
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel elastic rubbers from CO2-based polycarbonates†
Giulia
Chiarioni
a,
Martin
van Duin
ab and
Paolo P.
Pescarmona
 *a
*a
aChemical Engineering Group, Engineering and Technology Institute Groningen (ENTEG), University of Groningen, Nijenborgh 4, 9747 AG Groningen, The Netherlands. E-mail: p.p.pescarmona@rug.nl
bARLANXEO Performance Elastomers, Innovation, P.O. Box 1130, 6160 BC Geleen, The Netherlands
First published on 16th August 2023
Abstract
We report the fixation of carbon dioxide (CO2) into novel rubber polymers based on polycarbonate domains. Our strategy consisted in the atom-efficient terpolymerisation of CO2 with a long-alkyl-chain epoxide and an unsaturated epoxide to obtain polycarbonates with a glass transition temperature (Tg) below room temperature and with pendant double bonds along the backbone to enable peroxide-promoted cross-linking, thus generating an elastic rubber. First, a wide range of epoxides with long alkyl chains (C6–C12) were coupled with CO2 to give polycarbonates with high yields, using a binary catalytic system consisting of an aluminium amino-tris(phenolate) complex and bis(triphenylphosphoranylidene)ammonium chloride (PPNCl). The synthesised polycarbonates were characterised using FTIR and NMR spectroscopy to determine yields and selectivity, using DSC to measure the Tg, and using GPC to obtain the molecular weight distribution. Next, the terpolymerisation was carried out by including allyl glycidyl ether (AGE) in the reaction mixture together with a long-alkyl-chain epoxide and CO2. Almost complete epoxide conversions (81–100%) and extremely high selectivity (>97%) towards the desired polycarbonates were achieved, with only traces of the cyclic carbonate side-products. The obtained polycarbonates displayed a Tg < 0 °C and thus behave as low-viscosity fluids at room temperature. The pendant unsaturated groups introduced with the AGE monomers allowed cross-linking of the terpolymers with dicumyl peroxide, leading to an elastic rubber-like behaviour as witnessed by their markedly decreased solubility in gel-content tests and by their storage modulus, loss modulus, and Tg, which were determined by dynamic mechanical analysis (DMA). In summary, we have successfully demonstrated that the terpolymerisation of long-chain epoxides, AGE and CO2 yields polycarbonates that can be cross-linked to obtain elastic rubber properties, thus opening the prospects for a new range of applications for CO2-based green polycarbonates.
1. Introduction
