
Discovery and development of botanical natural products and their analogues as therapeutics for ovarian cancer
Brittney K.
Mize
 a,
Amrita
Salvi
b,
Yulin
Ren
a,
Joanna E.
Burdette
*b and
James R.
Fuchs
*a
a,
Amrita
Salvi
b,
Yulin
Ren
a,
Joanna E.
Burdette
*b and
James R.
Fuchs
*a
aDivision of Medicinal Chemistry and Pharmacognosy, College of Pharmacy, The Ohio State University, Columbus, Ohio, USA. E-mail: fuchs.42@osu.edu
bDepartment of Pharmaceutical Sciences, College of Pharmacy, University of Illinois at Chicago, Chicago, Illinois, USA
First published on 30th June 2023
Abstract
Covering: 2015 through the end of July 2022
Ovarian cancer is one of the most common cancers affecting the female reproductive organs and has the highest mortality rate among gynecological cancers. Although botanical drugs and their derivatives, namely members of the taxane and camptothecin families, represent significant therapeutics currently available for the treatment of ovarian cancer, new drugs that have alternative mechanisms of action are still needed to combat the disease. For this reason, many efforts to identify additional novel compounds from botanical sources, along with the further development of existing therapeutics, have continued to appear in the literature. This review is designed to serve as a comprehensive look at both the currently available small-molecule therapeutic options and the recently reported botanically-derived natural products currently being studied and developed as potential future therapeutics that could one day be used against ovarian cancer. Specifically, key properties, structural features, and biological data are highlighted that are important for the successful development of potential agents. Recently reported examples are specifically discussed in the context of “drug discovery attributes,” including the presence of structure–activity relationship, mechanism of action, toxicity, and pharmacokinetic studies, to help indicate the potential for future development and to highlight where these compounds currently exist in the development process. The lessons learned from both the successful development of the taxanes and camptothecins, as well as the strategies currently being employed for new drug development, are expected to ultimately help guide the future development of botanical natural products for ovarian cancer.
 Brittney K. Mize | Dr Brittney Mize obtained her Bachelor of Science in Biochemistry and Molecular Biology from Otterbein University in 2018. She obtained her PhD in Pharmaceutical Science under the supervision of Dr James Fuchs at The Ohio State University in 2023. She has focused her research on analogue development and target identification of natural products with anti-ovarian cancer activity. In addition, she has also designed and executed a synthesis of novel CDK9 degraders. She was awarded The Ohio State Presidential Fellowship in 2018 and has presented her research in several local and national conferences. |
 Amrita Salvi | Dr Amrita Salvi is a Research Assistant Professor in Dr Joanna Burdette's laboratory at the University of Illinois, Chicago. She received a Master's in Biotechnology from the University of Mumbai, India and her PhD in Cell and Molecular Biology from Nanyang Technological University, Singapore. With great interest in drug discovery and cancer biology, Dr Salvi joined the Burdette lab for her postdoctoral research in 2017. Her research focuses on understanding ovarian tumorigenesis and using natural compounds for drug discovery in ovarian cancer. Specifically, she has studied the mechanism of action of natural compounds from the Verticillin and Phyllanthusmin families. |
 Yulin Ren | Dr Yulin Ren received her PhD degree in Medicinal Chemistry and Pharmacognosy in 2001 (Peking Union Medical College) followed by training as a postdoctoral researcher in both Organic Synthesis and Biomedical Sciences in the United States. Her research interests focus on the design and discovery of natural product anticancer agents, and she identified the phyllanthusmin antitumor lead compounds from Phyllanthus poilanei and characterized their primary structure–activity relationship and molecular targets. Dr Ren has published over 70 peer-reviewed research and review articles and five book chapters. |
 Joanna E. Burdette | Dr Joanna E. Burdette is the Edward and Josephine Chair in Pharmacognosy and Medicinal Chemistry as well as the Associate Dean for Research and Graduate Studies in the College of Pharmacy. She serves as the co-director for the Cancer Biology Program in the UI Cancer Center and the co-director of the IRACDA program, which focuses on training postdoctoral fellows in research and teaching with an emphasis on diversity and inclusion. Her research has helped to develop three-dimensional models of the fallopian tube epithelium, which is thought to be the source for the deadliest type of ovarian cancer, high grade serous cancer. |
 James R. Fuchs | Dr James Fuchs obtained his B.S. in Chemistry from the University of Toledo in Ohio in 1998. He subsequently completed PhD studies at Penn State University in the area of natural product synthesis with Prof. Ray Funk in 2005. After completing an NIH postdoctoral fellowship at the Scripps Research Institute in the lab of Prof. Dale Boger, he took a faculty position in the College of Pharmacy at the Ohio State University in 2007. As a medicinal chemist, his research focuses on drug design and development of lead compounds, particularly natural products, in the areas of cancer and infectious disease. |
1. Introduction
Natural products have historically played an important role as therapeutics for a wide range of diseases. This utility can be traced back to the unique structural diversity and complexity of these molecules, as exemplified by their numerous rings, stereogenic centers, and functional groups. These structural motifs have been engineered by nature to efficiently interact with a myriad of biological targets in the environment. Throughout history, applications of these natural substances, and more recently their constituents, have been discovered in the context of human health. Improvements in biochemical assays and screening strategies, chemical synthesis, and drug formulation have facilitated the translation of many natural products and their semi-synthetic and synthetic derivatives to the clinic. In fact, natural products and their semi-synthetic derivatives make up or have inspired more than 35% of all of the FDA-approved therapeutics from 1981 to 2019 and over 50% of all small-molecule approvals.1 For cancer therapy the role of natural products is even more significant,2 with the number sometimes being quoted as over 60% of existing drugs.The four primary sources for these natural products can be broadly classified as animal, fungal, bacterial, and botanical. Of these sources, botanicals have been the most widely documented for their application to human health. Specifically, work with terrestrial plants has led to the discovery of some of the most significant cancer drug classes reported to date, including the vinca alkaloids, podophyllotoxins, taxanes, and camptothecins. With this in mind, this review will highlight the discovery and early development of the taxanes and camptothecins, two of these classes that have found application in the treatment of ovarian cancer, before shifting to cover the identification and development of additional botanical natural products and derivatives that have been studied for their activity against ovarian cancer as reported from 2015 to the time of publication. This latter section will specifically highlight critical drug properties for these compounds and the data that is necessary to support their advancement to the clinic. Together, these attributes help to define the potential of a particular compound for continued studies and whether these compounds are currently at an early or more advanced stage of development. In addition to botanically-derived therapies, the non-botanical therapeutics currently approved for use against ovarian cancer will also be mentioned to establish a comprehensive picture of the small molecule therapeutics available for ovarian cancer therapy.
2. Ovarian cancer
Ovarian cancer is a group of diseases originating in the ovaries and neighboring organs such as the fallopian tubes. It is one of the most common cancers affecting the female reproductive organs with the highest mortality rate among gynecological cancers.3 The average 5 year survival rate for women with ovarian cancer is ∼40%. According to the American Cancer Society an estimated 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 880 new cases and 12810 deaths will be caused by ovarian cancer in the year 2022.4 Ovarian cancer is deadly because it causes subtle symptoms in the early stages and lacks effective screening methods.5 It is typically diagnosed at a late stage after it has metastasized to distant organs.3
880 new cases and 12810 deaths will be caused by ovarian cancer in the year 2022.4 Ovarian cancer is deadly because it causes subtle symptoms in the early stages and lacks effective screening methods.5 It is typically diagnosed at a late stage after it has metastasized to distant organs.3
Epithelial ovarian cancer (EOC) is the most common subtype of ovarian cancer and accounts for ∼90% of cases. The four major histological subtypes of EOC are serous, endometrioid, clear cell, and mucinous carcinoma, of which serous accounts for ∼70% of EOCs. Serous carcinoma is categorized as high-grade and low-grade carcinoma. High grade serous ovarian cancer (HGSOC) is the most common (∼90% of serous cases) and lethal subtype of EOC and originates within the fallopian tube epithelium. It is often diagnosed at an advanced metastatic stage.6,7
2.1. Therapeutic options
Current treatment for ovarian cancer is debulking surgery followed by combination chemotherapy with cisplatin, a synthetic chemotherapeutic agent (Section 3.1), and the botanical natural product chemotherapeutic agents paclitaxel and camptothecin (Section 4 and 5, respectively). Following the first line of treatment, ovarian cancer patients are typically prescribed maintenance therapy such as poly ADP ribose polymerase (PARP) inhibitors and angiogenesis inhibitors (Section 3.2).8,9 Furthermore, the response to intraperitoneal (IP) chemotherapy has shown more benefits and this influences the importance of IP therapy in preclinical models of early drug testing. While first line treatments are efficacious in combating the disease, resistance to chemotherapy continues to be a major clinical challenge10 that necessitates the continued discovery and development of new agents with alternative mechanisms of action.2.2. Commonly studied cell lines
EOC is heterogenous and is accompanied by inter-patient and intra-tumoral heterogeneity.11 To identify new therapies and study their mechanism of action in ovarian cancer, it is crucial that well characterized and clinically relevant cell lines are used, taking into consideration the site of origin and mutations commonly observed in patients. Recent studies have summarized the frequently used cell lines and identified those with common features to ovarian tumors in terms of genetics, growth characteristics and tumor histology.11–14 Beaufort et al. characterized 39 ovarian cancer cell lines commonly used for in vitro studies and classified them in terms of subtypes (histological and morphological), clinical pathological characteristics and prognosis.11 Since HGSOC accounts for two-thirds of ovarian cancer deaths, it is the most extensively studied subtype and urgently requires novel effective therapies. Domcke et al. performed a thorough genetic analysis of 47 ovarian cancer cell lines and identified cell lines with the highest genetic similarity to ovarian tumors.12 This study ranked cell lines in terms of suitability as HGSOC models and characterized three major genomic features of HGSOC namely high copy number alterations (CNAs), universal TP53 (tumor protein P53) mutation and low frequency of somatic mutation in protein-coding regions.12 Mutations in TP53 can lead to 3 different phenotypes: loss-of-function (LOF), dominant-negative (DN) and gain-of-function (GOF). Loss of wild type TP53 function can be due to loss of DNA binding activity (LOF) of TP53 or a DN effect, wherein the mutated allele inhibits the function of the wild type allele. Most common missense mutations of TP53 (example: R273H, R175H, G245S, R249S, R273C) retain partial wild type TP53 activity. These mutants, however, acquire novel oncogenic functions and are referred to as GOF mutants. HGSOC shows >90% frequency of TP53 mutations, 80% of which are missense mutations located in the DNA binding domain. Thus, due to their frequency and clustering in the DNA binding domain, most TP53 mutations in ovarian cancer are referred to as GOF mutants.15–17 Another comprehensive analysis by Mitra et al. defined HGSOC cell lines which reliably formed intraperitoneal HGSOC histology. Six cell lines namely OVCAR8, OVCAR3, OVCAR4, OVCAR5, OVSAHO, CAOV3 reliably formed intraperitoneal HGSOC tumors, with only OVCAR8 model forming ascites.14 Further, these cell lines were characterized for common functional assays such as proliferation, clonogenicity, invasion, migration, EMT phenotype and cisplatin resistance and were demonstrated to have a range of phenotypic efficiencies.18 The tumor microenvironment of ovarian cancer has been demonstrated to have low immunogenic potential making HGSOC less responsive to immune therapies.19 Hence, it is crucial to use in vivo models using the correct cell of origin for studying immune response against HGSOC. Since most ovarian cancers originate in the fallopian tube, recent studies have employed syngeneic models using murine fallopian tube epithelial cells with common genetic alterations typical of HGSOC.20–22 These cell lines can be injected in immune intact mice to study the relationship between tumor genetic alterations and response to immune therapies. Previously, the Burdette lab has generated FTE derived syngeneic models with alterations in Pten, Trp53, KRAS and allografted these cell lines in mice to evaluate immune responses.20 In another comprehensive study by Iyer et al., the authors engineered a panel of murine fallopian tube cell lines with alterations in Trp53, Brca1, Pten, Nf1, Ccne1, Akt2, KRAS etc. providing a valuable tool for identifying novel immunotherapy targets in HGSOC.21 Additionally, drug resistant cells can prove to be a valuable tool to study acquired secondary mutations, predict response to therapies and devise strategies to overcome resistance. As an example, PEO1 is BRCA2 mutant cell line demonstrating sensitivity to cisplatin and PARP inhibitors, whereas PEO4 is the reverted BRCA2 proficient cell line derived from the same patient that demonstrates resistance to cisplatin and PARP inhibitors.23 Thus, using an appropriate cell model is not only important for translating in vitro findings into an in vivo model but is crucial in terms of clinical implications.3. Non-botanical derived therapeutics approved for use against ovarian cancer
3.1. Platinum derivatives
Cisplatin (1) represents the first platinum-based ovarian cancer therapeutic (Fig. 1).24 This synthetic chemotherapeutic agent was serendipitously discovered by Rosenberg25 in 1965 and gained approval for clinical use in the USA and Europe in the late 1970's. Cisplatin is composed of a central platinum II with a cis-relationship between both the chlorine and ammonia groups attached to the platinum. Cisplatin and its more recent FDA approved analogues, carboplatin (2) and oxaliplatin (3), are activated upon hydrolysis within the body and subsequently scavenge biological nucleophiles such as DNA. In addition, nedaplatin (4) has been approved for use in Japan, heptaplatin (5) for use in China, and lobaplatin (6) for use in South Korea.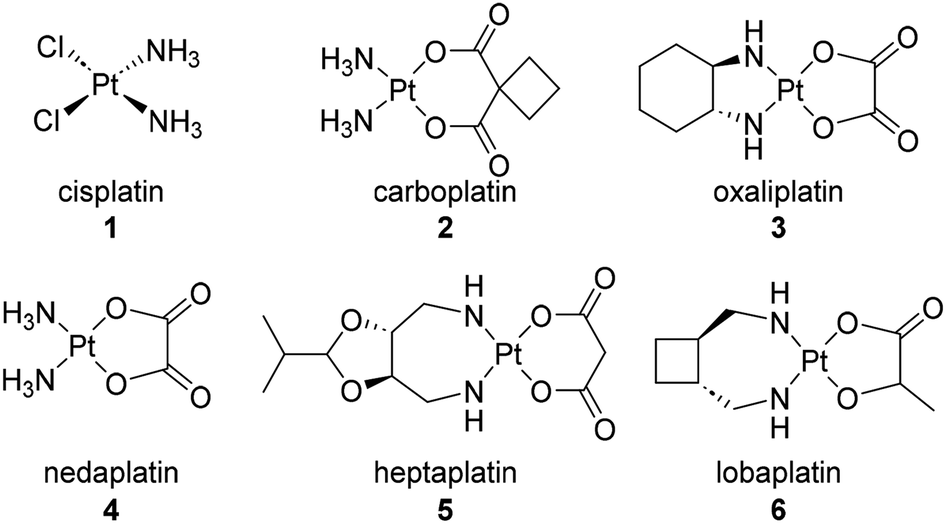 | ||
| Fig. 1 Platinum-based ovarian cancer therapeutics. | ||
Cisplatin is commonly mistaken for a DNA alkylating agent because it can cross-link DNA. However, unlike true DNA alkylating agents, it bears no alkyl functionality.24 Platinum based chemotherapies have been well investigated in their ability to elicit DNA damage and block DNA replication once bound to the purine bases. Other mechanisms of action include increasing overall cellular oxidative stress through the overproduction of mitochondrial reactive oxygen species and non-specific binding to proteins in essential pathways.24,26 While platinum based therapeutics have represented the first line of defense for several decades, their efficacy has come into question as side effects and increasing incidence of resistance have occurred. Known resistance mechanisms against platinum-based chemotherapies include decreased drug accumulation, increased efflux, factors preventing localization of the drug with its target, and cell repair mechanisms.24 One way to modulate identifiable platinum resistance is treating the interfering transporters or cell receptors with known efflux inhibitors. However, it is often the case that resistance mechanisms are hard to identify making the treatment less effective.
