
Catalytic active centers beyond transition metals: atomically dispersed alkaline-earth metals for the electroreduction of nitrate to ammonia†
Donghai
Wu
abd,
Peng
Lv
ad,
Jiarui
Wu
ad,
Bingling
He
ad,
Xue
Li
ad,
Ke
Chu
 e,
Yu
Jia
*acd and
Dongwei
Ma
*ad
e,
Yu
Jia
*acd and
Dongwei
Ma
*ad
aKey Laboratory for Special Functional Materials of Ministry of Education, School of Materials Science and Engineering, Henan University, Kaifeng 475004, China. E-mail: jiayu@henu.edu.cn; madw@henu.edu.cn; dwmachina@126.com
bHenan Key Laboratory of Nanocomposites and Applications, Institute of Nanostructured Functional Materials, Huanghe Science and Technology College, Zhengzhou 450006, China
cInternational Laboratory for Quantum Functional Materials of Henan, School of Physics, Zhengzhou University, Zhengzhou 450001, China
dJoint Center for Theoretical Physics, Center for Topological Functional Materials, Henan University, Kaifeng 475004, China
eSchool of Materials Science and Engineering, Lanzhou Jiaotong University, Lanzhou 730070, China
First published on 19th December 2022
Abstract
Alkaline-earth (AE) metals have rarely been reported to be the active centers in heterogeneous catalysis. However, in nature, Mg cofactors in enzymes exhibit super activity for biochemical reactions. Herein, taking the AE metal single-atom catalyst (AE-SAC) supported on graphene as a representative, we theoretically explore the feasibility of AE metals as active centers in heterogeneous catalysis for the electrocatalytic nitrate (NO3−) reduction reaction (eNO3RR) to produce NH3. Intriguingly, the AE metal active centers could strongly adsorb and effectively activate NO3−, and catalyze eNO3RR efficiently, similar to that of the transition-metal active centers. In particular, Ba and Sr SACs coordinated with three nitrogen and one carbon atoms exhibited super eNO3RR activity with an ultralow limiting potential of −0.05 V, which also showed desirable selectivity and good stability. Mechanistic investigations indicated that, although AE metal elements are highly ionic, they function as transmitters to assist the charge transfer between the catalyst support and NO3− as well as effective intermediates, which enables the effective activation of NO3−, via the “donation-back donation” mechanism, and further completes the catalytic cycle. Our work could broaden the ideas and lay a theoretical foundation for the development of heterogeneous catalysts with AE metals as active centers.
1. Introduction
Heterogenous catalysis, mainly based on metal and metal-oxides, plays an important role in modern industries and emerging renewable energy conversion and storage technology.1–3 Active centers of the vast majority of heterogeneous catalysts consist of d-block transition metals (TMs). The underlying physical mechanism can be traced back to the partially filled d orbitals, which enables the electron “donation-back donation” process for the molecule adsorption and activation, as described in the Blyholder model4 and the d-band center theory.5 Conversely, s-block metals, alkali, and alkaline-earth (AE) metals are generally thought to be chemically inert due to the lack of combined empty and occupied orbitals.6 It is known that they are generally prone to lose s valence electrons and form the cations in the compounds, which are hard to change the oxidation state rapidly and reversibly involved in elementary steps of a catalytic cycle. Thus, s-block metals are often considered to be catalytic inactive.7Nevertheless, nature has given us some inspiration.8–12 To be specific, magnesium (Mg), as a cofactor in enzymes, has been revealed to play key roles in the photosynthesis system and nucleic acid biochemistry.9,10 Mg2+ in a cofactor has a proper binding affinity for the oxygenated species, and thus, the cofactor can serve as a Lewis acid for the transfer of a phosphate intermediate from one compound to another or the activation of Mg-bound water molecules to a hydroxide ion.8 For example, due to the particularly right affinity of the O atom, Mg2+-bound DNA and RNA polymerases can participate in the neutralization of the polyanionic charge of the nucleic acid, so they work efficiently.9,10 Despite their unique and critical biochemical activities and even the widely used molecular catalysis,13 AE metals acting as active centers of the heterogeneous catalyst have been reported rarely.14–17 For instance, in 2020, Chen et al. first showed that the Mg atom coordinated with N and C atoms embedded in graphene exhibits an excellent four-electron oxygen reduction reaction activity.14 We found that Mg2+ in two-dimensionally layered Mg3(hexaiminotriphenylene)2 electrocatalyst serves as the active center to selectively produce H2O2 by electroreduction of O2.16
It is known that ammonia (NH3) is one of the most important chemicals in modern life, which has also been regarded as a potential clean energy carrier.18 In recent years, the electrocatalytic nitrogen reduction reaction (NRR) to synthesize NH3 under ambient conditions, driven by renewable energy, has gained considerable attention as an alternative to the traditional Haber–Bosch procedure.19–23 However, the low solubility and chemical inertness of N2 molecules lead to an unsatisfactory NH3 production rate and Faraday efficiency.24–26 In this case, very recently nitrate (NO3−), having a much weaker intramolecular chemical bond (176 KJ mol−1) than N2 (941 KJ mol−1), is emerging as a novel nitrogen resource for NH3 synthesis.27 Considering the ever-increasing concentration of NO3− can severely destroy ecological balance and threaten human health,19 electrocatalytic nitrate reduction reaction (eNO3RR) could be considered as a “one stone two bird” strategy for simultaneous nitrate pollution treatment and NH3 production.28,29 Therefore, it is full of importance and challenge to design efficient electrocatalysts with high stability for direct electroreduction of NO3− into NH3.30–32
Encouragingly, diverse catalysts, such as molecular solids,33 metallic alloys,29 metallic compounds,34,35 pure metals,36,37 and atomic catalysts38,39 have been proven to be efficient for eNO3RR, and all the reported catalysts have the TM atoms as active centers. On the other hand, single-atom catalysts (SACs) have gained significant attention in electrocatalysis due to their high efficiency and simple active sites, which makes SAC an ideal platform for studying catalysis mechanisms through theoretical calculations.40–42 For eNO3RR, several SACs have been reported experimentally32,38,43,44 or theoretically.45–52 Interestingly, for these SACs, it is predicted that the binding strength of NO3− could serve as a descriptor to predict the activity trend, and the active center with the adsorption-free energy for NO3− (ΔG(*NO3)) of around −2.0 eV exhibiting a high eNO3RR activity. Considering that AE ions have a unique affinity for the oxygenated species,14,16 the catalysts with AE ions as active sites may be able to bind NO3− effectively; moreover, by tuning the coordination environment or the metal center identities (Mg, Ca, Sr, and Ba), a suitable binding strength of NO3− and nice eNO3RR activity could be achieved.53,54 However, the direct electroreduction of NO3− into NH3 by the AE metal active centers in the electrocatalysts has not been explored and remains an open challenge.
In this work, taking AE metal single atom incorporated graphene as representatives, we comprehensively explored the feasibility of AE metals as active centers for eNO3RR based on the first-principles calculations. Interestingly, the AE metal active centers in the built AE metal single-atom catalyst (AE-SAC) can strongly adsorb and effectively activate NO3− ions, similar to the cases of the TM active centers. Intriguingly, by changing the active center identities and their coordination environment, the binding strength of NO3− can be tuned properly to realize high eNO3RR activity. Especially, Ba and Sr SACs coordinated with one C and three N atoms exhibit ultrahigh eNO3RR activity with near-zero limiting potential (UL) of −0.05 V, together with desirable selectivity and good stability, comparable or even better than the TM-based SACs reported so far. Furthermore, the underlying mechanism for the effective NO3− activation and the excellent eNO3RR activity on the AE metal active centers were explored.
2. Computational methods
All the spin-polarized density functional theory (DFT) calculations were performed based on the projector augmented wave (PAW)55 method as implemented in the Vienna ab initio simulation package (VASP).56 Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA) was employed to treat the exchange-correlation effect.57 The plane-wave cutoff energy was 450 eV. The van der Waals correction was performed using the DFT-D3 method.58 The energy and force convergence criteria for the structural optimization were set as 10−5 eV and 0.02 eV Å−1, respectively. The AE-SAC was built based on the 5 × 5 × 1 graphene supercell. The effect of relaxing lattice parameters on the adsorption stability was tested, and it was found that thanks to the large supercells, relaxing the lattice parameters has negligible influence on the adsorption energy (Fig. S1†). The Monkhorst–Pack scheme was used to sample the first Brillouin zone,59 and the k-points grids were 3 × 3 × 1 and 6 × 6 × 1 for the structural optimization and densities of states (DOS) calculation, respectively. We also tested the DOS calculated with the hybrid functional of Heyd–Scuseria–Ernzerhof (HSE06),60 and results indicate that the distributions of electronic states were very similar for PBE and HSE06 functionals (Fig. S2†). Given that all the AE-SACs studied in this work exhibited obviously metallic features without a band gap, thus the DOS calculated with the PBE functional was qualified. To avoid the interaction between the periodic units, a vacuum layer of ∼20 Å was added along the z direction. Ab initio molecular dynamics (AIMD) simulation was carried out to estimate the thermal stability with a time step of 2 fs.61To explore the accessibility of the experimental synthesis, the formation energy (Ef) was calculated using the following formula:62,63
| Ef = Etotal − EG + ∑niμi | (1) |
To estimate the binding strength between AE metal and the N-doped graphene substrate, the binding energy (Eb) is expressed as:
| Eb = Etotal − Esubstrate − EAE | (2) |
In addition, to further verify the thermodynamical stability of the AE-SAC systems, the energy difference (ΔEb) between Eb and Ecoh was calculated.64 In which, Ecoh is the cohesive energy of AE metal bulk and can be calculated using the formula of Ecoh = (Ebulk – nEAE)/n, where Ebulk is the total energy of the AE metal crystal and n is the number of metal atoms in the crystal. A negative ΔEb indicates that the AE metal single atom tends to remain isolated on the substrate rather than agglomerate, and a more negative ΔEb means higher thermodynamical stability.
