
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePyrrolidine-2,3-diones: heterocyclic scaffolds that inhibit and eradicate S. aureus biofilms†
M. Alejandro Valdes-Pena ac,
Andrew Ratchfordbc,
Minhua Nieac,
Lauren V. Schnabelbc and
Joshua G. Pierce*ac
ac,
Andrew Ratchfordbc,
Minhua Nieac,
Lauren V. Schnabelbc and
Joshua G. Pierce*ac
aDepartment of Chemistry and Integrative Sciences Initiative, NC State University, Raleigh, NC 27695, USA. E-mail: jgpierce@ncsu.edu
bDepartment of Clinical Sciences College of Veterinary Medicine, NC State University, 1060 William Moore Drive, Raleigh, NC 27607, USA
cComparative Medicine Institute, NC State University, Raleigh, NC 27695, USA
First published on 23rd September 2024
Abstract
The absence of novel antibiotic classes, coupled with the rising threat of antibiotic persistence and resistance, is pushing the world perilously close to a new pre-antibiotic era. Over 35![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people die every year in the US as a consequence of antimicrobial-resistant infections. Bacterial biofilms further complicate this scenario, as they are inherently more resistant to antibiotic treatments. Currently, there are no approved single agent or adjuvant small molecules for treating biofilm-complicated infections. Herein, we report the synthesis and microbiological evaluation of a novel library of 25+ monomeric and dimeric pyrrolidine-2,3-dione scaffolds. These compounds have displayed improved aqueous solubility, potent anti-biofilm properties, a low MBEC-to-MIC ratio, and synergism with FDA-approved antimicrobials against biofilm infections, constituting a promising technology as antimicrobial adjuvants.
000 people die every year in the US as a consequence of antimicrobial-resistant infections. Bacterial biofilms further complicate this scenario, as they are inherently more resistant to antibiotic treatments. Currently, there are no approved single agent or adjuvant small molecules for treating biofilm-complicated infections. Herein, we report the synthesis and microbiological evaluation of a novel library of 25+ monomeric and dimeric pyrrolidine-2,3-dione scaffolds. These compounds have displayed improved aqueous solubility, potent anti-biofilm properties, a low MBEC-to-MIC ratio, and synergism with FDA-approved antimicrobials against biofilm infections, constituting a promising technology as antimicrobial adjuvants.
According to the most recent WHO report, antimicrobial resistance has been listed as one of humanity's top 10 global health threats.1–3 Among the 12 highest-priority multidrug-resistant pathogens listed by the WHO in 2017, five belong to the ESKAPE pathogens group.4 Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and the Enterobacter species are the species making up the ESKAPE acronym. There are over 2.8 million antibiotic resistance-related infections each year in the US, and more than 10% of these infections result in death.2 One of the most significant contributors to the death toll continues to be methicillin-resistant S. aureus (MRSA), which results in over 10
000 deaths annually in the US alone.2 Furthermore, traditional antibiotic treatments are becoming ineffective against bacterial infections at an accelerated pace as antimicrobial resistance increases. The resistance-free life span of antibiotics have steadily declined over the years to the point that a resistant bacterial strain is identified almost immediately after a new antibiotic scaffold enters the clinic.5
In addition to traditional acquired resistance mechanisms, bacteria possess intrinsic biofilm defense pathways that directly contribute to antimicrobial tolerance. Bacterial biofilms are a surface-attached community of bacteria surrounded by a self-produced extracellular matrix of polysaccharides, proteins, and extracellular DNA (e-DNA).6 The biofilm provides a range of advantages for bacteria, including physical separation from antibiotics, reduction in metabolic activity,7 facile exchange of genetic information via e-DNA,8 and many others. This unique cellular environment within the biofilm allows most bacteria to overcome antimicrobial treatments, which often results in FDA-approved antibiotics being 10–1000 times less effective against biofilms compared with the treatment of planktonic bacteria.6,9–12 This is particularly concerning considering that no new classes of antibiotics have been approved in the last decades and that even fewer antibiofilm technologies proceed to the in vivo evaluation stage and ultimately reach clinical use.13 Due to the inherent connections between antimicrobial resistance generation and biofilm formation, it is imperative to continue to identify new antimicrobials and anti-biofilm scaffolds.