3.2. Other non-botanical therapeutics approved for the treatment of ovarian cancer
In addition to platinum-based compounds, other FDA approved small molecule therapies for ovarian cancer that were not derived from botanical natural products include the PARP inhibitors olaparib (7, Fig. 2), niraparib (8), and rucaparib (9), the fluorinated nucleoside prodrug gemcitabine (10), and the microbial anthracycline doxorubicin (11), which have previously been reviewed,27 and the organophosphorus compound thiotepa (12).28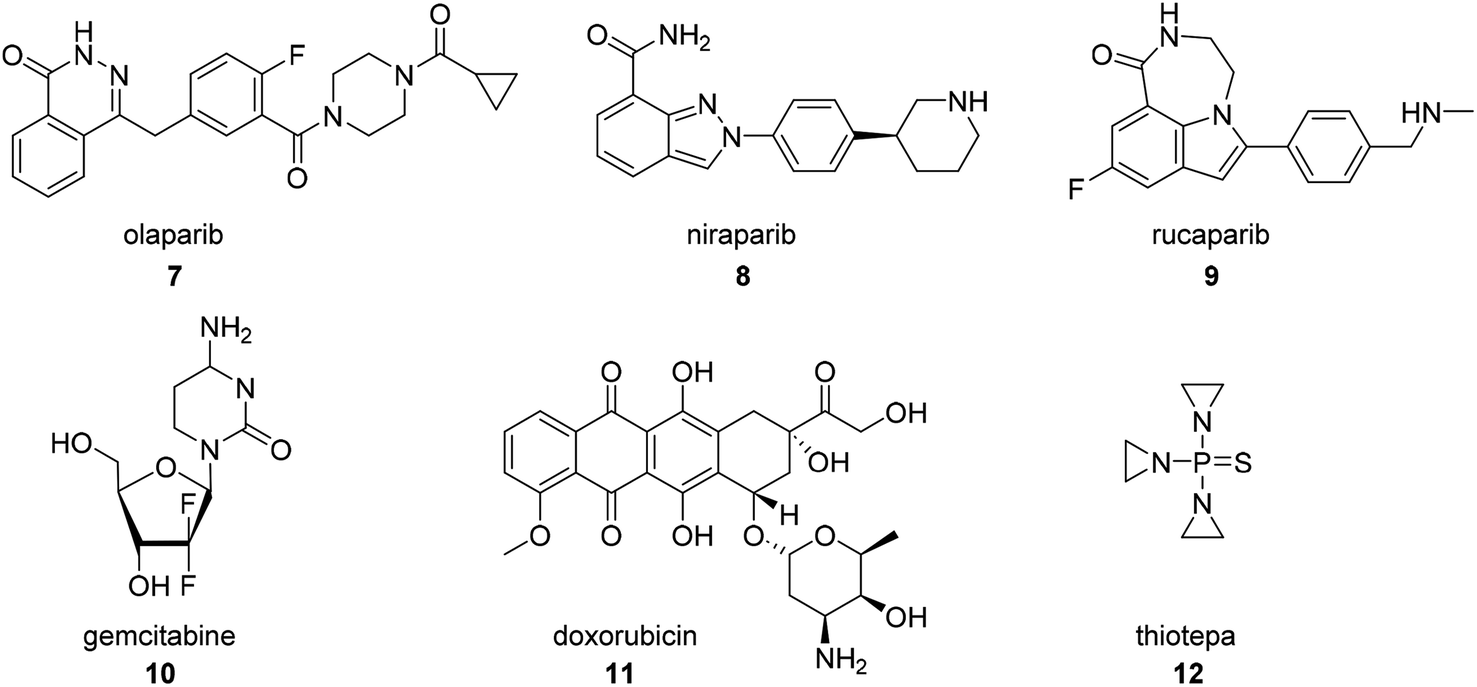 | ||
| Fig. 2 Structures of other non-botanical therapeutics approved for the treatment of ovarian cancer. | ||
4. Taxanes
In 1963, samples of the Pacific Yew tree, Taxus brevifolia, were collected by the United States Department of Agriculture in collaboration with the Cancer Chemotherapy National Service Center of the National Cancer Institute. Although the extracts of the bark sample were found to be cytotoxic against a keratin-forming tumor cell line (KB), they generated only modest initial interest in subsequent development as a number of labs were concerned about their potentiastion. This group was able to successfully isolate the active constituent, paclitaxel (13, Fig. 3), despite obtaining only a low 0.01% yield from the crude bark.29 Interestingly, although the purified compound was obtained and assigned a unique identifier within about a year of receiving the sample, structural elucidation proved to be exceedingly difficult based on the complexity of the ring system and determination of the position of the side chain. Ultimately, the structure was not reported until 1971.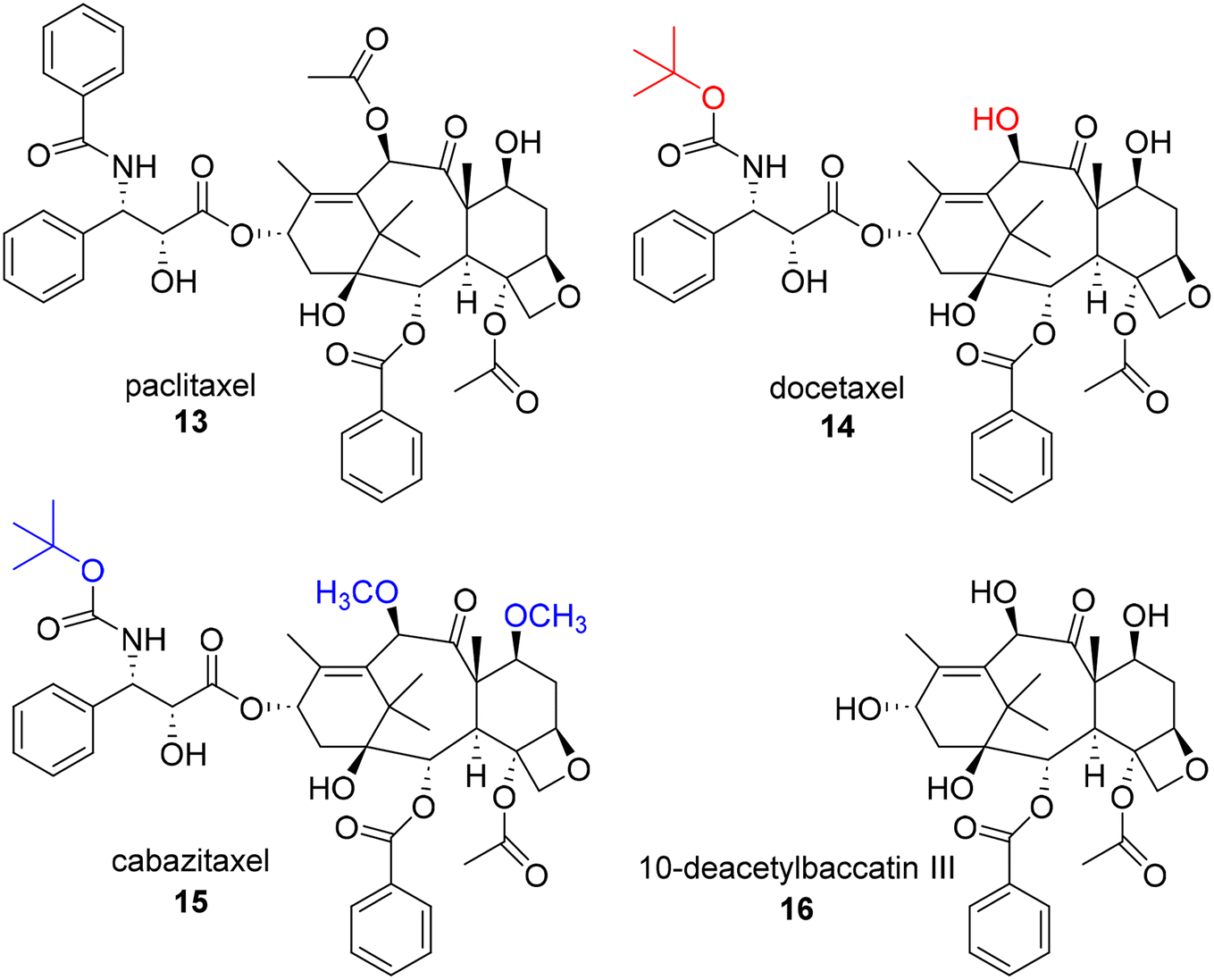 | ||
| Fig. 3 Structures of first and second generation taxanes and 10-deacetylbaccatin III. | ||
Despite this unique chemical structure, paclitaxel faced a number of challenges that needed to be overcome in order to advance to the clinic, including issues related to in vivo activity and drug properties. Surprisingly, in early in vivo assays, paclitaxel showed only moderate activity. Paclitaxel was only prioritized for further development due to promising results specifically obtained in a B16 mouse melanoma experiment, which was recognized as a useful model for both metastasis and solid tumor formation, and its progression benefitted greatly from the development of new xenograft models.30–32 Paclitaxel also demonstrated very poor water solubility, limiting its ability to be administrated effectively in vivo. This was overcome using a formulation of Cremaphor EL. In addition, there was limited knowledge of the mechanism of action of paclitaxel during early development, suggesting only that it inhibited proliferation at the G2-M phase in the cell cycle. Due to this common, resistance prone mechanism of action, excitement for the project once again wavered. Other G2-M modulators had also been found to potently kill cancer cells by “poisoning” the spindle fibers, but all accrued similar observable resistance over time. Fortunately, the NCI assigned a grant to Susan Horwitz to explore this mechanism in more detail. She found that paclitaxel had a unique mechanism of stabilizing microtubules which prevents the cellular structures from reverting back to tubulin, ultimately resulting in mitotic catastrophes and inhibiting mitosis.31,33 While the resistance was still a common issue among the G2-M modulators and paclitaxel, the discovery of paclitaxel's unique mechanism of action expedited its advancement into the clinic. In order to get the compound into large scale studies, however, the low availability of the compound from the bark of the slow growing tree also needed to be addressed. This was solved through the development of efficient semi-synthetic routes to paclitaxel from 10-deacetylbaccatin III (16) by Potier34 and Holton.35 Importantly, 16 is available from the needles of various Taxus species, making it much easier to obtain the required quantities of paclitaxel in a non-destructive, environmentally friendly manner and facilitating the preparation of novel paclitaxel analogues. For example, first generation taxane docetaxel (14) and second generation taxane cabazitaxel (15), are both semi-synthetic analogues derived from 16. These compounds possess t-butylcarbamate groups at the C3′ position rather than the amide found in paclitaxel and also lack the acetyl group at the C10 position. In addition, the secondary alcohols at positions C7 and C10 of cabazitaxel are methylated in comparison to 16. Notably, although nursery cultivation of Taxus species has remained an important source of 16 for the production of paclitaxel, additional methods, including cell culture, have also been explored and utilized for production.36
The first generation taxanes, including paclitaxel (FDA approval 1992), docetaxel (14, FDA approval 2004), and the second generation taxane cabazitaxel, possessing modifications at both the C3′ and C10 positions (15, FDA approval 2010) were in high demand for continued research despite causing undesirable side effects, possessing low aqueous solubility, and low natural abundance.37,38 Thus, in the 1980's and 90's, many research groups took on the task of synthesizing semisynthetic taxanes to address these issues. There are many additional individual accomplishments that collectively help paint the early SAR picture of paclitaxel and help to map out the key pharmacophore. A comprehensive outline of the SAR and second and third generation taxanes (characterized by additional structural modifications on C-2-benzoyl group) can be found in a review by Kingston and coworkers.30
To date, the only FDA approved taxanes are formulations, combinations, or delivery variations (i.e. nanoparticle) of the first and second generation taxanes (paclitaxel and doceltaxel, and cabazitaxel, respectively) listed above. While traditional SAR efforts have not yet led to a new FDA approved structural analogue, recent research is focused on binding differences between first and third generation taxoids with tubulin/microtubules. These differences point to promising effects on cytotoxicity, drug resistance, and tumor efficacay.37,38 Building on previously published work, studies into resistance to paclitaxel have implicated a number of contributing protein targets, including βIII tubulin expression,45–47 the efflux protein p-glycoprotein (P-gp),39 Aurora A kinase,40 and IKK-β.41 In addition, molecular dynamic studies have revealed some potential causes of resistance, thereby influencing current taxane SAR studies.42 Notably, members of the taxane class continue to be developed for ovarian cancer therapy and several examples of these recent advances are presented in Section 6.4 SAR and 6.6 Delivery/activity aids below.
