
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImproving time-resolution and sensitivity of in situ X-ray photoelectron spectroscopy of a powder catalyst by modulated excitation†
M.
Roger
ab,
L.
Artiglia
 *a,
A.
Boucly
a,
F.
Buttignol
ab,
M.
Agote-Arán
a,
J. A.
van Bokhoven
ac,
O.
Kröcher
ab and
D.
Ferri
*a
*a,
A.
Boucly
a,
F.
Buttignol
ab,
M.
Agote-Arán
a,
J. A.
van Bokhoven
ac,
O.
Kröcher
ab and
D.
Ferri
*a
aPaul Scherrer Institut, Forschungsstrasse 111, CH-5232 Villigen PSI, Switzerland. E-mail: luca.artiglia@psi.ch; davide.ferri@psi.ch
bÉcole Polytechnique Fédérale de Lausanne (EPFL), Institute for Chemical Sciences and Engineering, CH-1015, Lausanne, Switzerland
cDepartment of Chemistry and Applied Biosciences, Institute for Chemical and Bioengineering, ETH Zurich, 8093, Zurich, Switzerland
First published on 30th May 2023
Abstract
Ambient pressure X-ray photoelectron spectroscopy (APXPS) is a powerful tool to characterize the surface structure of heterogeneous catalysts in situ. In order to improve the time resolution and the signal-to-noise (S/N) ratio of photoemission spectra, we collected consecutive APXP spectra during the periodic perturbation of a powder Pd/Al2O3 catalyst away from its equilibrium state according to the modulated excitation approach (ME). Averaging of the spectra along the alternate pulses of O2 and CO improved the S/N ratio demonstrating that the time resolution of the measurement can be limited solely to the acquisition time of one spectrum. Through phase sensitive analysis of the averaged time-resolved spectra, the formation/consumption dynamics of three oxidic species, two metal species, adsorbed CO on Pd0 as well as Pdn+ (n > 2) was followed along the gas switches. Pdn+ and 2-fold surface PdO species were recognised as most reactive to the gas switches. Our approach demonstrates that phase sensitive detection of time-resolved XPS data allows following the dynamics of reactive species at the solid–gas interface under different reaction environments with unprecedented precision.
Introduction
X-ray photoemission spectroscopy (XPS) is a core-level, surface-sensitive technique.1,2 Due to the localization of the core level, XPS is element-specific and therefore well suited for studying chemical composition, local bonding and site-specific dynamics in the solid, liquid and gas phase as well as at the respective interfaces.3–5 Until recently, the main drawback of XPS was the necessity to work in high vacuum due to the low kinetic energy of the photoelectrons emitted from the first layers of the condensed material, which can be easily scattered inelastically in the gas phase. Significant technological improvements and scientific breakthroughs have been made in the past decades to bridge the gap between ultrahigh vacuum and elevated pressure conditions using ambient pressure X-ray photoelectron spectroscopy (APXPS).6–10 This represents a step toward the study of realistic catalysts, i.e. materials such as nanoparticles of catalytically relevant metals with low metal loadings dispersed upon metal oxides, in realistic environments. Efforts are still needed to further advance the performance of APXPS, including improvement of the signal-to-noise (S/N) ratio of photoemission spectra of realistic catalysts and of the time resolution of photoemission measurements.4 The S/N ratio can be improved by increasing the content of the active element or by averaging the signal over long or repeated acquisition times.4 By prolonged measurement time of the spectra of samples containing high levels of the catalytically relevant element, the time resolution of the measurement can be considerably decreased, but the high content may not represent the real catalytic system. Yet, improving the time resolution becomes especially important when the purpose is to detect active species whose concentrations are small and lifetime is limited.11 Indeed, the elementary steps of a reaction occur in the picosecond range while milliseconds to seconds are needed to follow structural surface changes of the catalyst, such as phase transitions, surface roughening and segregation.12The data quality of any spectroscopic measurement can be improved by the modulated excitation (ME) method,13 where a perturbation to the system (i.e. optical, pressure, or chemical modification) is introduced in order to force it away from its equilibrium state. The perturbation is chosen such that the species of interest, for example the catalytically active species, will respond selectively, if not exclusively. Under these conditions, the system can either return to its previous equilibrium state (reversible process) or move to a new equilibrium state (non-reversible process). The dynamic changes induced by the perturbation can be followed in a time-resolved manner using for example infrared spectroscopy,14,15 X-ray diffraction,16,17 and X-ray absorption spectroscopy.18–20 In the case of a reversible system, the perturbation can be applied periodically by sequences of alternated pulses, which allow spectral averaging along all pulses.21 This decreases the size of the dataset, improving the S/N ratio by the square root of the number of averaged cycles22 and enabling a time resolution equal to the acquisition duration of a single spectrum. For accurate data alignment and averaging, this method requires performing fast and reproducible perturbations, ideally without delay between the perturbation event and the time at which the sample experiences it. Recently, Knudsen et al.21 carried out in situ APXPS measurements with repeated gas switches over a Pd(100) single crystal achieving a time resolution of 60 ms. To overcome the difficulty of the alignment of the cycles due to the delay between the gas switch and the gas replacement in the experimental cell as well as pulse irregularities, a lock-in signal within the data was identified that characterised a reversible structural change and was used for data averaging. This approach requires a sufficiently high initial S/N ratio to find the lock-in signal using an image recognition algorithm, as well as a fully reversible process, which are both difficult to achieve with actual catalytic materials.
The averaged data obtained by following the ME protocol contain the response of the active species, the spectator species and the noise.23 Phase-sensitive detection (PSD, eqn (1); also named demodulation)22 can be then applied to the averaged data:
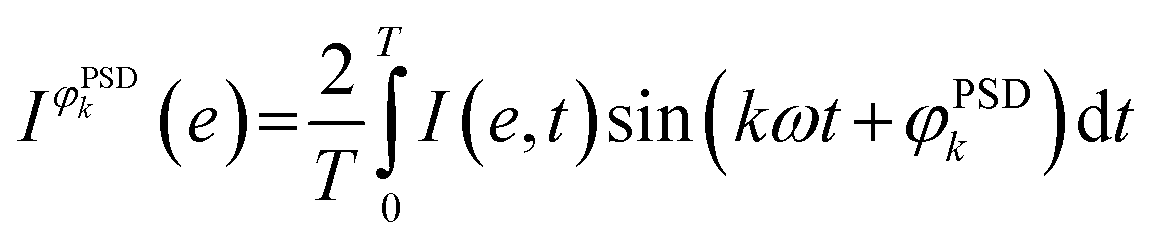 | (1) |
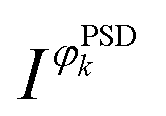 is the signal in the phase domain, T the modulation period, k the demodulation index, ω the stimulation frequency, and φPSDk the phase angle. PSD converts the time domain response (I(e,t)) to a phase domain response I(φPSD) wherein the signal from the active species affected by the perturbation remains, whereas the signal from the spectator species and the noise (unaffected by the perturbation) is suppressed. Hence, PSD allows to significantly improve the S/N ratio of the signals of species affected by the perturbation22 enabling capturing their dynamics (rate of consumption/formation) with increased sensitivity compared to what is achieved in the time domain.
is the signal in the phase domain, T the modulation period, k the demodulation index, ω the stimulation frequency, and φPSDk the phase angle. PSD converts the time domain response (I(e,t)) to a phase domain response I(φPSD) wherein the signal from the active species affected by the perturbation remains, whereas the signal from the spectator species and the noise (unaffected by the perturbation) is suppressed. Hence, PSD allows to significantly improve the S/N ratio of the signals of species affected by the perturbation22 enabling capturing their dynamics (rate of consumption/formation) with increased sensitivity compared to what is achieved in the time domain.
In this work, the Pd 3d photoemission signal of a commercial powder 5 wt% Pd/Al2O3 catalyst was followed by in situ APXPS while switching repeatedly between oxidising (O2) and reducing (CO) environments. The improved signal and time resolution of the APXPS experiment obtained by the modulated excitation experiment and PSD analysis revealed that Pdn+ (n > 2) and 2-fold surface oxide species on the catalyst are the most reactive toward CO oxidation.