The computational hydrogen electrode (CHE) mode65 was adopted to calculate the Gibbs free energy change (ΔG) for each elementary step as follows
| ΔG = ΔE +ΔEZPE − TΔS | (3) |
In the equation, ΔE is the electronic energy contribution directly obtained from DFT calculations. ΔEZPE and TΔS are the contributions of zero-point energy and entropy (at 298.15 K), respectively, which could be acquired by computing the vibrational frequency for intermediates and from the NIST database66 for free molecules (tabulated in Tables S1 and S2†). The limiting potential (UL) was formalized with UL = –ΔGmax/e, where ΔGmax is the maximum free energy change among all elementary steps along the lowest-energy pathway, i.e., the potential-determining step (PDS).67 In light of the difficulty of directly calculating the energy of charged NO3−, the adsorption free energy of NO3− (ΔG(*NO3)) was calculated with the assistance of the gaseous HNO3 (ref. 36 and 68) as follows
| ΔG(*NO3) = G(*NO3) − G(*) − [G(HNO3) − 0.5 × G(H2)] + ΔGcorrect | (4) |
3. Results and discussion
3.1. The properties of AE-SACs
With various merits, such as facile fabrication, excellent stability, and easily tunable electronic structure, graphene-based SACs have received considerable attention for various electrocatalytic reactions.70 Moreover, regulating the coordination environment of the active center can greatly affect its catalytic activity and selectivity.53,54 Herein, we constructed a series of AE-SACs, where the AE metal (Mg, Ca, Sr, Ba) single-atoms are embedded into graphene with single vacancy or double vacancy decorated by certain numbers of N atoms, as illustrated in Fig. 1. Considering that Be is extremely toxic71 and Ra is a typical radioactive element,72 they are not involved in this work. By adjusting the number of coordinated N atoms, seven DV-MNxC4–x and four SV-MNxC3–x configurations were established for each AE metal single atom, producing 44 SAC candidates. The key parameters of these systems are listed in Table S3,† including bond length, charge transfer, spin magnetic moment, formation energy, and binding energy.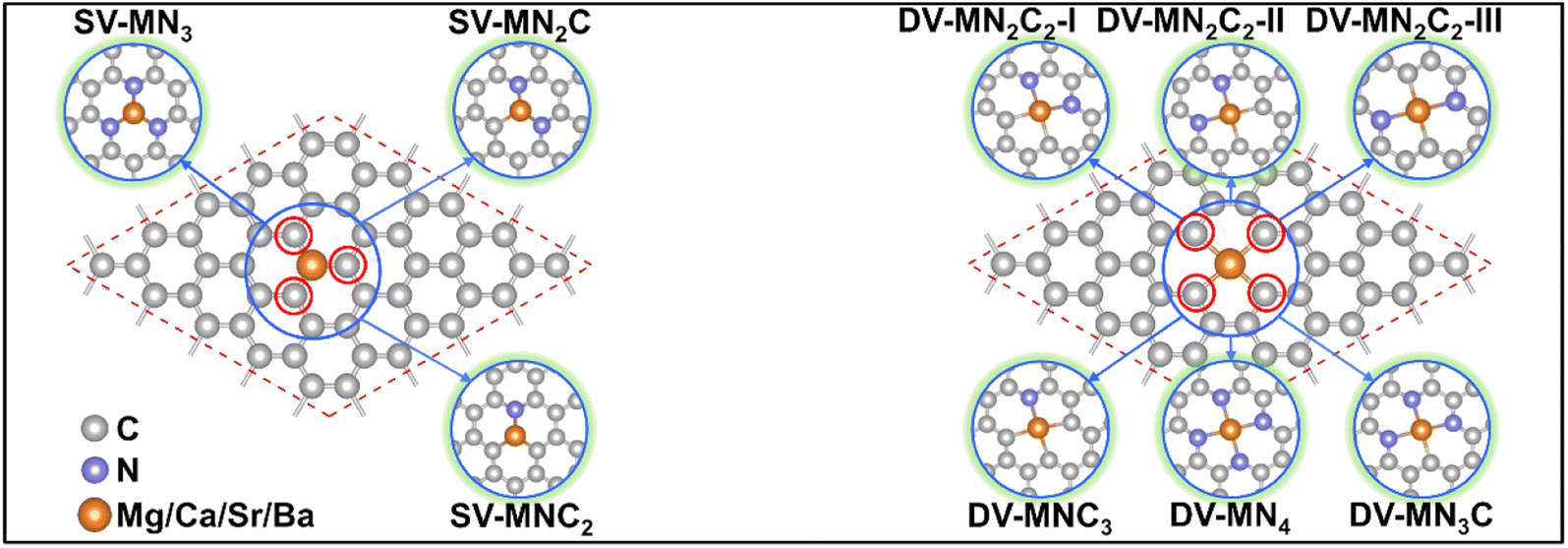 | ||
| Fig. 1 Schematic illustrations of the AE-SAC systems with single Mg/Ca/Sr/Ba atom anchoring on the graphene support with different coordination environments. | ||
The experimentally synthetic feasibility of the proposed AE-SACs can be estimated theoretically from the formation energy.62,63 Since MgN2C2-III was synthesized experimentally,14 was selected as a benchmark. It can be seen from Fig. 2a that the lower the number of coordinated N atoms, the higher the formation energy. More importantly, except DV-MNC3, DV-MC4, SV-MNC2, and SV-MC3, all the AE-SACs have lower or comparable formation energies compared with MgN2C2-III, strongly suggesting that they are able to be synthesized experimentally too. Furthermore, as shown in Fig. 2b, the significantly negative binding energies for all the AE-SACs (except DV-MgC4) indicate that there are strong chemical bonds between AE metals and their neighboring C or N atoms. Clearly, for all AE-SACs (except DV-MgC4 and SV-MgNC2), the energy differences (ΔEb) between Eb and Ecoh are less than zero (Fig. 2c), suggesting that when the AE-SACs are synthesized, the AE metals prefer to be dispersed on the supports instead of agglomerating into nanoparticles, which further demonstrates their high thermodynamical stability. Finally, the Bader charge analysis73 shows that the AE metals lose most of their valence electrons (∼1.5e) to graphene and form AE cations, similar to the AE elements in other compounds. Therefore, it is expected that the AE-SACs could bind NO3− effectively. In addition, the total DOS of all the AE-SAC systems (Fig. S4–S7†) indicate their metallic characteristics, which facilitate the charge transfer during eNO3RR.74
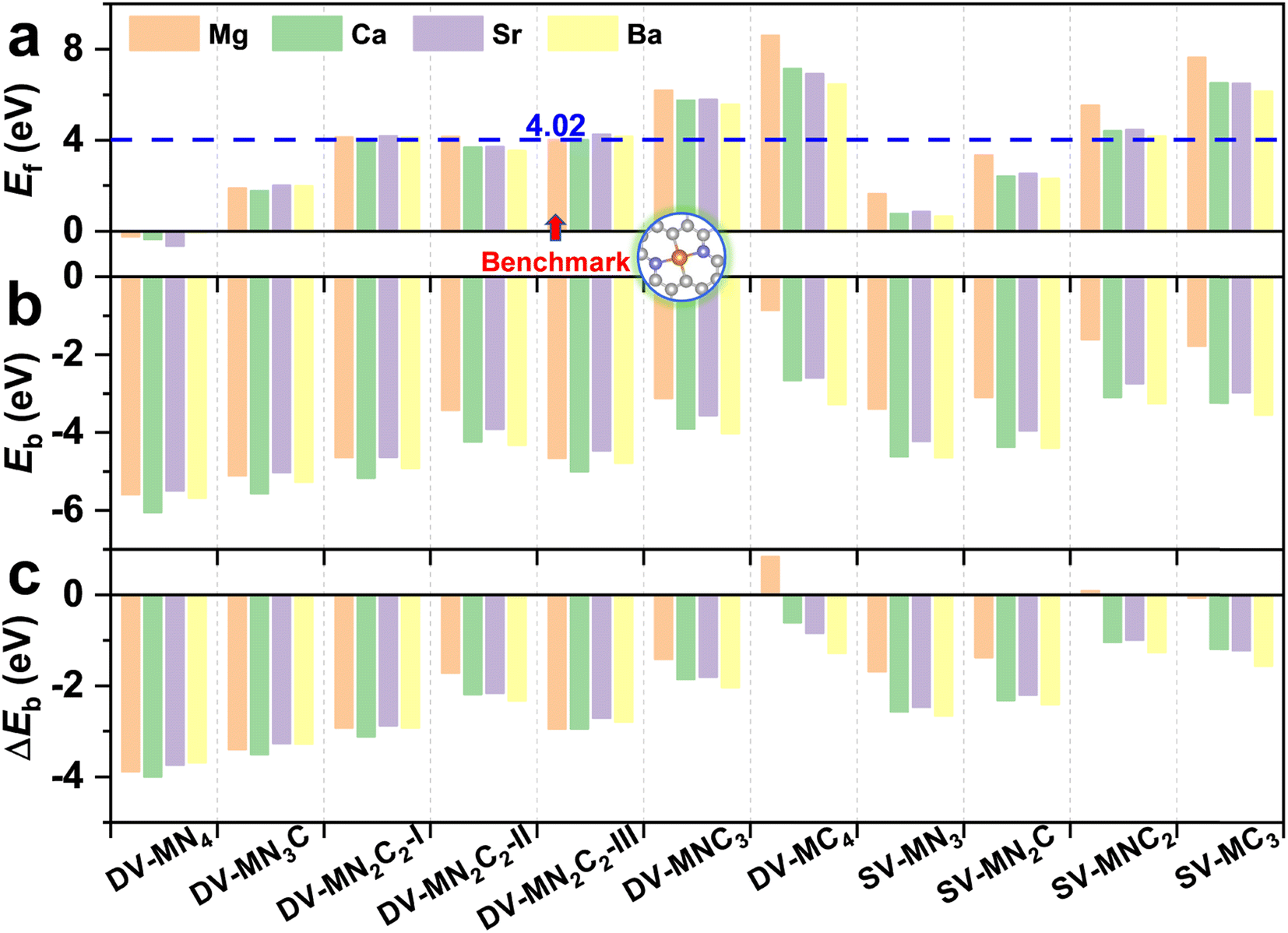 | ||
| Fig. 2 The calculated formation energy, Ef (a), binding energy, Eb (b), and the energy difference between Eb and Ecoh, ΔEb (c) for the AE-SAC systems. DV-MgN2C2-III is adopted as a reference for the formation energy. | ||
