
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe influence of exogenous amines on the electrochemical CO2 reduction activity of a cobalt–pyridyldiimine catalyst†
Piyush Kumar
Verma
 a and
Charles C. L.
McCrory
*ab
a and
Charles C. L.
McCrory
*ab
aDepartment of Chemistry, University of Michigan, Ann Arbor, Michigan 48109-1055, USA. E-mail: cmccrory@umich.edu
bMacromolecular Science and Engineering Program, University of Michigan, Ann Arbor, Michigan 48109-1055, USA
First published on 2nd July 2024
Abstract
Studying the interactions between CO2 sorbents and electrocatalysts for the electrochemical CO2 reduction reaction (e-CO2RR) can offer viable strategies to advance the development of the Reactive Capture of CO2 (RCC). In this report we studied the effect of amines on the performance of the [Co(PDI-Py)] catalyst for the e-CO2RR. The presence of amines shifts the onset potential for the e-CO2RR more positive and increases the catalytic activity while maintaining the high Faradaic efficiency (≥90%) for CO production.
CO2 capture from dilute sources such as flue gas and its fixation for later use is an important strategy in decarbonizing industry to minimize the net release of CO2 into atmosphere.1 In this carbon capture and utilization (CCU) process, CO2 is captured by sorbents in the form of sorbent–CO2 complexes and is used at later stages by reversing the capturing process upon thermal application. In essence, CCU enables the reuse or recycling of waste CO2 to its higher value forms such as CO, HCOOH and MeOH. However, a major limitation of this strategy is that thermal release and concentration of CO2 from sorbent–CO2 adducts is an energy intensive process that limits the overall viability of CCU for achieving net zero carbon emissions.2
In contrast, the reactive capture of CO2 (RCC) bypasses the need for CO2 release by reducing the CO2 while it is still adsorbed to the sorbent in its captured form, e.g. as a sorbent–CO2 adduct. Hence, RCC is considered as a more energy efficient strategy compared to CCU.3,4 RCC can describe three distinct processes: (a) the direct reduction of CO2 in its sorbent-bound form into various products; (b) the in situ desorption and subsequent reduction of proximal CO2 at the catalyst, and (c) the direct reduction of CO2 from dilute (<1 atm) streams. Several catalytic systems have been explored within this broad RCC framework in all areas of catalysis: thermal,5,6 photocatalytic,7,8 and electrocatalytic RCC.9,10 State-of-the-art homogeneous RCC processes are thermal processes where in situ generated transition-metal hydrides reduce captured-CO2 by amines/alkanolamines under an H2 atmosphere.5
Electrocatalytic RCC (e-RCC) is a promising alternative to thermal RCC because e-RCC allows for the reduction of captured CO2 under ambient conditions without high pressure H2. However, the implementation of e-RCC with molecular electrocatalysts remains relatively underexplored. A notable example of homogeneous e-RCC was reported using an iron–phosphine complex that is reported to reduce CO2 to methanol in the presence of diethylamine as co-catalyst.11 The diethylamine is reported to react with CO2 to form diethylammonium carbamate which is then electrochemically reduced to methanol with 68% Faradaic efficiency by the molecular catalyst. In another report, a Re–bipyridine complex is reported to reduce 1% CO2 gas to CO with 85% Faradaic efficiency in the presence of triethanolamine. The triethanolamine is reported to react with CO2 to form aminoethylcarbonate which coordinates to the Re-center and is subsequently reduced to generate CO.12 Other insightful studies on the interactive effects of amines, phenol, and ionic liquids on catalyst systems for the electrochemical CO2 reduction reaction (e-CO2RR) underline the mechanisms through which the sorbents can affect the course of reaction either by acting as a ligand or by affecting the kinetics of the protonation steps involved in the catalysis.13–16
In this work, we report a new system for e-RCC based on a cobalt pyridyldiimine complex [Co(PDI-Py)] (Scheme 1a). We have shown previously that [Co(PDI-Py)] is active and selective for the e-CO2RR to CO in CO2-saturated acetonitrile (MeCN) solutions containing H2O as a proton source.17 Herein, we show that the e-CO2RR activity of [Co(PDI-Py)] is enhanced when various aliphatic amines are added into the electrolyte solution (Scheme 1a). Specifically, adding primary or secondary aliphatic amines to a CO2-saturated MeCN electrolyte solutions containing [Co(PDI-Py)] results in a favorable positive shift in the e-CO2RR catalytic onset potential and an increase in the magnitude of the electrocatalytic current while maintaining high Faradaic efficiencies for CO production of FE(CO) ≥ 90%. The amines react with CO2 in solution to form different CO2 adducts that exist in equilibrium, including alkylammonium carbamate salts (Scheme 1b). However, attempts to directly reduce carbamate salts in the absence of added CO2 result in a lower Faradaic efficiency of FE(CO) ≈ 16%, possibly because the equilibration of carbamate salt results in the lower effective concentration of carbamate and substantial concentration of free amine which can suppress the overall catalytic activity. Our results offer crucial insights for the development of effective e-RCC catalytic systems based on amines, which are prominently used industrial sorbent.
