
Investigating the cut-off effect of n-alcohols on lipid movement: a biophysical study†
Michael H.L.
Nguyen
a,
Dominik
Dziura
 a,
Mitchell
DiPasquale
a,
Stuart R.
Castillo
a,
Elizabeth G.
Kelley
b and
Drew
Marquardt
*ac
a,
Mitchell
DiPasquale
a,
Stuart R.
Castillo
a,
Elizabeth G.
Kelley
b and
Drew
Marquardt
*ac
aDepartment of Chemistry and Biochemistry, University of Windsor, Windsor, Ontario, Canada
bNIST Center for Neutron Research, National Institute of Standards and Technology, Gaithersburg, MD, USA
cDepartment of Physics, University of Windsor, Windsor, Ontario, Canada. E-mail: drew.marquardt@uwindsor.ca; Tel: +1 519-253-3000 ext. 3537
First published on 14th June 2023
Abstract
Cellular membranes are responsible for absorbing the effects of external perturbants for the cell's survival. Such perturbants include small ubiquitous molecules like n-alcohols which were observed to exhibit anesthetic capabilities, with this effect tapering off at a cut-off alcohol chain length. To explain this cut-off effect and complement prior biochemical studies, we investigated a series of n-alcohols (with carbon lengths 2–18) and their impact on several bilayer properties, including lipid flip-flop, intervesicular exchange, diffusion, membrane bending rigidity and more. To this end, we employed an array of biophysical techniques such as time-resolved small angle neutron scattering (TR-SANS), small angle X-ray scattering (SAXS), all atomistic and coarse-grained molecular dynamics (MD) simulations, and calcein leakage assays. At an alcohol concentration of 30 mol% of the overall lipid content, TR-SANS showed 1-hexanol (C6OH) increased transverse lipid diffusion, i.e. flip-flop. As alcohol chain length increased from C6 to C10 and longer, lipid flip-flop slowed by factors of 5.6 to 32.2. Intervesicular lipid exchange contrasted these results with only a slight cut-off at alcohol concentrations of 30 mol% but not 10 mol%. SAXS, MD simulations, and leakage assays revealed changes to key bilayer properties, such as bilayer thickness and fluidity, that correlate well with the effects on lipid flip-flop rates. Finally, we tie our results to a defect-mediated pathway for alcohol-induced lipid flip-flop.
1 Introduction
The near ubiquity of n-alcohols in everyday human-life (seen in the pharmaceutical, food, cosmetic industries and more, as solvents, stabilizers, emulsifiers, etc.) and their ever presence in the environments of organisms necessitates their study in a wide range of circumstances. One popular effect of many n-alcohols stem from their anesthetic capabilities,1,2 in which numerous biochemical3–6 and biophysical studies7–9 have helped elucidate their anesthetic mechanism. Such studies hoped to uncover a generality innate in anesthetic agents, which can lead to the better design of these agents. Therefore, many studies focused on n-alcohols and their interactions with biological/synthetic membranes and their components, importantly lipids and proteins.3–12Membranes are the first point-of-contact for many drugs, additives and perturbants (e.g. n-alcohols). For a long time, two pathways in which these exogenous particles enact their activity onto cells were presented: (1) a direct mechanism where alcohols bind to proteins, disturbing the protein-conformation equilibria, and (2) an indirect and non-specific mechanism where alcohols primarily influence the lipid matrix, which can often lead to the reduction or obsoletion of protein function. Regardless of the preferred pathway, lipid–alcohol interactions are generally understood to be a cornerstone of the biological activity of n-alcohols, with specific protein interactions being another. Combining these two concepts, it is now more accepted that alcohol-induced changes to the properties of the lipid matrix can modulate protein function, and in turn cellular responses.3,13,14
It has been known that n-alcohols exhibit a cut-off effect in which their anesthetic capabilities are lost once a certain carbon chain length is reached (typically C5–12, depending on the protein and system4,15). In an attempt to explain the findings of these biochemical studies, numerous biophysical undertakings were performed. For example, it was found that n-alcohols typically localize in the transverse direction of the bilayer (normal to the bilayer plane) due to their aliphatic tails that penetrate into the membrane, situating themselves between the phospholipid acyl chains.8 This proximity can alter bilayer properties and organization, eliciting biophysical membrane changes.8 Further, small angle neutron scattering and diffraction showed that increasing n-alcohol chain length resulted in thickness increases in bilayers composed of di18:1PC (dioleoylphosphatidylcholine, DOPC),9 yet no cut-off in efficacy was observed. At present, the current results of lipid–alcohol interactions inadequately describe the complete mechanism at play.
An overlooked foundational aspect of membranes is the organization, transport, and movement of lipid molecules. With countless types of lipids, lipid membrane organization and its maintenance are crucial to proper membrane function and cell survival.16 In fact, the loss of transverse and lateral lipid organization are signals of apoptosis.17–20 Studies examining the effect of alcohol chain length on lipid dynamics are extremely lacking, with the vast majority focusing on lateral lipid diffusion.10,20 A few works have looked at the transverse diffusion (i.e. flip-flop) of lipids but used covalently-modified lipids, conjugated to a fluorophore, as reporters of lipid flip-flop.7 These reporter lipids can change native lipid properties, while their bulky fluorescent moiety undoubtedly affects how it traverses the bilayer.21
To this end, we apply a probe-free time-resolved small angle neutron scattering (TR-SANS) scheme22,23 to monitor the intervesicular exchange (i.e. lipid monomer diffusion between vesicles) and transverse diffusion (i.e. lipid flip-flop) of dimyristoylphosphatidylcholine (di14:0 PC, DMPC) molecules in the presence of n-alcohols of varying chain lengths (C2–C18) and concentrations. This scheme exploits the sensitivity of SANS between hydrogen and deuterium, allowing the time-dependent mixing to be tracked. It has been successfully employed to monitor lipid motions in lipid-additive systems that included methanol,24 proteins/peptides,25–28 therapeutic agents29 lipid nanodiscs,30 and oil–water emulsions.31 We complement the neutron scattering experiments with structural studies using small angle X-ray scattering and molecular dynamics simulations in two resolutions: all-atomistic and coarse-grained. To shed more light on alcohols and their effect on lipid flip-flop, we conducted calcein leakage assays. In the end, we combine the prevailing thoughts to provide a more coherent framework from which small molecules can affect the transport of phospholipids.
2 Methods and materials
2.1 Materials
1,2-Dimyristoyl-d54-sn-glycero-3-phosphocholine [14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0(d27)/14:0(d27) PC, d-DMPC], 1,2-dimyristoyl-sn-glycero-3-phosphocholine [14:0/14:0 PC, h-DMPC], 1,2-dimyristoyl-sn-glycero-3-phosphoglycerol [14:0/14:0 PG, DMPG] and cholesterol were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL) as powders and used as received. C2OH [ethanol] was ACS grade and diluted to 3.43 mol L−1 in water for manageable dispensing volumes. C6OH [1-hexanol], C10OH [1-decanol], C14OH [1-tetradecanol] and C18OH [1-octadecanol] were purchased from Sigma Aldrich (St. Louis, MO). C14OH and C18OH were kept as stock solutions in chloroform or chloroform/methanol (2:1) at 250 mmol L−1. D2O was purchased from Cambridge Isotope Laboratories, Inc. (Andover, MA, USA). Calcein assay materials include Triton X-100, Sephadex G-75, 2, 2, 2,2-[(3, 6-Dihydroxy-3-oxo-3H-spiro[2benzofuran-1,9-xanthene]-2, 7- diyl)bis(methylenenitrilo)]tetraacetic acid [Calcein], Ethylenediaminetetraacetic acid disodium salt dihydrate [EDTA], Tris hydrochloride [Tris-HCl], sodium chloride [NaCl] and sodium hydroxide [NaOH] were all purchased from Sigma-Aldrich (St. Louis, MO).
:0(d27)/14:0(d27) PC, d-DMPC], 1,2-dimyristoyl-sn-glycero-3-phosphocholine [14:0/14:0 PC, h-DMPC], 1,2-dimyristoyl-sn-glycero-3-phosphoglycerol [14:0/14:0 PG, DMPG] and cholesterol were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL) as powders and used as received. C2OH [ethanol] was ACS grade and diluted to 3.43 mol L−1 in water for manageable dispensing volumes. C6OH [1-hexanol], C10OH [1-decanol], C14OH [1-tetradecanol] and C18OH [1-octadecanol] were purchased from Sigma Aldrich (St. Louis, MO). C14OH and C18OH were kept as stock solutions in chloroform or chloroform/methanol (2:1) at 250 mmol L−1. D2O was purchased from Cambridge Isotope Laboratories, Inc. (Andover, MA, USA). Calcein assay materials include Triton X-100, Sephadex G-75, 2, 2, 2,2-[(3, 6-Dihydroxy-3-oxo-3H-spiro[2benzofuran-1,9-xanthene]-2, 7- diyl)bis(methylenenitrilo)]tetraacetic acid [Calcein], Ethylenediaminetetraacetic acid disodium salt dihydrate [EDTA], Tris hydrochloride [Tris-HCl], sodium chloride [NaCl] and sodium hydroxide [NaOH] were all purchased from Sigma-Aldrich (St. Louis, MO).
2.2 Formation of large unilamellar vesicles
Two separate lipid stocks are needed: one with h-DMPC and the other d-DMPC with chain perdeuteration. The protiated and deuterated lipid stocks were mixed prior to SANS measurements. Lipids in chloroform were aliquoted in specific quantities, with the chloroform evaporated off under a stream of nitrogen and placed under vacuum overnight. Due to their relative water insolubility, C10OH, C14OH and C18OH were added at the proper molar quantity to the chloroformic mixture prior to evaporation, while C2OH and C6OH were added to fully prepared liposomes. DMPG, a negatively-charged phospholipid, was incorporated at a lipid mol ratio of 5% to ensure the presence of unilamellar vesicles32,33 and to minimize the formation of multilamellar vesicles (MLVs), which can complicate data analysis.23 Subsequently, the sample films were gravimetrically measured and hydrated to a concentration of 17 mg mL−1 (or 21 mmol L−1) using a 45 mol% D2O solution – an appropriate contrast match point that accounts for the incorporation of DMPG. The pre-incorporation of long chain alcohols (C10–C18) at 30 mol% drastically changes the neutron scattering length density of the overall lipid vesicle and was thus compensated by adjusting the buffer to 34.6 mol% D2O. Film hydration was followed by a series of 5 freeze–thaws with intermittent vortexing to break up large aggregates. To form large unilamellar vesicles (LUV), the lipid suspension was extruded through 100 nm pore diameter polycarbonate membranes 31 times to yield a hydrodynamic radius of ≈70 nm, as determined by dynamic light scattering (DLS, Wyatt DynaPro NanoStar; Santa Barbara, CA, USA). Such extrusions were conducted at ≈ 35 °C, above the melting transition temperature of DMPC (25 °C). These steps produced a series of isotopic populations: h-LUVs and d-LUVs for protiated and deuterated, respectively.Directly before SANS measurements, h-LUVs and d-LUVs that were treated separately with n-alcohols (and ran on a lab tube roller for 1 hour to ensure equilibration) were mixed at a 1:1 molar ratio and loaded into 1 mm quartz banjo cells and immediately placed into the 37 °C neutron sample environment.