5. Camptothecins
Not only did Wani and Wall elucidate the structure of paclitaxel, but five years prior they also played a significant role in the discovery and development of camptothecin (17, Fig. 4) as an anticancer agent. After displaying potent activity against cancer cells, extracts from the wood and bark of C. acuminata were further fractionated to identify the active component in 1963. This compound, camptothecin, showed unusually potent activity in an L1210 mouse leukemia assay, pushing the project forward into clinical trials. Unfortunately, clinical trials could only be conducted with the salt form of the natural product, which proved to be more soluble, but ultimately less potent.43 Following clinical trial results, formulation efforts were made to achieve a balance in solubility and potency. In addition, the elucidation of the mechanism of action was prioritized given these outcomes. Yaw-Huei Hsiang and coworkers discovered that while camptothecin damages DNA, the effect was not through direct binding. Instead, they proposed that camptothecin was binding to human topoisomerase I (topo I) and stabilizing the covalent protein–DNA complex, causing an observable buildup of single stranded DNA breaks in an enzymatic assay.44 This event blocks the advancement of the replication fork, prevents DNA replication and signals downstream apoptotic events. This mechanism has subsequently been confirmed by other research groups and topo I is recognized as the target of camptothecin and its structural analogues.45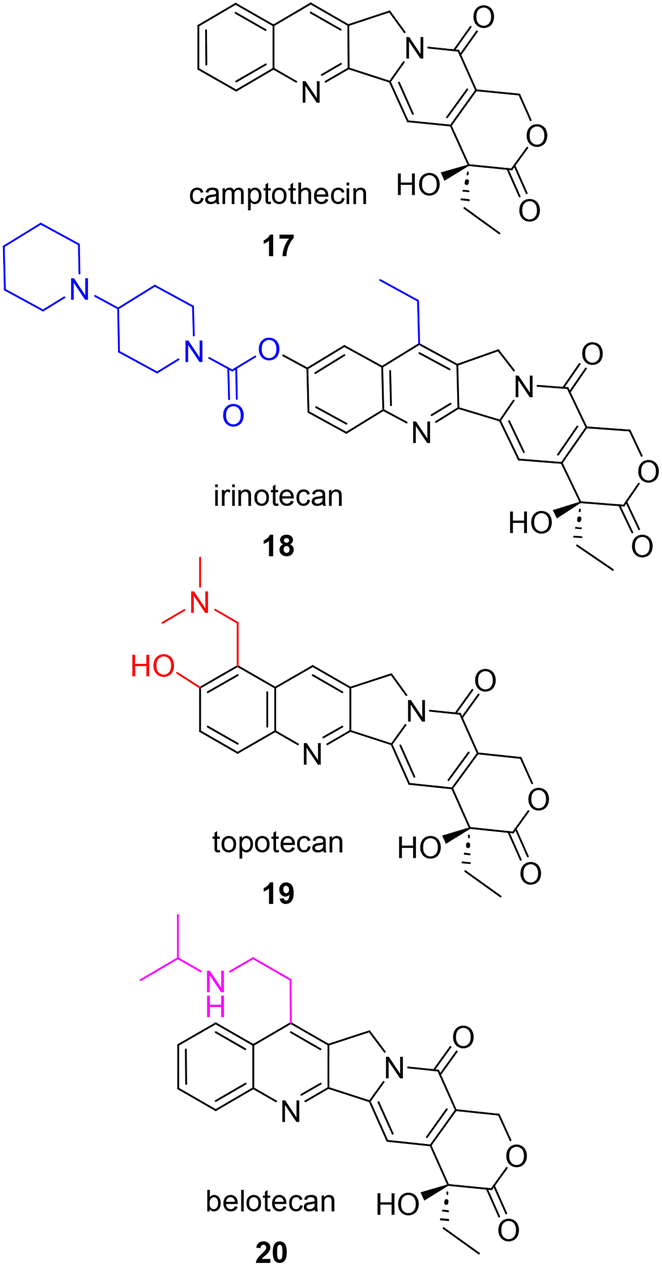 | ||
| Fig. 4 Structure of camptothecin and highlighted structural modifications on subsequent analogues approved for the treatment of ovarian cancer. | ||
The discovery of topo I as the target of camptothecin led to the pursuit of improving the bioavailability and effectiveness of this compound as a clinical agent. By the 1990's, two synthesized analogues, irinotecan (18), a prodrug version converted to the active camptothecin in vivo, and topotecan (19), which includes two additional polar, heteroatom-containing groups on the A ring, were FDA approved in 1996.46 Belotecan (20) is also approved for clinical use in South Korea for the treatment of small-cell lung cancer and ovarian cancer. Similar to topotecan, belotecan possesses an amine group on the A ring that leads to greater aqueous solubility when compared to that of camptothecin. Unfortunately, all camptothecins suffer from poor chemical stability due their quick inactivation at physiological pH by ring opening at the cyclic lactone.47 In addition, these clinically approved drugs come with their own set of side effects and are susceptible to resistance over time. The main side effects that have limited the use of camptothecins are hemorrhagic cystitis and severe bone-marrow suppression.48 A recent review by Verma and Hansch summarizes in great detail the three general mechanisms of camptothecin resistance: reduced cellular accumulation, alteration of structure of location of protein target (topo I), and alterations in the cellular response to the inhibited camptothecin-DNA complex.45
Over the years, efforts to synthesize new camptothecin analogues capable of combating the known side effects and acquired resistance have focused on modifications to the D-ring, the lactone moiety, and the planarity of the remainder of the compound.45,49 In addition, irinotecan has been studied as a combination therapy for various cancers with known anticancer therapeutics such as kinase inhibitors, 5-fluorouracil (5-FU), oxaliplatin, monoclonal antibodies, and cell cycle checkpoint inhibitors.50 Since the approval of irinotecan, topotecan, and belotecan, liposomal irinotecan injection (NaI-IRI) in combination with 5-FU and leuconorin was approved in 2015 to treat pancreatic ductal adenocarcinoma (PDAC). This PEGylated liposomal formulation encapsulates the active compound which in turn improves drug bioavailability and delays mononuclear phagocytic engulfment.50 As seen with the taxanes, development of the camptothecins has also appeared in the recent literature. These recent applications of camptothecins in the study of ovarian cancer are included in Section 6.5 PK/In vivo data and 6.6 Delivery/activity aids.
6. Recent studies on botanical NPs and associated drug development considerations
While the taxanes and camptothecins continue to play an important role in ovarian cancer therapy, the discovery and development of additional compounds with novel mechanisms of action are still needed to increase the number of therapeutic options available for treatment. As expected, natural products have remained an important source of hit and lead compounds for the discovery and development of these potential therapeutics. As evidence of the ongoing work in this area, more than 60 botanical natural products or classes of natural products have been reported and studied for their activities against ovarian cancer since 2015 (Table 1). These reported studies range from very early-stage discovery efforts using ovarian cancer cell lines as a primary assay for identification of compound activity to late-stage development efforts in which drug properties and delivery methods are carefully monitored to facilitate in vivo and clinical efficacy. In addition, both newly isolated compounds and existing drug classes, including both the taxanes and camptothecins, have been the subject of these research efforts.| Phytochemicalsa | Phytochemical Source | Class | Ovarian cancer cell line | In vitro effects (IC50 (μM) unless otherwise stated) | Drug discovery attributeb | Reference |
|---|---|---|---|---|---|---|
| a Numbers listed for phytochemicals refer to numbering of compounds in source publication. b P = Pains, MoA/BT = Mechanism of Action or Biological Target, T = Toxicity, SAR = Structure Activity Relationship, PK = Pharmacokinetics, V = in vivo, D/A = Delivery or Activity Aid. | ||||||
| (9βH)- and 17-Nor-pimaranes | Icacina oliviformis | Diterpenoids | OVCAR3 | 14α-Methoxyhumirianthol (6) = 6.3, annonalide (7) = 4.4 | J. Nat. Prod., 2021, 84, 949–955 | |
| 3α-O-(β-D-Glucopyranosyl)desoxypodophyllotoxin (1) and 4-O-(β-D-glucopyranosyl)dehydropodophyllotoxin (2) | Cleistanthus boivinianus | Lignans | A2780 | 1 = 0.033 ± 0.0036, 2 = 2.1 ± 0.3 | J. Nat. Prod 2015, 78, 7, 1543–1547 | |
| 7α-Hydroxyicacenone (5), and 2β-hydroxyhumirianthenolide C (6) | Icacina oliviformis | Pimarane derivatives | OVCAR3 | 5 = 7.53 6 = 3.23 | J. Nat. Prod, 2016, 79, 1815–1821 | |
| AD-1 and metabolites (M0–M16) | Panax notoginseng | Dammarane-type ginsenoside | HO-8901 | M6 = 2.086 | BT/MoA, SAR | Bioorganic chemistry, 2019, 88, 102961 |
| Amentoflavone | Selaginella tamariscina | Flavonoid | SKOV3 and OVCAR3 | AF exhibited dose- and time-dependent inhibition in both the cell lines | BT/MoA, V | Life sciences, 2017, 189, 96–105, https://doi.org/10.1016/j.lfs.2017.09.026 |
| Arborinine | Glycosmis pentaphylla | Alkaloid | OVCAR3 | GI50 = <10 (μg mL−1) | Journal of biologically Active products from nature, 2017, 7, 131–139, https://doi.org/10.1080/22311866.2017.1329666 | |
| Asiatic acid | Centella asiatica | Triterpenoid | SKOV3 and OVCAR3 | Concentration of 40 μg mL−1, asiatic acid caused about 50% reduction in the viability | BT/MoA | Pharmaceutical biology, 2016, 54, 2377–2382, https://doi.org/10.3109/13880209.2016.1156709 |
| Baicalein | Scutellaria baicalensis | Flavone | OVCAR5 | Activity is mediated through the glucocorticoid receptor | P, BT/MoA, V | Horm. Cancer, 2020, 11, 97–110 |
| Balanocarpol | Hopea dryobalanoides | Oligostilbeniod | A2780 | EC50 = 42.63 μM | P, T, D/A | Nanotechnology, 2020, 31, 195101 |
| Berbamine | Berberis amurensis | Bisbenzylisoquinoline alkaloid | SKOV3 and ES2 | (SKOV3 at 24 h) = 8.3 μg mL−1, (ES2 cells at 24 h) = 8.7 μg mL | BT/MoA | Acta Biochimica et Biophysica Sinica, 2018, 50, 532–539 |
| Berberine (BBR) | Multiple sources | Isoquinoline derivative alkaloid | OVCAR3, three patient-derived primary ovarian cancer cell lines, A2780, SKOV3 | BBR only = 99 ± 1.58 | P, D/A | Biological research, 2019, 52, 37. Cell. Physiol. Biochem. 2015, 36, 956–965. Cell Proliferation, 2017, 50, e12393 |
| Beta escin | Aesculus hippocastanum L. | Saponins | CaOV3, OVCAR5, OVKATE, Kuramochi, and Tyk-nu | EC50 (μM): (CaOV3) = 0.72, (OVCAR5) = 2.1, (OVKATE) = 0.75, (Kuramochi) = 2.4, (Tyk-nu) = 1.4 | BT/MoA, T, SAR, V | Cancers (basel), 2021, 13, 3931 |
| Betulinic acid (BA) | Betula pubescens | Pentacyclic triterpene | SKOV3 | 1.49 μg mL−1 (pH 6.5) and 3.41 μg mL−1 (pH 7.4) | BT/MoA, T, V, D/A | International Journal of Pharmaceutics, 2019, 571, 118751, https://doi.org/10.1016/j.ijpharm.2019.118751 |
| Boeravinones (ZML-1–24) | Boerhavia diffusa | Rotenoid | A2780 | ZML-23 = 5.12 ± 0.83 | SAR | Bioorganic chemistry, 2022, 122, 105747, https://doi.org/10.1016/j.bioorg.2022.105747 |
| B-type podolactones (1–4) | Podocarpus neriifolius | Diterpenes | OVCAR3 | 1 = 2.9, 3 = 5.2 | V | Nat. Prod. Bioprospect., 2019, 9, 157–163 |
| Celastrol | Tripterygium wilfordii and Tripterygium regelii | Pentacyclic nortriterpen quinone | SKVO3 | Celastrol without nanoparticle = 1.4 μg mL−1 | BT/MoA, D/A | Front. Chem., 2020, 8, 574614, https://doi.org/10.3389/fchem.2020.574614 |
| Colchicine (11), (−)-3-demethyldemecolcine (13) | Androcymbium palaestinum | Alkaloid | OVCAR3 | 11 = 0.023, 13 = 2.230 | Fitoterapia, 2020, 146, 104706, https://doi-org.proxy.lib.ohio-state.edu/10.1016/j.fitote.2020.104706 | |
| Colchicine Analogues | Liliaceae family | Alkaloid | SKOV3 | 7 (2018 report) = 0.0013 2 (2021 report) = 0.008 | BT/MoA, SAR, V | Med. Chem. Commun., 2018, 9, 1708–1714. Sci. Rep., 2021, 11, 9034 |
| Cryptocaryone | Cryptocarya spp. | Dihydrochalcone | SKOV3, TOV-21G, and TOV-112D | (TOV-21G) = 1.5, (SKOV3) = 3, (TOV-112D) = 9.5 | BT/MoA | Cells, 2022, 11, 641 |
| Cucurbitacin I | Cucurbitaceae | Tetracyclic triterpenoid | SKVO3 | 0.3 | BT/MoA | Mol. Medi. Rep., 2020, 22, 2545–2550 |
| Curcumin | Curcuma longa | Curcuminoid | Various: reference review | P, BT/MoA, SAR, V | Nutrition and cancer, 2022, 1–18, https://doi-org.proxy.lib.ohio-state.edu/10.1080/01635581.2022.2049321 | |
| Cycloviolacin O8 | Viola odorata | Anthelmintic cyclotie | OVCAR3 | 0.80 | Phytochemistry, 2018, 152, 61–70, https://doi.org/10.1016/j.phytochem.2018.04.014 | |
| Deoxyschizandrin | Schisandra chinensis | Lignan | A2780, SKOV3, and OVCAR3 | (A2780) = 27.81, (OVCAR3) = 70.34, (SKOV3) = 67.99 | BT/MoA | Nutrients, 2018, 10, 91 |
| Desmiflavasides | Desmidorchis flava | Pregnane glycosides | SKOV3 | Desmiflavaside C (1) = 64.5, desmiflavaside D (2) = 38.3, nizwaside (3) = 37.9 | BT/MoA | Bioorganic chemistry, 2016, 67, 95–104 |
| Digoxin | Digitalis lanata Ehrh. | Cardiac glycosides | OVCAR3 | 0.1 | BT/MoA, T, SAR | Molecules 2021, 26, 3672; J. Nat. Prod, 2020, 83, 638–648 |
| Dihydroartemisinin (DHA) | Artemisia annua | Terpenoid/artemisinin | SKOV3, HO8910-pm | 20 μM of DHA resulted in approximately 50% cell death in SKOV3-IP and HO8910-PM cells after 48 hours DHA treatment | P, BT/MoA | Cancer Medicine, 2018, 7, 5704–5715, https://doi.org/10.1002/cam4.1827 |
| Ellagic acid | Punira granalium | Polyphenol | A2780, ES-2 | Dose- and time-dependent responses were observed | P, BT/MoA, V | Cancer Biol. Ther., 2017, 18, 990–999 |
| Emodin | From rhizomes of Rhubarb, aloes, and other plants | Anthraquinone | A2780, SKOV3 | Significant inhibition at 40 μM and 80 μM | P, BT/MoA | BioMed research International, 2016, 2016, e6253280 |