The pyrrolidine-2,3-diones constitute an underexplored heterocyclic scaffold present in natural products such as leopolic acid A (1), phenopyrrozin (2), and p-hydroxyphenopyrrozin (3) (Fig. 1). This class of heterocycle has shown antimicrobial activity against Gram-positive pathogens, and has been recently reported to inhibit PBP3 in P. aureginosa.14 The total synthesis of leopolic acid A has been previously reported by Dallavalle and co-workers,15 and, more recently, by our group.16 We have reported that leopolic acid A, while displaying promising antimicrobial and antibiofilm activity, tolerated very few structural modifications without disrupting its antimicrobial activity. Additionally, our group has explored a family of unnatural pyrrolidine-2,3-dione scaffolds, which showed promising activity against planktonic bacteria and moderate biofilm inhibition properties (4–8).17,18 Most of these unnatural pyrrolidine-2,3-dione scaffolds displayed low aqueous solubility, posing a challenge to fully assess these compounds' antimicrobial potency. However, it has been well established that these types of pyrrolidine-2,3-diones display improved activity when bearing a 4-p-trifluoromethylphenyl- and a 5-ethyl moiety in its core, yet the N-substituent still requires further optimization to improve activity and physicochemical properties. Herein, we report the synthesis and antimicrobial assessment of a new library of pyrrolidine-2,3-dione monomers and dimers with enhanced aqueous solubility and improved antibiofilm properties.
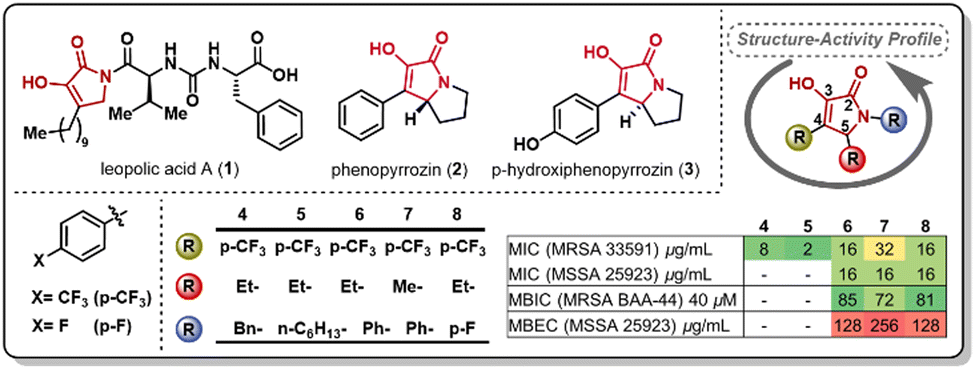 | ||
| Fig. 1 Examples of natural (1–3) and unnatural (4–8) reported pyrrolidine-2,3-diones and antimicrobial activity of selected unnatural scaffolds. | ||
Our initial strategy to overcome the limited aqueous solubility of the unnatural pyrrolidine-2,3-dione derivatives was to use diamines as the amine source for synthesizing these compounds. We have leveraged and optimized the use of a multicomponent reaction, which comprises a phenyl pyruvic ester derivative (13), an aldehyde (14), and an amine to yield pyrrolidine-2,3-diones in one pot, with exquisite C–C chemoselectivity (Fig. 2A).19 Our group has previously reported that using additional base such as trimethylamine results in C–O/C–C chemoselectivity issues20 however, in preliminary studies the use of diamines did not cause this issue.