Materials and methods
The powder 5 wt% Pd/γ-Al2O3 catalyst was available in its reduced form (type 324, Johnson Matthey). The presence of Pd0 was verified by the observation of a reflection at a 2 theta angle of 40.1° in the X-ray diffraction pattern of the sample (Fig. S1a†). The catalyst displayed a narrow particle size distribution, as measured by TEM (4.4 ± 1.1 nm; Fig. S1b and c†).Catalyst characterization
X-ray diffraction was measured using a D8 Advance Bruker instrument equipped with a 1D-LynxEye detector at a step size of 0.01° and an acquisition time of 2 s. Transmission electron microscopy images were obtained using a JEOL 2010 microscope equipped with a LaB6 cathode and operated at 200 keV. The images were recorded using a slow-scan CCD camera (4008 × 2672 pixels, Orius Gaten Inc.). The average particle size was obtained from counting 150 particles.X-ray photoelectron spectroscopy
The experiments were carried out at the In Situ Spectroscopy beamline (X07DB) at the Swiss Light Source of the Paul Scherrer Institute. The solid–gas interface endstation was connected to a differentially pumped Scienta R4000 HiPP-2 electron analyser to allow the manipulation of solid samples while dosing a gas mixture in the mbar range.24 The volume of the XPS cell (ca. 150 mL) is small enough to allow performing transient experiments with fast gas switches, while the evolution of a spectral line can be followed either in a swept mode or a fixed mode (also known as snapshot).4,25 In this work, the signals of Pd 3d and Al 2p were acquired with an excitation energy of 650 eV to generate Pd 3d photoelectrons with a kinetic energy of approximately 315–330 eV. A 20 eV kinetic energy window including the Pd 3d signal was acquired in fast scan (swept) mode in approximately 6 s. Linearly polarized light was used throughout the experiments. The spectra were processed and fitted using IgorPro and XPSPeak41 software. The energy scale of the Pd 3d core level spectra was calibrated using the Al 2p signal of Al2O3 as a reference (at 74.3 eV binding energy26). All spectra were fitted using a Shirley background, and Gaussian peaks.The catalyst pellet was prepared by pressing 5 mg of powder on a Ta mesh to decrease the surface charging and was then fixed to the sample holder using metal clips. The temperature was monitored with a Pt 100 sensor and the sample was heated using a tunable power IR laser (976 nm, maximum power 25 W) projected on the back of the sample holder. In the geometry adopted during the experiments, photoelectrons were detected at an angle of 30° with respect to surface normal.
The gas mixing setup was composed of two mass flow controllers, each connected to a solenoid valve (Series 9, Parker) enabling fast exchanges in gas phase composition. The valves were situated at the end of the gas line, directly before the experimental cell. Periodic switches between neat O2 (1 mbar) and neat CO (1 mbar) were carried out at 300 °C every 5 min for 6 h (T = 10 min in eqn (1)) while acquiring the Pd 3d core level and for 1 h while acquiring the Al 2p core level. Prior to the experiment, the pelletized catalyst was heated in O2 (1 mbar) to 300 °C to remove surface carbon contamination and avoid any structural change during measurements. A total of 36 full periods (5 min O2 + 5 min CO) were performed. The gas composition in the cell was followed using a quadrupole mass spectrometer (MS; RGA100, SRS) located in the second differential pumping stage of the analyser.
Results and discussion
The electron count of an X-ray photoemission spectrum of a catalyst with supported nanoparticles, such as the one used in this work, is considerably lower compared to that of a single crystal of the element representing the active phase when measurements are carried out at ambient pressure due to the collision with gas molecules in the experimental chamber.27 As a consequence, the quality (signal-to-noise (S/N) ratio) of a single Pd 3d fast scan of the 5 wt% Pd/Al2O3 catalyst is low despite the high Pd content (Fig. 1a). To distinguish the main features of the Pd 3d core level, 5 to 10 spectra have to be averaged, while 15 averaged spectra are required to achieve an acceptable S/N ratio for meaningful fitting of the raw data (Fig. 1). Because the acquisition time of a single spectrum with an energy window of 20 eV was ca. 6.20 s, ca. 93 s were required to measure 15 consecutive scans, which exceeds the time required to follow precisely surface processes occurring in the second to sub-second range. The acquisition time can be decreased either by narrowing the energy window and focusing for example, on the Pd 3d5/2 core level only or by using materials with higher Pd loading. Narrow energy windows would not allow for cross-checking the fitting with the spin–orbit-satellite signal, while increasing the Pd content implies working with unrealistic samples.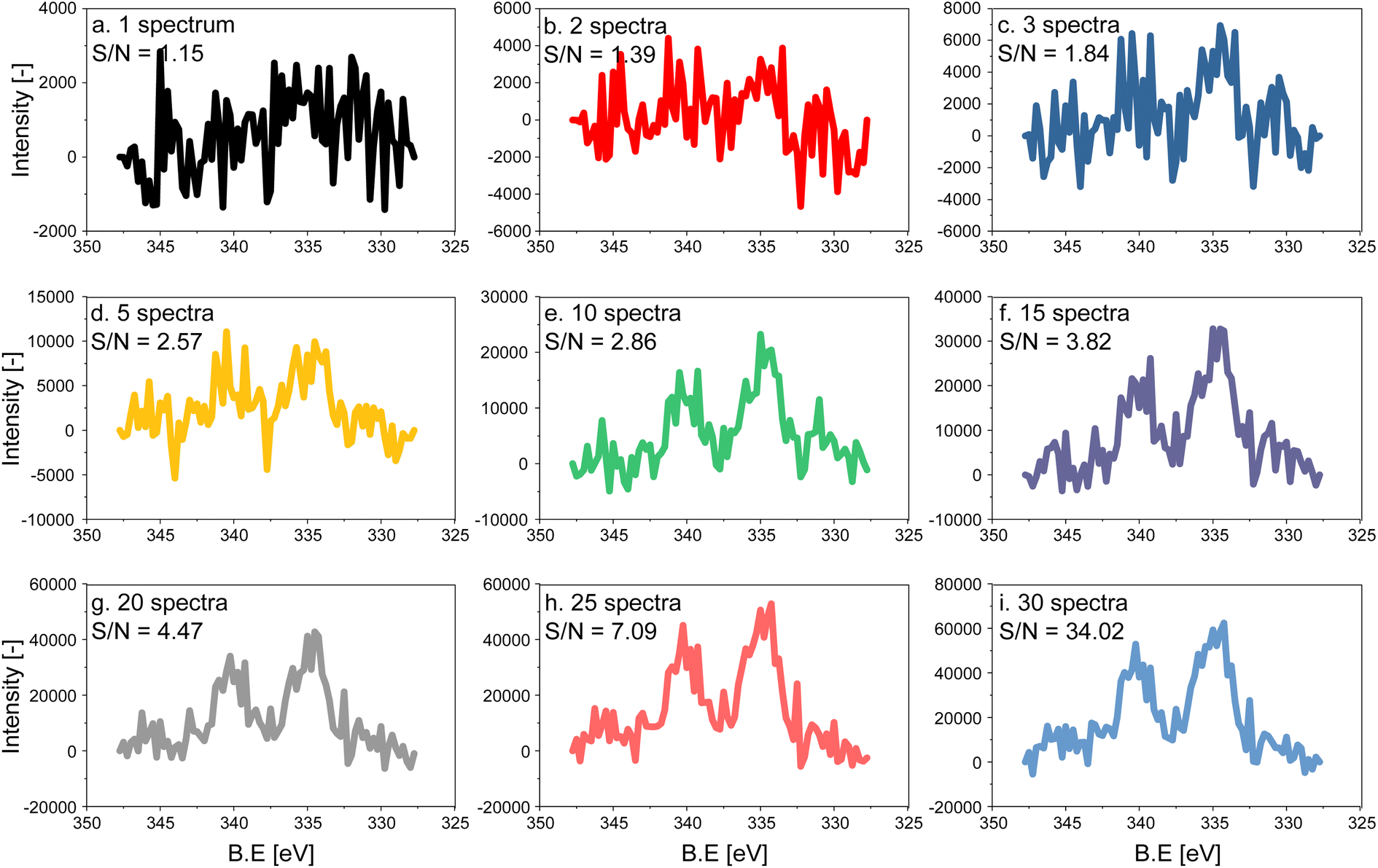 | ||
| Fig. 1 Effect of averaging on the quality of Pd 3d core level X-ray photoelectron spectra of 5 wt% Pd/Al2O3 acquired with a 300 eV kinetic energy: (a) one spectrum (ca. 6.20 s), (b) 2, (c) 3, (d) 5, (e) 10, (f) 15, (g) 20, (h) 25 and (i) 30 averaged spectra. S/N increases in the same order from 1.15 to 34.02. | ||
Modulated excitation experiment
