DOI:
10.1039/D3QO00314K
(Review Article)
Org. Chem. Front., 2023,
10, 2095-2114
Selective preparative ‘oxidase phase’ in sesquiterpenoids: the radical approach
Received
28th February 2023
, Accepted 16th March 2023
First published on 16th March 2023
Abstract
The necessity for highly selective oxidative transformations towards the synthesis of complex natural compounds such as sesquiterpenoids makes total synthesis a rich terrain of development for powerful widely applicable methods. Due to excessive oxidative decoration of sesquiterpenoids, which is traced back to the impressive performance of monooxygenases, synthetic efforts have been recently directed also towards more monooxygenase-like radical approaches to address the synthetic challenge of installing an ‘oxidase phase’ in their carbocycles. This review collects recent total syntheses of sesquiterpenoids relying mainly on radical reactions for allylic hydroxylations, hydrations and C–H oxidations.
 Maria Kourgiantaki | Maria Kourgiantaki graduated as a chemist in 2019 from Aristotle University of Thessaloniki (AUTH), where she also earned her M.Sc. in organic chemistry with a focus on natural product synthesis. In 2022 she started her Ph.D. at the same university under the supervision of Professor Alexandros L. Zografos. Her research aim focuses on the divergent total synthesis of highly oxygenated sesquiterpenoids. |
 Georgia G. Bagkavou | Georgia G. Bagkavou received her bachelor's degree from Aristotle University of Thessaloniki in 2020, spending the last semester as an exchange student at the University of Florence. In 2022, she obtained her master's degree from Aristotle University of Thessaloniki and currently she is pursuing her doctorate under the supervision of Prof. Christos Stathakis at the same university. Her research is focused on the synthesis of natural products utilizing transition metal catalytic processes. |
 Christos I. Stathakis | Christos I. Stathakis studied chemistry at Aristotle University of Thessaloniki (AUTH), where he also received his Ph.D. in organic chemistry in 2007. His postdoctoral experience (2008–2012) comprises appointments at the research groups of Prof. Snyder at Columbia University, Dr Vourloumis at NCSR Demokritos, and Prof. Knochel at Ludwig Maximillian University. For the next five years, Dr Stathakis worked as a research scientist at Pharmathen Pharmaceuticals, Greece. In 2018, he was elected as an assistant professor at the chemistry department of AUTH. The main interest of his research focuses on the total synthesis of constructively challenging molecules and the development of novel methodologies that promote structural complexity. |
 Alexandros L. Zografos | Alexandros L. Zografos graduated as a chemist in 1996 from National University of Athens. After earning his Ph.D. in 2001 at National Technical University of Athens, he pursued postdoctoral studies at The Scripps Research Institute and at Columbia University, before he moved back to Greece to work as a senior researcher first at National University of Athens and then at NCRS Demokritos Institute. In 2009, he began his independent career at Aristotle University of Thessaloniki. His group is working on the exploration of common synthetic strategies towards diverse natural molecular structures. |
1. Introduction
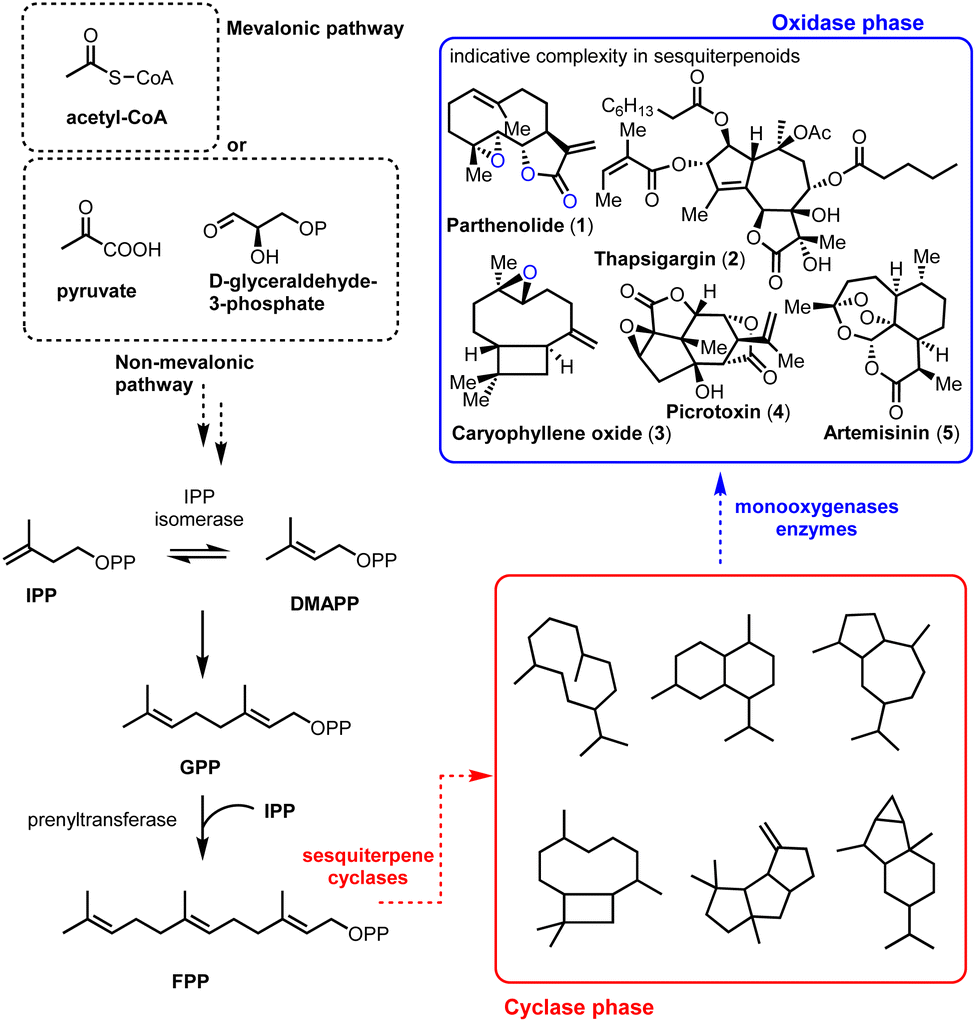
The impressive machinery of complexity as exemplified by the biosynthesis of terpenoids highlights the ability of Nature to perform highly selective transformations by utilizing enzymes and rather simple common scaffolds.1 This divergent protocol empowered by the iterative use of primary metabolism reactions (IPP, DMAPP, and cationic reactions) produces carbocyclic frameworks (cyclase phase) that finally become oxidative enzymes (oxidase phase) (Fig. 1). The common motifs of reactivity used by Nature permit, to some extent, the identification and application of selective biosynthetic-like reactions in modern total synthesis. Sesquiterpenoids play a pivotal role as model templates in biomimetic synthesis by sustaining the crucial balance between their observed molecular complexity and the straightforward link of how their selectivity might arise from their natural roots.
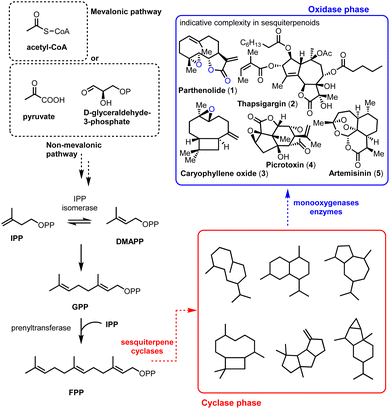 |
| | Fig. 1 Schematic representation of sesquiterpenoid biosynthesis. | |
Containing more than 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 members, sesquiterpenoids represent an active ‘X-ray scan’ of primitive mechanisms in biosynthesis, retaining their importance as biological probes. Compounds such as parthenolide (1), thapsigargin (2), picrotoxinin (4), and artemisinin (5) (Fig. 1, blue frame) are important pharmaceutical leads for anti-cancer, anti-malarial, anti-inflammatory agents etc. but most importantly they also serve as platforms to test the boundaries of total synthesis. A common molecular characteristic in many of the biologically important sesquiterpenoids is the richness of oxidative decoration that they possess. Methods to achieve selectivity in the oxidase phase, especially by their late-stage functionalization, are constantly gaining attention from the scientific community as powerful wide-scope prototypes for further applications.
000 members, sesquiterpenoids represent an active ‘X-ray scan’ of primitive mechanisms in biosynthesis, retaining their importance as biological probes. Compounds such as parthenolide (1), thapsigargin (2), picrotoxinin (4), and artemisinin (5) (Fig. 1, blue frame) are important pharmaceutical leads for anti-cancer, anti-malarial, anti-inflammatory agents etc. but most importantly they also serve as platforms to test the boundaries of total synthesis. A common molecular characteristic in many of the biologically important sesquiterpenoids is the richness of oxidative decoration that they possess. Methods to achieve selectivity in the oxidase phase, especially by their late-stage functionalization, are constantly gaining attention from the scientific community as powerful wide-scope prototypes for further applications.
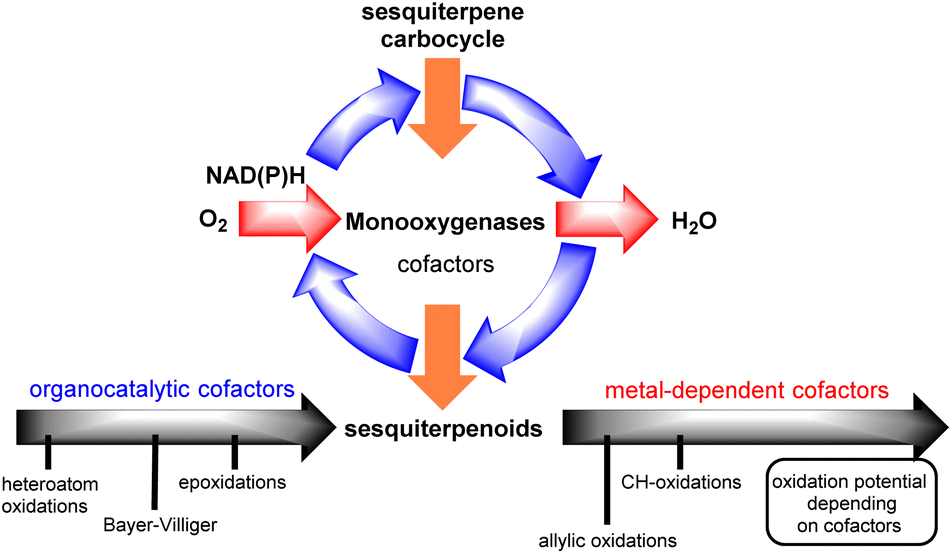
Interestingly, successful methods towards this goal are commonly the ones that unlock and harness the biosynthetic information of ‘natural-players’, namely monooxygenase enzymes (Fig. 2).2
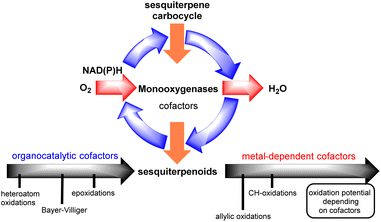 |
| | Fig. 2 Monooxygenase catalytic cycle for sesquiterpenoids. | |
Monooxygenases are common enzymes used in the oxidase phase of sesquiterpenoids. They are usually associated with co-reductase enzymes (NADH, NADPH) and co-factors such as hemes, flavins, pterins etc. for the activation of dioxygen which is utilized as the terminal oxidant of the process. During this process, depending on the associated co-factors, monooxygenases can achieve the reduction of dioxygen to organo- or alternatively metal-superoxy- and hydroperoxy-species to oxidize alkenes, allylic and/or C–H positions.
Depending on sesquiterpenoid complexity, one can recognize three sites for potential oxidation: (i) alkenes, (ii) allylic positions and (iii) unactivated C–H bonds, including those that are involved in lactone functionality formation. In all cases, Nature seems to follow the same one-electron, radical logic to access them, utilizing monooxygenases with co-factors possessing the appropriate oxidative potential for each purpose. On the other hand, in laboratory synthesis commonly the polar logic prevails for such transformations with those of peroxides being more applied for the epoxidation of alkenes3 and hydroboration for their hydration,4 and selenium dioxide metal-allylic activation for allylic oxidations, with only scarce success for selective polar C–H oxidations.5
In this review, we focus on methods that harness one-electron potential to achieve ‘oxidase’ functionalization. The review does not intend to be an exhaustive collection of total syntheses utilizing radicals, but instead a selection of synthetic routes bearing the most selective oxidative pathways on the way to the total synthesis of sesquiterpenoids.6 In this review no electrochemical methods and singlet oxygen oxidations are included and readers interested in these topics are advised to follow some excellent reviews.7,8
1.1. Monooxygenases and biomimetics in practice
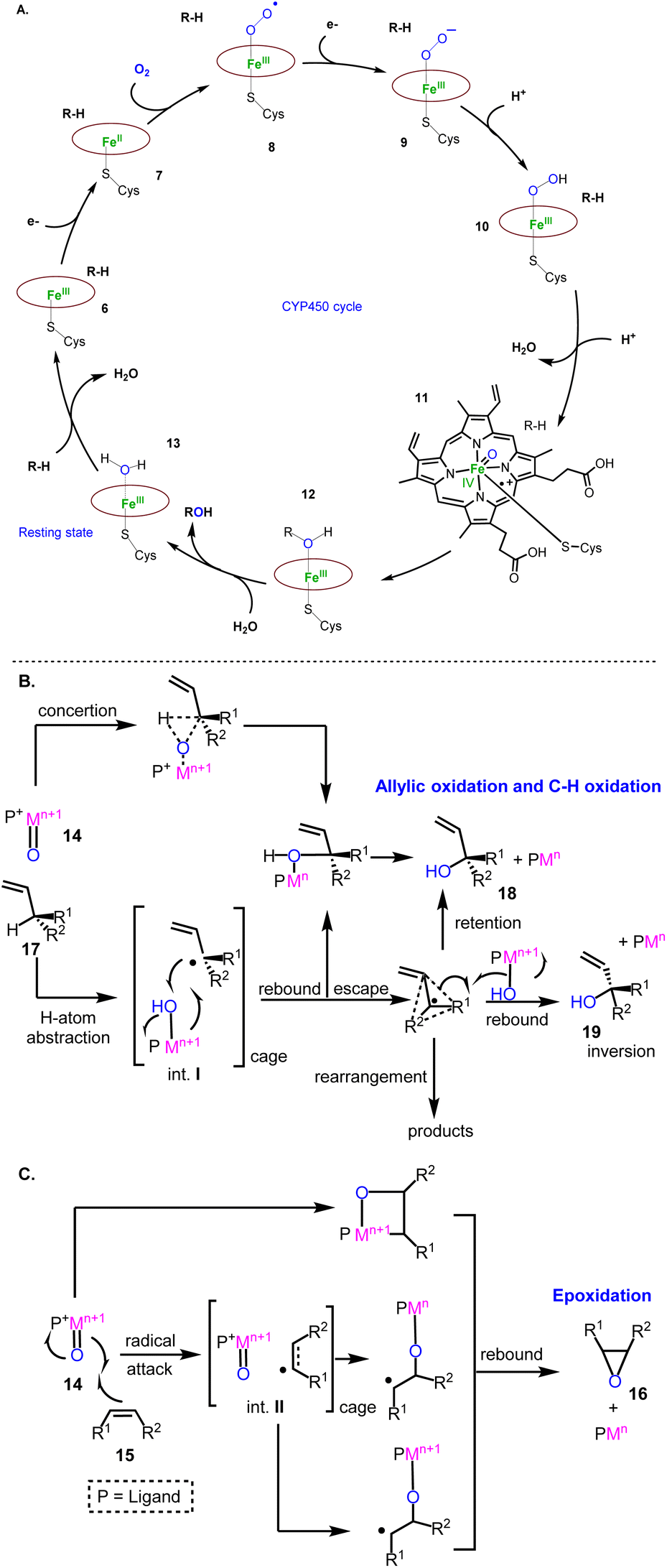
Cytochrome P450 monooxygenases (CYPs) are mostly involved in the oxidative decoration of sesquiterpenoid carbocyclic cores contributing significantly to their structural diversity. Therefore, a brief introduction of their mechanisms will help underline the evolution of the existing oxidative methods used to access sesquiterpenoids. Their mechanism of action has been extensively studied.9 It involves the initial binding of the substrate to the enzyme (6) followed by a one-electron transfer to the heme metal co-factor (7) (most commonly iron) independent of the class of CYPs used. The subsequent reduction of iron(III) to iron(II) initiates the activation of dioxygen to superoxide (8), followed by a second electron donation (9) and a double addition of protons to produce an iron(V) oxo-heme complex (11), responsible for the oxidation of the substrate (12) (Fig. 3A). The latter event is considered to involve, in its simplest form, the addition of the high valency metal-oxo PM![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species (14) to the alkene (15) (epoxidation, compound 16, Fig. 3C) or the abstraction of a proton (in the cases of allylic oxidation and C–H oxidation) to produce caged radical intermediates (I and II, Fig. 3B). For H-atom abstraction, the rebound of high valency hydroxyl complexes provides substrate oxidation with retention of the configuration or stereochemical epimerization (18 or 19) in the cases where the radical escapes the cage or if the rebound energy is high.
O species (14) to the alkene (15) (epoxidation, compound 16, Fig. 3C) or the abstraction of a proton (in the cases of allylic oxidation and C–H oxidation) to produce caged radical intermediates (I and II, Fig. 3B). For H-atom abstraction, the rebound of high valency hydroxyl complexes provides substrate oxidation with retention of the configuration or stereochemical epimerization (18 or 19) in the cases where the radical escapes the cage or if the rebound energy is high.
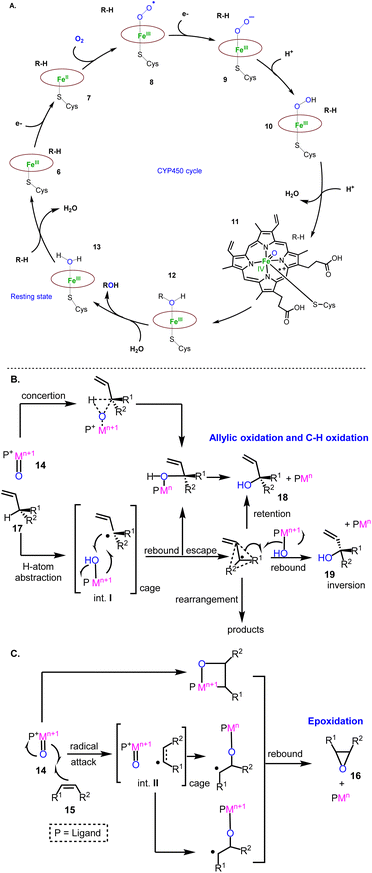 |
| | Fig. 3 General proposed mechanisms involved in monooxygenase oxidations. (A) Iron(v) oxo-heme complex; (B) Allylic and C-H oxidation; (C) Epoxidation mechanisms. | |
Taking into consideration this biosynthetic knowledge, many groups focused on the development of biomimetic variants harnessing the synthetic potential of radicals in their design.
Thus, the points of interest (alkenes, allylic positions and C–H bonds) were tested in radical reactions for their ability to selectively deliver the ‘oxidase phase’. The methods applied so far are not highly diverse and can be roughly divided into four classes: (i) photochemical Hofmann–Loffler–Freytag-type reactions and Suarez-modification; (ii) metal-catalyzed reactions; (iii) Mukaiyama-type hydrations; and (iv) stoichiometric metal oxidations.
2. Hofmann–Loffler–Freytag (HLF) reaction–Suarez modification
The Hofmann–Loffler–Freytag (HFL) reaction was invented in the late 19th century and involves the photochemical generation of nitrogen-centered radicals from N–X bonds.10 1,5-Hydrogen atom transfer (HAT) follows the radical generation to provide the requisite C–H activation for further functionalization. In its classic variant, the HFL reaction is used for the preparation of pyrrolidines and piperidines by the subsequent cyclization of halo-amines. The modification by Suarez enabled the reaction to be extended to O–X bonds by utilizing hypervalent iodine compounds followed by the photochemical cleavage of O–X bonds to deliver mostly ethers. The latter represents a powerful method for the C–H oxidation of sesquiterpenoids.