2.3 Time-resolved small angle neutron scattering
Time-resolved SANS measurements were conducted on the Very Small Angle Neutron Scattering (VSANS) instrument at the National Institute of Standards and Technology (NIST) Center for Neutron Research (NIST-CNR, MD, USA).34 Two detector carriages were set at sample-to-detector distances of 3.6 m and 15.3 m. The incident neutron wavelength was set at 5.3 Å to yield a q-range of 0.005 Å−1 to 0.2 Å−1. These experiments took advantage of a wide wavelength spread of 44% to maximize the overall signal intensity in order to minimize measurement times.24,27,35Samples were measured for 3 minutes at 37 °C and cycled repeatedly to obtain sufficient time-points to track the time-dependent mixing of the liposomal system. This DMPC mixing can be monitored due to the contrast match scheme employed22,24 and is displayed in Fig. 1. The 45% D2O background matches the scattering produced by equimolar LUVs of h-DMPC and d-DMPC, yielding a null scattering event. When the h-LUV and d-LUV populations are initially combined, the contrast (between LUVs and solvent) is at its greatest, producing a high intensity. As mixing proceeds, the overall system contrast decays, and therefore so does the signal. This scheme is depicted in Fig. 1. The vesicle integrity was monitored using DLS before and after alcohol treatment, as tabulated in Table S1 (ESI†).
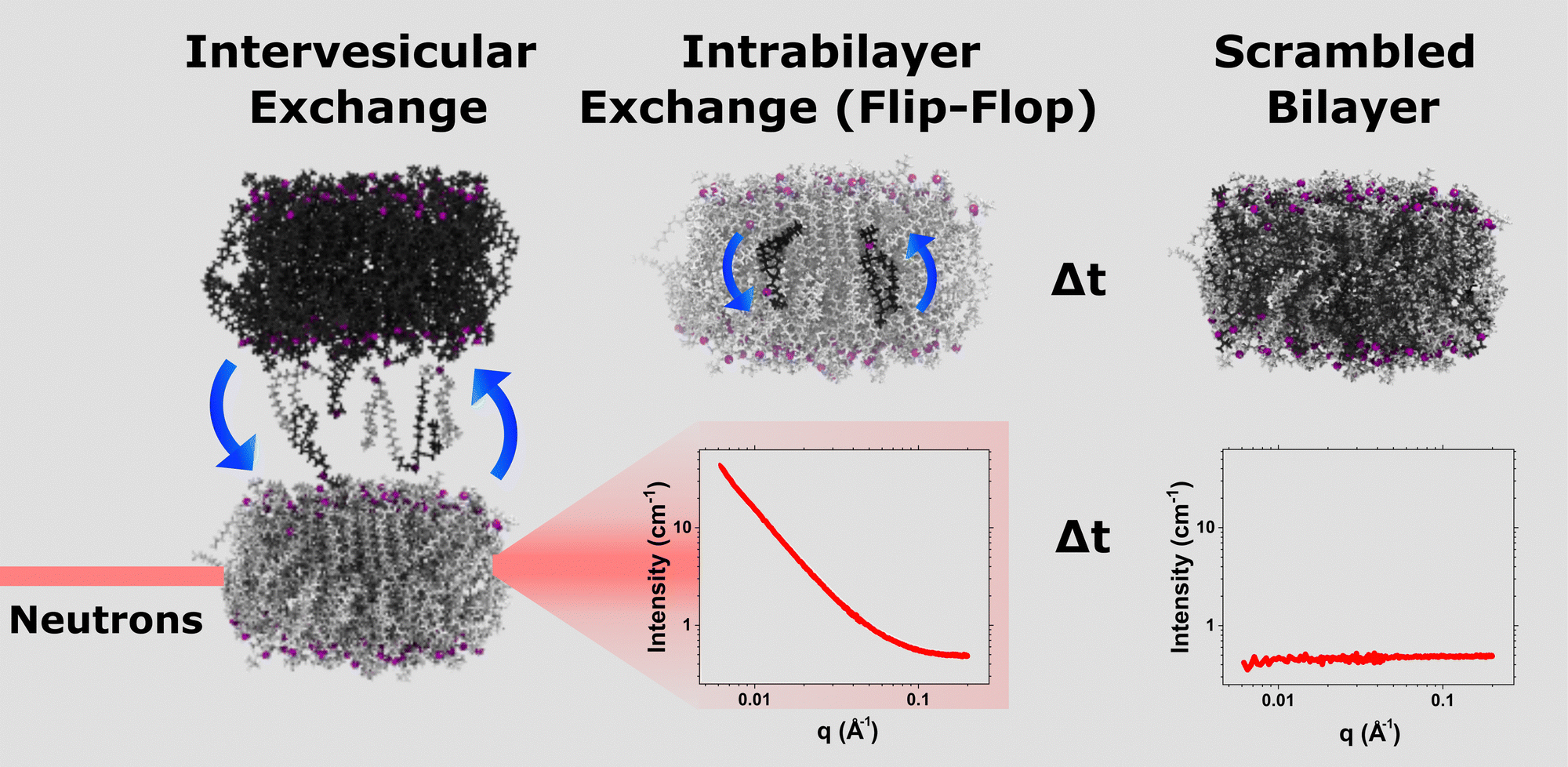 | ||
| Fig. 1 Schematic of the TR-SANS contrast matching scheme employed in the present study. Once one isotopic population is introduced to the other, monomeric lipid exchange occurs. These mixing samples are immediately placed into the SANS sample environment for neutron scattering measurements. At t0, i.e. the time mixing begins, the contrast or scattering intensity is at its greatest and can be viewed in the leftmost SANS I(q) plot. The exchanged lipid monomers can now undergo lipid flip-flop, diffusing from the outer leaflet towards the inner leaflet. As exchange and flip-flop proceed, the contrast decays as the scattering length density of the mixing system nears that of the D2O/H2O buffer background. Once the two isotopic populations completely mix into a single uniform population, the contrast is essentially null, as exemplified in the rightmost plot. Magenta beads represent phosphorus atoms. | ||
The resulting I(q) scattering curves obtained were corrected for instrument sensitivity and background using Igor Pro reduction software and VSANS macros provided by the NCNR.36 Post-reduction involved integrating the area beneath the I(q) curves at q < 0.1 Å−1 and plotting the integrated areas against the time of measurement. These measurements were normalized against their respective scrambled controls (Iinf), i.e. a system with equally mixed portions of d-DMPC and h-DMPC, and if applicable the fraction of alcohol doped) and the t0 measurement (I0). The normalization follows:
 | (1) |
 | (2) |
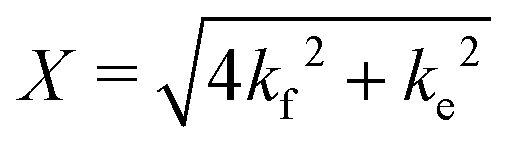 and Inorm is the normalized intensity decay.22 The standard error is derived from fits iterated by a Levenberg–Marquardt algorithm. Rate constants were converted to first order reaction half-times (t1/2) using:
and Inorm is the normalized intensity decay.22 The standard error is derived from fits iterated by a Levenberg–Marquardt algorithm. Rate constants were converted to first order reaction half-times (t1/2) using:| t1/2 = ln(2)/k | (3) |
2.4 Integrity analysis using small angle X-ray scattering
Small angle X-ray scattering (SAXS) analysis of the n-alcohol-liposomal samples were conducted to ensure the structural integrity of the lipid bilayers and vesicles were maintained over the course of SANS experimentation. SAXS was done on a Rigaku BioSAXS-2000 (Rigaku Americas, The Woodlands, TX) outfitted with a Pilatus 100 K detector and an HF007 rotating copper anode. Samples were measured several times (8–12) at 37 °C and assessed for radiation damage before averaging. Resulting SAXS curves were solvent (i.e. 45% D2O) background subtracted prior to model fitting.The Global Analysis Program (GAP) developed by Pabst and coworkers37 was used to analyze and fit X-ray scattering curves of the n-alcohol-treated LUVs. GAP works on a principle of fitting a three Gaussian model, two representing the electron-rich phosphate lipid headgroups and one representing the entire hydrocarbon chain region, onto the SAXS form factor. Based on the Gaussian locations and thicknesses, relevant bilayer parameters, such as the bilayer thickness (DHH), hydrocarbon thickness (2DC), and headgroup slab thickness (σHG) can be calculated. Because we did not add an additional Gaussian for the added n-alcohols electron density in the model, we release the σHG parameter, instead of the suggested 3 Å by the original authors,38 such that any changes to the headgroup region could also be measured alongside the bilayer thickness.
2.5 Molecular dynamics simulations
To provide a molecular description of the n-alcohol dependency of changes in phospholipid bilayer properties, molecular dynamics simulations in two resolutions were performed: All-atomistic (AA) and coarse-grained (CG). Each system's individual molecular make-up can be viewed in Table S2 (ESI†).2.6 All atomistic (AA) simulations and analysis
All AA systems were constructed using the CHARMM-GUI membrane builder.39 The resulting systems were formed with the CHARMM36 forcefield and contained 114 DMPC and 6 DMPG molecules, totaling 120 lipids, and hydrated with 7800 TIP3 water molecules. To offset the charges on the phospholipid headgroups and provide a physiologically relevant ionic solution (approximately 0.15 mol L−1 NaCl), 25 sodium and 19 chlorine ions were introduced. The alcohol-free system was energy minimized and equilibrated on the GROMACS version 2022 package40 using a series of steps and files provided by CHARMM-GUI. n-Alcohols were made using a CHARMM-GUI ligand reader module,41 where existing n-alcohol molecules available in the CHARMM force field library were used or, if not, were generated using CHARMM general force field. The n-alcohols were introduced into the equilibrated systems in Visual Molecular Dynamics (VMD)42 to avoid long simulation times that are typically required when introducing such molecules into the water in AA simulations. The new systems underwent a steepest descent minimization step followed by NVT and NPT ensemble equilibration steps. NVT and NPT runs used the velocity-rescale thermostat; the NPT runs employed the Parinello–Rahman barostat for semi-isotropic coupling to maintain system pressure at 1 bar, with a compressibility constant of 4.5 × 10−5 bar−1 and a time constant of 5 ps. Equilibrations ran for 1 ns long with a 2 fs timestep and hydrogen bonds constrained using the LINCS algorithm. All simulations were done at 37 °C to mimic the experimental data. The systems were visually inspected in VMD before the production runs which were simulated for 500 ns using the same conditions as the NPT runs. The final 250 ns were split into 5 blocks for error analysis and analyzed using a combination of in-house Python scripts, built-in gmx tools, and MEMBPLUGIN.43The area per lipid (ApL) and bilayer thicknesses (DHH and 2DC) were calculated using MEMBPLUGIN. In brief, the program assigns each lipid species’ phosphorus (P) atom as a point on an x and y plane. This plane is then broken down into polygons through a Voronoi diagram, resulting in areas for each polygon which in turn denotes each lipid's area. MEMBPLUGIN calculates membrane thicknesses based on a mass distribution of an assigned atom. Here, we used P to calculate DHH, which will allow us to directly compare these results to experimental SAXS membrane thickness results, where X-rays are most sensitive to the phosphates of the phospholipids. To calculate 2DC, we used the glycerol carbon C1 that is most closely embedded in the hydrocarbon core.
