
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tracing the origin of elevated springtime atmospheric sulfate on the southern Himalayan-Tibetan plateau†
Sanjeev
Dasari‡
 *a,
Guillaume
Paris
b,
Qiaomin
Pei
c,
Zhiyuan
Cong
c and
David
Widory
*d
*a,
Guillaume
Paris
b,
Qiaomin
Pei
c,
Zhiyuan
Cong
c and
David
Widory
*d
aInstitut des Géosciences de L'Environnement (IGE), University Grenoble Alpes, CNRS, IRD, Grenoble INP, Grenoble 38000, France
bUniversité de Lorraine, CNRS, CRPG, Nancy 54000, France
cKey Laboratory of Tibetan Environment Changes and Land Surface Processes, Institute of Tibetan Plateau Research, Chinese Academy of Sciences, Beijing 100101, China
dGEOTOP/ Université du Québec à Montréal, Montréal H3C 3P8, Canada. E-mail: widory.david@uqam.ca
First published on 5th June 2023
Abstract
The Himalayan Tibetan Plateau (HTP) is one of the world's most climate-sensitive regions outside the polar regions. Here, the climate–air quality–hydrological cycle affecting sulfate aerosols remains sparsely investigated, with their source origin(s) requiring further investigation. We tracked the evolution of sulfur mass-independent isotope fractionation [S-MIF i.e., Δ33S ≠ 0]—as a potential source tracer—in springtime aerosol sulfate over the southern HTP. In a first, at the southern HTP high-altitude receptor site Qomolangma-Mt. Everest station, QOMS, ∼4300 m a.s.l., elevated sulfate concentrations and S-MIF were both found to be associated with biomass burning aerosols (SO42− and Δ33S vs. K+: R2 = 0.92 [P < 0.001] and 0.61[P < 0.005], respectively). This is in stark contrast to Δ33S aerosol records from the central HTP and a downwind mountainous site wherein anomalous sulfur has been linked to stratospheric intrusions in the past, and geological lake records from the region which link the origin of modern sulfate on the HTP to the influx of mineral dust, respectively. The findings suggest that there are yet unknown biomass combustion-related processes (e.g., crop-residue and waste burning, wildfires) plausibly generating positive MIF in sulfur, which could have implications for historical S-isotope records. Comparing the triple-S-isotope imprint in aerosol sulfate on the HTP and its surrounding regions reveals the existence of spatial heterogeneity in the dominance of competing sulfate transport and formation processes with implications for the regional tropospheric chemical and radiation budgets.
Environmental significanceSulfate aerosols play an important role in Earth's climate, water cycle, and air quality. The Himalayan Tibetan Plateau (HTP), often dubbed as the ‘third pole’, is prone to sulfate-induced climatic effects. However, the likelihood of any diversity in sulfate transport and formation processes on the HTP remains less known. Here, in a first, using an intrinsic S-isotope characteristic as a potential source tracer we find that biomass burning in the global pollution hotspot Indo-Gangetic Plain likely influences sulfate sourced to the southern HTP region, whereas processes associated with stratospheric intrusions and influx of mineral dust influence sulfate on the central HTP. These findings suggest that sustainable practices and mitigation of open burning in South Asia could reduce the sulfate-induced complex environmental–ecological interactions on the southern HTP. |
Introduction
The Himalayan-Tibetan Plateau (HTP; 27–45° N, 70–105° E) is one of the most climatically sensitive regions in the world.1 Complex atmosphere–cryosphere–hydrosphere interactions in this region have strong environmental and ecological implications for the combined ∼2 billion population in East and South Asia.1,2 From a geographical standpoint, the HTP is vulnerable to both anthropogenic emissions from the world's most serious pollution hotspots in Asia, and naturally occurring dust from surrounding desert regions as well as high seasonal UV flux.3,4 Atmospheric aerosols transported to the HTP, by such means, have been found to affect cloud and radiation budget in the region — leading to rapid warming, intensified cryospheric melt and weaking of the Asian monsoon.3 While increasing attention has been paid towards the scientific underpinning of light-absorbing aerosols (e.g., black carbon, brown carbon) transported to/locally sourced on the HTP,5 a similar know-how is lacking for other climate-relevant species such as sulfate.6In general, sulfate forms the bulk of inorganic aerosol loadings on the HTP.3 Global chemistry-climate modeling studies have suggested that changes in sulfate loading pose an existential threat to the atmospheric warming and monsoonal rainfall (with implications on drought) in the region.7,8 These aspects are of relevance for the vastly temperature and rainfall-dependent agricultural economies of Asia, with effects on the livelihood and well-being of millions of people.7,8 A pertinent issue is the contrasting trends in the SO2 emissions (on a global scale ∼50% of natural and anthropogenically emitted SO2 is oxidized to sulfate) in East and South Asia,9–13 subsequently affecting the HTP sulfate burden. Growing evidence from both airborne platforms and bottom-up emission inventories suggests that rising SO2 emissions in South Asia are rapidly overtaking the decreasing SO2 emissions in East Asia.10–13 While air mass trajectory analysis has allowed for demarcating well-defined source regions affecting the HTP around the year (i.e., periods of transport from East vs. South Asia to the HTP),14–16 the dominant source(s) and origin of sulfate in such atmospheric transport regimes to the HTP remain unclear due to a paucity of observations6 as well as the inherent uncertainties associated with both these measurement techniques.10,13
The origin of sulfate can be linked to various sources.6 Sulfate from fossil fuel-based emissions, biomass burning-based emissions, and terrigenous and mineral-dust loadings are all likely contributors to the sulfate burden on the HTP.3,4,15–18 Along with this, sulfate sourced from the descendance of stratospheric air masses (potentially of mixed anthropogenic and natural including volcanic origin) is an additional possibility, as the HTP is a hotspot for stratospheric intrusions.19–21 In particular, the springtime period often referred to as ‘pre-monsoon’ (March–June) is when high-altitude stratospheric air masses arrive at the HTP.19–21 This period is also concomitant with pollutants arriving from the densely populated and highly polluted Indo-Gangetic Plain (IGP) in South Asia.15–18,22,23 The transitional changes in the atmospheric boundary layer over the IGP between winter and summer months offer a conduit for vertical convective lofting of pollutants to the free troposphere.22,23 The relevance of this ‘up-and-over’ transport pathway has been debated,24 however, growing evidence suggests it to be of significance for the transport of pollutants to the HTP (at the least for the HTP-IGP boundary).25 A lack of synoptic measurements (i.e., observational sites connected by air masses during dominant prevailing meteorology) during vertical transport regimes in the IGP, and S Asia in general, has greatly limited the understanding of sulfate formation/origin in the region. Overall, one or a combination of sources can lead to the distinct springtime sulfate peak in the HTP. Yet, the dominant source/origin of sulfate on the HTP remains debatable,15–18,26 prompting the need for further examination.