| Evodiamine | Tetradium | Dietary supplement | A2780 (180 microarray) | 2.18 | BT/MoA, V | PLOS one, 2015, 10, e0132579, https://doi.org/10.1371/journal.pone.0132579 |
| Galtonosides A–E (3–7) | Galtonia regalis | Cholestane glycosides | A2780 | 3 = 0.09 ± 0.01 | J. Nat. Prod, 2020, 83, 4, 1043–1050 | |
| Gedunin | Xylocarpus granatum | Tetranorterpenoid | OVCAR3, PA-1 | (OVCAR3) = 18, (PA-1) = 8.1 | BT/MoA | Apoptosis, 2020, 25, 481–499 |
| Ginkgolide B analogues (1–11) | Ginkgo biloba | Diterpenoid | SKVO3 | 2 = 16.05, 3 = 15.65, 6 = 32.00, 7 = 63.30, 10 = 23.20, and 11 = 31.10 | SAR | Med. Chem. Res., 2021, 30, 1265–1272 |
| Ginsenoside-Rh2 | Panax ginseng | Ginsenosides | SKVO3 | Dosage of 60 μM displayed inhibition of proliferation, migration and invasion of cancer cells | P, BT/MoA | Physiol. Res., 2016, 65, 1031–1037 |
| Harmine | Peganum harmala | Beta-carboline alkaloid | SKOV3 | 5 μM progressively inhibited the growth of SKOV3 cells | P, BT/MoA | Oncology reports, 2017, 38, 2927–2934, https://doi.org/10.3892/or.2017.5952 |
| Hederagenin | Sapindus saponaria | Pentacyclic triterpene | A2780 | EC50 values ranging from 1.1–19.9 μM | BT/MoA, SAR | European Journal of medicinal chemistry, 2015, 105, 57–62 |
| Icacinlactones A–G (1–7) | Tuber of Icacina trichantha | 17-Norpimarane and (9βH)-17-Norpimarane diterpenoids | OVCAR3 | 1 = 17.76, 6 = 10.5 | J. Nat. Prod, 2015, 78, 4, 789–796 | |
| Inumakilactone A (1–3) | Podocarpus neriifolius | B-type podolactones | OVCAR3 | 1 = 2.9, 3 = 5.2 | V | Nat. Prod. Bioprospect., 2019, 9, 157–163 |
| Irilone | Red clover (Trifolium pratense) | Flavone | PEO1 | Potentiated the effect of progesterone in both endometrial and ovarian cancer cell lines | D/A | J. Nat. Prod, 2021, 84, 12, 3090–3099 |
| Isoliquiritigenin | Glycyrrhiza spp. | Chalcones | OVCAR5, ES-2 | (OVCAR5) = 11, (Es-2) = 25 | P, BT/MoA | International Journal of molecular sciences, 2017, 18, 2025 |
| Kadsuphilactone B | Schisandra chinensis | Nortriterpenoid | A2780 | 23.25 ± 1.46 | BT/MoA | Arch. Pharmacal Res., 2017, 40, 500–508, https://link.springer.com/article/10.1007/s12272-017-0902-5 |
| Lignans (1–5) | Trachelospermum asiaticum | Lignans | SKOV3 | 1 = 63.3, 2 = 23.8, 3 = 72.7, 4 = 24.7, 5 = 23.3 | Biomolecules, 2020, 10, 378 | |
| Ligustrazine | Ligusticum wallichii | Alkaloid/pyrazine | SKOV3 and OVCAR3 | Increase the expression of miR-211 | BT/MoA | Bioscience reports, 2021, 41, BSR20200199 |
| Luteolin | Punira granalium | Flavonoid | A2780, ES-2 | Dose- and time-dependent responses were observed | P, BT/MoA, V | Cancer Biol. Ther., 2017, 18, 990–999 |
| Magnolol-sulforaphane hybrids | Magnolia officinalis and cruciferous vegetables | N/A | OVCAR3 | (OVCAR3) = 5.34 ± 0.28 | BT/MoA, V, d/A | European Journal of medicinal chemistry, 2020, 199, 112441, https://doi-org.proxy.lib.ohio-state.edu/10.1016/j.ejmech.2020.112441 |
| Methyl lucidone | Lindera erythrocarpa | Cyclopentenediones | OVCAR8, SKOV3 | (OVCAR-8) = 33.3–54.7, (SKOV-3) = 48.8–60.7 | P, BT/MoA | Pharm. Biol., 2020, 58, 51–59 |
| Other cardiac glycosides (1–3) | Bark of Streblus asper | Cardiac glycosides | OVCAR3 | 2 = 0.62 | BT/MoA, T | Bioorganic & medicinal chemistry, 2020, 28, 115301 |
| Pentacyclic triterpenoids (1–12) | Syzygium corticosum | Steroid | OVCAR3 | 1–12 greater than 10.0 | BT/MoA, SAR | Bioorganic & medicinal chemistry, 2018, 26, 4452–4460, https://doi-org.proxy.lib.ohio-state.edu/10.1016/j.bmc.2018.07.025 |
| Phloroglucinols dauphinols (1–7) | Garcinia dauphinensis | Phloroglucinols | A2780 | 1 = 4.5 ± 0.9, 2 = 15.2 ± 0.3, 3 = 7.0 ± 2.4, 6 = 6.4 ± 0.2 | J. Nat. Prod, 2019, 82, 3, 431–439 | |
| Phyllanthusmin (6–18) | Phyllanthus poilanei | Arylnaphthalene lignan lactones | OVCAR3 | 6a = 0.29 | SAR | Bioorg. Med. Chem., 2018, 26, 2354–2364 |
| Phyllanthusmin analogues (PHY-25, 30, 34) | Phyllanthus poilanei | Arylnaphthalene lignan lactones | OVCAR3 and OVCAR8 | PHY=34 (both cell lines) = 0.004 | SAR, BT/MoA, PK, V | Mol. Cancer Ther., 2018, 17, 2123–2135. Cell Death Dis., 2022, 13, 1–13 |
| Polyphyllin D (PD) | Paris polyphylla | Saponin | A panel of 20 OVCA cell lines | IC50 values ranging from 0.2 to 1.4 μM. PD treatment significantly decreased cisplatin IC50 when co-administered | D/A | Journal of cancer research and clinical Oncology, 2015, 141, 237–242 |
| Propolones A–D (1–4), propolonones A–C (5–7) | Brazilian red propolis | Flavonoid-derived dimers | NCI-ADR/RES | Total growth inhibition (TGI (μM)) 2 = 19.1 ± 2.4, 5 = 29.9 ± 3.4 | J. Nat. Prod, 2020, 83, 6, 1784–1793 | |
| Quercetin | Various | Dietary flavonoid | Various: reference review | BT/MoA, D/A, P | J. Ovarian Res., 2019, 12, 1–9 | |
| Secoiridoid glycosides (2–7) | Cornus officinalis | Secoiridoid | Ishikawa | Potentiated the effect of progesterone in the PRE/Luc assay | BT/MoA | J. Nat. Prod, 2021, 84, 2612–2616 |
| Secoiridoids, demethoxy-cornuside (1–7) | Cornus officinalis | Secoiridoid | Ishikawa | Four phytoprogestins (1, 2, 6, 7) potentiated the effect of progesterone in the PRE/Luc assay | BT/MoA | J. Nat. Prod, 2021, 84, 9, 2612–2616 |
| Silybin analogues (15a–k) | Silybum marianum | Flavonolignans | OV2008, A2780 | 15k(OV2008) = 0.8 ± 0.1; (A2780) = 1.0 ± 0.1 | BT/MoA, SAR | European Journal of medicinal chemistry, 2017, 133, 365–378, https://doi.org/10.1016/j.ejmech.2017.03.033 |
| Strebloside | Bark of Streblus asper | Cardiac glycosides | OVCAR3, OVCAR4, OVCAR5, OVCAR8, kuramoch, Ovsaho | (OVCAR3) = 0.134, (OVCAR4) = 0.457, (OVCAR5) = 0.541, (OVCAR8) = 0.091, (kuramochi) = 3.437, (Ovsaho) = 0.560 | BT/MoA, T | J. Nat. Prod, 2017, 80, 3, 659–669 |
| Sulforaphane | Cruciferous vegetables | Dietary isothiocyanate | OVCAR3, OVCAR4, OVCAR5, and SKOV3 | (OVCAR cell lines) = 6.2–6.3, (SKOV3) = 3.6 | BT/MoA | Food & Nutrition research, 2017, 61, 1368321, https://doi.org/10.1080/16546628.2017.1368321 |
| Tanshinone | Salvia miltiorrhiza | Diterpene quinone | TOV-21G and SKOV3 | (TOV-21G) = 4.9 ± 0.6, (SKOV3) > 30 | BT/MoA | Chem. Res. Toxicol., 2015, 28, 1574–1583 |
| Thymoquinone analogues (1–22) | Nigella sativa | Benzoquinone | A2780, OVCAR8, CIS-A2780 | 6(A2780) = 6.2 ± 0.2, (OVCSR8) = 5.6 ± 0.4, (CIS-A2780) = 4.7 ± 0.5 | SAR | Bioorganic & medicinal chemistry Letters, 2018, 28, 1219–1222, https://doi.org/10.1016/j.bmcl.2018.02.051 |
| Trihydroxyalkylcyclohexenones | Pleiogynium timoriense | Hydroxy alkylcyclohexenones | A2780 | 1 = 0.8 ± 0.4, 2 = 0.7 ± 0.3, 3 = 0.8 ± 0.5 | J. Nat. Prod., 2015, 78, 7, 1752–1755 | |
| Triptolide | Tripterygium wilfordii | Diterpene triepoxide | A2780 | Confirmed the prodrug was a substrate of alkaline phosphatase | BT/MoA, T, PK, V, D/A | J. Med. Chem., 2015, 58, 23, 9334–9344 |
| Vibsanin A (1–6) | Viburnum awabuki | Vibsanins | ES-2 | 1 = 2.9, 4 = 4.3, 5 = 7.5 | BT/MoA, SAR | Bioorganic & medicinal chemistry, 2020, 28, 115253, https://doi.org/10.1016/j.bmc.2019.115253 |
| Voacamine (VOA) | Peschiera fuchsiaefolia | Bisindole alkaloid | A2780 and A2780 DX | Sensitizing effect observed | BT/MoA, d/A | Toxicology and Applied Pharmacology, 2022, 434, 115816, https://doi.org/10.1016/j.taap.2021.115816 |
Natural product drug discovery campaigns generally follow a relatively standard procedure: bioactivity-guided isolation, structure elucidation and purification, compound activity confirmation, hit-to-lead analogue development and target identification, lead optimization to improve potency or ADME properties, in vitro and selectivity studies, and finally preclinical pk studies.51,52 The end goal for promising drug candidates is progression into clinical studies and drug approval, but there are numerous challenges and considerations facing drug discovery researchers in the development of new therapeutics. Many of these challenges are based on the overall properties of the compounds being discovered. These properties can frequently be traced back to structural features found in the molecules. In addition, many compounds fail to advance because key biological data is not successfully obtained to support continued development of a potential compound. With this in mind, key drug discovery features or development stages have been highlighted for each of the compounds in Table 1. The “drug discovery attributes” in the last column of the table is intended to indicate the status of compounds in the drug discovery process and help to indicate the potential for compounds to advance. This analysis is specific towards only reported ovarian cancer efforts and is based on work referenced or performed in the publications being reviewed. In addition, the table also includes the phytochemical source, class, and summarizes the anti-ovarian cancer in vitro or in vivo activity of each compound. The drug discovery attributes considered, including PAINS analysis, mechanistic studies, drug toxicity, structure–activity relationships, pharmacokinetic and in vivo studies, and delivery aids, are explained in detail below along with commentary on recent publications that are representative for each attribute.
6.1. PAINS (P)
Compounds that appear as frequent hitters in bioassays are sometimes referred to as “promiscuous binders”53 or alternatively as “privileged scaffolds”54 depending on whether they can effectively be developed as promising drug leads. For compounds that contain reactive functionality or interfere with the assays, the term Pan Assay Interference Compounds (PAINS) is often used. These compounds and motifs were first identified by Baell and coworkers in 2010 in response to a high throughput screen analysis where several chemical motifs gave consistent false positive results.55 Since the initial publication, Baell has authored another thorough review of PAINS motifs with a special emphasis on natural products.56 These motifs include, but are not limited to, catechols, quinones, phenolic Mannich bases, polyphenols, and other reactive groups. Baell does warn that applying the PAINS criteria to natural products may not be relevant in some cases given the current FDA approved drugs inspired by natural products (i.e. topotecan). He argues that the identification of PAINs is most relevant to hit-to-lead drug discovery efforts while also noting that some FDA approved natural products have been found to have very high activity and required little or no elaboration thereafter.Identification of commonly known PAINS-containing natural products may aid modern drug discovery efforts by increasing efficiency in the development process. Identifying a natural product ahead of biological testing as having the propensity to promiscuously bind could help researchers make more informed decisions about the subsequent assays to perform or the overall chance of the compound moving forward toward FDA approval. The identification of PAINS also suggests that biological data must be carefully analyzed before committing to advancement of a compound class. Some common PAINs natural products that are mentioned in Baell's review and have also recently been studied as potential hit or lead compounds against ovarian cancer are artemisinin,57 curcumin (recently reviewed by Mohamadian et al.),58 resveratrol,59 berberine,60–62 thymoquinone,63 and topotecan.64 While these recent studies have added to the anti-ovarian cancer landscape, their reputations as frequent hitters may suggest further evaluation prior to initiation of a drug development program.