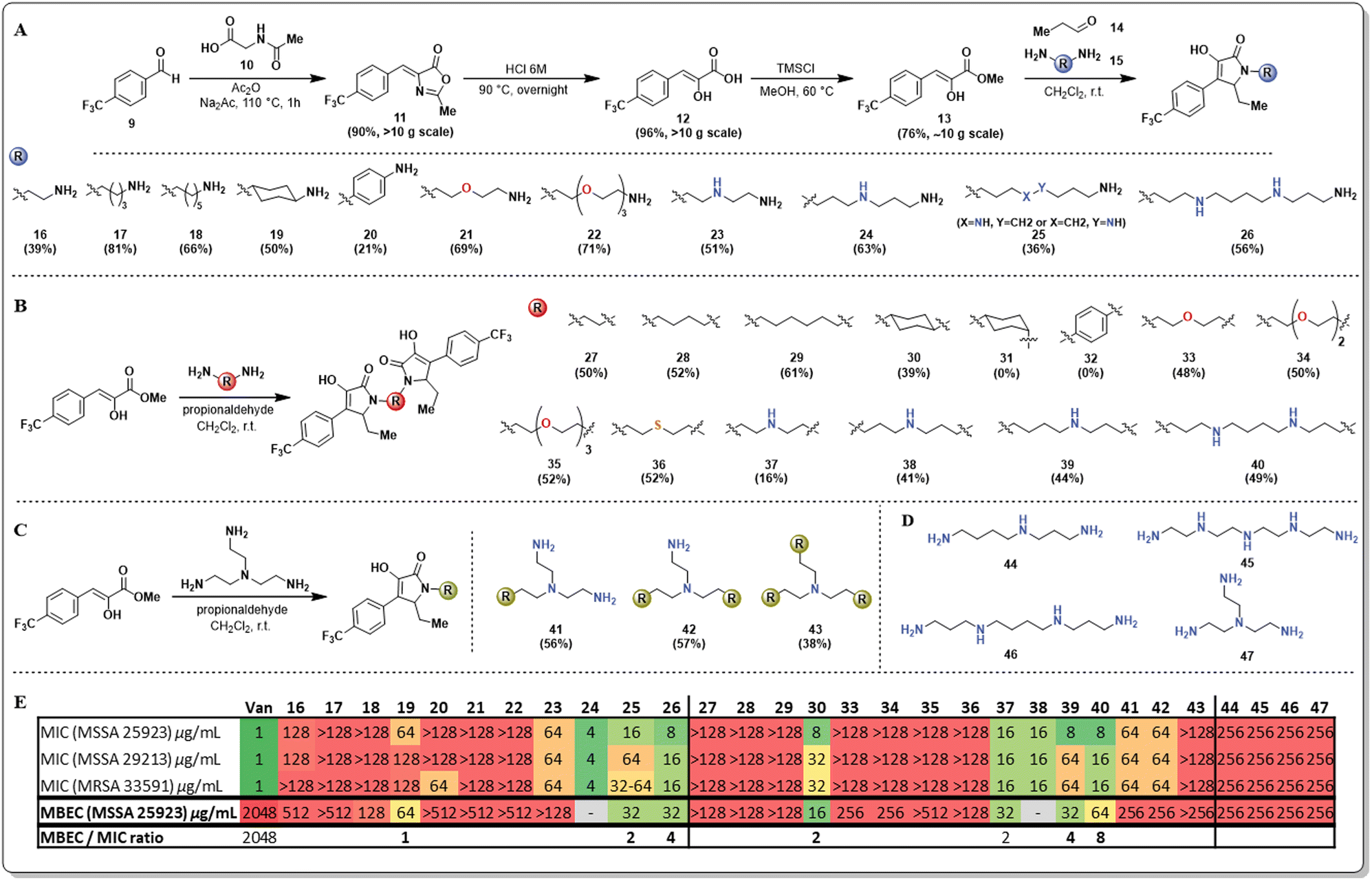 | ||
| Fig. 2 (A) Synthetic procedure to access pyrrolidine-2,3-dione monomer scaffolds through a multicomponent reaction. (B) Synthesis of pyrrolidine-2,3-dione dimeric scaffolds. (C) Synthesis of pyrrolidine-2,3-dione monomeric, dimeric and trimeric scaffolds. (D) Polyazadiamine linkers used in the multicomponent reaction. (E) Antimicrobial evaluation. *Microbiological assays not performed are denoted with “–“. | ||
An initial library of pyrrolidine-2,3-dione scaffolds was synthesized and the required structural features of the diamines to improve antimicrobial properties was explored (Fig. 2). Additional heteroatoms in the diamine (15) were crucial for turning on antimicrobial activity. The monomeric scaffolds bearing carbon atoms alone in the diamine, either linear (16–18), cyclic (19), or aromatic (20), resulted in entirely inactive compounds. Similarly, mono (21), or triethylene glycol (22) diamines yielded inactive compounds against both planktonic and biofilm phenotypes. The effect of aza-substituted diamines was also explored, and bis-propyltriamine and spermidine were used to access the corresponding pyrrolidine-2,3-dione monomers (23–25). Strikingly, the presence of the aza-moiety in the linker worked as a switch to turn on activity, while the length of the N-substituent appears to have a significant impact on activity. Some of these analogs presented single-digit MIC activity and, more importantly, displayed potent antibiofilm properties (MBEC = 32 μg mL−1). Compound 25 was obtained as an inseparable mixture of regio-isomers. To evaluate the effect of multiple nitrogen atoms in the n-alkyl chain of the diamine, the symmetric diamine spermine was selected to access monomer 26. The presence of an additional nitrogen atom did not improve the antimicrobial potency of 26, resulting in almost identical results to those obtained for compound 25.