Isotope analysis is a powerful tool for fingerprinting the source/origin of a targeted molecule/species.26–28 S-isotope analysis which historically has been limited to 34S/32S, has been extended to 33S/32S and 36S/32S to access ‘mass-independent fractionation’ (MIF) [e.g., Δ33S = ln(δ33S + 1) − 0.515 × ln(δ34S + 1), reported in ‰].29–32Δ33S ≠ 0, i.e., sulfur isotope anomaly, has proven to be a worthy geochemical tracer to decipher the origin of sulfate in both present and paleo-atmospheres.29,33–35 The origin of S-MIF in modern aerosols can be attributed to (i) photochemical processes in the presence of UV radiation: –SO2 absorption at 190–220 nm i.e., photolysis, and SO2 absorption at 250–350 nm i.e., photo-oxidation,36,37 (ii) combustion processes,35 (iii) heterogeneous reactions involving SO2 during the atmospheric processing of mineral dust.33,38 High UV flux is a characteristic of the HTP region during pre-monsoon through summer season owing to factors such as a pristine atmosphere, high altitudes with relatively low latitudes, variation in cloud cover and low columnar ozone amounts.3 While this could play a role in the production of photochemical S-MIF,30,31 it has been noted that the SO2 oxidation to sulfate through photochemical pathways is important and active mainly in the stratosphere, whereas reactions with oxidants such as OH. and O3 and with transition metal ions are dominant processes in the troposphere.32,36 All these tropospheric sulfate formation pathways have been found to be mass-dependent in nature (as shown recently for the HTP region as well).41 This is further supported by the fact that even in the event of an ozone hole e.g., over Antarctica which is an annual occurrence, no S-MIF has been observed in the tropospheric sulfate despite an expected increase in the influx of UV radiation.29 This suggests that S-MIF detected in tropospheric sulfate aerosols could be arising from the other processes mentioned above.33,44 While the S-MIF production mechanisms associated with photochemical processes are well researched and widely accepted,36,37 there is a lack of such understanding for the latter two processes. Nonetheless, the extent of isotope variations observed during combustion26,35 and mineral dust processing33 allows for harnessing the information associated with the S-MIF signal in modern aerosols. This, in combination with process-oriented chemical tracers, elemental ratios, and air mass analysis,39 can improve the feasibility for understanding the origin of sulfate. As such, here, the S-isotope anomaly is used for tracing the origin of the springtime sulfate sampled near the Mt. Everest region on the southern HTP. This scientific underpinning will aid in improving knowledge of source(s) contributing to the sulfate aerosol burden on the HTP and consequently towards model-based assessment of the sulfate-associated impacts for air quality, food, as well as water security in the climatically vulnerable ‘third pole’ and downwind regions of Asia.
Results
Air mass back trajectory and cluster analysis support that the notion that majority of air masses arriving at the southern HTP during spring were largely from the North Indian region and beyond, virtue of the up-and-over transport phenomenon14–18,22,23,40,41 (ESI Fig. S1†). The convective ‘pump’ during this season is rather reportedly centred around North India as well,42 with Delhi here being the most populous city and noted for its notorious air pollution record in the world.43 Given the sparse nature of aerosol Δ33S records in South Asia (to the best of our knowledge only one study, ref. 26 until 2021), we employed a synoptic sampling approach (samples collected in overlapping periods at different sites with a transport continuum existing during the period of collection)27,39 providing an opportunity to directly compare the S-isotope imprints between two meteorologically connected locations during springtime (also referred to as ‘pre-monsoon’ season).17,18,41 Aerosol sulfate concentrations and multi-S-isotope measurements were therefore conducted with high-volume sampling at the southern HTP-Mt. Everest observation site, QOMS (see Methods and Note S1;†Fig. 1) and, during overlapping periods (March–June of 2021), in the IGP megacity Delhi.33 Such synoptic comparisons have been made for carbonaceous aerosols in wintertime South Asia previously and proven immensely beneficial for decoupling of both source information as well as atmospheric processes.27,39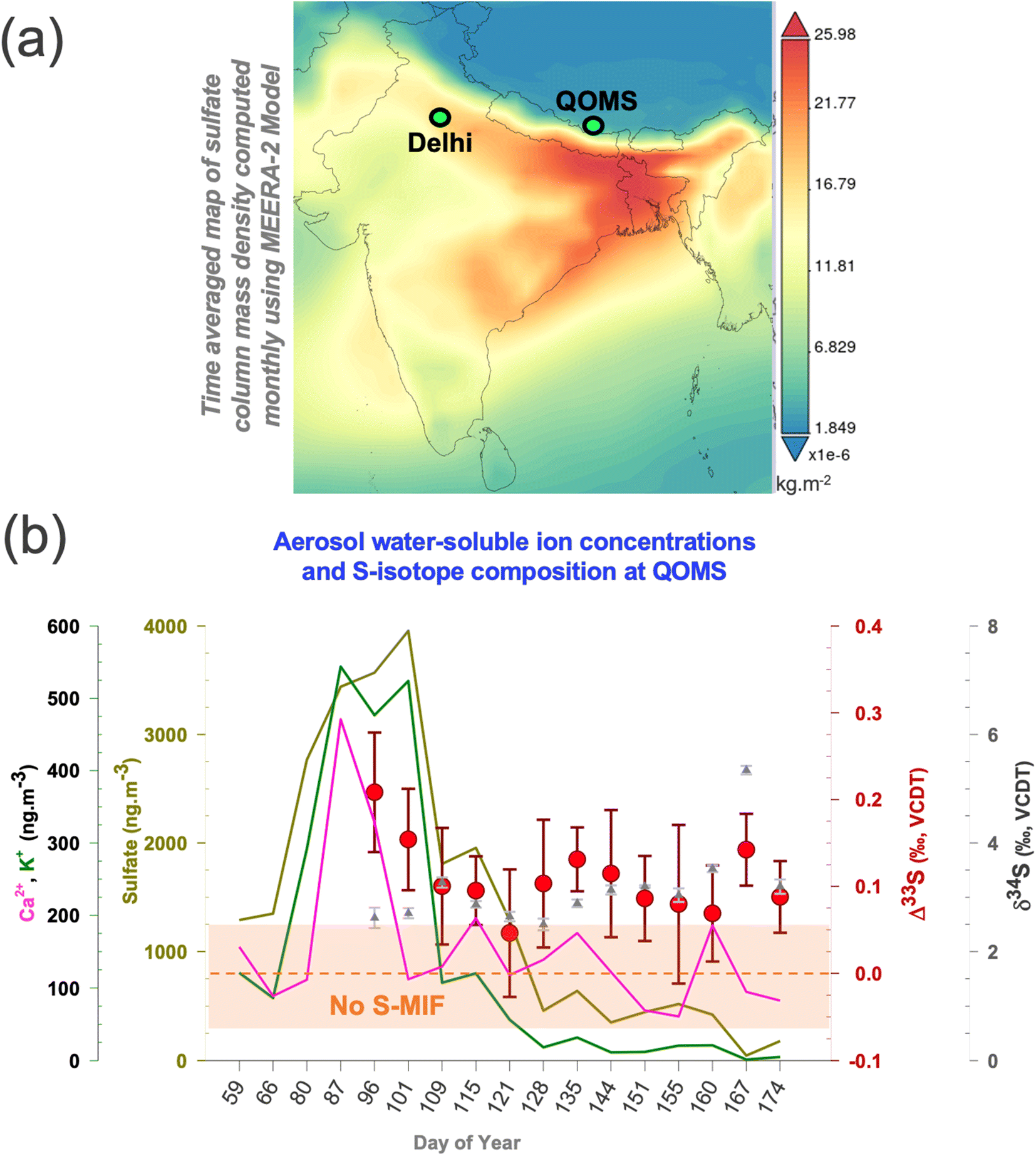 | ||