6.2. Mechanism of action/biological target (MoA/BT)
Identification of the molecular targets of a natural product can provide insight into the potential use of that compound as a therapeutic agent. Selectivity and inhibition of a novel target are important factors for anticancer drug discovery projects to advance into the clinic. While knowing the protein target of a compound is essential for late-stage drug discovery, the mechanism of action is frequently elucidated first. A recent review provides a summary of the primary mechanisms of action through which natural products cause anti-ovarian cancer effects. The review categorizes the functions and mechanism of natural botanical components applied in human ovarian cancer, including cytotoxic effects, inhibition of proliferation and promotion of apoptosis, suppression of cell migration and invasion, damage due to reactive oxygen species (ROS), cell cycle arrest, induction of autophagic cell death, inhibition of angiogenesis, interference with RNA expression, and promotion of DNA damage response.65 Once a biological function or mechanism is identified, target identification can then be narrowed down to the proteins involved in the pathways of interest. Discovering the broader mechanism of action is a beneficial first step in identifying an eventual protein target.One such example of the process from phenotypic observation to target elucidation is the report of the phyllanthusmin class of compounds by Kinghorn, Burdette, and Fuchs.66 Previously, the Kinghorn lab reported the isolation of two related series of arylnaphthalene lignan lactones, the phyllanthusmins and the acutissimalignans from Phyllanthus poilanei and Phyllanthus songboiensis, respectively.67,68PHY-34 (21) is the synthetic lead derived from the natural product hit Phylanthusmin D.66 The target elucidation story of PHY-34 began with sub-nanomolar in vitro potency observed in OVCAR3 and OVCAR8 cell lines.69 Encouraged by the initial potency, the Burdette lab found PHY-34 to be an autophagy modulator through LC3B puncta assay followed by an autophagic flux assay. Additive effects observed through co-administration of PHY-34 and late stage autophagy inhibitor Bafilomycin confirmed PHY-34 is a late stage autophagy inhibitor.69 Once the mechanism by which cell death was discovered, subsequent experiments were carried out to find the specific protein target within the autophagy pathway. Structurally, PHY-34 is similar to a Novartis compound, HTP-013, which had been published as a v-ATPase inhibitor.70PHY-34 displayed similar effects as HTP-013 in a series of cell lines expressing mutant ATP6V0A2 cDNA developed by Novartis.70 Mutation in ATP6V0A2 subunit reduced PHY-34 mediated cytotoxicity which suggests that PHY-34 interacts with the membrane-associated ATP6V0A2 subunit of the vacuolar ATPase (v-ATPase).71 Autophagy pathway is highly disrupted in ovarian cancer and studies have shown that patients with EOC develop resistance to PARP inhibitors due to elevated levels of autophagy.72–74 Hence, combining an autophagy inhibitor such as PHY-34 with existing chemotherapies may have tremendous potential to improve clinical outcomes for EOC patients.
For numerous natural products, however, these compounds may have a long history of biological response in the body that is still unattributed to specific protein targeting.75 The ruling out of a common anti-cancer pathway can be very useful in discovering a mechanism of action.76 For example, sulforaphane (22) is a well-known dietary isothiocyanate that is found in cruciferous vegetables and various epidemiological studies on the correlation between intake of cruciferous vegetables and reduced risk of particular cancers have been reported.75 Kim et al. dosed OVCAR3 MAPK pathway inhibitors to reveal that activation of MAPK pathways by sulforaphane is unlikely to mediate sulforaphane-induced growth inhibition. They also found that sulforaphane did not generate significant levels of intracellular ROS. While these specific pathways were ruled out as mechanisms of action, results of the ROS study led the researchers to investigate if thiol reducing agents can modulate anticancer effects in a dose-dependent manner. Ultimately, the researchers found that when co-dosing sulforaphane with thiol-reducing agents, the cytotoxic effects of sulforaphane diminished. They concluded that sulforaphane may act at least partly by interacting with protein thiols and altering the cellular thiol redox status. Therefore, the mechanism by which sulforaphane elicits its biological effects relies heavily on the preservation of the thiol pharmacophore.76
6.3. Toxicity (T)
Drug toxicity does not describe a singular toxic property of a potential drug but rather can encompass many mechanisms of toxicity at clinically relevant doses. The most common toxicities observed in the drug discovery process are cardiovascular and hepatic toxicity. There are five commonly accepted types of toxicity: on-target (mechanism based), hypersensitivity and immunological, off-target, biological activation (active metabolites), and idiosyncratic.77 Commonly, off-target toxicity such as hERG inhibition is assessed early on in drug discovery efforts since it has infamously halted drug discovery campaigns of the past. On the other hand, on-target toxicity is due to the desired target producing two responses, the anticipated pharmacological response and the toxic effects.77 Somewhere between these two definitions is the application of cardiac glycosides as anticancer therapeutics. While cardiac glycosides, such as the famous natural product digoxin (23, Fig. 5), have been used for centuries for the treatment of heart failure (with a well-documented mechanism of action), they are now being researched as broad spectrum immunogenic cell death inducers.78 Their original use for treatment of heart failure and cardiac arrhythmias requires a delicate balance in dose to which the dose is high enough to affect the Na+/K+-ATPase to stabilize the arrhythmias but not too much to cause global increase of intracellular calcium and subsequent heart failure.79 More recently, cardiac glycosides are being investigated for their anti-proliferative properties which may be due to either their ability to target Na+/K+-ATPase or an off target effect such as their ability to modulate N-glycosylation.78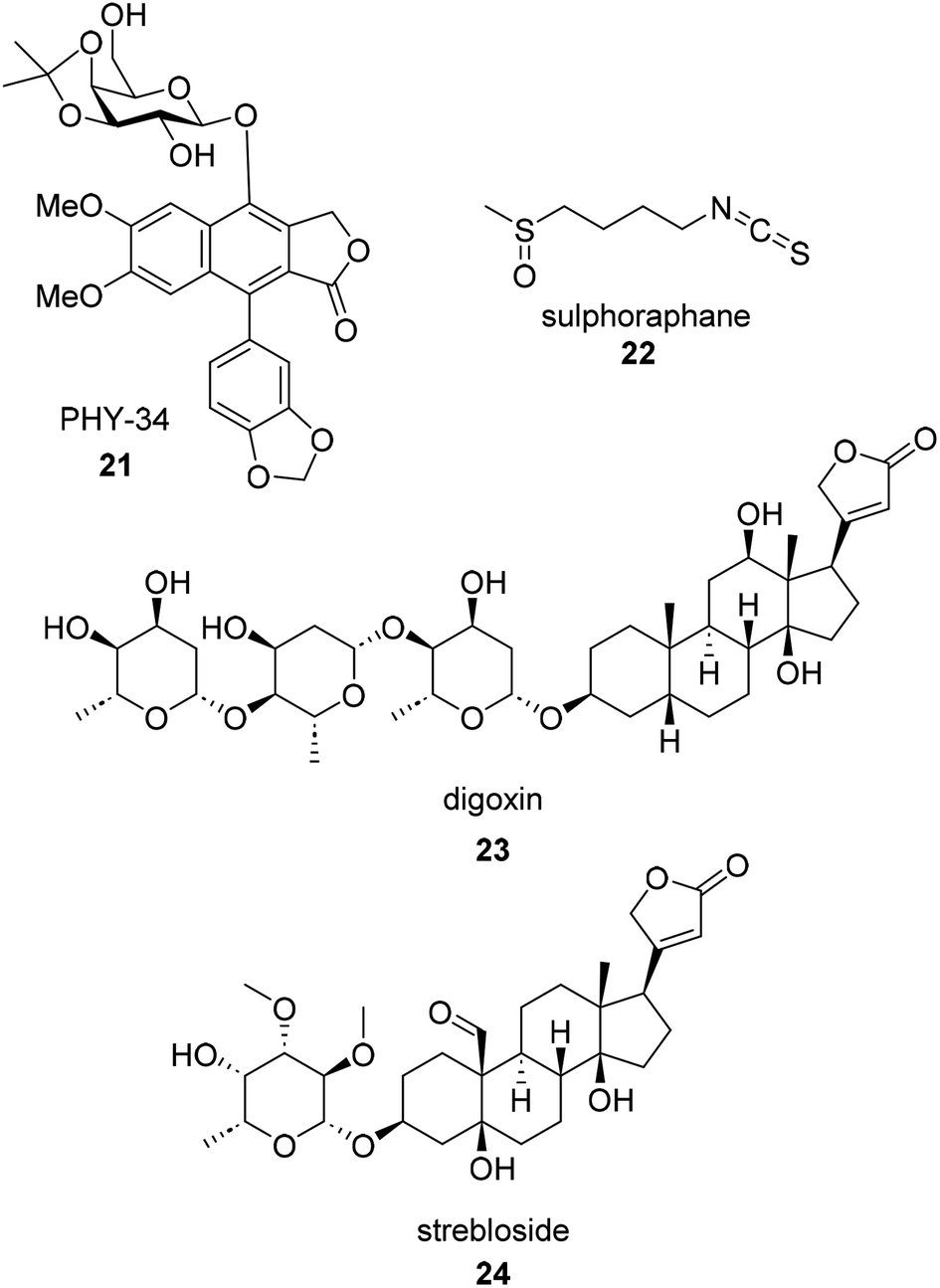 | ||
| Fig. 5 Examples of recently published botanical compounds with anti-ovarian cancer activity. | ||
A recent example of a natural product anti-ovarian cancer hit with known potential for toxicity is (+)-strebloside (24).80,81 (+)-Strebloside is a cardiac glycoside isolated from stem bark of S. asper. Its structure is similar to that of digoxin but possesses an uncharacteristic formyl group at the C-10 position. (+)-Strebloside is found to block Na+/K+-ATPase and inhibit cell proliferation in six high-grade serous ovarian cancer cell lines. It was found to have an IC50 value of 91.1 nM against OVCAR8 cells. While the authors of the recent work on (+)-strebloside found it to be a modulator of p53, NF-kB, binders of MDM2, and affect the mitotic cell cycle, they acknowledge that the key signaling events induced by (+)-strebloside are similar to that of other cardiac glycosides. They hypothesize that (+)-strebloside could suffer from similar side effects if the overall selectivity for cancer cells is not addressed through structural or delivery modifications.80–82 Despite more research needing to be done, strebloside may be a targeted therapeutic for HGSOC. TP53 mutations are observed in 96% of HGSOC tumors making a MDM2 inhibitor an attractive anti ovarian cancer strategy.83,84
6.4. SAR (SAR)
Structure–Activity Relationship (SAR) studies are an important step in most drug discovery campaigns. The “activity” being monitored in an SAR analysis can vary based on the optimizable property researchers desire, but is typically reported in the form of in vitro potency.85 Natural product hits with follow up SAR studies provide information about (1) the cell line's tolerance to structural modifications and (2) the potential pharmacophore of the hit compound. Once a pharmacophore is identified, other drug properties such as solubility and metabolic concerns can be more easily addressed through additional structural modifications. The synthesis and recent campaigns surrounding the taxanes exemplify in depth, methodical, and diverse SAR studies. As stated previously, the taxanes have a rich history of synthetic campaign to achieve readily available access to the natural products pharmacophore. Today, many SAR campaigns are still ongoing to address conferred resistance to the natural product and find solutions to solubility concerns. In addition to recent SAR campaigns pertaining to taxes, SAR studies on other natural products with activity against ovarian cancer cell lines are also included below.In 2018, Ma et al. published a series of C2- and C3′-modified semisynthetic analogues of docetaxel.86 This dual-purpose strategy aimed at enhancing the binding affinity for microtubules and improving the water solubility of docetaxel. Since it is well known that the C2 position is a critical site for taxane-microtubule binding, the authors sought to make substitutions based on previous reports.87–89 Their lead compound 25 (Fig. 6) was found to have a 64-fold water-solubility increase (0.019 mg mL−1) over paclitaxel and was able to overcome drug resistance in cultured P-gp-overexpressing tumor cells. 25 also showed greater activity than docetaxel against drug-resistant A2780/AD ovarian cancer xenografts in mice.86
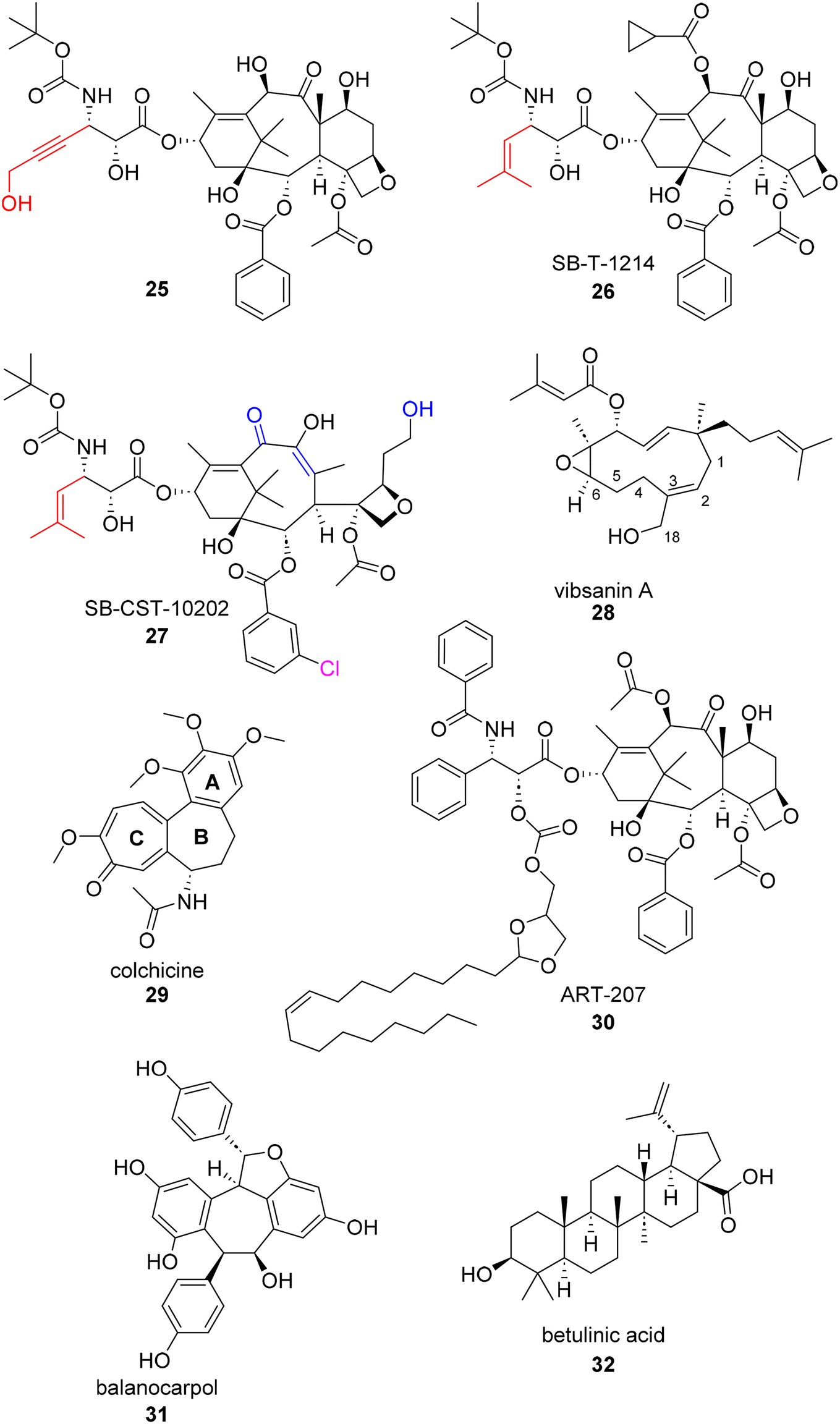 | ||
| Fig. 6 Additional examples of recently published botanical compounds with anti-ovarian cancer activity. | ||
“Next generation taxanes” (i.e. taxanes beyond first generation) such as SB-T-1214 and SB-CST-10202, 26 and 27, respectively, have been reported to possess high potency against paclitaxel-resistant cancer cell lines overexpressing βIII-tubulin and/or P-glycoprotein (P-gp).90–92 Recently, Horwitz and co-workers used photoaffinity labeling techniques to probe distinct inhibitory effects of these next generation taxanes on β-tubulin from different eukaryotic sources. They found that 26 and 27 can inhibit bovine brain tubulin (type βIII and βVI) by 38% and 32%, respectively, whereas 26 inhibited chicken erythrocyte tubulin (type βVI only) by 30% while 27 inhibited the same chicken erythrocyte tubulin by only 6%.93 This, along with a previous report,94 led the authors to conclude that the β-tubulin isotype composition can greatly influence taxane activity. They found that these two taxanes, particularly 26, are effective primarily against P-glycoprotein-overexpressing cells.93
Another SAR study carried out in an ovarian cancer cell line was centered around the natural product vibsanin A. Vibsanins (e.g.28) are botanical natural products isolated from Viburnum awabuki which is an evergreen shrub. There have been prior SAR studies for vibsanin A in acute monocytic leukemia THP-1 cells and Miura et al. have established a total synthesis for the natural product.95 They utilized the route to further test vibsanin A and subsequent analogues in several human cancer cell lines including ES-2. The analogues generated for this study focused on what effects ring opening and the oxidation of the atoms at or around the C2 and C3 positions would have on antiproliferative activity. It was globally concluded based off of activity in various cancer cell lines that the presence of a Michael acceptor on the 11 member ring was important for antiproliferative activity. Vibsanin A as well as the C18 analogues (compounds 4 and 5 in the original publication) displayed IC50s less than 8 μM in ES-2 cells. However, the Michael acceptor analogues point to a unique mechanism of action for this series by displaying HSP90 inhibition in vitro.95
Colchicine (29) is a botanical alkaloid typically isolated from plants within the Liliaceae family. Colchicine has been used for almost two centuries to treat inflammation, specifically inflammation as a result of gout. The mechanism of action for colchicine was found to be inhibition of microtubule polymerization in the 1970s, a known mechanism for anti-cancer therapeutics. As a result, several colchicine SAR studies have been published to overcome known toxicity and increase antiproliferative activity.96 Throughout the history of SAR studies surrounding colchicine, dozens of A, B, and C ring modification have been studied for their antiproliferative effects in various cancer cell lines such as lung, colon, colorectal, leukemia, breast, and ovarian (as reviewed by Ghawanmeh et al. in 2020).97 In Ghawanmeh's thorough review of colchicine SAR, they recapitulate that due to colchicine's low therapeutic index, SAR studies are vital to uncovering modifications that reduce unselective cytotoxicity and improve therapeutic activity. Generally, C-7 thio-derivatives, meta-nitro substituents in the benzylamide group, and addition of thiazole and triazole moieties at various positions show an increase in in vitro efficacy.97