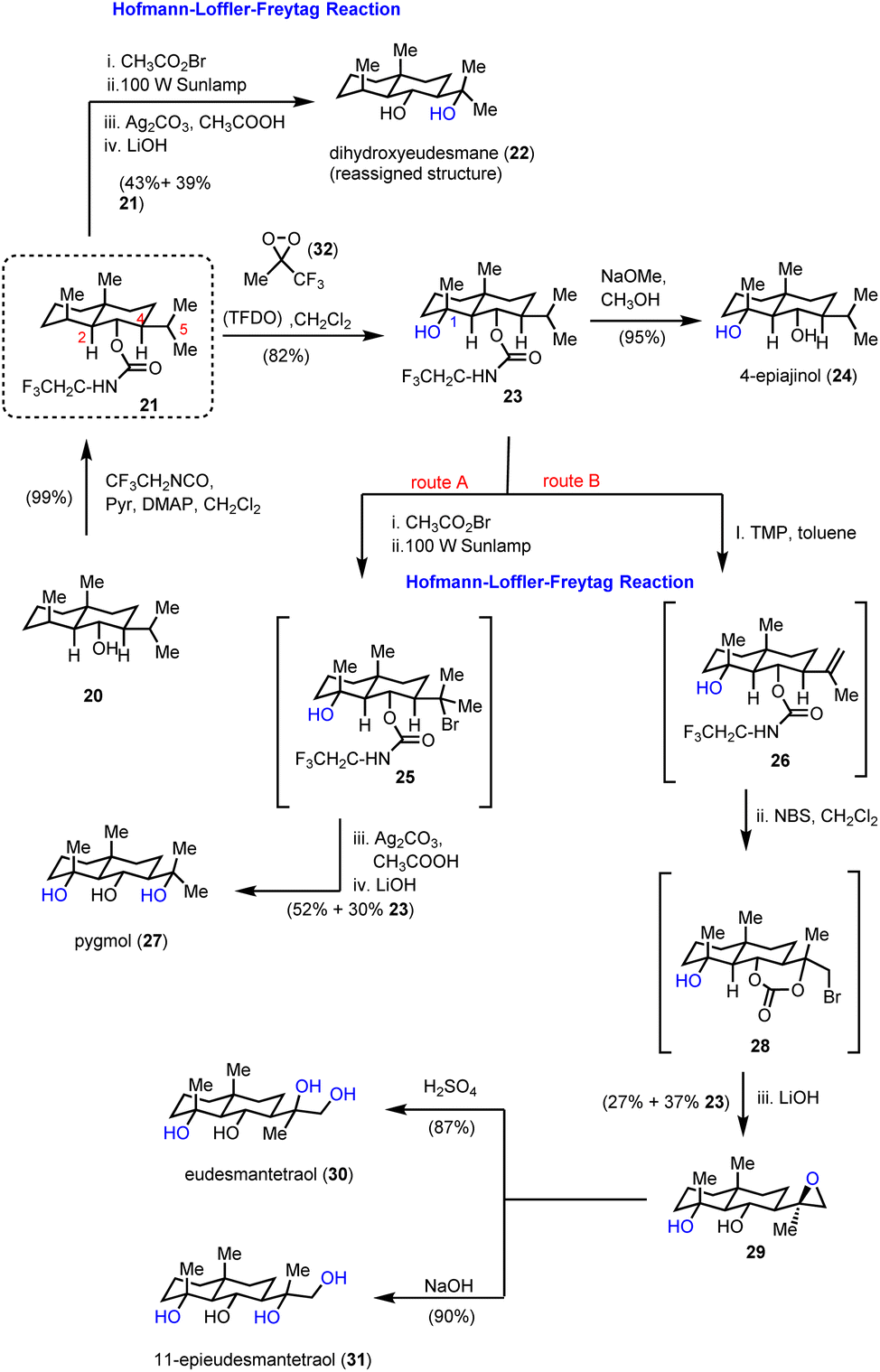
2.1. Eudesmanes (Baran's group)
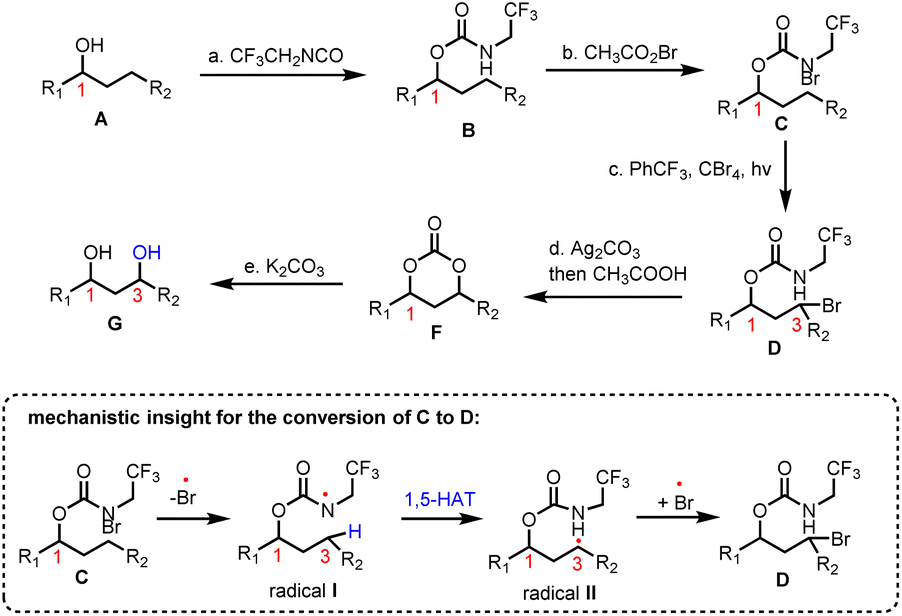
Inspired by the HLF reaction, Baran and co-workers developed a method to convert alcohols A to 1,3-diols Gvia a photochemically initiated intramolecular radical hydrogen atom transfer from an appropriately bromo activated trifluoroethyl-substituted carbamate (C) (Scheme 1).11 The latter allows the formation of iminocarbonates in the presence of Ag2CO3, which can be readily hydrolyzed by acetic acid and K2CO3 to provide 1,3-diols. This concept has been successfully applied to an impressive highly selective divergent synthesis of eudesmane sesquiterpenoids: 4-epiajanol (24), dihydroxyeudesmane (22), pygmol (27) and eudesmantetraol (30) (Scheme 2).12 Accordingly, dihydrojunenol (20), representing the end point of the cyclase phase and the common synthetic intermediate for this divergent synthesis, was accessed in a gram scale quantity via a 9 step sequence in 21% yield and was transformed to its trifluoroethyl carbamate counterpart 21 (Scheme 2).13 The latter, apart from its projected participation in the HFL-type reaction for the installation of hydroxyl moieties, served also the purpose of depleting the electron density from neighbouring carbon atoms (C-2 and C-4) to deactivate them during the electrophilic C–H activation event. The gram scale oxidation of 21 at C-1 to 23 was selectively achieved by Curci oxidation with TFDO in 82% yield.14 This impressive selectivity between the five electronically similar tertiary positions in this scaffold was explained on the basis of carbon nucleophilicity, steric strain and strain release. In particular, the observed non-expected selectivity between the C-5 position (more nucleophilic and less sterically strained) which is more prone to electrophilic oxidation and the obtained oxidation product at the C-1 position can be attributed to the high strain energy release of the latter in the transition state formation.15 The hydrolysis of trifluoroethyl carbamate in 23 allowed the total synthesis of 4-epiajanol (24). Demonstrating the power of their radical 1,3-diol protocol in both 21 and 23 allowed them to access dihydroxyeudesmane (22) and pigmol (27) in 43% and 52% yields, respectively, after photochemical bromination, iminocarbonate formation and hydrolytic cleavage. Additionally, further oxidative manipulations provided the total syntheses of tetraols eudesmanotetraol (30) and 11-epieudesmantetraol (31).
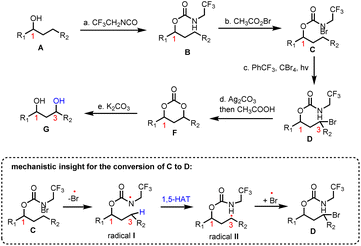 |
| | Scheme 1 Method to achieve 1,3-diols utilizing a modified HFL reaction. | |
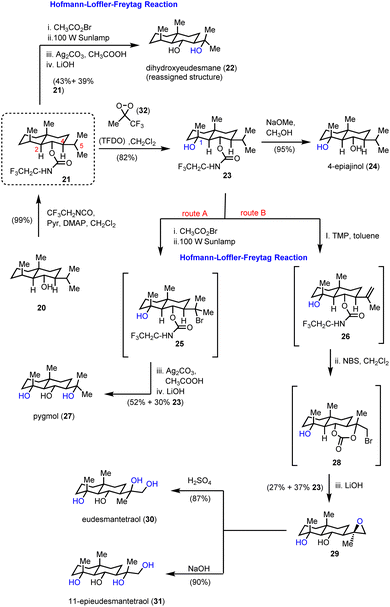 |
| | Scheme 2 Divergent synthesis of eudesmanes based on the HLF oxidation protocol (Baran's group). | |
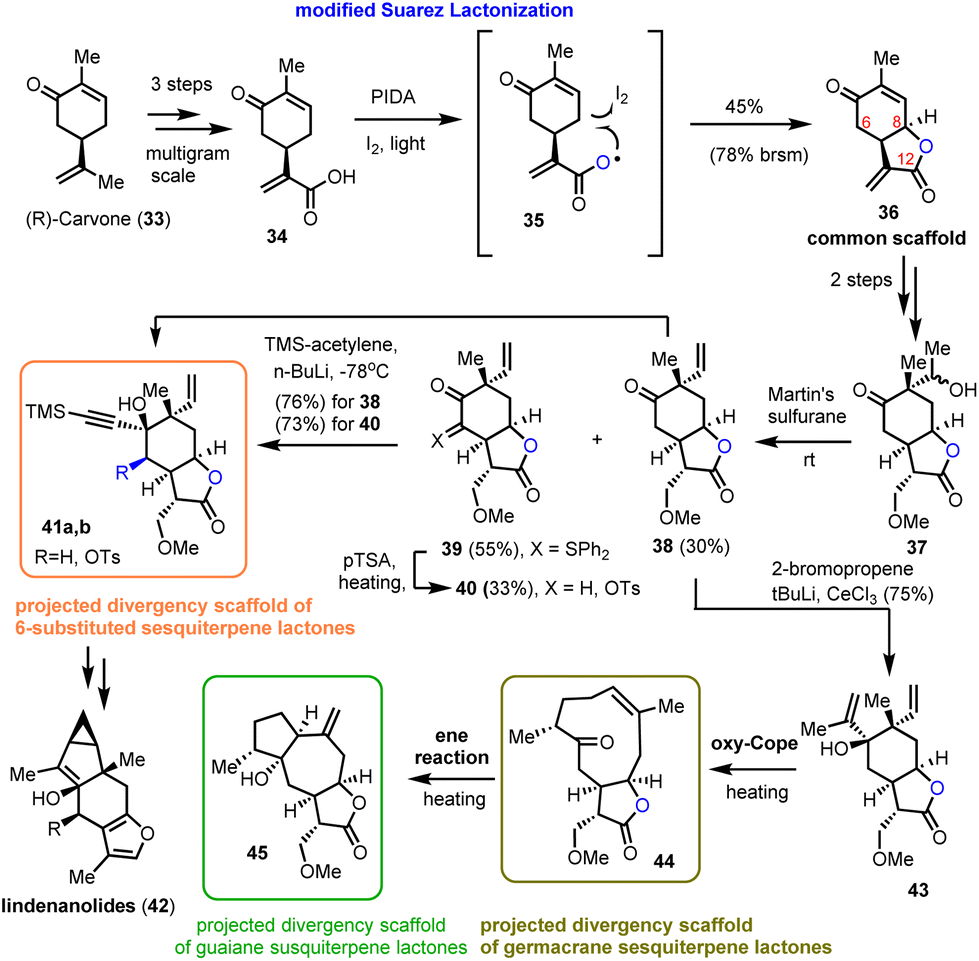
2.2. Asteraceae guaianolides (Zografos’ group)
In 2021, Zografos’ group demonstrated the ability of a modified Suarez reaction to elaborate the biologically important sesquiterpenoid scaffolds α-methylene-γ-lactones.16 The group highlighted the efficiency of the method in the synthesis of the common scaffold 36 that served as a multipurpose intermediate for total syntheses and for accessing sesquiterpenoid core structures (Scheme 3).17 More specifically, acrylic acid 34 reacts with PIDA and iodine to form intermediate 35, which can be photochemically cleaved to provide a long-lived carboxyl-radical able to initiate a 1,5-hydrogen atom transfer to the respective iodide (structure not shown). The latter is readily cyclized under the reaction conditions to allow the synthesis of 36 in 45% yield (78% brsm). Interestingly, despite the higher acidity of C-6 than that of C-8, only 8,12-lactone has been observed under these conditions. In contrast, α-ketone halogenation, performed under light or radical initiators, resulted only in the formation of 6,12-lactone, proving the essential participation of the carboxyl moiety in the radical event. Faster hydrogen atom transfer from the allylic position (lower energy of allylic radicals) ensures the chemoselectivity of the process. Compound 36 was then selectively alkylated to 37 followed by Martin's sulfurane dehydration to provide 38 and the sulfur-ylide 39. Subsequent alkylation of both resulted in 41a andb and 43 that served as points of divergence to access germacranolides and guaianolides through oxy–Cope reactions16 and furo sesquiterpenoids by cycloisomerizations.17 Worth mentioning is the ability of sulfur-ylides to serve as convenient precursors for C-6 oxidation.
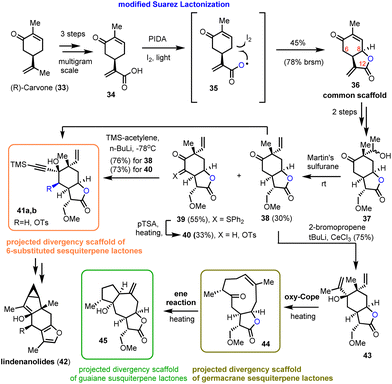 |
| | Scheme 3 Early lactonization with a modified Suarez reaction to access diversity in sesquiterpenoids (Zografos’ group). | |
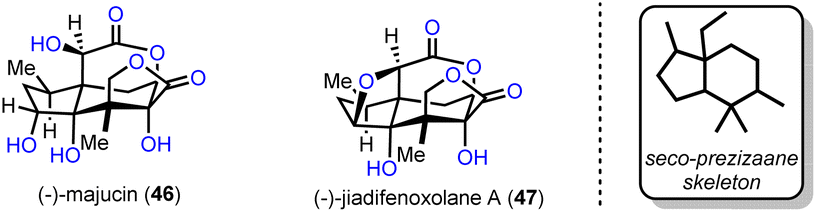
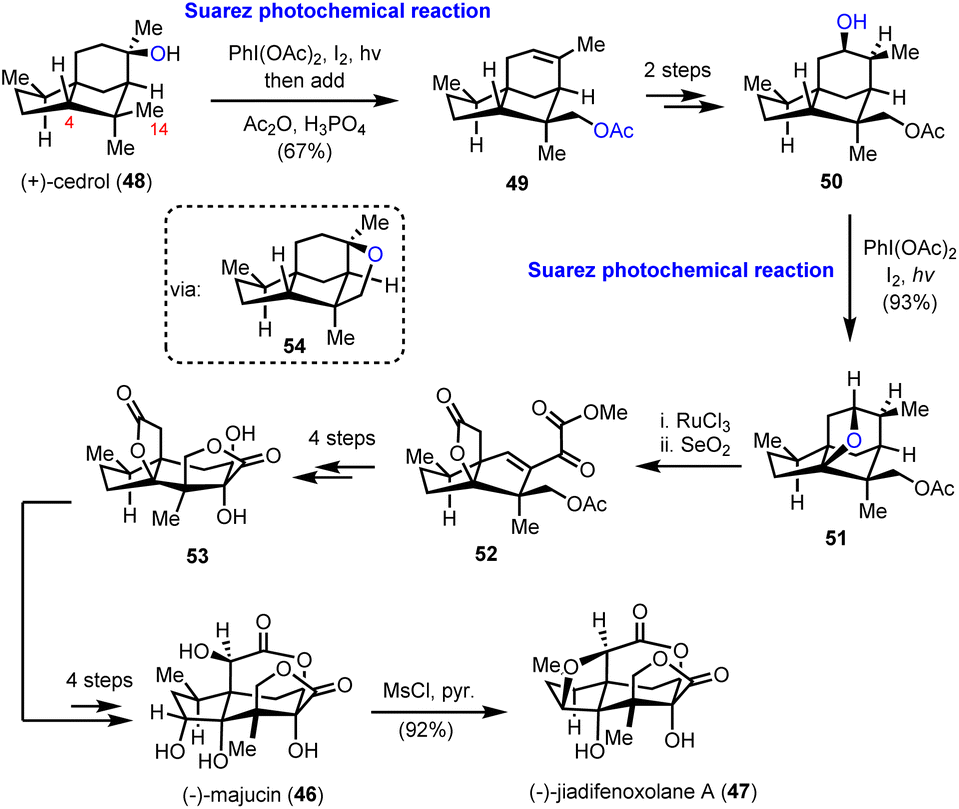
2.3. (−)-Majucin and (−)-jiadifenoxolane A (Maimone's group)
Two distinct Suarez reactions were among the ten net oxidation events involved in the chemical synthesis of (−)-majucin (46) and (−)-jiadifenoxolane A (47), reported by Maimone's group in 2017.18 The latter natural products are distinguished members of the seco-prezizaane subtype of Illicium sesquiterpene family, the key core of which is presented in Scheme 4. Specifically, (−)-majucin (46) is one of the most highly oxidized members, and although it was isolated about thirty years ago, its total synthesis had not been achieved before the impressive work by Maimone and his co-workers.19 Its sophisticated architecture features a bridging δ-lactone and a γ-lactone, in proximity with four other hydroxylated sites.
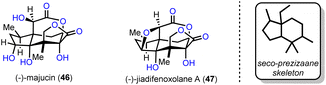 |
| | Scheme 4 Seco-prezizaane-type Illicium sesquiterpenoids. | |
The elegant synthetic endeavor was based on the easily accessible (+)-cedrol (48) as the feedstock material, which was subjected to Suarez photochemical conditions to activate selectively the equatorial methyl in the nearby gem-dimethyl moiety (intermediate 54, Scheme 5). The observed selectivity was attributed to the favorable positioning of the C-14 C–H bond with respect to the hydroxyl group, due to the favorable entropic factor of a 1,5-hydrogen atom transfer (1,5-HAT) compared to a 1,6-HAT event that implies the activation of hydrogen at C-4. Opening of the tetrahydrofuranyl ring in 54 led to alkene 49 in 67% combined yield for the two successive transformations. Working on alkene 49, in another two trivial chemical steps, alcohol 50, with a stereo-defined hydroxyl substituent on the six-membered ring, was obtained. The latter was a suitable substrate for a second Suarez oxidation event to selectively oxidize the C-4 position with impressive efficiency (93% yield) (Scheme 5). Again, the observed selectivity can be explained by the advantage of the 1,5-HAT event compared to the 1,6 one. To capitalize on this entropic difference between the two hydrogen transfers, the selection of the protection group on the C-14 hydroxyl group proved pivotal. As smoothly illustrated by Maimone's group in a subsequent report,20 protection of the C-14 hydroxyl group by a methyl ether resulted in a reversal of the preference due to the substantial weakening of the C–H bond in the α-position to an ether, compared to that of an ester. Additional oxidations including a ruthenium catalyzed scission of the six-membered ring led to intermediate 52, and then to lactone 53, after selective hydroxylation with DMDO and adjustment of the oxidation state of the molecule. Further delicate manipulations, involving, among others, stereoselective enolate hydroxylation, ruthenium catalyzed free alcohol isomerization and conventional OsO4 catalyzed dihydroxylation, led to (−)-majucin (46). (−)-Jiadifenoxolane A (47) was accessed from the latter, after careful activation of the less hindered secondary alcohol and intramolecular displacement by the α-hydroxyl group of δ-lactone in 92% yield.
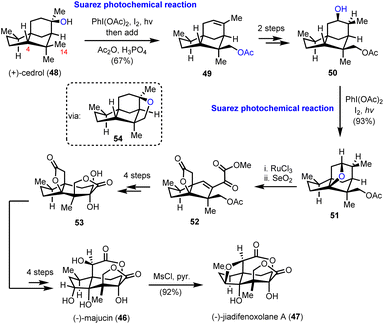 |
| | Scheme 5 Multiple oxidative manipulations to illicium sesquiterpenoids 46 and 47 (Maimone's group). | |
2.4. Seco-prezizaane type sesquiterpenes (Gademann's group)
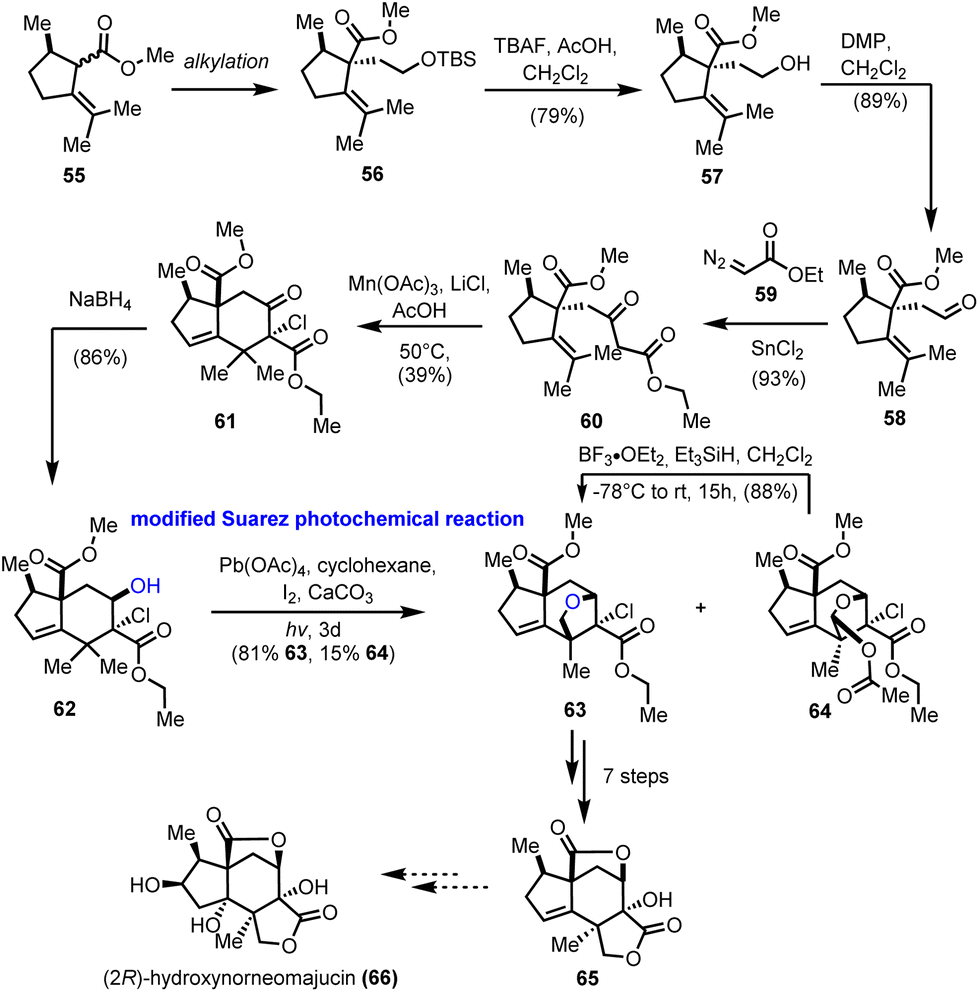
The seco-prezizaane sesquiterpenes have been the center of interest for many other synthetic endeavors. At around the same time as Maimone's group, the work of Gademann and his co-workers on the preparation of the main scaffold of seco-prezizaanes was presented (Scheme 6).21 Starting from enantiopure (R)-pulegone, intermediate 55 was obtained as a mixture of diastereomers by a known procedure.22 Alkylation of the latter, followed by typical FGI and Roskamp reaction23 led to enantiopure β-ketoester 60 in a high overall yield. Subsequent free radical cyclization to alkene 61 was realized with moderate efficiency using Mn(OAc)3 in the presence of LiCl. Diastereoselective reduction of the ketone functionality provided the desired stereoisomer of alcohol 62, bringing the hydroxyl group in proximity to one of the nearby methyl groups on the six-membered ring. At this point, the ground for a successful Suarez oxidation was laid, a transformation that was realized in excellent yield 81%, using modified conditions (Pb(OAc)4) for the generation of the initial O-centered radical.24 Along with the desired ether 63, an overoxidized protected acetal 64 was formed in 15% yield, which could, ideally, be converted reductively to ether 63, thus increasing the overall efficiency of the reaction. Applying conventional chemical transformations, compound 63 was converted to the advanced intermediate 65, which is regarded as a common precursor to various members of the seco-prezizaane sesquiterpene family. In particular, the authors suggested certain modifications on the five-membered ring of 65 to gain access to (2R)-hydroxynorneomajucin (66); however, no experimental proof has been provided.