In order to simultaneously improve the S/N ratio and the time resolution of the APXPS experiment without compromising the energy window, we perturbed the sample environment according to the modulated excitation (ME) method22 by changing the gas composition between neat O2 and CO every 5 min for 6 h while scanning the Pd 3d core level as mentioned in the previous section. The repeated pulses caused a temporary pressure increase in the experimental cell from 1 mbar up to 1.8 mbar (Fig. 2), which translated into a shift of the peak position by ca. 4 eV, as exemplified by the Al 2p core level (Fig. S2†). The position of this peak stabilized only once the pressure in the cell returned to ca. 1 mbar. Because this powder catalyst is an insulator, the sample accumulates surface charging. During measurements, the kinetic energy of photoemission peaks is affected by the partial pressure in the chamber and the type of gas. Indeed, ionization of gas molecules by the incident X-rays can generate charges that partially compensate for the charging at the solid surface.28 The higher the pressure, thus density, of gas around the sample, the more efficiently surface charging is removed. The change in pressure resulted in spikes in O2 (m/z = 32) and CO (m/z = 28) intensity measured by mass spectrometry (MS, Fig. 2 and S3a†), which were not observed in an identical off-line experiment with a conventional catalytic quartz plug-flow reactor loaded with the powder catalyst (Fig. S3b†). Hence, the geometry of the APXPS cell and/or the intrinsic properties of the gases were responsible for this artefact. If the sudden change in pressure was caused only by the geometry of the chamber, the increase in pressure upon the gas switch would have been the same for both CO and O2, but the effect was more pronounced at the O2 → CO switch. This can be explained by the fact that CO sticks more strongly than O2 to the metallic parts of the vacuum system and that the pumping efficiency (root pump downstream the cell) varies with the gas composition. Furthermore, Fig. 2 shows that the pressure rises to slightly different values when comparing two consecutive pulses of the same gas. At the same time, Fig. S3a† shows that the MS signals are similar when comparing consecutive gas pulses. Given that MS signals are measured at the sample site while the pressure is measured by a baratron on top of the analysis cell, we consider the reading from the MS to be more representative of the gas environment around the sample. We can conclude that gas switches lead to repeatable local gas environments.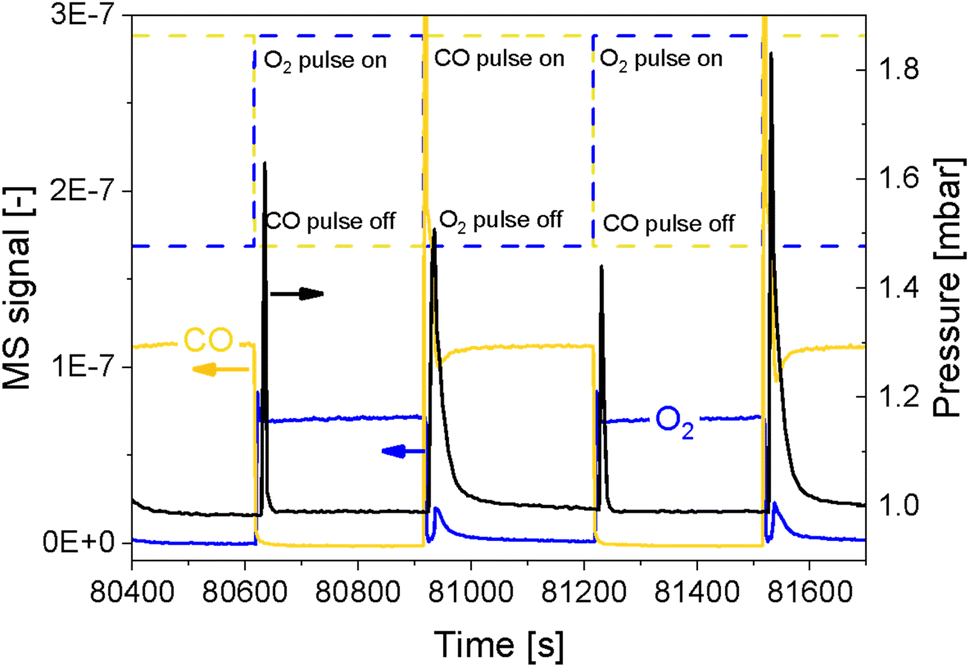 | ||
| Fig. 2 Temporal behaviour of CO (yellow) and O2 (blue) MS signals as well as of the pressure in the experimental cell (black) during alternate O2/CO pulsing. Pulses shape and timing is shown by the dashed lines. | ||
Gases in the mbar range such as O2 and CO used in these experiments affect differently the charging and work function of the sample by transferring electrons to holes in the solid.3 This was observed by the energy shift by 0.25 eV between the periods measured in O2 and CO once the pressure in the cell was stabilized at ca. 1 mbar (Fig. S2,† emphasized by the blue lines). In order to correct the shift caused by both the change in pressure and the sample charging, we used the 2p peak of Al from the alumina support (Fig. 3), under the assumption that the electronic state of Al2O3 is not affected by the reactive environment. The first step of the data treatment consisted of aligning each Al 2p spectrum measured during the experiment (Fig. 3d and e). This defined the extent of shift that had to be applied to the Pd 3d core level data at each point in time in order to align the spectra and to be able to observe only the effect of the occurring chemistry. In order to easily apply the shifts defined from the Al 2p data to the Pd 3d data set, the acquisition time (time resolution) of the Al and Pd spectral ranges has to be the same and the start of the pulses needs to be correctly synchronized with the spectra. We achieved this by carefully considering the status of the pulsed valves (on/off, with a time resolution of 1 s) as well as the acquisition time of each single scan. The second step of the data treatment consisted of applying the shifts evaluated for each Al 2p spectrum to the Pd 3d spectra making sure that the time and the period considered were the same. The difference in the Pd 3d core level before and after alignment is visible in the averaged spectra in the O2 or the CO half-periods (Fig. 3c, f and S4†). Without alignment, the difference in binding energy (ΔBE = 1.75 eV) between the two half-periods was larger than after alignment (1.5 eV), proving that the influence of different gas environments on the sample surface charging was correctly taken into consideration.
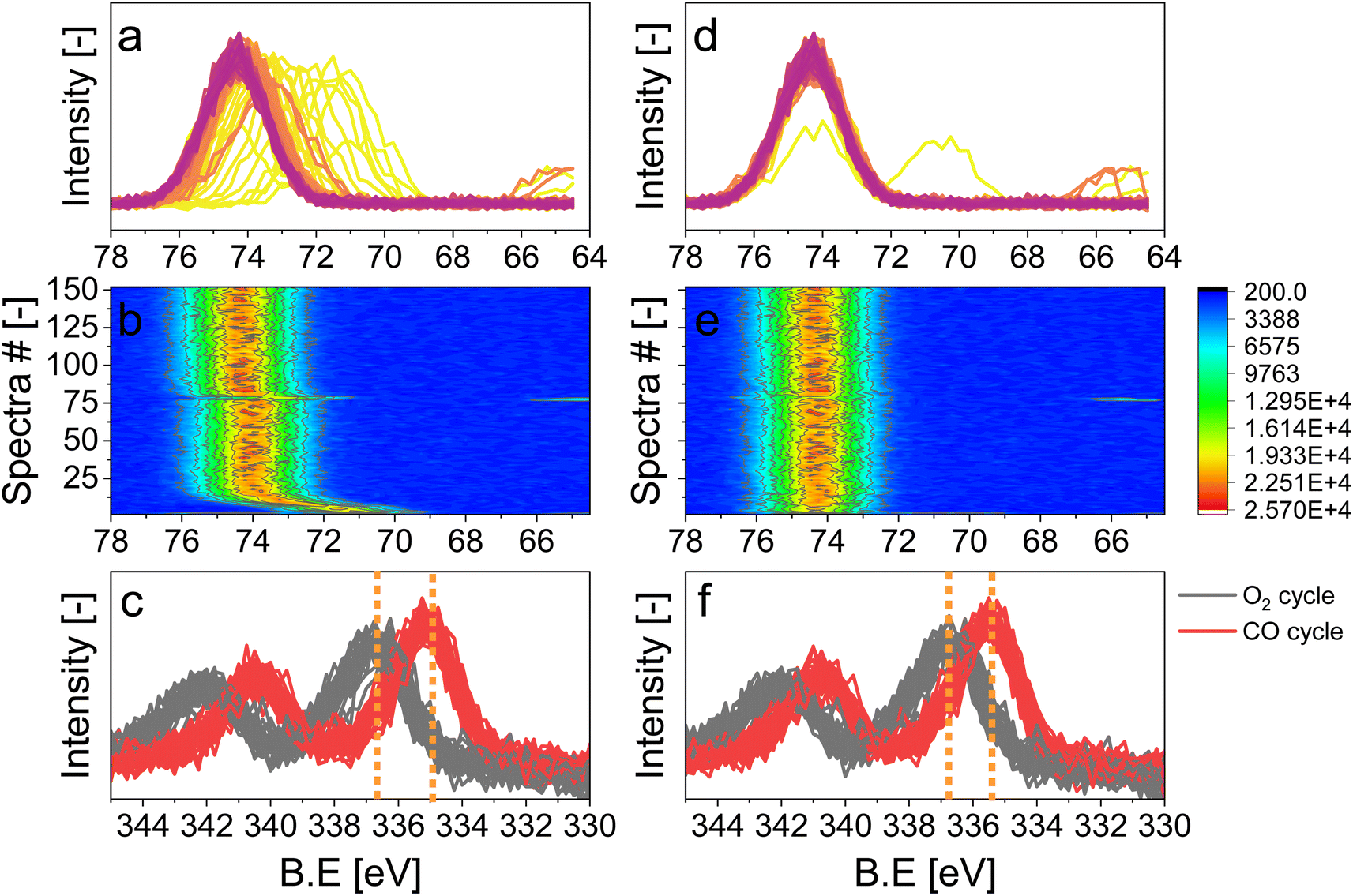 | ||
| Fig. 3 X-ray photoelectron spectra of 5 wt% Pd/Al2O3. (a and b) and (d and e) Al 2p core level with a 650 eV kinetic energy. (b and e) Heat map of Al 2p core level spectra acquired during the 6 h measurement. (c and f) Pd 3d core level with a 300 eV kinetic energy. (a–c) Non-aligned spectra; (d–f) aligned spectra. The vertical lines in panels (c) and (f) correspond to the maximum intensity of the Pd 3d core level features in the O2 and CO periods. | ||
