
Iridium(III)-catalyzed photoredox cross-coupling of alkyl bromides with trialkyl amines: access to α-alkylated aldehydes†
Gaurav
Shukla
,
Malkeet
Singh
,
Saurabh
Singh
and
Maya Shankar
Singh
 *
*
Department of Chemistry, Institute of Science, Banaras Hindu University, Varanasi-211005, India. E-mail: mayashankarbhu@gmail.com; mssingh@bhu.ac.in
First published on 23rd April 2024
Abstract
A C(sp3)–C(sp3) cross coupling approach based on an iridium-photocatalytic radical process has been developed enabling the synthesis of various α-alkylated aldehydes from easily available/synthesized alkyl bromides and trialkyl amines under mild photocatalytic conditions. The synthesized aldehydes are also explored as a functional handle for various useful products such as carboxylic acid, alcohol and N-heterocycle synthesis.
In the age of contemporary organic synthesis, cross coupling reactions enabling the formation of carbon–carbon bonds are some of the most crucial steps.1 These reactions are highly significant synthetic tools for the synthesis of several medicines and naturally occurring compounds.2 However, the majority of C–C bond forming cross-coupling methods involve the use of expensive metal complexes, strong oxidizing agents, high temperature and tedious work-up procedures.3 Hence, alternative techniques that could overcome most of the existing glitches are highly needed. In this context, over the past two decades, photo-organic synthesis has seen a rebirth in comparison to the traditional approaches, and has become a potent tool for the cross-coupling C–C bond forming reactions.4 Due to its unique capacity to produce extremely reactive radicals and radical ion intermediates by a direct single electron transfer (SET) event, photoredox methods have increased interest in recent years as a sustainable strategy for numerous syntheses.5 Inspired by this, significant progress has been achieved in the creation of artificial photoredox reactions for the formation of C–C bonds.6
Functionalized aldehydes have been often employed as precursors towards the syntheses of numerous significant molecules such as carboxylic acids, amines, amides, alcohols, nitriles, and many other carbocyclic as well as heterocyclic systems.7 They are also well-known vital constituents of many natural products and pharmaceutically important molecules.8 Functionalized aldehydes have also been utilized as new radical alkylating reagents,9 as tools for a variety of synthetic, biological and in materials-based applications.10 Over the past few years, most of the reported methods involve direct metal and chiral base-mediated arylation/alkylation of aldehydes under harsh reaction conditions.11 Recently, a few photocatalytic methods for the direct functionalization of α-CH of aliphatic aldehydes have been reported by some research groups.12 Although all given methods successfully achieved the functionalized aliphatic aldehydes, the use of a co-catalytic system and direct use of aldehydes make these protocols a bit complex because of cost-ineffectiveness and formation of various side products by the oxidation of aldehydes under the given catalytic systems. Hence, an alternative method involving cost-effective low catalytic loading, which could possibly suppress the formation of oxidized side products, is highly needed. Herein, we report the cross-coupling reactions of phenacyl bromides with alkyl amines under Ir(III)-catalyzed photoredox conditions, which successfully provided the functionalized aliphatic aldehydes in good to excellent yields.
Keeping all the challenges in mind, optimization studies were conducted using the combination of 2-bromo-1-phenylethan-1-one (1a; 0.5 mmol, 1.0 equiv.) and TEA (2a; 2.0 mmol, 4.0 equiv.) in the presence of (Ir[dF(CF3)ppy]2(dtbpy))PF6 (PC-1, 2.0 mol%) and TBAI (0.5 equiv.) in 1,4-dioxane (2.0 mL) under Kessil blue LED Light irradiation (blue; 40 W, 467 nm) at 30 °C for 5 h in an open atmosphere. The work-up of the reaction mixture provided the product 3a in 76% isolated yield (characterized as 4-oxo-2-(2-oxo-2-phenylethyl)-4-phenyl butanal by NMR and HRMS studies, Table 1, entry 1). After the successful synthesis of 3a, we moved to improve the reaction conditions for better outcomes. Using different transition metal photocatalysts such as PC-2 = Ir(ppy)2(dtbbpy)PF6 and PC-3 = [Ru(bpy)3]Cl2 could not provide better results, and 3a was obtained in a lower yield than the initial reaction conditions (Table 1, entries 2 and 3). Next, to check the efficiency of the organic photocatalyst towards the model reaction, we used eosin Y (PC-4) instead of PC-1. Unfortunately, with PC-4 we did not get the desired product 3a even in trace amounts (Table 1, entry 4). Next, additive TBAB was also tested instead of TBAI, and with TBAB a significant decrease in the yield of the desired product 3a was observed (Table 1, entry 5).