Carbon dioxide (CO2) emissions have strongly increased over the past two centuries as a consequence of human and industrial activities, resulting in an accumulation of 407 ppm (February 2020) of this compound in the atmosphere.1 CO2 is a well-known greenhouse gas and its increased concentration in the atmosphere has been correlated to worrisome environmental changes, such as global warming and increasing ocean water acidity.2,3 In this context, increasing scientific and technical efforts are being dedicated to mitigating the effect of CO2 emissions through carbon capture and utilisation techniques.4,5 The conversion of carbon dioxide into useful chemicals is also attractive because this C1-feedstock is abundant, non-toxic, inexpensive, inert and renewable.4–6 A broad range of products can be potentially obtained through the chemical conversion of CO2, ranging from bulk chemicals, such as methane and methanol, to higher value but smaller volume chemicals, such as cyclic carbonates or polycarbonates.7–9 The fixation of CO2 into valuable chemicals is not straightforward though. Since the CO2 molecule is thermodynamically highly stable, its conversion into other products requires a large energy input. A strategy to overcome this issue is to use a coupling reactant with a high free energy, such as hydrogen, small-ring heterocycles or amines.7–13 In this context, the 100% atom-efficient copolymerisation of CO2 with epoxides to produce polycarbonates is an attractive route to convert CO2 into valuable products (Fig. 1A).10,14–16 These polycarbonates have been successfully synthesised through this approach using a wide variety of epoxides and catalysts.10,14–20 So far, the obtained polymers have been studied for (potential) applications such as thermoplastics and thermoset resins or as polyols in the synthesis of polyurethanes.10,21–23 Cross-linking of polycarbonates with pendant double bonds was also attempted using thiol–ene click chemistry to form 3D-network materials.24,25,30,32 Moreover, polycarbonates can also be prepared by reacting CO2 with epoxides bearing a long alkyl chain, yielding polymers with Tg < 0 °C that are therefore in the rubbery state at room temperature.26–28 In this work, we conceived and investigated a novel strategy to prepare a new class of elastic rubber polycarbonates. To achieve this target, first we carried out the terpolymerisation of CO2 with two epoxides (Fig. 1B), one bearing a long alkyl chain (C6–C12) and the other with a pendant allyl or vinyl group, e.g. the bio-based allyl glycidyl ether (AGE) and vinylcyclohexene oxide (VCHO); see Fig. 2 for the full set of monomers. The strategy of using two epoxides with different pendant groups was designed to obtain a polycarbonate with the desired features to undergo cross-linking and generate an elastic rubber polymer (i.e. an elastomer):• The glass transition temperature (Tg) of the polymer should be below room temperature, which is essential to impart softness and flexibility to the final cross-linked material.27 For this purpose, we utilised long-alkyl-chain epoxides as monomers, because long pendant groups have been reported to limit the dipolar interactions between the polymer chains, which in turns leads to low Tg values.15,28,29