During the synthesis of these new pyrrolidine-2,3-dione monomers, we observed that the corresponding dimeric scaffolds formed readily in the reaction mixture, although in low yields. The optimization of the reaction allowed access to the corresponding pyrrolidine-2,3-dione dimers as major products, except when limited solubility of the corresponding monomers in dichloromethane prevented dimer formation. Alternative solvents such as methanol or acetonitrile were explored in those cases of limited monomer solubility; however, this did not result in an improved yield relative to the use of dichloromethane.
The strategy described allowed the synthesis of novel pyrrolidine-2,3-dione dimers (Fig. 2B, 27–40), and their antimicrobial activity was determined and compared with the activity of their corresponding monomers. The dimers bearing the n-alkyl diamine linker(s) (27–29) did not improve their antimicrobial activity. Strikingly, the trans-cyclohexyl dimer 30 had very potent antimicrobial activity, displaying single-digit MIC values against MSSA strains and significant antibiofilm activity, with an MBEC of 16 μg mL−1. To explore the impact of this compound's geometry on its antimicrobial activity, we aimed to access the cis-cyclohexyl dimer (31), yet despite several reaction conditions screened, this analog was not observed. We hypothesize that the steric constraints of the axial amino group are hindering imine formation, which is the first step of the multicomponent reaction, thus preventing the second cyclization from proceeding. Similarly, we sought to explore the effect of electronics on the cyclohexyl dimer's activity but were unsuccessful in accessing dimer 32 using p-aminoaniline. Once the monomeric pyrrolidine-2,3-dione (20) was formed, the reduced electron density on the aniline results in reduced reactivity. Nevertheless, the unexpectedly potent bioactivity of compound 30 set our expectations high for the other dimers bearing heteroatoms in the linker. Unfortunately, using polyethylene glycol diamines resulted in completely inactive compounds (33–35), and a similar result was obtained for a thio-substituted linker (36), rendering thio- and oxa-containing linkers inactive.
Additionally, a series of dimers containing aza- or polyaza-linkers (37–40) were synthesized and, as expected, these dimers were as active as their corresponding monomers. The length of the linker and the number of nitrogen atoms present mildly affect the antimicrobial activity against planktonic Gram-positive bacteria (MSSA and MRSA), as most MIC values fluctuate between 8–16 μg mL−1. However, a more significant difference was found in the activity against biofilm-embedded bacteria. The anti-biofilm activity is not as strong for short linkers (37) or longer linkers with two nitrogen atoms (40). Medium-sized linkers that are 7–8 atoms long and have a single nitrogen atom display the best activity. Considering the remarkable activity of dimer 30 in contrast with its corresponding monomer 19, we wanted to clarify if this activity enhancement would be transferred to a trimeric scaffold. We utilized tris-triamine (Fig. 2C, 41–43) as the linker to readily access all three monomeric, dimeric, and trimeric scaffolds. Surprisingly, monomer 41 and dimer 42 displayed inferior antimicrobial activity, and most surprising was the finding that the trimer 43 displayed no antimicrobial or antibiofilm activity. As a control, the antimicrobial activity of the linkers used for the aza- and polyaza monomers and dimers was evaluated to determine if the linker induced the antimicrobial activity we observed (Fig. 2D). Poor antimicrobial and antibiofilm activity (256 μg mL−1) was found for spermidine (44), tetraethylene pentamine (45), spermine (46), and 2,2′,2′′-triaminotriethylamine (47).