Structural modifications of the surface can be taken as reference events to align spectra to perform averaging over the repeated modulation periods. Here, we used the alternate gas switches as defined events to precisely average the periods. Valves installed on the chamber allowed performing equidistant and well-defined gas switches and by the sufficiently shorter delay between the switching time and the change in gas composition in the experimental cell measured by MS (ca. 4 s to exchange 95%; Fig. 2) than the time needed to acquire a single spectrum (6.20 s). The cell volume (ca. 150 mL), the flow configuration of the cell described above and the short distance between the valves and the cell allowed for such fast gas exchange. The rate of exchange of gas composition could be further improved by increasing the gas flow. The pressure, measured by a baratron head, was delayed by ca. 12 s compared to the gas switching event (Fig. 2). This is due to the location of the pressure sensor, on top of the cell, whereas the MS is situated in the second differential pumping stage of the electron analyser.4,24 Hence, in correspondence of a gas switch, it took a few seconds for the gases to diffuse to the top of the cell and to reach the baratron. Since the inlet gas feed and the MS are close to the sample, we assume that the gas composition around the sample was exchanged almost immediately at the gas switch. Therefore, with such a setup configuration, it is only necessary to synchronize the acquisition of the first spectrum used for the data processing with the time at which the first pulse occurs.
For the following phase sensitive analysis of the averaged data, we discarded the first ten periods because the response of the active species to the periodic stimulation needed several periods to attain a new equilibrium around which it oscillated reversibly at the same frequency as that of the stimulation (Fig. S5†). Changes occurring in the first ten periods can be evaluated separately and individually to analyze possible transitions of the structure from that before the ME experiment to that changing reversibly during the ME experiment. Fig. 4a shows the Pd 3d core level spectra averaged over 26 periods and corresponding to the middle of the CO and O2 half-periods (t = 150 s). Under oxidizing conditions (O2), the Pd 3d5/2 and Pd 3d3/2 signals are centred at ca. 337 and 342.25 eV, respectively. The spectral features shift to lower binding energy in the presence of CO (ca. 335.25 and 340.5 eV) and exhibit a narrower line shape compared to those in oxidizing conditions. The comparison between the two spectra demonstrates that the oxidation state of palladium changes reversibly upon the periodic modification of the reaction environment.
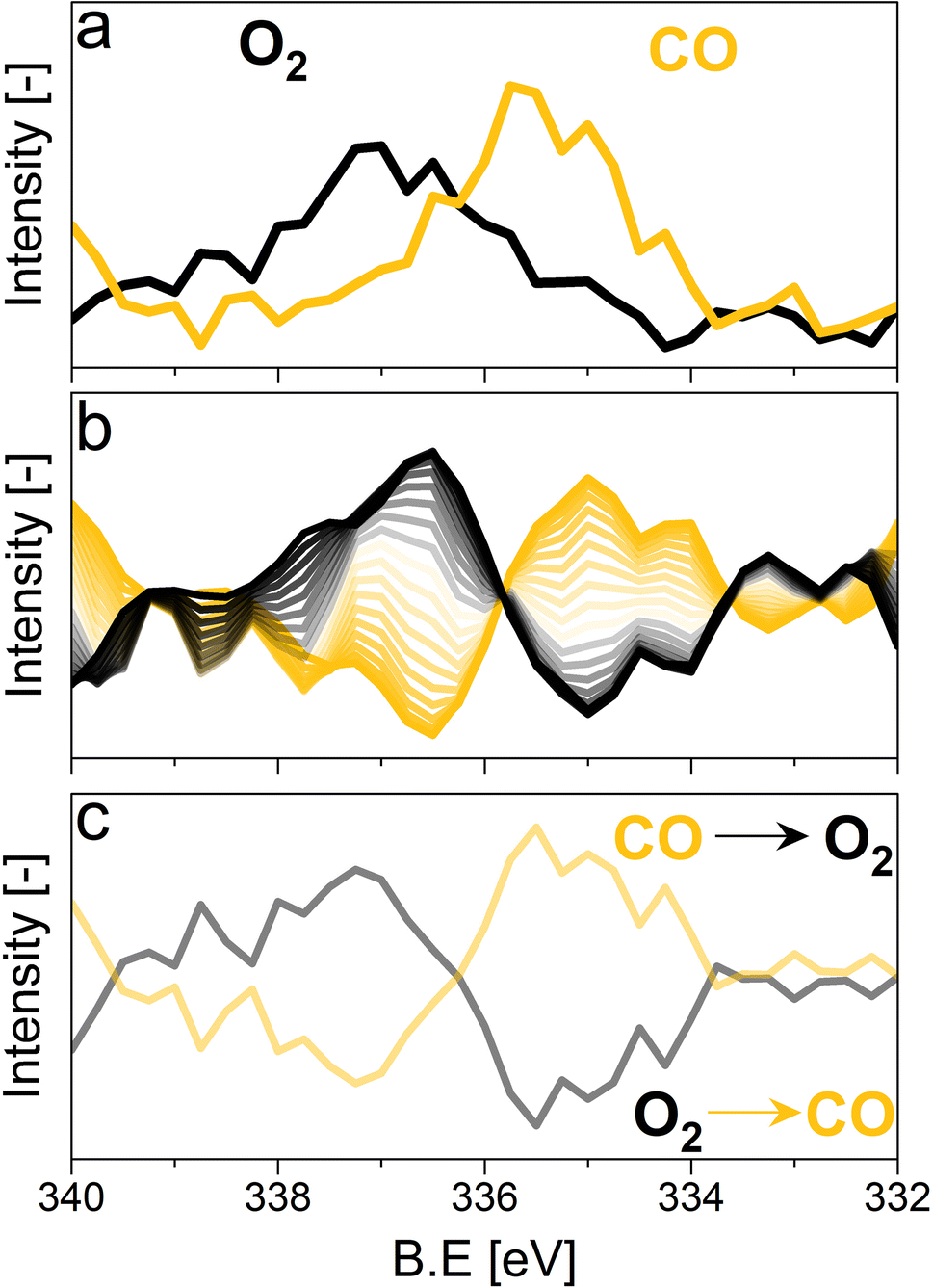 | ||
| Fig. 4 (a) Average X-ray photoelectron spectra of 5 wt% Pd/Al2O3 at the Pd 3d core level with a 300 eV kinetic energy obtained 150 s after changing to the reducing and to oxidizing conditions. (b) Phase domain analysis performed on the smoothed data (binomial 1). (c) Difference spectra between the two boundary time-resolved spectra shown in the panel a. Only the Pd 3d5/2 core level is shown. | ||
Identification of species on 5 wt% Pd/Al2O3
The repeated and alternated O2/CO pulses performed in the modulation sequence enable to increase the time resolution of the APXPS experiment and the S/N ratio. Fig. 4b depicts the spectra obtained after the phase-sensitive detection (PSD) algorithm was applied to the averaged and aligned data (Fig. S6a†). The spectral resolution (ca. 0.3 eV) is most likely responsible for the sharp features in the averaged aligned data and thus for what we consider the residual noise. To improve the readability of these spectra, the averaged aligned data were smoothed before performing PSD (binomial smoothing of degrees 1, 2, and 3; Fig. S6b–d† and Fig. 4). The resulting spectra displayed similar signal features to the aligned PSD data and were clearly distinct from the remaining noise (Fig. 4). Data smoothing has to be considered with care since it can blur the PSD data and affect fitting (see for example binomial smoothing 3 in Fig. S6d†). All the smoothed data were aligned prior to smoothing, but will only be referred to as smoothed data in the subsequent discussion.The phase-resolved data of Fig. 4b show the changes already visible in the time domain but with improved quality and sensitivity. Focusing on the Pd 3d5/2 peak, similar to the difference spectrum obtained subtracting the last time-resolved spectrum in the CO pulse from the last one in the O2 pulse (Fig. 4c), they show the presence of two main peaks of opposite sign centred at 335.0 and 337.5 eV representing the macroscopic changes induced by reduction in CO and oxidation in O2. Each peak is however clearly accompanied by at least two overlapping signals at 334.2 and 337.7 eV, respectively, that are visible in the phase domain but become difficult to identify in the time domain. The difficulty to evaluate the number of Pd species affected by the perturbation and contributing to the time-resolved signal as well as to follow their dynamics (consumption/formation) is overcome by meticulously evaluating and comparing the fits of the phase and of the time domains in order to manually cross-adjust