|
|
||
|---|---|---|
| Entry | Variation in optimized conditions | Yieldb (%) |
| a Reaction conditions: all reactions were performed with 1a (0.5 mmol, 1.0 equiv.), 2a (2.0 mmol, 4.0 equiv.), additive (0.5 equiv.) and PC (2.0 mol%) in solvent (2.0 mL) under an open atmosphere for 5 h at 30 °C. b Isolated yield of 3a. c Reaction time 24 h; PC = photocatalyst, TEA = triethyl amine, TBAI = tetra butyl ammonium iodide, TBAB = tetra butyl ammonium bromide, nd = not detected. | ||
| 1 | None | 76 |
| 2 | PC-2 instead of PC-1 | 70 |
| 3 | PC-3 instead of PC-1 | 67 |
| 4 | PC-4 instead of PC-1 | nd |
| 5 | TBAB instead of TBAI | 56 |
| 6 | Reaction in ACN | 45 |
| 7 | Reaction in DMF | 35 |
| 8 | Reaction under blue LED (1 W, 480 nm) | 40 |
| 9 | Reaction under green LED (1 W, 540 nm) | Trace |
| 10 | Reaction without PC-1 | nd |
| 11 | Reaction without light | nd |
| 12 | Under nitrogen atmosphere | Trace |
| 13 | Reaction at 40 °C without light | nd |
| 14 | Without TBAI | 61c |
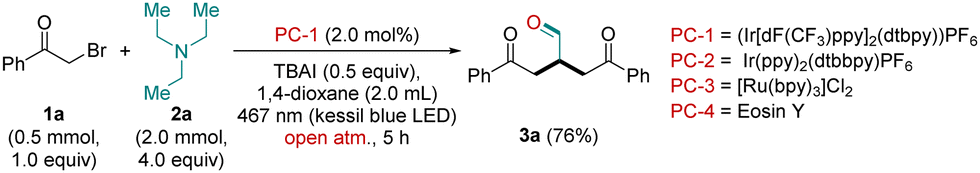
To check the effect of the solvent, we also carried out our model reaction in polar aprotic solvents like ACN and DMF. In both the solvents, we could not get any fruitful results (Table 1, entries 6 and 7). The reaction under different light sources (such as blue LED, 1 W, 480 nm; green LED, 1 W, 540 nm) did not provide better results (Table 1, entries 8 and 9). Next, to prove the role of the photocatalyst (PC-1) and the light source, we also performed our model reaction in the absence of PC-1 and light, separately. As anticipated, in both cases we did not get the desired aldehyde 3a even in a trace amount (Table 1, entries 10 and 11). Reaction under a N2 atmosphere yielded product 3a in a trace amount (Table 1, entry 12). Moreover, to prove the photo-mediated nature of the reaction, we carried out the model reaction at 40 °C in a dark chamber. The reaction at 40 °C failed to provide the product 3a even in a trace amount (Table 1, entry 13). Hence, after a careful survey of different reaction parameters, it was found that the use of 1a (0.5 mmol, 1.0 equiv.), 2a (2.0 mmol, 4.0 equiv.), PC-1 (2.0 mol%) and TBAI (0.5 equiv.) in 2.0 mL of 1,4-dioxane under blue light irradiation (Kessil blue; 40 W, 467 nm) in an open atmosphere for 5 h provided the desired aldehyde 3a in a better yield, is thus considered as the standard reaction conditions (76%; Table 1, entry 1; for detailed optimization of the catalysts, additives, solvents and light sources, see ESI†).
