
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnprecedented perspectives on the application of CinNapht fluorophores provided by a “late-stage” functionalization strategy†
Eléonore
Tacke
a,
Minh-Duc
Hoang
 a,
Kevin
Tatoueix
b,
Benoît
Keromnes
a,
Elsa
Van Eslande
a,
Philippe
Durand
a,
Gregory
Pieters
b and
Arnaud
Chevalier
*a
a,
Kevin
Tatoueix
b,
Benoît
Keromnes
a,
Elsa
Van Eslande
a,
Philippe
Durand
a,
Gregory
Pieters
b and
Arnaud
Chevalier
*a
aUniversité Paris-Saclay, CNRS, Institut de Chimie des Substances Naturelles, UPR 2301, 91198, Gif-sur-Yvette, France. E-mail: arnaud.chevalier@cnrs.fr
bDépartement Médicaments et Technologies pour la Santé (DMTS), SCBM, Université Paris-Saclay, CEA, INRAE, 91191 Gif-sur-Yvette, France
First published on 3rd May 2023
Abstract
A simple and easy-to-implement process based on a nucleophilic aromatic substitution reaction with a wide variety of nucleophiles on a fluorinated CinNapht is described. This process has the key advantage of introducing multiple functionalities at a very late stage, thus providing access to new applications including the synthesis of photostable and bioconjugatable large Stokes shift red emitting dyes and selective organelle imaging agents, as well as AIEE-based wash-free lipid droplet imaging in live cells with high signal-to-noise ratio. The synthesis of bench-stable CinNapht-F has been optimized and can be reproduced on a large scale, making it an easy-to-store starting material that can be used at will to prepare new molecular imaging tools.
Introduction
For decades, chemists and biochemists have been working on the development of new organic fluorophores by devising new luminescent aromatic structures. These small molecules are now commonly used for multiple applications, including fluorogenic labeling1 or as molecular probes dedicated to sensing.2 Nevertheless, we are still looking for the perfect dye that would meet all the criteria sought by scientists. This is a fact: the perfect dye does not exist and probably never will. Despite this, “tireless chemists” continue to devote themselves to the development of new fine-tuned structures that could provide precious help to biologists.1c It is, therefore, essential to encourage the development of synthetic methods that allow access to an ever-increasing variety of fluorophores. Despite recent improvements through the development of new near-infrared fluorophores or the emergence of biphotonic microscopy, there remains a real advantage in using large Stokes shift red-emitting fluorophores. They have the key advantage of avoiding spectral overlap, and therefore allow optimization of the acquisition parameters during imaging experiments. The synthesis of such fluorophores is being extensively studied, and several approaches have been elaborated, providing multiple original fluorophores.3 In this context, we have recently reported a naphthalimide–cinnoline hybrid dye called CinNapht which has proven to be useful in imaging experiments.4 We showed that the modification of the amine moiety allows the photophysical properties to be adjusted.5 This feature makes CinNaphts versatile platforms for biological or photophysical purposes. However, variation of the electron-donating aniline moiety in the fluorophore requires a complete re-synthesis of the dye, starting from the suitable pinacolborane meta-substituted aniline (Fig. 1, top), which often needs to be prepared. This is the main limitation of the previous synthetic approach, which resulted in a restriction in both the number and variety of accessible CinNapht analogues. In addition, the last step of the synthetic process, based on the SEAr (electrophilic aromatic substitution) type reaction of an intermediate diazonium salt, leads to the formation of ortho and fluorescent para regioisomers. Even though the para isomer was the major product, its isolation requires purification steps which inevitably causes reduced reaction yields.4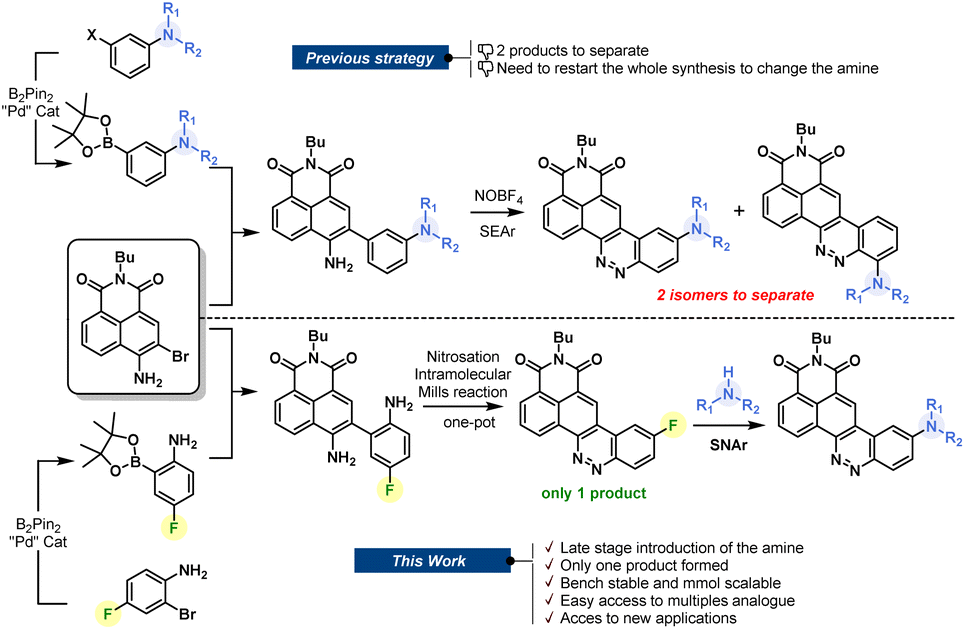 | ||
| Fig. 1 Description of the new CinNapht synthesis strategy and highlighting of improvements. | ||
Based on these considerations, we sought to design a robust and efficient synthesis procedure allowing the real potential of CinNaphts for fluorescence imaging to be unlocked. In this paper, we propose a synthetic pathway allowing various nucleophilic moieties to be introduced at the very end of the synthesis through nucleophilic aromatic substitution (SNAr). The late-stage strategy is an astute way to conveniently incorporate electron donors on a fluorescent scaffold. Already illustrated in the literature, the previously described examples are mostly based on a Buchwald palladium-catalyzed amination reaction on an aromatic triflate or halide.6 This strategy led to multiple examples of fluorescent probes with improved photophysical properties,7 providing a new perspective for bioanalytical purposes.8 Despite the fact that it avoids the use of metal, SNAr has rarely been used in fluorophore postamination strategies,9 and has never been considered for the final functionalization of CinNaphts. In this paper, we propose a robust synthetic route for the large-scale preparation of an aryl fluoride derivative of CinNapht that can undergo a nucleophilic substitution reaction with a wide variety of nucleophiles to provide simple access to a broad range of fluorophores, including bioconjugatable analogs, CinNaphts with improved photostability and unprecedented CinNapht-based organelle markers.
Results and discussion
Large scale synthesis of CinNapht-F and study of its reactivity through SNAr reactions
We started by designing a synthetic route for the preparation of a fluorinated CinNapht (CinNapht-F, 4). The major issue to be managed was the formation of the N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond constituting the cinnoline part (Fig. 1). In addition to the fact that it leads to the formation of two isomers, the strategy initially employed for the synthesis of these fluorophores uses an aromatic electrophilic substitution reaction requiring an enriched aromatic unit. As we had anticipated, the implementation of this strategy for the synthesis of a fluorinated fluorophore gave particularly poor results, probably due to the low electronic enrichment of the fluorinated aromatic moiety, which did not allow for a good reactivity with SEAr.
N double bond constituting the cinnoline part (Fig. 1). In addition to the fact that it leads to the formation of two isomers, the strategy initially employed for the synthesis of these fluorophores uses an aromatic electrophilic substitution reaction requiring an enriched aromatic unit. As we had anticipated, the implementation of this strategy for the synthesis of a fluorinated fluorophore gave particularly poor results, probably due to the low electronic enrichment of the fluorinated aromatic moiety, which did not allow for a good reactivity with SEAr.
We thus revised the synthetic route in order to allow the formation of azobenzene by an intramolecular nitrosation/Mills reaction one-pot process, which would therefore not require an electron-rich fluorinated aromatic ring (Fig. 2). Thus, the aromatic bis adduct 3 was prepared by a Suzuki–Miyaura cross-coupling reaction catalysed by Pd(PPh3)4 in very good yield of 92% starting from molecule 2. This intermediate was then treated with mCPBA to form the desired derivative in 70% yield. The structure of 4 was unambiguously confirmed by NMR and HRMS, and an X-ray diffraction pattern was also obtained. This reaction was shown to be reproducible and implementable on a large scale (2–3 mmol) making it a robust process for the synthesis of CinNapht-F. To our delight, this fluorinated dye was found to be highly stable over months, without any specific storage conditions. We then studied the reactivity of CinNapht-F in a model SNAr reaction using n-butylamine as nucleophile (Table 1).