each fit iteratively (Fig. S7†). The fitting was performed on selected phase domain spectra of the aligned and smoothed data (binomial 1; phase angles: 0°, 20°, and 40°) initially using two peaks (Fig. S7†). The peak features obtained from the phase domain fit (peak position and full width at half maximum, FWHM) were fixed to fit the time domain spectra, corresponding to the non-smoothed averaged and aligned data set taken at t = 150 s in the reducing and oxidising half-periods (Fig. 4). Fitting the time domain spectra with the parameters obtained in the phase domain (see the deconvolution of Pd 3d5/2 spectra in Fig. S7†) helps to visualize where a peak might be missing in the initial fit of the phase-resolved spectra. This information was used to iterate the fit in the phase domain spectra including the additional features missing from the time domain. The iterative fit started with two peaks (Fig. S7,† plots c and d top, left) and ended with seven peaks, which were needed to obtain a good correlation with the raw data (Fig. 5 and Table 1). The reverse strategy (fitting the data by identifying the missing peaks in the time domain and transferring it to the phase domain) was also carried out to validate the fits (Fig. S8†). It is important to highlight that initial fitting guesses were based on previous experiments performed at the same beamline on actual samples and more peaks were added based on literature reports (see below).29
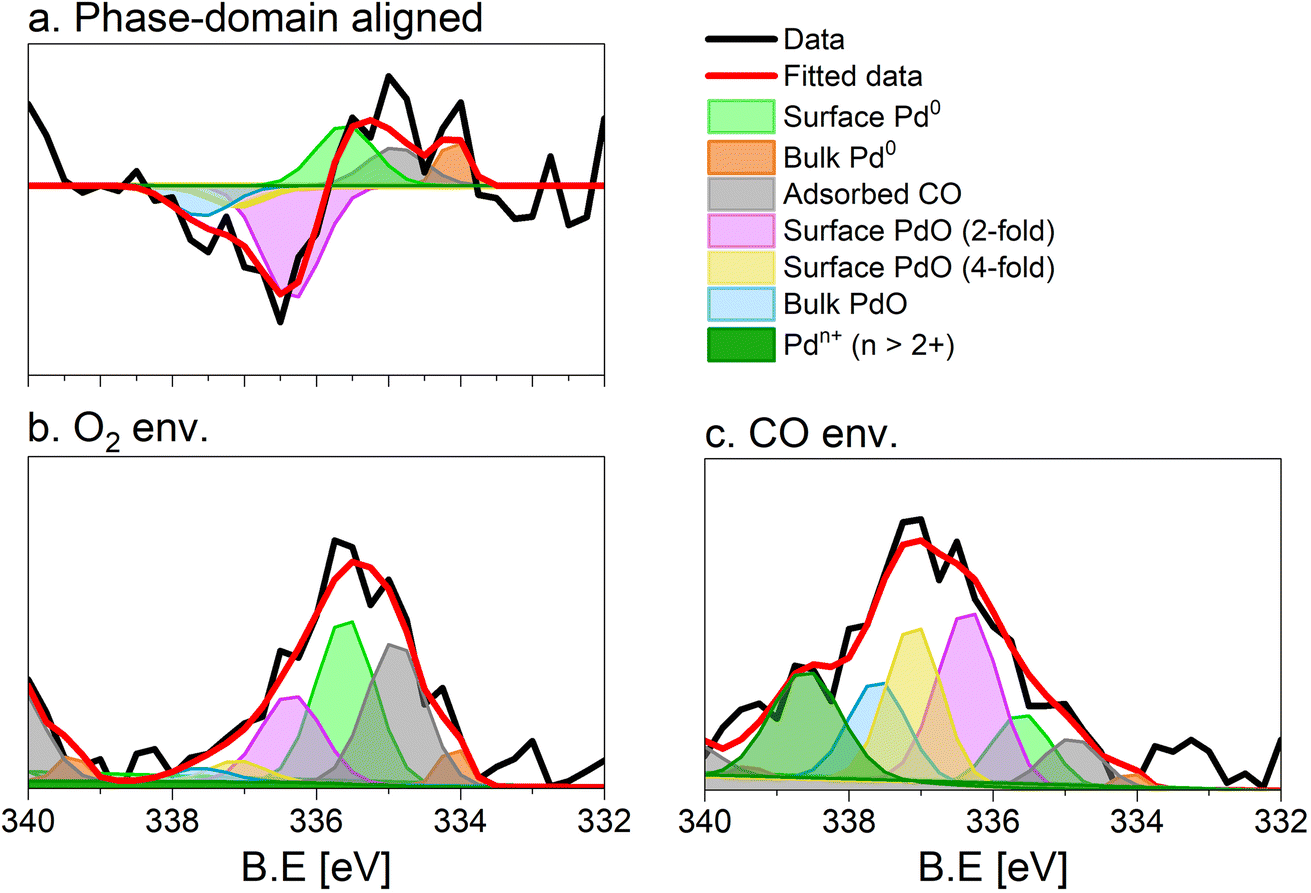 | ||
| Fig. 5 Fit of the (a) phase (0°) and (b and c) time domain spectra (t = 150 s), X-ray photoelectron spectra of 5 wt% Pd/Al2O3 acquired at the Pd 3d core level. (b) Averaged spectrum in oxidising conditions, (c) averaged spectrum in reducing conditions. Peaks are defined in Table 1. Only the Pd 3d5/2 core level is shown. | ||
| Binding energy [eV] | FWHMa [eV] | Assignment | |
|---|---|---|---|
| a Full width at half maximum. | |||
| Peak 1 | 334.1 | 0.4 | Surface Pd0 (ref. 30–32) |
| Peak 2 | 334.9 | 0.9 | Bulk Pd0 (ref. 30–32) |
| Peak 3 | 335.6 | 0.9 | Adsorbed CO30–32 |
| Peak 4 | 336.3 | 1 | Surface PdO (2-fold)31,33–40 |
| Peak 5 | 337.1 | 0.9 | Subsurface PdO (4-fold)31,33–40 |
| Peak 6 | 337.6 | 1 | Bulk PdO30,31 |
| Peak 7 | 338.6 | 0.8 | Pdn+ (n > 2+)27,41–53 |
This procedure enabled us to identify the following peaks that are assigned according to previous literature reports. The bulk Pd0 and the surface Pd0 components were detected at 334.9 eV, and 334.1 eV, respectively.30–32 The third Pd species centred at 335.6 eV is attributed to Pd0 with adsorbed CO. The CO molecules can adsorb at the hollow, bridge and on-top sites of the surface Pd0 but their very close binding energy values (within 0.7 eV) did not allow their separation.30,31,33 The literature suggests that under the experimental conditions adopted in this work (1 mbar, 300 °C) and the average Pd particle size (Fig. S1c†), CO is expected to adsorb almost quantitatively on bridge sites.32 The shift by +0.7 eV compared to bulk Pd0 is in good agreement with such an adsorption geometry, which has been observed on supported Pd catalysts.33
Regarding the oxidized Pd species, bulk PdO was found at 337.60 eV.30,33 In this work, we identified two additional Pd oxide species at 336.55 and 337.0 eV corresponding likely to a 2-fold and a 4-fold surface oxide, respectively arising from what was associated to the two-dimensional Pd5O4 phase.31,33,34 The 2-fold oxide species of Pd5O4 is an outmost surface species formed by chemisorbed oxygen coordinated to Pd and exposed to the solid–gas interface. The 4-fold Pd oxide species is considered to be the subsurface oxide of Pd5O4 and is regarded as a precursor to the PdO bulk.35,36 Both species are defined as sub-stoichiometric PdOx<1, which is considered an intermediate state between chemisorbed oxygen on Pd0 and bulk PdO. Such species were observed mainly on Pd(111) single crystals37,39 but also on an powder Pd/Al2O3 catalyst.40
The assignment of the last component characterized by the highest binding energy (338.6 eV) is more complex and debated.41–44 It can be related to cationic Pd species that are surface species (1) in an oxidation state higher than 2+45–47 and/or (2) possessing strong interaction with the alumina support48–52 as well as (3) replacing a cation in the metal oxide lattice (depending on the support).41,44,53 Recently, we assigned the component characterized by the peak at 338.6 eV to palladium adatoms on the surface of the alumina support.27
The fit of the averaged spectrum obtained at 150 s under oxidising conditions is dominated by cationic Pd species (Fig. 5a and Table 2). The contribution of Pd0 is low (7%), while surface Pd0 is absent. The fraction of Pd0 with adsorbed CO (11%) is not negligible and is attributed to the Pd surface poisoning by CO due to the strong Pd–CO bond. The poisoning effect of CO on Pd sites is supported by the very low levels of CO2 all along the O2 half-period in the MS data (Fig. S3†), after a less pronounced sharp peak than that detected in the CO half-period. The presence of the signals of surface and bulk Pd0 under oxidizing conditions suggests that the thickness of the oxide layer formed is smaller than the mean escape depth of photoelectrons and thus that metallic Pd is coated by a thin skin of oxide.