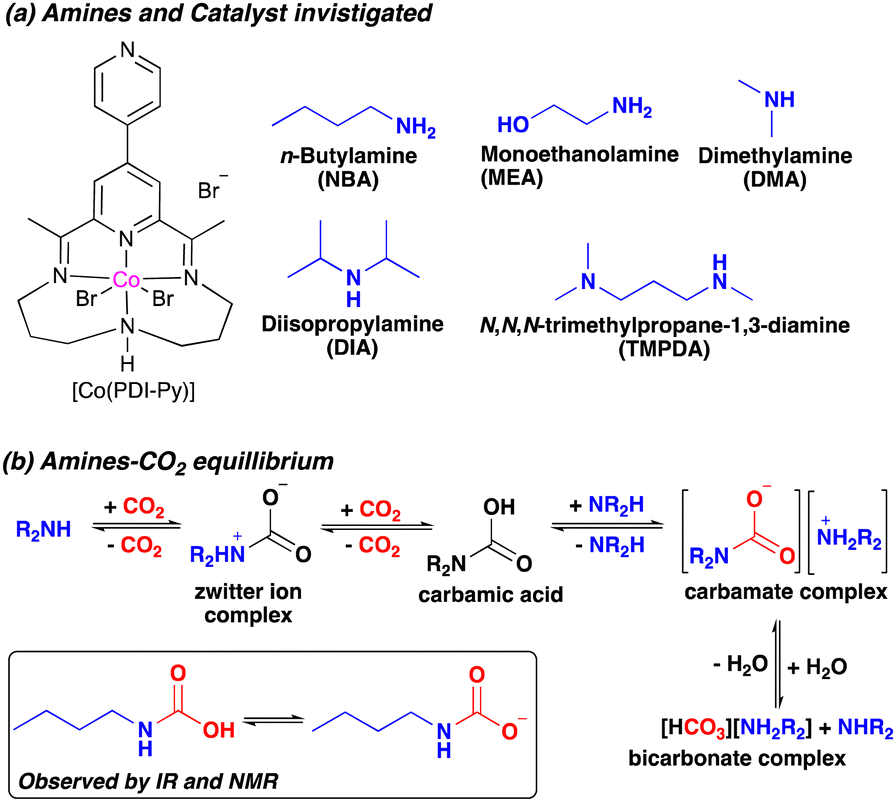 | ||
| Scheme 1 (a) Amines studied in this work and (b) amine-CO2 equilibrium. | ||
Cyclic voltammograms (CVs) of [Co(PDI-Py)] in Ar-sparged MeCN electrolyte show four reversible redox features assigned to two metal-based reductions (Co3+/2+ and Co2+/+) followed by two ligand-based reductions (PDI-Py/PDI-Py˙− and PDI˙−/PDI-Py2−) (Fig. S4 and Table S1, ESI†). When the electrolyte is sparged with CO2 and 11 M H2O is added, the reductive current increases, indicative of a catalytic process with a catalytic onset near the PDI-Py/PDI-Py˙− couple (Fig. 1). Presence of H2O enhances the catalytic rate for CO2RR and stability of the catalyst as we observed in our previous studies of [Co(PDI-Py)].17 After the amine NBA is added to the CO2-saturated electrolyte solution containing [Co(PDI-Py)] and 11 M H2O, we observe an increase in the magnitude of the catalytic current (ic,amine/ic = 1.78) and a ∼0.18 V positive shift in the catalytic onset potential (Fig. 1). Similar responses were also observed for the e-CO2RR by Co(PDI-Py) in presence of other amines (Fig. S5–S10, ESI†). The catalytic onset potential (Eonset) and ratio of the catalytic CO2RR peak current with amine present and without amine present (ic,amine/ic) are summarized in Table 1. The presence of amines resulted in a positive shift in Eonset by ∼0.18–0.14 V, and varying degrees of electrocatalytic enhancement from a high of ic,amine/ic = 1.78 for NBA to the lowest enhancement of ic,amine/ic = 1.23 for MEA.
