
Tailoring the electronic structure of In2O3/C photocatalysts for enhanced CO2 reduction†
Awu
Zhou
 ab,
Chen
Zhao
b,
Jianchi
Zhou
a,
Yibo
Dou
*a,
Jian-Rong
Li
b and
Min
Wei
a
ab,
Chen
Zhao
b,
Jianchi
Zhou
a,
Yibo
Dou
*a,
Jian-Rong
Li
b and
Min
Wei
a
aState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, Beijing 100029, P. R. China. E-mail: douyb@buct.edu.cn
bBeijing Key Laboratory for Green Catalysis and Separation and Department of Chemistry and Chemical Engineering, Beijing University of Technology, Beijing 100124, P. R. China
First published on 1st March 2023
Abstract
Photoreduction of CO2 into value-added fuels under mild conditions is a promising route to relieve the pressure from extensive CO2 emission and energy consumption, but the rational design of novel photocatalysts for efficient CO2 reduction remains a big challenge. Herein, a series of transition metal doped In2O3/C (M = Fe, Cu, and Zn) photocatalysts are prepared based on a bimetallic metal–organic framework template, where the rearrangement of electron density distribution is facilely achieved via doping metal atoms. Correspondingly, the broadened light-harvesting scope, charge transfer rate, and produced intermediates in photocatalytic CO2 reduction can be optimized. In particular, Cu–In2O3/C exhibits a largely improved CO yield with a high selectivity, which is superior to that of most of the previously reported photocatalysts derived from MOFs. This work thus provides an efficient approach for tailoring the electronic structure of photocatalysts, which shows promising applications in carbon cycling.
10th Anniversary statementTen years of Journal of Materials Chemistry A have seen many fascinating topics on materials for energy and sustainability arise, mature and succeed into practical application. This manuscript is dedicated to celebrating the 10th anniversary of the journal. Considering the challenge of exploring efficient catalysts for photocatalytic reduction of CO2 to valuable chemical feedstocks, we reported a general strategy for synthesizing various transition metal-doped In2O3/C nanotube (M–In2O3/C, M = Fe, Cu, and Zn) photocatalysts. The experimental results and theoretical simulation demonstrate that transition metals can affect photocatalytic activity and selectivity by modifying the doping state and electronic structure. The light-harvesting scope, charge transfer rate, and adsorption/desorption of intermediates are thus optimized. The present work involves a hot topic of functional materials used for solar-to-chemical energy conversion, which is very close to the theme of Journal of Materials Chemistry A. |
1 Introduction
The energy crisis and climate change are regarded as two major issues in the foreseeable future.1–3 In addition to the traditional technologies to achieve energy conservation and emission reduction, renewable energy and related development strategies are highly desirable.4–6 As an efficient way of utilizing carbon dioxide (CO2) to produce various chemical fuels, solar-driven CO2 reduction has gained a lot of attention.7–10 However, the photocatalytic efficiency remains unsatisfactory because the stubborn C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond in a nonpolar CO2 molecule requires high energy for activation, leading to sluggish kinetics issues.11–13 In light of this, great efforts have been devoted to optimizing the structure of photocatalysts so as to improve the conversion and selectivity of CO2 reduction.14–17 Up to now, a variety of photocatalysts with tailorable band structures have been explored, especially semiconductors (such as TiO2, Zn2CeO4, and C3N4).18–22 Despite great endeavours, the photocatalytic performance of conventional photocatalysts is still limited by their solar light harvesting ability, unsuitable band gaps, weak CO2 adsorption capacity, and/or low density of active sites. Thus, it is urgent to rationally design and modify the structure of photocatalysts to achieve efficient CO2 reduction.23–28
O double bond in a nonpolar CO2 molecule requires high energy for activation, leading to sluggish kinetics issues.11–13 In light of this, great efforts have been devoted to optimizing the structure of photocatalysts so as to improve the conversion and selectivity of CO2 reduction.14–17 Up to now, a variety of photocatalysts with tailorable band structures have been explored, especially semiconductors (such as TiO2, Zn2CeO4, and C3N4).18–22 Despite great endeavours, the photocatalytic performance of conventional photocatalysts is still limited by their solar light harvesting ability, unsuitable band gaps, weak CO2 adsorption capacity, and/or low density of active sites. Thus, it is urgent to rationally design and modify the structure of photocatalysts to achieve efficient CO2 reduction.23–28
Up to now, various impressive strategies have been proposed and implemented to improve photocatalytic activity. For instance, heterojunction construction, surface modification, doping and vacancy creation, and interface engineering strategies are widely investigated to improve the activity of photocatalysts.29–37 Specifically, the doping engineering strategy is capable of adjusting the band gap or electronic structure of photocatalysts. Correspondingly, the light absorption scope, electron–hole separation efficiency, and adsorption of CO2 and intermediates would be optimized.38–40 A recent investigation has reported that transition metal doped photocatalysts can significantly improve photocatalytic activity owing to their special d-band.41–45 It is well known that doping of transition metal ions into photocatalysts can introduce electron capture centers, resulting in defect formation and inhibition of electron/hole recombination to enhance catalytic performance. Moreover, transition metal atoms are highly effective as active catalytic species for the conversion of small molecule CO2. Previous studies have doped various transition metals in semiconductors for accelerating kinetics and profiling catalytic mechanisms in multi-electron CO2 reduction.44,45 However, it remains ambiguous whether any kind of transition metal doping is conducive to photocatalytic CO2 reduction.
Herein, we intend to demonstrate the effect of transition metal doping on CO2 reduction performance for semiconductor photocatalysts. As a proof of concept, various transition metal-doped In2O3/C photocatalysts are utilized to study the doping effects of metal species on the activity and selectivity of CO2 photoreduction (Scheme 1). To exclude other interference factors such as morphology structure and surface properties, transition metal-doped metal–organic frameworks (MOFs) were used as templates to fabricate various transition metal-doped In2O3/C nanotubes (M–In2O3/C, M = Fe, Cu, and Zn). The MOF-derived hollow structure is favorable for harvesting solar light and exposing the surface-active sites. Compared with unmodified In2O3, Cu–In2O3/C, Fe–In2O3/C, and Zn–In2O3/C exhibit an expanded photo-response range for light harvesting, and provide a strong driving force for the occurrence of CO2 activation. In particular, Cu–In2O3/C presents a largely improved CO yield (43.7 μmol g−1 h−1) with a high selectivity (78%), which is 11 times higher than that of pristine In2O3. The experimental results and theoretical calculation verify that the narrowed band gap generated by Cu doping causes the d-band center upshift, which facilitates the adsorption/activation of CO2 molecules and charge migration in photocatalysis.