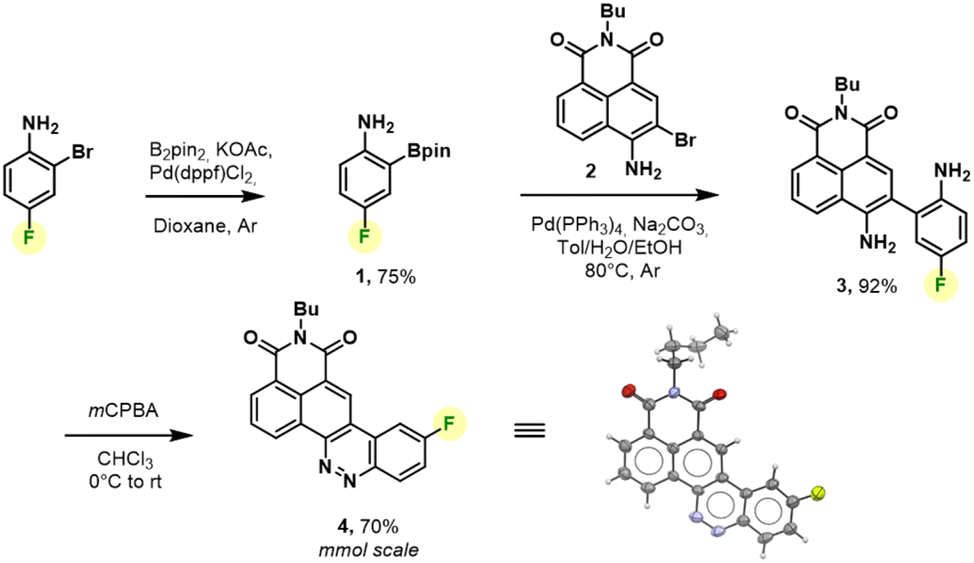 | ||
| Fig. 2 mmol scalable synthesis and X-ray diffraction structure of bench stable CinNapht-F 4. | ||
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solventa | Baseb | Temp. (°C) | Time (h) | HPLC yieldc |
| a Dry solvent was used. b 4.0 equivalents of base were used. c Calculated on a Max Plot PDA chromatogram. d Not determined due to lack of solubility. e Using 1.05 equivalents of nBuNH2. | |||||
| 1 | DMF | Na2CO3 | 100 | 24 | 95 |
| 2 | DMF | K2CO3 | 100 | 24 | 87 |
| 3 | DMF | Cs2CO3 | 100 | 24 | 68 |
| 4 | DMF | K3PO4 | 100 | 24 | 83 |
| 5 | DMF | K2HPO4 | 100 | 24 | 86 |
| 6 | DMF | NaHCO3 | 100 | 24 | 95 |
| 7 | DMF | KOH | 100 | 24 | 31 |
| 8 | DMF | AcOK | 100 | 24 | 80 |
| 9 | DMF | tBuOK | 100 | 24 | 13 |
| 10 | CH3CN | NaHCO3 | 80 | 24 | —d |
| 11 | DMSO | NaHCO3 | 100 | 24 | 91 |
| 12 | Dioxane | NaHCO3 | 100 | 24 | —d |
| 13 | NMP | NaHCO3 | 100 | 24 | 96 |
| 14 | DCE | NaHCO3 | 70 | 24 | 3 |
| 15 | Tol | NaHCO3 | 100 | 24 | —d |
| 16 | DMF | NaHCO3 | 60 | 24 | 60 |
| 17 | DMF | NaHCO3 | 25 | 24 | 0 |
| 18 | DMF | NaHCO3 | 100 | 24 | 85e |
| 19 | DMF | NaHCO3 | 100 | 0.5 | 80 |
| 20 | DMF | NaHCO 3 | 100 | 1 | 97 |
| 21 | DMF | NaHCO3 | 100 | 4 | 96 |
Butylamine was reacted with CinNapht-F 4 under a range of conditions. Lines 1 to 9 in Table 1 show that in DMF while heating, the reaction tolerates most of the bases we tried, with the exception of slightly stronger bases such as tBuOK and NaOH (lines 7 and 9). In the same way, the aprotic polar solvents allowing the solubilization of the starting CinNapht are on the whole usable with no particular variability for this reaction. The lack of conversions observed in some cases was mainly due to insolubility of the starting substrate (lines 10, 12, 14 and 15). We could observe that the reaction also works at 60 °C and after changing the number of equivalents and studying the reaction time, we finally established that, in the case of n-butylamine, 1 h of reaction at 100 °C in DMF using NaHCO3 as base provided the best result with 97% HPLC yield. We next conducted further studies to evaluate the scope of this reaction. A wide variety of nucleophiles, exhibiting different reactivities, were tested with variable isolated yields summarized in Fig. 3. Aliphatic (5a–5c) and benzylic amines (5d, 5e), whether primary or secondary, gave good results with isolated yields reaching up to 95%. α-Type nucleophiles were also investigated. While methylhydrazine showed good results with 80% isolated yield of CinNapht 5g, methoxyamine was surprisingly found to be unreactive. Other oxyamines were tested but none of them led to SNAr reaction products (data not shown).
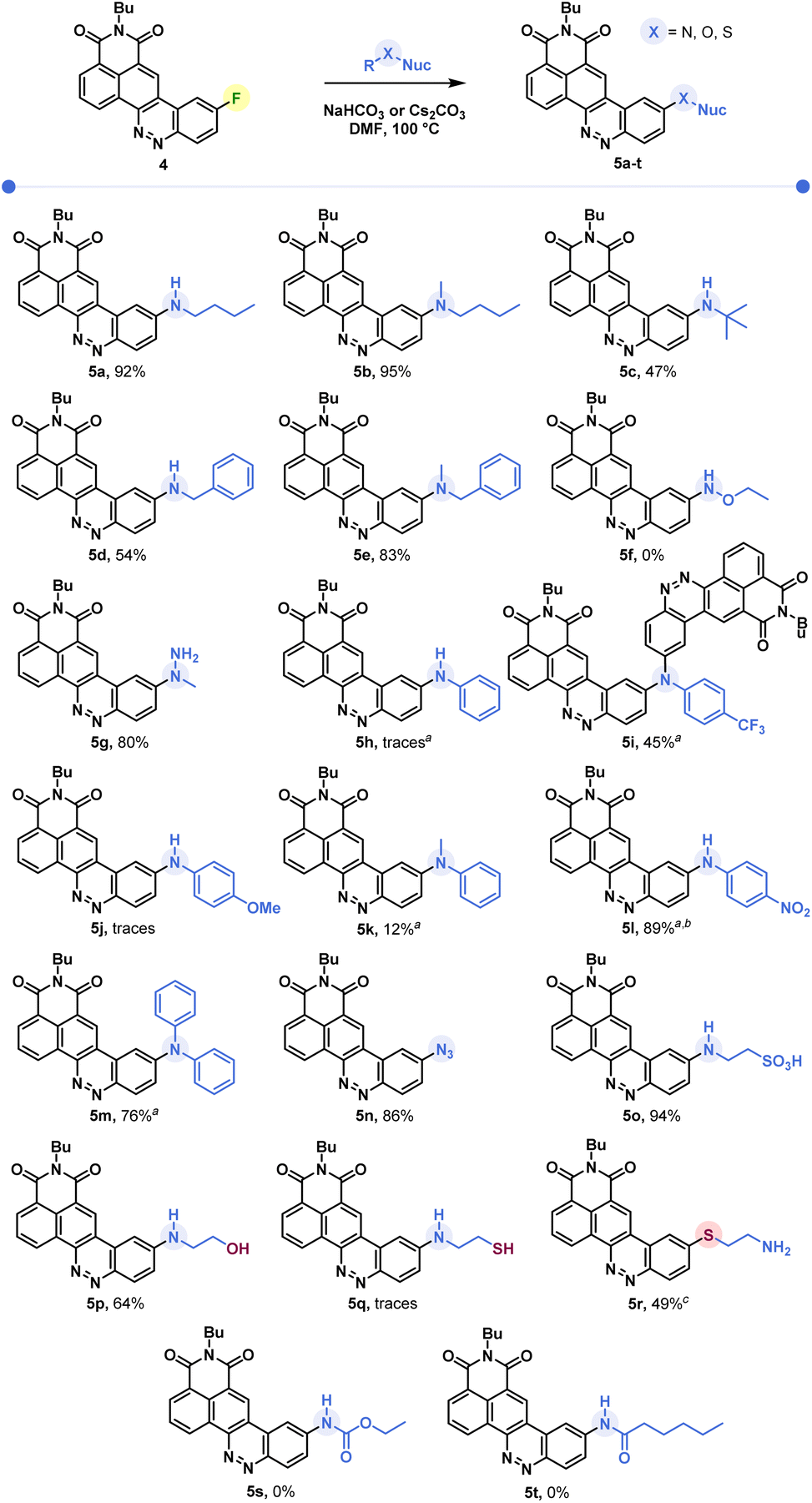 | ||
| Fig. 3 Scope of the reaction of SNAr on CinNapht-F. a Cs2CO3 used as a base. b HPLC yield based on a Max Plot chromatogram. c Isolated yields by reaction of thioethanolamine (nuc = nucleophile). | ||
The reactivity of aromatic amines was also studied (5h–5m). Either no reaction or very low conversion was observed using NaHCO3 as a base. Nevertheless, we were able to obtain the product of diphenylamine addition by changing the base to Cs2CO3. Interestingly, in the meantime no conversion was noticed with Na2CO3 and only poor conversion was noticed after one night using K2CO3 (Fig. S1†). In order to better understand this selectivity of caesium carbonate, we performed two additional experiments (Fig. S1†). The addition of crown ether (15-C-5) to the reaction in order to increase both the solubility and the basicity of the carbonate did not improve the reactivity of the diphenylaniline and no product was observed. In the meantime, the addition of 1 equivalent of CsF led to a very good conversion (see Fig. S1, line 5†). We also noticed that a supplementation with caesium fluoride using NaHCO3 led to a significant conversion, demonstrating that the caesium cation plays a key role in the improvement of aromatic amines' reactivity. Thus, using Cs2CO3, we studied the reactivity of aromatic aniline with variable electronic enrichments (5i to 5m). Good conversion was observed in most of the cases (5i, 5k, 5l and 5m) and pure CinNaphts were isolated with yields ranging from 12% to 77%. Despite its good reactivity (see the crude mixture chromatogram, Fig. S2†), we failed in trying to isolate CinNapht 5l as a pure compound, thus only HPLC conversion yield of 89% can be obtained. Unfortunately, despite the use of caesium carbonate neither of the CinNaphts 5h (aniline) or 5j (para-methoxyaniline) could be isolated, and only traces of the products were observed. Surprisingly, the reaction of para-trifluoromethylaniline led to the formation of a dimer 5i. This could be explained by the formation of a mono-addition product whose proton is acidic enough to allow deprotonation by caesium carbonate. This results in the formation of a much more reactive anion which will react preferentially to generate the dimer 5i. All together, these results show that the implementation of the reaction with aromatic amines using caesium carbonate as a base must be considered carefully. Sodium azide can also be used for this reaction providing azido CinNapht 5n with a good yield of 86%. This new derivative was thought to constitute a good candidate for fluorogenic click reactions,10 radiation activation or sensing of thiols11 for example. Unfortunately, compound 5n was found to be unstable, especially in an aqueous environment in which we could quickly observe the formation of the fluorescent reduction product (see Fig. S3†). These results were confirmed by the appearance of a non-specific fluorescent signal in cells during confocal microscopy experiments. Moreover, the product of SPAAC reaction between 5n and DBCO turned out to be not fluorescent, preventing its use in a fluorogenic click reaction (see Fig. S4†). This disappointing result leads us to warn against the use of the aryl azide 5n for fluorogenic applications in biological media. The competition between the amine and other nucleophilic functions such as an alcohol or a thiol was also studied. While exclusive amine addition was observed in the case of ethanolamine (5p), a preferred thiol addition was witnessed using thioethanolamine resulting in the formation of product 5q in line with the higher nucleophilicity of the thiol compared to the primary amine substructures. The product of amine addition may be observed in trace amounts, but exclusively under its oxidized disulphide form. This particular reactivity of thiols on aromatic fluorides of fluorophore derivatives could thus give access to sulfide derivatives of fluorophores which can be used for sensing ROS such as hypochloric acid. This result can thus represent a source of inspiration in this field.12 Finally, our attempts to introduce carbamides (5s) or primary amides (5u) were unsuccessful which further illustrates the limitation of this reaction, which nevertheless allows the introduction of a very broad range of amines.