| Fig. 1 (a) Map showing the modelled sulfate column density with the sites marked wherein a synoptic transport continuum27,39 exists during spring (see also ESI Fig. S1†). The data are obtained from giovanni.gsfc.nasa.gov/giovanni/ (https://giovanni.gsfc.nasa.gov/giovanni/). MERRA-2 tavgM_2d_aer_Nx: 2d, monthly mean, time-averaged, single-level, assimilation, aerosol diagnostics V5.12.4 (ref. 63). (b) Temporal changes in the springtime sulfate, calcium and potassium concentrations (solid lines), δ34S (triangles), and Δ33S (circles) for the samples collected at the QOMS site are shown. The same can be found in our previous publication for the Delhi site.33 The highlighted portion (light brown) shows the analytical uncertainty range (1σ) wherein S-MIF i.e., Sulfur Mass Independent Fractionation signal is not considered as ‘real’. | ||
Considerable variability was found in the sulfate concentrations and the corresponding isotope signals during the investigated period (Fig. 1). The total sulfate concentrations, δ34S, and Δ33S values at QOMS varied from 50 to 4000 ng m−3, from 2.5 ± 0.11‰ to 5.3 ± 0.08‰, and from +0.05 ± 0.07‰ to +0.21 ± 0.07‰, respectively (see Table S1†). A characteristic sulfate peak is observed for the Mar–Apr months in agreement with previous findings.6,17 A change in airborne sulfate concentrations can in fact occur from resuspended dust i.e., terrigenous sulfate.43,44 The HTP is affected by long-range transported dust,4 which could be resuspended along with soil particles due to the dominant wind patterns2–4 leading to a ‘locally-sourced’ springtime sulfate peak. However, the terrigenous sulfate fraction was found to only contribute on average 3% to the total sulfate fraction with the proceeding of the springtime (ESI Fig. S5†). Therefore, in line with previous studies,17 we corroborate that the characteristic springtime sulfate peak at QOMS is not a local phenomenon but more likely related to transported sulfate due to the prevailing meteorology. As such, the concomitant changes in the S-isotopes are likely also related to this transported sulfate.
In comparison, the multi-S isotope compositions from Delhi ranged from 0.70 ± 0.05 to 4.70 ± 0.02‰ for δ34S and from 0.20 ± 0.10 to 0.50 ± 0.08‰ for Δ33S for the corresponding period.33 The contribution of terrigenous sulfate fraction to total sulfate fraction at the IGP site varied from as much as 25% in April to as low as 3% in May of 2021.33 Unlike the IGP counterpart,33 the dual S-isotope characteristics on the southern HTP do not show any shift with the proceeding of the springtime and appear to be rather limited to a narrow δ34S range only relative to other atmospheric sulfate aerosol S-isotope records (ESI Fig. S2†). The observations here point to a potentially distinct origin of the isotopically elevated sulfate on the southern HTP during springtime, an aspect that hasn't yet been reported or explored.
Discussion
Spatially heterogeneous anomalous S-isotope records
Subtle differences were found when comparing the Δ33S records from the HTP and the surrounding region in immediate vicinity. First, the S-isotope anomalies in ambient aerosols in the southern HTP are comparable to but marginally higher than those previously reported from the central HTP26 and downwind Mt. Wuyi35 (which are likely sourced from subsidence of stratospheric air) (Fig. 2).26,35 However, they are found to be higher than in the central HTP sediment core record which did not show S-MIF in the Nam Co Lake deposits26 from the last decades (red diamonds in Fig. 2). Second, despite prevailing synoptic transport, springtime Δ33S values were restricted to < 0.2‰ at QOMS/southern HTP (average: +0.1 ± 0.05‰) but > 0.2‰ at Delhi/IGP (average: +0.3 ± 0.1‰).33 This is in contrast to previous studies having shown similar isotope imprint and dynamics in pollutants transported between the IGP and high-altitude sites in North India, including the southern HTP.25,40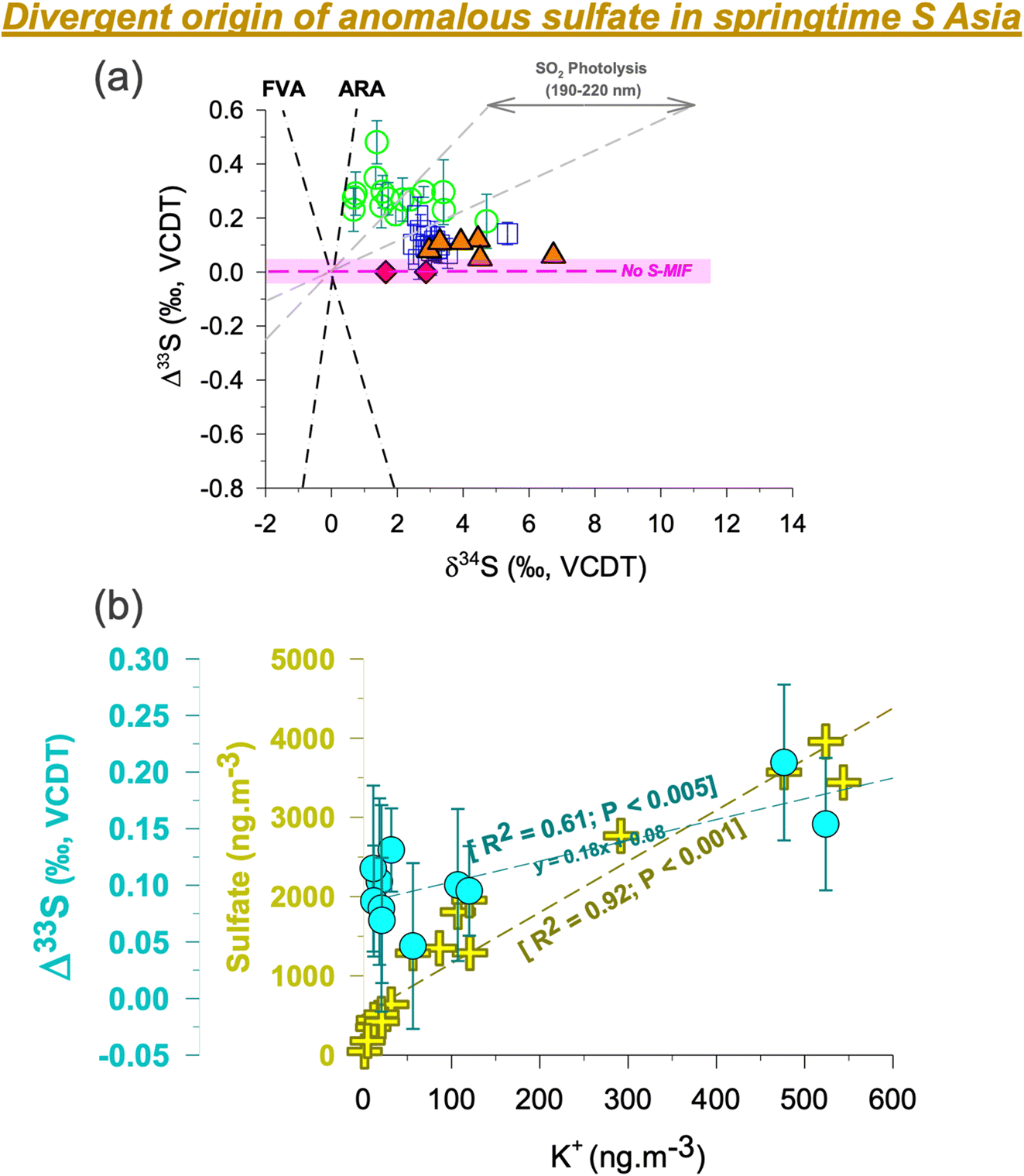 | ||