Acyl chain deuterium order parameters (SCD) measure the bilayer fluidity, which will give insight into how alcohols are influencing lipid transport. We opted for SCD as it can be compared to past works that used 2H NMR. Again, we relied on the built-in tools of MEMBPLUGIN to calculate these parameters. Here, the C–D bonds in the lipid acyl chains are very dynamic and this dynamism can be quantified using:
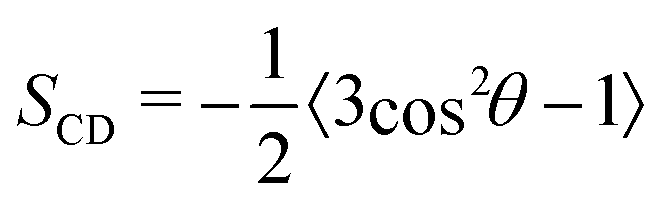 | (4) |
Lateral DMPC diffusion was measured using the supplied gromacs gmx msd module. In brief, the mean square displacement (MSD) was calculated for all 120 DMPC molecules. To avoid potential system drifting that can influence lateral diffusion calculations, we converted the trajectory file using gmx trjconv with the -pbc nojump and -center switches on. Center of mass (COM) drift was checked by following the COM of the bilayer system (all DMPC and DMPG molecules) along the x, y and z axis: no discernible drifting could be observed. The linear portion of the MSD curves (25–100 ns of the final 250 ns used for analysis as mentioned above) was analyzed by least-square fitting using:
 | (5) |
The error in DL was determined by splitting the total DMPC population into 6 equal subpopulations. The individual DL for all 6 was calculated just as the full population to yield the standard error.
The bending rigidity, κ (κBT), was calculated for each molecular system by applying a Real Space Fluctuations (RSF) computational method.44 RSF is suitable for smaller lipid systems of 100 lipids and larger, whereas other methods require 400 and more.44 Briefly, RSF assigns a vector to each lipid headgroup COM (i.e. P and C2 atoms) which is connected to the COM of the last three terminal carbons of each lipid acyl chain (i.e. C212, C213, C214, C312, C313 and C314). These directional lipid vectors are sampled against a field of vectors normal to the bilayer plane. Changes to the directional vector relative to the membrane vector informs on the lipids’ tilt and splay degrees of freedom. The distribution of these quantities can be used to calculate the tilt modulus (not included in the present work) and the bending rigidity. For a more detailed explanation, refer to the original manuscript.44
2.7 Coarse-grain (CG) simulations and analysis
To complement our AA MD simulations, we also performed CG MD simulations as it accurately portrays the properties of many fluid phase lipid bilayers.45 CG lipid bilayers were made using the CHARMM-GUI Martini maker module46 with the Martini 2.2 force field. Systems are composed of 486 DLPC/DMPC lipids and 26 DLPG/DMPG lipids and hydrated with 16110 W molecules. The pure lipid system was energy minimized and equilibrated using steps and protocols provided by CHARMM-GUI. In brief, a total of 7 minimization and equilibration runs were done with a velocity-rescale thermostat and Berendsen barostat, and as standard with increasing time steps from 2 fs to 20 fs for increased accuracy. CHARMM-GUI also provided a final pre-production run which ran with a velocity-rescale thermostat. Parinello–Rahman barostat was used with a coupling constant of 12 ps to maintain a pressure of 1 bar.
The Martini force field is based upon a building block philosophy, allowing the intuitive construction of molecules like the n-alcohols here.45 As seen in Fig. S1 (ESI†), C2OH is represented by a P2 bead and subsequent n-alcohols in the series, i.e. C6OH, C10OH, C14OH, and C18OH, contain an additional C1 bead per four carbons.45 Water molecules were replaced with a total of 57 alcohol molecules to yield 10 mol% of the final bilayer composition. These alcohol-doped systems were energy minimized and equilibrated using the same steps above. The final production runs were conducted for greater than 2 μs, depending on the time required for the alcohols to become embdedded into the bilayer (if ever), with a 20 fs time step. Again the velocity-rescale thermostat was applied alongside the Parinello–Rahman barostat. All equilibration and production runs were ran at 310 K to mirror the experimental data and using the GROMACS simulation package 2018.3.40
The final 1 μs of the production run was split into 5 blocks and used for analysis. The ApL and membrane thicknesses were calculated by MEMBPLUGIN.43 Here, the PO4 bead was taken as the atom of choice for both ApL and DHH calculations. Lateral diffusion coefficients were calculated using gmx msd provided by GROMACS. The final frame snapshots of the CG, as well as AA, can be seen in Fig. S1 (ESI†).
2.8 Preparation of calcein-filled DMPC LUVs
Stock lipid and alcohol solutions (≈ 20 mmol L−1, dissolved in a 2:1 chloroform to methanol solvent) were aliquoted to create a solution with the desired mole ratios of each component and a sample mass of 10 mg. The final alcohol:lipid molar ratio was ≈0.10. The majority of the organic solvent was first removed by evaporation under a steady stream of argon and then the samples were left in a vacuum oven overnight to remove any residual amounts of solvent. Samples were then hydrated with 70 mmol L−1 calcein (pH ≈ 7.4) to a final lipid concentration of 20 mg mL−1. These samples were then subjected to 5 freeze–thaw vortex cycles to facilitate their transformation from MLVs to LUVs. Next, the samples were extruded 31 times through a 100 nm pore diameter polycarbonate filter using an Avanti mini extruder (Avanti Lipids) at ≈35 °C. A size exclusion chromatography column, created with G-75 sephadex, was used to separate calcein-filled LUVs from free calcein that still remained in solution. The LUVs were eluted using a 0.5 mmol L−1 EDTA, 10 mmol L−1 Tris HCl, 150 mmol L−1 NaCl buffer (pH ≈ 7.4) through a 9 cm long column with a 1 cm diameter. The column was regenerated by running 0.1 mol L−1 NaOH through it, followed by the EDTA–Tris buffer. The eluted sample was then diluted to a total volume of 10 mL using the buffer, and kept in a cool water bath (≈5 °C) until samples were ready for measurement. All samples were created in this manner with the exception of the C2OH and C6OH samples, which were added to the liposomal sample following elution from the column.
2.9 Kinetic measurement of calcein leakage
Fluorescent measurements were collected using a SpectraMax M5e Multi-Mode Microplate Reader. The measurements were taken once every 15 seconds for 20 minutes at an excitation wavelength of 490 nm and emission wavelength of 520 nm. Using the temperature control of the plate reader, samples were incubated at 30 °C, 37 °C, and 40 °C during the measurements. Fluorescent data was collected in triplicate using a 96-well plate to measure the sample blank, the max fluorescence (where 2 μL of 0.1% w/v Triton X-100 was used for liposome solubilization) and the fluorescence of the sample which was able to release calcein due to the incubation. Three complete and independent trials were conducted for each lipid composition and temperature in order to calculate standard deviation and error bars for each sample measurement. Once the trial was completed the relative amount of calcein release was determined using eqn (6):| %RFT = 100(FT − Fo)/(FM − Fo) | (6) |
After the relative fluorescence changes were calculated for all the time intervals, the data was fitted according to first-order kinetics, eqn (7), in order to determine the rate constant (k) for each sample.
| %RFT = %RFM(1 − exp(−kt)) | (7) |
3 Results
Extensive studies focus on the structural perturbations caused by alcohols,9,20 yet few studies have looked at the organization11 and transport of lipids within these bilayers.7 This study aims to provide a more complete picture on how small molecules with increasing hydrophobic character, like n-alcohols, affects lipid mobility in model DMPC membranes. We employ a neutron contrast matching scheme in combination with TR-SANS to simultaneously measure the intervesicular exchange and transverse lipid diffusion of DMPC in the presence of primary alcohols with carbon lengths C2, C6, C10, C14, and C18. To explain our findings, we probe our systems at nanometer and atomic level resolutions using SAXS and molecular dynamics simulations, coupled with calcein leakage assays.Short chain n-alcohol promote lipid exchange and flip-flop at elevated concentrations