Synthesis of original CinNaphts for new applications
Considering this remarkable versatility of the SNAr reaction we studied the introduction of more complex amines in order to generate more sophisticated fluorophores, dedicated to new applications. Some chosen examples are presented in Fig. 4. First, we successfully introduced a difluoroazetidine moiety already reported to induce a slight hypsochromic shift of both absorption and emission wavelengths that we could verify also with our CinNapht.7a Then we incorporated chemical functions that could allow the bioconjugation of CinNaphts. Disappointingly, the addition of glycine or sarcosine to CinNapht-F 4 failed, preventing its functionalisation by a carboxylic acid. We countered this by resorting to isonipecotic acid and obtained the CinNapht 6b in a good yield of 60% without any prior protection of the carboxylic function. We also prepared a primary amine derivative 6d that presents the particularity of having a PEG-10 linker separating the CinNapht from the amine function. Note that, by reacting these two CinNaphts 6b and 6d using BOP reagent, we easily obtained the CinNapht dimer 7a (Fig. 4, bottom) by amide bond formation in a good isolated yield of 63%. This additional result confirmed the potential use of both the acidic derivative 6b and the amine derivative 6d in amidation type reactions. An alkyne derivative of CinNapht (6c) was also obtained in good yield and its reactivity with azide in “click” CuAAC reaction13 was demonstrated by reacting it with benzyl azide using copper sulfate (0.1 equiv.) and sodium ascorbate (0.2 equiv.). The 1,4-disubstituted triazole adduct 7b was isolated with 83% yield, confirming CinNapht 6c to be a potent dye for fluorescent labelling using CuAAC type click reaction. Keeping in mind the selectivity of the amine addition over the hydroxyl moiety (Fig. 3, 5p), we tried to use more complex poly-alcohol units such as sugars. Thus, a modified mannose was prepared by introducing a free amine triethylene glycol linker on the anomeric position (see compounds 8 and 9 in the ESI†). It was reacted with CinNapht-F 4 to obtain a CinNapht-mannose 6e derivative in 81% yield. It is noteworthy that no protection of the hydroxyl groups of the mannose was required for this reaction, which would never have been conceivable with the reported access to CinNaphts. The amino acid derivative of CinNapht (6f) was also prepared using Fmoc–Lys–OtBu as a nucleophile partner for the SNAr reaction followed by the removal of the Fmoc protecting group. The tBu protected CinNapht 6f was isolated in one step with a moderate yield of 27%. In order to reach new application in cell imaging, we thought that CinNapht-F could constitute an easy to functionalize building block, allowing the introduction of organelle targeting functions. We started by introducing a morpholine14 moiety that led to a lysosome accumulating CinNapht (6g) with 60% yield. The introduction of a zwitterionic lipophilic amine (cf. ESI,† compound 10) enables the CinNapht 6h to be obtained, for cell plasma membrane labelling, and isolated with 87% yield.15 We also introduced with good yield both a triphenylphosphonium (6i) or a pyridinium moiety (6j) with the idea of targeting mitochondria.16 The possibility of introducing sophisticated amines at a very late stage of the synthetic process led us also to consider the introduction of an amine derivative of Hoechst.17 This led to a CinNapht (6o) with 10% yield, designed for imaging the nucleus of the cells. Finally, we investigated the access to deuterated CinNaphts. This was motivated by recent works from the Lavis lab suggesting that the deuteration of the donating amines in fluorophores can improve their photophysical performances,7b and this was further confirmed by a recent study published by Lehmann, Broichhagen and coworkers.18 Thus, introduction of regioselectively α-deuterated pyrrolidine19 was performed and led to 6l with very good isolated yield of 92%. This came to complete a set of new fluorophores whose photophysical properties were investigated.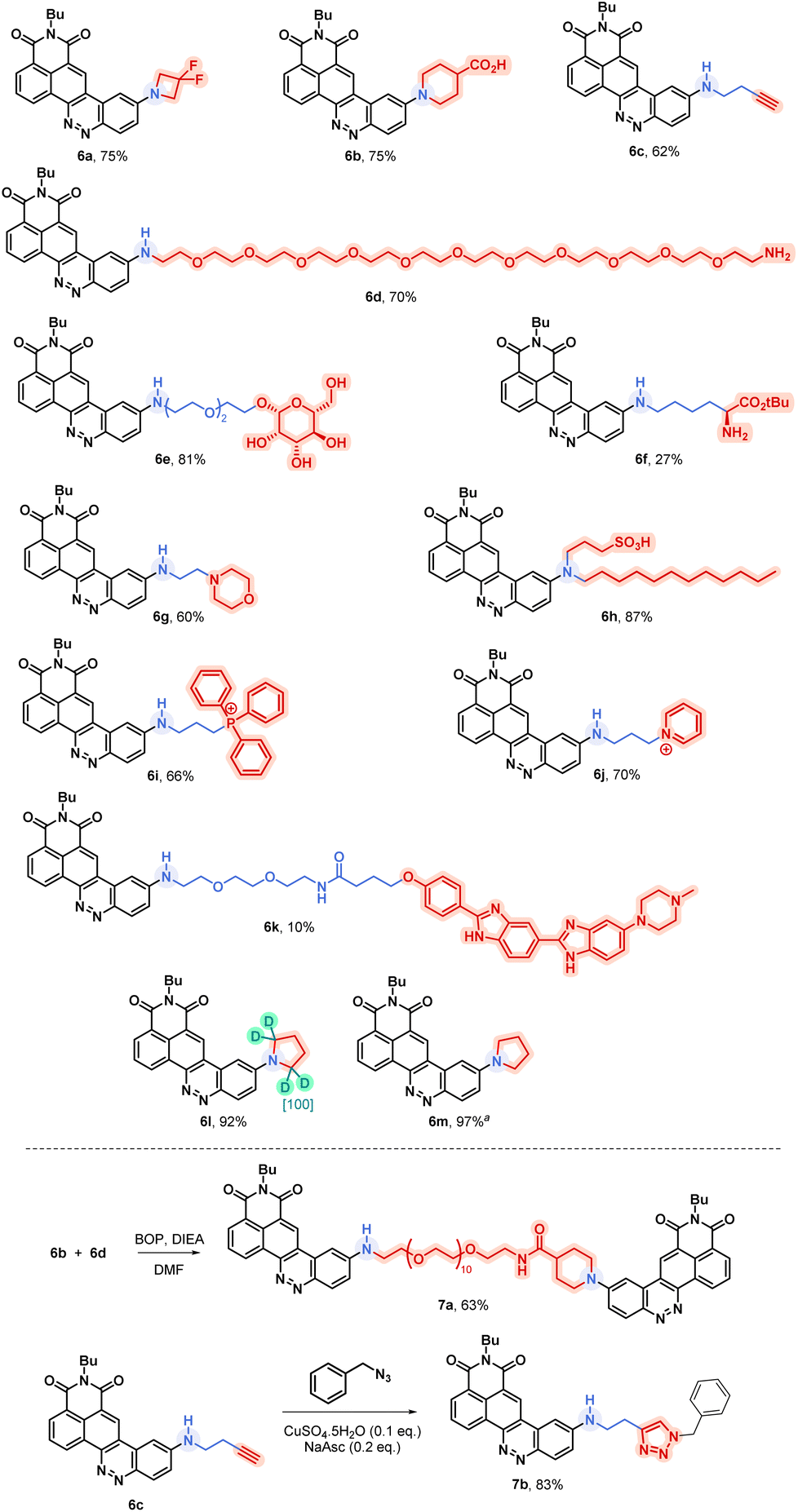 | ||
| Fig. 4 Examples of accessible original CinNaphts using SNAr late-stage functionalization with isolated yields. a Compound described previously5 and accessible thanks to the SNAr late-stage functionalization. | ||
Photophysical properties