The results obtained for the SAR studies indicated that we have accessed a class of compounds that were not as effective in killing bacteria in a planktonic state, yet were relatively potent at clearing preformed bacterial biofilms. Most FDA-approved antimicrobials lack antibiofilm properties; thus, the MBEC/MIC ratio tends to be hundreds or thousands (ciprofloxacin, 16; doxycycline, 512; linezolid, 512; vancomycin, 4096; Fig. S1, ESI†). Interestingly, these pyrrolidine-2,3-dione monomers and dimers had remarkable single-digit MBEC/MIC ratios (2–4, Fig. S1, ESI†), indicating that these compounds were able to eradicate bacterial biofilms (MBEC) at concentrations very close to the concentration at which they kill planktonic bacteria (MIC). To further explore the anti-biofilm capabilities of these compounds, we decided to perform an MBEC synergism assay between our lead dimer 30 and different FDA-approved antimicrobials possessing distinct mechanisms of action (Fig. 3). Interestingly, we found that dimer 30 only displayed synergism with vancomycin, causing a 4-fold reduction of vancomycin's MBEC value (2048 to 512 μg mL−1) when using a sub-MBEC of dimer 30 (8 μg mL−1). This result positions these pyrrolidine-2,3-dione scaffolds as potential adjuvants in antimicrobial therapies against biofilm-complicated infections, for which most antibiotics render ineffective.21–27
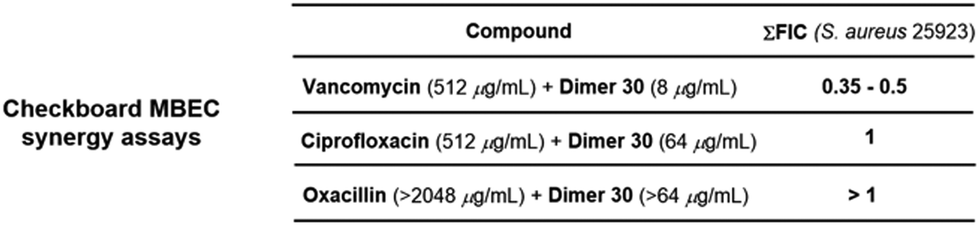 | ||
| Fig. 3 Minimum biofilm eradication concentration (MBEC) synergism assays between one of the 2,3-pyrrolidinedione leads and different FDA-approved antimicrobials. *ΣFIC: sum of fractional inhibitory concentration. | ||
At this point, we demonstrated that the presence of a nitrogen heteroatom contributed to improved activity, and the only active scaffold without a N-containing linker was dimer 30. To study the role of the enol moiety in the structure of the pyrrolidine-2,3-diones dimers, dimer 30 was methylated (Fig. 4, 48). This scaffold's antimicrobial and antibiofilm activity was determined, and it was found that the enol moiety is essential for antimicrobial activity. The methylation of dimer 48 entirely shut down any antimicrobial activity that dimer 30 displayed.
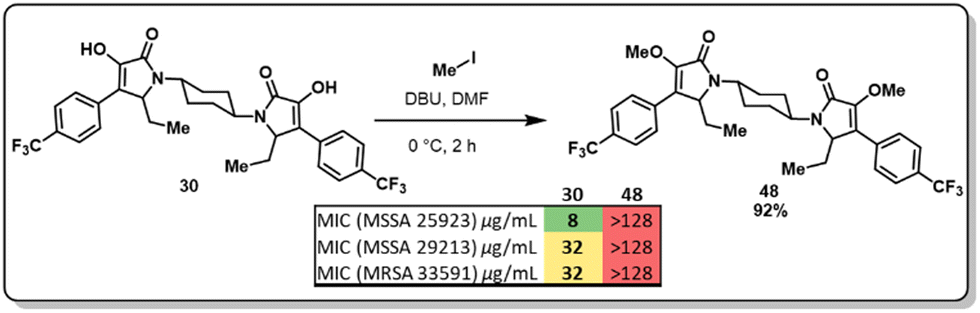 | ||
| Fig. 4 Synthesis and microbiological evaluation of methylated cyclohexyl dimer 30. | ||
To classify the broader inhibition pattern, the antimicrobial activity of dimer 30 was further investigated through time-dependent inhibition (Fig. 5). Vancomycin displayed its characteristic bactericidal activity by way of >2log10 reduction in CFU mL−1 after 24 hours.19 Despite exposure to the MIC of dimer 30, established by broth microdilution before the time-dependent inhibition, cells exposed to dimer 30 experienced significant recovery at some point between 12 and 24 hours post-exposure. It is currently unknown whether this result is due to a difference in the behavior of the bacteria due to increased culture volume, the compound's mechanism of action, or the stability of the compound in aqueous culture media. In addition to their synergistic relationship with vancomycin, the striking antibiofilm activity of these compounds relative to their reduced activity against planktonic cells warrants further investigation into the biological activity of pyrrolidine-2,3-diones before their potential as an anti-biofilm therapeutic can be fully evaluated.
