. Dixon, Chem. Sci., 2018, 9, 6653–6658 RSC.
- J. Davies, S. P. Morcillo, J. J. Douglas and D. Leonori, Chem. – Eur. J., 2018, 24, 12154–12163 CrossRef CAS PubMed.
- C. Pratley, S. Fenner and J. A. Murphy, Chem. Rev., 2022, 122, 8181–8260 CrossRef CAS PubMed.
- S. C. Cosgrove, J. M. C. Plane and S. P. Marsden, Chem. Sci., 2018, 9, 6647–6652 RSC.
- E. Falk, V. C. M. Gasser and B. Morandi, Org. Lett., 2021, 23, 1422–1426 CrossRef CAS PubMed.
- C. Pratley, S. Fenner and J. A. Murphy, Org. Lett., 2024, 26, 1287–1292 CrossRef CAS PubMed.
- M. P. Paudyal, A. A. M. Adebesin, S. R. Burt, D. H. Ess, Z. Ma, L. Kürti and J. R. Fack, Science, 2016, 353, 1144–1147 CrossRef CAS PubMed.
- J. J. Farndon, X. Ma and J. F. Bower, J. Am. Chem. Soc., 2017, 139, 14005–14008 CrossRef CAS PubMed.
- S. Sabir, G. Kumar and J. L. Jat, Org. Biomol. Chem., 2018, 16, 3314–3327 RSC.
- V. C. M. Gasser, S. Makai and B. Morandi, Chem. Commun., 2022, 58, 9991–10003 RSC.
- L. Legnani and B. Morandi, Angew. Chem., Int. Ed., 2016, 55, 2248–2251 CrossRef CAS PubMed.
- L. Legnani, G. Prina-Cerai, T. Delcaillau, S. Willems and B. Morandi, Science, 2018, 362, 434–439 CrossRef CAS PubMed.
- S. Makai, E. Falk and B. Morandi, J. Am. Chem. Soc., 2020, 142, 21548–21555 CrossRef CAS PubMed.
- V. Pozhydaiev, M. Vayer, C. Fave, J. Moran and D. Lebœuf, Angew. Chem., Int. Ed., 2023, 62, e202215257 CrossRef CAS PubMed.
- V. Pozhydaiev, A. Paparesta, J. Moran and D. Lebœuf, Angew. Chem., Int. Ed., 2024, 63, e202411992 Search PubMed.
- V. Pozhydaiev, M. Power, V. Gandon, J. Moran and D. Lebœuf, Chem. Commun., 2020, 56, 11548–11564 RSC.
- H. F. Motiwala, A. M. Armaly, J. G. Cacioppo, T. C. Coombs, K. R. K. Koehn, V. M. Norwood IV and J. Aubé, Chem. Rev., 2022, 122, 12544–12747 CrossRef CAS PubMed.
- M. Piejko, J. Moran and D. Lebœuf, ACS Org. Inorg. Au, 2024, 4, 287–300 CrossRef CAS PubMed.
- R. R. Anugu and J. R. Falck, Chem. Sci., 2022, 13, 4821–4827 RSC.
- S. Chatterjee, I. Harden, G. Bistoni, R. G. Castillo, S. Chabbra, M. van Gastel, A. Schnegg, E. Bill, J. A. Birrell, B. Morandi, F. Neese and S. DeBeer, J. Am. Chem. Soc., 2022, 144, 2637–2656 CrossRef CAS PubMed.
- S. Kim and J. Y. Do, J. Chem. Soc., Chem. Commun., 1995, 1607–1608 RSC.
- C. Morrill, J. E. Gillespie and R. J. Phipps, Angew. Chem., Int. Ed., 2022, 61, e202204025 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc04014g |
| This journal is © The Royal Society of Chemistry 2024 |