The photophysical properties of all the synthesized analogues have been investigated. The absorption, excitation and emission spectra were recorded at 25 °C in CHCl3. We determined the λmaxEm and λmaxAbs as well as the molar extinction coefficient (εmax) and the fluorescence quantum yields (QYFl). The results of this study are reported in Table 2. A moderate modulation of the absorption maxima, centred mainly between 465 and 500 nm, was observed depending on the nature of the amine. We can nevertheless observe that a methylation of the amine generates an expected bathochromic shift of the absorption maximum, which is located here at about 30 nm as can be seen by comparing the derivative 5a to the derivative 5b. This seems to be independent of the nature of the amine, since the same type of behaviour is observed for benzyl amine derivatives 5d and 5e. Arylation induces a slight bathochromic shift especially for the diphenylamine derivative 5m whose absorption maximum reaches 496 nm. Note also that the thioether derivative 5r displays a particularly blue-shifted absorption maximum. Regarding the fluorescent properties of these compounds, several variations can be highlighted. First of all, the use of a secondary amine does not seem to generate a particular shift of the emission wavelength with almost identical maxima for compounds 5a and 5b for instance. On the other hand, this substitution seems to provide an improvement of the fluorescence efficiency with a doubled fluorescence quantum yield for 5b compared to 5a, which is also observed for 5d and 5e. This may be attributed to the more hindered amine substituent preventing non-radiative deexcitation due to C–N bond rotation. We can thus suggest that the introduction of a secondary amine allows the fluorescence quantum yield to be improved, but induces a decrease of the Stokes shift. The use of an α-nucleophile or aromatic moiety seems to be deleterious for the fluorescence since negligible fluorescence was observed for the derivatives 5g and 5k. Also a complete absence of fluorescence for the azide compound 5n as well as the thioether 5r was noticed. The set of fluorophores (6a–n) that have been prepared for potential new applications, including use in biological systems, have also been characterized. For this, the set of measurements in CHCl3 was completed by performing additional measurements in a PBS buffer containing 5% BSA. The results obtained in CHCl3 are in most cases comparable with the results presented previously, i.e. an absorption centered between 470 and 500 nm and an emission observed around 580 nm. Only compound 6h displays a singular absorption maximum, slightly red-shifted and centered at 520 nm and an emission at 615 nm. Note that the ICT nature of the fluorescence mechanism involved for CinNaphts results in a pronounced bathochromic shift of the emission maximum in polar solvents.4 Thus for compounds 6b, 6j and 6k that were not soluble in chloroform, and for which we resorted to DMSO, we notice that emissions are particularly red-shifted. A significant example of this phenomenon is the isonipecotic acid derivative 6b which has an emission maximum measured at 682 nm for a Stokes shift of 6302 cm−1 and a QYFl of 0.17. Compound 6a incorporating a difluoroazetidine unit is distinct in that it was proposed to slightly blue-shift the absorption and emission maxima. The result is consistent with the literature as it shows the lowest maxima of all fluorophores (λmaxAbs/Em = 446/554 nm). The quantum yields of these fluorophores measured in organic solvents vary little with values close to 20% which is in accordance with the values previously described for this type of fluorophore.5 However, 6a showed an erosion of QYFl that had not been particularly observed in the literature. It seems that the introduction of a difluoroazetidine allows the hypsochromic tuning of the emission wavelength, but causes a decrease of the fluorescence efficiency for CinNapht dyes. The best values are obtained for the compounds 6l and 6m, with values above 30%, in accordance with those observed for the corresponding non-deuterated derivative. The weak solubility of CinNaphts in PBS buffer leads to a low fluorescence emission and did not allow the study of their photophysical properties. Nevertheless, by supplementing the PBS buffer with 5% BSA, the fluorescent behavior of CinNaphts in aqueous medium can be evaluated. In general, we also observed a drastic decrease of the fluorescence efficiency in the PBS + 5% BSA buffer with values mainly from 2 to 5%, which is expected and consistent with the lack of solubility that we noticed, also characterized by a low molar extinction coefficient. The set of CinNaphts proposed in this article exhibit promising photophysical properties which nevertheless remain below those of the equivalent commercially available ones (DCM,20 ATTO 490LS21 or DyLight 515-LS22). However, thanks to the wide variety of CinNapht structures, they constitute valuable tools that allow multiple applications to be considered.| Dye | Solvent | λ maxAbsa (nm) | ε max (M−1 cm−1) | λ maxEm (nm) | Stokes shiftc (cm−1) | QYFlc | Brightness (εmax × QYFl) |
|---|---|---|---|---|---|---|---|
| a Values corresponding to the S0–S1 transition, but a strong S0–S2 transition is also observed (see Fig. S5–S7). b Molar extinction coefficient related to the S0–S1 transition (see Fig. S8 and S9). c Relative QY determined at 25 °C using 4-(dicyanomethylene)-2-methyl-6-(4-dimethylaminostyryl)-4H-pyran, “DCM” (QY = 0.43 in EtOH).20 d Compound 6n was previously described,5 but photophysical characterization was reproduced using the same fluorimeter as for other CinNaphts in order to enable more accurate comparison. | |||||||
| 4 | CHCl3 | 359 | 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 292 292 |