| Fig. 2 (a) Triple s-isotope characteristics of sulfate aerosols in S Asia are shown from three different sites [Delhi33 – green circles; central HTP/NAM co26 – orange triangles; southern HTP/QOMS (this study) – blue squares]. The dataset for the central HTP includes previously published aerosol samples (orange triangles) and sediment core samples (red diamonds, shown here for the year 2000 and onwards).26 The geometry of the isotopic signatures in modern aerosols is different from either the ARA representing the Archean Reference Array based on the data from Neoarchean and Paleoarchean rocks in Australia and Africa, or the FVA representing the felsic volcanic array.63,64 The grey field represents experimental data from SO2 photolysis in the range of 190–220 nm (xenon arc lamp).30,36 (b) The correlations of the S-isotope anomaly and sulfate concentrations with the potassium concentrations at the QOMS site are shown. The two samples with negligible K+ concentrations <5 ng m−3 are not included. Even when considering these samples there is still a statistically significant dependence between Δ33S and K+. For ancillary correlations of other water-soluble ions, please refer to the ESI (Fig. S5).† The highlighted portion (light pink) shows the analytical uncertainty range (1σ) wherein S-MIF i.e., Sulfur Mass Independent Fractionation signal is not considered as ‘real’. | ||
The current view on the origin of MIF-carrying sulfate aerosols at high-altitude sites is that they are sourced from stratospheric intrusions.19–21,26,35 Studies using the cosmogenic radionuclide, 35S, have demonstrated a strong springtime stratosphere-to-troposphere transport at the Himalayas and beyond.19,20 The HTP is more prone to stratospheric air intrusion than other regions in the world.19–21 As such, subsidence of stratospheric air is likely to bring sulfate aerosols from higher altitudes to the HTP. While the S-MIF is much larger in the stratosphere, only a fraction of that is witnessed in the planetary boundary layer.37 Even 1 to 2% of stratospheric air intercepted in the boundary layer as constrained previously could indeed account for the sulfur isotope anomaly observed at the southern HTP site during spring. This could be applicable to the datapoint on day 167 in Fig. 1. However, multiyear record of radiogenic tracer 35S suggests otherwise with no spike witnessed in the 35S concentrations for such months (June-July).19,20 Furthermore, a recent study from the southern HTP on the isotope constraints on sulfate using anomalous oxygen (Δ17O) suggested a relatively lower influence of stratospheric air during spring.41 This can also be seen in the satellite-derived cloud properties (ESI Fig. S3†). The low cloud optical thickness and high cloud pressure on the southern HTP relative to the central and northern HTP imply a limited influence of stratospheric intrusions during spring at the QOMS site. As such, it is here ascertained that the S-MIF values reported at QOMS in this study are not likely to be of photochemical origin, as also reported elsewhere with regard to tropospheric sulfate.29 Rather, it is likely that other competing transport/formation processes lead to MIF behaviour in S isotopes on the southern HTP.
S-MIF could be occurring in heterogeneous reactions of SO2 with mineral dust.38,44 Our recent work has shown that one of the main reasons for the MIF behaviour in sulfate in urban aerosols could be pinned to the atmospheric processing of mineral dust.33 It can be expected that anomalous sulfate formed in this manner at Delhi should have been transported to higher altitudes (i.e., the QOMS site) during the corresponding springtime periods of deep convection and air mass transport (see ESI Fig. S1†), and intercepted on the southern HTP.22,23,25,41,42 However, satellite-based observations reveal lower influx of dust at QOMS compared to Delhi during spring (ESI Fig. S4†). This is also noticed from a poor association of SO42− and Ca2+ (a tracer for dust; R2 = 0.33, ESI Fig. S5†) at QOMS, together suggesting that MIF bearing sulfate (and sulfate in general) on this high-altitude southern HTP region is not modulated by mineral dust as in the central HTP,26 indicative of spatial heterogeneity in the origin(s) of sulfate on the HTP. A recent study conducted at QOMS41 found significant correlations between SO42− and Ca2+, however, the difference being that total suspended particles (TSP) were collected in that study. Influx of coarse mode mineral dust at the barren QOMS site and the ensuing sulfate production through heterogeneous reactions on the surface of such long-range transported dust are well known.4,33 It is therefore not surprising that such a correlation is viable for the TSP size fraction. In the present scenario, PM2.5 samples were only collected i.e., aerosols with aerodynamic size <2.5 μm or fine mode aerosols. As such, we do not find such a correlation with dust aerosols (ESI Fig. S5†). It is also possible that there were fewer dust events in 2021 than in 2015 when the Wang et al.40 study was conducted at QOMS (see also Fig. S4†). Coupling potential source markers with isotope signals might aid in understanding the dominant source(s) of springtime sulfate on the HTP.