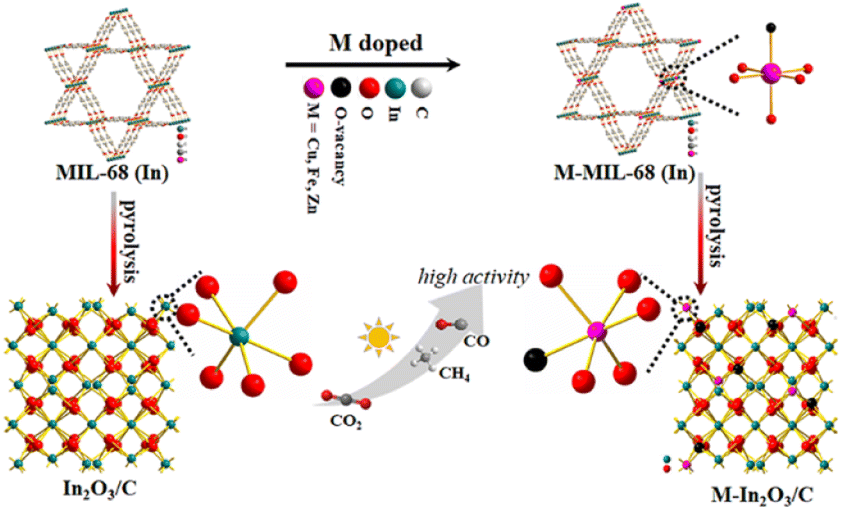 | ||
| Scheme 1 Schematic illustration of M–In2O3/C (M = Fe, Cu, and Zn) showed enhanced performance for solar light-driven CO2 reduction, compared with an In2O3/C photocatalyst. | ||
2 Results and discussion
2.1 Synthesis and structural characterization
A series of transition M–In2O3/C (M = Fe, Cu, and Zn) photocatalysts were prepared based on a bimetallic MOF template. First, the hexagonal M-MIL-68 (In) (M = Fe, Cu, and Zn) were synthesized via a solvothermal method.46,47 The powder X-ray diffraction (XRD) analysis shows that the resulting M-MIL-68 (In) is in good agreement with the bulk phase of MIL-68 (In) without any additional phase.48,49 As shown in Fig. 1A and B, the scanning electron microscopy (SEM) images illustrate that the Cu-MIL-68 (In) presents a uniform hexagonal prism-shaped morphology, similar to pristine MIL-68 (In). The combination of the XRD patterns (Fig. S1†) and SEM (Fig. S2 and S3†) analysis demonstrates that the M-MIL-68 (In) maintains the original structure, which is suitable as a template for synthesizing derivatives M–In2O3/C. As expected, the M–In2O3/C derived from M-MIL-68 (In) inherits the skeleton of M-MIL-68 (In) after pyrolysis. As shown in Fig. 1D, the SEM image displays the well-defined hexagonal Cu–In2O3/C nanotube, which is consistent with In2O3/C (Fig. 1C). Besides, the obvious contrast between the dark parts and the relatively bright parts in the transmission electron microscope (TEM) images (Fig. 1E and F) confirms the inherited hollow feature. The high-resolution TEM (HR-TEM) analysis further shows that the shell of In2O3/C nanotubes is assembled with small nanocrystals that are firmly interconnected with each other (Fig. S4†), and exhibits porous characteristics. As for the HR-TEM image (Fig. 1F, inset), the lattice fringe with an interlayer distance of 0.289 nm is assigned to the (222) crystal plane of cubic In2O3 (Fig. S4B†).47 The chemical composition and distribution of the Cu–In2O3/C nanotubes were also examined by elemental mappings (Fig. 1G) and energy dispersive X-ray (EDX) spectroscopy. The elemental mappings of a single Cu–In2O3/C nanotube show a high distribution of Cu, In, O, and C elements. Similarly, Zn–In2O3/C and Fe–In2O3/C (Fig. S5–S8†) nanotubes can be feasibly obtained by pyrolysis of the precursor Zn-MIL-68 (In) and Fe-MIL-68 (In), respectively. | ||
| Fig. 1 SEM images of (A) MIL-68 (In), (B) Cu-MIL-68 (In), (C) In2O3/C, and (D) Cu–In2O3/C. TEM images of (E) In2O3/C, and (F) Cu–In2O3/C. (G) TEM and EDX elemental mapping images of Cu–In2O3/C. | ||
Then, the chemical structure of the obtained photocatalysts was investigated. As shown in Fig. 2A, the XRD diffraction peaks of the Cu–In2O3/C, Zn–In2O3/C, and Fe–In2O3/C derived from M-MIL-68 (In) are in good agreement with the cubic phase of In2O3 (JCPDS card No. 060416).50,51 No other additional phase in XRD is observed, which confirms the high purity of the photocatalysts. As for the M–In2O3/C nanotubes, the peaks slightly shift to higher 2θ values relative to In2O3/C (Fig. 2A, inset) because the ionic radii of Fe3+ (0.69 Å), Zn2+ (0.74 Å), and Cu2+ (0.73 Å) are smaller than that of In3+ (0.80 Å).52–57 The result indicates that metal ions are indeed doped into the In2O3 lattice. In addition, the Raman peaks observed at 127, 302, 363, 492, and 621 cm−1 are characteristic peaks of body-centered-cubic (bcc) In2O3 (Fig. 2B).58 Peak E2g at 363 cm−1 reflects the content of oxygen vacancy (Ov). Under the same pyrolysis treatment, the relative intensity of peak E2g in Cu–In2O3/C is higher than those of Fe–In2O3/C and Zn–In2O3/C. The results illustrate that Cu–In2O3/C possesses the highest concentration of surface Ov. Besides, the Raman spectra of M–In2O3/C in Fig. 2B show a D band (1340 cm−1) from disordered or defect sp2 carbon and a G band (1591 cm−1) from graphitic sp2 carbon. Moreover, the low ID/IG (the intensity ratio of D to G bands) values manifest a distinct degree of graphitization in M–In2O3/C.