To measure the DMPC flip-flop and exchange rates in DMPC (doped with 5 mol% DMPG to promote unilamellarity32) systems, we employed a probe-free protocol: TR-SANS, as illustrated in Fig. 1. Briefly, two isotopically distinct vesicle populations are introduced together, and the time-dependent mixing is monitored by a neutron intensity decay. The integrated area of these intensity curves are then normalized and fitted using eqn (2) to yield the system's respective kf and ke. In this way, the alcohol-free control system yielded kf and ke of 8.6 ± 0.3 and 9.8 ± 0.1 × 10−3 min−1 or half-times of 81.0 ± 3.3 and 70.9 ± 0.7 min, respectively. These values are in good agreement with prior measurements on the same system at differing lipid concentrations.24,27 Because C2OH had the highest water solubility of the n-alcohols, we measured TR-SANS over an extensive ethanol concentration series spanning three magnitudes of concentration. Vesicle integrity was monitored using DLS (Table S1, ESI†). The normalized intensity decay which measures the amount of lipid transport can be viewed in Fig. 2a and b for C2OH and C6OH, respectively. Low ethanol concentrations (i.e. 2 and 9 mmol L−1) caused a rightward shift in the intensity decay curves, indicating slowing of the vesicle mixing. The double-exponential fit found in eqn (2) was again used to calculate the lipid kf and ke. Indeed, the slowing found in the decay curves was captured and are displayed in Fig. 2c and d. Meanwhile, concentrations of 25 mmol L−1 and above enhanced lipid mixing, consistent with previous n-alcohol studies on similar and pertinent lipid rate constants using TR-SANS24 and radioactively-labeled PC lipids.7 Extending the studies to C6OH, Fig. 2d shows that the concentration break-point between slowing and accelerating lipid mixing is between 2 and 9 mmol L−1. This same break-point is between 9 and 25 mmol L−1 for C2OH, signaling that at similar concentrations C6OH is more potent than C2OH at accelerating lipid mixing.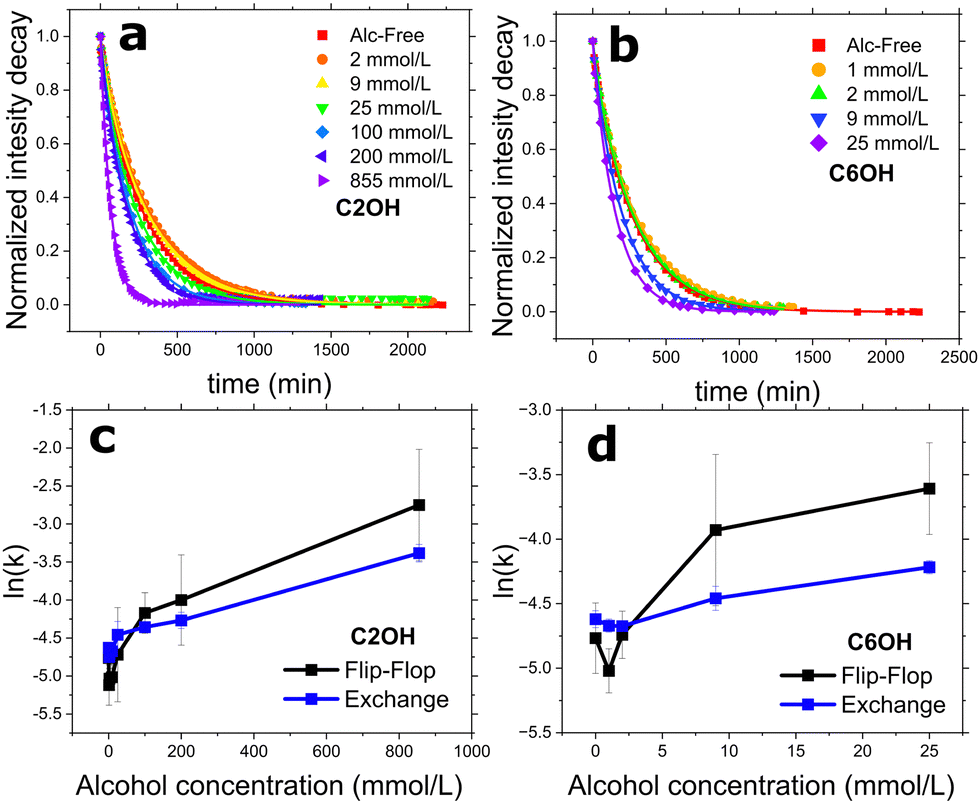 | ||
| Fig. 2 (a) and (b) Normalized intensity decay curves of DMPC vesicle mixing under the influence of short-chain alcohols: ethanol (C2OH) and 1-hexanol (C6OH), respectively. Symbols represent the normalized integrated area under each SANS curve at their corresponding time-point; lines are the fits using eqn (2) to the decay curves to extract out DMPC flip-flop and exchange rate constants, as seen in (c) and (d). All measurements were conducted at a temperature of 37 °C. | ||
Long chain 1-alkanols inhibits DMPC flip-flop
The longer chain alcohols, C10 up to C18, could not be added to externally to the already prepared lipid vesicle solutions. Therefore, we preincorporated them during the lipid film preparation stage at 10 and 30 mol% of the total bilayer entity, which corresponds to alcohol concentrations of ≈2 and ≈9 mmol L−1 with an overall lipid concentration of ≈21 mmol L−1. Fig. 3 represents how flip-flop and exchange rate constants were affected by increasing carbon length. A notable cut-off phenomena is not apparent at 2 mmol L−1 for either lipid rates. Nevertheless, a cut-off emerges at 9 mmol L−1 with regards to the kf. As previously mentioned, C6OH increases DMPC flip-flop rates at 9 mmol L−1. The cut-off appears in between C6 and C10OH with alcohol carbon lengths of greater than C6 causing a dramatic decrease in kf. C10OH, C14OH, and C18OH reduced the kf of DMPC by a factor of 5.6, 6.7, and 19.6 at 2 mmol L−1 and 11.3, 21.2, and 32.2 at 9 mmol L−1, respectively.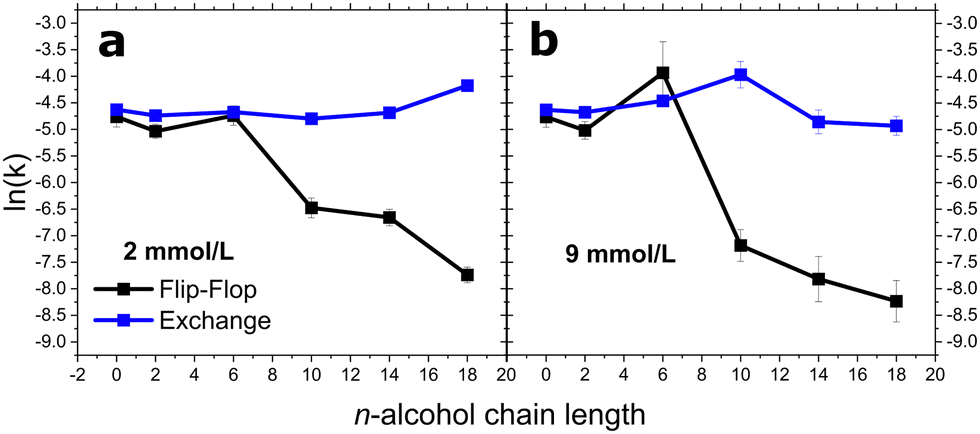 | ||
| Fig. 3 The alcohol chain length dependence of DMPC intervesicular exchange (blue) and flip-flop (black) at (a) 2 and (b) 9 mmol L−1 concentrations of alcohols. The standard error is derived from fits iterated by a Levenberg–Marquardt algorithm. | ||
DMPC exchange did not mirror the flip-flop results. At 2 mmol L−1, the exchange rate remained relatively constant until C18OH, where the rate increased by a factor of 1.6. These effects differ at 9 mmol L−1: the exchange rate accelerated with increasing alcohol chain length, with this effect at its maximal with C10OH. C14OH and C18OH caused the exchange to fall by a factor of 0.8 and 0.7.
SAXS to determine bilayer thickness
To provide an explanation for the observed lipid dynamics, we performed SAXS on the same DMPC vesicles doped with 5 mol% of DMPG and a series of n-alcohols. SAXS is sensitive to the phosphates present in phospholipid headgroups, allowing for the determination of headgroup-to-headgroup bilayer thickness (DHH). Alongside the DHH, we obtained the hydrocarbon thickness 2DC through fitting, as shown in Fig. 4. The values can be viewed in Table 1. The 2DC shows a similar trend as DHH, but is slightly thicker than DHH at C14OH and C18OH. The DHH of DMPC with 5 mol% DMPG produced a thickness of 36.3 ± 0.7 Å, similar to other experimentally-derived values for pure DMPC bilayers.49 We then introduced very high levels of n-alcohols – the concentrations are shown in Table 1 – due to previous SAXS studies of ours showing a lack of structural changes at lower concentrations despite an acceleration in lipid reorganization.24,50 Also, other studies looking at alternate bilayer features saw little to no changes in bilayer thickness; for example, hemi-fusion occurred before any thickness changes were observed by Chanturiya et al.51 | ||
| Fig. 4 (a) Small angle X-ray scattering curves of DMPC vesicles at 17 mg mL−1 (21 mmol L−1) in 45% D2O with the inclusion of high concentrations of n-alcohols. Open symbols are the background subtracted data obtained from SAXS; solid lines represent the fits obtained by GAP. (b) The electron density profile across the bilayer (z) provided by the GAP fit. Note, 0 is the bilayer centre. Relevant bilayer thickness, DHH and 2DC, are shown in (c). Experimental error is estimated to be ≤2% as per the scattering density profile model.38 All measurements were conducted at 37 °C. | ||
| n-Alcohol | Concentration (mmol L−1) | σ HG (Å) | D HH (Å) | 2DC (Å) |
|---|---|---|---|---|
| 0 | 0 | 3.3 | 36.3 ± 0.7 | 29.7 ± 0.6 |
| 2 | 200 | 3.3 | 36.2 ± 0.7 | 29.5 ± 0.6 |
| 6 | 100 | 3.4 | 34.3 ± 0.7 | 27.5 ± 0.5 |
| 10 | 25 | 3.4 | 36.5 ± 0.7 | 29.7 ± 0.6 |
| 14 | 25 | 3.0 | 46.3 ± 0.9 | 40.3 ± 0.8 |
| 18 | 25 | 2.8 | 47.1 ± 0.9 | 41.5 ± 0.8 |
At 200 mmol L−1 of C2OH, the DHH of DMPC remained constant at 36.2 ± 0.7 Å, whereas 100 mmol L−1 of C6OH reduced the thickness to 34.3 ± 0.7 Å. As expected from previous structural studies on closely related PC systems,9,20 the same concentration of the three longest chain increased the bilayer thickness by 0.2 Å, 10.0 Å, and 10.8 Å, respectively. The increase in bilayer thickness that occurs at C10OH, C14OH, and C18OH is corroborated by AA and CG simulation data, as will be shown below. The DHH values with C14OH and C18OH parallels closely to experimental DHH values of neat di16:0PC (dipalmitoyl-PC) and di18:1PC (dioleoyl-PC) bilayers.49
To view changes to the headgroup region, we freed the σHG parameter representing the headgroup slab thickness during the fitting process. The σHG in a pure lipid system is 3.3 Å and it expands to 3.4 Å with C6OH and C10OH. In contrast, C14OH and C18OH reduced the values to 3.0 and 2.8 Å, respectively. While there was a large alcohol concentration difference between the TR-SANS and SAXS studies, both indicate different behaviors for long and short- chain n-alcohols and suggest more detailed biophysical studies are needed to reveal potential origins of the cut off effect.