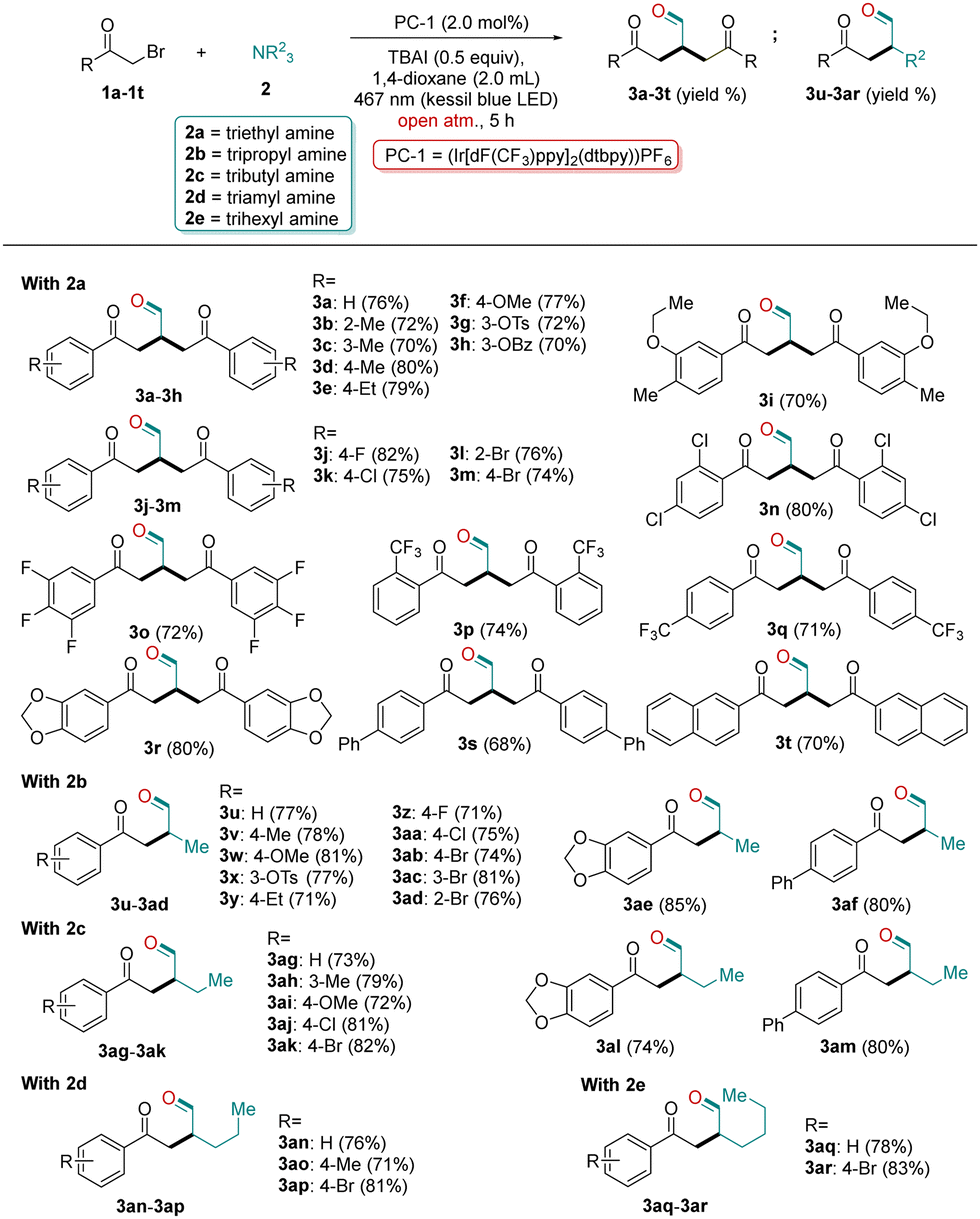
With the standard reaction conditions in hand (Table 1, entry 1), we investigated the scope of alkyl bromide 1 with TEA (2a) for cross-coupling to α-alkylated aldehydes 3a–3t. Various substituted alkyl bromides 1a–1t abided well under the optimized reaction conditions and provided aldehyde products 3a–3t in good to high yields (Table 2). This reaction tolerated a multitude of substrates with the electron-donating or electron-withdrawing substituents, located either at para-/meta-/ortho positions of the phenyl ring of 1a–1t (Table 2). The brominated compounds (1b–1e) containing electron-donating alkyl groups (Me & Et) at the o-, m- and p-positions of the phenyl ring were tolerated well under the optimized conditions affording the desired products in good to excellent yields (3b–3e; 70–80%, Table 2). Moreover, substrates with strong electron-donating groups such as 3-OMe (1f), 3-OTs (1g), 3-OBz (1h), & 3-O-pentyl (1i) at the meta-position were also tolerated well, and furnished the corresponding aldehydes in excellent yields (3f–3i; 65–81%, Table 2).