One recent colchicine SAR campaign that tests their analogues an in ovarian cancer cell lines is from El-Elimat and co-workers.98 El-Elimat and coworkers isolated (−)-colchicine in a new species, Androcymbium palaestinum. When tested for potency in OVCAR3 cells, they found the amino methylated naturally occurring analogue (IC50 = 77 nM) of colchicine to be 3 fold less potent than colchicine (IC50 = 23 nM) but 10 fold more potent than the de-methoxy naturally occurring analogue (IC50 = 2230 nM).98 Another SAR study tested in ovarian cancer cells SKOV3 found 7-deacetyl-10-alkyl thiochochicine derivatives displayed significantly better potency than the natural product. The C10 thiomethane derivative displayed the most potent activity in SKOV3 cells with an IC50 of 1.3 nM.99 Similarly, the amino acyl version of the C10 thiomethane derivative (tested in a different report) displayed an IC50 = 8 nM in SKOV3 cells.96
6.5. PK/in vivo data (PK,V)
As recommended by Chung et al., in vitro ADME (Absorption, Distribution, Metabolism, and Excretion) studies are critical for pre-clinical studies.100 These studies aid drug design and help better determine optimal lead drug candidates when paired with enzymatic assays that elucidate biological activity.100In vitro ADME studies help researchers evaluate Structure–Activity Relationships (SAR) and Structure Property Relationships (SPR), providing a more complete profile for drug candidates. These assays are relatively rapid when compared to in vivo testing and can serve as more cost-effective means to achieving preliminary data.100 In addition to early ADME studies, Lombardino and Lowe argue that performing animal testing alongside early in vitro testing can give powerful correlations between in vitro pharmacokinetics screens and in vivo data. This early correlation could provide natural products drug discovery campaigns the ability to overcome any innate deficiencies in pharmacokinetics early in the process.101 In a historical sense and in many industrial drug discovery applications, including in the discovery of paclitaxel, in vivo activity was prioritized in early stage lead discovery. In most modern drug discovery campaigns, however, the emphasis has shifted to more in vitro studies in the early stage. If this approach had been implemented for the paclitaxel campaign, the molecule may have been rejected and never progressed to being the blockbuster drug it is today. Unfortunately, due to the relative cost associated with PK studies, this data is frequently not obtained for newly discovered or reported compounds. In fact, of the compounds in Table 1, only the phyllanthusmins and triptolide had associated PK data reported.69,102As mentioned above in Section 6.2, PHY-34 (21, Fig. 5), the arylnaphthalene lignan lactone derived from phyllanthusmin D also represents an example of a compound that has undergone PK and in vivo testing.69 The solubility and maximum tolerated dose (MTD) of PHY-34 was determined prior to conducting in vivo studies. Based on the solubility studies, PEG300 was the solvent used for in vivo testing with the maximum soluble concentration found to be 27.6 mg mL−1. Observed MTDs of PHY-34 by various routes of administration were as follows: 0.6 mg kg−1 by intravenous administration, 1.8 mg kg−1 by intraperitoneal administration, and >30 mg kg−1 by oral administration. The observed intraperitoneal bioavailability (F) for PHY-34 was 56.6% and for the oral route of administration was 2.5%.69 No toxicity was found to be associated with oral administration of PHY-34. Systemic clearance (Cl) following oral dose was 194.1 L h−1 kg−1, roughly 40 times the mouse liver blood flow, suggesting significant extrahepatic clearance. The Tmax of PHY34 (0.25 hours) suggests very fast absorption of this compound orally. More importantly administering chemotherapy intraperitoneally has demonstrated significant advantage based on clinical studies in EOC patients.103 Based on the PK data using PHY-34, an intraperitoneal dose of 0.75 mg kg−1, less than half the MTD was used for in vivo testing. In addition to the mechanism of action studies described above, PHY-34 was also found to significantly reduce tumor burden in an OVCAR8 xenograft model that correlated with early PK/ADME data.69PHY-34 exhibited no gross toxicities when dosed in vivo and was able to significantly reduce tumor burden at a dose 6-fold smaller than that of positive control paclitaxel.69
For compounds that show promising efficacy and no observable gross toxicities in vivo, clinical evaluation is the next step in the drug discovery process. In 2021 an analysis of a 2014 clinical trial (NCT01630018) was conducted and found that the camptothecin analogue belotecan (20, Fig. 4) was not inferior to topotecan (19, Fig. 4) in terms of overall response for recurrent ovarian cancer. Specifically, the trial showed that there was no significant difference between overall response rate or progression free survival. However, the trial, which consisted of a total of 130 patients, did show an association between improved overall survival and belotecan treatment.64
6.6. Delivery/activity aids (D/A)
Xenobiotics are classified as anything non-native to the body. Some xenobiotics can be active and able to disperse through the body in a meaningful way without any modulator. Other xenobiotics need assistance to obtain similar effects.104 For the sake of this review, both assistance with xenobiotic delivery and activity will be summarized with one indicator on the chart, although the two categories “aid” quite differently. “Delivery aids” encompass structural modifications (i.e. prodrugs), additions of a vesical (i.e. micelle), and antibody drug conjugates, or co administration with a cell-modulator pertaining to drug localization (i.e. efflux pump inhibitor). “Activity aids” encompass studies that look at the synergistic effects between a natural product and a known anticancer therapeutic. This category was included for two reasons: (1) the natural products isolation community values synergistic effects and therefore the frequency of reporting is elevated105 and (2) combination formulations of known modulators are common in clinical trials. Combinations are often proposed to combat known resistance.106Haider et al. report the design and in vitro and in vivo testing of highly lipophilic, acid-labile paclitaxel-based conjugates for the tumor-selective delivery of paclitaxel. Their lead prodrug, a paclitaxel conjugate ART-207 (30) derived from oleic acid and racemic solketal, was incorporated in their TumorSelect® approach to minimize undesirable side effects of conventional chemotherapy. This approach tricks the body's cholesterol seeking cells (i.e. high turnover cancer cells) into taking in pseudo-LDL nanoparticles with the paclitaxel prodrug inside. When compared after 72 hours to the FDA approved paclitaxel formulation, Abraxane®, the EC50 of ART-207 with pseudo-LDL nanoparticle formulations was at least 3-fold more potent in an ovarian cancer cell line (SKOV3).107
A recent report by Colombo et al.108 describes a betulinic acid-based conjugate of cabazitaxel and other tubulin-binding drugs in an effort to increase the bioavailability of the native drugs. The conjugates self-assemble to form nanoparticles targeted to release the active drug in the body via a labile linker. While this is not the first attempt at synthesizing a taxane-based nanoparticle for ovarian cancer,109 it is the first report of self-assembled nanoparticles without the need for an additional anti-aggregate stabilizing compound. Both the conjugates as monomers and the relative nanoparticles were tested for their antiproliferative activity in ovarian cancer cells (A2780). While there was promising activity observed with the conjugate approach (GI50 = 0.35 μM), the nanoparticle showed somewhat lower potency (GI50 = 12 μM). In this study, the native drug remained the most potent compound examined (GI50 = 0.327 nM).108
In hopes of minimizing the toxicity of camptothecin, various clinical trials have been undertaken to assess the selectivity of a nanoparticle–drug conjugate of camptothecin.48,110–112 These trials have been reassessing the efficacy of a novel cyclodextrin-containing polymer conjugate of camptothecin either by itself or in combination with other known anti-cancer therapeutics (bevacizumab, olaparib, and paclitaxel) against ovarian cancer patients. Overall, the nanoparticle conjugate was found to be a clinically effective and tolerable topoisomerase I inhibitor and could be safely co-administered with the tested anti-cancer therapeutics. Each clinical trials reports optimism of this particular nanoparticle conjugate progressing forward into more trials.
In addition to camptothecin, which was recently reformulated as a nanoparticle, another recent example of nanoparticle technology being used to enhance the activity of a botanical natural product is with balanocarpol (31, Fig. 6). Balanocarpol, a multi-aromatic fused-ring compound has poor bioavailability as well poor aqueous solubility. Therefore, Obeid et al. improved the characteristics of balanocarpol and increased its anticancer activity through its encapsulation in a bilayer structure of a lipid-based nanoparticle drug delivery system.113 While this project is far from a clinical trial, the application of nanotechnology to aid in the delivery of a non-native compound is exemplified. Similarly, betulinic acid (32) was used as a chemosensitizer in combination with doxorubicin in a recent study published by Jin et al.114 Betulinic acid acts on tumor mitochondria, decreasing the supply of ATP in tumor efflux pumps, and promoting doxorubicin uptake, working synergistically to affect cancer cell growth. The research team set out to construct an all-in-one multifunctional multidrug pH-sensitive targeting delivery system for the synergistic co-delivery of doxorubicin and betulinic acid using a peptide micelle. Not only did the micelle display enhanced anti-tumor effects because of its ability to deliver doxorubicin/betulinic acid at the tumor location, the synergistic relationship between doxorubicin/betulinic acid was maintained in the dual delivery system. The researchers also found that doxorubicin/betulinic acid micelles elicited fewer toxic effects on cardiac function in SKOV3 subcutaneous xenograft models.97
7. Conclusions and perspectives
The success stories of efficacious, botanically derived, therapeutics such as paclitaxel and the camptothecin analogues topotecan and irinotecan point to the potential for the discovery of additional natural products for the treatment of ovarian cancer. Their discoveries and eventual FDA approvals continue to inspire the next generation of botanist, biologist, chemist, and clinician to pursue botanical sources for the treatment of the disease. Interestingly, however, it is noteworthy that the path toward clinical success for both the taxanes and the camptothecins required significant problem solving in addition to the careful consideration of physicochemical properties and biological data.New technologies, along with an estimated 15–21% of the world's flowering plants yet to be characterized,115,116 continue to provide abundant opportunities for current researchers to discover novel bioactive secondary metabolites with anticancer activity. In general, while natural product drug discovery in this area remains strong and numerous active molecules are reported as hits against ovarian cancer and other diseases, current drug development efforts require an even greater focus on drug parameters to propel them into the clinic. This development process can be aided by modern drug discovery and development techniques, including through the use of predictive tools, computational docking and screening methods, high throughput assays to aid in the identification of more potent and synthetically accessible compounds, a focus on drug properties and features, and modern drug delivery methodology.117,118 It is hoped that in addition to the application of modern techniques, a careful analysis of the development processes currently being employed for the advancement of new compounds from botanical sources, coupled with the historical knowledge of the challenges faced in the development of compounds like the taxanes and camptothecins, will lead to improved efficiency in advancing natural products or their derivatives to the clinic. Ultimately, the identification of novel compounds with in vitro activity against an ovarian cancer cell line can be a good starting point for a drug development campaign, but a sustained effort focused on multiple development parameters, data points, and milestones is required to successfully move new compounds into therapeutic relevance.
8. Author contributions
Conceptualization: JB and JF; data curation: BM and YR; writing – original draft: BM, AS, and JF; writing – review and editing: BM, AS, YR, JB, and JF.9. Conflicts of interest
There are no conflicts to declare.10. Acknowledgements
This research was supported by P01 CA125066 (to JEB: Core 1 and JRF: Core 2) and the National Center for Complementary & Alternative Medicine (R01 AT008824).11. References
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- G. M. Cragg and J. M. Pezzuto, Med Princ Pract., 2016, 25(Suppl 2), 41–59 CrossRef PubMed.
- R. L. Siegel, K. D. Miller, H. E. Fuchs and A. Jemal, Ca-Cancer J. Clin., 2022, 72, 7–33 CrossRef PubMed.
- American Cancer Society|Cancer Facts & Statistics, http://cancerstatisticscenter.cancer.org/, accessed November 29, 2022.