| Binding energy [eV] | O2 [%] | CO [%] | |
|---|---|---|---|
| Surface Pd0 | 334.1 | 1 | 4 |
| Bulk Pd0 | 334.9 | 7 | 32 |
| Adsorbed CO | 335.6 | 11 | 36 |
| Surface PdO (2-fold) | 336.35 | 26 | 20 |
| Subsurface PdO (4-fold) | 337.1 | 14 | 4 |
| Bulk PdO | 337.6 | 22 | 4 |
| Pdn+ (n > 2) | 338.6 | 19 | 0 |
Under reducing conditions, the fit of the averaged spectrum at 150 s (Fig. 5b and Table 2) shows predominantly peaks corresponding to metallic Pd species as well as Pd0 with adsorbed CO together with minor contributions from bulk and surface PdO but no signal of Pdn+. The poor contribution from the surface Pd0 (4%) is justified by the presence of adsorbed CO molecules.
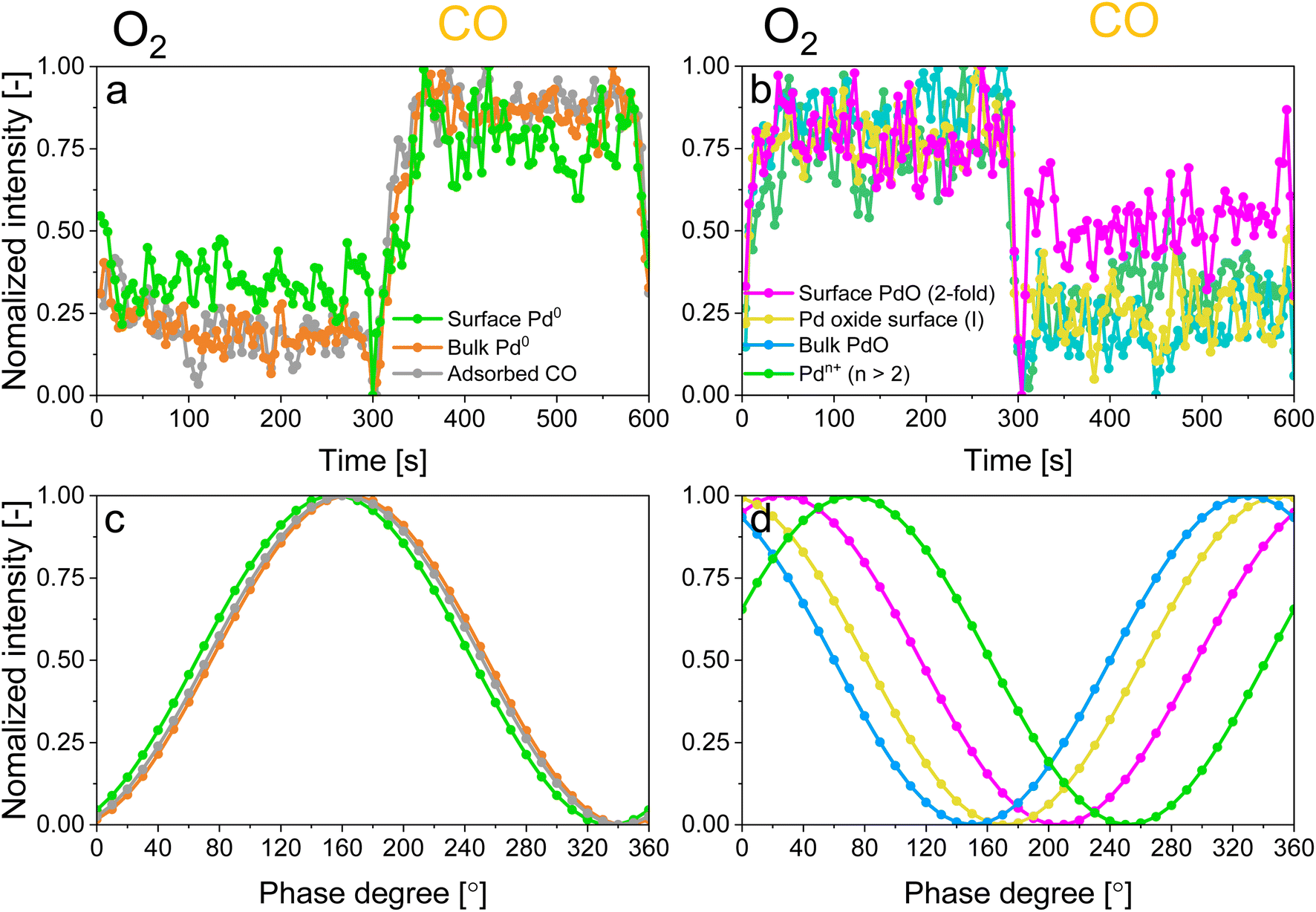 | ||
| Fig. 6 Behaviour of normalised (a and c) time domain and (c and d) phase domain intensities of (a and c) all reduced Pd species and (b and d) all oxidized Pd species from the aligned and smoothed (binomial 1) data set. | ||
Dynamics and mechanism
The identification of the various signals in the fit of the phase-resolved data allowed to select their position in the time-resolved spectra and to plot their temporal behaviour. Fig. 6a and b shows that changes are well recognizable in the time domain within an O2/CO period and that regions where no photoelectron signal is expected (such as at 330.5 and 333 eV in Fig. S9†) are not affected by the repeated gas switches. However, it is also clear that in the time domain no differences in the dynamics of the various reduced and oxidized species could be distinguished. In contrast, the phase domain data delivered important information on the dynamics of the species affected by the perturbation with greater details.The behaviour of reduced and oxidized Pd species in the phase domain of the smoothed data (binomial 1) is presented in Fig. 6c and d, respectively. Based on the idea that a shift of the sinusoidal response of a species to higher phase angles implies its faster evolution over the complete O2/CO period,54 the phase behaviour of the smoothed data (binomial 1) suggests a different (faster) response of bulk Pd0 compared to surface Pd0 and Pd0 with adsorbed CO (Fig. 6c). This is emphasized when looking at the aligned data (Fig. S10c†).
In contrast to the reduced species, significant differences are visible in the behaviour of the oxidized Pd species. Pdn+ species, represented by the binding energy at 338.6 eV, respond faster than the others to the O2/CO modulation, followed in the order by the 2-fold surface PdO species, the 4-fold subsurface PdO and the bulk PdO species. Here no significant difference can be observed between aligned and smoothed data (Fig. 6 and S10†) likely because it is already significant in the aligned data in contrast to the behaviour of the reduced species.