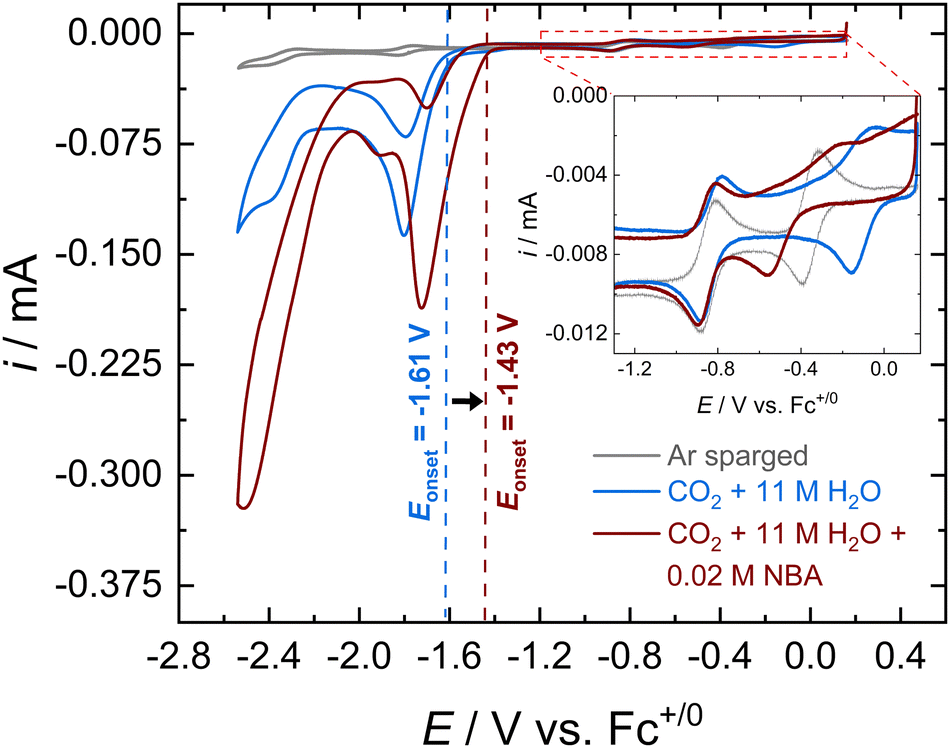 | ||
| Fig. 1 Representative CVs of 0.3 mM Co(PDI-Py) in MeCN with 0.1 M nBu4NPF6 at a scan rate of 0.05 V s−1. CVs were taken in Ar-sparged solution (gray line), CO2-saturated solution containing 11 M H2O (red line), and CO2-saturated solution containing 11 M H2O and 0.2 M NBA (blue line). The inset is a zoom in of the region in the dashed orange box showing the Co3+/2+ and Co2+/+ peaks. | ||
| Added amine | E 1/2(Co3+/2+)/V vs. Fc+/0 | E 1/2(Co2+/1+)/V vs. Fc+/0 | E onset/V vs. Fc+/0 | I c,amine/ic |
|---|---|---|---|---|
| a CV experiments were performed with 0.3 mM [Co(PDI-Py)] in CO2-saturated solution of 0.1 M nBu4NPF6 and 11 M H2O in MeCN with a scan rate of 0.05 V s−1. For experiments with amines, the concentration of the amine was 0.2 M. | ||||
| No amine | −0.07 | −0.85 | −1.61 | — |
| NBA | −0.46 | −0.85 | −1.43 | 1.78 |
| MEA | −0.39 | −0.85 | −1.46 | 1.23 |
| DMA | −0.43 | −0.87 | −1.47 | 1.69 |
| DIA | −0.41 | −0.85 | −1.47 | 1.62 |
| TPDA | −0.36 | −0.86 | −1.46 | 1.34 |
| Aniline | −0.09 | −0.85 | −1.61 | 1.0 |
Note that the addition of NBA to the CO2-saturated solution with [Co(PDI-Py)] and 11 M H2O also results in a −0.39 V shift in the Co3+/2+ couple, but no observable shift in the Co2+/+ couple as seen in the Fig. 1, inset for NBA, and for all amines in Fig. S5–S10 (ESI†). The resulting E1/2 for the Co3+/2+ couples and the unchanged Co2+/+ couple in the CO2-saturated solutions with 11 M H2O are summarized in Table 1. These results suggest coordination of the carbamate species