• No or a low degree of crystallinity is preferred, for the same reasons mentioned above. CO2-based polycarbonates prepared from long-alkyl-chain epoxides are suitable for this purpose as they are typically amorphous.15
• The polycarbonate should contain pendant groups to enable the cross-linking of the polymer chains and thus achieve elasticity and improved solvent resistance. For this purpose, epoxides with a terminal unsaturation were utilised in the terpolymerisation. The pendant double bonds can be used for post-synthetic cross-linking using peroxides.34 Dicumyl peroxide was chosen as the curing agent due to its high half degradation time, which enables the easy preparation of cross-linked rubbers via a solution route.
• A high molecular weight (Mw) is desirable for achieving a cross-linked polymeric network with a minimum of loose ends and sol fraction, in order to have optimum elasticity, elongation at break and mechanical strength of the final cross-linked rubber products.33,34
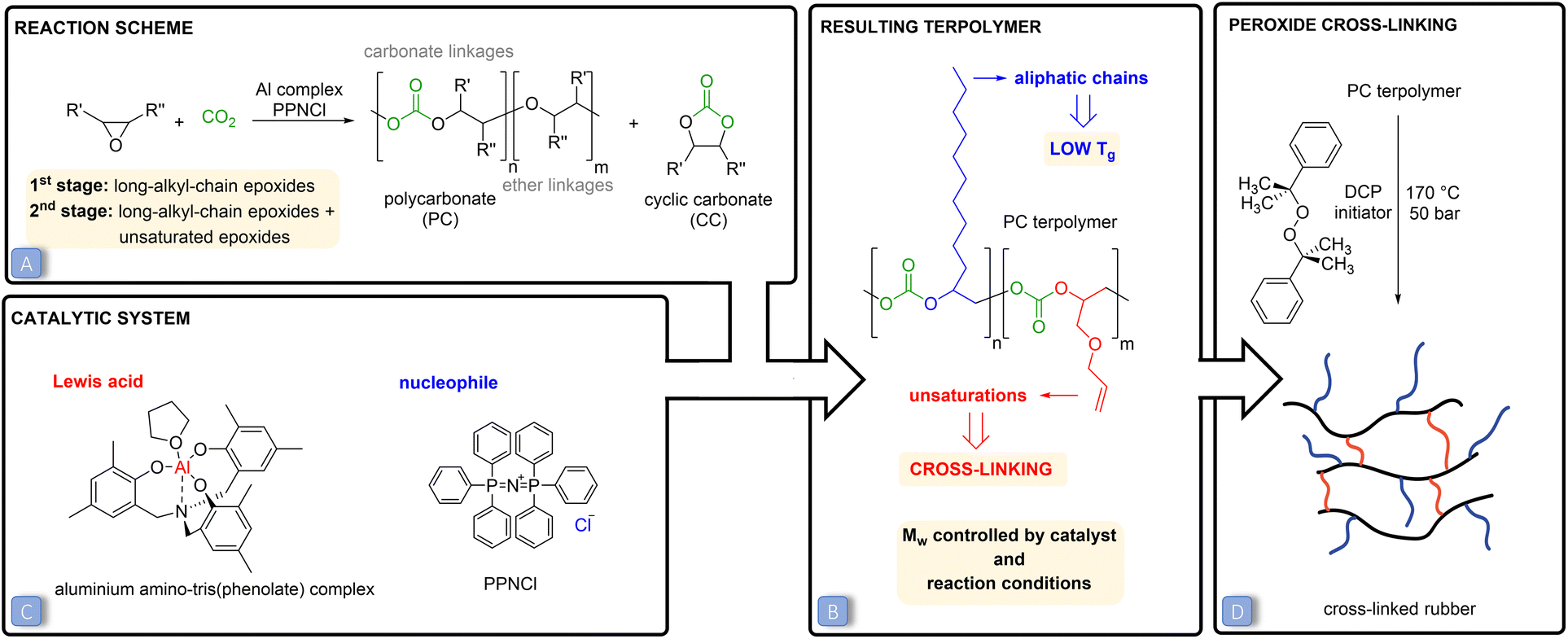 | ||
| Fig. 1 (A) The reaction between CO2 and epoxides yielding polycarbonates (and cyclic carbonates as possible side products). The first stage involves the copolymerisation of CO2 with long-alkyl-chain epoxides (copolymerisation), while in the second stage unsaturated epoxides are added to the reaction mixture (terpolymerisation). (B) The features of the prepared CO2-based polycarbonates that make them suitable for application as rubbers. (C) The binary catalytic system used in this work, consisting of an aluminium tris(phenolate) complex (acting as Lewis acid) and bis(triphenylphosphoranylidene)ammonium chloride (PPNCl, acting as nucleophile). (D) Peroxide cross-linking of the polycarbonate terpolymers to achieve an elastic rubber. | ||
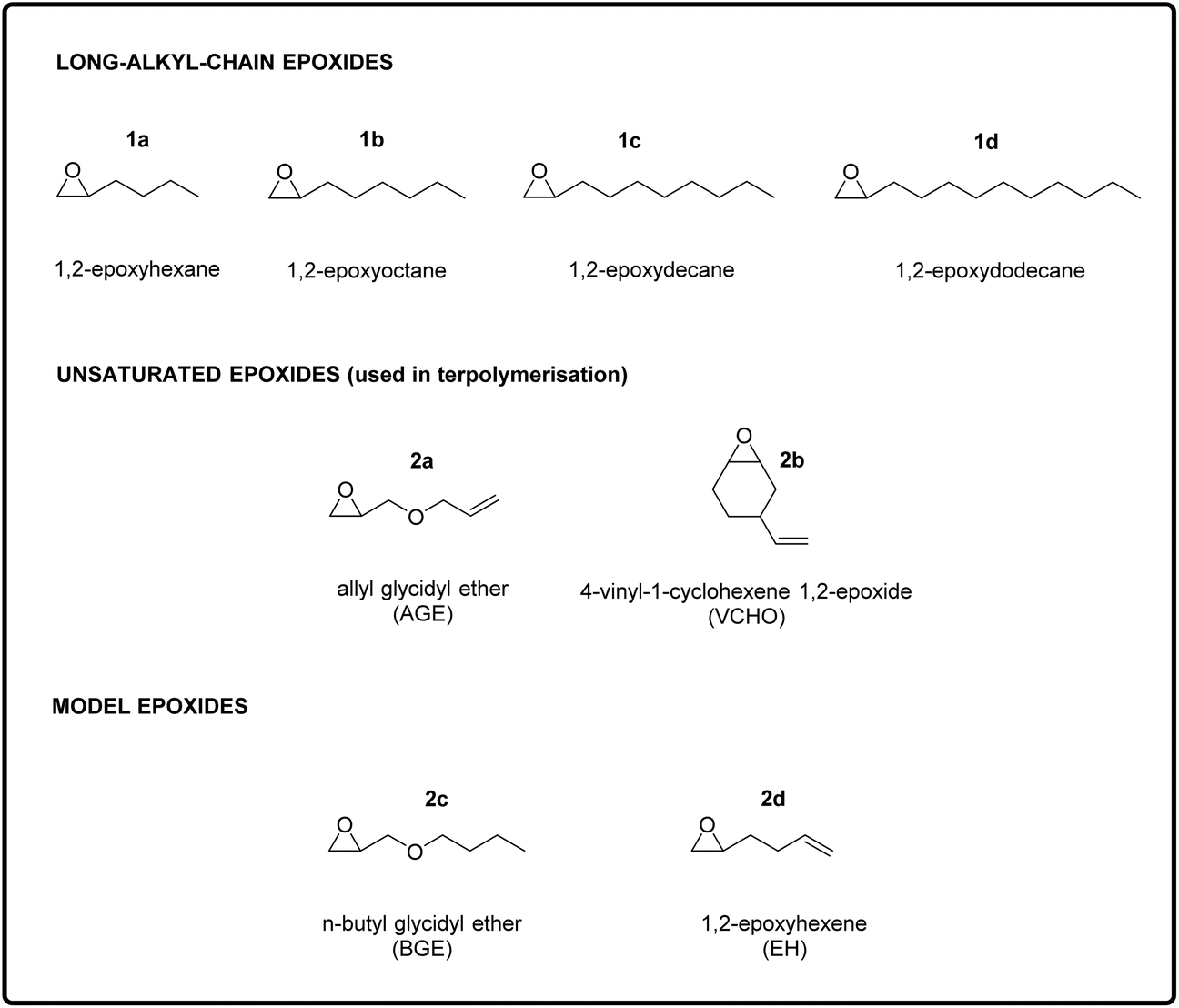 | ||
| Fig. 2 Substrates utilised for the atom efficient polymerisation of epoxides and CO2. Top: long-chain epoxides, used to impart low Tg to the polycarbonates. From left to right: 1,2-epoxyhexane (1a), 1,2-epoxyoctane (1b), 1,2-epoxydecane (1c), and 1,2-epoxydodecane (1d). Middle: unsaturated epoxides used as terpolymerisation monomers to allow the incorporation of pendant double bonds, which were then used to cross-link the polycarbonate chains to generate elastic rubbers. These epoxides are allyl glycidyl ether (AGE, 2a) and 4-vinyl-cyclohexene 1,2-epoxide (VCHO, 2b). Bottom: n-butyl glycidyl ether (BGE, 2c) and 1,2-epoxyhexene (EH, 2d), which were chosen as model epoxides to rationalise the terpolymerisations results. | ||