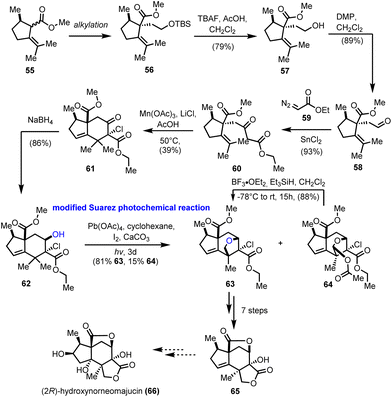 |
| | Scheme 6 Synthesis of core structure of seco-prezizaane-type sesquiterpenoids (Gademann's group). | |
3. Metal catalyzed C–H oxidations
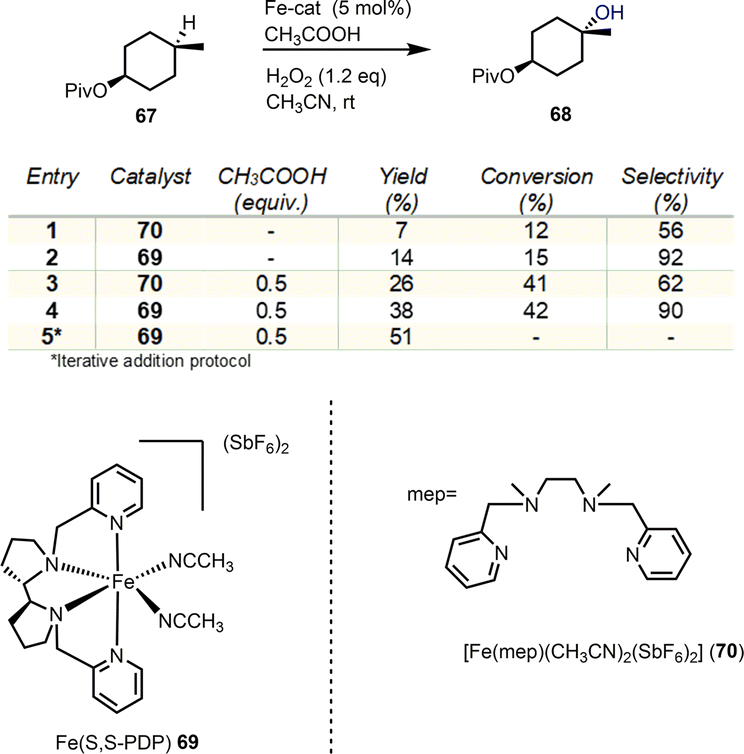
Selective oxidation of C–H bonds is a major challenge in organic synthesis. The multitude of applications and selectivity provided by natural oxygenases, especially in the cyclase phase of terpenoids, has been the determinant for the development of biomimetics. Non-heme iron and manganese catalysts bearing tetradentate N4 ligands are of exceptional importance as biomimetics allowing the efficient, stereospecific hydroxylation of C–H bonds by utilizing H2O2 as the oxidant in the process.25 The mechanisms of these processes collectively follow the heme-iron mechanism (see Fig. 3A), utilizing high valency metal-oxo species to perform hydrogen abstraction from the hydrocarbon with the incipient alkyl radical to rebound with the nascent Fe–OH moiety to provide the hydroxylated product.26 In 2007, a seminal report by White's group described the non-heme iron complex 69 of the tetradentate PDP ligand as an efficient, selective and most importantly predictable catalyst for the hydroxylation of hydrocarbons (Scheme 7).27
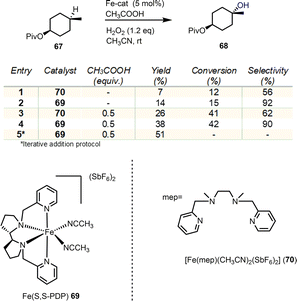 |
| | Scheme 7 Seminal report by White's group for the preparative C–H oxidation of alkanes. | |
In their report, they demonstrate that the electronic and steric properties of C–H bonds can effectively provide prediction for the sites of oxidation, regarding any directing groups. Later, Baran and Eschenmoser added the strain release in the transition state formation of the C–O bond as an effective factor for C–H oxidation prediction.15 Additionally, under this catalytic protocol, White's group demonstrated the utilization of carboxyl groups as directing groups to allow the predictable formation of lactones under mild conditions. Based on these findings, several similar catalysts have been developed utilizing the same electrophilicity logic to direct oxidations in more nucleophilic, less sterically hindered positions. In all cases, equatorial hydrogens are more amenable for abstraction and C–H activation due to their enhanced strain release in the transition state formation.
3.1. Illisimonin A (Rychnovsky's group)
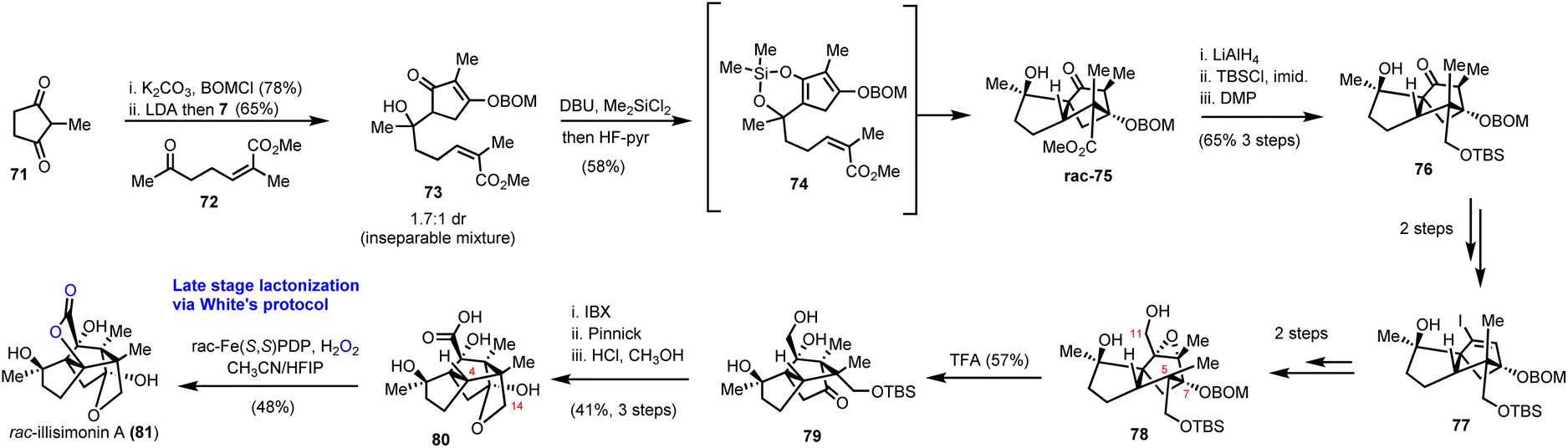
Illicium sesquiterpenoids are common templates in modern organic synthesis and challenge chemists’ creativity. Illisimonin A is a member of the class isolated from Illicium simonsii with an unprecedented tricyclo[5.2.1.01.6] decane carbon framework.28,29 Illisimonin A displays neuroprotective effects against oxygen-glycose deprivation-induced cell injury in SH-SY5Y cells.28 The first total synthesis of illisimonin A, reported by Rychnovsky's group, relied on a highly diastereoselective intramolecular Diels–Alder (IMDA) reaction for the synthesis of the congested carbocycle 75 from the in situ prepared silacycle 74 (Scheme 8).28 The latter was readily prepared from an aldol reaction between protected 71 and 72 followed by silylation. The silacycle which stabilizes the enol-diene was warmed to 40 °C to initiate the IMDA reaction producing the five stereocenters and two rings of 75 in 58% yield in a single synthetic step. Functional group interconversions (FGI) followed by introduction of C-11 and epoxide formation led to compound 78. An impressive semipinacol rearrangement follows after acidic deprotection to produce 79 bearing the correct trans-pentalene core for the natural product. Sequential oxidation of the primary alcohol to the acid followed by deprotection led to 80. At this point, a lactone moiety at C-4 was missing to address the natural complexity. This especially difficult challenge was addressed in 48% yield by late stage functionalization utilizing White's catalyst Fe(S,S)(PDP) 69 with the aid of hydrogen peroxide as the terminal oxidant of the process. The reaction was performed in an acetonitrile/HFIP mixture to avoid the overoxidation to lactol at C-14 through hydrogen bonding deactivation of the alcohol by HFIP.
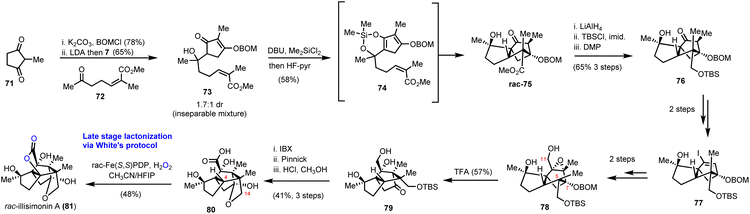 |
| | Scheme 8 Total synthesis of illisimonin A (Rychnovsky's group). | |
3.2. Gochnatiolides A–C and ainsliadimer B (Lei's group)
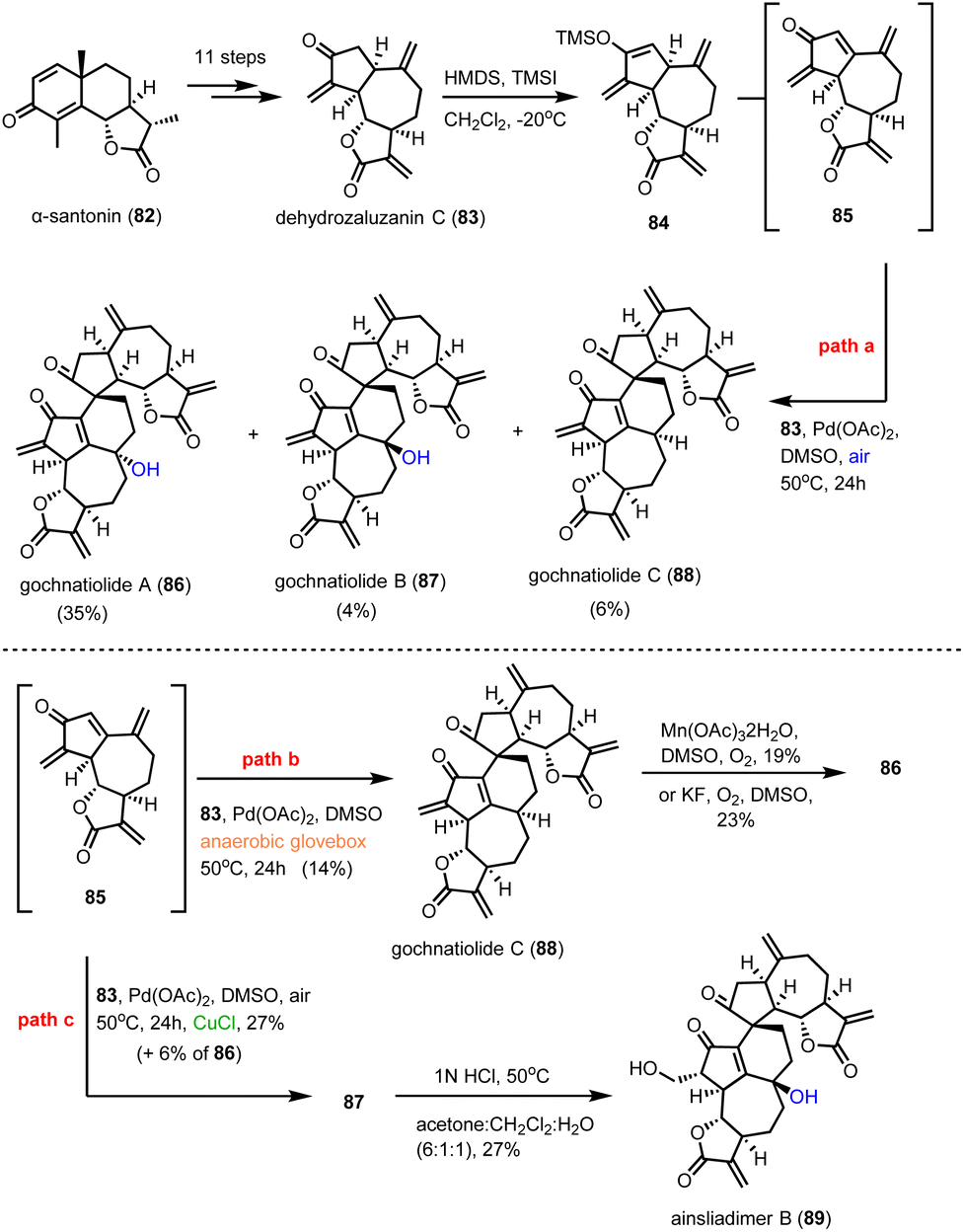
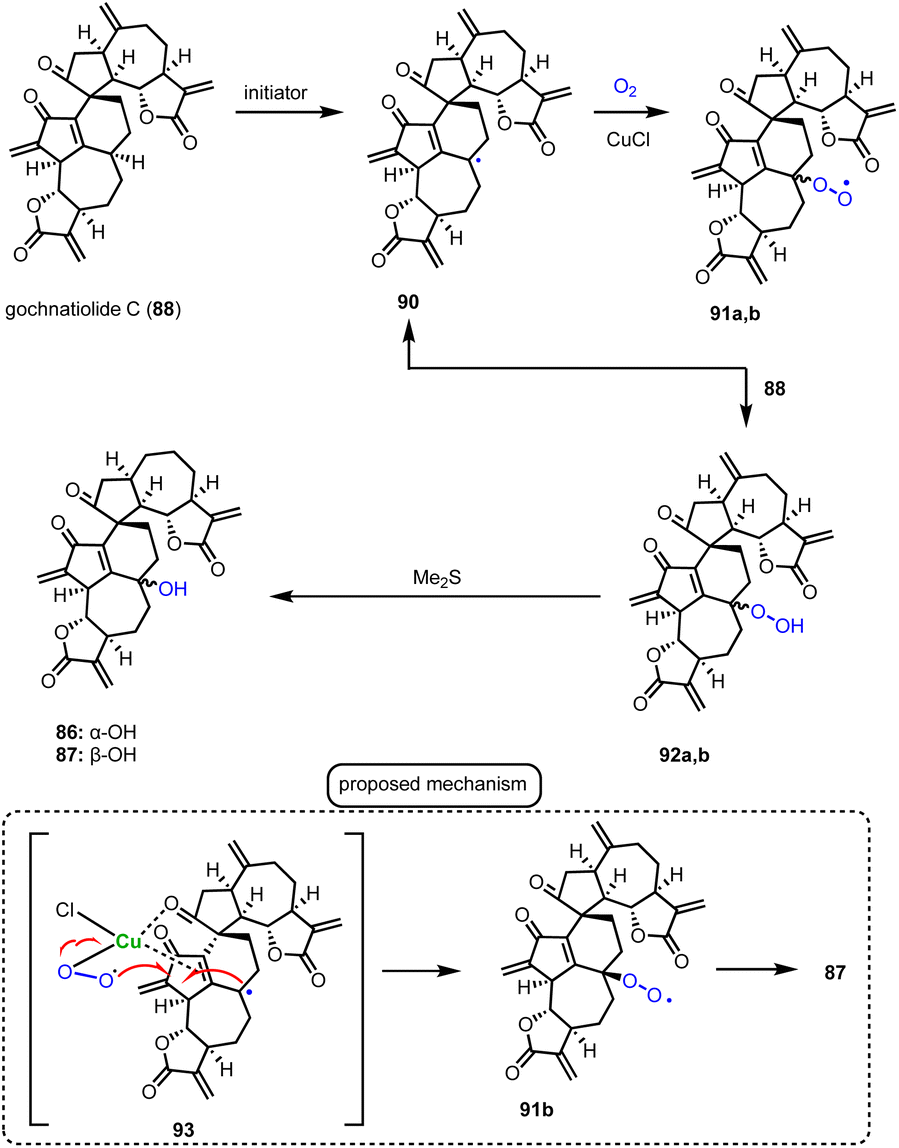
Guainolide dimers represent a highly complex class of sesquiterpenoid natural products. Members of this class exhibit potent biological profiles as anticancer, anti-inflammatory and anti-HIV agents.30 Gochnatiolide A–C (86–88) and ainsliadimer B (89) are characteristic natural products of this class and despite the rather obvious Diels–Alder reaction as the origin of their dimerization, significant challenges still exist in exploring their biomimetic routes (Scheme 9). In 2012, Lei's group developed a biomimetic divergent plan for their synthesis starting from the proposed natural monomeric congener 83 (Scheme 9).31 The monomer was previously synthesized in an 11-step sequence starting from α-santonin.32 Based on previous studies of the same group demonstrating the BINOL-promoted homodimerization of 83 to (+)-ainsliadimer A,32 they explored the feasibility of a biomimetic cross-coupling between 83 and its oxidized congener 85, formed by the Saegusa oxidation. In situ preparation of 85 from silyl enol 84 with Pd(OAc)2 in DMSO and air in the presence of an excess of 83 to avoid the homodimerization of 85 was selected, to provide 86–88 in 35%, 4% and 6% yields, respectively. Interestingly, strict exclusion of dioxygen from the reaction mixture resulted only in the formation of 88 in 14% yield, suggesting the participation of dioxygen for the late stage allylic oxidation event. Indeed, when 88 was allowed to react under radical conditions either with Mn(OAc)3 or KF in the presence of dioxygen it led to the selective formation of 86 in 19% and 23% yields, respectively. On the other hand, when the oxidation of 85 was performed in the presence of a catalytic amount of CuCl2, a preference for the formation of gochnatiolide B was observed delivering 87 in 27% yield and reversed ratio of products 86:87 from 6.6:1 (KF) to 1:4.5 (CuCl2). For the latter, a monomeric Cu(II)-peroxo-species was proposed to interact with carbonyl at C-3′ to deliver selectively the hydroperoxy-group from the β-face (see transition state 93) (Scheme 10). Finally, hydration of 87 by 1 N HCl in a 6:1:1 acetone/DCM/H2O solvent mixture allowed the synthesis of ainsliadimer B (89) in 27% yield.