|
Next, a few halogenated substrates were also engaged for the synthesis of aldehydes under the optimized photoredox conditions. The compounds containing F, Cl and Br atoms at ortho-, meta- and para-positions of the phenyl ring (1j–1m) successfully converted to their corresponding desired products in good yields, without demonstrating any obvious electronic effect of halogens towards the yield of products (3j–3m; 74–82%, Table 2). After the successful conversion of mono-halogenated substrates to their corresponding aldehydes, next we employed di- and tri-halogenated substrates (1n & 1o) with triethyl amine 2a under the optimized conditions. The above two substrates were successfully converted to their desired products in excellent yields (3n & 3o; 80% & 72%, respectively; Table 2). To further check the tolerance of other groups under the optimized conditions, we employed o-CF3 and p-CF3 containing substrates (1p & 1q). To our pleasure, both the substrates were converted to their corresponding aldehydes in excellent yields (3p & 3q; 74% & 71%, respectively; Table 2). Furthermore, a 3,4-methylenedioxy containing substrate (1r) also furnished the desired product in a good yield (3r, 80%). A few extended aromatic substrates (1s & 1t) were also tested under the optimized conditions, and provided the desired products in good yields (3s & 3t; 68% & 70%, respectively, Table 2).
Next, we planned to explore some other trialkyl amines (2b–2e) for the synthesis of aldehydes under standard conditions. Using tri-propyl amine 2b, surprisingly, yielded a new product 3u (77%). Comprehensive NMR and HRMS studies of 3u indicated that the product formation involved mono-functionalization of 2b, unlike product 3a, which involved bi-functionalization of 2a. This unprecedented result might be attributed to the steric hindrance of the methyl group of 2b, which inhibits the attack of the second molecule of 1a to 3u. With this unique result in hand, next, we explored the substrate scope of 1 with 2b. A variety of differently substituted alkyl bromides were tolerated well under the optimized conditions furnishing the desired products 3u–3af in excellent yields (Table 2). The substrates with electron-donating groups (Me, OMe, OTs and Et) as well as with electron-withdrawing groups (F, Cl and Br) at their respective positions (ortho, meta, para) were tolerated well with 2b under the optimized photoredox conditions and furnished the desired α-functionalized aldehydes in good to excellent yields (3v–3ad, 69–81%; Table 2). Moreover, 3,4-methylenedioxy and extended aromatic substituted substrates were also treated with 2b, which provided the corresponding desired products in a good yield (3ae & 3af, 85% & 80%, respectively Table 2). Next, to introduce a few long-chain aldehydes into the list, we performed our reaction with other trialkyl amines such as tri-butyl amine 2c, tri-amyl amine 2d and tri-hexyl amine 2e. To our pleasure, all tested amines worked well under the optimized photoredox conditions and furnished their corresponding long chain α-functionalized aldehydes in good to excellent yields (3ag–3ar, 71–83%; Table 2). However, the use of alkyl bromides such as bromoethane, 1-bromobutane and 1-bromopentane could not provide the desired product under the established protocols exhibiting the limitation of this protocol. The desired product could not be obtained even with secondary amines, and it afforded a simple nucleophilic substitution product.