- I. J. Jacobs, U. Menon, A. Ryan, A. Gentry-Maharaj, M. Burnell, J. K. Kalsi, N. N. Amso, S. Apostolidou, E. Benjamin, D. Cruickshank, D. N. Crump, S. K. Davies, A. Dawnay, S. Dobbs, G. Fletcher, J. Ford, K. Godfrey, R. Gunu, M. Habib, R. Hallett, J. Herod, H. Jenkins, C. Karpinskyj, S. Leeson, S. J. Lewis, W. R. Liston, A. Lopes, T. Mould, J. Murdoch, D. Oram, D. J. Rabideau, K. Reynolds, I. Scott, M. W. Seif, A. Sharma, N. Singh, J. Taylor, F. Warburton, M. Widschwendter, K. Williamson, R. Woolas, L. Fallowfield, A. J. McGuire, S. Campbell, M. Parmar and S. J. Skates, Lancet, 2016, 387, 945–956 CrossRef PubMed.
- R. J. Kurman, Ann. Oncol., 2013, 24, x16–x21 CrossRef PubMed.
- Morphologic, Immunophenotypic, and Molecular Features of Epithelial Ovarian Cancer, https://www.cancernetwork.com/view/morphologic-immunophenotypic-and-molecular-features-epithelial-ovarian-cancer, accessed November 1, 2022.
- J. A. Ledermann, Lancet Oncol., 2019, 20, 470–472 CrossRef PubMed.
- R. L. Coleman, G. F. Fleming, M. F. Brady, E. M. Swisher, K. D. Steffensen, M. Friedlander, A. Okamoto, K. N. Moore, N. Efrat Ben-Baruch, T. L. Werner, N. G. Cloven, A. Oaknin, P. A. DiSilvestro, M. A. Morgan, J.-H. Nam, C. A. Leath, S. Nicum, A. R. Hagemann, R. D. Littell, D. Cella, S. Baron-Hay, J. Garcia-Donas, M. Mizuno, K. Bell-McGuinn, D. M. Sullivan, B. A. Bach, S. Bhattacharya, C. K. Ratajczak, P. J. Ansell, M. H. Dinh, C. Aghajanian and M. A. Bookman, N. Engl. J. Med., 2019, 381, 2403–2415 CrossRef CAS PubMed.
- T. J. Herzog and B. J. Monk, Gynecologic Oncology Research and Practice, 2017, 4, 13 CrossRef PubMed.
- C. M. Beaufort, J. C. A. Helmijr, A. M. Piskorz, M. Hoogstraat, K. Ruigrok-Ritstier, N. Besselink, M. Murtaza, W. F. J. van IJcken, A. A. J. Heine, M. Smid, M. J. Koudijs, J. D. Brenton, E. M. J. J. Berns and J. Helleman, PLoS One, 2014, 9, e103988 CrossRef PubMed.
- S. Domcke, R. Sinha, D. A. Levine, C. Sander and N. Schultz, Nat. Commun., 2013, 4, 2126 CrossRef PubMed.
- H. W. Cheung, G. S. Cowley, B. A. Weir, J. S. Boehm, S. Rusin, J. A. Scott, A. East, L. D. Ali, P. H. Lizotte, T. C. Wong, G. Jiang, J. Hsiao, C. H. Mermel, G. Getz, J. Barretina, S. Gopal, P. Tamayo, J. Gould, A. Tsherniak, N. Stransky, B. Luo, Y. Ren, R. Drapkin, S. N. Bhatia, J. P. Mesirov, L. A. Garraway, M. Meyerson, E. S. Lander, D. E. Root and W. C. Hahn, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 12372–12377 CrossRef CAS PubMed.
- A. Mitra, D. A. Davis, S. Tomar, L. Roy, H. Gurler, J. Xie, D. D. Lantvit, H. Cardenas, F. Fang, Y. Liu, E. Loughran, J. Yang, M. S. Stack, R. E. Emerson, K. D. C. Dahl, M. Barbolina, K. P. Nephew, D. Matei and J. E. Burdette, Gynecol. Oncol., 2015, 138, 372 CrossRef CAS PubMed.
- Y. Zhang, L. Cao, D. Nguyen and H. Lu, Transl. Cancer Res., 2016, 5, 650–663 CrossRef CAS PubMed.
- M. Tuna, Z. Ju, K. Yoshihara, C. I. Amos, J. L. Tanyi and G. B. Mills, Br. J. Cancer, 2020, 122, 405–412 CrossRef CAS PubMed.
- A. J. Cole, T. Dwight, A. J. Gill, K.-A. Dickson, Y. Zhu, A. Clarkson, G. B. Gard, J. Maidens, S. Valmadre, R. Clifton-Bligh and D. J. Marsh, Sci. Rep., 2016, 6, 26191 CrossRef CAS PubMed.
- J. Haley, S. Tomar, N. Pulliam, S. Xiong, S. M. Perkins, A. R. Karpf, S. Mitra, K. P. Nephew and A. K. Mitra, Oncotarget, 2016, 7, 32810–32820 CrossRef PubMed.
- G. M. Rodriguez, K. J. C. Galpin, C. W. McCloskey and B. C. Vanderhyden, Cancers, 2018, 10, 242 CrossRef PubMed.
- S. L. Eddie, S. M. Quartuccio, E. Ó. hAinmhire, G. Moyle-Heyrman, D. D. Lantvit, J.-J. Wei, B. C. Vanderhyden and J. E. Burdette, Oncotarget, 2015, 6, 20500–20512 CrossRef PubMed.
- S. Iyer, S. Zhang, S. Yucel, H. Horn, S. G. Smith, F. Reinhardt, E. Hoefsmit, B. Assatova, J. Casado, M.-C. Meinsohn, M. I. Barrasa, G. W. Bell, F. Pérez-Villatoro, K. Huhtinen, J. Hynninen, J. Oikkonen, P. M. Galhenage, S. Pathania, P. T. Hammond, B. G. Neel, A. Farkkila, D. Pépin and R. A. Weinberg, Cancer Discovery, 2021, 11, 384–407 CrossRef CAS PubMed.
- C. Yee, Front. Bioeng. Biotechnol., 2022, 10(116), 35223797 Search PubMed.
- W. Sakai, E. M. Swisher, C. Jacquemont, K. V. Chandramohan, F. J. Couch, S. P. Langdon, K. Wurz, J. Higgins, E. Villegas and T. Taniguchi, Cancer Res., 2009, 69, 6381–6386 CrossRef CAS PubMed.
- S. Rottenberg, C. Disler and P. Perego, Nat. Rev. Cancer, 2021, 21, 37–50 CrossRef CAS PubMed.
- B. Rosenberg and L. VanCamp, Cancer Res., 1970, 30(6), 1799–1802 CAS.
- J. Mikuła-Pietrasik, A. Witucka, M. Pakuła, P. Uruski, B. Begier-Krasińska, A. Niklas, A. Tykarski and K. Książek, Cell. Mol. Life Sci., 2019, 76, 681–697 CrossRef PubMed.
- D. Sarvepalli, M. U. Rashid, H. Zafar, S. Jehanzeb, E. Zahid and S. Ahmad, in Overcoming Drug Resistance in Gynecologic Cancers, ed. R. Basha and S. Ahmad, Academic Press, 2021, vol. 17, pp. 283–301 Search PubMed.
- M. E. Gordinier, A. P. Kudelka, J. J. Kavanagh, J. T. Wharton and R. S. Freedman, J. Gynecol. Cancer, 2002, 12, 710–714 CrossRef CAS.
- M. C. Wani, H. L. Taylor, M. E. Wall, P. Coggon and A. T. McPhail, J. Am. Chem. Soc., 1971, 93, 2325–2327 CrossRef CAS PubMed.
- D. G. I. Kingston, Phytochemistry, 2007, 68, 1844–1854 CrossRef CAS PubMed.
- M. Suffness and M. E. Wall, Taxol: Science and Applications, CRC Press, Inc, Boca Raton, FL, 1995 Search PubMed.
- M. I. Nicoletti, V. Lucchini, G. Massazza, B. J. Abbott, M. D'Incalci and R. Giavazzi, Ann. Oncol., 1993, 4, 151–155 CrossRef CAS PubMed.
- C. Amaya, S. Luo, J. Baigorri, R. Baucells, E. R. Smith and X.-X. Xu, BMC Cancer, 2021, 21, 1–13 CrossRef PubMed.
- J. N. Denis, A. E. Greene, D. Guenard, F. Gueritte-Voegelein, L. Mangatal and P. Potier, J. Am. Chem. Soc., 1988, 110, 5917–5919 CrossRef CAS.
- R. A. Holton, C. Somoza, H. B. Kim, F. Liang, R. J. Biediger, P. D. Boatman, M. Shindo, C. C. Smith and S. Kim, J. Am. Chem. Soc., 1994, 116, 1597–1598 CrossRef CAS.
- W. C. Liu, T. Gong and P. Zhu, RSC Adv., 2016, 6, 48800–48809 RSC.
- I. Ojima, X. Wang, Y. Jing and C. Wang, J. Nat. Prod., 2018, 81, 703–721 CrossRef CAS PubMed.
- J. Škubník, V. Pavlíčková, T. Ruml and S. Rimpelová, Plants, 2021, 10, 569 CrossRef PubMed.
- V. Němcová-Fürstová, D. Kopperová, K. Balušíková, M. Ehrlichová, V. Brynychová, R. Václavíková, P. Daniel, P. Souček and J. Kovář, Toxicol. Appl. Pharmacol., 2016, 310, 215–228 CrossRef PubMed.
- N. Yang, C. Wang, J. Wang, Z. Wang, D. Huang, M. Yan, M. Kamran, Q. Liu and B. Xu, J. Cell. Mol. Med., 2019, 23, 6442–6453 CrossRef CAS PubMed.
- Y. Huang, G. Chen, Y. Wang, R. He, J. Du, X. Jiao and Q. Tai, Biochem. Biophys. Res. Commun., 2018, 503, 1035–1041 CrossRef CAS PubMed.
- D. J. McGrail, N. N. Khambhati, M. X. Qi, K. S. Patel, N. Ravikumar, C. P. Brandenburg and M. R. Dawson, Sci. Rep., 2015, 5, 9529 CrossRef PubMed.
- Discovery of Camptothecin and Taxol - National Historic Chemical Landmark, https://www.acs.org/content/acs/en/education/whatischemistry/landmarks/camptothecintaxol.html, accessed June 30, 2022.
- Y. H. Hsiang, R. Hertzberg, S. Hecht and L. F. Liu, J. Biol. Chem., 1985, 260, 14873–14878 CrossRef CAS PubMed.
- R. P. Verma and C. Hansch, Chem. Rev., 2009, 109, 213–235 CrossRef CAS PubMed.
- K. E. Hevener, T. A. Verstak, K. E. Lutat, D. L. Riggsbee and J. W. Mooney, Acta Pharm. Sin. B, 2018, 8, 844–861 CrossRef PubMed.
- Y. Pommier, E. Leo, H. Zhang and C. Marchand, Chem. Biol., 2010, 17, 421–433 CrossRef CAS PubMed.
- G. J. Weiss, J. Chao, J. D. Neidhart, R. K. Ramanathan, D. Bassett, J. A. Neidhart, C. H. J. Choi, W. Chow, V. Chung, S. J. Forman, E. Garmey, J. Hwang, D. L. Kalinoski, M. Koczywas, J. Longmate, R. J. Melton, R. Morgan, J. Oliver, J. J. Peterkin, J. L. Ryan, T. Schluep, T. W. Synold, P. Twardowski, M. E. Davis and Y. Yen, Invest. New Drugs, 2013, 31, 986–1000 CrossRef CAS PubMed.
- N. Khaiwa, N. R. Maarouf, M. H. Darwish, D. W. M. Alhamad, A. Sebastian, M. Hamad, H. A. Omar, G. Orive and T. H. Al-Tel, Eur. J. Med. Chem., 2021, 223, 113639 CrossRef CAS PubMed.
- C. Bailly, Pharmacol. Res., 2019, 148, 104398 CrossRef CAS PubMed.
- J. Hughes, S. Rees, S. Kalindjian and K. Philpott, Br. J. Pharmacol., 2011, 162, 1239–1249 CrossRef CAS PubMed.
- B. E. Blass, Basic Principles of Drug Discovery and Development, Elsevier, 2015 Search PubMed.
- R. F. Bruns and I. A. Watson, J. Med. Chem., 2012, 55, 9763–9772 CrossRef CAS PubMed.
- E. K. Davison and M. A. Brimble, Curr. Opin. Chem. Biol., 2019, 52, 1–8 CrossRef CAS PubMed.
- J. B. Baell and G. A. Holloway, J. Med. Chem., 2010, 53, 2719–2740 CrossRef CAS PubMed.
- J. B. Baell, J. Nat. Prod., 2016, 79, 616–628 CrossRef CAS PubMed.
- Y. Liu, S. Gao, J. Zhu, Y. Zheng, H. Zhang and H. Sun, Cancer Med., 2018, 7, 5704–5715 CrossRef CAS PubMed.
- M. Mohamadian, A. Bahrami, M. Moradi Binabaj, F. Asgharzadeh and G. A. Ferns, Nutr. Cancer, 2022, 1–18 Search PubMed.
- X.-L. Xu, S.-L. Deng, Z.-X. Lian and K. Yu, Antioxidants, 2021, 10, 1718 CrossRef CAS PubMed.
- Q. Chen, R. Qin, Y. Fang and H. Li, Chem. Pharm. Bull., 2015, 36, 956–965 CAS.
- L. Liu, J. Fan, G. Ai, J. Liu, N. Luo, C. Li and Z. Cheng, Biol. Res., 2019, 52, 37 CrossRef PubMed.
- Y. Zhao, L. Cui, Y. Pan, D. Shao, X. Zheng, F. Zhang, H. Zhang, K. He and L. Chen, Cell Proliferation, 2017, 50, e12393 CrossRef PubMed.
- O. R. Johnson-Ajinwo, I. Ullah, H. Mbye, A. Richardson, P. Horrocks and W.-W. Li, Bioorg. Med. Chem. Lett., 2018, 28, 1219–1222 CrossRef CAS PubMed.
- H. S. Kim, S.-Y. Park, C.-Y. Park, Y. T. Kim, B.-J. Kim, Y. J. Song, B.-G. Kim, Y. B. Kim, C.-H. Cho, J.-H. Kim and Y. S. Song, Br. J. Cancer, 2021, 124, 375–382 CrossRef CAS PubMed.
- J. Wu, T. Zhou, Y. Wang, Y. Jiang and Y. Wang, Molecules, 2021, 26, 5949 CrossRef CAS PubMed.