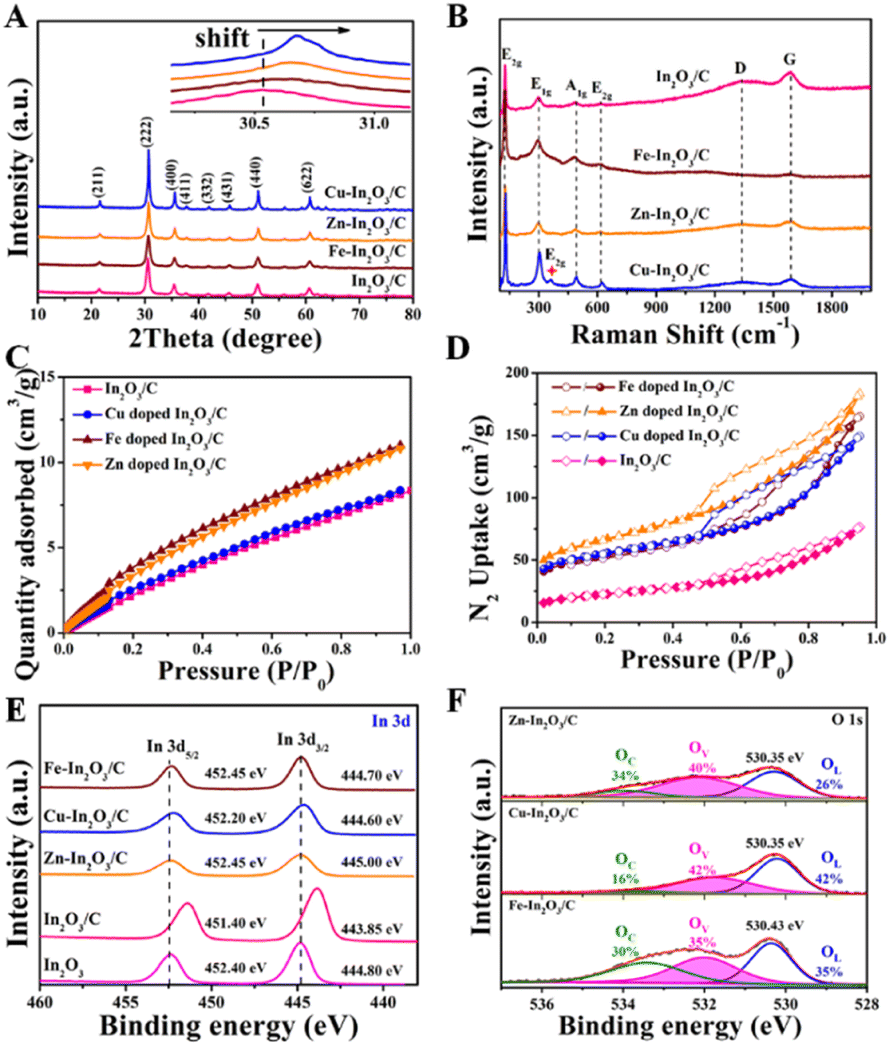 | ||
| Fig. 2 (A) XRD patterns, (B) Raman spectra, (C) CO2 adsorption isotherms and (D) N2 adsorption–desorption isotherms of In2O3/C, Cu–In2O3/C, Zn–In2O3/C, and Fe–In2O3/C. (E) High-resolution In 3d XPS spectra of In2O3, In2O3/C, Cu–In2O3/C, Zn–In2O3/C, and Fe–In2O3/C. (F) High-resolution O 1s XPS spectra of Cu–In2O3/C, Zn–In2O3/C, and Fe–In2O3/C. | ||
After characterization of the morphology and chemical structure of the obtained M–In2O3/C, we investigated their porous structure and CO2 adsorption capacity by gas adsorption/desorption measurements. As shown in Fig. S9,† a type-I isotherm curve is observed for MIL-68 (In), confirming a typical microporous structure for the precursor (Fig. S10†). After pyrolysis of the MIL-68 (In), the N2 adsorption–desorption measurements show that In2O3/C, Cu–In2O3/C, Zn–In2O3/C, and Fe–In2O3/C nanotubes maintain a high Brunauer–Emmet–Teller (BET) surface area (Fig. 2D). The N2 adsorption–desorption isotherms with a type IV behavior show an H3-type hysteresis loop, indicating the existence of mesopores in M–In2O3/C. The corresponding Barrett–Joyner–Halenda analysis for the In2O3/C nanotubes illustrates that the majority of the pores are around 13 nm (Fig. S11†). Moreover, CO2 adsorption measurements demonstrate the advantages of such porous properties in improving CO2 adsorption capacity. And In2O3/C, Cu–In2O3/C, Zn–In2O3/C, and Fe–In2O3/C nanotubes exhibit a high CO2 uptake at 25 °C (Fig. 2C), respectively, which is probably inherited from the high CO2 adsorption properties of MIL-68 (In) (Fig. S12†). The above results thus confirm that M–In2O3/C derived from M-MIL-68 (In) retains the MOF skeleton and porous structure, which is conducive to the exposure of surface sites for the adsorption of CO2.
To further demonstrate the influence of M doping on the electronic structure of M–In2O3/C, X-ray photoelectron spectroscopy (XPS) characterization was conducted. The C 1s spectrum of In2O3/C is coherently fitted by four peaks centered at 284.8, 285.5, 286.7, and 288.9 eV (Fig. S13†), which are ascribed to CC, C–C, C–O, and CO bonds, respectively. High-resolution XPS of In 3d exhibits two prominent bands of In 3d3/2 and In 3d5/2 at 444.8 and 452.4 eV, respectively (Fig. 2E). After the introduction of carbon, the two In 3d peaks of In2O3/C decrease to 443.85 and 451.4 eV, confirming the electronic coupling interaction between In2O3 and C. As shown in Fig. S14–S16,† the obtained high-resolution XPS spectra of Cu, Zn and Fe illustrate that the transition metals have been introduced into M–In2O3/C. Most importantly, the In 3d5/2 and In 3d3/2 peaks in the M–In2O3/C nanotubes show an obvious positive shift. The results thus confirm that M doping could modify the electronic structure of In2O3/C. In addition, the high-resolution XPS spectrum of O 1s (Fig. 2F and S17†) displays a broad asymmetric curve, which is deconvoluted into three contributions including a distinct peak and two shoulder peaks. The distinct peak is attributed to the typical lattice oxygen (OL). The two shoulder peaks are assigned to Ov and dissociated/chemisorbed oxygen (OC), respectively. The ratios of Ov for In2O3/C, Cu–In2O3/C, Zn–In2O3/C and Fe–In2O3/C are about 26%, 42%, 40% and 35%, respectively. Moreover, the ratio of Ov for Fe–In2O3/C is lower than that for Cu–In2O3/C. Compared with high valent Fe3+/Fe2+ in Fe–In2O3/C, the low valent Cu2+/Cu+ in Cu–In2O3/C can elongate and weaken the M–O bonds, hence facilitating the generation of more Ov. This comparison thus illustrates that the ratio of the generated Ov is highly dependent on the doped element M.
2.2 Evaluation of photocatalytic CO2 reduction performance
Encouraged by the above characterization results, the photocatalytic CO2 reduction performance of M–In2O3/C was evaluated. As shown in Fig. 3A and B, the production rates of CO and CH4 for M–In2O3/C are following the sequence Cu–In2O3/C (43.7 and 15.9 μmol g−1 h−1) > Zn–In2O3/C (32.1 and 11.4 μmol g−1 h−1) > Fe–In2O3/C (21.9 and 6.3 μmol g−1 h−1), which are all higher than that of pristine In2O3 (3.9 and 1.4 μmol g−1 h−1) and In2O3/C (9.1 and 2.4 μmol g−1 h−1). As for the CO and CH4 production rates, Cu–In2O3/C exhibits a ∼11-fold and 7-fold higher rate than that of pristine In2O3. The comparison illustrates that Cu doping in the structure of In2O3 enables facilitation of the improvement of CO2 activation. Besides the products of