to the charged Co(III)-complex, but that the extent of coordination may not be as significant in the Co(II)-complex. Note that noncatalytic CVs of [Co(PDI-Py)] conducted in Ar-sparged CH3CN electrolytes showed no significant change in peak heights or peak potentials upon the addition of amines (Fig. S11–S15 and Table S2, ESI†), suggesting that the coordination to the Co-complex observed in the CO2-saturated solution is from the carbamate formed from the reaction of CO2 and amine, and not from the free amine.
The tolerance of an electrocatalyst to higher concentration of sorbent is an important prerequisite for the practicality of an e-RCC system. It is expected that increasing the concentration of sorbent in solution should proportionally increase the partitioning of CO2 into the solution, therefore increasing the activity of the catalytic system. We investigated the e-CO2RR activity for [Co(PDI-Py)] in CO2-saturated MeCN electrolyte solutions as a function of NBA concentration (Fig. 2). Most noticeably, there is a proportional increase in the magnitude of the catalytic peak for competitive HER at ∼−2.4 V vs. Fc+/0. When we zoom into the catalytic peak for the CO2RR at ∼−1.6 V vs. Fc+/0, we observe a decrease in activity with increasing NBA concentration (Fig. 2, inset). We observe a similar trend of decreasing CO2RR activity with increasing amine concentration for the other amines investigated (Fig. S16–S19, ESI†). We attribute this decrease in CO2RR activity to the possible coordination of free amines in solution to [Co(PDI-Py)].