3.2. Adsorption of NO3− on AE-SACs
As an initial and pivotal step for the eNO3RR process, NO3− should be adsorbed on the active center effectively. As illustrated in Fig. S8a,† all the possible NO3− adsorption configurations have been taken into account, including those with one O atom or two O atoms binding, and the parallel pattern. In addition, we have also considered several possible adsorption positions around the AE metal active centers for *NO3 and the other key reaction intermediates (Fig. S8b†). The most stable adsorption configurations for *NO3 are shown in Fig. S9,† meanwhile, some important parameters (bond length, charge transfer, and ΔG(*NO3)) are displayed in Table S4.† We can see that for all the cases the configuration with two O atoms binding with the AE metal atom is the most stable. Intriguingly, the values of ΔG(*NO3) are all less than −1.0 eV (ranging from −1.18 to −3.26 eV), indicating that the active site consisting of AE metal can strongly adsorb NO3−, similar to the TM active site.33,45 However, contrary to TM active sites, for all the systems, a considerable amount of charge transfers (∼1.6e) from the catalysts to NO3− can be observed, suggesting that the binding of NO3− mainly relied on the ionic bond interaction, for which the detailed analysis will be presented below.It is known that hydrogen evolution reaction (HER) is the main competing reaction against eNO3RR mainly because protons can also bind with the active sites.33 To effectively inhibit this disadvantage, active sites should be preferentially occupied by NO3− instead of H atoms, which requires strong adsorption of NO3− compared with H.45,51 The adsorption configurations for *H are displayed in Fig. S10,† meanwhile, some important parameters (bond length, charge transfer, and ΔG(*H)) are compiled in Table S5.† As shown in Fig. 3a, for all the systems, ΔG(*NO3) is negative, while ΔG(*H) is positive (except SV-MgN3), indicating that the adsorption of NO3− is much stronger than that of H atoms. Therefore, eNO3RR will dominate and the HER can be effectively inhibited on these SACs. By analyzing the charge transfer (Fig. 3b), it is found that, for all the cases, NO3− gains more electrons from SACs than H atoms, and the NO3− bonded AE metal atoms carry more positive charges than the H-bonded ones, which could be responsible for the much stronger binding of NO3−.
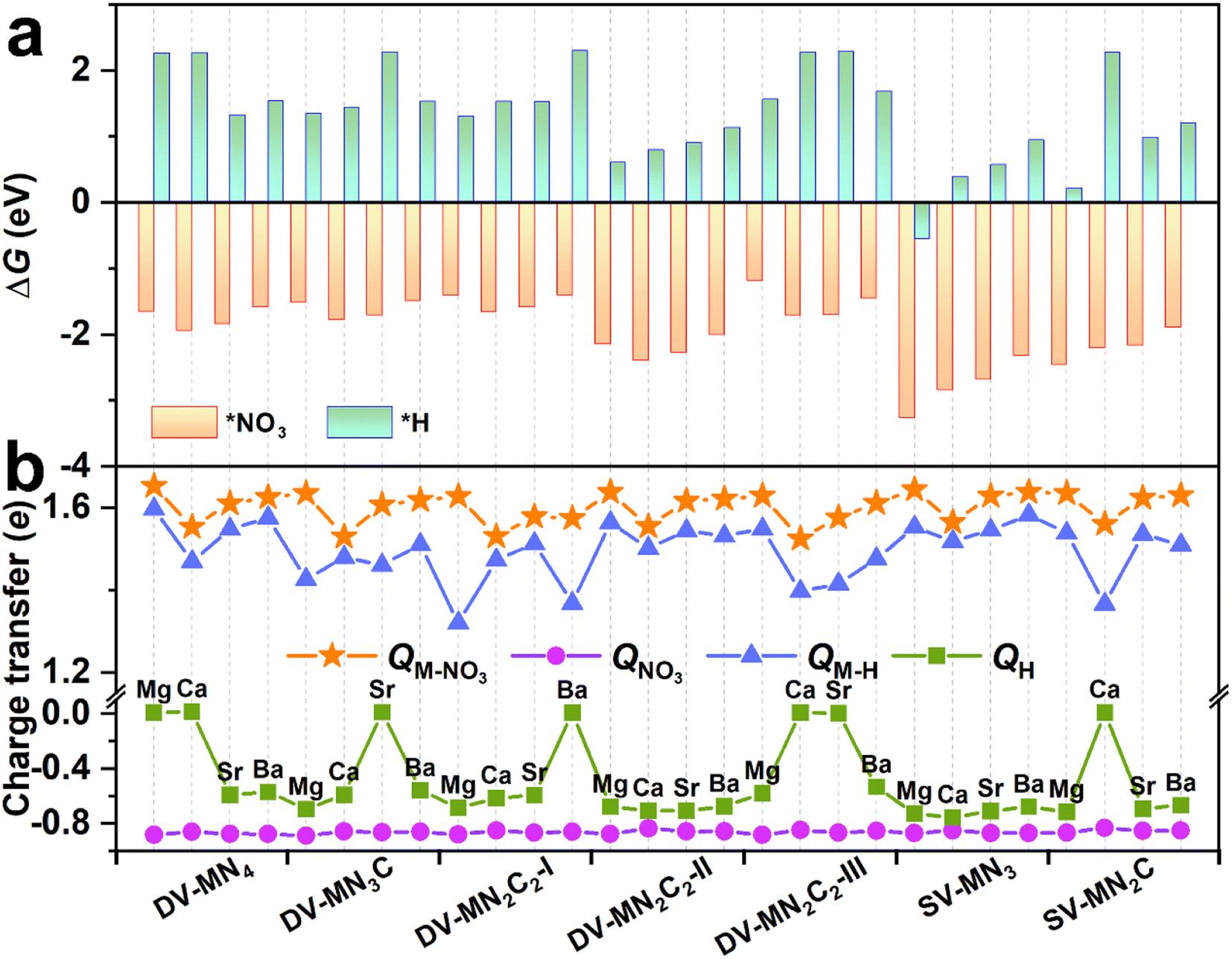 | ||
| Fig. 3 Adsorption free energy (ΔG) (a) and charge transfer (Q) (b) for NO3− and H adsorption on the considered AE-SAC systems. QM–NO3 and QM–H denote the charge transfer of AE metals, and QNO3 and QH are the charge transfer of *NO3 and *H, respectively. The positive and negative values represent the electron loss and electron gain, respectively. | ||
3.3. Mechanism of eNO3RR on AE-SACs
From the previously reported results, the binding strength of NO3− can serve as the activity descriptor for eNO3RR, and for various kinds of SACs, the active sites with ΔG(*NO3) of around −2 eV show high eNO3RR activity.45–48 Above results show that by changing the AE metal active center and its coordination environment, ΔG(*NO3) values can be adjusted from −1.18 to −3.26 eV. Therefore, it is expected that the AE-SACs can achieve high catalytic activity for eNO3RR. Despite the complex eNO3RR process with multiple products, according to the Pourbaix diagram of nitrogen species, NH3/NH4+ has been identified as the most stable product in thermodynamics under negative electrode potential.45,75 Therefore, it is essential to make a thorough investigation of the reaction mechanism of the selective electroreduction of NO3− to NH3. Fig. 4a displays the exhaustive reaction pathways of eNO3RR, where all the possible configurations of *HNO3, *NO2, and *HNO2 have been considered. Given the multiple configurations and hydrogenation positions of intermediates during the eNO3RR process, to economically and effectively obtain the optimal pathway, only the most stable structure in each elementary step is chosen to further study the subsequent reaction. Generally, UL has been used to evaluate the catalytic activity of an electrocatalytic reaction,65 and the calculated UL for these AE-SACs are shown in Fig. 4b and tabulated in Table S6.