— | — | — | NA |
| 5a | CHCl3 | 466 | 8111 | 581 | 4248 | 0.15 | 1217 |
| 5b | CHCl3 | 498 | 12099 |
582 | 2898 | 0.29 | 3509 |
| 5c | CHCl3 | 471 | 6731 | 576 | 3870 | 0.15 | 1010 |
| 5d | CHCl3 | 456 | 11495 |
576 | 4569 | 0.11 | 1264 |
| 5e | CHCl3 | 484 | 9180 | 570 | 3117 | 0.21 | 1928 |
| 5g | CHCl3 | 470 | 12916 |
583 | 4124 | <0.01 | ND |
| 5i | CHCl3 | 476 | 12217 |
554 | 2958 | 0.08 | 977 |
| 5k | CHCl3 | 482 | 11055 |
630 | 4874 | <0.01 | ND |
| 5m | CHCl3 | 496 | 15078 |
665 | 5124 | 0.04 | 603 |
| 5n | CHCl3 | 378 | 8148 | — | — | — | NA |
| 5o | DMSO | 485 | 3143 | 665 | 5581 | 0.05 | 157 |
| 5p | CHCl3 | 459 | 11808 |
590 | 4837 | 0.15 | 1771 |
| 5r | CHCl3 | 403 | 8312 | — | — | — | NA |
| 6a | CHCl3 | 446 | 4765 | 554 | 4371 | 0.05 | 238 |
| PBS + 5% BSA | 471 | ND | 560 | ND | <0.01 | ND | |
| 6b | DMSO | 477 | 4961 | 682 | 6302 | 0.17 | 843 |
| PBS + 5% BSA | 508 | 3615 | 610 | 3292 | 0.02 | 072 | |
| 6c | CHCl3 | 456 | 9374 | 570 | 4386 | 0.09 | 844 |
| PBS + 5% BSA | 490 | 8223 | 608 | 3961 | 0.02 | 164 | |
| 6d | CHCl3 | 470 | 9193 | 603 | 4693 | 0.19 | 1747 |
| PBS + 5% BSA | 493 | 7280 | 611 | 3917 | 0.02 | 146 | |
| 6e | CHCl3 | 471 | 4866 | 592 | 4340 | 0.17 | 827 |
| DMSO | 486 | 6441 | 655 | 5309 | 0.18 | 1159 | |
| PBS + 5% BSA | 479 | 5088 | 585 | 3783 | 0.02 | 102 | |
| 6f | CHCl3 | 468 | 11265 |
586 | 4303 | 0.19 | 2140 |
| PBS + 5% BSA | 477 | 9970 | 584 | 3841 | 0.02 | 199 | |
| 6g | CHCl3 | 465 | 2144 | 570 | 3962 | 0.10 | 214 |
| DMSO | 485 | 2729 | 659 | 5444 | 0.13 | 355 | |
| PBS + 5% BSA | 485 | 2177 | 575 | 3227 | 0.02 | 44 | |
| 6h | CHCl3 | 520 | 3069 | 615 | 2971 | 0.32 | 982 |
| DMSO | 511 | 4639 | 677 | 4798 | 0.28 | 1299 | |
| PBS + 5% BSA | 513 | 4374 | 620 | 3364 | 0.05 | 219 | |
| 6i | CHCl3 | 487 | 2155 | 594 | 3699 | 0.23 | 496 |
| DMSO | 484 | 3432 | 651 | 5300 | 0.11 | 377 | |
| PBS + 5% BSA | 485 | 2977 | 593 | 3755 | 0.02 | 60 | |
| 6j | DMSO | 480 | 9017 | 652 | 5496 | 0.16 | 1443 |
| PBS + 5% BSA | 493 | 7946 | 619 | 4129 | 0.02 | 159 | |
| 6k | DMSO | 486 | 4763 | 660 | 5425 | 0.18 | 857 |
| PBS + 5% BSA | 491 | 3026 | ND | ND | <0.01 | ND | |
| 6l | CHCl3 | 502 | 10926 |
590 | 2971 | 0.33 | 3606 |
| PBS + 5% BSA | 515 | 6830 | 628 | 3494 | 0.03 | 205 | |
| 6m | CHCl3 | 502d | 9296 | 592d | 3028c | 0.33c,d | 3068 |
| PBS + 5% BSA | 515d | 3646d | 612d | 3078d | 0.04d | 146 | |
| 7a | CHCl3 | 470 | 26337 |
584 | 4153 | 0.12 | 3160 |
| DMSO | 485 | 20611 |
667 | 5626 | 0.14 | 2885 | |
| 7b | CHCl3 | 463 | 5209 | 584 | 4475 | 0.11 | 573 |
| DCM | EtOH | 467 | 42000 |
622 | 5336 | 0.43 | 18060 |
| ATTO 490LS | PBS buffer | 495 | 40000 |
658 | 5004 | 0.30 | 12000 |
| DyLight 515-LS | 515 | 50000 |
650 | 4032 | — | — | |
TD-DFT calculations
In order to better understand the variability of photophysical properties observed with these novel CinNaphts, DFT and TD-DFT calculations were conducted. The ground state geometries of representative compounds (5a, 5b, 5d, 5e, 5m, 5n, 5r, 6l) were first optimized using the B3LYP-D3(BJ)/6-311G(d,p)/IEF-PCM(CHCl3) level of calculation followed by a frequency calculation to confirm the convergence to a local minimum. Then, TD-DFT calculations were performed at the PBE1PBE/6-311G(d,p) level of theory with an IEF-PCM solvation model for CHCl3. The absorption energies and oscillator strengths of their first 12 vertical excitations were calculated and used to simulate the UV spectra (ESI pages 112–116†). The use of the PBE1PBE/6-311G(d,p) level of theory with the IEF-PCM solvation model has allowed relatively accurate estimation of the λmaxAbs (Fig. 5) and simulated UV spectra to be furnished in very good agreement with the experimental ones (table in Fig. 5). The observed redshift of the λmaxAbs of 5b compared to 5a is rather well reproduced by the calculation (experimental: 32 nm; calculated: 21 nm) and is in line with the difference in terms of HOMO–LUMO gaps calculated for both molecules (3.05 eV for 5a and 2.92 eV for 5b). As expected, for all of the emissive CinNaphts, the first excitation mainly involves a HOMO–LUMO π–π* transition (contribution of 99%). The frontier orbital distributions for all the emissive compounds confirm the charge transfer character of the first excited state with the HOMO coefficients located around the alkylamine substituent whereas the LUMO coefficients are closer to the naphthalimide substructure (see ESI pages 113–116 for FMO distributions†).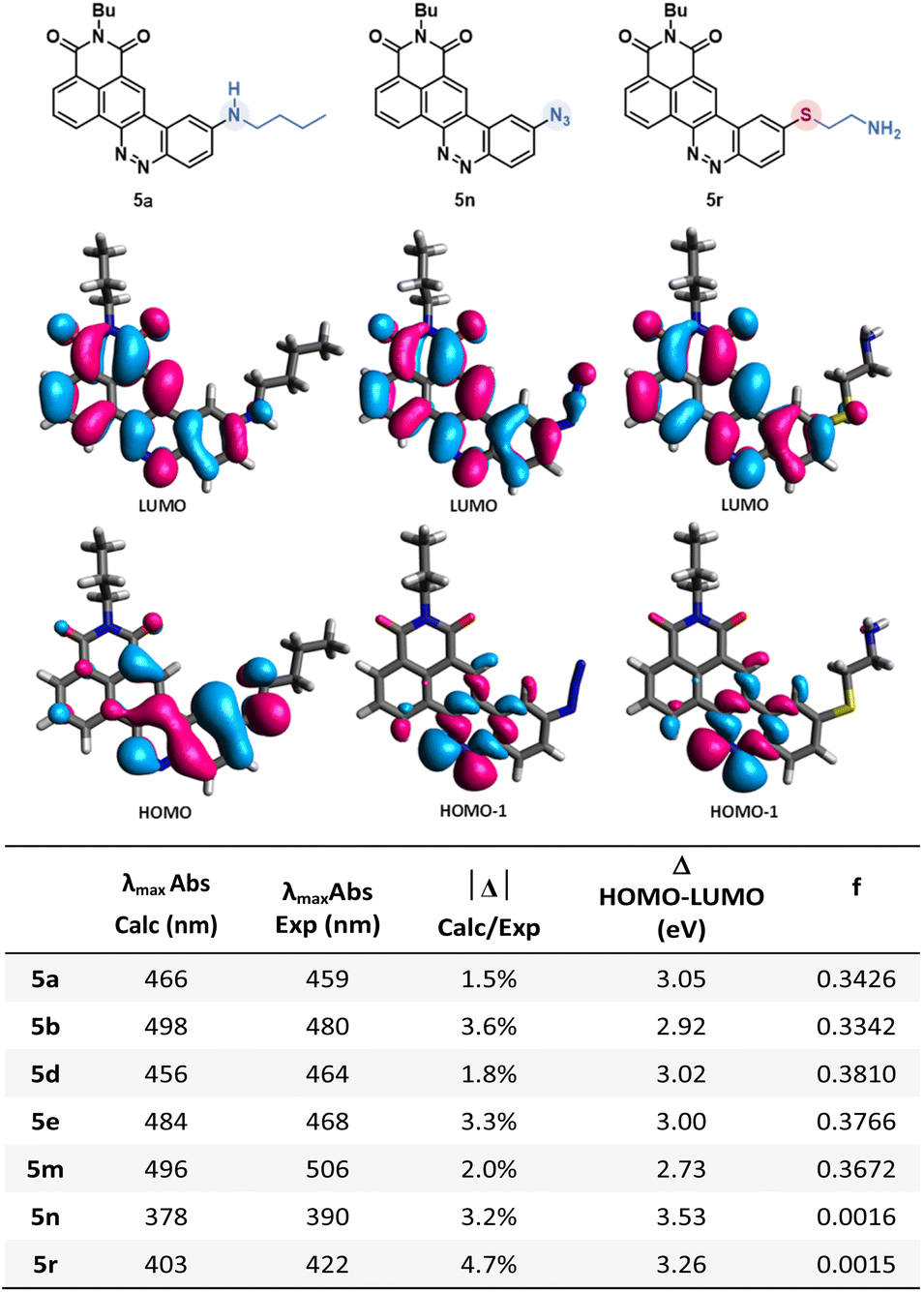 | ||
| Fig. 5 Rationalization through quantum mechanical calculations. Frontier molecular orbitals of 5a, 5n and 5r highlighting the HOMO nature difference. Table: Comparison between experimental and theoretical results including oscillator strength (f). | ||
Interestingly, for non-emissive CinNaphts 5n and 5r, the TD-DFT calculations predict a first excitation involving mainly a HOMO−1 → LUMO transition, corresponding to an n–π* type of transition, the HOMO−1 being located mainly on the nitrogen atoms of the cinnoline moiety (see ESI page 115†). This result may explain the absence of fluorescence for CinNaphts 5n and 5r.