CO and CH4, other by-products including C2H4 and H2 are also detected (Fig. 3C). The comparison illustrates that CO is the main carbonous product. It should be noted that the selectivity is highly dependent on the doping elements in In2O3/C (Fig. 3D). The comparison displays that Cu doping contributes higher selectivity of CO than Fe or Zn doping. The obtained Cu–In2O3/C exhibits a high selectivity of 78% for CO2 reduction towards CO relative to the main carbon product (CO, C2H4, and CH4). In addition, the obtained Cu–In2O3/C and In2O3/C exhibit a CO selectivity of ∼50% and ∼59%, respectively. Of note, the CO yield of Cu–In2O3/C is ∼4.8 times higher than that of pristine In2O3/C. Compared with the high catalytic efficiency, the difference in catalytic selectivity between Cu–In2O3/C and In2O3/C is not more obvious. Of note, the apparent quantum efficiency (AQE) of Cu–In2O3/C, Zn–In2O3/C, and Fe–In2O3/C for CO production is about 2 × 10−3%, 1 × 10−3% and 1 × 10−3% at 380 nm, respectively. The photocatalytic results thus confirm that In2O3 doped with transition metals can enhance the photocatalytic performance for CO2 reduction by regulating the internal electronic structure. Most importantly, the activation of CO2 is highly dependent on the doped metal. And, Cu is confirmed as an optimized candidate doped into In2O3 because of the best photocatalytic CO2 reduction performance (Fig. 3A and B). Compared with previously reported MOF derivative-based photocatalysts, Cu–In2O3/C displays higher efficiency in photocatalytic CO2 reduction (Table S1†).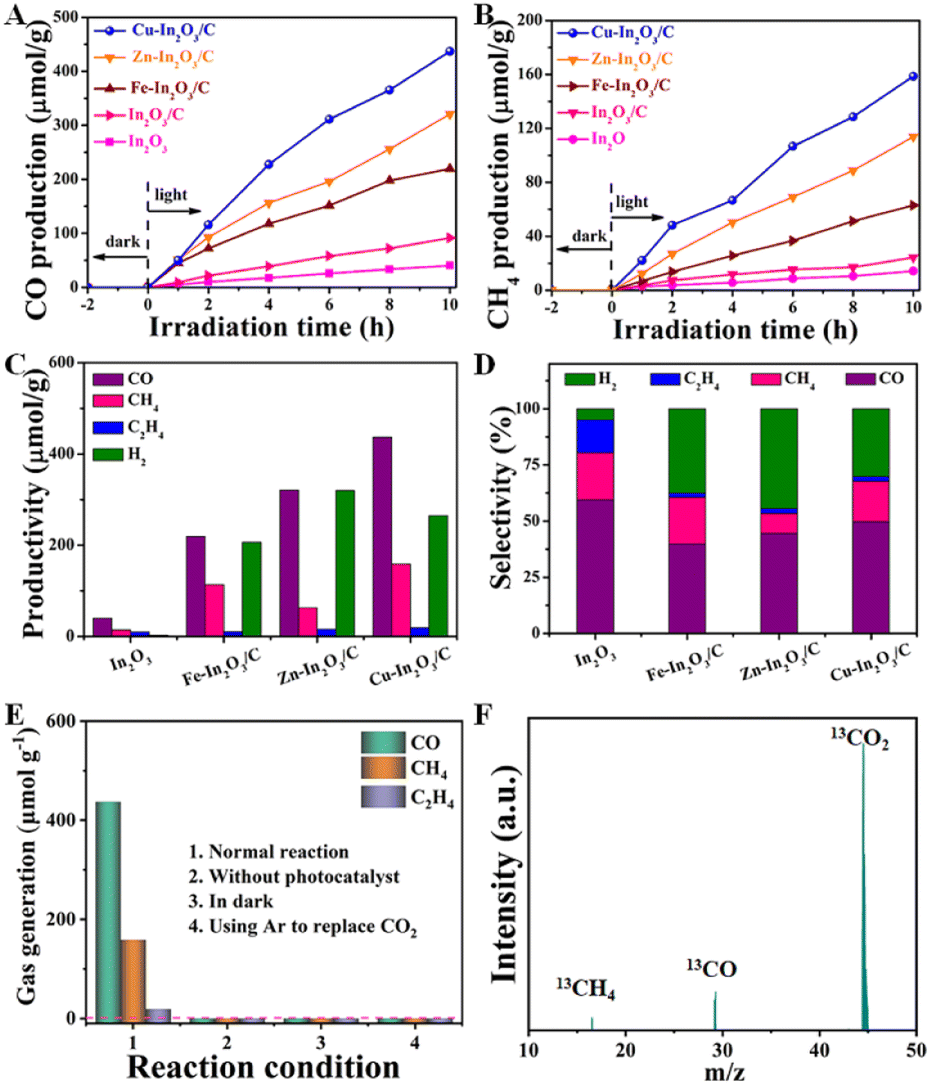 | ||
| Fig. 3 Time-production plots of (A) CO and of (B) CH4 over In2O3/C, Cu–In2O3/C, Zn–In2O3/C, and Fe–In2O3/C nanotubes. (C) Productivity and (D) selectivity of different products over In2O3/C, Cu–In2O3/C, Zn–In2O3/C, and Fe–In2O3/C nanotubes. (E) Photocatalytic CO2 reduction performance of Cu–In2O3/C under different conditions. (F) Result of mass spectra analysis from the 13CO2 isotope-labeling experiment for Cu–In2O3/C. | ||
In addition, a series of control experiments were conducted to investigate the source of CO and CH4 over Cu–In2O3/C (Fig. 3E). There is no detectable product when the photocatalytic CO2 reduction reaction is carried out in an Ar atmosphere, under dark conditions, or without catalysts (Fig. 3E). To further verify the origin of the as-produced CH4 and CO, isotopic 13CO2 is used as a reactant for CO2 photoreduction. The obtained products were examined by using the mass spectrum (Fig. 3F). The corresponding signal at m/z = 29 and 17 can be ascribed to 13CO and 13CH4, respectively, which confirms that the products CH4 and CO originate from the photocatalytic reduction of CO2. In addition, the cycling experiment (Fig. S18†) and the corresponding SEM image (Fig. S19†) after 30 h for the CO2 reduction reaction were recorded. The results show that the morphology and photoactivity of Cu–In2O3/C do not change significantly, hence exhibiting high photocatalytic stability.
2.3 Investigation of photocatalytic properties
To illustrate the effect of M doping on enhancement of photocatalytic CO2 reduction, the photocatalytic properties of M–In2O3/C photocatalysts are investigated. The optical absorption of the as-obtained M–In2O3/C nanotubes was measured by UV-vis diffuse reflectance spectroscopy (UV-DRS). It is observed that all samples M–In2O3/C have strong absorption in the UV-vis range (Fig. 4A). Compared with the M-MIL-68 (In) (200 nm–320 nm) and M–In2O3 (200 nm–500 nm) (Fig. S20 and S21†), M–In2O3/C presents a broad light absorption region towards visible light due to the existence of abundant Ov and graphitic carbon. According to the obtained spectra, the band gap (Eg) is calculated by using the Kubelka–Munk theorem. Compared with the Eg of In2O3 (3.2 eV), narrow Eg values of Cu–In2O3 (3.0 eV), Fe–In2O3 (3.1 eV), and Zn–In2O3 (3.1 eV) are observed (Fig. S22–S25†). Based on the Mott–Schottky analysis (Fig. S26–S29†), the ECB values of In2O3/C, Cu–In2O3/C, Zn–In2O3/C and Fe–In2O3/C are calculated to be −0.75, −0.66, −0.70 and −0.69 eV vs. the normal hydrogen electrode (NHE), respectively. According to the formula: EVB = ECB + Eg, the EVB of In2O3/C, Cu–In2O3/C, Zn–In2O3/C and Fe–In2O3/C is calculated to be 2.55, 2.44, 2.40 and 2.31 eV vs. NHE (Fig. S30†), respectively. The reason is that M doping and Ov in M–In2O3/C are capable of modifying the electronic structure, resulting in