Based on the behaviour of the reduced and oxidized species in the phase domain we can propose an explanation of the process captured by APXPS. Taking into account that PdO reduction by CO proceeds through oxygen diffusion from the bulk to the surface according to the diffusion-controlled reduction model,55,56 the data of Fig. 6 suggest that during reduction bulk Pd0 grows first and is followed by the simultaneous formation of surface Pd0 and CO adsorption. The 10-fold faster reactivity of the Pdn+ species towards CO oxidation compared to PdO nanoparticles at room temperature47 explains the early change in the signal of Pdn+ in the phase domain. The observation that the 2-fold surface PdO species are more reactive towards CO oxidation than bulk PdO and 4-fold subsurface PdO species33–35 due to the direct accessibility of the CO binding sites by the surface oxides sites,35,36,57 rationalises the observed order in the phase domain of these species. If we consider that Pd oxidation follows the Cabrera-Mott (CM) model58 for a surface oxide layer below 3 nm and then the 3D diffusion model,59 the initial formation of Pd surface species (Pdn+ and 2-fold surface PdO) upon oxidation is coherent with the formation of a thin, highly defective oxide film across which electrons diffuse from the metallic core to ionize adsorbed oxygen. The 4-fold subsurface PdO species exhibits a faster initial formation rate than that of bulk PdO,35,36,55,60 supporting the hypothesis that it acts as a precursor of bulk PdO. The defectiveness of the surface oxide layer predicted by the CM model56 possibly explains why APXPS detects the consumption of the fraction of bulk Pd0 prior to that of the Pd0 surface.
The results obtained in this work demonstrate that PSD offers the unique possibility to extrapolate the kinetics of species formation/consumption from in situ time-resolved photoelectron spectroscopy acquired on a realistic catalyst, which is very challenging to observe otherwise, e.g. plotting the species evolution in the time domain. The reaction mechanism of CO oxidation can be assessed through additional sets of experiments where the conditions, such as temperature and nature of the pulse sequences are varied, which goes beyond the scope of this work. Such a technique can be used in other facilities, as the main requirement is a precise control of the reaction phases (e.g. achievable by means of switching valves) and their synchronization with spectroscopy. Results obtained with time-resolved in situ XPS experiments can provide complementary information to those achievable by means of other spectroscopy tools, because the technique is sensitive to interfaces, where catalytic reactions take place.
Conclusions
Time-resolved in situ X-ray photoelectron measurements were performed on a powder 5 wt% Pd/Al2O3 catalyst, while repeatedly switching between oxidative (O2) and reducing (CO) environment in a typical modulated excitation (ME) experiment. All the suitable modulation periods were averaged into one period based on the gas switching events since the setup used allowed for rapid gas switches without significant delays between the pulsing event and the change in gas composition in the experimental cell. By averaging the modulation periods both the signal-to-noise ratio of a photoemission spectrum and the time resolution of the measurement, which was now only limited to the acquisition time of one spectrum, were improved. By comparative fitting of phase and time domain spectra, a total of seven Pd species were detected and their evolution monitored during the gas switches: three reduced Pd species (bulk and surface Pd0, and adsorbed CO on Pd0), and four oxidized species (bulk, 2-fold surface PdO, 4-fold subsurface PdO as well as Pdn+ species). The phase domain data obtained from the PSD algorithm allowed following the dynamic evolution of these species in great detail. It was found that the most susceptible species towards the alternate CO and O2 pulses are Pdn+ species and 2-fold surface PdO species.The work presented here paves the way for significant advances in the field of time-resolved ambient pressure X-ray photoemission spectroscopy applied on powder catalysts with low metal loadings of practical relevance, especially considering that the proposed approach is fairly simple and can be used to process data acquired at different facilities. Further improvements of the spectral resolution of the photoemission spectra could allow the use the ME approach to understand more complex or novel systems.
Data availability
Data will be made available upon request to the corresponding authors.Author contributions
M. R.: conceptualization, methodology, validation, formal analysis, investigation, data curation, writing – original draft, writing – review & editing, visualization. L. A.: methodology, validation, investigation, resources, writing – review & editing, visualization. A. B.: methodology, validation, investigation, writing – review & editing preparation. F. B: methodology, validation, investigation, writing – review & editing. M. A.: methodology, validation, investigation, writing – review & editing. J. van B.: writing – review & editing. O. K.: writing – review & editing, supervision. D. F.: conceptualization, methodology, validation, investigation, writing – review & editing, visualization, supervision, funding acquisition.Conflicts of interest
The authors have no competing interests to declare that are relevant to the content of this article.Acknowledgements
We acknowledge the financial support from the Swiss National Science Foundation (SNF, project no. 200021-175786), the In Situ Spectroscopy beamline of the Swiss Light Source at PSI for beamtime allocation, Dr I. Alxneit for the STEM images, Dr M. K. Ghosalya for the discussions in the early stage of the project, M. Guo for useful discussion on data alignment and fitting.References
- S. Hagström, C. Nordling and K. Siegbahn, Z. Phys., 1964, 178, 439–444 CrossRef.
- M. P. Seah, Surf. Interface Anal., 1980, 2, 222–239 CrossRef CAS.
- A. R. Head, Advances in Solid/Gas Interface Studies with Ambient Pressure X-ray Photoelectron Spectroscopy, 2021 Search PubMed.
- K. Roy, L. Artiglia and J. A. vanBokhoven, ChemCatChem, 2018, 10, 666–682 CrossRef CAS.
- M. Salmeron, Top. Catal., 2018, 61, 2044–2051 CrossRef CAS.
- J. A. Rodriguez, J. C. Hanson and P. J. Chupas, In situ Characterization of Heterogeneous Catalysts, Wiley, 2013 Search PubMed.
- F. Aksoy, M. E. Grass, S. H. Joo, N. Jabeen, Y. P. Hong, Z. Hussain, B. S. Mun and Z. Liu, Nucl. Instrum. Methods Phys. Res., Sect. A, 2011, 645, 260–265 CrossRef CAS.
- H. Bluhm, J. Electron Spectrosc. Relat. Phenom., 2010, 177, 71–84 CrossRef CAS.
- E. J. Crumlin, Z. Liu, H. Bluhm, W. Yang, J. Guo and Z. Hussain, J. Electron Spectrosc. Relat. Phenom., 2015, 200, 264–273 CrossRef CAS.
- D. E. Starr, Z. Liu, M. Hävecker, A. Knop-Gericke and H. Bluhm, Chem. Soc. Rev., 2013, 42, 5833–5857 RSC.
- L. Artiglia, F. Orlando, K. Roy, R. Kopelent, O. Safonova, M. Nachtegaal, T. Huthwelker and J. A. vanBokhoven, J. Phys. Chem. Lett., 2017, 8, 102–108 CrossRef CAS PubMed.
- K. F. Kalz, R. Kraehnert, M. Dvoyashkin, R. Dittmeyer, R. Gläser, U. Krewer, K. Reuter and J. D. Grunwaldt, ChemCatChem, 2017, 9, 17–29 CrossRef CAS PubMed.
- P. Müller and I. Hermans, Ind. Eng. Chem. Res., 2017, 56, 1123–1136 CrossRef.
- T. Bürgi and A. Baiker, J. Phys. Chem. B, 2003, 106, 10649–10658 CrossRef.
- R. Kydd, D. Ferri, P. Hug, J. Scott, W. Y. Teoh and R. Amal, J. Catal., 2011, 277, 64–71 CrossRef CAS.
- D. Ferri, M. A. Newton, M. DiMichiel, G. L. Chiarello, S. Yoon, Y. Lu and J. Andrieux, Angew. Chem., Int. Ed., 2014, 53, 8890–8894 CrossRef CAS PubMed.
- D. Chernyshov, W. vanBeek, H. Emerich, M. Milanesio, A. Urakawa, D. Viterbo, L. Palin and R. Caliandro, Acta Crystallogr., Sect. A: Found. Crystallogr., 2011, 67, 327–335 CrossRef CAS PubMed.
- D. Ferri, S. K. Matam, R. Wirz, A. Eyssler, O. Korsak, P. Hug, A. Weidenkaff and M. A. Newton, Phys. Chem. Chem. Phys., 2010, 12, 5634–5646 RSC.
- D. Ferri, M. A. Newton and M. Nachtegaal, Top. Catal., 2011, 54, 1070–1078 CrossRef CAS.
- C. F. J. König, J. A. vanBokhoven, T. J. Schildhauer and M. Nachtegaal, J. Phys. Chem. C, 2012, 116, 19857–19866 CrossRef.