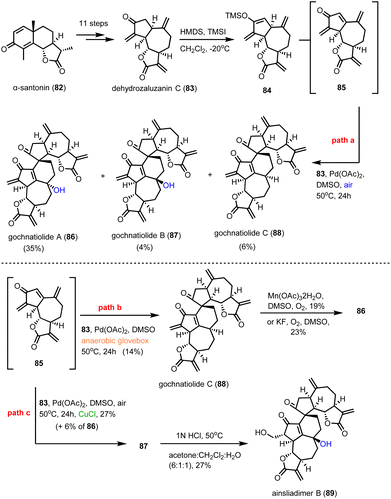 |
| | Scheme 9 Biomimetic synthesis of gochnatiolides (Lei's group). | |
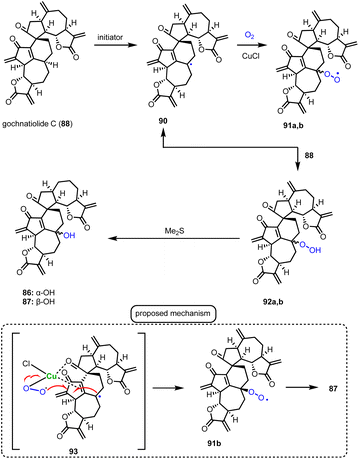 |
| | Scheme 10 Postulated mechanism for aerobic oxidation. | |
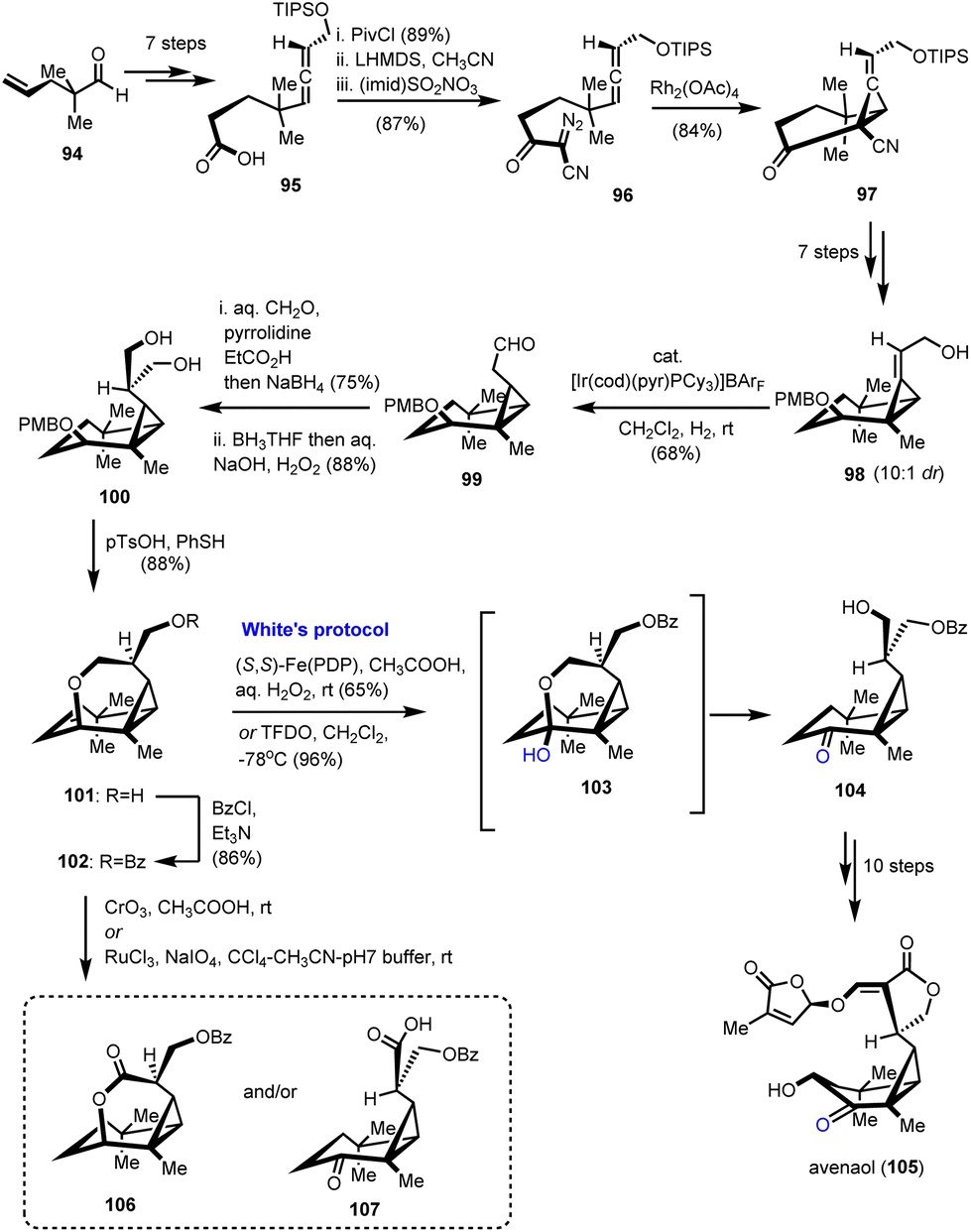
3.3. Total synthesis of avenaol (Takemoto's group)
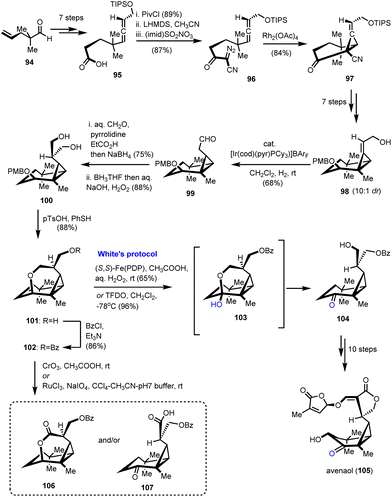
Strigolactones are sesquiterpenoid plant hormones that play a pivotal role in the response of plants to various biotic and abiotic stresses.33 Avenaol (105) which was isolated from the allelopathic plant black oat is the first C20 germination stimulant that was found to be related to strigolactones (Scheme 11).34 As part of the extreme synthetic challenge of the target, Takemoto's group utilized C–H oxidation as a way to differentiate similar hydroxyl moieties in their synthetic endeavour.35 According to them, avenaol possessing a challenging all-cis-cyclopropane ring and six stereogenic centers was derived from the substituted diazo-allene compound 96 through rhodium-catalyzed carbene cyclopropanation (Scheme 11). This method was selected on the basis of all-cis-cyclopropane sensitivity to decomposition and easy formation of caged-type compounds. Precursor 96 was prepared from 95, which in turn was delivered in a 7-step sequence from a known aldehyde 94. Reaction of 95 to the mixed anhydride followed by the nucleophilic attack by acetonitrile resulted in β-cyanoketone amenable for diazo-introduction. The latter was achieved using the imidazole-1-sulfonyl-azide hydrochloride diazo-transfer reagent to provide compound 96. Cyclopropanation with the aid of Rh2(OAc)4 followed to deliver 97 in 84% yield. Attempts to prepare the all-cis cyclopropane by direct reduction of the alkene moiety failed and the authors appropriately transformed 97 to 98 to succeed in their goal by hydroxyl-directed iridium-catalyzed isomerization of cyclopropyl alkene to compound 99. With all-cis cyclopropane in hand, the next challenge was to install the lactone ring. Further FGI led to the introduction of the hydroxymethyl group to the aldehyde to form compound 100. The diol functionalization using 100 was envisioned to give the ideal precursor of the lactone moiety if the differentiation of the hydroxyl groups were feasible. To address this challenge, the pendant hydroxyl was cyclized to the six-member ring of 101 followed by a subsequent protection of the free hydroxyl to 102. A C–H oxidation was designed to cleave the pyrane ring back to the free alcohol. Several C–H oxidations were tried to deliver in most cases byproducts 106 and 107. Interestingly, stoichiometric utilization of White's catalyst in the presence of hydrogen peroxide resulted in the selective formation of 104 in 65% yield. Further improvement of reaction yield using TFDO led to 104 in 96% yield. Ten reactions that followed completed the first total synthesis of avenaol (105).
 |
| | Scheme 11 Total synthesis of avenaol by C–H assisted desymmetrization of a diol (Takemoto's group). | |
4. Mukaiyama hydration
A common way to circumvent the challenging C–H oxidation in unsaturation-rich substrates such as sesquiterpenoids is by the direct oxidation of alkene moieties. Epoxidations and hydrations are part of the synthetic routine in terpenoid total synthesis. Sharpless epoxidation3 and Brown hydroboration4 provide reliable access to oxidized compounds but in some cases, further synthetic steps are needed to introduce the requisite hydroxyl functionality. Direct functionalizations allow for simpler retrosyntheses avoiding the protection–deprotection stages. Toward this end, use and advancement of radical hydrations was recently reconsidered in complex molecular settings. Radical Markovnikov additions, based on the seminal reports of the Mukaiyama hydration protocol,36 provide a reliable access to the hydroxyl functionality. The method relies on the formation of metal hydrides (usually Co(II)) for initial alkene hydrometallation followed by dioxygen oxidation. The method has inspired several approaches for hydrogen atom transfer-initiated C–C formation,37 alkene transposition38 and multi-oxidation processes.
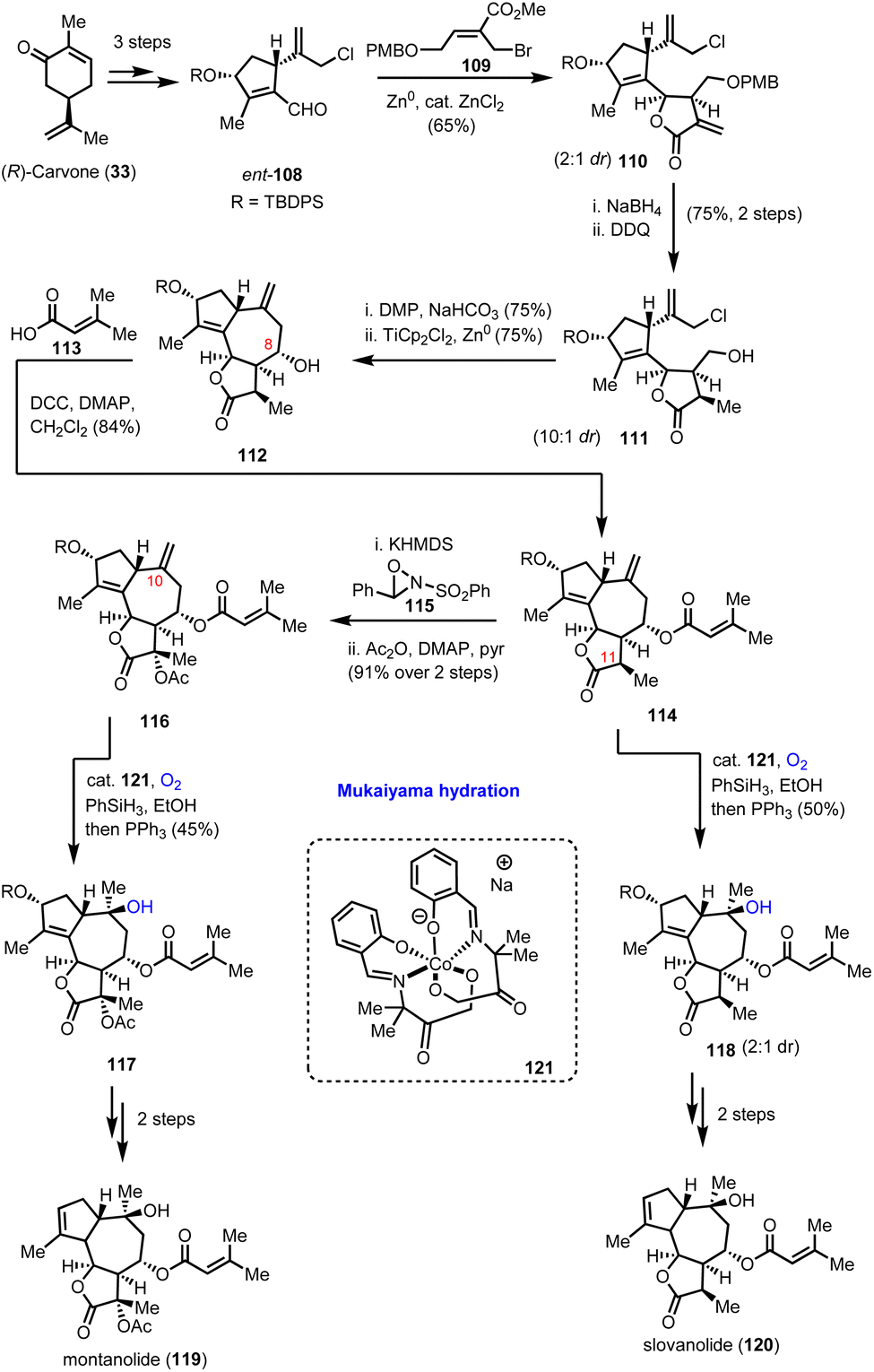
4.1. Montanolide and slovanolide (Maimone's group)
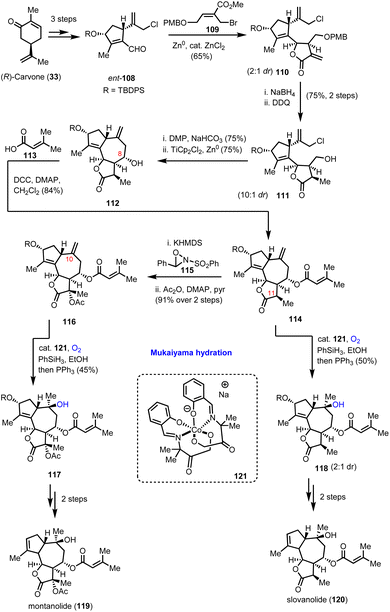
In 2019, Maimone's group reported a series of total synthesis of complex guaianolides from Apiaceae and Asteraceae families.39 The divergent synthetic routes heavily relied on radical Mukaiyama-type chemistry for the introduction of their ‘oxidase-phase’ complexity. Synthesis of highly functionalized cyclopentene 108 and its enantioisomer ent-108 from carvone enantiomers in gram scale quantities in 3 synthetic steps served as the starting point of their divergent plan (Scheme 12). A double reductive allylation protocol was found amenable for accessing the rich stereoselectivity possessed by guaianolide cores in both Asteraceae and Apiaceae families by just altering the allylation conditions between aldehydes 108 and appropriately substituted allylbromides of type-109. In Apiaceae, Zn0-mediated allylation was selected followed by ZnCl2 to provide 65% yield of 110 as a 2:1 diastereoisomeric mixture favouring the desired stereochemistry for the synthesis of slovanolide (120) and montanolide (119) natural products (Scheme 12). The presence of catalytic ZnCl2 was found essential to promote cyclization to the requisite lactone. Alkene reduction, followed by deprotection of the PMB group and subsequent oxidation set the stage for the second reductive allylation event which was accomplished by TiCp2Cl2 and Zn0 to result in compound 112. Extensive experimentation was required to develop the appropriate conditions to achieve the correct stereochemistry at C-8 in 75% yield and 10:1 diastereoisomeric ratio. Acylation provided the complete carbocycle 114 for both natural products, where the oxidative decoration is partially missing. To address it, the group selected Mukaiyama hydration for the hydroxylation of the C-10 position, utilizing catalyst 121 in the presence of PhSiH3 under dioxygen. Compound 118 was delivered in 50% yield and 2:1 diastereoisomeric ratio. Subsequent deprotection of the secondary alcohol followed by reductive transposition of the alkene furnished slovanolide (120). The more oxidized congener montanolide (119) was achieved under similar conditions after the introduction of the acetyl functionality at C-11 with Davis oxaziridine.
 |
| | Scheme 12 Total syntheses of montanolide and slovanolide guaianolides which relied on Mukaiyama hydration (Maimone's group). | |
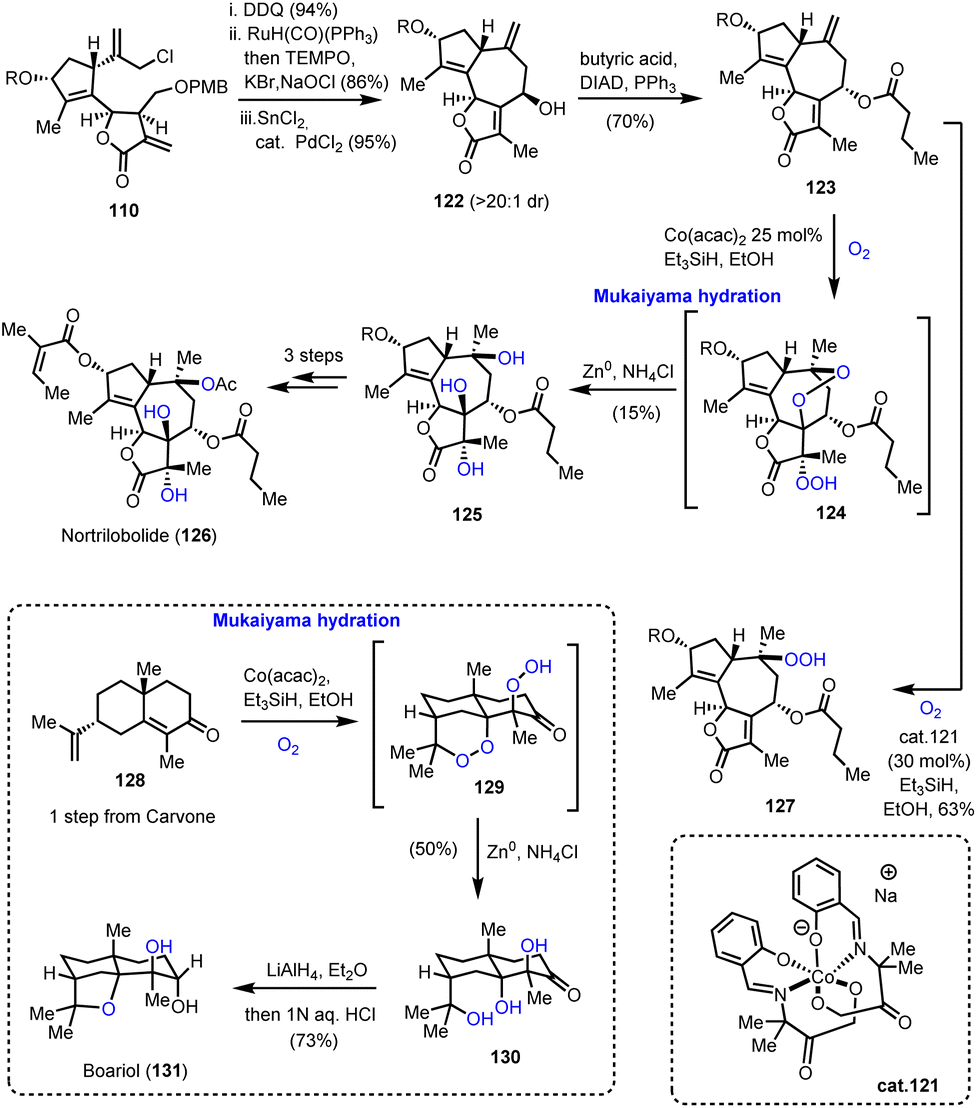
4.2. Notrilobolide, boariol (Maimone's group)
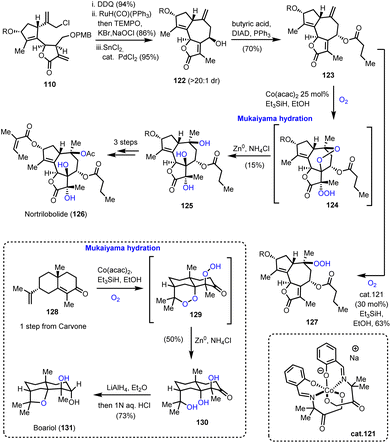
Utilizing the same common scaffold 110, Maimone's group also succeeded in the synthesis of nortrilobolide 126 (Scheme 13).39 The exocyclic double bond was isomerized using RuH(CO)(PPh)3 following a similar reductive allylation to furnish 122 in high diastereoselectivity. Mitsunobu epimerization and acylation provided compound 123 which served as an ideal template for a multi-oxidation event producing the correct stereochemistry for three tertiary hydroxyls. In accordance with that, when 123 was allowed to react under the classic Mukaiyama conditions, a cascade was initiated to produce a diperoxy-intermediate 124 which was reductively cleaved to produce 125 as a single diastereoisomer. The event invoked the HAT from the [Co–H] complex to initiate a radical at C-10 followed by dioxygen introduction. The formed superoxide cyclized to C-7 to form a peroxy-bridge and a radical that was trapped by dioxygen. Despite its low yield (15%), this impressive transformation unlocks a potential biosynthetic pathway towards these polyoxidized natural products. Screening of different conditions failed to optimize the yield but provided interesting insights on the reaction mechanism. Interestingly, when cobalt complex 121 was used, compound 127 in 63% yield (Scheme 13) was obtained.
 |
| | Scheme 13 Mukaiyama-type hydration cascade for the synthesis of notrilobolide and boariol (Maimone's group). | |
These insights allowed the three-step total synthesis of boariol (131) from compound 128. Mukaiyama hydration at 128 resulted in compound 130, which after reduction provided the natural product stereoselectively.