a reduced Eg. It should be noted that Cu doping largely narrows the Eg from 3.2 to 3.0 eV. The result thus indicates that doping of transition metals can tailor the band structure, hence promoting solar light harvesting and electron–hole separation capability of the photocatalysts. Besides, it is proved that M doping can affect the lifetime of charge migration for various M–In2O3/C. As shown in Fig. 4B, a weaker photoluminescence (PL) emission of Cu–In2O3/C than that of In2O3/C or In2O3 is observed. Meanwhile, Cu–In2O3/C has longer average decay times (∼1.5 ns), compared with In2O3/C (∼0.6 ns) or In2O3 (∼0.5 ns) (Fig. 4C). The result illustrates that doping of M acts as a trapping site for photogenerated electrons, which can increase the lifetime of charge carriers.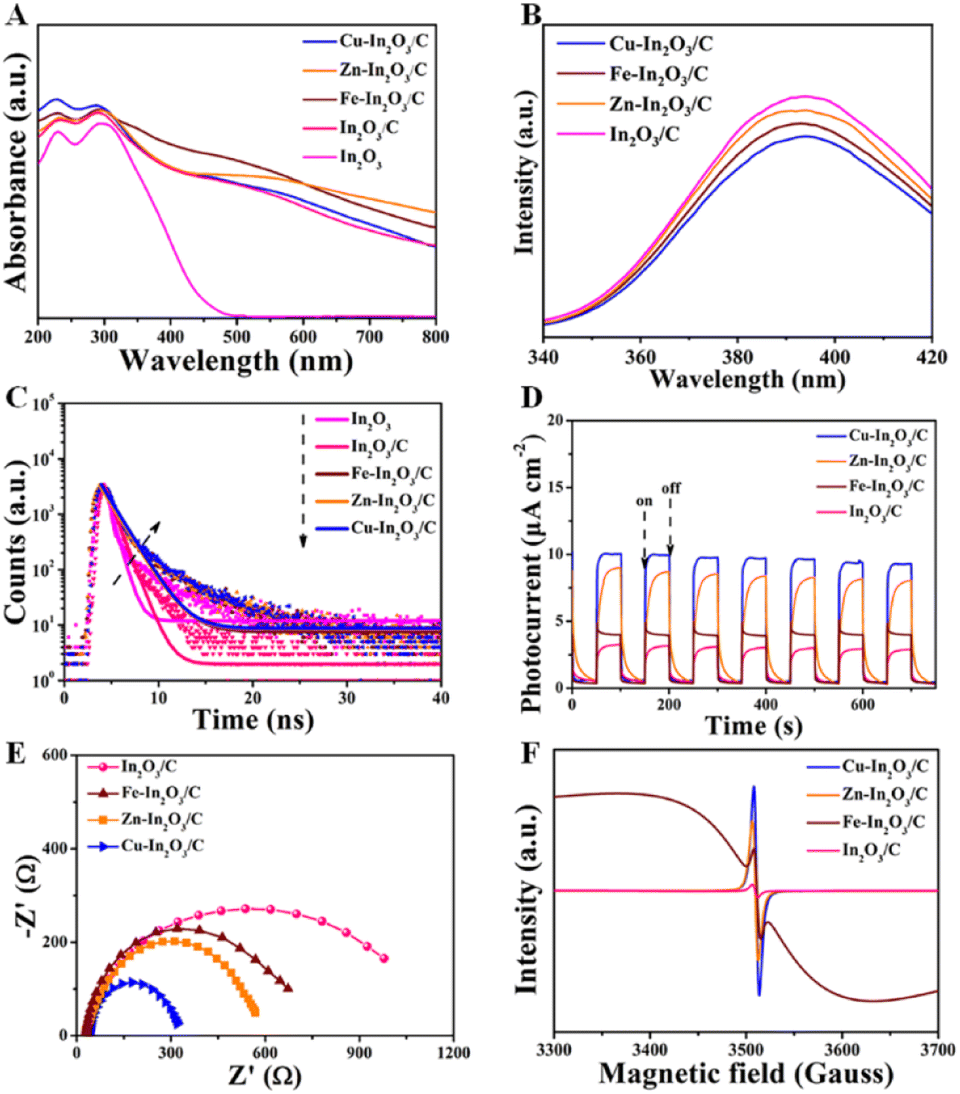 | ||
| Fig. 4 (A) UV-DRS of In2O3, In2O3/C, Cu–In2O3/C, Zn–In2O3/C, and Fe–In2O3/C nanotubes. (B) Steady-state PL spectra, (C) time-resolved PL decay spectra, (D) transient photocurrent responses, (E) EIS Nyquist plots, and (F) EPR spectra of In2O3/C, Cu–In2O3/C, Zn–In2O3/C, and Fe–In2O3/C nanotubes. | ||
To evaluate the charge transfer efficiency, the photocurrent density of In2O3/C and various M–In2O3/C nanotubes was measured under light and in the dark with 50 s intervals. As shown in Fig. 4D, the sequence of average photocurrent densities follows the order Cu–In2O3/C > Zn–In2O3/C > Fe–In2O3/C > In2O3/C. Meanwhile, the recorded electrochemical impedance spectrum (EIS) of Cu–In2O3/C shows a smaller semicircle in the Nyquist plot than those of In2O3/C, Zn–In2O3/C, and Fe–In2O3/C (Fig. 4E). These results thus confirm a higher separation efficiency of photogenerated electron–hole pairs and that a faster interfacial charge transfer rate occurred on Cu–In2O3/C. In addition, the electron paramagnetic resonance (EPR) spectra normally used for detecting unpaired electrons was obtained for In2O3/C and various M–In2O3/C nanotubes. According to previously reported work,33 the symmetry EPR signals at g = 2.003 are ascribed to the unpaired electrons trapped on the surface Ov. As shown in Fig. 4F, the EPR signals are significantly increased after M-doping, demonstrating an increased Ov concentration. Owing to the existence of abundant Ov for Cu–In2O3/C, more electrons can be captured at the surface, which can effectively modify the surface electronic state for accelerating charge transfer. Besides, the broadness of the EPR resonance signal for Fe–In2O3/C is generated by the random orientation of the ferromagnetic Fe, which scatters in the directions of the anisotropic field. As a result, the shape of the EPR curve would change. The above results therefore illustrate that the photocatalytic properties including light harvesting capacity, electro-hole separation efficiency and the surface charge transfer rate of Cu–In2O3/C are obviously higher than those of other M–In2O3/C and In2O3/C.
Then, we performed in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurements to monitor the reaction intermediates in the CO2 reduction process. DRIFTS of the Cu–In2O3/C photocatalyst collected without CO2 in the dark is selected as the background (Fig. 5). After adsorbing CO2 gas and H2O vapor for 25 minutes, monodentate carbonate (m-CO32−, located at 1537, 1518, 1469, 1178, and 1154 cm−1), bidentate carbonate (b-CO32−, located at 1502 and 1355 cm−1) and bicarbonate (HCO3−, located at 1452 cm−1) are visible. According to previous reports, *HCOO and COO− species, and *COH3 and CH3O− groups are intermediate species in CO and CH4 generation, respectively. It is most interesting that, the peak intensity of intermediate species increases along with the light irradiation time. It suggests that CO2 is continuously adsorbed and converted into carbon-active species. In the comparison, the intensity of peaks for intermediates on In2O3/C (Fig. S31†) is lower than that of Cu–In2O3/C. Most importantly, a new infrared peak at 1651 cm−1 in the Cu–In2O3/C system appears and it gradually increases with extension of the irradiation time. The peak is ascribed to COOH*, which is generally regarded as the crucial intermediate during CO2 reduction to CO.9,37 In addition, a remarkable signal at 2077 cm−1 ascribed to the CO* absorption band is also detected (Fig. S32†). The intensity gradually increases with extension of the irradiation time, hence accounting for the high CO selectivity of the Cu–In2O3/C photocatalyst.