Deciphering putative sources of anomalous sulfate on the southern HTP
There is a conspicuous difference in the triple-S-isotope geometry of signals observed at QOMS and its surrounding region (Fig. 2). With stratospheric intrusions and heterogeneous reactions on mineral dust being less likely as possible contributors of anomalous sulfate sampled at QOMS (discussed above), a third putative possibility i.e., ‘combustion-derived’ anomalous sulfur exists as a potential source. Given the anthropogenically dominated aerosol regime in S Asia,13,27 both fossil fuel and biomass-based combustion processes could indeed be linked to sulfate on this HTP site.Coal combustion was initially perceived as a source of anomalous sulfur.45 However, recent laboratory-scale investigation has found no evidence of S-MIF in representative coal samples from different coal mines from China putting an end to the coal hypothesis.35 We note that other fossil fuels tested in this regard had also not shown any evidence of S-MIF.46 As such, fossil fuel combustion can be omitted as a potential source, leaving biomass/biofuel burning as a plausible contender for the observed S-isotope signal at QOMS. However, there is a degree of ambiguity linked with the interpretation of the biomass burning-derived anomalous sulfur signals. A chamber-based study in the past, which included the use of savannah grass, rice straw and hay as ‘representative’ samples, did not find any evidence of S-MIF.46 We stress that a large variability in isotope signatures has been observed when measured systematically across various stages of biomass combustion,47,48 which were not fully tested for all samples in this study (ref. 46). Also, combustion materials such as dung cakes, fuelwood, wood pallets, and agricultural waste (such as bagasse), commonly burnt in S Asia,13,27 remain yet to be tested for their respective S-isotopic signatures and any related MIF occurrences. More recently, Δ33S values as low as −0.66‰ have been observed during winter in the heavy pollution events in Beijing and speculated to be arising from residential biofuel usage.45 Based on modelling and isotope mass-balance approaches a first-order biomass-burning end member of Δ33S was estimated to be −0.67 ± 0.20‰,26 however this value remains poorly constrained. Furthermore, other S-containing reservoirs have been suggested as a source of such negative Δ33S anomalies, thereby refuting the claim for them to be associated with biomass/biofuel combustion.49 Given the lack of any existing solid physico-chemical evidence for S-MIF from biomass-based combustion processes, the magnitude and even ‘sign’ of isotope anomalies associated with such processes remain currently unclear.26,35,45,46,49 Coupling isotope anomalies with appropriate chemical marker(s) can be attempted to better link source(s) to the origin of S-MIF.35,50
Linking chemical markers and S-isotope fingerprints
K+ is often used as a chemical marker for biomass burning and waste incineration, emitted mainly in the form of KCl, K2SO4 and KNO3, although other origins are possible.51–56 A pertinent issue using this chemical marker, in the context of studying the source of S-MIF, has been the ‘over association’ of K+ with several source origins.50 Particularly, in a city environment the source origins of K+ have been known to be much more complicated.56 We emphasize that the inability to demarcate a clear source origin of K+ could have hindered establishing a clear correlation between Δ33S and K+ in the past,50 among other aspects such as multiple origins of S-MIF in a city environment.33,44Being at a pristine high elevation site (QOMS), in this study we have a favourable opportunity where such issues are less likely to influence the findings. We try to categorically understand the associations and disassociations involving K+: first, we find a poor correlation between K+ and Ca2+ (ESI Fig. S5†) suggesting that it is less likely to be of mineral dust origin. Likewise, we also find a poor association between SO42− and Ca2+ implying limited influence from mineral dust for sulfate sampled at QOMS (see discussion above, ESI Fig. S5†). Second, we find a strong correlation between SO42− and K+ (R2 = 0.92; Fig. 2) pointing to the commonality of source origins. Such a correlation at QOMS during pre-monsoon has been reported in the past17 as well as in a recent study when the ‘non-dust’ sulfate fraction was apportioned.41 This combined with the observed strong association between K+ and Cl− (R2 = 0.92; ESI Fig. S5†) further suggests that the transported sulfate at QOMS during spring of 2021 is more likely linked to biomass burning origin. It should be noted that the ratios of non-sea-salt (nss)-SO42− to total SO42− and nss-K+ to total K+ were 99 ± 1% and 99 ± 4%, respectively (ESI Fig. S6†), suggest a peripheral contribution from marine sources. Taken together, this supports the observation that K+ at QOMS during spring 2021 was largely related to biomass burning. The influx of biomass burning aerosols during springtime is corroborated also by previous studies from the southern HTP using a wide array of chemical, optical, remote sensing and aerosol mass spectrometry modules.15–18
Indeed, widespread open burning in S Asia during the spring of 2021 is revealed by satellite imagery (ESI Fig. S7†). During this period extensive pre-monsoon crop-residue burning and forest fires happen to be common events in the IGP.15,51 Additionally, garbage burning is also widely employed as a cheap measure for waste disposal in the region due to socio-economic constraints.52 These activities together represent the biomass-combustion pool of sources that could likely be associated with positive MIF-carrying sulfate. Interestingly, a positive correlation was in fact established also between Δ33S and K+ (Fig. 2), further suggesting that anomalous sulfur at the high-altitude site is likely sourced from such biomass-combustion activities. A narrow range in δ34S (as seen in Fig. 2) further corroborates this fact that a single source (i.e., biomass burning) is indeed linked to the springtime sulfate at QOMS. More studies from other regions are warranted to better understand the magnitude of S-MIF from such combustion processes.
Delhi being surrounded by such activities should have also displayed a positive correlation between Δ33S and K+ as observed at QOMS. However, we note there was only a merely significant correlation at Delhi (see ESI† of Dasari et al., 2022). One plausible reason is the role of dual S-MIF generating processes: influx of open burning aerosols and mineral-dust processed aerosols. We find that the latter processes displayed significantly larger S-MIF and as such the abundance of mineral dust was a key factor for not having found a strong correlation between MIF and K+ at Delhi. At QOMS, these dual processes are not acting together. Rather, abundance of dust is found to be lower by an order of magnitude (ESI Fig. S4†) and as such the correlation between Δ33S and K+ becomes evident.