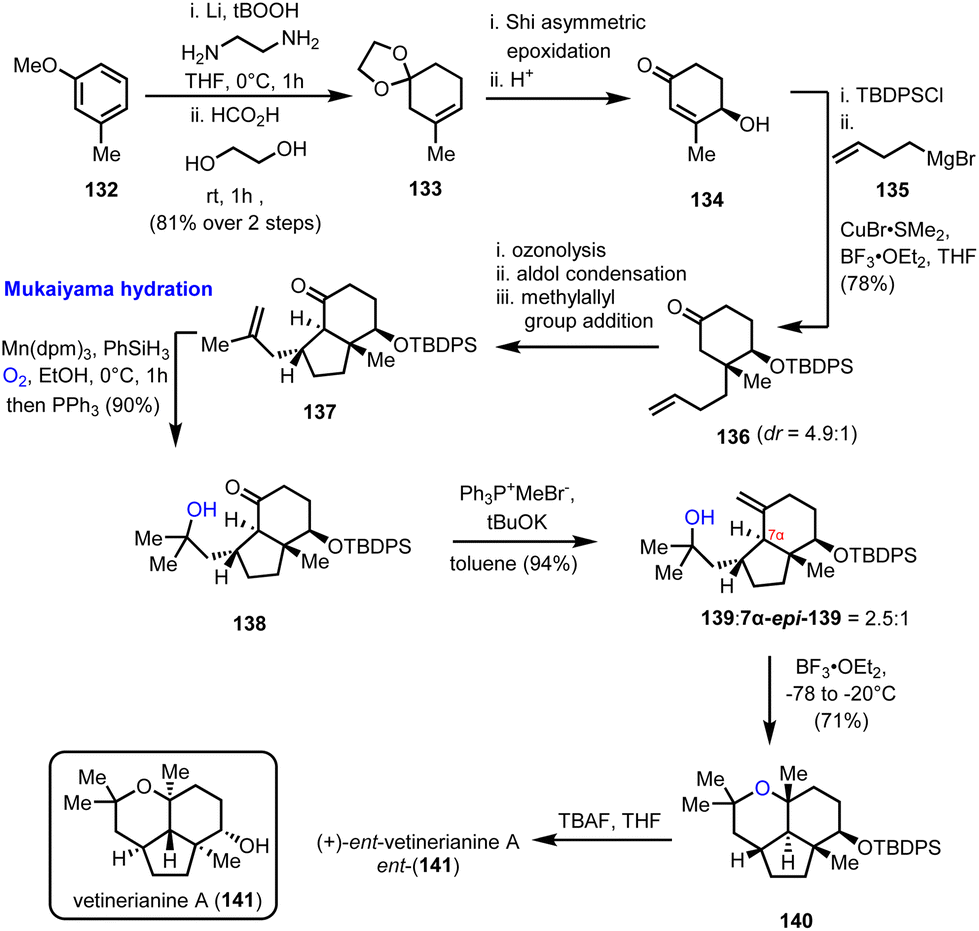
4.3. (+)-ent-Vetiverianine A (Sugita's group)
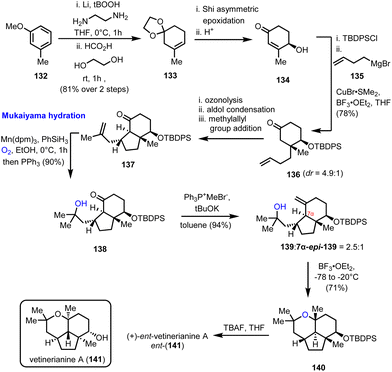
Mukaiyama hydration is the protocol of choice for the formation of a critical advanced intermediate en route to the 2,2,6,6 tetrasubstituted tetrahydropyrane ring found in vetiverianine A (141) (Scheme 14). Vetiverianines are novel sesquiterpenoids isolated in 2016 from vetiver oil, obtained via steam distillation of Vetiveria zizanioides roots, and exhibit cytotoxic activity against HL-60 cells and their full biological profile is still under investigation.40 Their distinctive 5/6/6 tricyclic ring system that encompasses a highly congested tetrahydropyrane makes their total synthesis challenging. The first synthesis of the enantiomer of the natural product (ent-141) to be reported was by Sugita's group in 2022, which made use of the inexpensive 3-methoxytoluene (132) as the starting material (Scheme 14).41 Birch reduction and acetalization led to trisubstituted alkene 133 and from there to enantioenriched cyclohexenone 134, employing a Shi asymmetric epoxidation and acidic treatment of the resulting epoxide. Steric interactions drove the subsequent conjugated addition of γ-butenyl Grignard reagent stereoselectively from the α-face of the protected enone giving ketone 136. Ozonolysis and aldol condensation led to the formation of the second ring of the system, while it also set the stage for a second stereo-controlled conjugated addition of the methylallyl group with the correct relative configuration. The product of this sequence of events, intermediate 137, was the substrate for a Mukaiyama hydration, which was realized in excellent yield (90%), using the sterically hindered Mn(dpm)3 as the catalyst of choice, instead of the conventional Co(acac)3 catalyst, providing access to the tertiary alcohol 139. The critical cyclization to the tetrasubstituted tetrahydropyranyl ring, which completed the synthesis of the enantiomer of the natural product (after deprotection), was accomplished using BF3·OEt2 at a low temperature.
 |
| | Scheme 14 Total synthesis of ent-vetiverianine A (141) (Sugita's group). | |
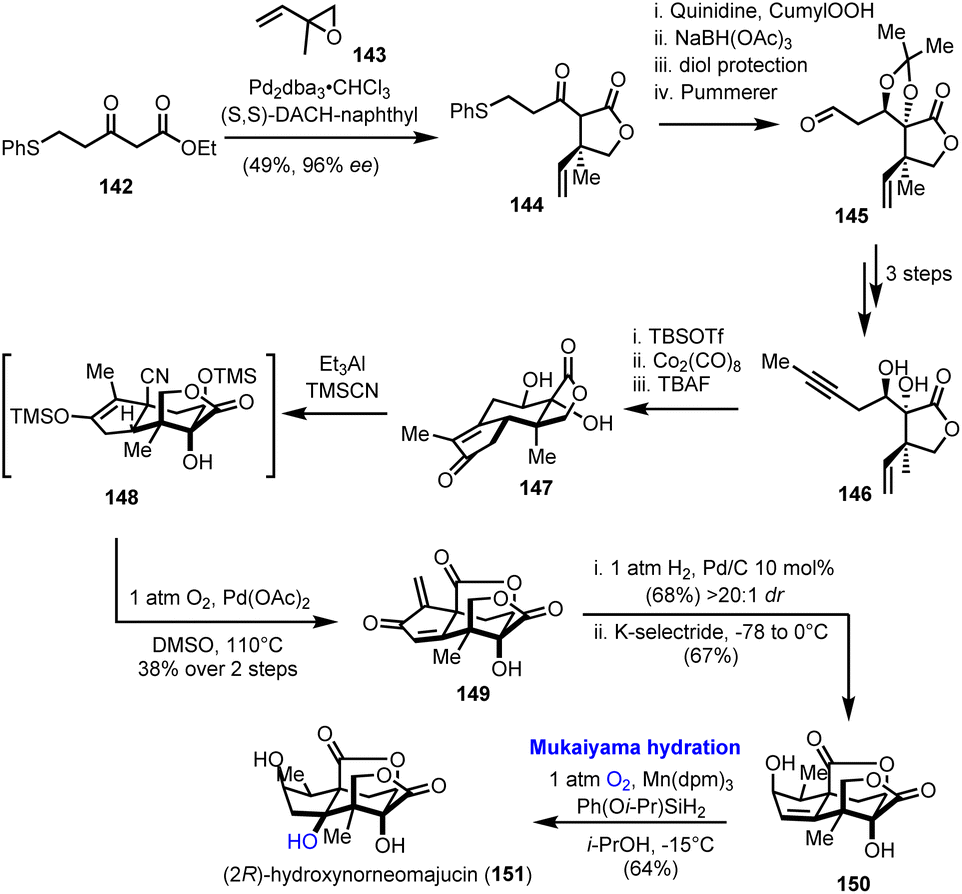
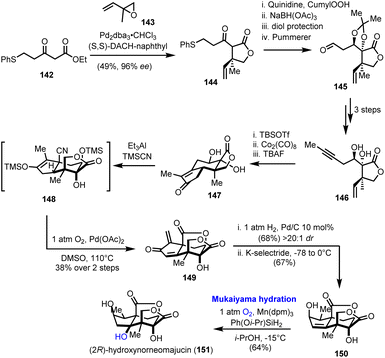
4.4. (2R)-Hydroxynorneomajucin (Rychnovsky's group)
A regio- and stereoselective Mukaiyama hydration was the final step in the first total synthesis of (2R)-hydroxynorneomajucin (151), reported by Rychnovsky's research team in 2022 (Scheme 15).42 The latter sesquiterpene belongs to the seco-prezizaane family of terpenes derived from the Illicium genus of flowering plants. Among other interesting highly oxidized natural products of the same origin, many of which are presented in the context of this review, (2R)-hydroxynorneomajucin holds a special position as its structure remained resistant to chemical synthesis although it had been known for more than ten years.43 Some early studies from Gademann and his co-workers on the seco-prezizaane scaffold, analysed in Section 2.4 of this manuscript, remained incomplete.21
 |
| | Scheme 15 Total synthesis of (2R)-hydroxynorneomajucin (141) (Rychnovsky's group). | |
In Rychnovsky's asymmetric synthesis, which assumes prominent importance for the introduction of chirality, a palladium catalyzed asymmetric allylation of β-ketoester 142 afforded γ-lactone 144 in excellent enantioselectivity (96% ee), using Trost's ligand (Scheme 15).44 A sequence of stereoselective hydroxylation and reduction of ketone, protection and a Pummerer rearrangement led to aldehyde 145 and eventually to alkyne 146, after a Seyferth–Gilbert homologation of aldehyde to alkyne, methylation and acetal cleavage. A Pauson–Khand reaction on protected 146 formulated the tricyclic enone 147. Stereochemical considerations on the manipulation of the five-membered carbocycle in 147 proved quite interesting.
Direct hydrocyanation on the enone moiety resulted in the wrong configuration of the α-methyl group, an issue that was elegantly addressed employing a Segusha–Ito oxidation to exomethylenic ketone 149 and subsequent catalytic hydrogenation to install the methyl group from the desired face of the ring. Stereoselective 1,2-reduction of the produced enone generated the corresponding alcohol leaving behind trisubstituted alkene 150. Despite the increased complexity of the latter, the anticipated Markovnikov hydration of the double bond was performed in satisfactory yield, 64%, and with excellent regio- and stereoselectivity applying Mn(dpm)3/Ph(Oi-Pr)SiH2 as the catalytic system. The role of the selected reductant was essential for the successful outcome of the conversion and the completion of the synthesis.
5. Stoichiometric metal oxidants
The ability of certain non-precious metals to trigger radical generation upon organic molecules has offered valuable solutions for the synthetic community, especially, those involved in the synthesis of terpene natural products. Mn and Cr, in various oxidation states, hold a special position, and their reagents have proved to be unique in succeeding where other protocols have failed. Despite their high toxicity, mainly of the Cr(VI) based oxidants, their stoichiometric use in various synthetic endeavors appeared indispensable, as it is clearly demonstrated in the following examples.
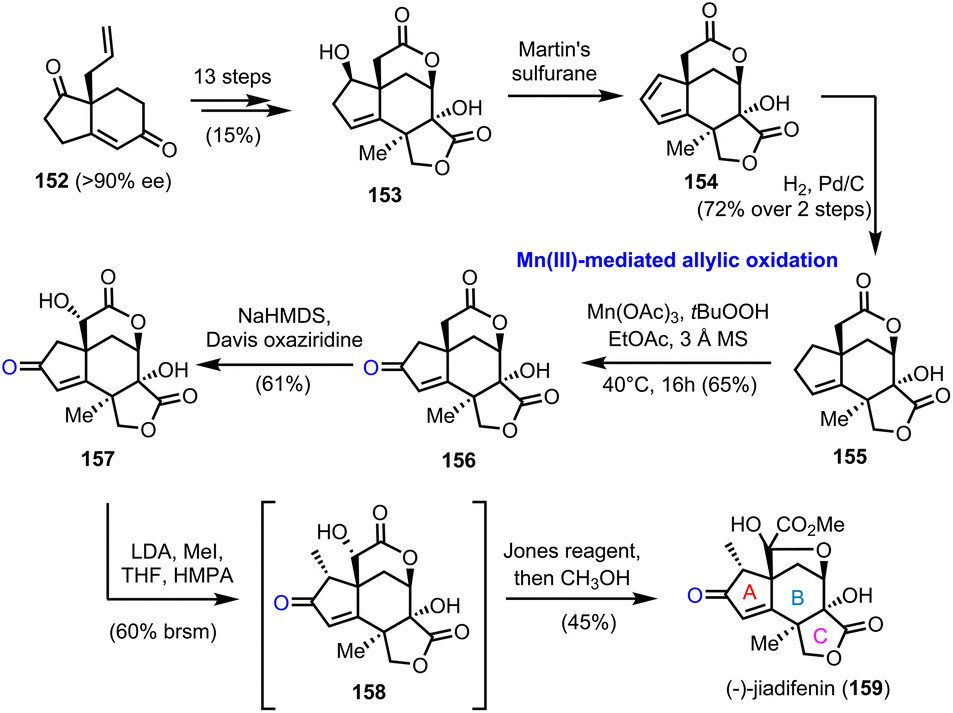
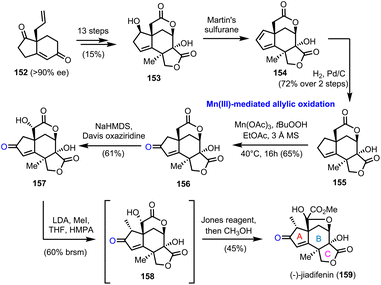
5.1. (−)-Jiadifenine (Theodorakis’ group)
In 2011 Theodorakis’ group reported the first enantioselective route to (−)-jiadifenin (159) (Scheme 16), a potent neurotrophic agent isolated from Illicium jiadifengpi.45 Their synthetic approach was based on an advanced intermediate, tetracyclic lactone 153, which was prepared in the context of jiadifenolide's (not shown) asymmetric synthesis, especially designed to offer diversification to other members of the family.46 Compound 153 was prepared in 13 steps and 15% overall yield from bicyclic “Hajos-Parrish-like” diketone 152, accessed by an asymmetric Robinson annulation in 90% ee (Scheme 16). Martin's sulfurane dehydration and selective hydrogenation of the more accessible double bond of the resulting diene afforded lactone 155, the allylic oxidation of which was critical for the appropriate decoration of the A ring of the natural product. Several conditions were tested, including SeO2, Cr(VI) based oxidants, hypervalent iodine reagents or transition metal catalysis (Pd and Rh), all of which were unable to bring about the desired oxidation. Gratefully, Mn(III) acetate proved to be unique for the selective activation of the allylic position and oxidized it in the presence of TBHP, providing enone 156 in 65% yield. The latter was converted to 159 after a regio- and stereoselective hydroxylation of the α-lactone position, which is more prone to enolization, methylation of the α-enone and Jones oxidation.
 |
| | Scheme 16 First asymmetric synthesis of (−)-jiadifenin (159) (Theodorakis’ group). The unique action of Mn(III) based reagents for the allylic oxidation of the A ring. | |
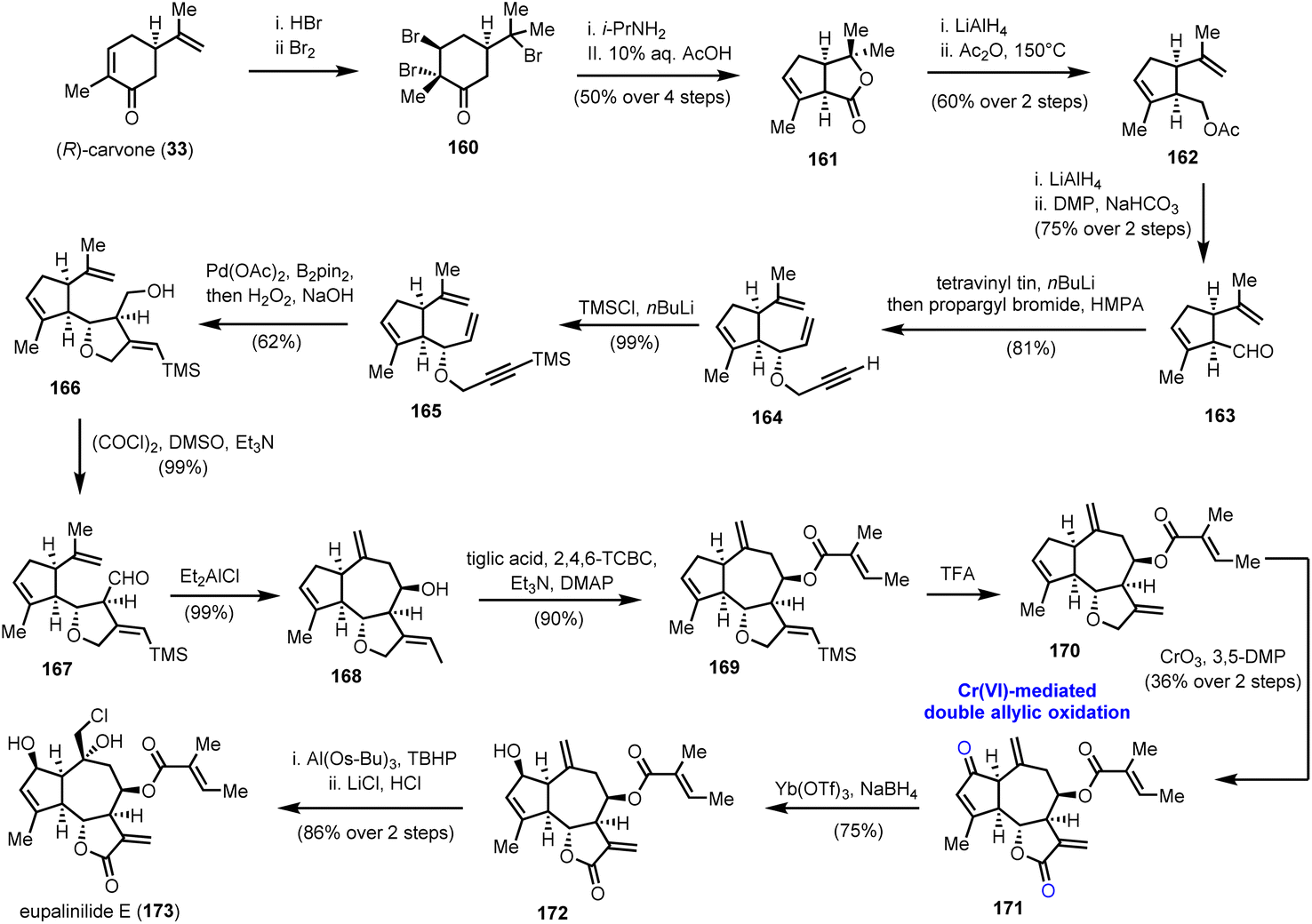
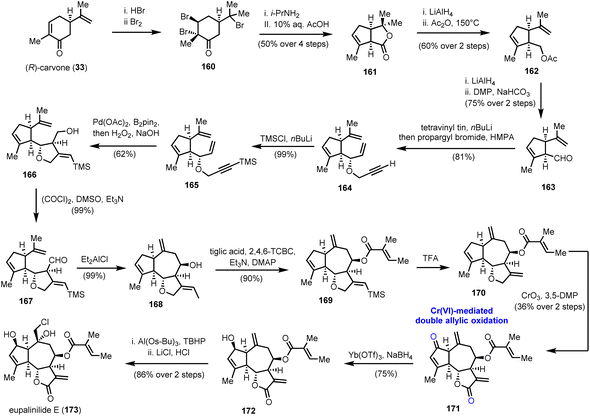
5.2. Eupalinilide E (Siegel's group)
A double allylic oxidation event using the CrO3/3,5-DMP (3,5-dimethylpyrazole) combination was among the key transformations in the enantioselective synthesis of eupalinilide E (173) (Scheme 17), a sesquiterpenoid lactone isolated from Eupatorium lindleyanum.47 The special attention paid to this natural product is attributed to its unique ability to expand hematopoietic stem and progenitor cells (HSPCs) and inhibit differentiation. In fact, eupalinilide E was found to be the most potent agent, among 704 pure natural products of variable origin from Novartis’ natural product collection, screened for that purpose.48 Despite its complex oxidative decoration, its elegant synthesis was accomplished by Siegel and co-workers in 2016,49 providing access to material (>400 mg) for further biological investigations.
 |
| | Scheme 17 Double allylic oxidation of the guaianolide skeleton en route to eupalinilide E (173) (Siege's group). | |
Chemistry wise, the enantioselective construction of the basic guaianolide framework was accomplished in 13 steps from (R)-carvone (33) and 11% overall yield, providing 12 g of the advanced intermediate 168 (Scheme 17). Noteworthy features of this array of transformations are a Favorskii ring contraction, to access substituted cyclopentene 161, and a Pd catalyzed cycloisomerization event on 1,6-enyne 165, followed by transmetallation to boron and oxidation to formulate the five-membered ring that serves as a precursor to the γ-lactone ring of the natural product. Alcohol 166, thus produced, was oxidized to aldehyde 167, which was subjected to a reductive allylation protocol to generate the seven-membered ring and complete the guaianolide skeleton. We have witnessed a similar strategy in Maimone's syntheses of montanolide and slovanolide, discussed earlier in this review.39 With an adequate amount of compound 168 in hand, and after a Yamaguchi esterification and TMS group removal, intermediate 170, the substrate for the highly anticipated double oxidation was prepared (Scheme 17). A thorough investigation of conditions to bring about the said transformation led to the CrO3/3,5-DMP combination as the one to provide an optimal yield (36% over two steps) of enone 171. Importantly, the authors highlight that this late-stage manipulation was crucial for the success of the synthesis due to the incompatibility of the chemistry of the early stages with the carbonyl groups. A separate report by the same team sheds light on previous unsuccessful efforts performed on pre-oxidized substrates.50 From the remaining steps to the completion of the synthesis, it is worthy to comment that the site-selectivity in the epoxidation of 172, where the desired product, is not thermodynamically favoured. The use of a bulky aluminium based Lewis acid was critical to achieve high levels of selectivity in favour of the exomethylenic double bond against the more reactive trisubstituted double bond.