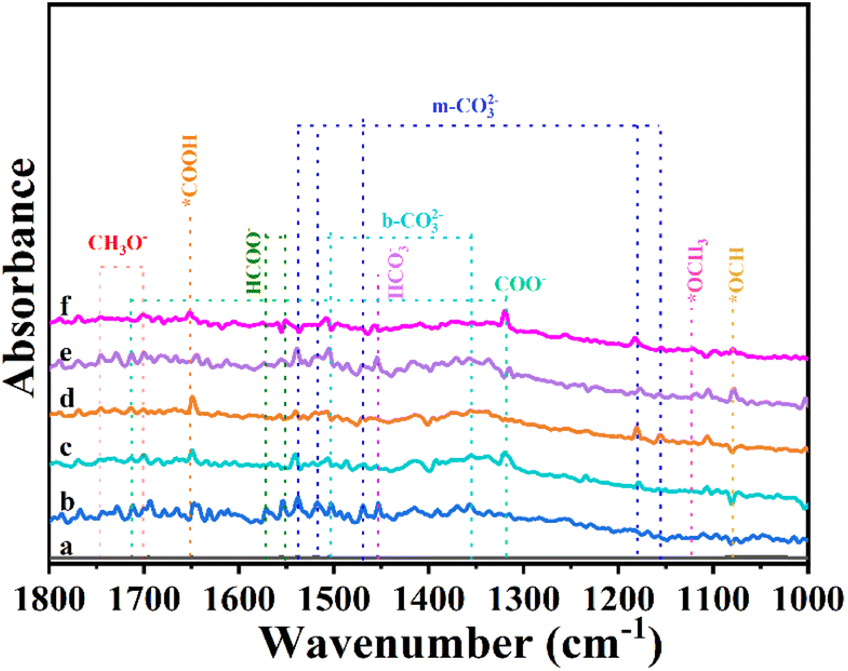 | ||
| Fig. 5 In situ DRIFT spectra of Cu–In2O3/C (a) in the dark, and under visible light irradiation for (b) 5, (c) 10, (d) 15, (e) 20, and (f) 25 min in the photocatalytic CO2 reduction process. | ||
2.4 Investigation of the theoretical mechanism
Density functional theory (DFT) calculations were carried out to elucidate the electronic structure and disclose the catalytic mechanism in the multi-electron CO2 reduction reaction for Cu–In2O3 and In2O3. As shown in Fig. 6A and B, the calculated partial density of states (PDOS) curves indicate that both Cu–In2O3 and In2O3 exhibit typical semiconductor features. As for pure In2O3, the Eg is calculated to be ∼3.1 eV. After introducing Cu atoms into In2O3, the Eg is narrowed, which is consistent with the experimental result of the UV-vis spectrum (Fig. S23†). The result illustrates that doping Cu into In2O3 is favorable for enhancing electrical conductivity and facilitating electron–hole separation, which is in good agreement with the photocurrent responsive test and EIS results (Fig. 4D and E). To gain atomic-level insight into the Cu doping effect, the electronic density difference of Cu–In2O3 was calculated. The result shows the obvious charge density difference on Cu sites (Fig. 6C). The reason is that Cu sites can modify the local electronic environment of In and O sites, thereby adjusting the band structure and improving photocatalytic activity of Cu–In2O3.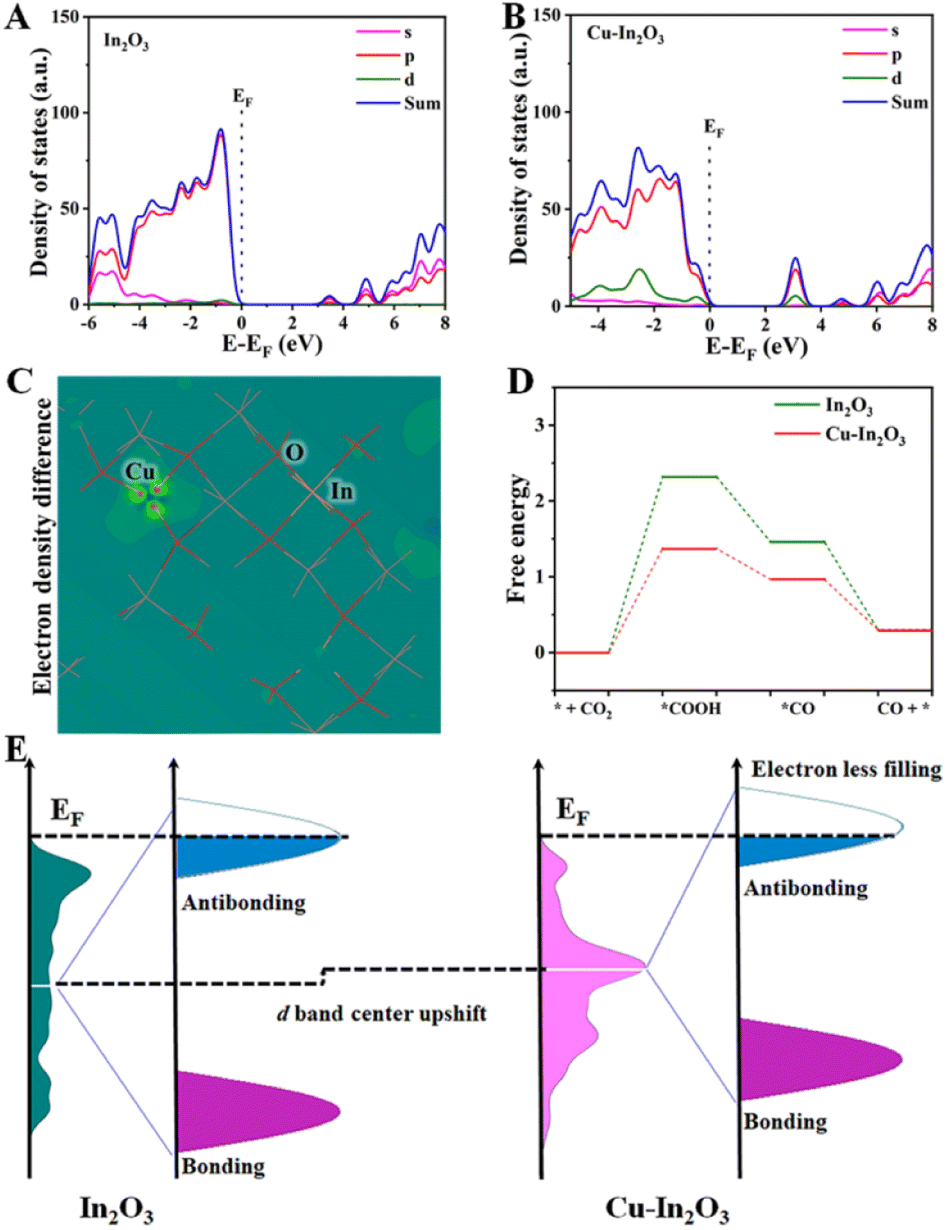 | ||
| Fig. 6 PDOS curves of (A) In2O3 and (B) Cu–In2O3. (C) Model of electronic density difference for Cu–In2O3. (D) Calculated free energy for the photocatalytic CO2 reduction reaction over Cu–In2O3 and In2O3. (E) d-band center of Cu–In2O3 and In2O3 indicated by the white line (upshifted d-band center of Cu–In2O3 compared to that of In2O3). | ||
The thermodynamic mechanisms for photocatalytic CO2 reduction towards the main product CO are then compared. The corresponding Gibbs free energy change (ΔG) for possible reduction pathways of CO2 → *COOH → *CO → CO (* is the reaction site) is shown in Fig. 6D. For both Cu–In2O3 and In2O3, the rate-determining step is the hydrogenation of CO2. Compared with In2O3 (ΔG = 2.3 eV), CO2 hydrogenation to *COOH requires 1.4 eV over Cu–In2O3. In addition, the formation of *CO is also easier for the Cu–In2O3 system compared with the In2O3 system. The results indicate that Cu doping enables tailoring the electronic structure of In2O3. As a result, faster kinetics of Cu–In2O3 is achieved for efficiently driving CO2 reduction, which is consistent with the photocatalytic evaluation results (Fig. 3A and B). In addition, the d-band structure affected by Cu sites is further analyzed (Fig. 6E) to demonstrate the improved photocatalytic CO2 reduction performance. After doping Cu, Cu–In2O3 exhibits an upward shift compared to In2O3. Normally the occupancy of anti-bonding states is highly deepened on the d-band position to the Fermi level (EF).59–61 Based on the d-band center theory, the upward shift of Cu–In2O3 would exhibit a strong binding strength of electrons on CO2. Correspondingly, Cu–In2O3 displays less filling and decreased occupation of anti-bonding states, which is favorable for adsorption/activation of CO2. Therefore, the above theoretical calculations combined with experiment results illustrates that Cu doping into In2O3 can feasibly optimize the electronic structure, thereby contributing to largely improved photocatalytic performance.