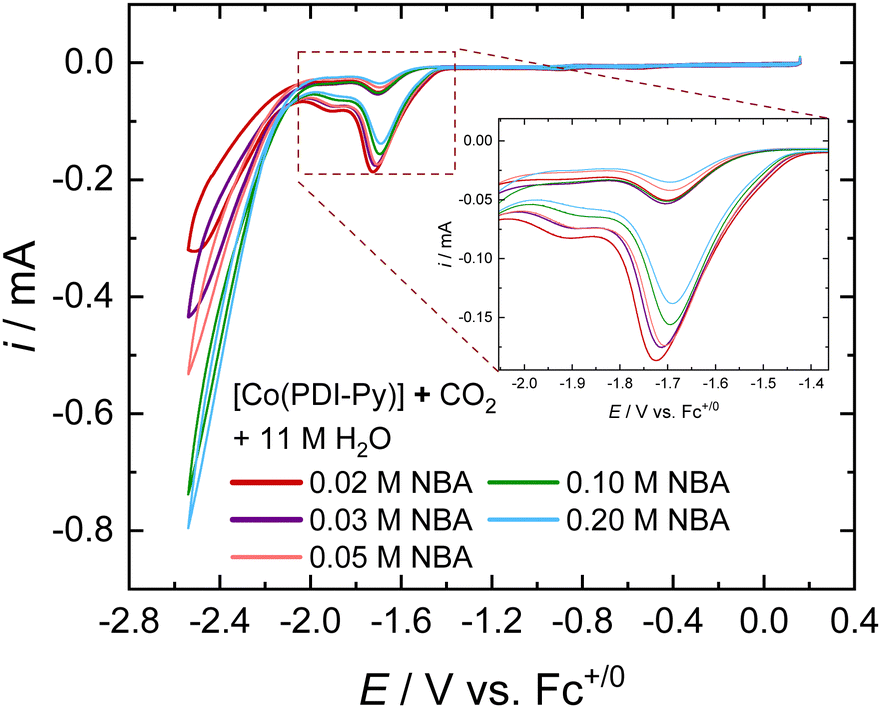 | ||
| Fig. 2 Representative CVs of 0.3 mM Co(PDI-Py) in MeCN with 0.1 M nBu4NPF6 and 11 M H2O at scan rate of 0.05 V s−1 with varying concentrations of NBA. The inset is the zoomed in region in the dashed orange box showing the e-CO2RR catalytic response. | ||
To confirm that the catalytic peak at ∼−1.6 V vs. Fc+/0 is due to the e-CO2RR to CO, we conducted controlled potential electrolysis (CPE) experiments at −1.69 V vs. Fc+/0 in previously described gastight, two-chamber H-cells (ESI,† Experimental methods).17 Representative current vs. time plots of the CPE experiments in the presence of 0.05 M of each amine studied are shown in Fig. S20–S25 (ESI†), and the results from the CPE experiments are summarized in Table 2. The Faradaic efficiency for CO production is FECO ≥ 95% regardless of the nature of the amine present. The highest activity is observed in the presence of NBA based on the charge passed during the CPE (Table 2) and the ic, amine/ic ratio (Table 1). However, there is no clear trend between the size of the added amine and the measured CO2RR activity. For comparison, CPEs conducted of [Co(PDI-Py)] in CO2-saturated solutions with 11 M H2O but no amine present also showed only CO production with FECO ≥ 90%, but with much less charge passed during the electrolyses. Post-CPE analysis of the working electrode by SEM-EDS experiments showed little precipitation of cobalt onto the electrode surface, suggesting there is minimal catalyst decomposition during the CPE experiments (Table 2, Fig. S26–S31, ESI†). Note that CPEs conducted at the more negative potential of −2.34 V vs. Fc+/0 in identically prepared CO2-saturated solutions with 11 M H2O, 0.05 M NBA, and 0.03 M [Co(PDI- Py)] show primarily H2 production with FEH2 ∼84% and FECO ∼12% (Fig. S32 and Table S3, ESI†), consistent with our previous studies.17
| Amine | Charge/C | FE(CO)/% | FE(H2)/% | Cobalt weight % |
|---|---|---|---|---|
| a All CPE experiments were conducted at −1.69 V vs. Fc+/0 for 30 min in a gas-tight, two-chamber H-cell. The working compartment contained the working electrode and reference electrode submerged in a CO2-saturated solution containing 0.3 mM of [Co(PDI-Py)], 0.1 M nBu4NPF6, and 11 M H2O in MeCN. For experiments with added amines, the concentration of the amine was 0.05 M. The auxiliary compartment contained the auxiliary electrode submerged in the same solution but with 5 mM Fc added as a sacrificial reductant. | ||||
| No amine | −2.6 ± 0.2 | 90 ± 11 | Trace | 0.13 ± 0.06 |
| NBA | −8.2 ± 0.4 | 99 ± 12 | 2.9 ± 0.9 | 0.17 ± 0.06 |
| MEA | −5.8 ± 0.6 | 98 ± 3 | 1.7 ± 0.5 | 0.07 ± 0.06 |
| DMA | −5.2 ± 0.7 | 98 ± 7 | 1.6 ± 0.4 | 0.20 ± 0.20 |
| DIA | −5.5 ± 0.4 | 97 ± 2 | 1.4 ± 0.2 | 0.33 ± 0.06 |
| TMPDA | −4.9 ± 0.8 | 95 ± 6 | 2.8 ± 1.6 | 0.03 ± 0.05 |