- J. Knudsen, T. Gallo, V. Boix, M. D. Strømsheim, G. D'Acunto, C. Goodwin, H. Wallander, S. Zhu, M. Soldemo, P. Lömker, F. Cavalca, M. Scardamaglia, D. Degerman, A. Nilsson, P. Amann, A. Shavorskiy and J. Schnadt, Nat. Commun., 2021, 12, 1–8 CrossRef PubMed.
- D. Baurecht and U. P. Fringeli, Rev. Sci. Instrum., 2001, 72, 3782 CrossRef CAS.
- A. Urakawa, T. Bürgi and A. Baiker, Chem. Eng. Sci., 2008, 63, 4902–4909 CrossRef CAS.
- F. Orlando, A. Waldner, T. Bartels-Rausch, M. Birrer, S. Kato, M. T. Lee, C. Proff, T. Huthwelker, A. Kleibert, J. vanBokhoven and M. Ammann, Top. Catal., 2016, 59, 591–604 CrossRef CAS.
- J. Gong, J. Pihl, D. Wang, M. Y. Kim, W. P. Partridge, J. Li, M. Cunningham, K. Kamasamudram, N. Currier and A. Yezerets, Catal. Today, 2021, 360, 294–304 CrossRef CAS.
- J. A. Rotole and P. M. A. Sherwood, Surf. Sci. Spectra, 2021, 5, 18 CrossRef.
- A. Boucly, L. Artiglia, M. Roger, M. Zabilskiy, A. Beck, D. Ferri and J. A. vanBokhoven, Appl. Surf. Sci., 2022, 606, 154927 CrossRef CAS.
- L. Trotochaud, A. R. Head, O. Karslıoğlu, L. Kyhl and H. Bluhm, J. Phys.: Condens. Matter, 2017, 29, 053002 CrossRef PubMed.
- X. Li, X. Wang, K. Roy, J. A. van Bokhoven and L. Artiglia, ACS Catal., 2020, 10(10), 5783–5792 CrossRef CAS.
- H. Kondoh, R. Toyoshima, Y. Monya, M. Yoshida, K. Mase, K. Amemiya and B. S. Mun, Catal. Today, 2016, 260, 14–20 CrossRef CAS.
- R. Toyoshima, M. Yoshida, Y. Monya, K. Suzuki, B. S. Mun, K. Amemiya, K. Mase and H. Kondoh, J. Phys. Chem. Lett., 2012, 3, 3182–3187 CrossRef CAS PubMed.
- S. Blomberg, U. Hejral, M. Shipilin, S. Albertin, H. Karlsson, C. Hulteberg, P. Lömker, C. Goodwin, D. Degerman, J. Gustafson, C. Schlueter, A. Nilsson, E. Lundgren and P. Amann, ACS Catal., 2021, 11, 9128–9135 CrossRef CAS PubMed.
- R. Toyoshima, M. Yoshida, Y. Monya, Y. Kousa, K. Suzuki, H. Abe, B. S. Mun, K. Mase, K. Amemiya and H. Kondoh, J. Phys. Chem. C, 2012, 116, 18691–18697 CrossRef CAS.
- R. Westerström, M. E. Messing, S. Blomberg, A. Hellman, H. Grönbeck, J. Gustafson, N. M. Martin, O. Balmes, R. vanRijn, J. N. Andersen, K. Deppert, H. Bluhm, Z. Liu, M. E. Grass, M. Hävecker and E. Lundgren, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 115440 CrossRef.
- H. Gabasch, W. Unterberger, K. Hayek, B. Klötzer, E. Kleimenov, D. Teschner, S. Zafeiratos, M. Hävecker, A. Knop-Gericke, R. Schlögl, J. Han, F. H. Ribeiro, B. Aszalos-Kiss, T. Curtin and D. Zemlyanov, Surf. Sci., 2006, 600, 2980–2989 CrossRef CAS.
- D. Zemlyanov, B. Aszalos-Kiss, E. Kleimenov, D. Teschner, S. Zafeiratos, M. Hävecker, A. Knop-Gericke, R. Schlögl, H. Gabasch, W. Unterberger, K. Hayek and B. Klötzer, Surf. Sci., 2006, 600, 983–994 CrossRef CAS.
- H. Gabasch, W. Unterberger, K. Hayek, B. Klötzer, G. Kresse, C. Klein, M. Schmid and P. Varga, Surf. Sci., 2006, 600, 205–2018 CrossRef CAS.
- E. Lundgren, G. Kresse, C. Klein, M. Borg, J. N. Andersen, M. DeSantis, Y. Gauthier, C. Konvicka, M. Schmid and P. Varga, Phys. Rev. Lett., 2002, 88, 246103 CrossRef CAS PubMed.
- M. A. VanSpronsen, J. W. M. Frenken and I. M. N. Groot, Chem. Soc. Rev., 2017, 46, 4347–4374 RSC.
- K. Zorn, S. Giorgio, E. Halwax, C. R. Henry, H. Grönbeck and G. Rupprechter, J. Phys. Chem. C, 2011, 115, 1103–1111 CrossRef CAS.
- M. M. Khader, M. J. Al-Marri, S. Ali and A. G. Abdelmoneim, Catalysts, 2018, 8, 66 CrossRef.
- G. Chen, Y. Yang, Z. Guo, D. Gao, W. Zhao, H. Yan, W. W. Wang, C. J. Jia and G. Sun, Catal. Sci. Technol., 2018, 8, 4413–4419 RSC.
- J. Kappler, N. Bârsan, U. Weimar, A. Dièguez, J. L. Alay and A. Romano-Roclriguez, Fresenius. J. Anal. Chem., 1998, 361, 110–114 CrossRef CAS.
- S. Fazlikeshteli, X. Vendrell and J. Llorca, Reactions, 2022, 2, 30–42 CrossRef.
- L. S. Kibis, A. I. Titkov, A. I. Stadnichenko, S. V. Koscheev and A. I. Boronin, Appl. Surf. Sci., 2009, 255, 9248–9254 CrossRef CAS.
- F. Yin, S. Ji, P. Wu, F. Zhao and C. Li, J. Catal., 2008, 257, 108–116 CrossRef CAS.
- L. S. Kibis, A. I. Stadnichenko, S. V. Koscheev, V. I. Zaikovskii and A. I. Boronin, J. Phys. Chem. C, 2012, 116, 19342–19348 CrossRef CAS.
- A. S. Ivanova, E. M. Slavinskaya, R. V. Gulyaev, V. I. Zaikovskii, O. A. Stonkus, I. G. Danilova, L. M. Plyasova, I. A. Polukhina and A. I. Boronin, Appl. Catal., B, 2010, 97, 57–71 CrossRef CAS.
- A. Aznárez, A. Gil and S. A. Korili, RSC Adv., 2015, 5, 82296–82309 RSC.
- B. Ealet and E. Gillet, Surf. Sci., 1993, 281, 91–101 CrossRef CAS.
- B. Ealet and E. Gillet, Surf. Sci., 1996, 367, 221–230 CrossRef CAS.
- K. Sun, J. Liu, N. Nag and N. D. Browning, Catal. Lett., 2002, 843, 193–199 CrossRef.
- A. J. Hill, C. Y. Seo, X. Chen, A. Bhat, G. B. Fisher, A. Lenert and J. W. Schwank, ACS Catal., 2020, 10, 1731–1741 CrossRef CAS.
- V. Marchionni, D. Ferri, O. Kröcher and A. Wokaun, Anal. Chem., 2017, 89, 5801–5809 CrossRef CAS PubMed.
- G. Ketteler, D. F. Ogletree, H. Bluhm, H. Liu, E. L. D. Hebenstreit and M. Salmeron, J. Am. Chem. Soc., 2005, 127, 18269–18273 CrossRef CAS PubMed.
- K. Reuter and M. Scheffler, Appl. Phys. A, 2004, 78, 793–798 CrossRef CAS.
- H. Gabasch, A. Knop-Gericke, R. Schlögl, M. Borasio, C. Weilach, G. Rupprechter, S. Penner, B. Jenewein, K. Hayeka and B. Klötzer, Phys. Chem. Chem. Phys., 2007, 9, 533–540 RSC.
- N. Cabrera and N. F. Mott, Rep. Prog. Phys., 1949, 12, 163–184 CrossRef CAS.
- A. Khawam and D. R. Flanagan, J. Phys. Chem. B, 2006, 110, 17315–17328 CrossRef CAS PubMed.
- N. Schmahl and E. Minzl, Z. Phys. Chem., 1975, 47, 547 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3sc01274c |
| This journal is © The Royal Society of Chemistry 2023 |