Influence of deuteration on photobleaching resistance
Encouraged by the results published by Lavis' group,7b we investigated the influence of the deuteration of the donating amine on the photophysical properties of our compounds. First, the emission spectra, quantum yields and molar absorbance coefficients were determined for compounds 6l (deuterated) and 6m (not deuterated). The values obtained and reported in Fig. 6d indicate a negligible influence on both the absorption and emission maxima, which is consistent with the literature. On the other hand, while previous works demonstrate a clear benefit on the quantum yield, no obvious improvement has been noticed in our case. This leads us to carefully consider the deuteration strategy when it comes to optimizing the fluorescence efficiency. Nonetheless, absorption coefficient values were found to be significantly enhanced in favour of the deuterated compound, which is a trend also observed by Lavis' group. We then performed bleaching experiments to evaluate the photochemical resistance of our compounds. Both of the two CinNaphts were irradiated at 470 nm using a Teleopto device (see the ESI, p. 45†), and recording of both absorbance and emission spectra was performed every 30 min. Every experiment was performed in triplicate. Fig. 6b depicts a decrease of the emission intensity concomitant with a slight hypsochromic shift of the emission maxima. This evolution is significantly slower for 6l than for 6m testifying to a substantial improvement of the photochemical resistance brought about by the regioselective insertion of deuterium atoms into the pyrrolidine moiety. The slight hypsochromic shift in both the emission spectrum and the absorption band over time (Fig. 6b and c) suggests a photochemical degradation partly characterized by dealkylation of the pyrrolidine nitrogen atom.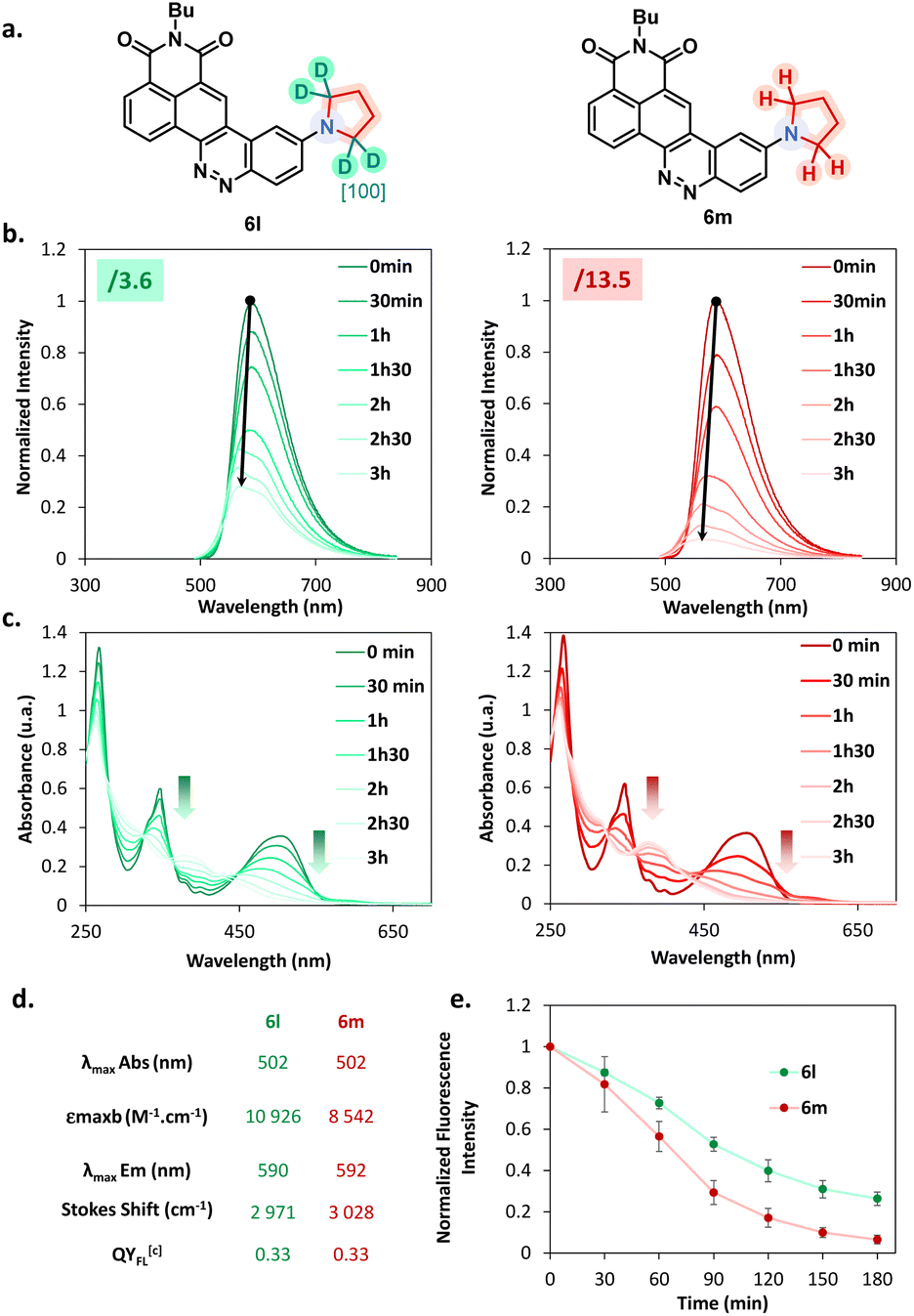 | ||
| Fig. 6 Influence of deuteration of pyrrolidine. (a) Structures of deuterated CinNapht 6l and non-deuterated CinNapht 6m. (b) Normalized fluorescence spectra of compounds 6l and 6m in CHCl3 at 25 °C every 30 min over 3 h of irradiation at 470 nm. (c) Absorbance spectra of compounds 6l and 6m in CHCl3 at 25 °C every 30 min over 3 h of irradiation at 470 nm. (d) Table of comparison of photophysical characteristics of CinNaphts 6l and 6m. (e) Comparison of fluorescence intensity decline over time after irradiation at 470 nm. | ||
This is in accordance with Grimm and co-workers' proposal7b on the basis of which we proposed a similar degradation mechanism (see Fig. S10†). The LCMS analyses performed during the photobleaching experiment demonstrate the appearance of intermediates over time which supports the suggested mechanism. Thus, we can reasonably argue that a primary kinetic isotope effect induced by the incorporation of deuterium atoms in the α-position of the nitrogen (the main site of the photobleaching degradation) is the crucial point in the increased stability of CinNapht 6m. This difference leads to a more difficult homolytic rupture of the C–D bond as a result of the photo-oxidation step, and consequently to a slower photochemical degradation of the deuterated dye. The introduction of a deuterated amine at the very end of the reaction is therefore an obvious advantage in the quest for photostable fluorophores. Considering the high cost of commercial deuterated amines, this strategy allows an undeniable cost economy compared to the use of deuterated compounds at the beginning of the synthesis which would require more material. We also studied the behavior of these two fluorophores during confocal microscopy experiments, but did not observe any obvious gain from the deuteration of our CinNaphts (data not shown). Therefore, we can state that in the case of our CinNaphts, alpha deuteration improves the absorption coefficient and is beneficial for the resistance to photobleaching, but does not allow any other improvement, either in terms of fluorescence efficiency or cell imaging.
Imaging of organelles in A549 living cells
A set of fluorophores was incubated with living A549 cells and then imaged with confocal microscopy experiments. We first looked at the model fluorophore 5a and noticed that the dyes enter efficiently into the cell without needing any pre-treatment.The images depicted in Fig. 7a also indicated that no significant compartmentalization of the dye was observed, which is consistent with the structure of the dye. However, the introduction of a specific chemical moiety led to the vectorization and accumulation of the CinNapht in specific cell organelles. Fig. 7b evidently shows that the intracellular compartmentalization of the CinNaphts 6g, 6h and 6k is different, so colocalization experiments were then conducted. We could easily observe a strong correlation between the fluorescence signal of the CinNapht 6g and the lysosome tracker LysoView™ 405 confirming the accumulation of the CinNapht 6g within the lysosome (Pearson Correlation Coefficient (PCC) = 0.91). The same conclusion was drawn when comparing the localisation of the CinNapht 6k signal with a plasma cell membrane selective dye (CellBrite® Green) with a PCC of 0.88. Finally, nuclear labeling experiments performed with compound 6k revealed a lack of signal homogeneity from one cell to another. Nevertheless, and regardless of the signal intensity, we exclusively observe nuclear labelling. This was confirmed with localization experiments showing a strong spatial correspondence with NucSpot® Live 488 Nuclear Stain and a PCC of 0.89. Note that despite strong efforts devoted to finding appropriate conditions for cell imaging using CinNaphts 6i and 6j, by changing dye concentration or incubation time, we could not achieve selective mitochondria imaging. The images presented in Fig. S11† seem to indicate that both of these dyes are sequestered in either endosomes or membranes and do not properly enter into the cells, and as a consequence cannot reach the mitochondria. This is important information confirming that the introduction of a cationic moiety, either pyridinium or triphenylphosphonium, does not constitute a systematic and safe way to target mitochondria. In closing, while the former strategy required the complete resynthesis of fluorophores in order to produce fluorescent markers for a given organelle, the late stage functionalization proposed in this paper allows the use of a single step that can be considered, at will, only a few days before the imaging experiment.