To understand the CO2-reactive species and its interaction with the Co-catalyst, we performed a series of spectroscopic studies. To determine whether this coordination persists for more reduced complexes, we studied the interaction of carbamates with the [Co(PDI-Py)]2+ complex by UV-Vis experiments. In our electrocatalytic experiments, the Co(III) complex [Co(PDI-Py)]3+ is first reduced to the Co(II) complex [Co(PDI-Py)]2+in situ to enter the catalytic cycle.17 The UV-Vis spectra of independently prepared [Co(PDI-Py)Br2] complex in N2-sparged electrolyte solution (Fig. 3, blue line) shows three absorption features at 550 nm, 465 nm, 365 nm. Upon saturating this solution with CO2, there is no change in the absorption peaks from the N2-sparged solution, confirming that CO2 does not coordinates with [Co(PDI-Py)Br2] (Fig. 3, purple line). We next collected a UV-Vis spectrum of the [Co(PDI-Py)Br2] complex in a N2-sparged solution containing NBA (Fig. 3, red line). We observe a blue shift in the absorption spectrum compared to the parent system without NBA present, and we interpret this shift as a result of coordination of NBA to the [Co(PDI-Py)]2+ complex. Sparging the NBA-containing solution with CO2 results in a noticeable change in the UV-Vis spectrum, with a disappearance of the absorption peak at ∼480 nm and the emergence of a new absorption peak at ∼350 nm (Fig. 3, green line). We interpret this change in the spectrum to indicate that CO2 is reacting with NBA in solution forming n-butylcarbamate (NBA-CO2), and this NBA-CO2 is then coordinating to the [Co(PDI-Py)]2+ active catalyst (Fig. S33, ESI†). The formation of NBA-CO2 in CO2-sparged MeCN containing NBA was confirmed through NMR and FTIR studies (Fig. S34 and S35, ESI†). The key result from these studies is that the carbamate formed in situ coordinates to the [Co(PDI-Py)]2+ complex, something that was not observable from the CVs.
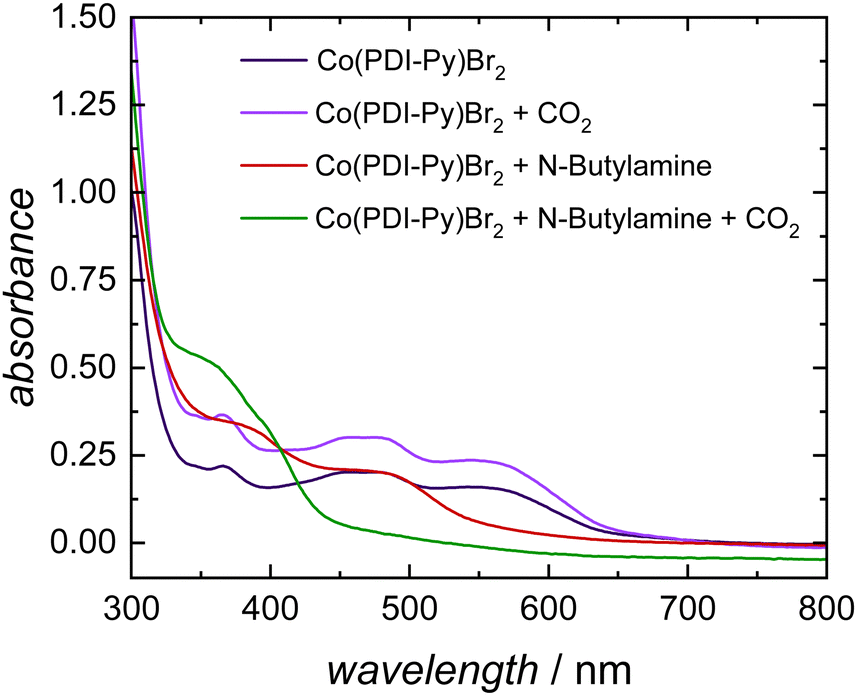 | ||
| Fig. 3 Absorption measurements of 0.1 mM [Co(PDI-Py)Br2] in MeCN solutions containing 0.03 M nBu4NPF6 and 11 M H2O in N2-sparged solution (blue line), CO2-saturated solution (purple line), N2-sparged solution with 6.6 mM NBA (red line), and CO2-saturated solution with 6.6 mM NBA (green line). | ||
Motivated by our spectroscopy results, we conducted CPE experiments of [Co(PDI)Py] in Ar-sparged MeCN electrolyte solution with 11 M H2O containing the commercially-available carbamate salt dimethylammonium dimethylcarbamate (DMA-CO2) as our sole CO2 source. Under these conditions, we observed lower selectivity and activity for CO formation: FECO ≈ 16% with only 1.9 C of charge passed (Table S4, ESI†). This lower activity for the carbamate compared to the CO2-saturated amine solution could be a consequence of the equilibria processes in Scheme 1b: without additional CO2 present in the system, the added carbamate may dissociate to form free DMA and the corresponding bicarbonate compound.