In the past decade several studies have employed multiple stable S-isotopes for source fingerprinting.35,44,45,50 While a clear correlation between Δ36S and biomass burning tracer K+ has never been found, apparent relationships for the same with other tracers such as levoglucosan and mannosan have been reported.50 However, such correlations with Δ33S have not been found at all leading to the notion that Δ33S is less influenced by biomass burning processes than Δ36S. There is a possibility that such an observation might in fact have been related to the aspect of an unresolved ‘mixed’ K+ source signal. The detection and measurement of 36S isotopes remain limited to only one of the currently employed three methods for S-isotope analysis involving spectrometric (involving e.g., the IRMS and MC-ICP-MS) and spectroscopic (involving e.g., the cavity ring-down spectroscopy, CRDS) techniques, which makes S-isotope based source fingerprinting challenging.33,37,58 There is thus a need to identify whether other stable S-isotopes can be employed as source tracers for biomass burning.
In fact, the findings in the present work address this knowledge gap suggesting that Δ33S can indeed be used for this purpose in pristine locations (where influence from aspects such as stratospheric intrusions and other combustion sources is peripheral). Contrary to the long-held notion, we emphasize that it is plausible that there are yet unknown mechanisms in combustion processes that lead to anomalous sulfur production in modern aerosols generating Δ33S. While recombination reactions and symmetry effects involving elemental sulfur have been argued as likely mechanisms to produce MIF in combustion,26,57,58 unambiguous ‘proof-of-concept’ probing remains at large with the reported values in the literature26,35 yet to be quantitatively validated by laboratory experiments. The elucidation of the physico-chemical aspect of elementary reactions, involving a wide array of representative materials for both garbage and biofuel, is much needed for delineating the associated S-MIF magnitudes. Despite these shortcomings, our observations are supportive of the fact that different processes generating S-MIF in urban and remote sites have distinct isotopic imprints which can be decoupled.26,33,35,44,45
Competing processes and implications for the sulfate burden on the HTP
The debate on the importance of stratospheric intrusions vs. biomass burning as potential pathways for the pollutant (e.g., ozone) transport to the HTP has been ongoing.21,59–61 It is possible that spatial variability in the extent of influence of stratospheric intrusions21,41 is a likely factor leading to the observed significant association of S-MIF with biomass burning. This along with reports on high spatial variability of biomass burning aerosols on the HTP62 further corroborates the findings here.Biomass burning emissions (from e.g., crop-residue and garbage burning, wildfires) have largely been neglected in global chemistry-climate model-based assessments of rising sulfate levels in S Asia.7,8 As such, future predictions of sulfate induced burden on air quality, food security and climate remain largely uncertain for the region including the HTP. The elucidation of stratosphere–troposphere exchange processes (which remains complicated in the vicinity of Mt. Everest) and inclusion of such biomass burning processes are therefore much needed in models, to better understand the consequences on the sulfate aerosol burden on the HTP in turn affecting the chemical and radiation budgets and the multitude of effects on the cryosphere-hydrosphere and the biosphere of this climatically vulnerable region.
Materials and methods
Aerosol PM2.5 samples were collected using a high-volume aerosol sampler, operated at 1 m3 min−1 with a sampling duration of 96 h. Sample collection was made between March and June 2021 at the QOMS site which has a history of long-term sampling campaigns and measurements.15–17 More details of the site and instrumentation can be found elsewhere.15–17 Based on the meteorological parameters, mainly precipitation, it has often been a convention to characterize four seasons at the Mt.Everest-QOMS region e.g.:17,18,41 pre-monsoon (March to June), followed by monsoon (July to Aug), post-monsoon (September to November) and winter (December to February) season, respectively, with the demarcation between the end of the pre-monsoon and beginning of the monsoon season varying annually. For the sake of convenience, we consider the samples collected at QOMS during the 2021 field campaign (see Table S1†) to be representative of the pre-monsoon season, here referred to as ‘springtime’ throughout the manuscript. Sampling information for the IGP site has been well documented in our recent publication.33The concentrations of water-soluble ions and metals, as well as the multiple S-isotope compositions, were measured using an ion chromatograph Metrohm IC (Professional 850), a triple quadrupole, a multi-collector Inductively coupled plasma mass spectrometer [QQQ-ICP-MS (Agilent 8900), and a MC-ICP-MS (Thermo Fisher Scientific Neptune Plus)] instrument, respectively. Measurements were carried out at the Centre de Recherches Pétrographiques et Géochimiques, CRPG (for S-isotopes) in France, and the Nanjing University of Information Science and Technology, NUIST, China for water-soluble ions and Université du Québec à Montréal, UQAM Canada for metals, respectively. Further details on analysis procedures, quality control and the dataset are provided in the ESI (ESI Note S1 and Table S1, respectively).†
Data availability
All data are included in the manuscript and/or ESI.†Author contributions
S. D. conceived the study; S. D. and D. W. designed research; Z. C. led the fieldwork; S. D., Q. P., and G. P. performed laboratory analysis; S. D. performed data analysis and wrote the paper with input from all co-authors.Conflicts of interest
The authors declare no competing interest.Acknowledgements
This work was supported by the French National CNRS-INSU program LEFE (Les Enveloppes Fluides et l'Environnement; grant awarded to G.P). D. W. and Z. C. acknowledge support from the Université du Québec à Montréal and the Chinese Academy of Sciences, respectively. The authors gratefully acknowledge the NOAA Air Resources Laboratory (ARL) for the provision of the HYSPLIT transport and dispersion model and/or READY website (https://www.ready.noaa.gov) used in this publication. We also acknowledge the use of data and/or imagery from NASA's Fire Information for Resource Management System (FIRMS) (https://earthdata.nasa.gov/firms), part of NASA's Earth Observing System Data and Information System (EOSDIS). Analyses and visualizations used in this study were produced with the Giovanni online data system, developed and maintained by the NASA GES DISC.References
- G. Wu, A. Duan, Y. Liu, J. Mao, R. Ren, Q. Bao, B. He, B. Liu and W. Hu, Natl. Sci. Rev., 2015, 2, 100–116 CrossRef.
- C. Zhao, Y. Yang, H. Fan, J. Huang, Y. Fu, X. Zhang, S. Kang, Z. Cong, H. Letu and M. Menenti, Natl. Sci. Rev., 2020, 7, 492–495 CrossRef CAS PubMed.
- A. Dahlback, N. Gelsor, J. J. Stamnes and Y. Gjessing, J. Geophys. Res.: Atmos., 2007, 112, D09308 CrossRef.
- D. Zhao, S. Chen, Y. Chen, Y. Gong, G. Lou, S. Cheng and H. Bi, Front. Environ. Sci., 2022, 10, 563 Search PubMed.
- C. Li, F. Yan, S. Kang, C. Yan, Z. Hu, P. Chen, S. Gao, C. Zhang, C. He, S. Kaspari and A. Stubbins, Envi. Int., 2021, 146, 106281 CrossRef CAS PubMed.