Besides the above-discussed effect of the nature of the epoxides, the properties of low-Tg polycarbonates can be controlled by tuning the type and amount of catalyst and the reaction conditions. In this study, a binary catalytic system (Fig. 1C) discovered by Kleij and co-workers31 and consisting of an aluminium amino-tris(phenolate) complex in combination with bis(triphenylphosphoranylidene)ammonium chloride (PPNCl) was chosen to prepare the desired polycarbonates. This catalytic system has several assets, as it relies on a non-toxic, abundant metal, Al, and has proved to be robust and efficient in promoting the copolymerisation of CO2 with a broad range of epoxides (see Fig. S1† for the proposed reaction mechanism).10,30,31 The Mw of the prepared polycarbonates was maximised by carrying out the polymerisations in supercritical CO2 (p > 73.8 bar) and at a relatively low temperature (45 or 60 °C). Low temperatures were applied to maximise the selectivity towards the kinetically-favoured polycarbonate products over cyclic carbonates (Fig. 1A), which are the thermodynamically-favoured products. To further prevent the formation of cylic carbonates, a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio between the Al complex and PPNCl was used, as previous studies have demonstrated that an equimolar amount or excess of the metal relative to the nucleophile prevents the displacement of the carbonate intermediate (Fig. S1†), thus inhibiting the ring closure that would yield the cyclic carbonate.14,15,35 High pressures were applied to promote the insertion of CO2 into the polymeric chain and to decrease the occurrence of ether linkages (see Fig. 1A).11,36
:1 molar ratio between the Al complex and PPNCl was used, as previous studies have demonstrated that an equimolar amount or excess of the metal relative to the nucleophile prevents the displacement of the carbonate intermediate (Fig. S1†), thus inhibiting the ring closure that would yield the cyclic carbonate.14,15,35 High pressures were applied to promote the insertion of CO2 into the polymeric chain and to decrease the occurrence of ether linkages (see Fig. 1A).11,36
Our strategy allowed the preparation of low-Tg terpolymers with different ratios of pendant unsaturations, which enabled cross-linking and thus led to the formation of a new class of elastic rubber polymers. The obtained cross-linked polycarbonates are a promising and green alternative to commercially available petrol-based elastomers,34 since they are prepared using renewable monomers such as CO2 and bio-based epoxides with a pendant double bond. Additionally, these materials have the potential to become even greener by utilising bio-based long-chain epoxides instead of the fossil-based ones used in this work.