† Surprisingly, in contrast with the traditional view that the AE metal active centers are catalytically inactive, most of the AE-SACs exhibit a high eNO3RR activity with UL lower than −0.6 V. Especially, DV-SrN3C and DV-BaN3C possess the highest eNO3RR activity with an ultralow UL of −0.05 V. This value is much lower than those of the previous predicted TM-based SACs. For example, UL of Ti/g-CN,45 Hf/g-C3N4,46 Ru/g-C3N4,47 V/g-C3N4,48 Os/GDY,49 V/h-BP,51 and Fe/NG52 are −0.39, −0.11, −0.34, −0.25, −0.37, −0.22, and −0.38 V, respectively. Therefore, the catalysts with active sites consisting of AE metal could be even superior to those consisting of TM.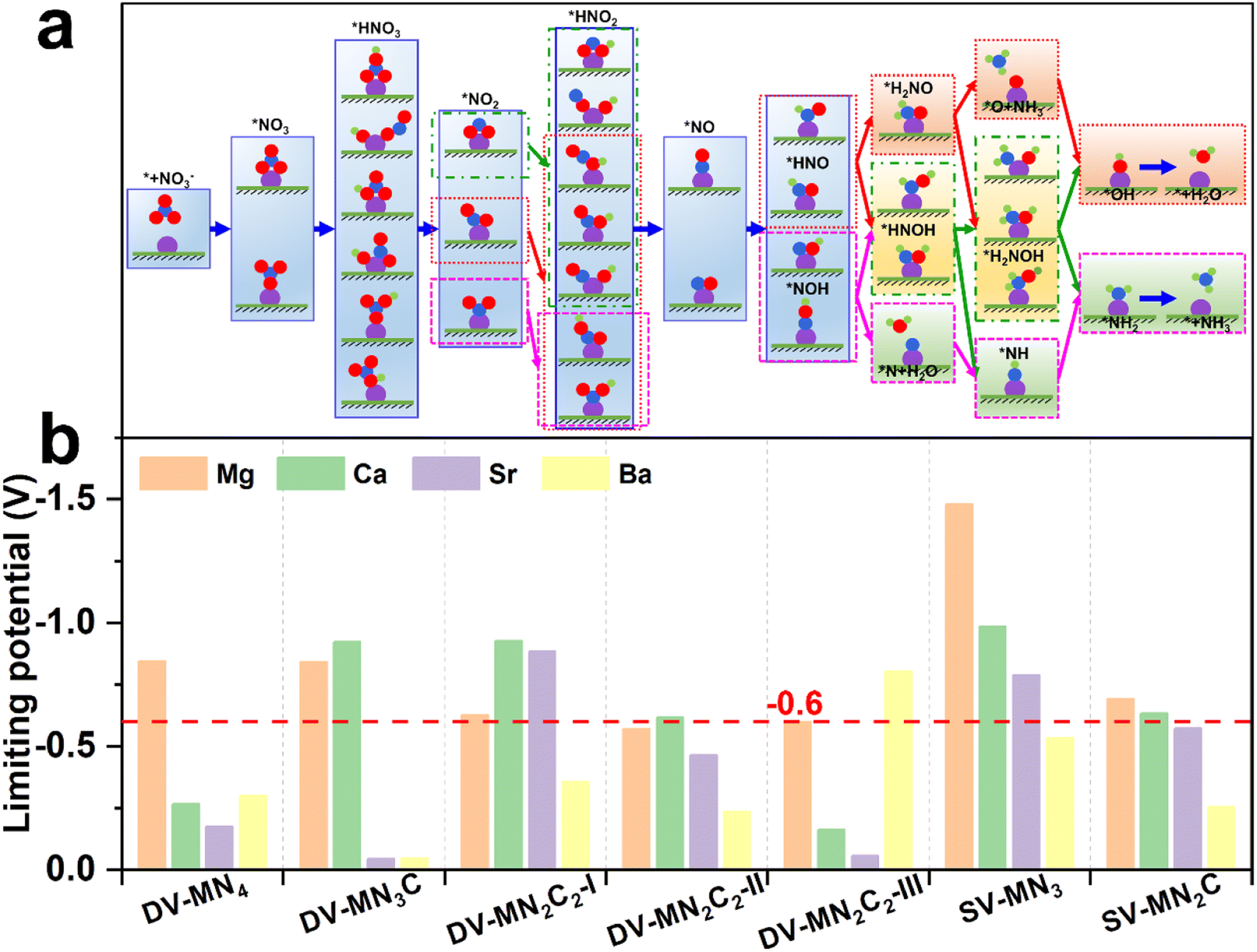 | ||
| Fig. 4 (a) Schematic diagram of possible eNO3RR pathways. (b) Summary of the limiting potential of the AE-SACs for eNO3RR through the minimum energy pathway. | ||
The calculated UL as a function of ΔG(*NO3) is presented in Fig. 5, interestingly, which also exhibits a prototypical volcano-shaped correlation, similar to the case of SACs with TM as the active center.45,47–49,52 One can see that, on the left branch of the volcano diagram, too strong adsorption of NO3− often leads to the last hydrogenation step (*OH → * + H2O) as PDS, while the relatively weak adsorption of NO3− results in the early hydrogenation step (*NO3 → *OH + *NO2/*NO2 → *OH + *NO) as PDS. Note that the optimal systems herein, DV-SrN3C and DV-BaN3C are exactly located around the peak of the volcano diagram with ΔG(*NO3) of −1.71 and −1.48 eV, respectively. In addition to DV-SrN3C and DV-BaN3C, there are also a large number of other AE-SAC systems (highlighted by the green background in Fig. 5) possessing acceptable UL, which are comparable to the reported TM catalysts, marked by the purple circle in Fig. 5.45–50 Furthermore, it is noted that ΔG(*NO3) of the above AE-SACs stand in the range from −1.3 to −2.5 eV, similar to the systems with TM active centers.45–50
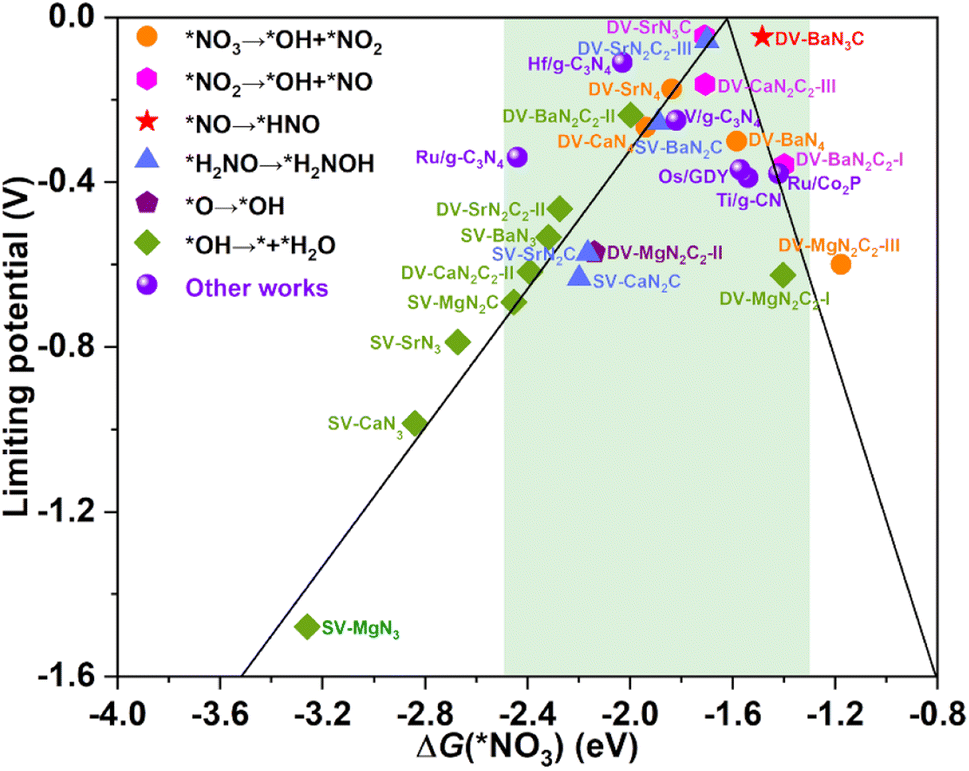 | ||
| Fig. 5 The eNO3RR volcano diagram of the considered AE-SACs with the descriptor of ΔG(*NO3). The purple spheres denote other works for comparison45–50 The green background encloses those AE-SACs with acceptable limiting potentials. | ||
To get more insight into the reaction mechanism, the eNO3RR pathways on DV-BaN3C and DV-SrN3C are plotted in Fig. 6, and those for other systems are depicted in Fig. S11–S16.† Interestingly, on DV-BaN3C, the first hydrogenation step of *NO3 occurs at the O site bonded to the Ba atom, generating the co-adsorbed *OH and *NO2 with a free energy downhill of 0.40 eV (Fig. 6a), which is completely different from previous works that the proton–electron (H+ + e−) pair firstly attacks the terminal O of *NO3.45,51 The second hydrogenation step results in the release of the first H2O molecule with the free energy downhill of 0.87 eV, leaving the NO2 asymmetric adsorption on DV-BaN3C via one Ba–O bond and one Ba–N bond. Similar to the first hydrogenation step, the third H+ + e− pair also prefers to attack the proximal O atom (bonded to Ba) of *NO2, breaking another N–O bond to form *OH + *NO with the free energy uphill of only 0.02 eV. Then, the second water molecule is formed exothermically. The *NO trends to adopt the end-on pattern with the N atom bonding to the Ba atom, similar to the cases of TM active centers.45,51 Then, the fifth H+ + e− pair prefers to be added on the N atom of *NO, turning the end-on adsorption pattern to a side-on adsorption pattern, which is also identified as the PDS with a free energy uphill of 0.05 eV. The subsequent electroreduction processes are all exothermic. In detail, the hydrogenation of *HNO follows the pathway, *HNO → *H2NO → *O + NH3 (g) → OH* → * + H2O (l), to recover the active center.