- J. L. Woodard, A. C. Huntsman, P. A. Patel, H.-B. Chai, R. Kanagasabai, S. Karmahapatra, A. N. Young, Y. Ren, M. S. Cole, D. Herrera, J. C. Yalowich, A. D. Kinghorn, J. E. Burdette and J. R. Fuchs, Bioorg. Med. Chem., 2018, 26, 2354–2364 CrossRef CAS PubMed.
- Y. Ren, C. Yuan, Y. Deng, R. Kanagasabai, T. N. Ninh, V. T. Tu, H.-B. Chai, D. D. Soejarto, J. R. Fuchs, J. C. Yalowich, J. Yu and A. Douglas Kinghorn, Phytochemistry, 2015, 111, 132–140 CrossRef CAS PubMed.
- Y. Ren, D. D. Lantvit, Y. Deng, R. Kanagasabai, J. C. Gallucci, T. N. Ninh, H.-B. Chai, D. D. Soejarto, J. R. Fuchs, J. C. Yalowich, J. Yu, S. M. Swanson and A. D. Kinghorn, J. Nat. Prod., 2014, 77, 1494–1504 CrossRef CAS PubMed.
- A. N. Young, D. Herrera, A. C. Huntsman, M. A. Korkmaz, D. D. Lantvit, S. Mazumder, S. Kolli, C. C. Coss, S. King, H. Wang, S. M. Swanson, A. D. Kinghorn, X. Zhang, M. A. Phelps, L. N. Aldrich, J. R. Fuchs and J. E. Burdette, Mol. Cancer Ther., 2018, 17, 2123–2135 CrossRef CAS PubMed.
- A. C. Wang, H. T. Pham, J. M. Lipps, S. M. Brittain, E. Harrington, Y. Wang, F. J. King, C. Russ, X. Pan, D. Hoepfner, J. Tallarico, Y. Feng, R. K. Jain, M. Schirle and J. R. Thomas, ACS Chem. Biol., 2019, 14, 20–26 CrossRef CAS PubMed.
- A. Salvi, A. N. Young, A. C. Huntsman, M. R. Pergande, M. A. Korkmaz, R. A. Rathnayake, B. K. Mize, A. D. Kinghorn, X. Zhang, K. Ratia, M. Schirle, J. R. Thomas, S. M. Brittain, C. Shelton, L. N. Aldrich, S. M. Cologna, J. R. Fuchs and J. E. Burdette, Cell Death Dis., 2022, 13, 1–13 Search PubMed.
- J. R. Delaney, C. B. Patel, K. M. Willis, M. Haghighiabyaneh, J. Axelrod, I. Tancioni, D. Lu, J. Bapat, S. Young, O. Cadassou, A. Bartakova, P. Sheth, C. Haft, S. Hui, C. Saenz, D. D. Schlaepfer, O. Harismendy and D. G. Stupack, Nat. Commun., 2017, 8, 14423 CrossRef CAS PubMed.
- J. Wang and G. S. Wu, J. Biol. Chem., 2014, 289, 17163–17173 CrossRef CAS PubMed.
- J. M. Santiago-O’Farrill, S. J. Weroha, X. Hou, A. L. Oberg, E. P. Heinzen, M. J. Maurer, L. Pang, P. Rask, R. K. Amaravadi, S. E. Becker, I. Romero, M. J. Rubio, X. Matias-Guiu, M. Santacana, A. Llombart-Cussac, A. Poveda, Z. Lu and R. C. Bast Jr, Cancer, 2020, 126, 894–907 CrossRef PubMed.
- Cruciferous Vegetables and Cancer Prevention - NCI, https://www.cancer.gov/about-cancer/causes-prevention/risk/diet/cruciferous-vegetables-fact-sheet, accessed November 1, 2022.
- S. C. Kim, B. Choi and Y. Kwon, Food Nutr. Res., 2017, 61, 1368321 CrossRef PubMed.
- F. P. Guengerich, Drug Metab. Pharmacokinet., 2011, 26, 3–14 CrossRef CAS PubMed.
- J. I. Ayogu and A. S. Odoh, ACS Comb. Sci., 2020, 22, 543–553 CrossRef CAS PubMed.
- E. D. Cummings and H. D. Swoboda, in StatPearls, StatPearls Publishing, Treasure Island (FL), 2022 Search PubMed.
- Y. Ren, W.-L. Chen, D. D. Lantvit, E. J. Sass, P. Shriwas, T. N. Ninh, H.-B. Chai, X. Zhang, D. D. Soejarto, X. Chen, D. M. Lucas, S. M. Swanson, J. E. Burdette and A. D. Kinghorn, J. Nat. Prod., 2017, 80, 648–658 CrossRef CAS PubMed.
- W.-L. Chen, Y. Ren, J. Ren, C. Erxleben, M. E. Johnson, S. Gentile, A. D. Kinghorn, S. M. Swanson and J. E. Burdette, J. Nat. Prod., 2017, 80, 659–669 CrossRef CAS PubMed.
- Y. Ren, S. Wu, S. Chen, J. E. Burdette, X. Cheng and A. D. Kinghorn, Molecules, 2021, 26, 5675 CrossRef CAS PubMed.
- C. Makii, K. Oda, Y. Ikeda, K. Sone, K. Hasegawa, Y. Uehara, A. Nishijima, K. Asada, T. Koso, T. Fukuda, K. Inaba, S. Oki, H. Machino, M. Kojima, T. Kashiyama, M. Mori-Uchino, T. Arimoto, O. Wada-Hiraike, K. Kawana, T. Yano, K. Fujiwara, H. Aburatani, Y. Osuga and T. Fujii, Oncotarget, 2016, 7, 75328–75338 CrossRef PubMed.
- Nature , 2011, 474, 609–615, DOI:10.1038/nature10166.
- A. M. Wassermann, M. Wawer and J. Bajorath, J. Med. Chem., 2010, 53, 8209–8223 CrossRef CAS PubMed.
- Y.-T. Ma, Y. Yang, P. Cai, D.-Y. Sun, P. A. Sánchez-Murcia, X.-Y. Zhang, W.-Q. Jia, L. Lei, M. Guo, F. Gago, H. Wang and W.-S. Fang, J. Nat. Prod., 2018, 81, 524–533 CrossRef CAS PubMed.
- C. Coderch, Y. Tang, J. Klett, S.-E. Zhang, Y.-T. Ma, W. Shaorong, R. Matesanz, B. Pera, A. Canales, J. Jiménez-Barbero, A. Morreale, J. Fernando Díaz, W.-S. Fang and F. Gago, Org. Biomol. Chem., 2013, 11, 3046–3056 RSC.
- D. G. I. Kingston, A. G. Chaudhary, M. D. Chordia, M. Gharpure, A. A. L. Gunatilaka, P. I. Higgs, J. M. Rimoldi, L. Samala, P. G. Jagtap, P. Giannakakou, Y. Q. Jiang, C. M. Lin, E. Hamel, B. H. Long, C. R. Fairchild and K. A. Johnston, J. Med. Chem., 1998, 41, 3715–3726 CrossRef CAS PubMed.
- D. G. I. Kingston and D. J. Newman, Curr. Opin. Drug Discovery Dev., 2007, 10, 130–144 CAS.
- I. Ojima, J. C. Slater, E. Michaud, S. D. Kuduk, P.-Y. Bounaud, P. Vrignaud, M.-C. Bissery, J. M. Veith, P. Pera and R. J. Bernacki, J. Med. Chem., 1996, 39, 3889–3896 CrossRef CAS PubMed.
- I. Ojima, J. Chen, L. Sun, C. P. Borella, T. Wang, M. L. Miller, S. Lin, X. Geng, L. Kuznetsova, C. Qu, D. Gallager, X. Zhao, I. Zanardi, S. Xia, S. B. Horwitz, J. M.-St. Clair, J. L. Guerriero, D. Bar-Sagi, J. M. Veith, P. Pera and R. J. Bernacki, J. Med. Chem., 2008, 51, 3203–3221 CrossRef CAS PubMed.
- A. Pepe, L. Sun, I. Zanardi, X. Wu, C. Ferlini, G. Fontana, E. Bombardelli and I. Ojima, Bioorg. Med. Chem. Lett., 2009, 19, 3300–3304 CrossRef CAS PubMed.
- C.-P. H. Yang, C. Wang, I. Ojima and S. B. Horwitz, J. Nat. Prod., 2018, 81, 600–606 CrossRef CAS PubMed.
- C.-P. H. Yang, E.-H. Yap, H. Xiao, A. Fiser and S. B. Horwitz, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11294–11299 CrossRef CAS PubMed.
- K. Miura, W. Matsuki, A. Ogura, K. Takao and S. Simizu, Bioorg. Med. Chem., 2020, 28, 115253 CrossRef CAS PubMed.
- J. Kurek, K. Myszkowski, I. Okulicz-Kozaryn, A. Kurant, E. Kamińska, M. Szulc, B. Rubiś, M. Kaczmarek, P. Ł. Mikołajczak and M. Murias, Sci. Rep., 2021, 11, 9034 CrossRef CAS PubMed.
- A. A. Ghawanmeh, H. M. Al-Bajalan, M. M. Mackeen, F. Q. Alali and K. F. Chong, Eur. J. Med. Chem., 2020, 185, 111788 CrossRef CAS PubMed.
- T. El-Elimat, M. B. Alhawarri, J. Rivera-Chávez, J. E. Burdette, A. Czarnecki, M. Al-Gharaibeh, A. H. Al Sharie, A. Alhusban, F. Q. Alali and N. H. Oberlies, Fitoterapia, 2020, 146, 104706 CrossRef CAS PubMed.
- J. Kurek, P. Kwaśniewska-Sip, K. Myszkowski, G. Cofta, M. Murias, P. Barczyński, B. Jasiewicz and R. Kurczab, Med. Chem. Commun., 2018, 9, 1708–1714 RSC.
- T. D. Y Chung, D. B. Terry and L. H Smith, In Vitro and In Vivo Assessment of ADME and PK Properties During Lead Selection and Lead Optimization – Guidelines, Benchmarks and Rules of Thumb, The Assay Guidance Manual, 2004 Search PubMed.
- J. G. Lombardino and J. A. Lowe, Nat. Rev. Drug Discovery, 2004, 3, 853–862 CrossRef CAS PubMed.
- S. Patil, L. G. Lis, R. J. Schumacher, B. J. Norris, M. L. Morgan, R. A. D. Cuellar, B. R. Blazar, R. Suryanarayanan, V. J. Gurvich and G. I. Georg, J. Med. Chem., 2015, 58, 9334–9344 CrossRef CAS PubMed.
- W. J. van Driel, S. N. Koole, K. Sikorska, J. H. Schagen van Leeuwen, H. W. R. Schreuder, R. H. M. Hermans, I. H. J. T. de Hingh, J. van der Velden, H. J. Arts, L. F. A. G. Massuger, A. G. J. Aalbers, V. J. Verwaal, J. M. Kieffer, K. K. Van de Vijver, H. van Tinteren, N. K. Aaronson and G. S. Sonke, N. Engl. J. Med., 2018, 378, 230–240 CrossRef CAS PubMed.
- N. R. Srinivas, Eur. J. Drug Metab. Pharmacokinet., 2011, 36, 49–59 CrossRef CAS PubMed.
- L. K. Caesar and N. B. Cech, Nat. Prod. Rep., 2019, 36, 869–888 RSC.
- C. J. Paller, E. P. Huang, T. Luechtefeld, H. A. Massett, C. C. Williams, J. Zhao, A. E. Gravell, T. Tamashiro, S. A. Reeves, G. L. Rosner, M. A. Carducci, L. Rubinstein and S. P. Ivy, Front. Med., 2019, 6 DOI:10.3389/fmed.2019.00122.
- S. Haider, P. Penfornis, P. P. Claudio, J. D. McChesney and A. G. Chittiboyina, Eur. J. Med. Chem., 2022, 227, 113891 CrossRef CAS PubMed.
- E. Colombo, L. Polito, M. Biocotino, P. Marzullo, M. Hyeraci, L. D. Via and D. Passarella, ACS Med. Chem. Lett., 2020, 11, 895–898 CrossRef CAS PubMed.
- S. M. Ansell, S. A. Johnstone, P. G. Tardi, L. Lo, S. Xie, Y. Shu, T. O. Harasym, N. L. Harasym, L. Williams, D. Bermudes, B. D. Liboiron, W. Saad, R. K. Prudhomme and L. D. Mayer, J. Med. Chem., 2008, 51, 3288–3296 CrossRef CAS PubMed.
- Ellipses Pharma, Phase 2 Study to Evaluate the Safety & Efficacy of EP0057 in Combination With Olaparib in Advanced Ovarian Cancer Patients Who Have: Cohort 1 - Platinum Resistant Disease; Cohort 2 – Had at Least 1 Prior Line of Therapy Which Must Include at Least 1 Line of Platinum-based Chemotherapy Followed by PARP Inhibitor Maintenance, clinicaltrials.gov, 2022.
- L. R. Duska, C. N. Krasner, D. M. O'Malley, J. L. Hays, S. C. Modesitt, C. A. Mathews, K. N. Moore, P. H. Thaker, A. Miller, C. Purdy, W. C. Zamboni, A. T. Lucas, J. G. Supko and R. J. Schilder, Gynecol. Oncol., 2021, 160, 688–695 CrossRef CAS PubMed.
- C. N. Krasner, S. M. Campos, C. L. Young, K. R. Chadda, H. Lee, M. J. Birrer, N. S. Horowitz, P. A. Konstantinopoulos, A. M. D'Ascanio, U. A. Matulonis and R. T. Penson, Gynecol. Oncol., 2021, 162, 661–666 CrossRef CAS PubMed.
- M. A. Obeid, S. A. S. Gany, A. I. Gray, L. Young, J. O. Igoli and V. A. Ferro, Nanotechnology, 2020, 31, 195101 CrossRef PubMed.
- X. Jin, J. Zhou, Z. Zhang and H. Lv, Int. J. Pharm., 2019, 571, 118751 CrossRef CAS PubMed.
- L. N. Joppa, D. L. Roberts, N. Myers and S. L. Pimm, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13171–13176 CrossRef CAS PubMed.
- W. F. Laurance and D. P. Edwards, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 12971–12972 CrossRef CAS PubMed.
- A. G. Atanasov, S. B. Zotchev, V. M. Dirsch and C. T. Supuran, Nat. Rev. Drug Discovery, 2021, 20, 200–216 CrossRef CAS PubMed.
- J. J. Kellogg, M. F. Paine, J. S. McCune, N. H. Oberlies and N. B. Cech, Nat. Prod. Rep., 2019, 36, 1196–1221 RSC.
| This journal is © The Royal Society of Chemistry 2023 |