Because of inferior activity for CO production using DMA-CO2 as the sole CO2 source, we wanted to determine whether the in situ formation of a carbamate is important for the observed catalytic activity. We studied the electrocatalytic activity for CO production by [Co(PDI-Py)] in CO2 saturated solutions with 11 M H2O using aniline as the amine source. Aniline does not form stable carbamate species upon reaction with CO2 due to aniline's comparatively low nucleophilicity.18 CVs of [Co(PDI-Py)] in the presence of 0.02 M aniline and CO2 saturated electrolyte solution show no shift in redox potentials for the Co3+/2+ and Co2+/+ compared to CVs of [Co(PDI-Py)] in aniline-free solvent (Fig. S10, ESI† and Table 1). Similarly, Eonset is the same in the absence or presence of aniline, and no increase in catalytic activity was observed (ic,amine/ic = 1). Overall, these results suggest that in situ carbamate production by amine-CO2 reaction plays an important role in enhancing catalytic activity for the e-CO2RR by [Co(PDI-Py)] catalyst. The carbamate species is likely acting as an active substrate for CO2RR to CO.12,19,20 However, we cannot entirely rule out the possibility that CO2 is the active species for reduction to CO, and that the in situ formed carbamate is acting as a delivery mechanism for CO2 to the catalyst.21
In conclusion, we have shown that adding amines to CO2-saturated MeCN solutions containing the catalyst [Co(PDI-Py)] and 11 M H2O as a proton source leads to an increase in the e-CO2RR activity for CO production. CO2 reacts with the amines in solution, generating in situ alkyl-carbamate species. UV-Vis and electrochemical studies suggest that this carbamate species interacts with the [Co(PDI-Py)] complex, and is likely the active substrate for CO production. Aniline which cannot form carbamate salts, doesn’t enhance the catalytic activity. Overall, these findings can be a useful starting point for the development of efficient electrocatalytic systems for electrochemical reactive capture of CO2.
This work was supported as part of the Center for Closing the Carbon Cycle, an Energy Frontier Research Center funded by the U. S. Department of Energy, Office of Science, Basic Energy Sciences, under Award Number DE-SC0023427.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Notes and references
- M. Bui, C. S. Adjiman, A. Bardow, E. J. Anthony, A. Boston, S. Brown, P. S. Fennell, S. Fuss, A. Galindo, L. A. Hackett, J. P. Hallett, H. J. Herzog, G. Jackson, J. Kemper, S. Krevor, G. C. Maitland, M. Matuszewski, I. S. Metcalfe, C. Petit, G. Puxty, J. Reimer, D. M. Reiner, E. S. Rubin, S. A. Scott, N. Shah, B. Smit, J. P. M. Trusler, P. Webley, J. Wilcox and N. Mac Dowell, Energy Environ. Sci., 2018, 11, 1062–1176 RSC.