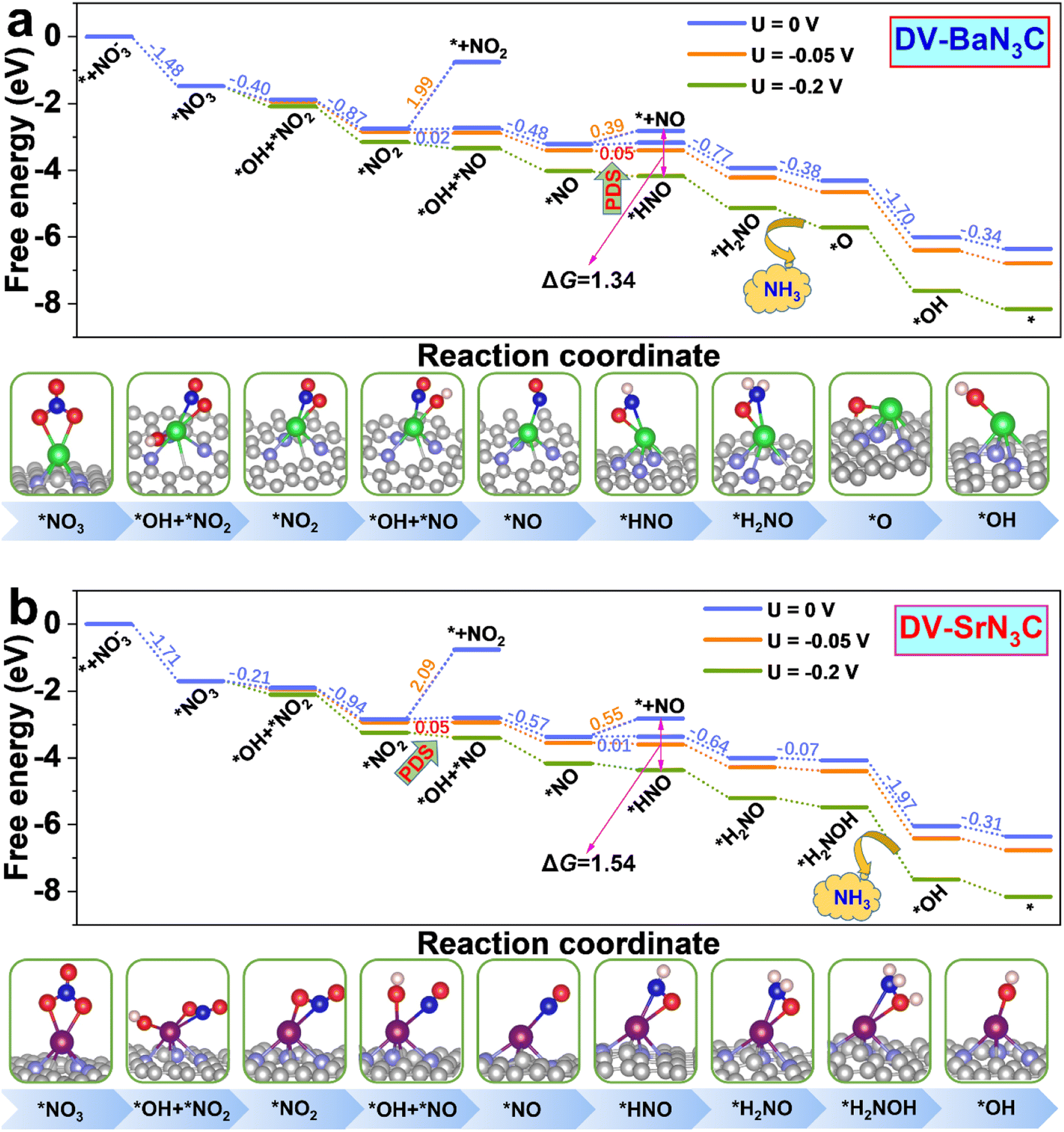 | ||
| Fig. 6 Free energy diagrams together with corresponding intermediate configurations for eNO3RR on DV-BaN3C (a) and DV-SrN3C (b) under the potentials of 0, –0.05, and −0.2 V. | ||
Overall, the eNO3RR pathway on DV-SrN3C (Fig. 6b) is almost the same as that on DV-BaN3C, and the only difference is that the seventh hydrogenation step occurs on the O atom to form *H2NOH, and NH3 is released after the eighth hydrogenation step (*H2NOH → *OH + NH3 (g)). It is noted that all the reaction steps are exothermic on DV-SrN3C, except the third (*NO2 → *OH + *NO) and fifth (*NO → *HNO) hydrogenation steps with the free energy uphill of 0.05 and 0.01 eV, respectively.
Moreover, we also calculated the adsorption-free energy of *H2O (ΔG*H2O), and the reaction-free energy of the hydrogenation of *OH to *H2O (ΔG*OH→*H2O) for the studied AE-SAC systems. As seen in Table S7,† the negative ΔG*OH→*H2O for most of the AE-SAC systems indicate that the hydrogenation of *OH to form *H2O is spontaneous and quite easy, thereby, avoiding the *OH poisoning. It is worth mentioning that the SV-MgN3, SV-CaN3, and SV-SrN3 systems with large ΔG*OH→*H2O exhibit poor catalytic activity, which requires a high potential to remove *OH. Moreover, the ΔG*H2O values for all AE-SAC systems are larger than −0.6 eV, indicating the weak adsorption of H2O in the AE-SAC systems. Combining the aforementioned stronger adsorption of *NO3 than *H, it can be concluded that the *H, *OH, and *H2O will not poison the proposed catalysts (except SV-MgN3, SV-CaN3, and SV-SrN3).
For the reaction selectivity, besides the aforementioned HER, other byproducts, mainly NO2, NO, N2O, and N2, during eNO3RR may be produced. It can be seen from Fig. 6 that direct desorption of *NO2 from DV-BaN3C and DV-SrN3C is extremely difficult because large energy barriers as high as ∼2 eV are needed to overcome. Although the desorption energy barriers of *NO are not particularly high as NO2 (0.39 and 0.55 eV), they are much higher than those for further hydrogenation (0.05 and 0.01 eV). Especially, when a tiny potential was applied, the further hydrogenation of *NO would be much more favorable thermodynamically. For instance, with an applied potential of −0.2 V, the free energy difference between the NO desorption and hydrogenation will exceed 1.3 eV. Therefore, *NO will further undergo electrochemical hydrogenation on DV-BaN3C and DV-SrN3C. On the other side, the NO dimer (N2O2) with cis O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–NO structure is the vital precursor to form N2O and N2, which could be formed by the coupling of two NO.49,76,77 Obviously, the isolated single-atom active site cannot accommodate two NO3− simultaneously, and thus Langmuir–Hinshelwood mechanism for the formation of NO dimer can be excluded.78 For the Eley–Rideal mechanism, the *NO needs to combine with a solvated NO molecule to form a NO dimer.79 However, due to that *NO will be further hydrogenated rather than desorbed, the Eley–Rideal mechanism for NO dimer formation is also infeasible. Hence, the formation of NO2, NO, N2O, and N2 will be effectively inhibited. Moreover, the desorption of the target product NH3 is exothermic and spontaneous, further confirming high eNO3RR selectivity toward NH3 on DV-BaN3C and DV-SrN3C.
N–NO structure is the vital precursor to form N2O and N2, which could be formed by the coupling of two NO.49,76,77 Obviously, the isolated single-atom active site cannot accommodate two NO3− simultaneously, and thus Langmuir–Hinshelwood mechanism for the formation of NO dimer can be excluded.78 For the Eley–Rideal mechanism, the *NO needs to combine with a solvated NO molecule to form a NO dimer.79 However, due to that *NO will be further hydrogenated rather than desorbed, the Eley–Rideal mechanism for NO dimer formation is also infeasible. Hence, the formation of NO2, NO, N2O, and N2 will be effectively inhibited. Moreover, the desorption of the target product NH3 is exothermic and spontaneous, further confirming high eNO3RR selectivity toward NH3 on DV-BaN3C and DV-SrN3C.
The electrode potential could significantly affect the selectivity of eNO3RR against HER.36 To be specific, the more negative electrode potential can bring more positive ΔG(*NO3) but more negative ΔG(*H), so the adsorption of NO3− will become less favorable, whereas, the adsorption of the H atom may be dominant, exacerbating the selectivity problem. As shown in Fig. S17,† the adsorption of NO3− is still stronger than that of H on DV-BaN3C and DV-SrN3C even after applying a potential as large as −1.0 V. Therefore, the robust adsorption of NO3− could completely suppress the HER under the limiting potential, again affirming the high eNO3RR selectivities of DV-BaN3C and DV-SrN3C.