Following the successful photocatalytic synthesis of differentially substituted α-functionalized aldehydes, our attention turned to converting these molecules to additional derivatives, including carboxylic acids, alcohols and N-heterocycles. Thus, when the aldehyde derivatives were subjected to oxidation in the presence of oxone under open atmospheric conditions, the corresponding acid derivatives were obtained in good yields (Scheme 1a). Additionally, under reductive conditions (NaBH4 + MeOH), we successfully converted the products 3 to their corresponding primary alcohol derivative 6 and 1,4-diol derivative 7 in good yields (Scheme 1b). The synthesized aldehyde derivatives 3 have two carbonyl groups (one keto and one aldehyde), which could provide good reaction sites for the synthesis of heterocycles. Keeping this idea in mind, when we treated compound 3 with 4-tert-butylaniline in methanol at room temperature, the pyrrole derivatives (8 & 9) were obtained in good yields [Scheme 1c (i)]. Similarly, a few functionalized aldehydes effectively gave their respective pyridazine derivatives in high yields, when treated with hydrazine hydrochloride in the presence of NaHCO3 in ACN at room temperature [Scheme 1c (ii)].
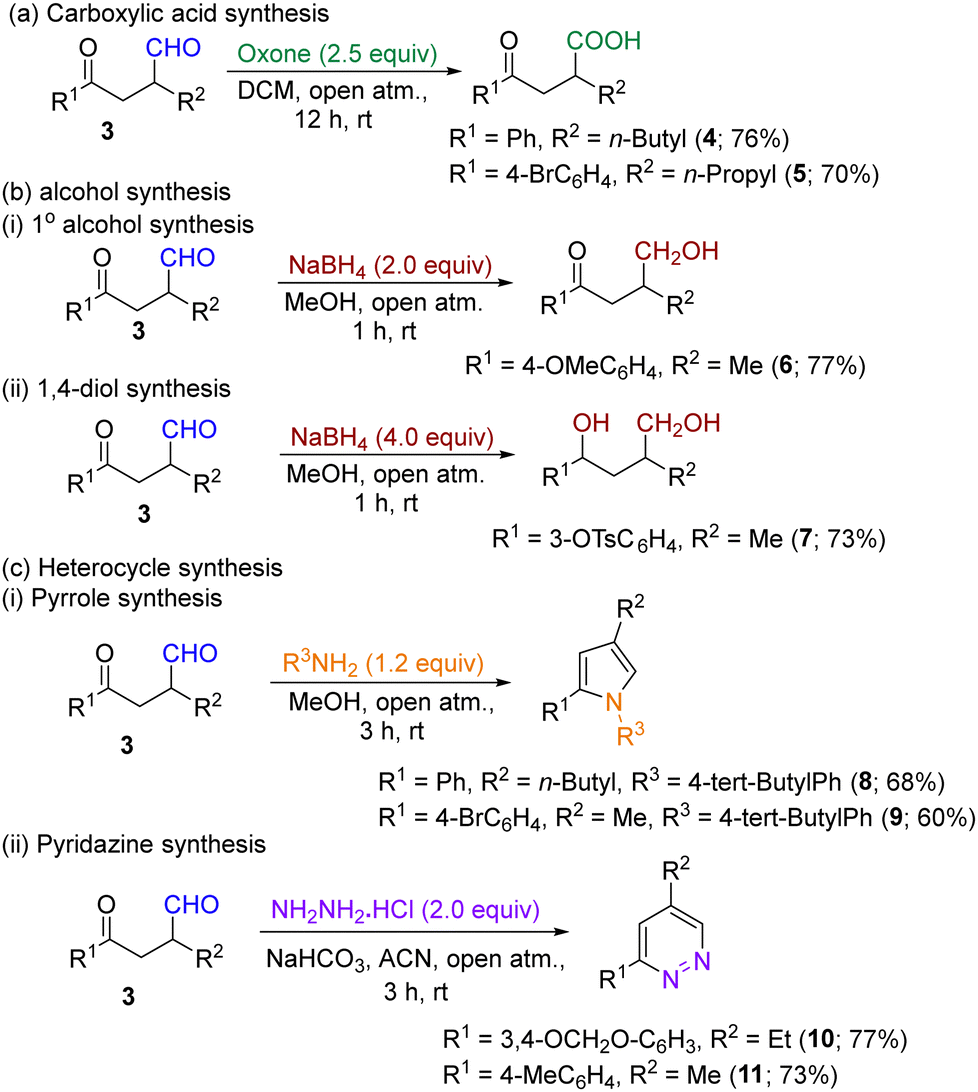 | ||
| Scheme 1 Synthetic applications of the aldehydes. | ||
Based on previous reports,12,13 and data collected from cyclic voltammetry and control experiments (see Scheme S3 in ESI† for details), we proposed a tentative mechanistic pathway for the formation of functionalized aldehydes by the cross-coupling of alkyl bromides 1 with alkyl amines 2 under Ir(III)-catalyzed photoredox conditions (Scheme 2). The first step of the reaction involved reductive quenching to generate N-centred radical cation A of alkyl amine 2 (Eox = +0.98 V for 2a, Eox = +1.02 V for 2d and Eox = +0.91 V for 2e; see ESI† for detail CV experiments) by excited Ir(III)* photocatalyst (E1/2*III/II = +1.21 V vs. SCE).13 This radical cation by the elimination of a hydrogen radical generated iminium intermediate B. Intermediate B on