- E. Sanchez Fernandez, E. L. V. Goetheer, G. Manzolini, E. Macchi, S. Rezvani and T. J. H. Vlugt, Fuel, 2014, 129, 318–329 CrossRef CAS.
- R. E. Siegel, S. Pattanayak and L. A. Berben, ACS Catal., 2023, 13, 766–784 CrossRef CAS.
- A. M. Appel and J. Y. Yang, ACS Energy Lett., 2024, 9, 768–770 CrossRef CAS.
- S. Kar, A. Goeppert and G. K. S. Prakash, Acc. Chem. Res., 2019, 52, 2892–2903 CrossRef CAS PubMed.
- S. Sun, H. Sun, P. T. Williams and C. Wu, Sustainable Energy Fuels, 2021, 5, 4546–4559 RSC.
- S. Wang and X. Wang, Angew. Chem., Int. Ed., 2016, 55, 2308–2320 CrossRef CAS PubMed.
- E. Fujita, D. C. Grills, G. F. Manbeck and D. E. Polyansky, Acc. Chem. Res., 2022, 55, 616–628 CrossRef CAS PubMed.
- I. Sullivan, A. Goryachev, I. A. Digdaya, X. Li, H. A. Atwater, D. A. Vermaas and C. Xiang, Nat. Catal., 2021, 4, 952–958 CrossRef CAS.
- S. E. Jerng and B. M. Gallant, iScience, 2022, 25, 104558 CrossRef CAS PubMed.
- J. Bi, P. Hou, F.-W. Liu and P. Kang, ChemSusChem, 2019, 12, 2195–2201 CrossRef CAS PubMed.
- H. Kumagai, T. Nishikawa, H. Koizumi, T. Yatsu, G. Sahara, Y. Yamazaki, Y. Tamaki and O. Ishitani, Chem. Sci., 2019, 10, 1597–1606 RSC.
- D. C. Grills, Y. Matsubara, Y. Kuwahara, S. R. Golisz, D. A. Kurtz and B. A. Mello, J. Phys. Chem. Lett., 2014, 5, 2033–2038 CrossRef CAS PubMed.
- M. Bhattacharya, S. Sebghati, R. T. VanderLinden and C. T. Saouma, J. Am. Chem. Soc., 2020, 142, 17589–17597 CrossRef CAS PubMed.
- C. G. Margarit, N. G. Asimow, C. Costentin and D. G. Nocera, ACS Energy Lett., 2020, 5, 72–78 CrossRef CAS.
- J. B. Jakobsen, M. H. Rønne, K. Daasbjerg and T. Skrydstrup, Angew. Chem., Int. Ed., 2021, 60, 9174–9179 CrossRef CAS PubMed.
- W. Nie, D. E. Tarnopol and C. C. L. McCrory, J. Am. Chem. Soc., 2021, 143, 3764–3778 CrossRef CAS PubMed.
- P. V. Kortunov, M. Siskin, L. S. Baugh and D. C. Calabro, Energy Fuels, 2015, 29, 5919–5939 CrossRef CAS.
- A. Khurram, L. Yan, Y. Yin, L. Zhao and B. M. Gallant, J. Phys. Chem. C, 2019, 123, 18222–18231 CrossRef CAS.
- G. Lee, Y. C. Li, J.-Y. Kim, T. Peng, D.-H. Nam, A. Sedighian Rasouli, F. Li, M. Luo, A. H. Ip, Y.-C. Joo and E. H. Sargent, Nat. Energy, 2021, 6, 46–53 CrossRef CAS.
- G. Leverick, E. M. Bernhardt, A. I. Ismail, J. H. Law, A. Arifutzzaman, M. K. Aroua and B. M. Gallant, ACS Catal., 2023, 13, 12322–12337 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc02709d |
| This journal is © The Royal Society of Chemistry 2024 |