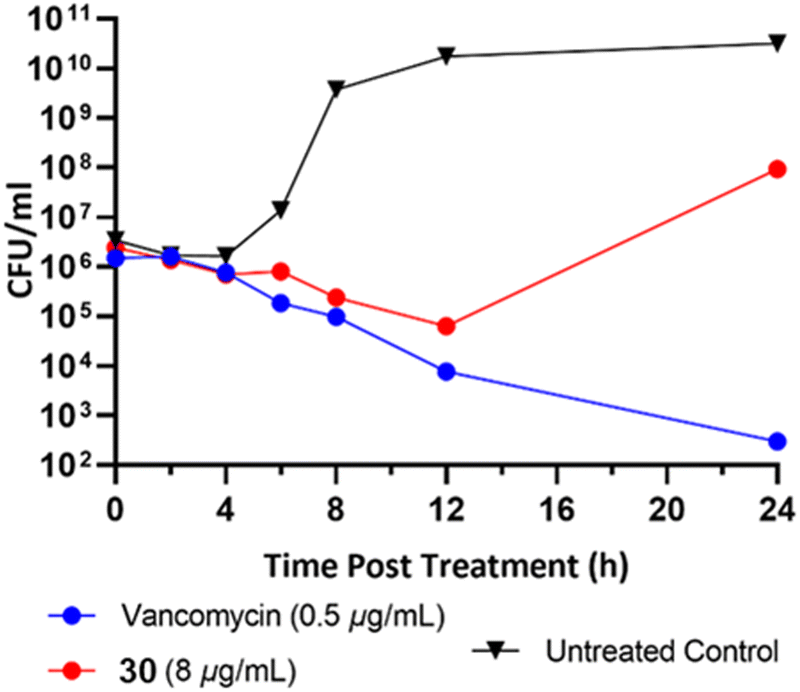 | ||
| Fig. 5 Time-dependent inhibition of planktonic S. aureus USA300 LAC at the MIC. Viable bacteria were quantified as colony-forming units (CFU) per mL up to 24 hours post-exposure. The lower limit of detection was 100 CFU mL−1. | ||
In summary, we have synthesized a novel library of pyrrolidine-2,3-dione monomers and dimers with improved antimicrobial activity, aqueous solubility, and anti-biofilm properties. More than 25 analogs were synthesized, and the requirements of the linker have been established. It has been determined that linkers between 5 and 8 atoms in length, bearing one heteroatom, display the best antimicrobial activity. The trans-1,4-cyclohexyldiamine linker was the only example of a potent analog with no heteroatom in its structure. Non-nitrogen heteroatoms, such as oxygen or sulfur, did not appear to enhance the antimicrobial properties of these compounds. The enol moiety was essential for antimicrobial activity, and dimeric forms were preferred for antibiofilm activity (Fig. 6). This work sets the stage for leveraging these compounds for the development of novel antimicrobials and further efforts toward this goal will be reported in due course.