3.1 Area per lipid and bilayer thicknesses via MD simulations
Because the experimental method of n-alcohol addition differed for the short and long chain alcohols, we employed an unbiased means, i.e. MD simulations, to investigate the validity and presence of this cut-off effect. To do so, we generated DMPC molecular systems, doped with 5 mol% DMPG and 5–10 mol% n-alcohol, in the CHARMM36 force field to mimic our experimental samples (at 1–2 mmol L−1). We then calculated important bilayer properties, such as the area per lipid (ApL) and bilayer thicknesses (phosphate–phosphate distance, DHH and hydrocarbon thickness, 2DC), which are presented in Table 2. The ApL, DHH and 2DC results are plotted as a function of alcohol chain length in Fig. 5a and b. Fig. 5c is a three-dimensional representation of the atomic distribution of the DMPC phosphorous and C1 glycerol carbon. From the AA simulations, regardless of concentration (5–10 mol%), there is no clear relation between ApL of DMPC and n-alcohol chain length. In Fig. 5a, at 5 mol%, there is an initial increase in ApL at C6OH that subsequently subsides as the carbon number increases. When the n-alcohol concentration is doubled, the effects observed become more pronounced with an additional observation that the ApL remains constant at longer chain lengths (C10–C18). The CG simulations ApL results show a concomitant increase in ApL with n-alcohol carbon number until a plateau at alcohol carbon number C10 and higher. This cut-off was not observed for simulations looking at C8OH and longer on di18:1PC,12 which found the lateral area of the unit cell (AUC) increased with all alcohols using SANS and CG simulations. The AUC here includes both lipid and alcohol, whereas in the present study both were separately calculated. Also, this difference could be due to the lipid's longer acyl chain and extra degree of unsaturation, both of which allow the longer chain alcohols to be better accommodated.| Resolution | System | X alc | ApL (Å2) | D HH (Å) | 2DC (Å) | D L (10−7 cm2 s−1) | κ (κBT) |
|---|---|---|---|---|---|---|---|
| All-atomistic | Alc-Free | 0 | 60.3 ± 0.2 | 36.3 ± 0.1 | 32.4 ± 0.1 | 0.90 ± 0.12 | 47.1 ± 1.7 |
| C2OH | 0.05 | 60.3 ± 0.5 | 36.3 ± 0.3 | 32.5 ± 0.3 | 1.34 ± 0.21 | 47.5 ± 1.7 | |
| 0.1 | 60.5 ± 0.5 | 36.2 ± 0.2 | 32.4 ± 0.2 | 1.66 ± 0.16 | 45.8 ± 1.8 | ||
| C6OH | 0.05 | 60.8 ± 0.4 | 36.4 ± 0.2 | 32.5 ± 0.2 | 1.42 ± 0.14 | 49.7 ± 2.0 | |
| 0.1 | 62.4 ± 0.2 | 36.1 ± 0.1 | 32.2 ± 0.1 | 1.83 ± 0.09 | 40.7 ± 1.3 | ||
| C10OH | 0.05 | 60.5 ± 0.3 | 36.6 ± 0.1 | 32.8 ± 0.1 | 2.04 ± 0.21 | 46.7 ± 1.6 | |
| 0.1 | 61.1 ± 0.3 | 36.9 ± 0.1 | 33.1 ± 0.1 | 1.26 ± 0.21 | 50.2 ± 1.9 | ||
| C14OH | 0.05 | 60.3 ± 0.5 | 36.9 ± 0.2 | 33.0 ± 0.2 | 2.11 ± 0.71 | 49.5 ± 1.8 | |
| 0.1 | 60.9 ± 0.7 | 37.4 ± 0.3 | 33.5 ± 0.3 | 1.23 ± 0.04 | 48.0 ± 1.6 | ||
| C18OH | 0.05 | 60.5 ± 0.3 | 37.0 ± 0.2 | 33.1 ± 0.2 | 1.39 ± 0.08 | 49.4 ± 2.1 | |
| 0.1 | 61.0 ± 0.4 | 37.7 ± 0.2 | 33.9 ± 0.2 | 1.54 ± 0.11 | 55.2 ± 2.2 | ||
| Coarse-grained | Alc-Free | 0 | 60.3 ± 0.1 | 34.6 ± 0.1 | 26.9 ± 0.1 | 2.12 ± 0.10 | — |
| C2OH | 0.1 | 60.5 ± 0.3 | 34.6 ± 0.1 | 26.9 ± 0.1 | 2.18 ± 0.06 | — | |
| C6OH | 0.1 | 62.1 ± 0.2 | 34.4 ± 0.1 | 26.7 ± 0.1 | 2.32 ± 0.13 | — | |
| C10OH | 0.1 | 63.0 ± 0.3 | 34.5 ± 0.1 | 26.8 ± 0.1 | 2.29 ± 0.23 | — | |
| C14OH | 0.1 | 62.9 ± 0.2 | 34.8 ± 0.1 | 27.2 ± 0.1 | 2.18 ± 0.25 | — | |
| C18OH | 0.1 | 63.1 ± 0.2 | 35.1 ± 0.1 | 27.5 ± 0.1 | 2.30 ± 0.09 | — | |
 | ||
| Fig. 5 (a) Area per lipid of DMPC calculated from AA (solid) and CG (transparent) MD simulations in alcohol-free and alcohol-doped conditions (i.e. 5 and 10 mol% for AA, open black circles and blue X, respectively, and only 10 mol% for CG, solid squares). (b) Plots of bilayer thicknesses from the phosphate peak-to-peak distance (DHH, black) and the glycerol backbone (2DC, blue). 5 mol% (open circle) and 10 mol% (X) are shown. (b) Inset: DHH and 2DC from CG (10 mol% only). The bilayer thicknesses is visually displayed with an xyz axis in (c), showing the density of phosphorus and glycerol carbon atoms that is used to calculate the bilayer thicknesses. The bending rigidity, κ, of the AA systems are shown in (d and e) as a function of alcohol chain length and mole fraction, respectively. The plot in (f) shows a best fit linear correlation between DHH and κ as a function of ln(kf) and a 95% confidence interval. | ||
The bilayer thickness results can be viewed in Fig. 5b and Table 2. An increase in the n-alcohol chain length is accompanied by an almost linear increase in the AA bilayer thicknesses at 5 mol%. At 10 mol%, the AA thicknesses decrease and then a sharp increase occurs at C10OH and above. Again, a sharp change appears between C6OH and C10OH. The DHH and 2DC trends mirror each other almost exactly at both concentrations. CG simulations results for DHH and 2DC are shown in the inset of Fig. 5b and show a similar trend to the AA results: an initial thinning of the bilayer followed by a thickness increase with alcohol length. With only C10OH showing a slight deviation between the two resolutions, the AA and CG thickness results are otherwise consistent. Interestingly, the trends in the AA and CG bilayer thickness differ from those found by other groups where they observe thickness of di18:1 PC bilayers to increase monotonically with C8-16OH.9,12 The cut-off effect must be dependent not only on alcohol chain length but on the lipid acyl chain length and unsaturation level as well, tying closely the idea of membrane thickness and alcohol potentiation/inhibition activity.52
3.2 Bending rigidity
The softness and undulations of a bilayer is generally dictated by its component lipid packing. As seen from the order parameter results above, the chain and conversely bilayer order is closely tied to the alcohol chain length. It is well established that short chain alcohols, such as methanol and ethanol, decreases the bending rigidity,51 however the effect of longer alcohols have yet to be explored in detail. To this end, we calculate the bending rigidity of our AA DMPC systems53 with increasing alcohol chain lengths.The bending modulus trends at 5 and 10 alcohol mol% are listed in Table 2 and can be viewed in Fig. 5d. The bending modulus is relatively unchanged at low alcohol concentrations. When the concentration is doubled, the membrane softens dramatically at C6OH. These effects are not apparent in the measured flip-flop and exchange rates (Fig. 3a) but sheds light on potential mechanisms at the molecular level. The membrane begins to rigidify as C10OH, C14OH, and C18OH and their tails incorporate deeper into the membrane interior. Fig. 5e illustrates the bending modulus and its dependence on alcohol concentration, where values above the grey dashed line signifies a more rigid membrane and vice versa. Notably, C10OH and C18OH caused greater membrane rigidity at higher concentrations (Xalc = 0.10) while C14OH did not follow this trend. The effects by C14OH may be muted by virtue of having similar biophysical characteristics as DMPC (e.g. alkyl chain length), essentially imitating DMPC in these undulating bilayers.
The main goal of the simulations was to extract those parameters that would correlate with the dramatic slowdown in DMPC's flip-flop rate as alcohol of increasing lengths was incorporated into DMPC membranes. The simulations were done at low alcohol concentrations, with 2 mmol L−1 (as in Fig. 3a) corresponding to 10 mol% relative to the lipid content. As shown in Fig. 5f, we found that the simulated bilayer thickness correlates with the flip-flop rates within a 95% confidence interval. The membrane rigidity is also found to correlate with the flip-flop rate trends but with less confidence.
3.3 Alcohol location within the bilayer
To determine the alcohols’ location within the bilayer, we calculated the atom number density profile (NDP) for a number of atoms and molecules within the all atomistic systems. First, we present the DMPC NDP in Fig. 6a. With increasing alcohol chain length, the atom density of DMPC deep within the hydrophobic core subsides. This can easily be interpreted two-folds: (1) the “growing” alcohol alkyl chain is embedding deeper in the bilayer core and (2) that the bilayer is thickening as indicated by the peak regions of DMPC splitting farther apart. In Fig. 6b, C2OH only weakly localizes to the water–lipid interface. A greater carbon number results in the alcohol mass density (primarily the alkyl tail) residing deeper within the bilayer core, mirroring the trends seen with n-alcohol and membrane partitioning coefficients.54 The polar hydroxyl moiety of the alcohols continuously reside near the carbonyl-phosphate backbone, as has been observed in CG simulations of DOPC systems,9,12 multi-component domain-forming bilayers,11 and in 2 H-NMR experiments on saturated lipids.55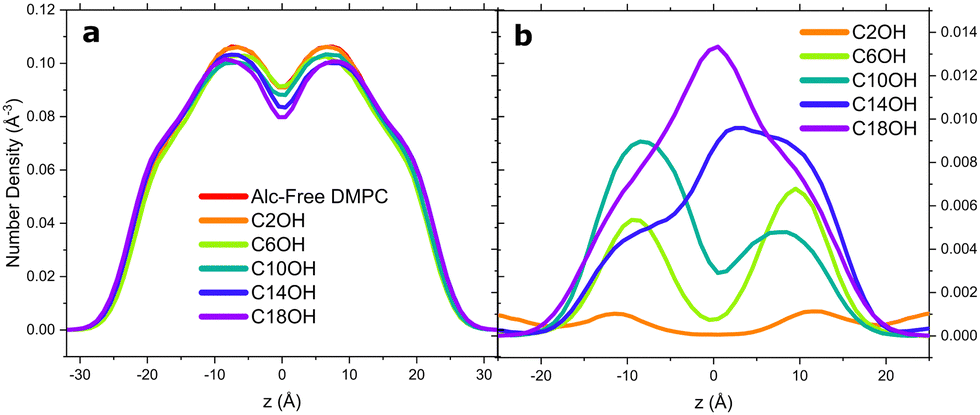 | ||
| Fig. 6 Calculations from all atomistic molecular dynamics simulations are presented here to show the (a) number density profile of DMPC under the influence of 10 mol% of increasing alcohol chain length. (b) Number density profile of the alcohols as calculated by MEMBPLUGIN. | ||
C18OH is interesting because its chain is four carbons longer than the alkyl tails of DMPC. This manifested itself with a significant density being situated within the bilayer core, causing the measured density to be at a maximum at z = 0 Å. Viewing the trajectory snapshots, this increase in density stems from the added terminal methyl and methylene groups of the increasing alcohol chain length, and not because of the whole alcohol molecule migrating to the bilayer core as has been observed with n-alkanes.56 This effect by the longer chain alcohols unsurprisingly caused the leaflet lipid interdigitation to decrease (Fig. S2, ESI†) and the bilayer thickness to increase. Here, interdigitation is calculated from the fraction of inter-leaflet contacts43 and not necessarily the formation of a distinct interdigitated phase; though, alcohol–lipid membranes have been observed to form such a phase10 but that is beyond the scope of the present work.