 | ||
| Fig. 7 Confocal microscopy images of live A549 cancer cell treated CinNaphts. (a) Imaging of CinNapht 5a using a 63× oil immersion objective; the nucleus was stained with 1 μg mL−1 Hoechst 33342 for 20 min at 37 °C (λExc: 405 nm, λEm: 425 to 500 nm), and CinNapht 5a was incubated at 1 μM for 3 h at 37 °C (λExc: 470 nm, λEm: 550 to 700 nm). Scale bar: 20 μM. (b) Subcellular localization of CinNaphts using a 63× oil immersion objective; CinNaphts were incubated at 1 μM for 3 h at 37 °C: 6g (λExc: 470 nm, λEm: 500 to 700 nm), 6h (λExc: 490 nm, λEm: 510 to 720 nm), 6k (λExc: 470 nm, λEm: 500 to 700 nm). Scale bar: 50 μM. (c) Colocalization experiments. CinNaphts were incubated at 1 μM for 3 h at 37 °C, then organelle trackers were added using a procedure suggested by the Biotium company (cf. ESI†). 6g (λExc: 470 nm, λEm: 550 to 700 nm) colocalized with LysoView™ 405 (λExc: 405 nm, λEm: 425 to 500 nm), 6h (λExc: 470 nm, λEm: 550 to 700 nm) colocalized with CellBrite® Green (Neuro-DiO) (λExc: 470 nm, λEm: 490 to 530 nm), 6k (λExc: 470 nm, λEm: 550 to 700 nm) colocalized with NucSpot® Live 488 Nuclear Stain (λExc: 470 nm, λEm: 490 to 530 nm). The Pearson Correlation Coefficients (PCC) were calculated using the JACoP plugin of ImageJ software. Scale bars: 10 μM. | ||
Investigation and use of AIEE behaviour-exhibiting CinNapht 5m for “wash-free” selective imaging of lipid droplets
The introduction of a diphenylamine moiety as a donating group in ICT type fluorophores often leads to a change of behavior through aggregation.23 Thus, we looked at the emission spectra of the CinNapht 5m in different mixtures in which the percentage of water was regularly increased in a THF/H2O mixture. We noticed a significant emission intensity in THF that brutally dropped when 20% water was added (Fig. 8a). Interestingly, the intensity increased from 80% water reaching its maximum in 99% water. This phenomenon is also evidenced by a substantial increase of the fluorescence quantum yield from QYFl = 0.02 in THF to QYFl = 0.04 in H2O. This typical “Aggregation-Induced Emission Enhancement” (AIEE) behavior is well illustrated by the picture reported in Fig. 8b, and to the best of our knowledge, this is the first example of such behavior with CinNapht dyes.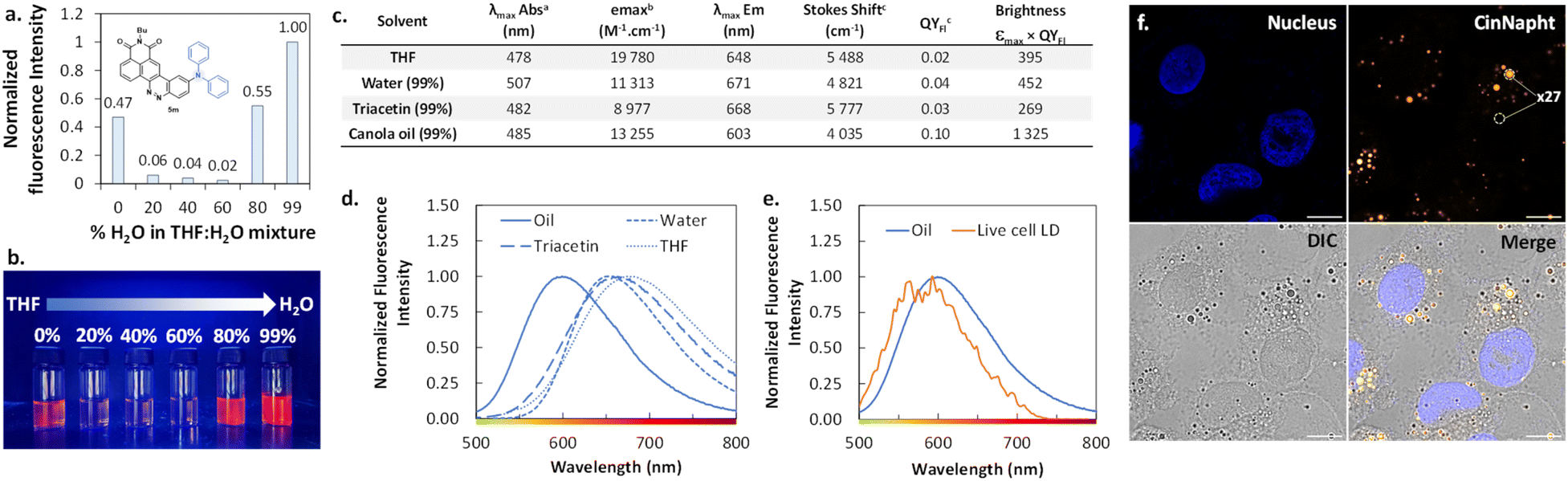 | ||
| Fig. 8 AIEE behaviour-exhibiting triphenylamine analogue of CinNapht 5m. (a) Fluorescence intensity of CinNapht 5m in solution at 10−5 M in different THF/H2O mixtures with different percentages of H2O, normalized to the maximum sample intensity (λExc: 490 nm). (b) Photograph of the 10−5 M CinNapht 5m solutions under illumination at 365 nm. (c) Table showing the photophysical properties of CinNapht 5m in different media. (d) Superposition of normalized emission spectra of CinNapht 5m in different media. (e) Emission spectrum recorded in living cell lipid droplets. (f) Imaging of CinNaphts 5a using a 63× oil immersion objective; the nucleus was stained with 1 μg mL−1 Hoechst 33342 for 20 min at 37 °C (λExc: 405 nm, λEm: 425 to 500 nm), and CinNapht 5m was incubated at 5 μM for 3 h at 37 °C (λExc: 470 nm, λEm: 550 to 700 nm). The turn-on factor represents the average of n = 20 measurements. Scale bar: 10 μM. | ||
We then incubated the CinNapht 5m at 5 μM with A549 living cells and studied the intracellular biodistribution of the dye. CinNapht 5m accumulates selectively in lipid droplets (LD) as a result of the local concentration increase leading to the aggregation of the dye resulting in an orange emission appearance as can be seen in Fig. 8f.24 What is important to note is that 5m exhibits a very high signal-to-noise ratio (calculated to be ×27), and this without needing any washing steps. Note that LD labelling could also be observed with lower concentration (up to 0.2 μM), showing nonetheless a lower signal-to-noise ratio. We performed photophysical characterization of compound 5m in different media including triacetin and oil in order to mimic the lipid droplet content (Fig. 8c and d). The best brightness was measured in canola oil. This is coherent with the observations made showing a high signal selectively localized in LDs. Moreover, the measurement of the emission spectrum in the living cell LDs shows an emission centred around 580–590 nm which is close to the values measured in canola oil (Fig. 8e). Altogether, these data demonstrate that CinNapht 5m constitutes a powerful example of an orange-emitting wash-free probe for lipid droplet imaging in living cells that was easily obtained in only one step.
Conclusions
To summarize, we propose a new and much more efficient method of accessing CinNaphts that unlocks the potential of theses dyes as imaging tools. This strategy is based on the late stage introduction of the donor amine by aromatic nucleophilic substitution on a highly stable fluorinated CinNapht which can be synthesized on a large scale. This allows us to store it for a considerable period of time and to utilize it for the insertion of a wide variety of nucleophiles as we have demonstrated with the scope of the reaction. We have succeeded in developing a number of new CinNaphts that have offered access to new and more efficient organelle markers as well as photostable regioselectively deuterated derivatives. We believe that this will allow us in the future to consider more sophisticated CinNapht based imaging tools dedicated to the study of more complex biological problems. More generally, this strategy should be applicable to other families of fluorophores, thus this work could inspire the development of a number of fluorophores based on other fluorescent polyaromatic skeletons.Author contributions
Chemical synthesis and characterization were performed by E. T. and M.-D. H. with participation of K. T. for deuteration and B. K. for mannose functionalization. E. T. analyzed the photophysical properties, and E. V.-E. performed single crystal X-ray analysis and interpreted the structural data. Theoretical calculation and interpretation of the data were performed by G. P. who also participated in the writing of the manuscript. Fluorescence microscopy and cell culture were performed by A. C. who also supervised the project and wrote the manuscript. All authors commented on the manuscript and gave approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This project has received funding by the French National Research Agency under the program CHARMMMAT ANR-11-LABX-0039-grant and was supported by the “Chemistry Graduate School of Université Paris-Saclay. We also thank the Institut de Chimie des Substances Naturelles for their financial support. The present work has benefited from the Imagerie-Gif light microscopy core facility supported by the French National Research Agency (ANR-11-EQPX-0029/Morphoscope, ANR-10-INBS-04/FranceBioImaging; ANR-11-IDEX-0003-02/Saclay Plant Sciences). Université Paris-Saclay and the CNRS are acknowledged for funding. GP thanks CEA, SCBM and the European Union's Horizon 2020 research and innovation program under the FET-OPEN Grant Agreement No. 862179 for funding K. T. This work was granted access to the CCRT High-Performance Computing (HPC) facility under the Grant CCRT2023-gpieters awarded by the Fundamental Research Division (DRF) of CEA.Notes and references
- (a) C. P. Toseland, J. Chem. Biol., 2013, 6, 85 CrossRef PubMed; (b) M. S. T. Gonçalves, Chem. Rev., 2009, 109, 190 CrossRef PubMed; (c) J. B. Grimm and L. D. Lavis, Nat. Methods, 2022, 19, 149 CrossRef CAS PubMed.
- (a) J. Zhou and H. Ma, Chem. Sci., 2016, 7, 6309 RSC; (b) L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142 CrossRef CAS PubMed; (c) J. Chan, S. C. Dodani and C. J. Chang, Nat. Chem., 2012, 4, 973 CrossRef CAS PubMed; (d) J. B. Grimm, L. M. Heckman and L. D. Lavis, Prog. Mol. Biol. Transl. Sci., 2013, 113, 1 CAS.
- (a) M. Más-Montoya, M. F. Montenegro, A. Espinosa Ferao, A. Tárraga, J. N. Rodríguez-López and D. Curiel, Org. Lett., 2020, 22, 3356 CrossRef PubMed; (b) E. M. Santos, W. Sheng, R. Esmatpour Salmani, S. Tahmasebi Nick, A. Ghanbarpour, H. Gholami, C. Vasileiou, J. H. Geiger and B. Borhan, J. Am. Chem. Soc., 2021, 143, 15091 CrossRef CAS PubMed; (c) A. Gandioso, R. Bresolí-Obach, A. Nin-Hill, M. Bosch, M. Palau, A. Galindo, S. Contreras, A. Rovira, C. Rovira, S. Nonell and V. Marchán, J. Org. Chem., 2018, 83, 1185 CrossRef CAS PubMed; (d) J.-A. Richard, M. Massonneau, P.-Y. Renard and A. Romieu, Org. Lett., 2008, 10, 4175 CrossRef CAS PubMed; (e) L. Zhou, Q. Wang, Y. Tan, M. J. Lang, H. Sun and X. Liu, Chem.–Eur. J., 2017, 23, 8736 CrossRef CAS PubMed; (f) X. Zhang, J. Wang, F. Yu, X. Huang, N. Wang, T. Wang and H. Hao, Cryst. Growth Des., 2022, 22, 3198 CrossRef CAS; (g) I. Likhotkin, R. Lincoln, M. L. Bossi, A. N. Butkevich and S. W. Hell, J. Am. Chem. Soc., 2023, 145, 1530 CrossRef CAS PubMed; (h) A. Gandioso, R. Bresoli-Obach, A. Nin-Hill, M. Bosch, M. Palau, A. Galindo, S. Contreras, A. Rovira, C. Rovira, S. Nonell and V. Marchan, J. Org. Chem., 2018, 83, 1185 CrossRef CAS PubMed; (i) L. Yuan, W. Lin, S. Zhao, W. Gao, B. Chen, L. He and S. Zhu, J. Am. Chem. Soc., 2012, 134, 13510 CrossRef CAS PubMed.