elimination of a proton generated suitable intermediate C. On the other hand, reductant Ir(II) carried out the reduction of alkyl bromide (Ered = −1.73 V for 1a) to generate alkyl radical D (R = PhCO; trapped by BHT radical scavenger and detected in HRMS analysis; see ESI† for the detailed experiment), completing the iridium photoredox cycle. Next, the radical intermediate D reacted with C generating another radical intermediate E. The radical intermediate E upon reaction with hydroxyl radicals (which could possibly be generated by the reaction of singlet oxygen with hydrogen radicals followed by its reaction with H2O) produced intermediate F. Intermediate F on elimination of a secondary amine (detected in HRMS analysis; see ESI† for detail) provided aldehyde products (3u–3ar). In the case of TEA (2a), the radical intermediate E again undergoes hydrogen radical elimination to generate intermediate G. Intermediate G could follow the same mechanistic pathway (as followed by intermediate C) and produced the desired aldehyde products (3a–3t) upon elimination of a secondary amine.
 | ||
| Scheme 2 Tentative mechanistic pathway. | ||
We gratefully acknowledge the financial support from the IoE Incentive grants, BHU (Scheme No. 6031) and JC Bose National Fellowship (JCB/2020/000023), New Delhi. The authors (GS, MS and SS) are thankful to SERB-JCB & UGC, New Delhi for the fellowship.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) D. Ravelli, S. Protti and M. Fagnoni, Chem. Rev., 2016, 116, 9850–9913 CrossRef CAS PubMed; (b) N. G. Schmidt, E. Eger and W. Kroutil, ACS Catal., 2016, 6, 4286–4311 CrossRef CAS PubMed; (c) Q. Yang, X. Guo, Y. Liu and H. Jiang, Int. J. Mol. Sci., 2021, 22, 1890 CrossRef CAS PubMed.
- (a) Z. Huang, S. Tang and A. Lei, Sci. Bull., 2015, 60, 1391–1394 CrossRef CAS; (b) O. Kose and S. Saito, Org. Biomol. Chem., 2010, 8, 896–900 RSC; (c) H. Yu, J. Mo, X. Niu, D. Luo and G. Che, Adv. Synth. Catal., 2023, 365, 3118–3128 CrossRef CAS; (d) X. Qi, L.-B. Jiang, H.-P. Li and X.-F. Wu, Chem. – Eur. J., 2015, 21, 17650–17656 CrossRef CAS PubMed.