Finally, the effect of the solvation on eNO3RR was investigated for DV-BaN3C and DV-SrN3C. One can see from the free energy diagrams in Fig. S18† that the overall reaction pathway and PDS were unchanged when considering the solvation correction. In addition, UL values are slightly decreased by 0.05 and 0.06 V for DV-BaN3C and DV-SrN3C with solvation correction, respectively, similar to other works.39,48,49
3.4. Activity origin of eNO3RR on AE-SACs
As the binding strength of NO3− can well predict the activity trend of eNO3RR, the investigation of its activation mechanism and binding strength change, based on the electronic structure analysis, could give us insight into the activity origin. For comparison, DV-BaN3C and SV-MgN3 were considered, and they exhibited the highest (UL = −0.05 V) and lowest (UL = −1.48 V) eNO3RR activity, respectively. Firstly, the charge density difference (CDD) is displayed in Fig. 7a and b show that considerable electrons are transferred from the embedded AE metals to the graphene support, and the AE metal atoms should be significantly positively charged, which is in agreement with the Bader charge analysis (1.42e for Ba and 1.20e for Mg). Consequently, the AE metals are stabilized by mainly ionic bond interaction, as verified by electron localization function (ELF) analysis. The positively-charged AE metals are responsible for the stable adsorption of the oxygenated species. For the adsorption of NO3− on DV-BaN3C and SV-MgN3, Bader charge analysis gives that *NO3 gains 0.86 and 0.87e from the support, respectively. NO3− adsorbed Ba and Mg were positively charged by 1.62 and 1.64, respectively. Therefore, the adsorption of NO3− is mainly through the ionic bond interaction. However, from Fig. 7c and d, ELF results show that there is a certain charge overlap between *NO3 and its bonded AE metal atoms, and especially, more electrons are located between *NO3 and Mg than *NO3 and Ba, indicating stronger covalent interaction between *NO3 and Mg. It is worth noting that the charge enclosed on the Ba atom of the DV-BaN3C system is an indicator of its lone-pair electrons. Importantly, CDD for NO3 adsorbed systems indicates that charge accumulation and depletion occur simultaneously on *NO3. Especially, the electrons obtained by *NO3 mainly occupy its antibonding orbitals (positive section around the periphery of O atoms of *NO3, as shown in Fig. 7c and d), while *NO3 also loses some charges from its bonding orbitals (negative section on the N–O bonds of *NO3, as shown in Fig. 7c and d). Therefore, NO3− can be effectively activated by the AE metal active center, through the “donation-back donation” mechanism, similar to the cases of TM active center,51,52 the reason for which will be discussed later.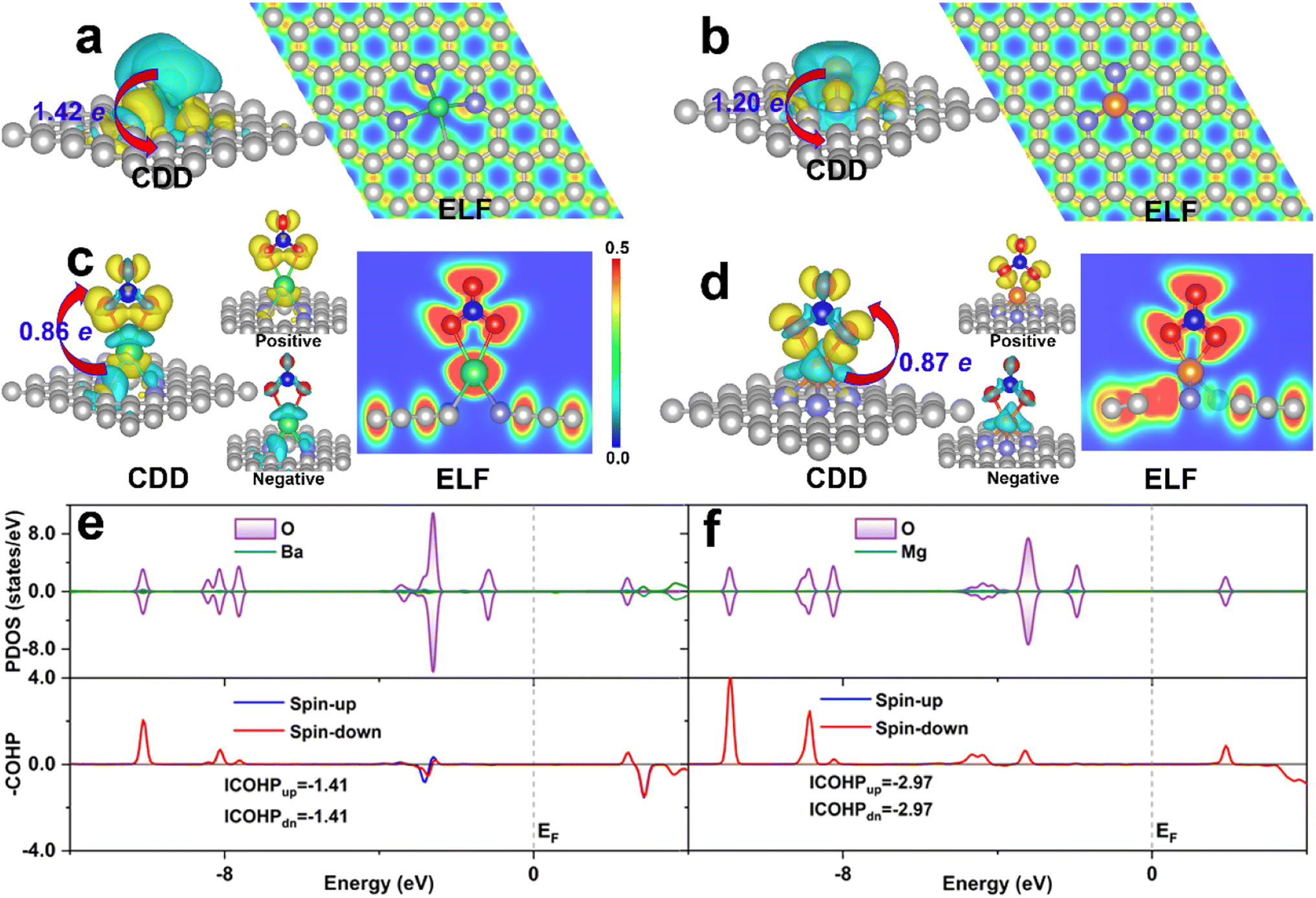 | ||
| Fig. 7 The charge density difference (CDD) and electron localization function (ELF) for the bare DV-BaN3C (a), bare SV-MgN3 (b), NO3 adsorbed DV-BaN3C (c) and NO3 adsorbed SV-MgN3 (d). Projected densities of states (PDOS) and COHP for the NO3 adsorbed DV-BaN3C (e) and NO3 adsorbed SV-MgN3 (f). For CDD, the yellow and cyan regions denote electron accumulation and depletion, respectively, and the isosurface value was set to 0.002 e/bohr−3. In PDOS and COHP, the Fermi level (EF) is labeled by a vertical dash line and set to 0 eV. COHP was calculated for the interaction between NO3− and Ba (Mg). | ||
The above picture of NO3− adsorption can be supported by the DOS analysis. From Fig. 7e and f, we can see that a large number of electronic states of the O atoms in *NO3, which are bonded with the AE metal atom, are located below the Fermi level, while the electronic states of the Ba and Mg atoms are very few. This result is in line with the ionic bond nature discussed above. Furthermore, crystal orbital Hamilton population (COHP)80 analyses are shown in Fig. 7e and f indicate that there are more bonding states (positive value) between *NO3 and Mg than those between *NO3 and Ba. In addition, there are also some antibonding states occupied between *NO3 and Ba (negative value). Integrated COHP (ICOHP) can quantitatively characterize the covalent interaction strength, that is, a more negative ICOHP signifies a stronger chemical bond interaction. The ICOHP (−5.94 eV) for the Mg–O bond is much more negative than that for the Ba–O bond (−2.82 eV). This verifies the stronger covalent interaction between *NO3 and Mg than *NO3 and Ba. Indeed, ΔG(*NO3) was as high as −3.26 eV, for SV-MgN3, which resulted in a large energy barrier (1.48 eV) in the last step of *OH → * + H2O (Fig. S11c†). In contrast, the moderate chemical activity of the Ba active center in DV-BaN3C endows it with ultrahigh eNO3RR activity.
In order to further understand the excellent catalytic activity of the AE active center for eNO3RR, the charge variations along with the reaction pathway for DV-BaN3C and DV-SrN3C were analyzed by subdividing the systems into three moieties, i.e., the N-doped graphene (moiety1), AE metal atom (moiety2), and adsorbate (moiety3), as illustrated in Fig. S19.† The detailed charge variations of the three moieties are presented in Fig. 8a and b, where steps 0 and 1 represent the charge distribution of the bare support and the NO3− adsorbed support, and steps 2–9 are the subsequent hydrogenation steps. Concretely, during the eNO3RR reaction processes, the net charge of the Ba and Sr atoms remains almost unchanged (∼1.50e), and the charge change trends are similar for DV-BaN3C and DV-SrN3C. Meanwhile, the charges of the support (moiety1) dynamically change, which exchange charges with the intermediates indirectly with the AE ion as transmitters. Especially, the electron reservoir nature of the support enables the activation of NO3− following the charge “donation-back donation” mechanism, mimicking the behavior of TM active centers.51,52
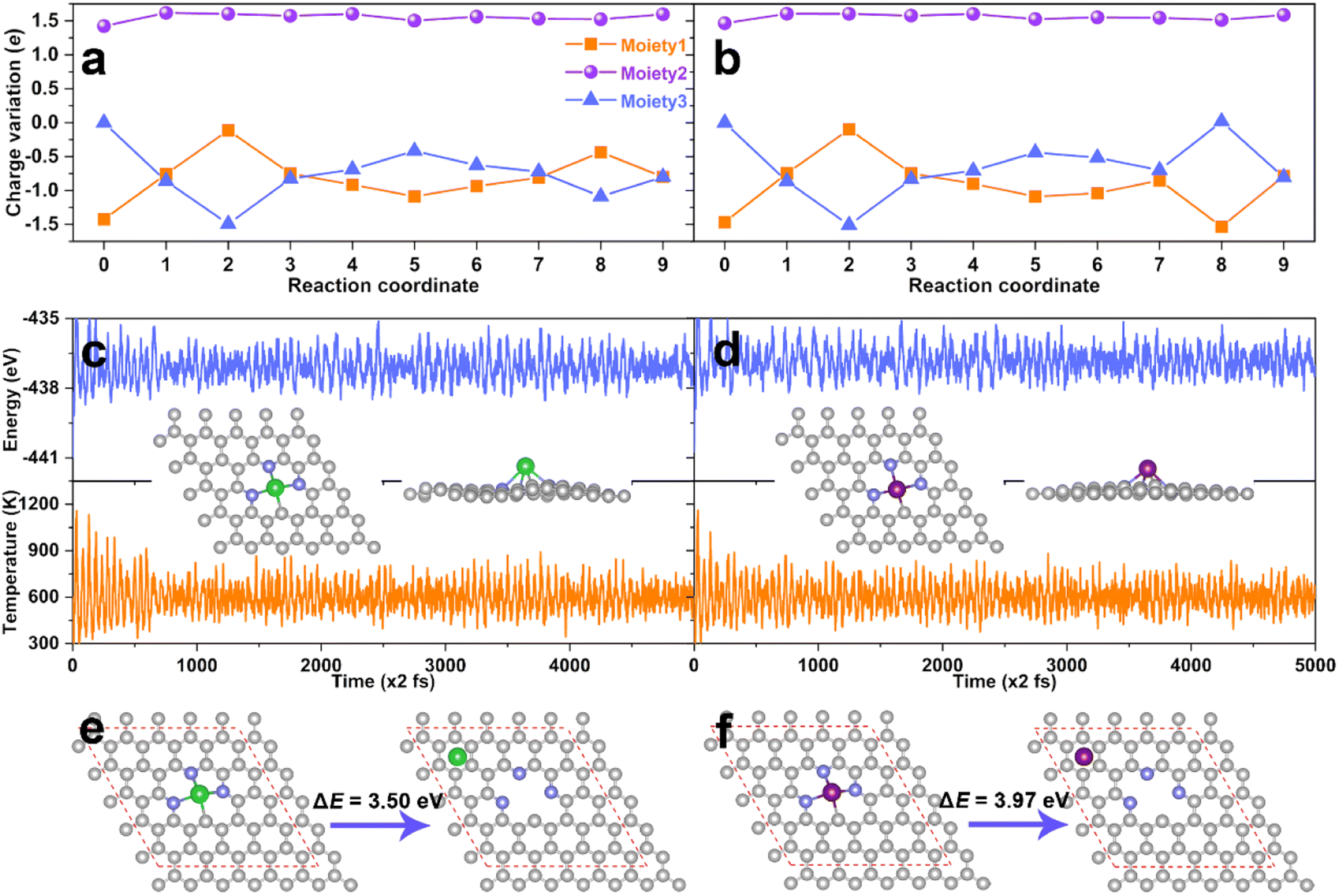 | ||
| Fig. 8 The charge variation during the eNO3RR process on DV-BaN3C (a) and DV-SrN3C (b). Energy and temperature evolution during the AIMD simulation for DV-BaN3C (c) and DV-SrN3C (d). Insets are the top and side views of the snapshots after 10 ps. The most stable configuration and second stable one with the Ba (e) and Sr (f) near the hollow sites, where ΔE is their energy difference. | ||