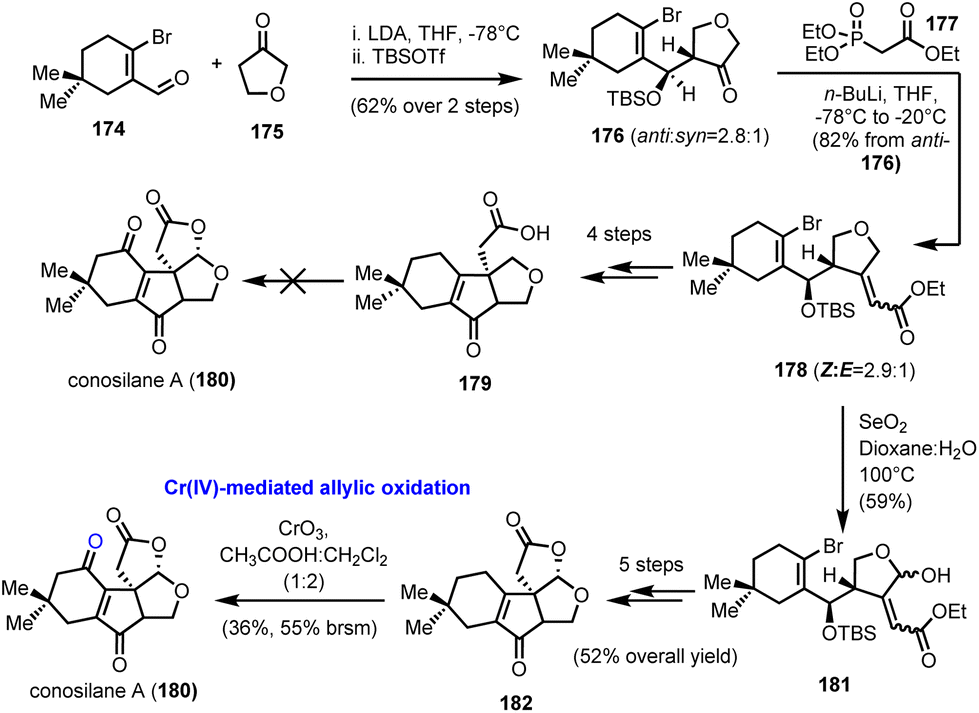
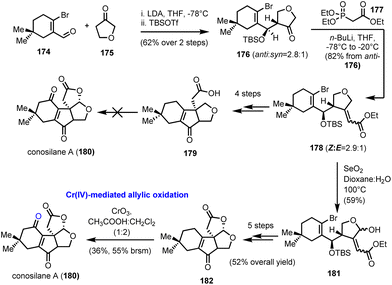
5.3. Conosilane A (Liu's group)
Another success story for the chromium based stoichiometric oxidation of allylic C–H bonds is demonstrated in the racemic synthesis of nonisoprenoid tremulane-type sesquiterpene conosilane A (180, Scheme 18), from the culture of mushroom C. siliginea, reported by Liu and co-workers in 2018.51 In fact, the authors described a preliminary unsuccessful study that relied largely on a final stage lactonization by the activation of a C–H bond in the α-position to an oxygen heteroatom (in Scheme 18, conversion of 179 to 180). Although such a tactic has been effectively applied to numerous cases, including some described previously in this review,16,28 on substrate 179 it did not deliver the desired product. As an alternative, Liu's team selected to facilitate the lactonization step by converting the tetrahydrofuranyl ring into the respective hemiacetal (SeO2 oxidation of common intermediate 178 to hemiacetal 181). After some typical chemical transformations, the natural product precursor 182 was afforded in a good overall yield of 52%. The concluding allylic oxidation of the cyclohexene ring was realized in a moderate yield (36%), using CrO3 in a 1:2 CH3COOH:CH2Cl2 mixture at ambient temperature. Although the conversion seems ineffective, it was the only one that could deliver productive amounts of the natural product, among several other tested, including the “privileged” combination of CrO3 with 3,5-DMP. Transition metal catalysis, as well as direct electrophilic oxidation by DMDO, afforded only a trace amount of conosilane A.
 |
| | Scheme 18 Total synthesis of conosilane A (180) which relied on CrO3/AcOH oxidation (Liu's group). | |
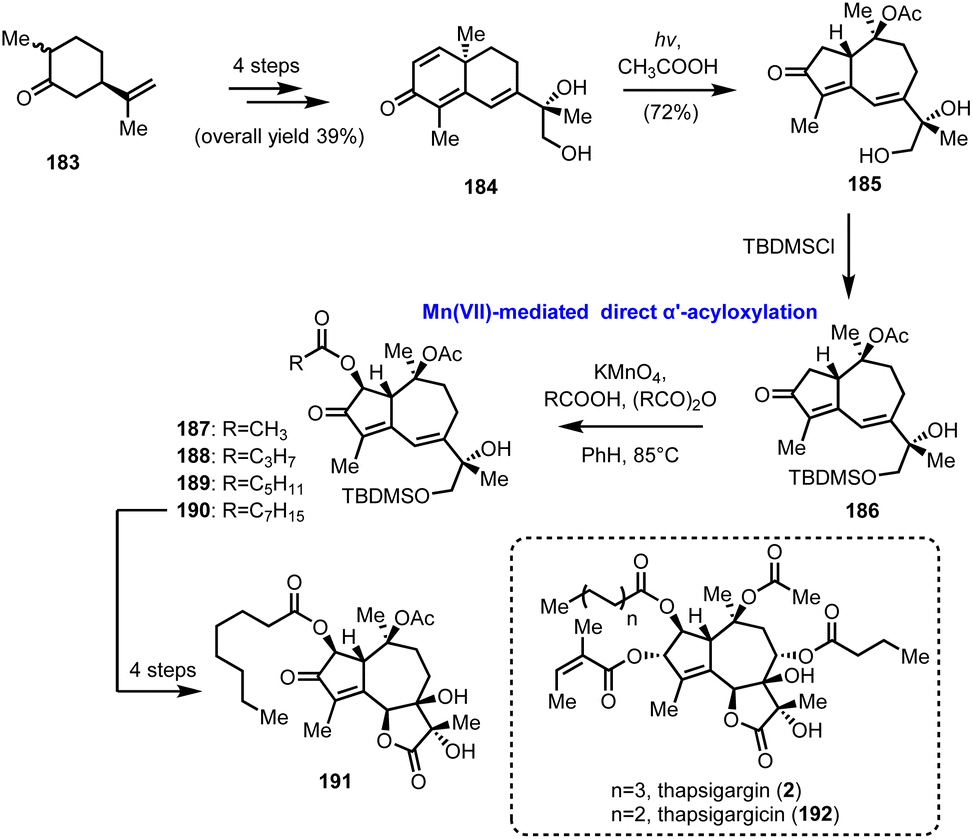
5.4. KMnO4 direct α′-acyloxylation of enones in guaianolides (Moreno-Dorado's and Guerra's groups)
In 1989 Watt and his co-workers reported the Mn(OAc)3 mediated oxidative acyloxylation of enones at the α′-position in the presence of an appropriate carboxylic acid.52 Despite the increased interest in such a transformation, due to the direct access to a structural motif profusely found in biologically important compounds, its establishment as a routinely used synthetic tool never materialized, due, mainly, to reproducibility and practicality issues. Subsequent improvements by Demir et al., where KMnO4 was used for the in situ generation of Mn(OAc)3, showed the way for more controllable and predictable reactivity.53 In 2014 Moreno-Dorado and Guerra published their thorough study on the optimization of the reaction conditions using a computationally aimed design of experiment (DoE) approach, which attests the robustness of the methodology.54 Several experimental variables have been fine-tuned and the optimum reaction conditions were tested on various substrates, including guaianes and guaianolides, the structural precursors of thapsigargin (2) and thapsigargicin (192), the structures of which are shown in Scheme 19. In particular, compound 186 was synthesized in 6 steps and 28% overall yield from partially hydrogenated (R)-carvone 183, utilizing the photochemical rearrangement of “santonine-like” intermediate 184 to guaiane derivative 185 as the key transformation (Scheme 19). Next, the critical α′-acyloxylation was performed achieving good yields (56–98%) with an array of linear carboxylic acids, compounds 187–190, demonstrating the reliability of the method. Finally, advanced intermediate 190 was converted to thapsigargin analogue (191) in 3 additional manipulations, involving silyl group removal, site- and stereo selective dihydroxylation and oxidation of primary alcohol to carboxylic acid followed by spontaneous lactonization.
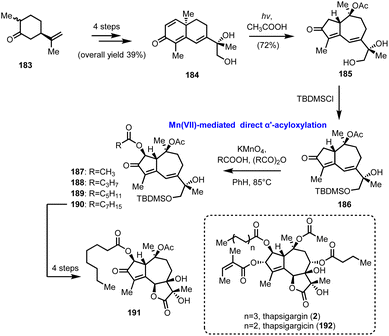 |
| | Scheme 19 Direct α′-acyloxylation of a guaianolide type enone based on the KMnO4/CH3COOH oxidation system (Moreno-Dorado's and Guerra's groups). | |
6. Multi oxidation processes
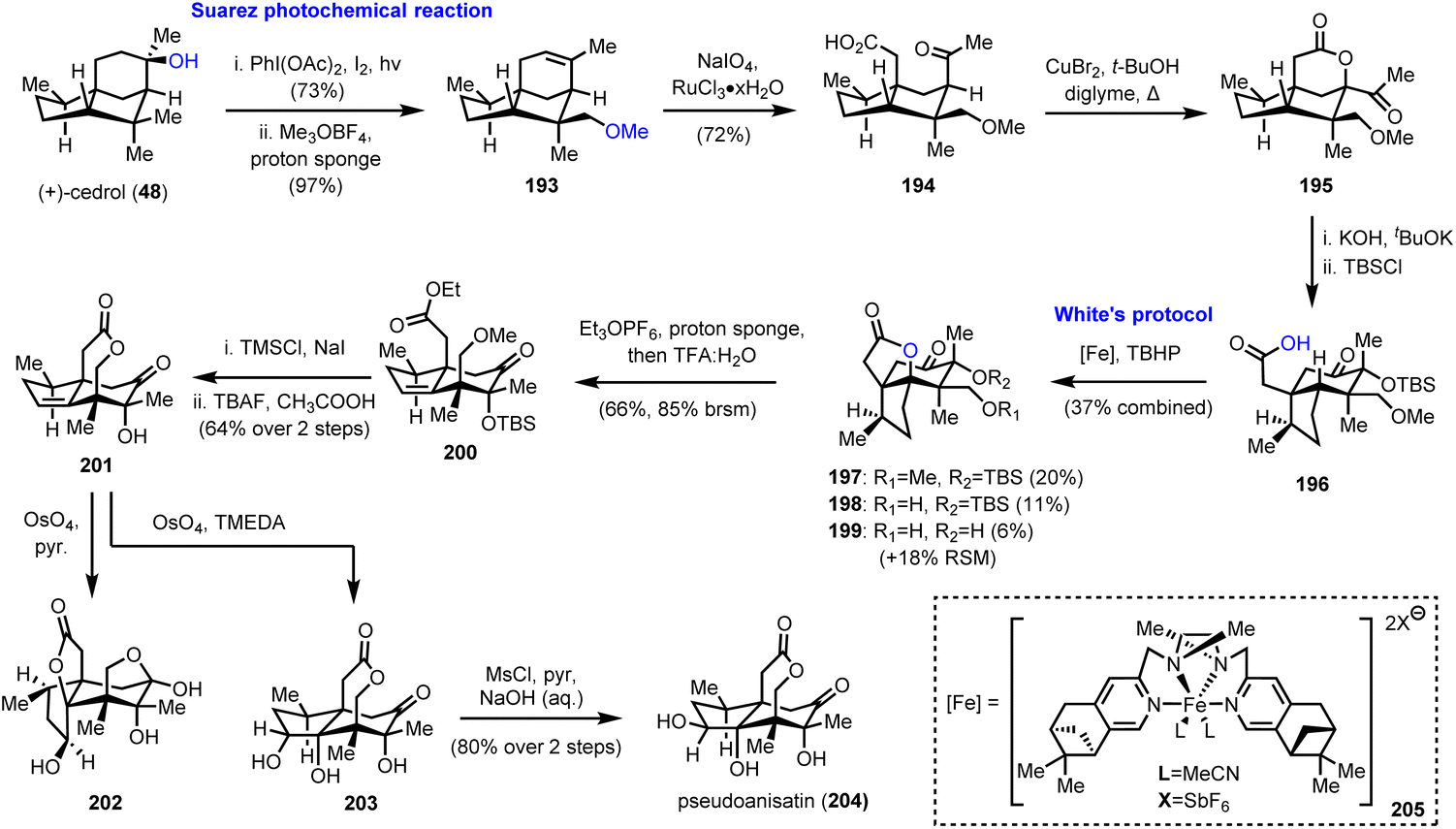
6.1. (+)-Pseudoanisatin (Maimone's group)
As has been easily understood so far, polyhydroxylated seco-prezizaane sesquiterpenoids represent a fertile ground for the exploration of the limits of C–H bond oxidation to act as a direct means to introduce substituent complexity. The case was magnificently demonstrated by Maimone and his group, by the first chemical synthesis of (+)-pseudoanisatin (204, Scheme 20),55 a sesquiterpene of the Illicium family, known since 1968,56 yet never previously synthesized in the lab. His elegant synthetic studies relied largely on radical-based C–H oxidation methods, properly designed to bring about important structural arrangements.
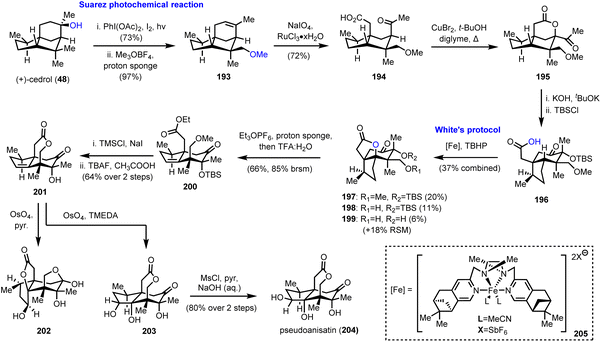 |
| | Scheme 20 Total synthesis of (+)-pseudoanisatin (204) (Maimone's group). | |
In the same vein as the synthesis of (−)-majucin (46), reported by the same team and discussed earlier in Section 2.3 of this review,18 (+)-cedrol (48) was subjected to a Suarez photochemical reaction to selectively oxidize the nearby equatorial methyl group (Scheme 20). Indicative of the power and the robustness of the method is the scale of the reaction that was as large as 50 mmol. Thus, the produced alkene 193 was further elaborated to intermediate 195 by ruthenium catalyzed eruption of the double bond and CuBr2 mediated lactonization at the α-position of methyl ketone 194. A sophisticated base-induced α-ketol rearrangement was used to convert the 5/5 bicyclic system found in cedrol to the desired 5/6 ring system of the Illicium sesquiterpenes; thus carboxylic acid 196 was afforded after silicon-based protection of the hydroxyl group. Preparation of the latter set the stage for the decisive C–H activation of the adjacent tertiary methine group at the bridgehead position. Selectivity issues impose a tremendous challenge in the specific structural setup, and Maimone and his team took up the gauntlet employing nonheme iron catalysis. Building upon existing knowledge from White's57 and other groups’ observations,58 they discovered that a complex of iron with pinene-based mepp ligands (205 see the inset of Scheme 20) could attain decent levels of selectivity, essentially capitalizing on the directing ability of the carboxylic group. Due to partial deprotection of either the silyl or the methyl ether a mixture of usable products 197–199 was obtained in 37% combined yield, along with 18% recovered starting material. Working with the fully protected lactone 197, treatment with Meerwein's type salt and then acidic conditions, alkene 200 was produced whereas unsaturated lactone 201 was afforded after two deprotections and spontaneous lactonization. Interestingly, the attempted dihydroxylation of the latter using OsO4 and pyridine as the base took place from the undesired face of the trisubstituted double bond (compound 202, Scheme 20), revealing certain facial preference. Fortunately, by switching the base selection from pyridine to TMEDA, a strong directing effect of the homoallylic hydroxyl group was operative and capable of overcoming the stereochemical bias, leading to triol 203. Inversion of the stereochemistry of the secondary hydroxyl group by selective mesylation and NaOH hydrolysis completed the synthesis of (+)-pseudoanisatine (204).
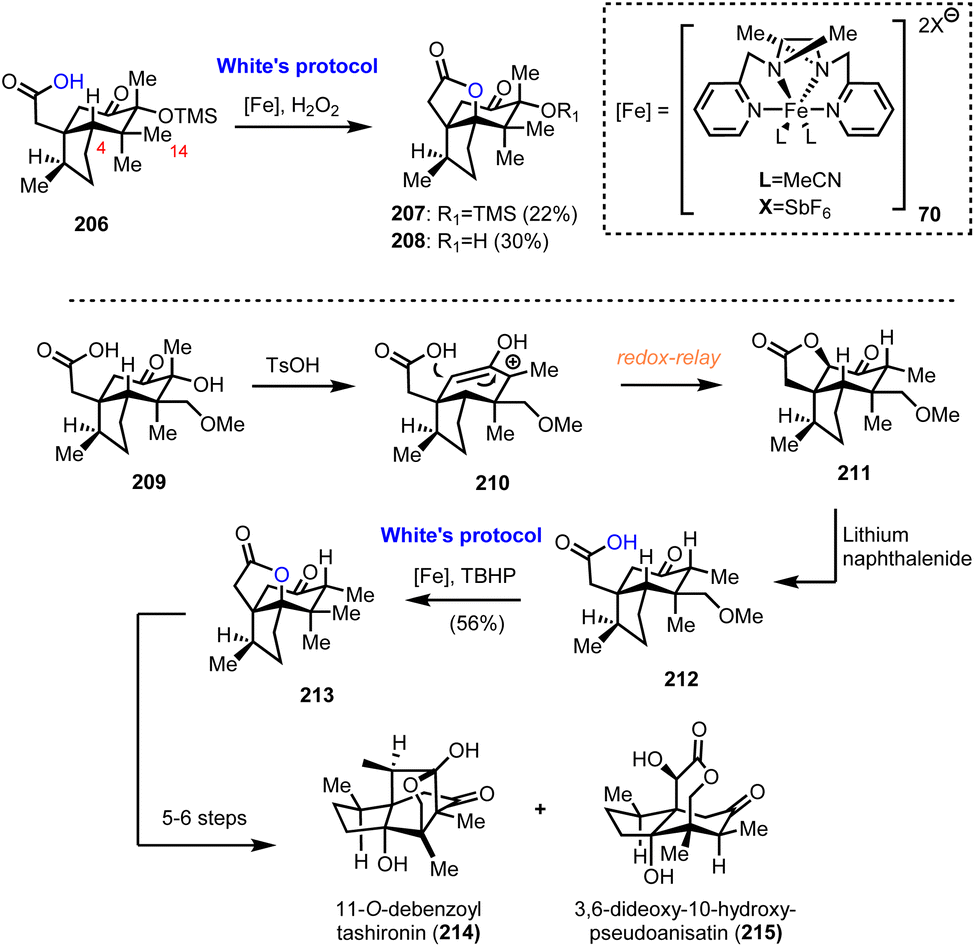
6.2. Collective synthesis of Illicum sesquiterpenoids (Maimone's group)
Numerous interesting observations have emerged during the synthetic studies of pseudoanisatin and related natural products by Maimone's group (Scheme 21). A quite enlightening account, offering fascinating details of C–H oxidation in complex organic settings, have been reported recently.20 In this saga in the field of effective terpene oxidations, (+)-cedrol (48) was the protagonist and Maimone explored all the possible transformations of its framework to access a plethora of scaffolds found within the illicium sesquiterpene family. This effort has provided a wealth of substrates where radical C–H oxidation has been tested. On the way to (+)-pseudoanisatin (204), compounds 206 and 212 were explored as suitable substrates for C–H activation induced lactonization during initial studies (Scheme 21). The former was explored in the context of a C-14 late-stage oxidation strategy and represented the first breakthrough of non-heme iron success in C-4 methine selective C–H activation. Furthermore, while working with the C-14 oxidized substrate 209, a TsOH induced redox-relay cascade was discovered that led to lactone 211 and then to carboxylic acid 212 after lithium naphthalenide reduction. Once again, iron catalysis was effective and provided the respective lactone in 56% yield, thus allowing the access to sesquiterpenes 214 and 215, through a reported sequence.59
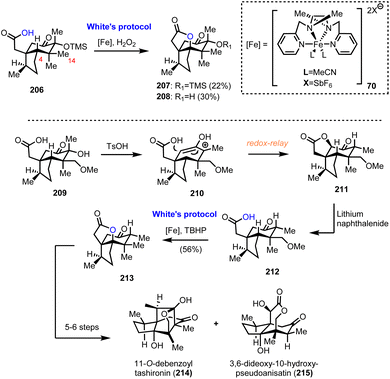 |
| | Scheme 21 Substrates for successful implementation of non-heme iron catalysis (Maimone's group). | |
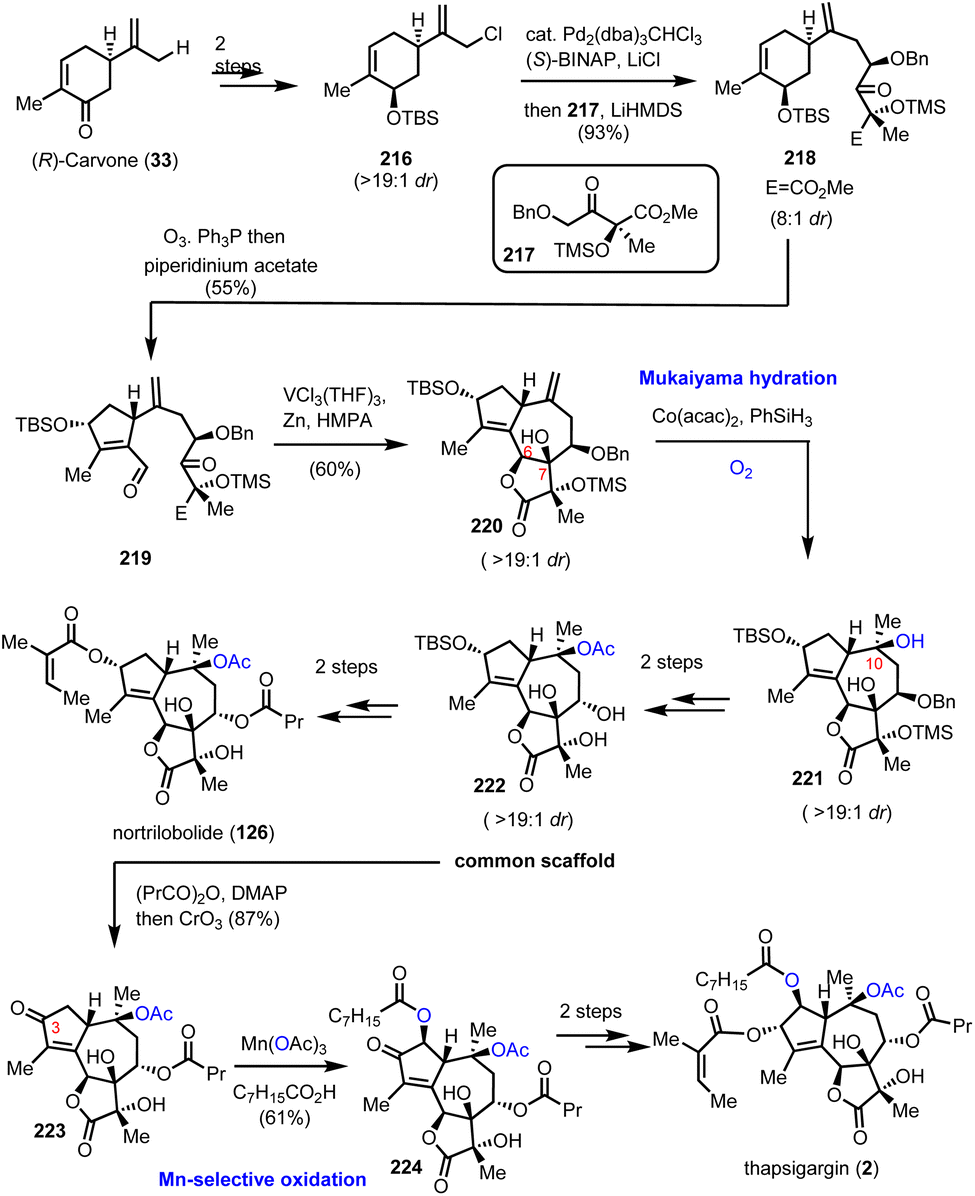
6.3. Nortrilobolide and thapsigargin (Evans’ group)
Thapsigargin (2) is among the most prominent structures for potent new anticancer agents. The high level of selectivity as an inhibitor of intracellular calcium ion transport enzymes60 and the high induction of cell apoptosis due to calcium signals61 has led to the development of novel cancer theurapeutics.62 In 2007, Evans’ group reported a concise total synthesis of thapsigargin (2) and nortrilobolide (126) by utilizing the common synthetic scaffold 222 (Scheme 22).63 The divergent plan of Evans’ group was based on bioinspired retrosynthesis to form the C-6–C-7 bond as the last step for the synthesis of the guaianolide core. The synthesis started by the asymmetric alkylation of carvone derivative 216, readily available from (R)-carvone in 2 steps, and derivative 217 available from methylerythritol under Pd2(dba)3CHCl3 and (S)-BINAP catalysis to furnish 218 in 93% yield (Scheme 22). Selective ozonolysis of the cyclohexene moiety followed by organocatalytic aldol reaction provided compound 219 in gram scale quantity and 55% yield. A pinacol coupling/lactonization cascade resulted from the reaction of 219 with the [V2Cl3(THF)6]2[Zn2Cl6] complex, prepared in situ in the presence of hexamethylphosphoramide (HMPA). The sequence allowed the concise synthesis of the complete carbocyclic core of the guaianolide in a high overall yield and diastereoselectivity (19:1). From this point, the quest to introduce the rest of the oxidase phase began. A Mukaiyama hydration with the aid of Co(acac)2 and PhSiH3 was selected for C-10 hydroxylation to provide 221 in 79% yield and high diastereoselectivity (19:1). FGI for the introduction of the requisite acyl groups followed for the synthesis of nortrilobolide (126), and with subsequent oxidation of C-3 to ketone for the synthesis of thapsigargin (Scheme 22). The latter was used as a handle for α-hydroxylation with the aid of Mn(OAc)3 and octanoic acid to provide compound 224, which was readily reduced and acylated to furnish thapsigargin (2).