3 Conclusions
In summary, a feasible bimetallic MOF template is developed for the synthesis of a series of transition metal-doped In2O3/C photocatalysts. The electronic structure can be adjusted by selecting a suitable doping transition metal, which is an effective way to optimize the photocatalytic properties, such as accelerating charge transfer rates, exposing active sites, and enhancing solar light absorption. In the comparison, the resultant Cu–In2O3/C exhibits largely improved photocatalytic performance for CO2 reduction. The experiment results and theoretical calculation verify that the narrow Eg generated by the Cu doping effect is favorable for adsorption/desorption of CO2 and charge transfer, thereby contributing to efficient CO2 conversion towards CO. This present work therefore offers a new perspective for modulating the structure of doped photocatalysts by the MOF template strategy, which shows promising applications in solar-to-chemical energy conversion.Author contributions
A. Zhou performed the experiments and data analysis, and wrote the manuscript. Y. Dou performed the theoretical calculations, and gave specific guidance and revision in the experimental and manuscript preparation stages. J. Zhou and C. Zhao synthesized and characterized the photocatalysts, and tested the photocatalytic performance. J.-R. Li and M. Wei directed and edited the manuscript. All authors read, revised, and approved the submission of the final manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work was supported by the National Natural Science Foundation of China (22278029), the Fundamental Research Funds for the Central Universities (BUCTRC202203), the National Key R&D Program of China (2021YFC2103500), and the Open Research Fund Program of the Key Laboratory of Cleaner Production and Integrated Resource Utilization of China National Light Industry, the Cultivating Fund of Faculty of Environment and Life, BJUT (PY202302).References
- B. C. Qiu, M. M. Du, Y. X. Ma, Q. H. Zhu, M. Y. Xing and J. L. Zhang, Energy Environ. Sci., 2021, 14, 5260 RSC
.
- C. Wang, Z. Sun, Y. Zheng and Y. H. Hu, J. Mater. Chem. A, 2019, 7, 865 RSC
- P. De Luna, C. Hahn, D. Higgins, S. A. Jaffer, T. F. Jaramillo and E. H. Sargent, Science, 2019, 364, 350 CrossRef PubMed
- J. Pei, T. Wang, R. Sui, X. Zhang, D. Zhou, F. Qin, X. Zhao, Q. Liu, W. Yan, J. Dong, L. Zheng, A. Li, J. Mao, W. Zhu, W. Chen and Z. Zhuang, Energy Environ. Sci., 2021, 14, 3019 RSC
- H. S. Feng, H. Ding, P. N. He, S. Wang, Z. Y. Li, Z. K. Zheng, Y. S. Yang, M. Wei and X. Zhang, J. Mater. Chem. A, 2022, 10, 18803 RSC
- Z. Y. Li, Y. S. Yang and M. Wei, Acta Chim. Sinica, 2022, 80, 199 CrossRef CAS
- D. Mao, S. Yang, Y. Hu, H. He, S. Yang, S. Zheng, C. Sun, Z. Jiang, X. Qu and P. K. Wong, Appl. Catal., B, 2023, 321, 122031 CrossRef CAS
- Y. Zhang, X. Zhi, J. Harmer, H. Xu, K. Davey, J. Ran and S. Qiao, Angew. Chem., Int. Ed., 2022, 134, 202212 Search PubMed
- Q. Bi, M. Wang, M. S. Riaz, X. Du, G. Li and F. Q. Huang, J. Mater. Chem. A, 2022, 10, 23854 RSC
- A. Zhou, Y. Dou, C. Zhao, J. Zhou, X.-Q. Wu and J.-R. Li, Appl. Catal., B, 2020, 264, 118519 CrossRef
- Y. Dou, A. Zhou, Y. Yao, S. Y. Lim, J.-R. Li and W. Zhang, Appl. Catal., B, 2021, 286, 119876 CrossRef CAS
- L. Xiao, C. Yuan, P. Chen, Y. Liu, J. Sheng, S. Zhang, F. Dong and Y. Sun, ACS Sustainable Chem. Eng., 2022, 10, 11902 CrossRef CAS
- J. Li, H. Huang, W. Xue, K. Sun, X. Song, C. Wu, L. Nie, Y. Li, C. Liu, Y. Pan, H.-L. Jiang, D. Mei and C. Zhong, Nat. Catal., 2021, 4, 719 CrossRef CAS
- X. Li, Y. Sun, J. Xu, Y. Shao, J. Wu, X. Xu, Y. Pan, H. Ju, J. Zhu and Y. Xie, Nat. Energy, 2019, 4, 690 CrossRef CAS
- M. Schreier, F. Heroguel, L. Steier, S. Ahmad, J. S. Luterbacher, M. T. Mayer, J. Luo and M. Gratzel, Nat. Energy, 2017, 2, 17087 CrossRef CAS
- C. S. Diercks, Y. Liu, K. E. Cordova and O. M. Yaghi, Nat. Mater., 2018, 17, 301 CrossRef CAS PubMed
- N. Sadeghi, S. Sharifnia and T. O. Do, J. Mater. Chem. A, 2018, 6, 18031 RSC
- L. Wei, C. Yu, Q. Zhang, H. Liu and Y. Wang, J. Mater. Chem. A, 2018, 6, 22411 RSC
- R. Malik, V. K. Tomer, N. J. Joshi, T. Dankwort, L. Lin and L. Kienle, ACS Appl. Mater. Interfaces, 2018, 10, 22372 CrossRef PubMed
- H. J. Lu, J. Tournet, K. Dastafkan, Y. Liu, Y. H. Ng, S. K. Karuturi, C. Zhao and Z. Y. Yin, Chem. Rev., 2021, 121, 10271 CrossRef CAS PubMed
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783 RSC
- J. Zhu, W. Shao, X. Li, X. Jiao, J. Zhu, Y. Sun and Y. Xie, J. Am. Chem. Soc., 2021, 143, 18233 CrossRef CAS PubMed