3.4 S CD order parameters
To view the overall membrane fluidity, we show the simulated SCD and Sseg values for the control and alcohol systems of AA and CG, respectively, in Fig. 7 (left). The right of the figure show order parameter differences between the alcohol systems and the control. For the sn-1 chain, C2OH lowers SCD, C6OH slightly increases SCD, and C10OH, C14OH and C18OH dramatically increase chain order. These trends are reflected with the sn-2 chain save for C6OH where the regions closest and farthest from the DMPC headgroup are unaffected or fluidized. Carrying these points to the CG system, we see that in Fig. 7e and f, inclusion of the hydroxyl moiety of C6OH to C18OH into the CG bilayer causes a reduction in Sseg of NC3-PO4 and PO4-GL1, indicating a destabilization of the order in the headgroup region. Towards the segments pertaining to the lipid chains, we observe trends that directly mirror the lipid flip-flop cut-off, i.e. C2OH and C6OH decreased the CG segmental order, while C10OH, C14OH, and C18OH increased the order. Generally, beyond C6OH, the longer the alcohol alkyl chain, the greater the ordering effect. These results mirror those conducted on similar bilayer MD simulations where C10OH, C12OH, and C14OH ordered longer saturated chain lipids.11 Interestingly, this effect can also be dependent on the degree of lipid unsaturation.9,11 The order parameter findings sheds light that the alcohol alkyl tails indeed extend towards the bilayer core; however, with sufficient concentrations the shorter alcohols do not extend fully and leave voids within the bilayer core, destabilizing the bilayer interior. | ||
| Fig. 7 (a) and (c) Deuterium order parameter of the DMPC alkyl sn-1 and sn-2 tails as a function of n-alcohol chain length. The corresponding (b) and (d) plots are the order parameter differences between undoped and doped sn-1 and sn-2 tails, respectively. (e) Segmental order parameters calculated from CG DLPC (same as DMPC using Martini beads) systems. (f) Solid lines and filled symbols are the segmental order parameter difference of sn-1 chains, while transparent dashed lines and symbols are that of sn-2 chains. | ||
3.5 Lateral lipid diffusion
Because lipid flip-flop occurs slowly, magnitudes longer than can be surveyed by AA simulations, we looked towards other dynamical processes such as lateral lipid diffusion and the membrane bending rigidity of our AA models to determine if this cut-off was present as well. Fig. 8a and b show the measured MSD curves of AA and CG systems, respectively. Such curves were fitted according to eqn (5) to obtain a set of DL values presented in Table 2 and Fig. 8c. The calculated lateral diffusion of DMPC is 0.9 ± 0.12 × 10−7 cm2 s−1 mirroring closely to experimental values10 and other simulations.57 Due to bead smoothing and relatively shorter acyl chain length associated with the CG system, the DL of DLPC (same as DMPC in Martini) is about twice as fast as the AA value at 2.12 ± 0.10 × 10−7 cm2 s−1. Interestingly, as shown in Fig. 8c, we found that the lateral diffusion of DMPC was not significantly affected by varying the alcohol chain length.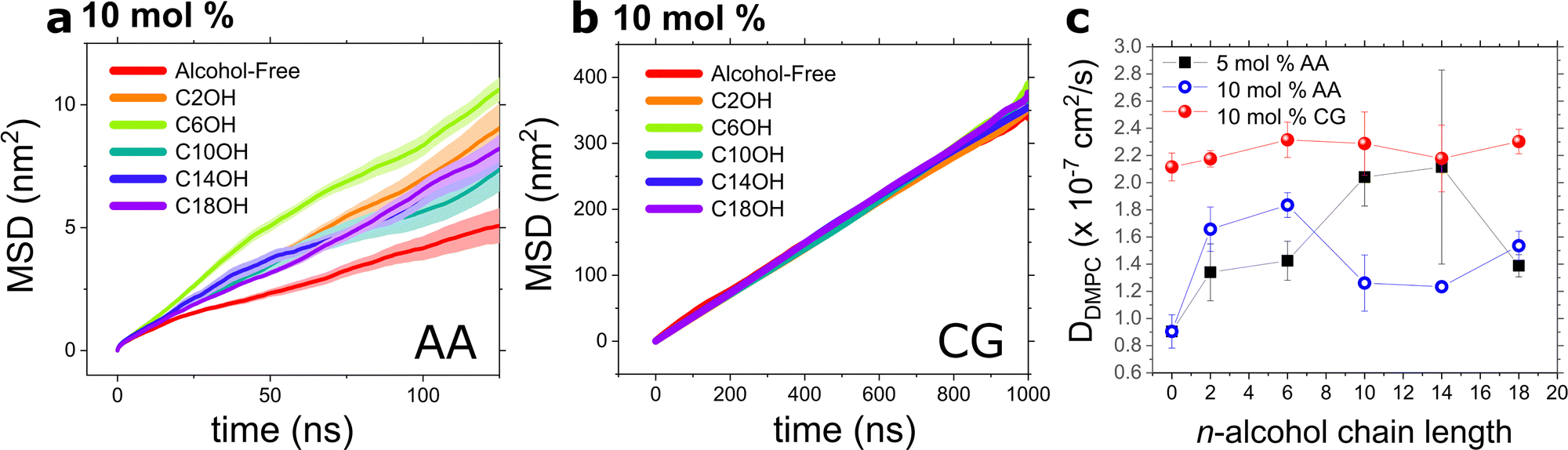 | ||
| Fig. 8 Mean square displacement plots calculated from (a) AA and (b) CG simulations of DMPC model bilayers doped with 5 or 10 mol% n-alcohols at 37 °C. Linear fits were performed on linear portions of the MSD curves and the resulting lateral DMPC diffusion coefficients within the plane of the lipid bilayer, i.e. perpendicular to the bilayer normal, are presented in (c). Errors are estimated from six sub-populations of DMPC molecules for AA values. | ||
3.6 Calcein leakage
With bilayer thinning, greater membrane disorder and fluidity, the previous results hint at a defect-mediated pathway enacted by the short chain n-alcohols. To investigate this premise, we conducted calcein leakage studies that measure the overall vesicle and membrane stability and general porosity. To our knowledge, calcein leakage studies done on DMPC-alcohol systems do not exist. There have, however, been several adjacent studies that looked at chemically-modified lipids,58 longer chained PC lipids,58 or naturally extracted lipids looking at Na+ leakage59 in the presence of some alcohols. C2OH was introduced at 9 mmol L−1 and 200 mmol L−1 and measured at several temperatures (30°, 37°, and 45°). But because it was added externally, this introduced a hump to the otherwise normal leakage plots (Fig. S3, ESI†). Such plots show a two-process leakage: (1) an initial bursting of calcein-filled vesicles due to the introduction of ethanol, and (2) a slower consistent leakage from the remaining vesicles. This two process was not observed in longer chain lipids which are typically more stable against leakage than DMPC.58 C6OH was measured at 1 mmol L−1 and 9 mmol L−1 and increased the rate of leakage so dramatically that it was immeasurable minutes after introduction. The results from the alcohol-free system and samples doped with C10OH and above can be viewed in Fig. 9. Like the above results, C10OH slowed the leakage of calcein with this effect plateauing upon longer aliphatic chain lengths, C14OH and C18OH. Interestingly, leakage in liposomes with C14OH was the slowest of all the systems measured, albeit by a small margin. Despite, the complications involved with the short chain alcohols, it was observed that C6OH is still the most potent, which may highlight the link between calcein-leakage, pore defects, and phospholipid flip-flop. | ||
| Fig. 9 Natural logarithm of the rate of leakage as a function of the temperature reciprocal. DMPC vesicles, with 5 mol% DMPG and enclosing fluorescent calcein, was doped with a series of longer chain alcohols at 9 mmol L−1 or 10 mol%: C10OH, C14OH, and C18OH. Three temperature points were measured at 30 °C, 37 °C, and 40 °C. | ||
4 Discussion
The lipid bilayer forms the first line of defense for organisms against the harsh external environment. Its interaction with small molecules from the outside dictates the responses the cell ultimately mounts. Here, we present a comprehensive study that details the role of primary alcohols, with increasing alkyl chain length, on influencing the different lipid motions and the bilayer properties of model membranes. Surmisings on changes in classical bilayer properties to translating that into changes in lipid mobility is the basis of this paper.A protein–alcohol pathway would suggest alcohols bind to ion channels involved with neurotransmission to enact their anesthetic activity. It can be explained that as the chain length increases, the cut-off arises from when the alcohol size surpasses the protein's binding pocket which reduces the anesthetic activity. However, this cut-off point ranges widely, depending on the protein examined: for example, the cut-off that appeared for neuronal nicotinic acetylcholine receptors was reached at only C5OH,4 while N-methyl-D-aspartate receptors at C6-8OH.5 This starkly contrasts with others seen to exhibit cut-offs at C10OH and above.6 Because of these discrepancies, we looked toward alcohol effects on membrane properties that are known to modulate protein activity. More recently, several groups have tied the effects of alcoholic anesthetics to lipid mixing within the bilayer plane11,60,61 and directly to changes to the critical temperature for lipid phase separation rather than the anesthetic's overall hydrophobicity. These changes are predicted to disturb the protein-lipid raft equilibria that is considered to be pivotal for signal transduction. This idea of entangling lipid phase separation and anesthetic effect on proteins can be extended more generally to lipid organization as well.
Albeit at lower concentrations we did not observe changes in bilayer structure using SAXS. We previously explained this to be due to the action of certain alcohols to be on very short-ranged and short-lived length- and time-scales.24 These actions may be too short to be surveyed using techniques that measure global sample averages, such as SAXS and DLS. For this reason, here we increased the alcohol concentrations in the present SAXS work to elicit tangible structural changes, as well as employed MD to probe at shorter length- and time-scales. As a result, the short chain alcohols, C2OH and C6OH, were observed to induce greater bilayer disorder, enhanced membrane bending fluctuations, bilayer thinning, and an increase in the area occupied per lipid, all of which are hallmarks and/or precursors to bilayer defects. Such defects can be defined as any discontinuity occurring along or perpendicular to the bilayer plane and they have been proposed to be at the heart of lipid flip-flop.62,63
By intercalating in between phospholipid headgroups, the shorter chain alcohols (in particular C6OH) disperse the lipid network, effectively thinning the bilayer as shown by the SAXS and in silico data (Fig. 4 and 5b). They also induce voids where typically the lipid acyl chains would reside.12 This void can be filled by adjacent lipid acyl chains, the bulk phase or, more pertinently, filled by a nearby lipid headgroup, all of which increasing the odds of a lipid flip-flop event.63 Though we do not show such deformations, it has been observed numerous times before.12,62,63 All these points indicate a flip-flop pathway that becomes more favorable with the introduction of short chain alcohols. This is corroborated by the rapid leakage of calcein with C6OH which occurs when pores, large enough to allow calcein passage out of the vesicle lumen, appear. These pores have been postulated to be a key source of observed accelerated lipid flip-flop, as has been demonstrated by Monte Carlo simulations on substrate-supported bilayers62 and TR-SANS.64 More investigations are required however for C2OH due to the potential emergence of two leakage-causing processes, arising from its external addition. Through these pores, lipids are able to laterally diffuse along the lipid-lining directly to the apposing leaflet, circumventing the energetically unfavourable headgroup desolvation and passage through the hydrophobic interior entirely.