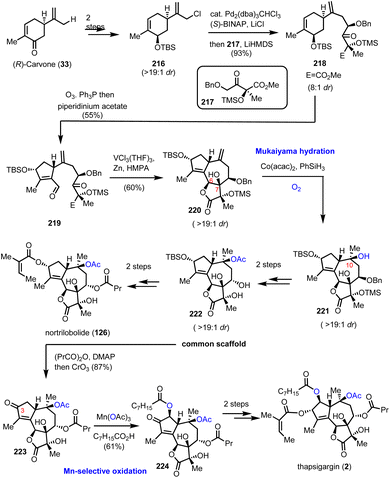 |
| | Scheme 22 Total synthesis of the thapsigargin and nortrilobolide oxidase phase relying on Mukaiyama hydration (Evans’ group). | |
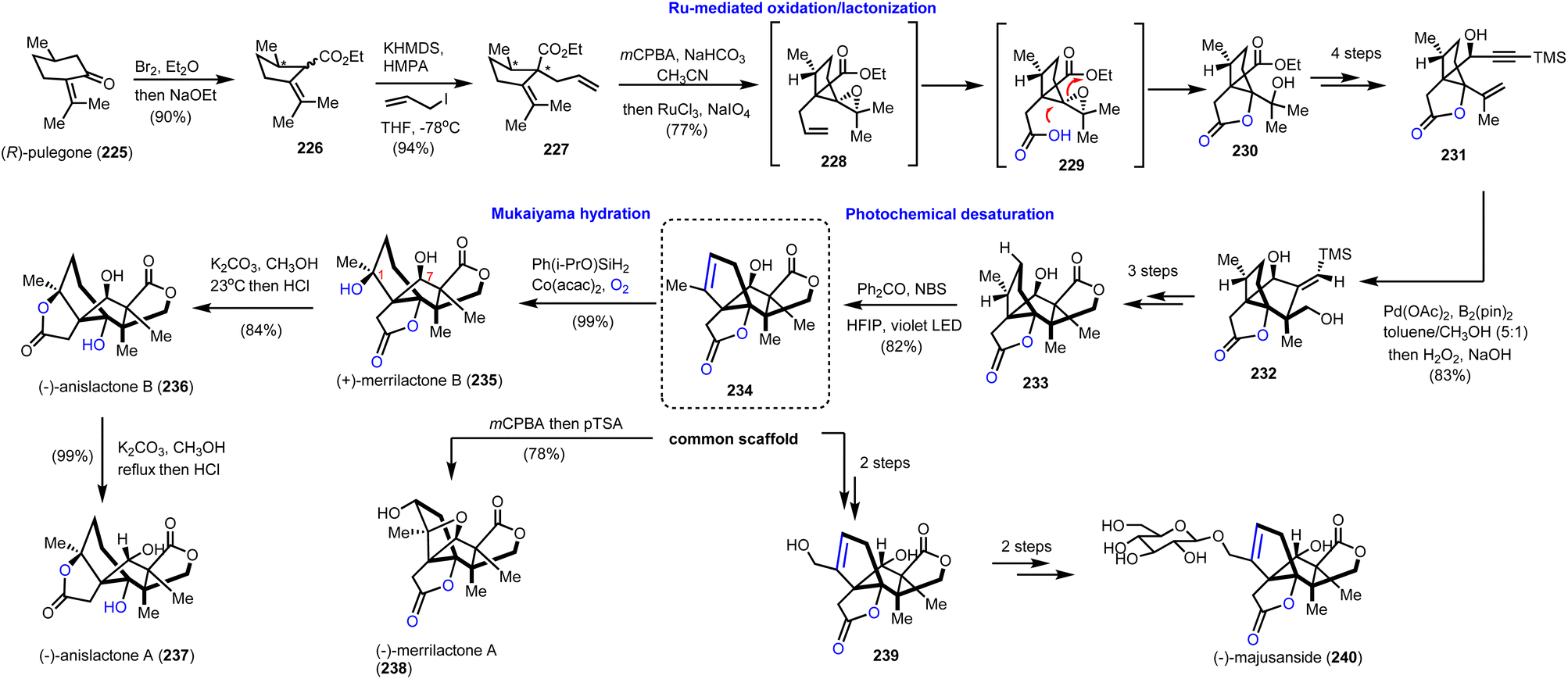
6.4. Merrilactone B, anislactone A, B and majusanside (Zhang's and Zhang's groups)
To take advantage of the synthetic potential of alkenes in stereoselective oxidations, Zhang's and Zhang's groups developed a novel aliphatic desaturation protocol able to work under complex molecular settings.64 To reach Illicium complexity authors realized that they can use the appropriately substituted alkene 234 (Scheme 23, dashed frame) as a common synthetic scaffold for the divergent total synthesis of merrilactone A (238) and B (235), anislactone A (237) and B (236) and majusanside (240). The synthesis began with the transformation of (R)-pulegone (225) to 226 through dibromination and Favorskii rearrangement. Stereoselective introduction of an allyl chain followed by mCPBA epoxidation and ruthenium chloride, sodium hypoiodide alkene cleavage/oxidation furnished lactone 230. FGI and subsequent introduction of the acetylene moiety allowed a palladium(II)-promoted cyclization-borylation of enyne to 232, which was three steps away from precursor 233. At this point, the authors explored the possibility of an unsaturation event to 234. To achieve it a photochemical bromination was considered. Molecular calculations on BDE values of the C–H cleavage on 233 revealed C-1 and C-7 as more prone positions for an electrophilic cleavage. Bromination with the aid of benzophenone under violet light in HFIP resulted in alkene 234 in 82% yield. HFIP served as an efficient hydrogen donor to the hydroxyl moiety altering the BDE value at the C-7 position and masking it. Having in hand compound 234 in multigram quantities allowed them to complete the total synthesis of merrilactone A (238) by m-CPBA epoxidation followed by intramolecular epoxide opening (Scheme 23). Mukaiyama hydration of 234 provided stereoselectively merrilactone B (235) in 99% yield by using Co(acac)2 and Ph(i-PrO)SiH2 under dioxygen. Translactonization of the latter with potassium methoxide formed anislactone B (236) and anislactone A (237) after epimerization. Finally, epimerization of C-7 followed by selenium dioxide allylic oxidation completed the total synthesis of the aglycon of (−)-majusanside (240).
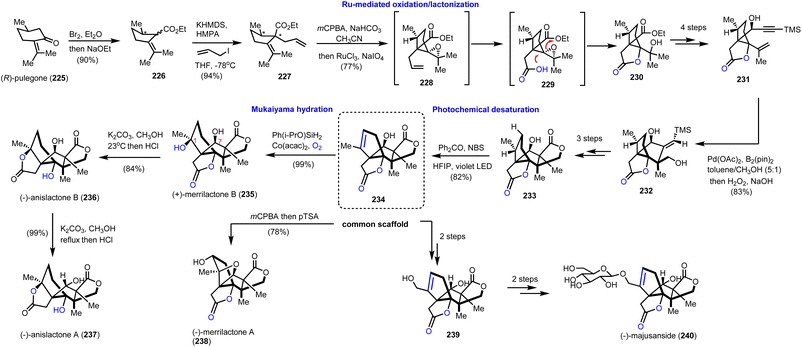 |
| | Scheme 23 Divergent synthesis of merrilactone B, anislactone A, B and majusanside based on a ‘desaturation’ protocol (Zhang's and Zhang's groups). | |
7. Miscellaneous
7.1.
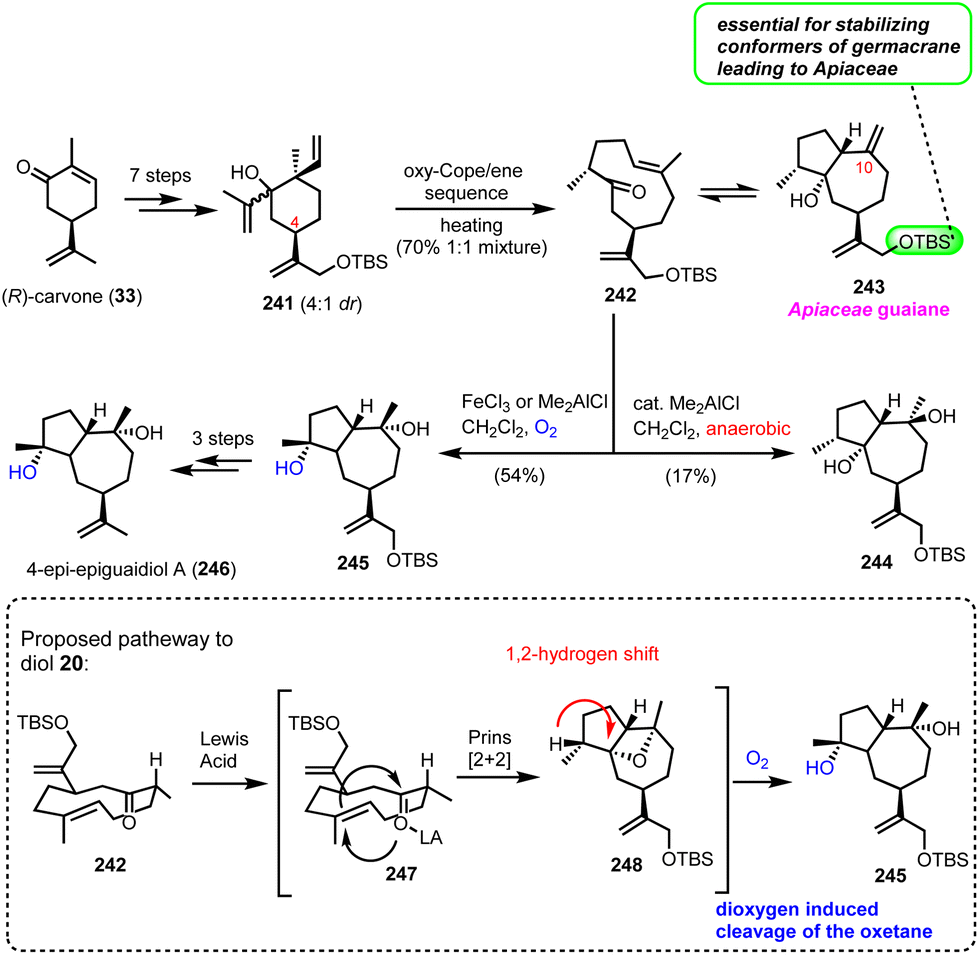
epi-Epiguaianol A (Zografos’ group)
Recently, Zografos’ group reported an interesting dioxygen-induced C–H oxidation for the synthesis of epi-epiguaianol (246).65 Based on earlier findings of the group, non-natural elemane-type sesquiterpenoids can serve as ideal precursors to access germacranolides and guaianolides through an oxy–Cope-(ene) reaction (cascades).17 Applying an oxy–Cope/ene sequence to a diasteroisomeric mixture of 241 (anti:syn = 4:1), readily accessible from (R)-carvone (33) in 7 steps, led to germacrane 242 and guaiane 243 as a 50:50 mixture in 70% yield (Scheme 24). Interestingly, the isopropenyloxy-TBS substituent at the 4-position was found essential to lock the desired chair conformations for the oxy–Cope event to deliver only the correct stereochemistry for Apiaceae guaiane sesquiterpenoids (Scheme 24). The evidence of a thermal equilibrium between 242 and 243 allowed further elaboration of germacrane 242 to oxidized members of the family. Thus, when 242 was treated with Lewis acids in the presence of dioxygen (FeCl3 or Me2AlCl), an impressive highly stereoselective oxidation was realized to produce 245 in 54% yield. On the other hand, treating 242 with catalytic Me2AlCl in the absence of dioxygen provided diol 244, possessing the diastereomeric alcohol at C-10 in 17% yield. Based on the different behaviour of 242 when dioxygen was absent, the authors proposed a Prins-[2 + 2]-type reaction for the formation of the congested oxetane ring 248, during the Lewis-acid reaction, followed by dioxygen mediated cleavage of the oxetane ring and hydroxyl migration for compound 245. FGI that followed provided the synthesis of epi-epiguaianol A (246).
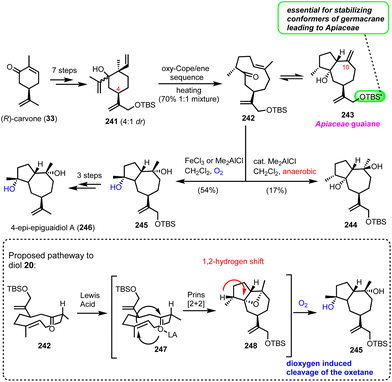 |
| | Scheme 24 Stereoselective oxidation for the synthesis of 4-epi-epiguaianol based on dioxygen induced oxetane cleavage (Zografos’ group). | |
8. Conclusions
The impressive transformations highlighted in this review underline the power of selected radical reactions to achieve selectivity where polar reactions may fail. The uniqueness of the Hoffmann–Loffler–Freytag variants and P450-like metal oxidations paves the way for further development of the field, indicating the possibility of achieving highly selective transformations in a non-enzyme environment. We strongly believe that, in the future, further development will be achieved in organocatalytic radical reactions that will manage to harness the inherent activity of sesquiterpenoid scaffolds for oxidation.
Conflicts of interest
There are no conflicts to declare.
References
-
(a) E. E. Anagnostaki and A. L. Zografos, “Common synthetic scaffolds” in the synthesis of structurally diverse natural products, Chem. Soc. Rev., 2012, 41, 5613 RSC;
(b) K. Gennaiou, A. Kelesidis, M. Kourgiantaki and A. L. Zografos, Combining the best of both worlds: radical-based divergent total synthesis, Beilstein J. Org. Chem., 2023, 19, 1–26 CrossRef CAS PubMed.
- M. Petsi and A. L. Zografos, Advances in Catalytic Aerobic Oxidations by Activation of Dioxygen-Monooxygenase Enzymes and Biomimetics, Synthesis, 2018, 50, 4715 CrossRef CAS.
-
(a) T. Yamada, K. Imagawa, T. Nagata and T. Mukaiyama, Enantioselective Epoxidation of Unfunctionalized Olefins with Molecular Oxygen and Aldehyde Catalyzed by Optically Active Manganese(III) Complexes, Chem. Lett., 1992, 21, 2231 CrossRef;
(b) T. Yamada, K. Imagawa and T. Mukaiyama, β-Selective Epoxidation of Cholesterol Derivatives with Molecular Oxygen and Aldehyde Catalyzed by Manganese(II) Complex, Chem. Lett., 1992, 21, 2109 CrossRef;
(c)
R. A. Johnson and K. B. Sharpless, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Elsevier Science Ltd, 1991, 7, ch. 3.2, pp. 389–436 Search PubMed;
(d) M. G. Finn and B. Sharpless, Mechanism of asymmetric epoxidation. 2. Catalyst structure, J. Am. Chem. Soc., 1991, 113, 113 CrossRef CAS;
(e) T. Katsuki and K. B. Sharpless, The first practical method for asymmetric epoxidation, J. Am. Chem. Soc., 1980, 102, 5974 CrossRef CAS;
(f) Y. Gao, R. M. Hanson, J. M. Klunder, S. Y. Ko, H. Masamune and K. B. Sharpless, Catalytic asymmetric epoxidation and kinetic resolution: modified procedures including in situ derivatization, J. Am. Chem. Soc., 1987, 109, 5765 CrossRef CAS;
(g) W. Zhang, J. L. Loebach, S. R. Wilson and E. N. Jacobsen, Enantioselective epoxidation of unfunctionalized olefins catalyzed by salen manganese complexes, J. Am. Chem. Soc., 1990, 112, 2801 CrossRef CAS;
(h) R. Irie, K. Noda, Y. Ito, N. Matsumoto and T. Katsuki, Catalytic asymmetric epoxidation of unfunctionalized olefins, Tetrahedron Lett., 1990, 31, 7345 CrossRef CAS;
(i) E. N. Jacobsen, W. Zhang, A. R. Muci, J. R. Ecker and L. Deng, Highly enantioselective epoxidation catalysts derived from 1,2-diaminocyclohexane, J. Am. Chem. Soc., 1991, 113, 7063 CrossRef CAS;
(j) I. Triantafillidi, M. G. Kokotou, D. Lotter, C. Sparr and C. G. Kokotos, Aldehyde-catalyzed epoxidation of unactivated alkenes with aqueous hydrogen peroxide, Chem. Sci., 2021, 12, 10191 RSC;
(k) G. Peris, C. E. Jacobsche and S. J. Miller, Aspartate-catalyzed asymmetric epoxidation reactions, J. Am. Chem. Soc., 2007, 129, 8710 CrossRef CAS PubMed;
(l) D. Limnios and C. G. Kokotos, 2,2,2-Trifluoroacetophenone: an organocatalyst for an environmentally friendly epoxidation of alkenes, J. Org. Chem., 2014, 79, 4270 CrossRef CAS PubMed;
(m) C. E. Jacobsche, G. Peris and S. J. Miller, Functional analysis of an aspartate-based epoxidation catalyst with amide-to-alkene peptidomimetic catalyst analogues, Angew. Chem., Int. Ed., 2008, 47, 6707 CrossRef PubMed;
(n) I. Triantafillidi, D. I. Tzaras and C. G. Kokotos, Green organocatalytic oxidative methods using activated ketones, ChemCatChem, 2018, 10, 2521 CrossRef.
-
(a) S. J. Geier, C. M. Vogels, J. A. Melanson and S. A. Westcott, The transition metal-catalyzed hydroboration reaction, Chem. Soc. Rev., 2022, 51, 8877 RSC;
(b)
H. C. Brown, Hydroboration, W. A. Benjamin, New York, NY, 1962 Search PubMed;
(c) E. F. Knights and H. C. Brown, 9-Borabicyclo[3.3.1]nonane as a convenient selective hydroborating agent, J. Am. Chem. Soc., 1968, 90, 5281 CrossRef CAS;
(d) E. F. Knights and H. C. Brown, Cyclic hydroboration of 1,5-cyclooctadiene. A simple synthesis of 9-borabicyclo[3.3.1]nonane, an unusually stable dialkylborane, J. Am. Chem. Soc., 1968, 90, 5280 CrossRef CAS.
-
(a) M. A. Warpehoski, B. Chabaud and K. B. Sharpless, Selenium dioxide oxidation of endocyclic olefins. Evidence for a dissociation-recombination pathway, J. Org. Chem., 1982, 47, 2897 CrossRef CAS;
(b) M. A. Umbreit and K. B. Sharpless, Allylic oxidation of olefins by catalytic and stoichiometric selenium dioxide with tert-butyl hydroperoxide, J. Am. Chem. Soc., 1977, 99, 5526 CrossRef CAS;
(c) M. S. Chen and M. C. White, A Sulfoxide-Promoted, Catalytic Method for the Regioselective Synthesis of Allylic Acetates from Monosubstituted Olefins via C–H Oxidation, J. Am. Chem. Soc., 2004, 126, 1346 CrossRef CAS PubMed;
(d) C. C. Patillo, I. I. Strambeanu, P. Calleja, N. A. Vermeulen, T. Mizuno and M. C. White, Aerobic Linear Allylic C–H Amination: Overcoming Benzoquinone Inhibition, J. Am. Chem. Soc., 2016, 138, 1265 CrossRef PubMed;
(e) D. Wang, A. B. Weinstein, P. B. White and S. S. Stahl, Ligand-Promoted Palladium-Catalyzed Aerobic Oxidation Reactions, Chem. Rev., 2018, 118, 2636 CrossRef CAS PubMed;
(f) C. V. Kozack, J. A. Sowin, J. N. Jaworski, A. V. Iosub and S. S. Stahl, Aerobic Acyloxylation of Allylic C−H Bonds Initiated by a Pd0 Precatalyst with 4,5-Diazafluoren-9-one as an Ancillary Ligand, ChemSusChem, 2019, 12, 3003 CrossRef CAS PubMed;
(g) T. H. El-Assaad, J. Zhu, A. Sebastian, D. V. McGrath, I. Neogi and K. N. Parida, Dioxiranes: a half century journey, Org. Chem. Front., 2022, 9, 5675 RSC.