- Q. Pei, E. Saikawa, S. Kaspari, D. Widory, C. Zhao, G. Wu, M. Loewen, X. Wan, S. Kang, X. Wang and Y. L. Zhang, Earth-Sci. Rev., 2021, 220, 103753 CrossRef CAS.
- S. Fadnavis, R. Müller, G. Kalita, M. Rowlinson, A. Rap, J. L. F. Li, B. Gasparini and A. Laakso, Atmos. Chem. Phys., 2019, 19, 9989–10008 CrossRef CAS.
- S. Fadnavis, T. P. Sabin, C. Roy, M. Rowlinson, A. Rap, J. P. Vernier and C. E. Sioris, Sci. Rep., 2019, 9, 1–11 CrossRef CAS PubMed.
- Global SO2 Emission Hotspots Database: Ranking the World'S 309 Worst Sources of SO2 Pollution, http://www.greenpeace.org/static/planet4-india-stateless/2019/08/204126d3-global-hotspot-and-emission-sources-for-so2_19th_august-2019.pdf, accessed May 2023 Search PubMed.
- Z. Qu, D. K. Henze, C. Li, N. Theys, Y. Wang, J. Wang, W. Wang, J. Han, C. Shim, R. R. Dickerson and X. Ren, J. Geophys. Res.: Atmos., 2019, 124, 8336–8359 CrossRef CAS PubMed.
- C. Li, C. McLinden, V. Fioletov, N. Krotkov, S. Carn, J. Joiner, D. Streets, H. He, X. Ren, Z. Li and R. R. Dickerson, Sci. Rep., 2017, 7, 1–7 CrossRef PubMed.
- J. Kurokawa and T. Ohara, Atmos. Chem. Phys., 2020, 20, 12761–12793 CrossRef CAS.
- C. Venkataraman, M. Brauer, K. Tibrewal, P. Sadavarte, Q. Ma, A. Cohen, S. Chaliyakunnel, J. Frostad, Z. Klimont, R. V. Martin and D. B. Millet, Atmos. Chem. Phys., 2018, 18, 8017–8039 CrossRef CAS PubMed.
- J. Zhang, X. Wu, S. Liu, Z. Bai, X. Xia, B. Chen, X. Zong and J. Bian, Environ. Res. Lett., 2019, 14, 124068 CrossRef CAS.
- X. Zhang, J. Xu, S. Kang, Y. Liu and Q. Zhang, Atmos. Chem. Phys., 2018, 18, 4617–4638 CrossRef CAS.
- J. Xu, A. P. S. Hettiyadura, Y. Liu, X. Zhang, S. Kang and A. Laskin, J. Geophys. Res.: Atmos., 2020, 125, 2019JD031226 Search PubMed.
- Z. Cong, S. Kang, K. Kawamura, B. Liu, X. Wan, Z. Wang and P. Fu, Atmos. Chem. Phys., 2015, 15, 1573–1584 CrossRef CAS.
- M. Pokharel, J. Guang, B. Liu, S. Kang, Y. Ma, B. N. Holben, X. A. Xia, J. Xin, K. Ram, D. Rupakheti and X. Wan, J. Geophys. Res.: Atmos., 2019, 124, 13357–13374 CrossRef CAS.
- A. Priyadarshi, J. Hill-Falkenthal, M. Thiemens, Z. Zhang, M. Lin, C. Y. Chan and S. Kang, J. Geophys. Res.: Atmos., 2014, 119, 4125–4135 CrossRef CAS.
- M. Lin, Z. Zhang, L. Su, J. Hill-Falkenthal, A. Priyadarshi, Q. Zhang and M. H. Thiemens, J. Geophys. Res.: Atmos., 2016, 121, 439–456 CrossRef CAS.
- M. Lin, K. Wang, S. Kang, Y. Li, Z. Fan and M. H. Thiemens, Sci. Bull., 2021, 66, 323–326 CrossRef CAS PubMed.
- V. S. Nair, S. S. Babu, M. M. Gogoi and K. K. Moorthy, Geophys. Res. Lett., 2016, 43, 11–453 CrossRef.
- M. M. Gogoi, N. B. Lakshmi, V. S. Nair, S. K. Kompalli, K. K. Moorthy and S. S. Babu, J. Earth Syst. Sci., 2019, 128, 1–22 CrossRef CAS.
- Q. Li, N. Wang, C. Barbante, S. Kang, P. Yao, X. Wan, E. Barbaro, M. D. C. V. Hidalgo, A. Gambaro, C. Li and H. Niu, Sci. Total Environ., 2018, 612, 1340–1347 CrossRef CAS PubMed.
- J. Liu, M. Chang, Z. Cheng, S. Zhu, P. Ding, F. Liu and G. Zhang, Environ. Sci. Technol. Lett., 2021, 8, 1026–1031 CrossRef CAS.
- M. Lin, S. Kang, R. Shaheen, C. Li, S. C. Hsu and M. H. Thiemens, Proc. Natl. Acad. Sci., 2018, 115, 6964–6969 CrossRef CAS PubMed.
- S. Dasari, A. Andersson, A. Stohl, N. Evangeliou, S. Bikkina, H. Holmstrand, K. Budhavant, A. Salam and O. Gustafsson, Environ. Sci. Technol., 2020, 54, 11771–11779 CrossRef CAS PubMed.
- S. Dasari, A. Andersson, M. E. Popa, T. Rockmann, H. Holmstrand, K. Budhavant and O. Gustafsson, Environ. Sci. Technol., 2021, 56, 165–174 CrossRef PubMed.
- S. Ishino, S. Hattori, J. Savarino, M. Legrand, E. Albalat, F. Albarede, S. Preunkert, B. Jourdain and N. Yoshida, Sci. Rep., 2019, 9, 12378 CrossRef PubMed.
- S. Ono, B. Wing, D. Johnston, J. Farquhar and D. Rumble, Geochim. Cosmochim. Acta, 2006, 70, 2238–2252 CrossRef CAS.
- S. Ono, Annu. Rev. Earth Planet. Sci., 2017, 45, 301–329 CrossRef CAS.
- S. Dasari, G. Paris, B. Saar, Q. Pei, Z. Cong and D. Widory, Environ. Sci. Technol. Lett., 2022, 9, 604–610 CrossRef CAS.
- S. Dasari, G. Paris, J. Charreau and J. Savarino, Proc. Natl. Acad. Sci. U. S. A., 2022, 1, 170 Search PubMed.
- M. Lin, X. Zhang, M. Li, Y. Xu, Z. Zhang, J. Tao, B. Su, L. Liu, Y. Shen and M. H. Thiemens, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 8541–8546 CrossRef CAS PubMed.