- M.-D. Hoang, J.-B. Bodin, F. Savina, V. Steinmetz, J. Bignon, P. Durand, G. Clavier, R. Méallet-Renault and A. Chevalier, RSC Adv., 2021, 11, 30088 RSC.
- M.-D. Hoang, F. Savina, P. Durand, P. R. Méallet-Renault, G. Clavier and A. Chevalier, ChemPhotoChem, 2022, 6, e202200138 CAS.
- (a) J. B. Grimm, A. J. Sung, W. R. Legant, P. Hulamm, S. M. Matlosz, E. Betzig and L. D. Lavis, ACS Chem. Biol., 2013, 8, 1303 CrossRef CAS PubMed; (b) S. M. Hickey, S. O. Nitschke, M. J. Sweetman, C. J. Sumby, D. A. Brooks, S. E. Plush and T. D. Ashton, J. Org. Chem., 2020, 85, 7986 CrossRef CAS PubMed; (c) J. B. Grimm and L. D. Lavis, Org. Lett., 2011, 13, 6354 CrossRef CAS PubMed; (d) X. Jin, C. Uttamapinant and A. Y. Ting, ChemBioChem, 2011, 12, 65 CrossRef CAS PubMed; (e) T. Peng and D. Yang, Org. Lett., 2010, 12, 496 CrossRef CAS PubMed; (f) K. N. Hearn, T. D. Nalder, R. P. Cox, H. D. Maynard, T. D. M. Bell, F. M. Pfeffer and T. D. Ashton, Chem. Commun., 2017, 53, 12298 RSC.
- (a) J. B. Grimm, A. K. Muthusamy, Y. Liang, T. A. Brown, W. C. Lemon, R. Patel, R. Lu, J. J. Macklin, P. J. Keller, N. Ji and L. D. Lavis, Nat. Methods, 2017, 14, 987 CrossRef CAS PubMed; (b) J. B. Grimm, L. Xie, J. C. Casler, R. Patel, A. N. Tkachuk, N. Falco, H. Choi, J. Lippincott-Schwartz, T. A. Brown, B. S. Glick, Z. Liu and L. D. Lavis, JACS Au, 2021, 1, 690 CrossRef CAS PubMed; (c) A. N. Butkevich, M. L. Bossi, G. Lukinavičius and S. W. Hell, J. Am. Chem. Soc., 2019, 141, 981 CrossRef CAS PubMed.
- (a) J. Bucevičius, G. Kostiuk, R. Gerasimaitė, T. Gilat and G. Lukinavičius, Chem. Sci., 2020, 11, 7313 RSC; (b) C. Deo, A. S. Abdelfattah, H. K. Bhargava, A. J. Berro, N. Falco, H. Farrants, B. Moeyaert, M. Chupanova, L. D. Lavis and E. R. Schreiter, Nat. Chem. Biol., 2021, 17, 718 CrossRef CAS PubMed; (c) T. Takagi, T. Ueno, K. Ikawa, D. Asanuma, Y. Nomura, S.-n. Uno, T. Komatsu, M. Kamiya, K. Hanaoka, C. Okimura, Y. Iwadate, K. Hirose, T. Nagano, K. Sugimura and Y. Urano, Sci. Adv., 2021, 7, eabg8585 CrossRef CAS PubMed; (d) J. M. Meinig, L. Fu and B. R. Peterson, Angew. Chem., Int. Ed., 2015, 54, 9696 CrossRef CAS PubMed.
- (a) W. Shi, J. Li, X. He, S. Zhou, H. Sun and H. Wu, Org. Lett., 2022, 24, 3368 CrossRef CAS PubMed; (b) M. Motoyama, T.-H. Doan, P. Hibner-Kulicka, R. Otake, M. Lukarska, J.-F. Lohier, K. Ozawa, S. Nanbu, C. Alayrac, Y. Suzuki and B. Witulski, Chem.–Asian J., 2021, 16, 2087 CrossRef CAS PubMed; (c) G. Zhang, M. Wang, P. Bobadova-Parvanova, F. R. Fronczek, K. M. Smith and M. G. H. Vicente, Chem.–Eur. J., 2022, 28, e202200421 CAS; (d) S. Zheng, G. Lingyue, M. J. H. Ong, D. Jacquemin, A. Romieu, J.-A. Richard and R. Srinivasan, Org. Biomol. Chem., 2019, 17, 4291 RSC; (e) G. Vives, C. Giansante, R. Bofinger, G. Raffy, A. D. Guerzo, B. Kauffmann, P. Batat, G. Jonusauskas and N. D. McClenaghan, Chem. Commun., 2011, 47, 10425 RSC; (f) M. Kotaskova, O. Osman Oglou and M. Helm, Org. Biomol. Chem., 2014, 12, 3816 RSC; (g) A. Chevalier, K. Renault, F. Boschetti, P.-Y. Renard and A. Romieu, Eur. J. Org. Chem., 2015, 2015, 152 CrossRef CAS.
- (a) H.-R. Jia, Y.-X. Zhu, Q.-Y. Duan and F.-G. Wu, Chem. Soc. Rev., 2021, 50, 6240 RSC; (b) P. Shieh, V. T. Dien, B. J. Beahm, J. M. Castellano, T. Wyss-Coray and C. R. Bertozzi, J. Am. Chem. Soc., 2015, 137, 7145 CrossRef CAS PubMed.
- (a) H. A. Henthorn and M. D. Pluth, J. Am. Chem. Soc., 2015, 137, 15330 CrossRef CAS PubMed; (b) V. S. Lin, W. Chen, M. Xian and C. J. Chang, Chem. Soc. Rev., 2015, 44, 4596 RSC.
- G. Zhou, S. Hou, N. Zhao, N. Finney and Y. Wang, Dyes Pigm., 2022, 204, 110394 CrossRef CAS.
- (a) V. V. Rostovtsev, L. G. Green, V. V. Fokin and K. B. Sharpless, Angew. Chem., Int. Ed., 2002, 41, 2596 CrossRef CAS; (b) C. W. Tornøe, C. Christensen and M. Meldal, J. Org. Chem., 2002, 67, 3057 CrossRef PubMed.
- (a) L. Wang, Y. Xiao, W. Tian and L. Deng, J. Am. Chem. Soc., 2013, 135, 2903 CrossRef CAS PubMed; (b) H. Yu, Y. Xiao and L. Jin, J. Am. Chem. Soc., 2012, 134, 17486 CrossRef CAS PubMed; (c) Q. Wan, S. Chen, W. Shi, L. Li and H. Ma, Angew. Chem., Int. Ed., 2014, 53, 10916 CrossRef CAS PubMed.
- D. I. Danylchuk, P.-H. Jouard and A. S. Klymchenko, J. Am. Chem. Soc., 2021, 143, 912 CrossRef CAS PubMed.
- (a) Z. Xu and L. Xu, Chem. Commun., 2016, 52, 1094 RSC; (b) Roopa, N. Kumar, V. Bhalla and M. Kumar, Chem. Commun., 2015, 51, 15614 RSC; (c) S. Samanta, Y. He, A. Sharma, J. Kim, W. Pan, Z. Yang, J. Li, W. Yan, L. Liu, J. Qu and J. S. Kim, Chem, 2019, 5, 1697 CrossRef CAS; (d) J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Chem. Rev., 2017, 117, 10043 CrossRef CAS PubMed; (e) R. A. J. Smith, C. M. Porteous, A. M. Gane and M. P. Murphy, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 5407 CrossRef CAS PubMed.
- (a) A. Nakamura, K. Takigawa, Y. Kurishita, K. Kuwata, M. Ishida, Y. Shimoda, I. Hamachi and S. Tsukiji, Chem. Commun., 2014, 50, 6149 RSC; (b) C. K. Spahn, M. Glaesmann, J. B. Grimm, A. X. Ayala, L. D. Lavis and M. Heilemann, Sci. Rep., 2018, 8, 14768 CrossRef PubMed.
- K. Roßmann, K. C. Akkaya, P. Poc, C. Charbonnier, J. Eichhorst, H. Gonschior, A. Valavalkar, N. Wendler, T. Cordes, B. Dietzek-Ivanšić, B. Jones, M. Lehmann and J. Broichhagen, Chem. Sci., 2022, 13, 8605 RSC.
- E. Levernier, K. Tatoueix, S. Garcia-Argote, V. Pfeifer, R. Kiesling, E. Gravel, S. Feuillastre and G. Pieters, JACS Au, 2022, 2, 801 CrossRef CAS PubMed.
- K. Rurack and M. Spieles, Anal. Chem., 2011, 83, 1232 CrossRef CAS PubMed.
- https://www.atto-tec.com/product-141-142.html?language=de) .
- https://www.thermofisher.com/order/catalog/product/fr/fr/82491) .
- (a) D. Wang and B. Z. Tang, Acc. Chem. Res., 2019, 52, 2559 CrossRef CAS PubMed; (b) Y. Lin, Y. Song, Y. Jin, B. Wang and C. Fan, Dyes Pigm., 2020, 183, 108711 CrossRef CAS; (c) K. Zhang, J. Shu, W. Chu, X. Liu, B. Xu and W. Jiang, Dyes Pigm., 2021, 185, 108898 CrossRef CAS.
- L. Wang, X. Chen, X. Ran, H. Tang and D. Cao, Dyes Pigm., 2022, 203, 110332 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2221167. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3sc01365k |
| This journal is © The Royal Society of Chemistry 2023 |