- For similar review topics please see:
(a) V. C. S. Santana, M. C. V. Fernandes, I. Cappuccelli, A. C. G. Richieri and E. C. de Lucca Jr., Metal-Catalyzed C–H Bond Oxidation in the Total Synthesis of Natural and Unnatural Products, Synthesis, 2022, 54, 5337 CrossRef CAS;
(b) Y. Qiu and S. Gao, Trends in applying C–H oxidation to the total synthesis of natural products, Nat. Prod. Rep., 2016, 33, 562 RSC;
(c) D. J. Abrams, P. A. Provencher and E. J. Sorensen, Recent applications of C–H functionalization in complex natural product synthesis, Chem. Soc. Rev., 2018, 47, 8925 RSC.
- L. F. T. Novaes, J. Liu, Y. Shen, L. Lu, J. M. Meinhardt and S. Lin, Electrocatalysis as an enabling technology for organic synthesis, Chem. Soc. Rev., 2021, 50, 7941 RSC.
-
(a) P. R. Ogilby, Singlet oxygen: there is indeed something new under the sun, Chem. Soc. Rev., 2010, 39, 3181 RSC;
(b) M. N. Alberti and M. Orfanopoulos, Unraveling the Mechanism of the Singlet Oxygen Ene Reaction: Recent Computational and Experimental Approaches, Chem. – Eur. J., 2010, 16, 9414 CrossRef CAS PubMed;
(c) T. Montagnon, M. Tofi and G. Vassilikogiannakis, Using Singlet Oxygen to Synthesize Polyoxygenated Natural Products from Furans, Acc. Chem. Res., 2008, 41, 1001 CrossRef CAS PubMed.
-
(a) D. E. T. Pazmiňo, M. Winkler, A. Glieder and M. W. Fraaije, Monooxygenases as biocatalysts: Classification, mechanistic aspects and biotechnological applications, J. Biotechnol., 2010, 146, 9 CrossRef PubMed;
(b) R. Bernhart, Cytochromes P450 as versatile biocatalysts, J. Biotechnol., 2006, 124, 128 CrossRef PubMed.
-
(a) A. W. Hofmann, Ueber die Einwirkung des Broms in alkalischer Lösung auf die Amine, Ber. Dtsch. Chem. Ges., 1883, 16, 558 CrossRef;
(b) K. Loffler and C. Freytag, Über eine neue Bildungsweise von N-alkylierten Pyrrolidinen, Ber. Dtsch. Chem. Ges., 1909, 42, 3427 CrossRef;
(c) E. J. Corey and W. R. Hertler, A Study of the Formation of Haloamines and Cyclic Amines by the Free Radical Chain Decomposition of N-Haloammonium Ions (Hofmann-Löffler Reaction), J. Am. Chem. Soc., 1960, 82, 1657 CrossRef CAS;
(d) P. L. Gkizis, Photochemical cross-coupling enables sp3-C bond formation by C-H homolytic cleavage: new developments and perspectives, Eur. J. Org. Chem., 2022, e202201139 CAS.
- K. Chen, J. M. Richter and P. S. Baran, 1,3-Diol Synthesis via Controlled, Radical-Mediated C–H Functionalization, J. Am. Chem. Soc., 2008, 130, 7247 CrossRef CAS PubMed.
- K. Chen and P. S. Baran, Total synthesis of eudesmane terpenes by site-selective C-H oxidations, Nature, 2009, 459, 824 CrossRef CAS PubMed.
- K. Chen, Y. Ishihara, M. M. Galán and P. S. Baran, Total synthesis of eudesmane terpenes: cyclase phase, Tetrahedron, 2010, 66, 4738 CrossRef CAS.
- R. Mello, M. Fiorentino, C. Fusco and R. Curci, Oxidations by methyl(trifluoromethyl)dioxirane. 2. Oxyfunctionalization of saturated hydrocarbons, J. Am. Chem. Soc., 1989, 111, 6749 CrossRef CAS.
- K. Chen, A. Eschenmoser and P. S. Baran, Strain Release in C-H Bond Activation?, Angew. Chem., Int. Ed., 2009, 48, 9705 CrossRef CAS PubMed.
-
V. Demertzidou and A. L. Zografos, Exploiting the Limits of Divergency Scaffolds in Natural Product Synthesis: The Case Study of Sesquiterpenoids, ChemRxiv, 2021, DOI:10.26434/chemrxiv.14554794.v1.
-
(a) E. E. Anagnostaki and A. L. Zografos, Non-natural Elemane as the “Stepping Stone” for the Synthesis of Germacrane and Guaiane Sesquiterpenes, Org. Lett., 2013, 15, 152 CrossRef CAS PubMed;
(b) E. E. Anagnostaki, V. P. Demertzidou and A. L. Zografos, Divergent pathways to furosesquiterpenes: first total syntheses of (+)-zedoarol and (Rac)-gweicurculactone, Chem. Commun., 2015, 51, 2364 RSC;
(c) V. P. Demertzidou and A. L. Zografos, Platinum-catalyzed cycloisomerizations of a common enyne: a divergent entry to cyclopropane sesquiterpenoids. Formal synthesis of sarcandralactone A, Org. Biomol. Chem., 2016, 14, 6942 RSC;
(d) V. P. Demertzidou, M. Kourgiantaki and A. L. Zografos, A multifunctional divergent scaffold to access the formal syntheses of various sesquiterpenoids, Org. Biomol. Chem., 2021, 19, 8687 RSC.
- M. L. Condakes, K. Hung, S. J. Harwood and T. J. Maimone, Total Syntheses of (−)-Majucin and (−)-Jiadifenoxolane A, Complex Majucin-Type Illicium Sesquiterpenes, J. Am. Chem. Soc., 2017, 139, 17783 CrossRef CAS PubMed.
- C.-S. Yang, I. Kouno, N. Kawano and S. Sato, New anisatin-like sesquitepene lactones from pericarps of illicium majus, Tetrahedron Lett., 1988, 29, 1165 CrossRef CAS.
- K. Hung, M. L. Condakes, L. F. T. Novaes, S. J. Harwood, T. Morikawa, Z. Yang and T. J. Maimone, Development of a Terpene Feedstock-Based Oxidative Synthetic Approach to the Illicium Sesquiterpenes, J. Am. Chem. Soc., 2019, 141, 3083 CrossRef CAS PubMed.
- J. Gomes, C. Daeppen, R. Liffert, J. Roesslein, E. Kaufmann, A. Heikinheimo, M. Neuburger and K. Gademann, Formal Total Synthesis of (−)-Jiadifenolide and Synthetic Studies toward seco-Prezizaane-Type Sesquiterpenes, J. Org. Chem., 2016, 81, 11017 CrossRef CAS PubMed.
- J. Wolinsky and D. Chan, The Favorskii Rearrangement of Pulegone Dibromide, J. Org. Chem., 1965, 30, 41 CrossRef CAS.
- C. R. Holmquist and E. J. Roskamp, Tin(II) chloride catalyzed addition of diazo sulfones, diazo phosphine oxides, and diazo phosphonates to aldehydes, Tetrahedron Lett., 1992, 33, 1131 CrossRef CAS.
- G. Cainelli, M. Lj. Mihailovic, D. Arigoni and O. Jeger, Über Steroide und Sexualhormone. 211. Mitteilung. Direkte Einführung einer Sauerstoffunktion in die Methylgruppe C-18 im intakten Steroidgerüst, Helv. Chim. Acta, 1959, 42, 1124 CrossRef CAS.
-
(a) M. Costas, K. Chen and L. Que Jr., Biomimetic nonheme iron catalysts for alkane hydroxylation, Coord. Chem. Rev., 2000, 200–202, 517 CrossRef CAS;
(b) K. Chen and L. Que Jr., Stereospecific Alkane Hydroxylation by Non-Heme Iron Catalysts: Mechanistic Evidence for an FeVO Active Species, J. Am. Chem. Soc., 2001, 123, 6327 CrossRef CAS PubMed;
(c) G. J. P. Britovsek, J. England and A. J. P. White, Non-heme iron(II) complexes containing tripodal tetradentate nitrogen ligands and their application in alkane oxidation catalysis, Inorg. Chem., 2005, 44, 8125 CrossRef CAS PubMed;
(d) Y. Mekmouche, S. Ménage, C. Toia-Duboc, M. Fontecave, J.-B. Galey, C. Lebrun and J. Pécaut, H2O2-Dependent Fe-Catalyzed
Oxidations: Control of the Active Species, Angew. Chem., Int. Ed., 2001, 40, 949 CrossRef CAS;
(e) A. Company, L. Gómez, M. Güell, X. Ribas, J. M. Luis, L. Que Jr. and M. Costas, Alkane Hydroxylation by a Nonheme Iron Catalyst that Challenges the Heme Paradigm for Oxygenase Action, J. Am. Chem. Soc., 2007, 129, 15766 CrossRef CAS PubMed.
- M. Merkx, D. A. Kopp, M. H. Sazinsky, J. L. Blazyk, J. Müller and S. J. Lippard, Dioxygen Activation and Methane Hydroxylation by Soluble Methane Monooxygenase: A Tale of Two Irons and Three Proteins A list of abbreviations can be found in Section 7, Angew. Chem., Int. Ed., 2001, 40, 2782 CrossRef CAS PubMed.
-
(a) M. S. Chen and M. C. White, A predictably selective aliphatic C-H oxidation reaction for complex molecule synthesis, Science, 2007, 318, 783 CrossRef CAS PubMed;
(b) M. A. Bigi, S. A. Reed and M. C. White, Directed Metal (Oxo) Aliphatic C−H Hydroxylations: Overriding Substrate Bias, J. Am. Chem. Soc., 2012, 134, 9721 CrossRef CAS PubMed.
- A. S. Burns and S. D. Rychnovsky, Total Synthesis and Structure Revision of (−)-Illisimonin A, a Neuroprotective Sesquiterpenoid from the Fruits of Illicium simonsii, J. Am. Chem. Soc., 2019, 141, 13295 CrossRef CAS PubMed.
- S.-G. Ma, M. Li, M.-B. Lin, L. Li, Y.-B. Liu, J. Qu, Y. Li, X.-J. Wang, R.-B. Wang, S. Xu, Q. Hou and S.-S. Yu, Illisimonin A, a Caged Sesquiterpenoid with a Tricyclo[5.2.1.01,6]decane Skeleton from the Fruits of Illicium simonsii, Org. Lett., 2017, 19, 6160 CrossRef CAS PubMed.
- Z.-J. Zhan, Y.-M. Ying, L.-F. Ma and W.-G. Shan, Natural disesquiterpenoids, Nat. Prod. Rep., 2011, 28, 594 RSC.
- C. Li, L. Dian, W. Zhang and X. Lei, Biomimetic Syntheses of (−)-Gochnatiolides A–C and (−)-Ainsliadimer B, J. Am. Chem. Soc., 2012, 134, 12414 CrossRef CAS PubMed.
- C. Li, X. Yu and X. Lei, A Biomimetic Total Synthesis of (+)-Ainsliadimer A, Org. Lett., 2010, 12, 4284 CrossRef CAS PubMed.
- V. Dell'Oste, F. Spyrakis and C. Prandi, Strigolactones, from Plants to Human Health: Achievements and Challenges, Molecules, 2021, 26, 4579 CrossRef PubMed.
- H. I. Kim,
et al., Avenaol, a germination stimulant for root parasitic plants from Avena strigosa, Phytochemistry, 2014, 103, 85 CrossRef CAS PubMed.
- M. Yasui, R. Ota, C. Tsukano and Y. Takemoto, Total synthesis of Avenaol, Nat. Commun., 2017, 8, 674 CrossRef PubMed.
- For seminal references, see the following:
(a) S. Isayama and T. Mukaiyama, A New Method for Preparation of Alcohols from Olefins with Molecular Oxygen and Phenylsilane by the Use of Bis(acetylacetonato) cobalt(II), Chem. Lett., 1989, 18, 1071 CrossRef;
(b) T. Mukaiyama, S. Isayama, S. Inoki, K. Kato, T. Yamada and T. Takai, Oxidation-Reduction Hydration of Olefins with Molecular Oxygen and 2-Propanol Catalyzed by Bis(acetylacetonato)cobalt(II), Chem. Lett., 1989, 18, 449 CrossRef;
(c) S. Inoki, K. Kato, T. Takai, S. Isayama, T. Yamada and T. Mukaiyama, Bis(trifluoro-acetylacetonato)cobalt(II) Catalyzed Oxidation-Reduction Hydration of Olefins Selective Formation of Alcohols from Olefins, Chem. Lett., 1989, 18, 515 CrossRef.
-
(a) S. W. M. Crossley, R. M. Martinez, C. Obradors and R. A. Shenvi, Mn, Fe, and Co-catalyzed radical hydrofunctionalization of olefins, Chem. Rev., 2016, 116, 8912 CrossRef CAS PubMed;
(b) B. Gaspar and E. M. Carreira, Mild cobalt- catalyzed hydrocyanation of olefins with tosyl cyanide, Angew. Chem., Int. Ed., 2007, 46, 4519 CrossRef CAS PubMed;
(c) J. C. Lo, Y. Yabe and P. S. Baran, A practical and catalytic reductive olefin coupling, J. Am. Chem. Soc., 2014, 136, 1304 CrossRef CAS PubMed;
(d) J. C. Lo, J. Gui, Y. Yabe, C.-M. Pan and P. S. Baran, Functionalized olefin cross-coupling to construct carbon-carbon bonds, Nature, 2014, 516, 343 CrossRef CAS PubMed.
-
(a) S. W. M. Crossley, F. Barabé and R. A. Shenvi, Simple, Chemoselective, Catalytic Olefin Isomerization, J. Am. Chem. Soc., 2014, 136, 16788 CrossRef CAS PubMed;
(b) S. A. Green, T. R. Huffman, R. O. McCourt, V. van der Puyl and R. A. Shenvi, Hydroalkylation of Olefins To Form Quaternary Carbons, J. Am. Chem. Soc., 2019, 141, 7709 CrossRef CAS PubMed.
- X. Hu, A. J. Musacchio, X. Chen, Y. Tao and T. J. Maimone, Allylative Approaches to the Synthesis of Complex Guaianolide Sesquiterpenes from Apiaceae and Asteraceae, J. Am. Chem. Soc., 2019, 141, 14904 CrossRef CAS PubMed.
- Y. Matsuo, S. Maeda, C. Ohba, H. Fukaya and Y. Mimaki, Vetiverianines A, B, and C: Sesquiterpenoids from Vetiveria zizanioides Roots, J. Nat. Prod., 2016, 79, 2175 CrossRef CAS PubMed.
- T. Mashiko, E. Nagata, H. Sakate, S. Kamo and K. Sugita, Total synthesis of (+)-ent-vetiverianine a via Lewis acid-mediated cyclization, Org. Chem. Front., 2022, 9, 6849 RSC.
- C. J. Dooley III and S. D. Rychnovsky, Asymmetric Total Synthesis of (2R)-Hydroxynorneomajucin, a Norsesquiterpene from Illicium jiadifengpi, Org. Lett., 2022, 24, 3411 CrossRef CAS PubMed.
- M. Kubo, K. Kobayashi, J.-M. Huang, K. Harada and Y. Fukuyama, The first examples of seco-prezizaane-type norsesquiterpenoids with neurotrophic activity from Illicium jiadifengpi, Tetrahedron Lett., 2012, 53, 1231 CrossRef CAS.
- B. M. Trost and C. Jiang, Pd-Catalyzed Asymmetric Allylic Alkylation. A Short Route to the Cyclopentyl Core of Viridenomycin, Org. Lett., 2003, 5, 1563 CrossRef CAS PubMed.
- L. Trzoss, J. Xu, M. H. Lacoske, W. C. Mobley and E. A. Theodorakis, Enantioselective Synthesis of (−)-Jiadifenin, a Potent Neurotrophic Modulator, Org. Lett., 2011, 13, 4554 CrossRef CAS PubMed.
- J. Xu, L. Trzoss, W. K. Chang and E. A. Theodorakis, Enantioselective Total Synthesis of (−)-Jiadifenolide, Angew. Chem., Int. Ed., 2011, 50, 3672 CrossRef CAS PubMed.
- J. Huo, S.-P. Yang, J. Ding and J.-M. Yue, Cytotoxic sesquiterpene lactones from Eupatorium lindleyanum, J. Nat. Prod., 2004, 67, 1470 CrossRef CAS PubMed.
- L. de Lichtervelde, A. E. Boitano, Y. Wang, P. Krastel, F. Petersen, M. P. Cooke and P. G. Schultz, Eupalinilide E inhibits erythropoiesis and promotes the expansion of hematopoietic progenitor cells, ACS Chem. Biol., 2013, 8, 866 CrossRef CAS PubMed.
- T. C. Johnson, M. R. Chin, T. Han, J. P. Shen, T. Rana and D. Siegel, Synthesis of Eupalinilide E, a Promoter of Human Hematopoietic Stem and Progenitor Cell Expansion, J. Am. Chem. Soc., 2016, 138, 6068 CrossRef CAS PubMed.
- T. C. Johnson, M. R. Chin and D. Siegel, Synthetic Route Development for the Laboratory Preparation of Eupalinilide E, J. Org. Chem., 2017, 82, 4640 CrossRef CAS PubMed.
- Z. Yuan, X. Hu, H. Zhang, L. Liu, P. Chen, M. He, X. Xie, X. Wanga and X. She, Total synthesis of conosilane A via a site-selective C–H functionalization strategy, Chem. Commun., 2018, 54, 912 RSC.
- A. S. Demir, A. Jeganathan and D. S. Watt, Synthesis of alpha’-acyloxy enones from enones using manganese(III) acetate in combination with manganese(II) carboxylates or carboxylic acids, J. Org. Chem., 1989, 54, 4020 CrossRef CAS.
- A. S. Demir, O. Reis and C. Igdir, Reinvestigation of the synthetic and mechanistic aspects of Mn(III) acetate mediated oxidation of enones, Tetrahedron, 2004, 60, 3427 CrossRef CAS.
- R. Marín-Barrios, A. L. García-Cabeza, F. J. Moreno-Dorado, F. M. Guerra and G. M. Massanet, Acyloxylation of Cyclic Enones: Synthesis of Densely Oxygenated Guaianolides, J. Org. Chem., 2014, 79, 6501 CrossRef PubMed.
- K. Hung, M. L. Condakes, T. Morikawa and T. J. Maimone, Oxidative Entry into the Illicium Sesquiterpenes: Enantiospecific Synthesis of (+)-Pseudoanisatin, J. Am. Chem. Soc., 2016, 138, 16616 CrossRef CAS PubMed.
- K. Yamada, S. Takada, S. Nakamura and Y. Hirata, The structures of anisatin and neoanisatin: Toxic sesquiterpenes from Illicium Anisatum L., Tetrahedron, 1968, 24, 199 CrossRef CAS.
- M. C. White and J. Zhao, Aliphatic C−H Oxidations for Late-Stage Functionalization, J. Am. Chem. Soc., 2018, 140, 13988 CrossRef CAS PubMed.
-
(a) C. M. Rasik and M. K. Brown, Total Synthesis of Gracilioether F: Development and Application of Lewis Acid Promoted Ketene–Alkene [2 + 2] Cycloadditions and Late-Stage C-H Oxidation, Angew. Chem., Int. Ed., 2014, 53, 14522 CrossRef CAS PubMed;
(b) M. E. McCallum, C. M. Rasik, J. L. Wood and M. K. Brown, Collaborative Total Synthesis: Routes to (±)-Hippolachnin A Enabled by Quadricyclane Cycloaddition and Late-Stage C–H Oxidation, J. Am. Chem. Soc., 2016, 138, 2437 CrossRef CAS PubMed;
(c) C.-L. Sun, B.-J. Li and Z.-J. Shi, Direct C−H Transformation via Iron Catalysis, Chem. Rev., 2011, 111, 1293 CrossRef CAS PubMed.
- M. Ohtawa, M. J. Krambis, R. Cerne, J. M. Schkeryantz, J. M. Witkin and R. A. Shenvi, Synthesis of (−)-11-O Debenzoyltashironin: Neurotrophic Sesquiterpenes Cause Hyperexcitation, J. Am. Chem. Soc., 2017, 139, 9637 CrossRef CAS PubMed.
- O. Thastrup, P. J. Cullen, B. K. Drøbak, M. R. Hanley and A. P. Dawson, Thapsigargin, a tumor promoter, discharges intracellular Ca2+stores by specific inhibition of the endoplasmic reticulum Ca2(+)-ATPase, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 2466 CrossRef CAS PubMed.
- M. Treiman, C. Caspersen and S. B. Christensen, A tool coming of age: thapsigargin as an inhibitor of sarco-endoplasmic reticulum Ca(2+)-ATPases, Trends Pharmacol. Sci., 1998, 19, 131 CrossRef CAS PubMed.
- S. B. Christensen, D. M. Skytte, S. R. Denmeade, C. Dionne, J. V. Moller, P. Nissen and J. T. Isaacs, A Trojan horse in drug development: targeting of thapsigargins towards prostate cancer cells, Anti-Cancer Agents Med. Chem., 2009, 9, 276 CrossRef CAS PubMed.
- D. Chen and P. A. Evans, A Concise, Efficient and Scalable Total Synthesis of Thapsigargin and Nortrilobolide from (R)-(-)-Carvone, J. Am. Chem. Soc., 2017, 139, 6046 CrossRef CAS PubMed.
- Y. Shen, L. Li, X. Xiao, S. Yang, Y. Hua, Y. Wang, Y.-w. Zhang and Y. Zhang, Site-Specific Photochemical Desaturation Enables Divergent Syntheses of Illicium Sesquiterpenes, J. Am. Chem. Soc., 2021, 143, 3256 CrossRef CAS PubMed.
- M. Kourgiantaki, V. Demertzidou and A. L. Zografos, Short Scalable Route to Apiaceae Sesquiterpene Scaffolds: Total Synthesis of 4- epi-Epiguaidiol A, Org. Lett., 2022, 24, 8476 CrossRef CAS PubMed.
|
| This journal is © the Partner Organisations 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence * and
Alexandros L.
Zografos
* and
Alexandros L.
Zografos