- A. R. Whitehill, B. Jiang, H. Guo and S. Ono, Atmos. Chem. Phys., 2015, 15, 1843–1864 CrossRef.
- S. Hattori, J. A. Schmidt, M. S. Johnson, S. O. Danielache, A. Yamada, Y. Ueno and N. Yoshida, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17656–17661 CrossRef CAS PubMed.
- A. C. Lasaga, T. Otake, Y. Watanabe and H. Ohmoto, Earth Planet. Sci. Lett., 2008, 268, 225–238 CrossRef CAS.
- S. Dasari, A. Andersson, S. Bikkina, H. Holmstrand, K. Budhavant, S. Satheesh, E. Asmi, J. Kesti, J. Backman, A. Salam and D. S. Bisht, Sci. Adv., 2019, 5, eaau8066 CrossRef PubMed.
- P. Chen, S. Kang, C. Li, L. Tripathee, M. Rai, T. Pu, X. Yin and Ö. Gustafsson, Geosci. Front., 2022, 14, 101516 CrossRef.
- K. Wang, S. Hattori, M. Lin, S. Ishino, B. Alexander, K. Kamezaki, N. Yoshida and S. Kang, Atmos. Chem. Phys., 2021, 21, 8357–8376 CrossRef CAS.
- J. P. Vernier, T. D. Fairlie, M. Natarajan, F. G. Wienhold, J. Bian, B. G. Martinsson, S. Crumeyrolle, L. W. Thomason and K. M. Bedka, J. Geophys. Res.: Atmos., 2015, 120, 1608–1619 CrossRef CAS PubMed.
- S. M. Hama, P. Kumar, R. M. Harrison, W. J. Bloss, M. Khare, S. Mishra, A. Namdeo, R. Sokhi, P. Goodman and C. Sharma, Sustain. Cities Soc., 2020, 54, 102003 CrossRef.
- D. Au Yang, P. Cartigny, K. Desboeufs and D. Widory, Atmos. Chem. Phys., 2019, 19, 3779–3796 CrossRef.
- X. Han, Q. Guo, H. Strauss, C. Liu, J. Hu, Z. Guo, R. Wei, M. Peters, L. Tian and J. Kong, Environ. Sci. Technol., 2017, 51, 7794–7803 CrossRef CAS PubMed.
- C. C. W. Lee, J. Savarino, H. Cachier and M. H. Thiemens, Tellus, 2002, 54, 193–200 Search PubMed.
- S. Kato, H. Akimoto, T. Röckmann, M. Bräunlich and C. A. M. Brenninkmeijer, Atmos. Environ., 1999, 33, 4357–4362 CrossRef CAS.
- C. Qiu, The Isotopic Signatures of CO in Biomass Burning Emission, Institute of Marine and Atmospheric Research Utrecht, Utrecht University, 2019 Search PubMed.
- I. Genot, D. Au Yang, E. Martin, P. Cartigny, E. Legendre and M. De Rafelis, Atmos. Chem. Phys., 2020, 20, 4255–4273 CrossRef CAS.
- B. Yin, X. Yu, X. Lin, Z. Zhang, Y. Zhang and M. Lin, ACS Earth Space Chem., 2023, 7, 800–811 CrossRef CAS.
- R. Kumar, M. Naja, S. K. Satheesh, N. Ojha, H. Joshi, T. Sarangi, P. Pant, U. C. Dumka, P. Hegde and S. Venkataramani, J. Geophys. Res.: Atmos., 2011, 116, D19302 CrossRef.
- E. Saikawa, Q. Wu, M. Zhong, A. Avramov, K. Ram, E. A. Stone, C. E. Stockwell, T. Jayarathne, A. K. Panday and R. J. Yokelson, Environ. Sci. Technol., 2020, 54, 9928–9938 CrossRef CAS PubMed.
- M. O. Andreae, Sci, 1983, 220, 1148–1151 CrossRef CAS PubMed.
- B. Vincenti, E. Paris, M. Carnevale, A. Palma, E. Guerriero, D. Borello, V. Paolini and F. Gallucci, Int. J. Environ. Res. Public Health, 2022, 19, 4387 CrossRef CAS PubMed.
- C. Li, S. Kang, Q. Zhang and S. Kaspari, Atmos. Res., 2007, 85, 351–360 CrossRef CAS.
- J. Yu, C. Yan, Y. Liu, X. Li, T. Zhou and M. Zheng, Aerosol Air Qual. Res., 2018, 18, 2447–2459 CrossRef CAS.
- D. Babikov, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 3062–3067 CrossRef CAS PubMed.
- J. Chaillot, S. Dasari, H. Fleurbaey, M. Daeron, J. Savarino and S. Kassi, Envi. Sci. Adv., 2023, 2, 78–86 RSC.
- J. Yang, K. Wang, M. Lin, X. Yin and S. Kang, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2211002119 CrossRef CAS PubMed.
- J. Peischl, I. Bourgeois, S. S. Brown and J. A. Neuman, Proc. Natl. Acad. Sci. U. S. A., 2022, 119, e2212326119 CrossRef CAS PubMed.
- I. Bourgeois, J. Peischl, J. A. Neuman, S. S. Brown, C. R. Thompson, K. C. Aikin, H. M. Allen, H. Angot, E. C. Apel, C. B. Baublitz and J. F. Brewer, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2109628118 CrossRef CAS PubMed.
- C. S. Zhu, Y. Qu, Z. S. Zhang, T. Zhang, W. T. Dai and J. J. Cao, Earth's Fut., 2022, 10, e2021EF002214 CAS.
- Goddard Earth Sciences Data and Information Services Center (GES DISC), MERRA-2 tavgM_2d_aer_Nx: 2d,Monthly mean,Time-averaged,Single-Level,Assimilation,Aerosol Diagnostics V5.12.4, Greenbelt, MD, USA, Goddard Earth Sciences Data and Information Services Center (GES DISC), https://disc.gsfc.nasa.gov/datasets/M2TMNXAER_5.12.4/summary, DOI:10.5067/FH9A0MLJPC7N, accessed: 20 May 2023.
- M. A. Mast, J. T. Turk, G. P. Ingersoll, D. W. Clow and C. L. Kester, Atmos. Environ., 2001, 35, 3303–3313 CrossRef CAS.
- P. Philippot, M. Van Zuilen and C. Rollion-Bard, Nat. Geosci., 2012, 5, 668–674 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3va00085k |
| ‡ Present address: Department of Earth Sciences, University of Oxford, Oxford OX1 3AN, UK, E-mail: sanjeev.dasari@earth.ox.ac.uk |
| This journal is © The Royal Society of Chemistry 2023 |