With regards to the long chain alcohols (C10OH, C14OH and C18OH), they induced greater bilayer order, reduced membrane bending fluctuations and bilayer thickening. These are indicators of potential changes in the fluid-to-gel phase transition (Tm) as shown by Klacsova and et al. in similar DMPC-alcohol systems.65 The bilayer phase state has tremendous influence on the rate of phospholipid flip-flop.62 It would stand to reason then the observed drops in kf at longer alcohol chain lengths is due to the appearance of a gel phase, occurring at 37 °C – a temperature much greater than the neat DMPC Tm (24 °C). However, this explanation is not completely satisfactory as Klacsova et al. showed DMPC-C10OH bilayers only demonstrated modest changes in Tm (Tm ≤ 30 °C).65 Despite this, several groups showed similar reasoning in several systems that 1-alcohols induced changes in the liquid-ordered and liquid-disordered phase transition temperature in domain-forming lipid mixtures.11,60,61,66,67 These details point to alcohol-induced reorganization of membranes may be causing the dramatic reduction in lipid flip-flop observed here. Investigations are currently underway to elucidate these details.
As this alcohol carbon number increases, however, the growing aliphatic tail fills these voids, where they become less water-filled, overall making this pathway less favorable. Once the chain length reaches a cut-off point, seen here at roughly C10–C14, the alcohols stop acting as a structural perturbant but as a sturdy structural component instead. For example, the bilayer packing becomes more ordered, making the bilayer less permeable to solutes, opposite to short chain alcohols,68 and the overall bilayer thickness expands. Bilayer thickness has always been closely tied to the degree of lipid flip-flop. This is because thickness changes can increase the distance the polar headgroup must travel in the hydrophobic core, reducing the overall number of flip-flop events occurring.21 Further, the longer acyl tails results in stronger van der Waals interactions increasing the already great enthalpic barrier.22,23,27,69–71 Ultimately, this switch from a lipid perturbant to a structural component aligns well with the current cut-off points defined and is likely to be dependent on the lipid types, specifically, the lipid fatty acyl chain length of the membranes. This would help explain the disparity between cut-off points found in biochemical studies which often use different biomimetic membranes for protein reconstitution.
One would expect the chain-length dependency of the flip-flop results to translate closely to intervesicular exchange, yet it did not, indicating these two lipid dynamical processes occur under different mechanisms. At 2 mmol L−1 the exchange was largely undisturbed until C18OH. At elevated concentrations (9 mmol L−1 or 30 mol%), the cut-off appears at C14OH, in contrast to C10OH for flip-flop. We suspect that this feature is based on an interplay between the bilayer thickness, membrane fluidity, and to the degree of lipid interdigitation that occurs between the two bilayer leaflets. At lower concentrations, these long chain alcohols only slightly thicken and stiffen the membrane that should inhibit exchange, however, our results do not show that. We presume this is due to the accompanied decrease in interdigitation with longer alcohol chain length. This property induces motional restrictions that can easily be translated to a multitude of lipid dynamics.72 Thus, one can presume less interdigitated acyl chains will allow lipid exchange to proceed less impeded. Another point to include is that the ordering effect of the long chain alcohols seen in the simulations do not significantly translate to slowing of DMPC lateral diffusion coefficients nor intervesicular exchange, which hints at a potential tie between lateral and intervesicular diffusion. Further research on this interplay must still be done to elucidate these initial findings however.
An interesting result stemming from this work shows an initial slowing of membrane dynamics upon exposure to C2OH and C6OH at low concentrations. A possible explanation may be related to the membrane partition coefficient (Kp) of these alcohols. The Kp increases exponentially with increasing alcohol chain length; for example, Westh and coworkers reported Kp values for C3OH to C8OH partitioned into DMPC bilayers at 30 °C, with a Kp ≈ 26 for C6OH.68 Extrapolating these values for C10OH yields a Kp ≈ 4.3 × 103, close to the reported value of C10OH (≈ 3.9 × 103) in another model system.73 Therefore, the real concentration of CnOH intercalated in the lipid bilayer depends on n. The present MD simulations reinforces this as C2OH only weakly associates at a concentration of 2 mmol L−1. This weak association may cause a degree of hydrogen bonding required to slow lipid dynamics, an effect mitigated with greater alcohol concentrations; these ideas require further investigation though. The partitioning explanation could also help to explain the weak cut-off effect observed in Fig. 3b. In summary, due to the large differences in the partitioning of the short (n = 2, 6) and long homologues (n > 8) of CnOH, the weak effect would become more significant when considering the real concentration of short chain alcohols (C2OH, C6OH) in the lipid bilayer.
An area of study that requires finer investigation is with regards to lipid flip-flop and the membrane's bending rigidity, which molecular theory has suggested.74 Specifically, the authors calculated that by restricting the bilayer's ability to rearrange its lipid components the bending elastic constant of a bilayer increases – even accounting for the area per lipid. Here, we present experimental and simulation data to support this idea. With greater lipid flip-flop, the bilayer became softer and more fluid as shown in Fig. 5f. Now, which property leads to the other is up for debate, but it can be supposed that flip-flop destabilizes, even if momentarily, the lipid network, effectively softening it.
The results shown here are attributed to the alcohols. In the relevant lipid concentrations, lipid flip-flop and exchange has not demonstrated a lipid concentration dependence in the measured regimes.24,27 Also, in Table S1 (ESI†) DLS data showed that the hydrodynamic particle diameter stayed relatively constant at 140 nm before and after TR-SANS measurements, with only a slight increase in the polydispersity index after 48–72 hours. Further, the lack of correlation peaks in the SANS and SAXS curves informs that intervesicular interactions are negligible at these low liposomal concentrations. With these details, we can omit the potential of vesicle hemi/-fusion and attribute our observations to alcohol-specific effects.
5 Conclusions
This study used complementary biophysical techniques to monitor lipid dynamics and bilayer structure of biomimetic DMPC membranes. TR-SANS revealed C2OH and C6OH accelerated flip-flop and exchange at elevated concentrations, while C10OH, C14OH, and C18OH slowed DMPC flip-flop at all measured concentrations. SAXS and MD simulations showed that the short chain alcohols diminished the membranes structural integrity, thinning, and fluidizing the membrane and the opposite for C10 and longer. The former aspects hinted at a defect-mediated pathway which was corroborated with calcein leakage assays that showed the shorter chained alcohols induced pores that not only allows cargo passage, but the diffusion of lipids within the bilayer leaflets.All in all, short chain alcohols act as perturbing agents, pushing the lipid headgroups farther apart and allowing lipid flip-flop and exchange to occur less impeded. Meanwhile, the addition of longer chain alcohols partition into these membranes and act as structural components instead of as perturbants, filling in voids and mitigating the appearance of defects. The disturbance that some small anesthetic molecules, like short chain n-alcohols, cause to lipid organization and bilayer integrity would no doubt influence the proteins that rely on it as a platform for activity.
Author contributions
M. H. L. N. and D. M. conceptualized and designed all of the experiments. M. H. L. N. and D. D. carried out experimentation, data acquisition, and analysis. E. G. K. and M. D. provided expert help on VSANS data acquisition and analysis. M. H. L. N. conducted all simulations and the subsequent analysis. M. H. L. N. and D. D. wrote the original manuscript. All authors reviewed and edited the original manuscript. D. M. acquired the funds for this study.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work acknowledges support from the Natural Sciences and Engineering Research Council (NSERC) of Canada (funding reference number RGPIN-2018-04841, D. M.), the NSERC Canada Graduate Scholarship-Doctoral award (CGS-D, M. H. L. N.) and the Canadian Institutes of Health Research CGS-D award (CIHR CGS-D, M. D.). Access to the VSANS Instrument was provided by the Center for High Resolution Neutron Scattering, a partnership between NIST and the NSF under Agreement No. DMR-2010792. The identification of commercial products of trade names does not imply endorsement or recommendation by the National Institute of Standards and Technology. The authors would like to thank Dr. Haden L. Scott for running the SAXS samples.Notes and references
- J. W. Dundee, M. Isaac and R. S. J. Clarke, Anesth. Analg., 1969, 48, 665–669 CrossRef CAS.
- S. M. Wong, E. Fong, D. L. Tauck and J. J. Kendig, Eur. J. Pharmacol., 1997, 329, 121–127 CrossRef CAS.
- S. N. Treistman and G. E. Martin, Trends Neurosci., 2009, 32, 629–637 CrossRef CAS.
- Y. Zuo, G. L. Aistrup, W. Marszalec, A. Gillespie, L. E. Chavez-Noriega, J. Z. Yeh and T. Narahashi, Mol. Pharmacol., 2001, 60, 700–711 CAS.
- R. W. Peoples and F. F. Weight, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 2825–2829 CrossRef CAS.
- M. J. Wick, S. J. Mihic, S. Ueno, M. P. Mascia, J. R. Trudell, S. J. Brozowski, Q. Ye, N. L. Harrison and R. A. Harris, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 6504–6509 CrossRef CAS.
- C. Schwichtenhövel, B. Deuticke and C. W. Haest, Biochim. Biophys. Acta, Biomembr., 1992, 1111, 35–44 CrossRef.
- N. Kucerka, T. Kondela, J. Gallová, T. Hauß, S.-J. Marrink and J. Barnoud, Molecules, 2017, 22, 2078 CrossRef.
- T. Kondela, J. Gallová, T. Hauß, O. Ivankov, N. Kucerka and P. Balgavý, Eur. Pharm. J., 2017, 64, 4–8 CrossRef CAS.
- S. Rifici, G. D’Angelo, C. Crupi, C. Branca, V. Conti Nibali, C. Corsaro and U. Wanderlingh, J. Phys. Chem. B, 2016, 120, 1285–1290 CrossRef CAS.
- P. A. Zapata-Morin, F. J. Sierra-Valdez and J. C. Ruiz-Suárez, Biochim. Biophys. Acta, Biomembr., 2020, 101, 107732 CAS.
- M. Klacsová, M. Bulacu, N. Kucerka, D. Uhríková, J. Teixeira, S. J. Marrink and P. Balgavý, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2136–2146 CrossRef.
- D. C. Mitchell, J. T. R. Lawrence and B. J. Litman, J. Biol. Chem., 1996, 271, 19033–19036 CrossRef CAS PubMed.
- H. I. Ingólfsson and O. S. Andersen, Biophys. J., 2011, 101, 847–855 CrossRef.
- T. Horishita and R. A. Harris, J. Pharmacol. Exp. Ther., 2008, 326, 270–277 CrossRef CAS PubMed.
- K. Simons and J. L. Sampaio, Cold Spring Harbor Perspect. Biol., 2011, 3, a004697 Search PubMed.
- Y. C. Li, M. J. Park, S.-K. Ye, C.-W. Kim and Y.-N. Kim, Am. J. Pathol., 2006, 168, 1107–1118 CrossRef CAS.
- P. M. Bratton, V. A. Henson, D. R. Fadok, P. A. Voelker, J. J. Campbell, D. L. Cohen, V. A. Fadok, D. R. Voelker, P. A. Campbell, J. J. Cohen, D. L. Bratton and P. M. Henson, J. Immunol., 1992, 148, 2207–2216 CrossRef.