As discussed above, the thermodynamic stability of DV-BaN3C and DV-SrN3C was confirmed. Next, the thermal stability was further checked through AIMD simulation. As plotted in Fig. 8c and d, the temperature and energy curves oscillate near the equilibrium states, and the DV-BaN3C and DV-SrN3C almost retain their intact geometric structures after 10 ps AIMD simulations under 600 K, implying their high thermal stability. Kinetically, Fig. 8e and f show that the energy differences between the most stable configuration and the second stable one with the Ba and Sr near the hollow sites are as large as 3.50 and 3.97 eV, respectively. Such high energy barriers mean that the diffusion of Ba or Sr atoms is unlikely on the graphene support, confirming the kinetic stability. Finally, the electrochemical stability was also evaluated by calculating the dissolution potential (Udiss).67 The obtained Udiss are −0.90, −1.26, and −1.27 V for DV-MgN2C2-III, DV-SrN3C, and DV-BaN3C systems, respectively. Considering that DV-MgN2C2-III SAC was experimentally synthesized and used in the oxygen reduction reaction,14 DV-SrN3C and DV-BaN3C SACs are also stable enough under the electrochemical conditions due to their comparable Udiss values compared with the experimentally synthesized MgN2C2-III system.
4. Conclusions
In summary, based on the first-principles calculations, the feasibility of AE metals as active centers for eNO3RR to produce NH3 was systematically investigated by taking a series of AE-SACs as representatives, which were constructed by embedding AE metal (Mg, Ca, Sr, Ba) single atom into the N-doped graphene. Surprisingly, similar to the TM active sites, it was found that the AE metal active centers could also strongly adsorb and effectively activate NO3−, to achieve excellent eNO3RR activity. By thoroughly investigating the possible reaction pathways, a volcano diagram was established with ΔG(*NO3) as the descriptor. The DV-BaN3C and DV-SrN3C systems were exactly located around the peak of the volcano diagram, exhibiting the highest activity for eNO3RR to produce NH3 with near-zero UL of −0.05 V, comparable and even better than the TM-based SACs. With good stability, both systems also exhibited excellent selectivity against the generation of the byproducts, including H2, NO2, NO, N2O, and N2. Moreover, the electronic structure analysis indicates that although AE metal elements are highly ionic during the reaction, the support can indirectly exchange charge via the AE cations with NO3− and intermediates effectively, which enables the effective activation of NO3−, via the charge “donation-back donation” mechanism, and further completes the catalytic cycle. On the one hand, these findings propose a novel kind of electrocatalyst for eNO3RR, which may be also suitable for other reactions. Moreover, this work lays a theoretical foundation for the development of heterogeneous catalysts with AE metal as active centers, and emphasizes the great potential of AE metals, beyond transition metals, as active centers in heterogeneous catalysis.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant No. 12074099) and the Program for Science & Technology Innovation Talents in Universities of Henan Province (Grant No. 20HASTIT028).References
- R. Schlögl, Angew. Chem., Int. Ed., 2015, 54, 3465–3520 CrossRef PubMed.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- B. W. J. Chen, L. Xu and M. Mavrikakis, Chem. Rev., 2021, 121, 1007–1048 CrossRef CAS PubMed.
- G. Blyholder, J. Phys. Chem., 1964, 68, 2772–2777 CrossRef CAS.
- B. Hammer, Y. Morikawa and J. K. Nørskov, Phys. Rev. Lett., 1996, 76, 2141–2144 CrossRef CAS PubMed.
- M.-A. Légaré, G. Bélanger-Chabot, D. Dewhurst Rian, E. Welz, I. Krummenacher, B. Engels and H. Braunschweig, Science, 2018, 359, 896–900 CrossRef PubMed.
- B. Rösch, T. X. Gentner, J. Langer, C. Färber, J. Eyselein, L. Zhao, C. Ding, G. Frenking and S. Harder, Science, 2021, 371, 1125–1128 CrossRef PubMed.
- J. A. Cowan, BioMetals, 2002, 15, 225–235 CrossRef CAS PubMed.
- C. Sissi and M. Palumbo, Nucleic Acids Res., 2009, 37, 702–711 CrossRef CAS PubMed.
- S.-L. Yean, G. Wuenschell, J. Termini and R.-J. Lin, Nature, 2000, 408, 881–884 CrossRef CAS PubMed.
- M. F. Dunn, J. A. Ramírez-Trujillo and I. Hernández-Lucas, Microbiology, 2009, 155, 3166–3175 CrossRef CAS PubMed.
- W. Guo, H. Nazim, Z. Liang and D. Yang, Crop J., 2016, 4, 83–91 CrossRef.
- M. S. Hill, D. J. Liptrot and C. Weetman, Chem. Soc. Rev., 2016, 45, 972–988 RSC.
- S. Liu, Z. Li, C. Wang, W. Tao, M. Huang, M. Zuo, Y. Yang, K. Yang, L. Zhang, S. Chen, P. Xu and Q. Chen, Nat. Commun., 2020, 11, 938 CrossRef CAS PubMed.
- Z. Lin, H. Huang, L. Cheng, W. Hu, P. Xu, Y. Yang, J. Li, F. Gao, K. Yang, S. Liu, P. Jiang, W. Yan, S. Chen, C. Wang, H. Tong, M. Huang, W. Zheng, H. Wang and Q. Chen, Adv. Mater., 2021, 33, 2107103 CrossRef CAS PubMed.
- K. Dong, J. Liang, Y. Wang, L. Zhang, Z. Xu, S. Sun, Y. Luo, T. Li, Q. Liu, N. Li, B. Tang, A. A. Alshehri, Q. Li, D. Ma and X. Sun, ACS Catal., 2022, 12, 6092–6099 CrossRef CAS.
- Q. Wang, K. Liu, J. Fu, C. Cai, H. Li, Y. Long, S. Chen, B. Liu, H. Li, W. Li, X. Qiu, N. Zhang, J. Hu, H. Pan and M. Liu, Angew. Chem., Int. Ed., 2021, 60, 25241–25245 CrossRef CAS.
- D. Bao, Q. Zhang, F.-L. Meng, H.-X. Zhong, M.-M. Shi, Y. Zhang, J.-M. Yan, Q. Jiang and X.-B. Zhang, Adv. Mater., 2017, 29, 1604799 CrossRef.
- N. Lehnert, H. T. Dong, J. B. Harland, A. P. Hunt and C. J. White, Nat. Rev. Chem., 2018, 2, 278–289 CrossRef CAS.
- K. Chu, Y. Luo, P. Shen, X. Li, Q. Li and Y. Guo, Adv. Energy Mater., 2022, 12, 2103022 CrossRef CAS.
- X. Li, P. Shen, Y. Luo, Y. Li, Y. Guo, H. Zhang and K. Chu, Angew. Chem., Int. Ed., 2022, 61, e202205923 CAS.
- J. Zhao and Z. Chen, J. Am. Chem. Soc., 2017, 139, 12480–12487 CrossRef CAS PubMed.
- C. Ling, Y. Ouyang, Q. Li, X. Bai, X. Mao, A. Du and J. Wang, Small Methods, 2019, 3, 1800376 CrossRef.
- B. H. R. Suryanto, H.-L. Du, D. Wang, J. Chen, A. N. Simonov and D. R. MacFarlane, Nat. Catal., 2019, 2, 290–296 CrossRef CAS.
- P. Shen, X. Li, Y. Luo, Y. Guo, X. Zhao and K. Chu, ACS Nano, 2022, 16, 7915–7925 CrossRef CAS PubMed.
- Y. Luo, Q. Li, Y. Tian, Y. Liu and K. Chu, J. Mater. Chem. A, 2022, 10, 1742–1749 RSC.
- P. H. van Langevelde, I. Katsounaros and M. T. M. Koper, Joule, 2021, 5, 290–294 CrossRef.