- (a) J. Shin, S. Gwon, S. Kim, J. Lee and K. Park, J. Am. Chem. Soc., 2020, 142, 4173–4183 CrossRef CAS PubMed; (b) I. Kanwal, A. Mujahid, N. Rasool, K. Rizwan, A. Malik, G. Ahmad, S. A. A. Shah, U. Rashid and N. M. Nasir, Catalysts, 2020, 10, 443 CrossRef CAS; (c) M. A. Andrade and L. M. D. R. S. Martins, Molecules, 2020, 25, 5506 CrossRef CAS PubMed; (d) J. Sherwood, J. H. Clark, I. J. S. Fairlamb and J. M. Slattery, Green Chem., 2019, 21, 2164–2213 RSC; (e) J. García-Álvarez, E. Hevia and V. Capriati, Eur. J. Org. Chem., 2015, 6779–6799 CrossRef.
- (a) J. A. Milligan, J. P. Phelan, S. O. Badir and G. A. Molander, Angew. Chem., Int. Ed., 2019, 58, 6152–6616 CrossRef CAS PubMed; (b) S. Kim and F. D. Toste, J. Am. Chem. Soc., 2019, 141, 4308–4315 CrossRef CAS PubMed; (c) M. Yuan, Z. Song, S. O. Badir, G. A. Molander and O. Gutierrez, J. Am. Chem. Soc., 2020, 142, 7225–7234 CrossRef CAS PubMed; (d) H. Huang, Y. Zhang, P. Ji, P. S. Mariano and W. Wang, Green Synth. Catal., 2012, 2, 27–31 CrossRef; (e) J. D. Tibbetts, H. E. Askey, Q. Cao, J. D. Grayson, S. L. Hobson, G. D. Johnson, J. C. Turner-Dore and A. J. Cresswell, Synthesis, 2023, 3239–3250 CrossRef CAS.
- (a) E. Hola and J. Ortyl, Eur. Polym. J., 2021, 150, 110365 CrossRef CAS; (b) L. H. M. de Groot, A. Ilic, J. Schwarz and K. Wärnmark, J. Am. Chem. Soc., 2023, 145, 9369–9388 CrossRef CAS PubMed; (c) M. González-Esguevillas, D. F. Fernández, J. A. Rincón, M. Barberis, O. de Frutos, C. Mateos, S. García-Cerrada, J. Agejas and D. W. C. MacMillan, ACS Cent. Sci., 2021, 7, 1126–1134 CrossRef PubMed; (d) F. Zhao, W. Zhou and Z. Zuo, Adv. Synth. Catal., 2022, 364, 234–267 CrossRef CAS; (e) B. Lu, W.-J. Xiao and J.-R. Chen, Molecules, 2022, 27, 517 CrossRef CAS PubMed.
- (a) N. Rodríguez and L. J. Goossen, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC; (b) Z. Zuo, D. T. Ahneman, L. Chu, J. A. Terrett, A. G. Doyle and D. W. C. MacMillan, Science, 2014, 345, 437–440 CrossRef CAS PubMed; (c) J. Luo and J. Zhang, ACS Catal., 2016, 6, 873–877 CrossRef CAS; (d) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
- (a) L. Vanoye and A. Favre-Réguillon, React. Chem. Eng., 2023, 8, 1043–1050 RSC; (b) X. Zhang, C. Yang, H. Gao, L. Wang, L. Guo and W. Xia, Org. Lett., 2021, 23, 3472–3476 CrossRef CAS PubMed; (c) X.-D. An and S. Yu, Org. Lett., 2015, 17, 5064–5067 CrossRef CAS PubMed; (d) Y. Zhou, H. Zhou and J. Xu, J. Org. Chem., 2022, 87, 3677–3685 CrossRef CAS PubMed.