- V. A. Fadok and P. M. Henson, Curr. Biol., 1998, 8, R693–R695 CrossRef CAS.
- B. Griepernau, S. Leis, M. F. Schneider, M. Sikor, D. Steppich and R. A. Böckmann, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 2899–2913 CrossRef CAS.
- J. Liu and J. C. Conboy, Biophys. J., 2005, 89, 2522–2532 CrossRef CAS.
- M. Nakano, M. Fukuda, T. Kudo, H. Endo and T. Handa, Phys. Rev. Lett., 2007, 98, 238101 CrossRef PubMed.
- M. Nakano, M. Fukuda, T. Kudo, N. Matsuzaki, T. Azuma, K. Sekine, H. Endo and T. Handa, J. Phys. Chem. B, 2009, 113, 6745–6748 CrossRef CAS PubMed.
- M. H. Nguyen, M. DiPasquale, B. W. Rickeard, C. B. Stanley, E. G. Kelley and D. Marquardt, Biophys. J., 2019, 116, 755–759 CrossRef CAS PubMed.
- T. Sugiura, C. Takahashi, Y. Chuma, M. Fukuda, M. Yamada, U. Yoshida, H. Nakao, K. Ikeda, D. Khan, A. H. Nile, V. A. Bankaitis and M. Nakano, Biophys. J., 2019, 116, 92–103 CrossRef CAS PubMed.
- M. Kaihara, H. Nakao, H. Yokoyama, H. Endo, Y. Ishihama, T. Handa and M. Nakano, Chem. Phys., 2013, 419, 78–83 CrossRef CAS.
- M. H. L. Nguyen, M. DiPasquale, B. W. Rickeard, C. G. Yip, K. N. Greco, E. G. Kelley and D. Marquardt, New J. Chem., 2021, 45, 447–456 RSC.
- J. E. Nielsen, V. A. Bjørnestad and R. Lund, Soft Matter, 2018, 14, 8750–8763 RSC.
- S. R. Castillo, B. W. Rickeard, M. DiPasquale, M. H. L. Nguyen, A. Lewis-Laurent, M. Doktorova, B. Kav, M. S. Miettinen, M. Nagao, E. G. Kelley and D. Marquardt, Mol. Pharmaceutics, 2022, 19, 1839–1852 CrossRef CAS.
- M. Nakano, M. Fukuda, T. Kudo, M. Miyazaki, Y. Wada, N. Matsuzaki, H. Endo and T. Handa, J. Am. Chem. Soc., 2009, 131, 8308–8312 CrossRef CAS PubMed.
- Y.-T. Lee, D. S. Li and L. D. Pozzo, Langmuir, 2019, 35, 15204–15213 CrossRef CAS PubMed.
- H. L. Scott, A. Skinkle, E. G. Kelley, M. N. Waxham, I. Levental and F. A. Heberle, Biophys. J., 2019, 117, 1381–1386 CrossRef CAS PubMed.
- S. R. Castillo, M. H. Nguyen, M. DiPasquale, E. G. Kelley and D. Marquardt, Biophys. J., 2023, 122, 2353–2366 CrossRef PubMed.
- J. Barker, J. Moyer, S. Kline, G. Jensen, J. Cook, C. Gagnon, E. Kelley, J. P. Chabot, N. Maliszewskyj, C. Parikh, W. Chen, R. P. Murphy and C. Glinka, J. Appl. Crystallogr., 2022, 55, 271–283 CrossRef CAS.
- M. DiPasquale, M. H. Nguyen, B. W. Rickeard, N. Cesca, C. Tannous, S. R. Castillo, J. Katsaras, E. G. Kelley, F. A. Heberle and D. Marquardt, Biochim. Biophys. Acta, Biomembr., 2020, 1862, 183189 CrossRef CAS.
- S. R. Kline, J. Appl. Crystallogr., 2006, 39, 895–900 CrossRef CAS.
- G. Pabst, R. Koschuch, B. Pozo-Navas, M. Rappolt, K. Lohner and P. Laggner, J. Appl. Crystallogr., 2003, 36, 1378–1388 CrossRef CAS.
- P. Heftberger, B. Kollmitzer, F. A. Heberle, J. Pan, M. Rappolt, H. Amenitsch, N. Kucerka, J. Katsaras and G. Pabst, J. Appl. Crystallogr., 2014, 47, 173–180 CrossRef CAS.
- E. L. Wu, X. Cheng, S. Jo, H. Rui, K. C. Song, E. M. Dávila-Contreras, Y. Qi, J. Lee, V. Monje-Galvan, R. M. Venable, J. B. Klauda and W. Im, J. Comput. Chem., 2014, 35, 1997–2004 CrossRef CAS.
- M. J. Abraham, T. Murtola, R. Schulz, S. Páll, J. C. Smith, B. Hess and E. Lindah, SoftwareXBiochim. Biophys. Acta, Biomembr., 2015, 1–2, 19–25 Search PubMed.
- S. Kim, J. Lee, S. Jo, C. L. Brooks, H. S. Lee and W. Im, J. Comput. Chem., 2017, 38, 1879–1886 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS.
- R. Guixà-González, I. Rodriguez-Espigares, J. M. Ramírez-Anguita, P. Carrió-Gaspar, H. Martinez-Seara, T. Giorgino and J. Selent, Bioinformatics, 2014, 30, 1478–1480 CrossRef.
- M. Doktorova, D. Harries and G. Khelashvili, Phys. Chem. Chem. Phys., 2017, 19, 16806–16818 RSC.
- S. J. Marrink, H. J. Risselada, S. Yefimov, D. P. Tieleman and A. H. de Vries, J. Phys. Chem. B, 2007, 111, 7812–7824 CrossRef CAS.
- Y. Qi, H. I. Ingólfsson, X. Cheng, J. Lee, S. J. Marrink and W. Im, J. Chem. Theory Comput., 2015, 11, 4486–4494 CrossRef CAS.
- T. Shimanouchi, P. Walde, J. Gardiner, Y. R. Mahajan, D. Seebach, A. Thomae, S. D. Krämer, M. Voser and R. Kuboi, Biochim. Biophys. Acta, Biomembr., 2007, 1768, 2726–2736 CrossRef CAS.
- B. Maherani, E. Arab-Tehrany, A. Kheirolomoom, D. Geny and M. Linder, Biochimie, 2013, 95, 2018–2033 CrossRef CAS PubMed.
- N. Kucerka, M. P. Nieh and J. Katsaras, Biochim. Biophys. Acta, Biomembr., 2011, 1808, 2761–2771 CrossRef CAS.
- M. H. L. Nguyen, M. DiPasquale, B. W. Rickeard, M. Doktorova, F. A. Heberle, H. L. Scott, F. N. Barrera, G. Taylor, C. P. Collier, C. B. Stanley, J. Katsaras and D. Marquardt, Langmuir, 2019, 35, 11735–11744 CrossRef CAS PubMed.
- A. Chanturiya, E. Leikina, J. Zimmerberg and L. V. Chernomordik, Biophys. J., 1999, 77, 2035–2045 CrossRef CAS PubMed.
- C. Yuan, R. J. O'Connell, A. Wilson, A. Z. Pietrzykowski and S. N. Treistman, J. Biol. Chem., 2008, 283, 5090–5098 CrossRef CAS PubMed.
- F. W. Stetter and T. Hugel, Biophys. J., 2013, 104, 1049–1055 CrossRef CAS PubMed.
- E. S. Rowe, Alcohol.: Clin. & Exp. Res., 1981, 5, 259–263 CrossRef CAS PubMed.
- P. Westerman, J. Pope, N. Phonphok, J. Doane and D. Dubro, Biochim. Biophys. Acta, Biomembr., 1988, 939, 64–78 CrossRef CAS PubMed.
- H. Usuda, M. Hishida, E. G. Kelley, Y. Yamamura, M. Nagao and K. Saito, Phys. Chem. Chem. Phys., 2020, 22, 5418–5426 RSC.
- R. Abedi Karjiban, N. S. Shaari, U. V. Gunasakaran and M. Basri, J. Chem., 2013, 2013, 1–6 CrossRef.
- N. Maurer, K. F. Wong, H. Stark, L. Louie, D. McIntosh, T. Wong, P. Scherrer, S. C. Semple and P. R. Cullis, Biophys. J., 2001, 80, 2310–2326 CrossRef CAS.
- E. Disalvo, Biochim. Biophys. Acta, Biomembr., 1987, 905, 9–16 CrossRef CAS.
- B. B. Machta, E. Gray, M. Nouri, N. L. McCarthy, E. M. Gray, A. L. Miller, N. J. Brooks and S. L. Veatch, Biophys. J., 2016, 111, 537–545 CrossRef CAS.
- C. E. Cornell, N. L. McCarthy, K. R. Levental, I. Levental, N. J. Brooks and S. L. Keller, Biophys. J., 2017, 113, 1200–1211 CrossRef CAS.
- D. Marquardt, F. A. Heberle, T. Miti, B. Eicher, E. London, J. Katsaras and G. Pabst, Langmuir, 2017, 33, 3731–3741 CrossRef CAS.
- N. Sapay, W. F. D. Bennett and D. P. Tieleman, Soft Matter, 2009, 5, 3295 RSC.
- B. Wah, J. M. Breidigan, J. Adams, P. Horbal, S. Garg, L. Porcar and U. Perez-Salas, Langmuir, 2017, 33, 3384–3394 CrossRef CAS.
- M. Klacsová, A. Bóta, P. Westh, S. de Souza Funari, D. Uhríková and P. Balgavý, Phys. Chem. Chem. Phys., 2021, 23, 8598–8606 RSC.
- S. L. Veatch and S. L. Keller, Biophys. J., 2003, 85, 3074–3083 CrossRef CAS.
- J. Ludwig and L. Maibaum, Chem. Phys. Lipids, 2020, 233, 104986 CrossRef CAS.
- T. H. Aagaard, M. N. Kristensen and P. Westh, Biophys. Chem., 2006, 119, 61–68 CrossRef CAS.
- J. S. Allhusen and J. C. Conboy, Acc. Chem. Res., 2017, 50, 58–65 CrossRef CAS PubMed.
- T. C. Anglin, M. P. Cooper, H. Li, K. Chandler and J. C. Conboy, J. Phys. Chem. B, 2010, 114, 1903–1914 CrossRef CAS PubMed.
- R. Homan and H. J. Pownall, Biochim. Biophys. Acta, Biomembr., 1988, 938, 155–166 CrossRef CAS PubMed.
- J. Boggs, G. Rangaraj and A. Watts, Biochim. Biophys. Acta, Biomembr., 1989, 981, 243–253 CrossRef CAS PubMed.
- N. P. Franks and W. R. Lieb, Proc. Natl. Acad. Sci. U. S. A., 1986, 83, 5116–5120 CrossRef CAS.
- I. Szleifer, D. Kramer, A. Ben-Shaul, W. M. Gelbart and S. A. Safran, J. Chem. Phys., 1990, 92, 6800–6817 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2sm01583h |
| This journal is © The Royal Society of Chemistry 2023 |