- X. Lu, H. Song, J. Cai and S. Lu, Electrochem. Commun., 2021, 129, 107094 CrossRef CAS.
- Y. Wang, A. Xu, Z. Wang, L. Huang, J. Li, F. Li, J. Wicks, M. Luo, D.-H. Nam, C.-S. Tan, Y. Ding, J. Wu, Y. Lum, C.-T. Dinh, D. Sinton, G. Zheng and E. H. Sargent, J. Am. Chem. Soc., 2020, 142, 5702–5708 CrossRef CAS PubMed.
- R. Jia, Y. Wang, C. Wang, Y. Ling, Y. Yu and B. Zhang, ACS Catal., 2020, 10, 3533–3540 CrossRef CAS.
- X. Zhang, Y. Wang, C. Liu, Y. Yu, S. Lu and B. Zhang, Chem. Eng. J., 2021, 403, 126269 CrossRef CAS.
- F.-Y. Chen, Z.-Y. Wu, S. Gupta, D. J. Rivera, S. V. Lambeets, S. Pecaut, J. Y. T. Kim, P. Zhu, Y. Z. Finfrock, D. M. Meira, G. King, G. Gao, W. Xu, D. A. Cullen, H. Zhou, Y. Han, D. E. Perea, C. L. Muhich and H. Wang, Nat. Nanotechnol., 2022, 17, 759–767 CrossRef CAS PubMed.
- G.-F. Chen, Y. Yuan, H. Jiang, S.-Y. Ren, L.-X. Ding, L. Ma, T. Wu, J. Lu and H. Wang, Nat. Energy, 2020, 5, 605–613 CrossRef CAS.
- J. Liang, B. Deng, Q. Liu, G. Wen, Q. Liu, T. Li, Y. Luo, A. A. Alshehri, K. A. Alzahrani, D. Ma and X. Sun, Green Chem., 2021, 23, 5487–5493 RSC.
- T. Mou, J. Liang, Z. Ma, L. Zhang, Y. Lin, T. Li, Q. Liu, Y. Luo, Y. Liu, S. Gao, H. Zhao, A. M. Asiri, D. Ma and X. Sun, J. Mater. Chem. A, 2021, 9, 24268–24275 RSC.
- J.-X. Liu, D. Richards, N. Singh and B. R. Goldsmith, ACS Catal., 2019, 9, 7052–7064 CrossRef CAS.
- Y. Lin, J. Liang, H. Li, L. Zhang, T. Mou, T. Li, L. Yue, Y. Ji, Q. Liu, Y. Luo, N. Li, B. Tang, Q. Wu, M. S. Hamdy, D. Ma and X. Sun, Mater. Today Phys., 2022, 22, 100611 CrossRef CAS.
- Z.-Y. Wu, M. Karamad, X. Yong, Q. Huang, D. A. Cullen, P. Zhu, C. Xia, Q. Xiao, M. Shakouri, F.-Y. Chen, J. Y. Kim, Y. Xia, K. Heck, Y. Hu, M. S. Wong, Q. Li, I. Gates, S. Siahrostami and H. Wang, Nat. Commun., 2021, 12, 2870 CrossRef CAS PubMed.
- P. Lv, D. Wu, B. He, X. Li, R. Zhu, G. Tang, Z. Lu, D. Ma and Y. Jia, J. Mater. Chem. A, 2022, 10, 9707–9716 RSC.
- H.-Y. Zhuo, X. Zhang, J.-X. Liang, Q. Yu, H. Xiao and J. Li, Chem. Rev., 2020, 120, 12315–12341 CrossRef CAS PubMed.
- Y. Wang, M. Wang, Z. Lu, D. Ma and Y. Jia, Nanoscale, 2021, 13, 13390–13400 RSC.
- J. Zhang, H. Zhang, Y. Wu, C. Liu, Y. Huang, W. Zhou and B. Zhang, J. Mater. Chem. A, 2022, 10, 5743–5757 RSC.
- P. Li, Z. Jin, Z. Fang and G. Yu, Energy Environ. Sci., 2021, 14, 3522–3531 RSC.
- J. Li, Y. Zhang, C. Liu, L. Zheng, E. Petit, K. Qi, Y. Zhang, H. Wu, W. Wang, A. Tiberj, X. Wang, M. Chhowalla, L. Lajaunie, R. Yu and D. Voiry, Adv. Funct. Mater., 2022, 32, 2108316 CrossRef CAS.
- H. Niu, Z. Zhang, X. Wang, X. Wan, C. Shao and Y. Guo, Adv. Funct. Mater., 2021, 31, 2008533 CrossRef CAS.
- L. Yang, S. Feng and W. Zhu, J. Phys. Chem. Lett., 2022, 13, 1726–1733 CrossRef CAS PubMed.
- L. Lv, Y. Shen, J. Liu, X. Meng, X. Gao, M. Zhou, Y. Zhang, D. Gong, Y. Zheng and Z. Zhou, J. Phys. Chem. Lett., 2021, 12, 11143–11150 CrossRef CAS PubMed.
- N. Sathishkumar, S.-Y. Wu and H.-T. Chen, Appl. Surf. Sci., 2022, 598, 153829 CrossRef CAS.
- M. Yang, Z. Wang, D. Jiao, G. Li, Q. Cai and J. Zhao, Appl. Surf. Sci., 2022, 592, 153213 CrossRef CAS.
- J. Wu and Y.-X. Yu, Catal. Sci. Technol., 2021, 11, 7160–7170 RSC.
- J. Wu, J.-H. Li and Y.-X. Yu, J. Phys. Chem. Lett., 2021, 12, 3968–3975 CrossRef CAS PubMed.
- Y. Wang, D. Wu, P. Lv, B. He, X. Li, D. Ma and Y. Jia, Nanoscale, 2022, 14, 10862–10872 RSC.
- X. Li, H. Rong, J. Zhang, D. Wang and Y. Li, Nano Res., 2020, 13, 1842–1855 CrossRef CAS.
- J. Zhang, H. Yang and B. Liu, Adv. Energy Mater., 2021, 11, 2002473 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed.
- J. P. Perdew, J. A. Chevary, S. H. Vosko, K. A. Jackson, M. R. Pederson, D. J. Singh and C. Fiolhais, Phys. Rev. B: Condens. Matter Mater. Phys., 1992, 46, 6671–6687 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188–5192 CrossRef.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- G. J. Martyna, M. L. Klein and M. Tuckerman, J. Chem. Phys., 1992, 97, 2635–2643 CrossRef.
- D. Ma, Y. Wang, L. Liu and Y. Jia, Phys. Chem. Chem. Phys., 2021, 23, 4018–4029 RSC.
- C. Ling, X. Niu, Q. Li, A. Du and J. Wang, J. Am. Chem. Soc., 2018, 140, 14161–14168 CrossRef CAS PubMed.
- S. Ghoshal, A. Ghosh, P. Roy, B. Ball, A. Pramanik and P. Sarkar, ACS Catal., 2022, 12, 15541–15575 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- http://webbook.nist.gov/chemistry/ .
- X. Guo, J. Gu, S. Lin, S. Zhang, Z. Chen and S. Huang, J. Am. Chem. Soc., 2020, 142, 5709–5721 CrossRef CAS PubMed.
- F. Calle-Vallejo, M. Huang, J. B. Henry, M. T. M. Koper and A. S. Bandarenka, Phys. Chem. Chem. Phys., 2013, 15, 3196–3202 RSC.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- Y. Peng, B. Lu and S. Chen, Adv. Mater., 2018, 30, 1801995 CrossRef PubMed.
- C. Strupp, Ann. Occup. Hyg., 2010, 55, 43–56 Search PubMed.
- Y.-J. Shi and W. J. Swiatecki, Phys. Rev. Lett., 1985, 54, 300–301 CrossRef CAS PubMed.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- D. Wu, B. He, Y. Wang, P. Lv, D. Ma and Y. Jia, J. Phys. D: Appl. Phys., 2022, 55, 203001 CrossRef.
- S. Garcia-Segura, M. Lanzarini-Lopes, K. Hristovski and P. Westerhoff, Appl. Catal., B, 2018, 236, 546–568 CrossRef CAS.
- Z. Wang, J. Zhao, J. Wang, C. R. Cabrera and Z. Chen, J. Mater. Chem. A, 2018, 6, 7547–7556 RSC.
- H. Niu, Z. Zhang, X. Wang, X. Wan, C. Kuai and Y. Guo, Small, 2021, 17, 2102396 CrossRef CAS PubMed.
- R. J. Baxter and P. Hu, J. Chem. Phys., 2002, 116, 4379–4381 CrossRef CAS.
- Y. Wang and M. Shao, ACS Catal., 2022, 12, 5407–5415 CrossRef CAS.
- V. L. Deringer, A. L. Tchougréeff and R. Dronskowski, J. Phys. Chem. A, 2011, 115, 5461–5466 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TDOS of AE-SACs; possible adsorption configurations of *NO3 on AE-SACs; most stable configurations and corresponding adsorption-free energies of *NO3 and *H on AE-SACs; free energy diagrams and corresponding intermediate configurations on other AE-SAC systems; adsorption free energies of *NO3 and *H versus electrode potential on DV-BaN3C and DV-SrN3C; free energy diagrams and corresponding intermediate configurations on DV-BaN3C and DV-SrN3C with consideration of the solvation effect; schematic diagram of three moieties; the key parameters of the AE-SAC systems; the key parameters of *NO3 and *H adsorption on the AE-SAC systems; the PDS and corresponding UL on the AE-SAC systems; the ΔEZPE and TΔS for intermediates and free molecules. See DOI: https://doi.org/10.1039/d2ta08027c |
| This journal is © The Royal Society of Chemistry 2023 |