- (a) M. Passiniemi and A. M. P. Koskinen, Beilstein J. Org. Chem., 2013, 9, 2641–2659 CrossRef PubMed; (b) C. Gampe and V. A. Verma, J. Med. Chem., 2020, 63, 14357–14381 CrossRef CAS PubMed; (c) J. Chen, Z.-C. Geng, N. Li, X.-F. Huang, F.-F. Pan and X.-W. Wang, J. Org. Chem., 2013, 78, 2362–2372 CrossRef CAS PubMed; (d) Z. Wang, Molecules, 2019, 24, 3412 CrossRef PubMed; (e) M. A. Donega, S. C. Mello, R. M. Moraes, S. K. Jain, B. L. Tekwani and C. L. Cantrell, Planta Med., 2014, 80, 1706–1711 CrossRef CAS PubMed.
- (a) L. Li, H. Zheng, F. Guo, Z. Fang, Q. Sun, J. Li, Q. Gao, T. Zhanga and L. Fang, Chem. Commun., 2023, 59, 3910–3913 RSC; (b) W.-C. Yang, J.-G. Feng, L. Wu and Y.-Q. Zhang, Adv. Synth. Catal., 2019, 361, 1700–1709 CrossRef CAS.
- (a) R. Tayebee, A. Hosseini-nasr, N. Zamand and B. Maleki, Polyhedron, 2015, 102, 503–513 CrossRef CAS; (b) C. Negrell, C. Voirin, B. Boutevin and V. Ladmiral, Eur. Polym. J., 2018, 109, 544–563 CrossRef CAS; (c) G. S. Heo, S. Cho and K. L. Wooley, Polym. Chem., 2014, 5, 3555–3558 RSC; (d) A. M. Kunjapur and K. L. J. Prather, Appl. Environ. Microbiol., 2015, 81, 1892–1901 CrossRef CAS PubMed; (e) E. Istif, D. Mantione, L. Vallan, G. Hadziioannou, C. Brochon, E. Cloutet and E. Pavlopoulou, ACS Appl. Mater. Interfaces, 2020, 12, 8695–8703 CrossRef CAS PubMed.
- (a) K. Yang, Q. Li, Y. Liu, G. Li and H. Ge, J. Am. Chem. Soc., 2016, 138, 12775–12778 CrossRef CAS PubMed; (b) K. Yang, Z. Li, C. Liu, Y. Li, Q. Hu, M. Elsaid, B. Li, J. Das, Y. Dang, D. Maiti and H. Ge, Chem. Sci., 2022, 13, 5938 RSC; (c) C. Dong, L. Wu, J. Yao and K. Wei, Org. Lett., 2019, 21, 2085–2089 CrossRef CAS PubMed; (d) S. Vera, A. Landa, A. Mielgo, I. Ganboa, M. Oiarbide and V. Soloshonok, Molecules, 2023, 28, 2694 CrossRef CAS PubMed; (e) J. Blom, G. J. Reyes-Rodríguez, H. N. Tobiesen, J. N. Lamhauge, M. V. Iversen, C. L. Barløse, N. Hammer, M. Rusbjerg and K. A. Jørgensen, Angew. Chem., Int. Ed., 2019, 58, 17856–17862 CrossRef CAS PubMed.
- (a) D. A. Nicewicz and D. W. C. Macmillan, Science, 2008, 322, 77–80 CrossRef CAS PubMed; (b) E. Arceo, I. D. Jurberg, A. Álvarez-Fernández and P. Melchiorre, Nat. Chem., 2013, 5, 750–756 CrossRef CAS PubMed.
- (a) J. M. R. Narayanam, J. W. Tucker and C. R. J. Stephenson, J. Am. Chem. Soc., 2009, 131, 8756–8757 CrossRef CAS PubMed; (b) D. A. Nagib, M. E. Scott and D. W. C. MacMillan, J. Am. Chem. Soc., 2009, 131, 10875–10877 CrossRef CAS PubMed; (c) L. Furst, J. M. R. Narayanam and C. R. J. Stephenson, Angew. Chem., Int. Ed., 2011, 50, 9655–9659 CrossRef CAS PubMed; (d) C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322–5363 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4cc01043d |
| This journal is © The Royal Society of Chemistry 2024 |