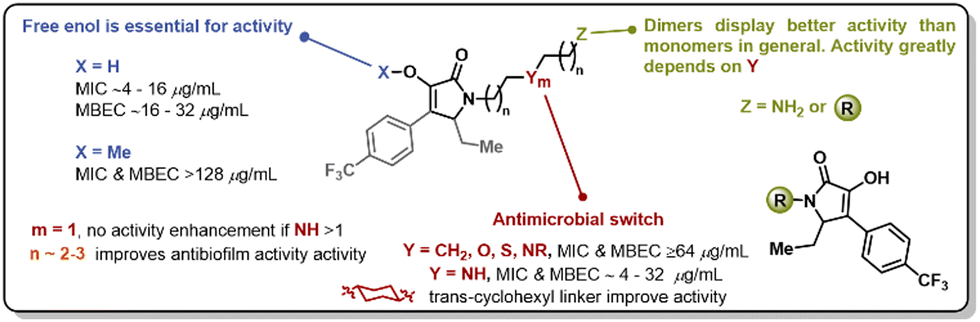 | ||
| Fig. 6 Summary of the SAR study performed for 2,3-pyrrolidiendione scaffolds. | ||
We are grateful to the NIH (R35GM139583) for generous support of this work and to NC State University for support of our program. Mass spectrometry data, NMR data and X-ray data were obtained at the NC State Molecular, Education, Technology and Research Innovation Center (METRIC).
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
J. G. P. is the founder of Synoxa Sciences, Inc.References
- M. A. Cook and G. D. Wright, Sci. Transl. Med., 2022, 14(657), eabo7793 CrossRef CAS PubMed.
- World Health Organization. 10 Global Health Issues to Track in 2021. 2020. 2024.
- Organization, W. H. Ten threats to global health in 2019 https://www.who.int/news-room/fact-sheets/detail/antibiotic-resistance accessed Apr 10, 2021.
- CDC. Antibiotic Resistance Threats in the United States, 2019, Atlanta, GA, U.S. Department of Health and Human Services, CDC, 2019.
- M. A. Valdes-Pena, et al., Acc. Chem. Res., 2021, 54, 1866–1877 CrossRef CAS PubMed.
- K. M. Craft, et al., MedChemComm, 2019, 10, 1231–1241 RSC.
- D. Sharma, et al., Antim. Resist Infect Control, 2019, 8, 76 CrossRef.
- S. D. Goodman and L. O. Bakaletz, Microorganisms, 2022, 10, 466 CrossRef CAS.
- P. S. Stewart and J. W. Costerton, Lancet, 2001, 358, 135–138 CrossRef CAS.
- A. Brauner, et al., Nat. Rev. Microbiol., 2016, 14, 320–330 CrossRef CAS.
- R. M. Donlan and J. W. Costerton, Clin. Microbiol. Rev., 2002, 15, 167–193 CrossRef CAS.
- D. Davies, Nat. Rev. Drug Discovery, 2003, 2, 114–122 CrossRef CAS.
- S. Cascioferro, et al., ChemMedChem, 2021, 16, 65–80 CrossRef CAS PubMed.
- A. López-Pérez, et al., Antibiotics, 2021, 10, 529 CrossRef.
- A. A. Dhavan, et al., Org. Chem., 2016, 12, 1624–1628 CAS.
- J. L. Breunig, et al., ACS Bio Med Chem Au, 2024, 4, 95–99 CrossRef CAS.
- A. Q. Cusumano and J. G. Pierce, Bioorg. Med. Chem. Lett., 2018, 28, 2732–2735 CrossRef CAS PubMed.
- M. Nie, et al., Bioorg. Med. Chem. Lett., 2024, 99, 129609 CrossRef CAS PubMed.
- S. V. Ryabukhin, et al., ACS Comb. Sci., 2012, 14, 631–635 CrossRef CAS PubMed.
- N. V. Shymanska and J. G. Pierce, Org. Lett., 2017, 19, 2961–2964 CrossRef CAS PubMed.
- R. J. Melander and C. Melander, ACS Infect. Dis., 2017, 3, 559–563 CrossRef CAS.
- G. Dhanda, et al., ACS Omega, 2023, 8, 10757–10783 CrossRef CAS PubMed.
- Y. Liu, et al., Crit. Rev. Microbiol., 2019, 45, 301–314 CrossRef CAS PubMed.
- V. Kumar, et al., Front. Cell. Infect. Microbiol., 2023, 13, 1293633 CrossRef CAS PubMed.
- R. J. Melander and C. Melander, Top. Med. Chem., 2017, 89–118 Search PubMed.
- R. Dey, et al., J. Chem. Sci., 2023, 15, 259–270 Search PubMed.
- N. Pant and D. P. Eisen, Antibiotics, 2021, 10, 1060 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc02708f |
| This journal is © The Royal Society of Chemistry 2024 |