- X. Li, J. Yu and M. Jaroniec, Chem. Soc. Rev., 2016, 45, 2603 RSC
- T. Zhang and W. Lin, Chem. Soc. Rev., 2014, 43, 5982 RSC
- Z. H. Yan, M. H. Du, J. Liu, S. Jin, C. Wang, G. L. Zhuang, X. J. Kong, L. S. Long and L. S. Zheng, Nat. Commun., 2018, 9, 3353 CrossRef PubMed
- T. Luo, J. Zhang, W. Li, Z. He, X. Sun, J. Shi, D. Shao, B. Zhang, X. Tan and B. Han, ACS Appl. Mater. Interfaces, 2017, 9, 41594 CrossRef CAS PubMed
- J. Yang, L. Jing, X. Zhu, W. Zhang, J. Deng, Y. She, K. Nie, Y. Wei, H. Li and H. Xu, Appl. Catal., B, 2023, 320, 122005 CrossRef CAS
- M. Chu, Y. Li, X. Chen, G. Hou, Y. Zhou, H. Kang, W. Qin and X. Wu, J. Mater. Chem. A, 2022, 10, 23666 RSC
- X. Zhao, J. Guan, J. Li, X. Li, H. Wang, P. Huo and Y. Yan, Appl. Surf. Sci., 2021, 537, 147891 CrossRef CAS
- X. Zhang, J. Yan, F. Zheng, J. Zhao and L. Y. S. Lee, Appl. Catal., B, 2021, 286, 119879 CrossRef CAS
- G. Wang, R. Huang, J. Zhang, J. Mao, D. Wang and Y. Li, Adv. Mater., 2021, 33, 2105904 CrossRef CAS PubMed
- Y. Zhao, Y. Zhao, R. Shi, B. Wang, G. I. N. Waterhouse, L.-Z. Wu, L.-Z. Tung and T. Zhang, Adv. Mater., 2019, 31, 1806482 CrossRef PubMed
- B. Wang, X. Wang, L. Lu, C. Zhou, Z. Xin, J. Wang, X.-K. Ke, G. Sheng, S. Yan and Z. Zou, ACS Catal., 2017, 8, 516 CrossRef
- J. Low, B. Dai, T. Tong, C. Jiang and J. Yu, Adv. Mater., 2019, 31, 1802981 CrossRef PubMed
- P. Zhou, J. Yu and M. Jaroniec, Adv. Mater., 2014, 26, 4920 CrossRef CAS PubMed
- A. Nakada, R. Kuriki, K. Sekizawa, S. Nishioka, J. J. M. Vequizo, T. Uchiyama, N. Kawakami, D. Lu, A. Yamakata, Y. Uchimoto, O. Ishitani and K. Maeda, ACS Catal., 2018, 8, 9744 CrossRef CAS
- W. Li, D.-K. Ma, X. Hu, F. Gou, X. Yang, W. MacSwain, C. Qi and W. Zheng, J. Catal., 2022, 415, 77 CrossRef CAS
- A. Zhang, Y. Liang, H. Zhang, Z. Geng and J. Zeng, Chem. Soc. Rev., 2021, 50, 9817 RSC
- G. S. Shanker, B. Tandon, T. Shibata, S. Chattopadhyay and A. Nag, Chem. Mater., 2015, 27, 892 CrossRef CAS
- H. Wang, L. Zhang, K. Wang, X. Sun and W. Wang, Appl. Catal., B, 2019, 243, 771 CrossRef CAS
- X. Ding, B. Yu, B. Han, H. Wang, T. Zheng, B. Chen, J. Wang, Z. Yu, T. Sun, X. Fu, D. Qi and J. Jiang, ACS Appl. Mater. Interfaces, 2022, 14, 8048 CrossRef CAS PubMed
- M. Surowka, M. Kobielusz, M. Trochowski, M. Buchalska, K. Kruczala, P. Bros and W. Macyk, Appl. Catal., B, 2019, 247, 173 CrossRef CAS
- K. Maeda, Adv. Mater., 2019, 31, 1808205 CrossRef PubMed
- G. Colón, M. Maicu, M. C. Hidalgo and J. A. Navío, Appl. Catal., B, 2006, 67, 41 CrossRef
- L. Tan, S.-M. Xu, Z. Wang, X. Hao, T. Li, H. Yan, W. Zhang, Y. Zhao and Y.-F. Song, Cell Rep. Phys. Sci., 2021, 2, 100322 CrossRef CAS
- C. Zhao, A. Zhou, Y. Dou, J. Zhou, J. Bai and J.-R. Li, Chem. Eng. J., 2021, 416, 129155 CrossRef CAS
- S. Wang, B. Y. Guan and X. W. D. Lou, J. Am. Chem. Soc., 2018, 140, 5037 CrossRef CAS PubMed
- R. Li, L. Sun, W. Zhan, Y.-A. Li, X. Wang and X. Han, J. Mater. Chem. A, 2018, 6, 15747 RSC
- S. Wang, B. Y. Guan, Y. Lu and X. W. D. Lou, J. Am. Chem. Soc., 2017, 139, 17305 CrossRef CAS PubMed
- H. Xie, S. Chen, F. Ma, J. Liang, Z. Miao, T. Wang, H. L. Wang, Y. Huang and Q. Li, ACS Appl. Mater. Interfaces, 2018, 10, 36996 CrossRef CAS PubMed
- F. Zhang, X. Li, Q. Zhao and D. Zhang, ACS Sustainable Chem. Eng., 2016, 4, 4554 CrossRef CAS
- R. K. Singhal, A. Samariya, S. Kumar, S. C. Sharma, Y. T. Xing, U. P. Deshpande, T. Shripathi and E. Saitovitch, Appl. Surf. Sci., 2010, 257, 1053 CrossRef CAS
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751 CrossRef
- H. Yang, S. Wang and Y. Yang, Crystengcomm, 2012, 14, 1135 RSC
- A. Shokohmanesh and F. Jamali-Sheini, Sens. Actuators, A, 2017, 265, 246 CrossRef CAS
- J. He, S. F. Xu, Y. K. Yoo, Q. Z. Xue, H. C. Lee, S. F. Cheng, X. D. Xiang, G. F. Dionne and I. Takeuchi, Appl. Phys. Lett., 2005, 86, 052503 CrossRef
- X. Li, C. Xia, G. Pei and X. He, J. Phys. Chem. Solids, 2007, 68, 1836 CrossRef CAS
- G. Fubo, L. Chunju, H. Dongmei and W. Zhihua, ACS Appl. Mater. Interfaces, 2018, 10, 933 CrossRef PubMed
- D. Zhou, Z. Cai, Y. Jia, X. Xiong, Q. Xie, S. Wang, Y. Zhang, W. Liu, H. Duan and X. Sun, Nanoscale Horiz., 2018, 3, 532 RSC
- J. Wang, S. Xin, Y. Xiao, Z. Zhang, Z. Li, W. Zhang, C. Li, R. Bao, J. Peng, J. Yi and S. Chou, Angew. Chem., Int. Ed., 2022, 61, 202202518 Search PubMed
- N. Zhang, C. Wang, J. Chen, C. Hu, J. Ma, X. Deng, B. Qiu, L. Cai, Y. Xiong and Y. Chai, ACS Nano, 2021, 15, 8537 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2ta09236k |
| This journal is © The Royal Society of Chemistry 2023 |