2. Results and discussion
The aim of this study was to design and synthesise new rubber polymers based on a wide range of green polycarbonates, which were produced by the terpolymerisation of two types of epoxides (one with a long alkyl chain and the other with a pendant double bond) with CO2. In order to display the desired rubbery properties, the synthesised polymers should have a low Tg and have no or hardly any crystallinity, should possess a high Mw, and should contain groups allowing easy cross-linking. To reach this target, we envisaged a stepwise strategy, consisting of three stages: (i) the synthesis and characterisation of polycarbonates with low Tg and high Mw prepared by the co-polymerisation of CO2 with long-alkyl-chain epoxides; (ii) the terpolymerisation of CO2 with one of the long-alkyl-chain epoxides selected in the first step and with an epoxide with a pendant double bond; and (iii) the synthesis of cross-linked elastic rubbers through the reaction of the double bonds of the optimum terpolymers identified in the second step using a peroxide as cross-linking agent. The choice of using epoxides with a long alkyl chain group as monomers to achieve polycarbonates with low Tg stems from previous reports, which proved that increasing the length of the aliphatic side chain of the epoxide monomer leads to a monotonic decrease of the Tg of the polymer obtained upon copolymerisation with CO2.15,28 This trend has been attributed to the higher free volume available in such polycarbonates, which is caused by a reduction in dipolar interactions between polymer chains.29 Therefore, long-chain aliphatic epoxides, such as 1,2-epoxydodecane, 1,2-epoxydecane, 1,2-epoxyoctane and 1,2-epoxyhexane (1a–d, Fig. 2), were chosen as substrates for the copolymerisation reaction with CO2.2.1. Copolymerisation of CO2 with long-alkyl-chain epoxides
The first challenge of this study was to achieve an efficient coupling reaction between CO2 and long-alkyl-chain epoxides (1a–d in Fig. 2). For this purpose, we prepared and tested the well-known catalytic system consisting of an aluminium amino-tris(phenolate) complex and of PPNCl,31 under reaction conditions previously reported for the co-polymerisation of CO2 with other epoxides (80 bar CO2, 18 h at 60 °C).24 The chosen catalytic system proved efficient in achieving the reaction of CO2 with a representative long-alkyl-chain epoxide, namely 1,2-epoxyoctane (Table 1, entry 1), with very high conversion (97%). However, the selectivity towards the polycarbonate product was only 46%, with the cyclic carbonate being the main and only other product under these conditions. The formed polycarbonate also contained a fraction of ether linkages (25%), caused by the insertion of two consecutive epoxide units in the growing polymer chain. In order to increase the selectivity towards the thermodynamically less favourable but kinetically more accessible polycarbonate product, we lowered the reaction temperature from 60 to 45 °C (and increased the reaction time from 18 to 24 h). The strategy proved successful and allowed improving dramatically the selectivity towards poly(1,2-octene carbonate) (85%), with only a minor decrease in epoxide conversion (Table 1, entry 3). Under these improved reaction conditions, we also tested other aliphatic epoxides (1,2-epoxyhexane, 1,2-epoxydecane and 1,2-epoxydodecane). Despite the mild reaction temperature, high conversion was achieved with all the tested long-alkyl-chain epoxides (Table 1, entries 2–5), with a lower value (52%) only in the case of 1,2-epoxydodecane, i.e. the epoxide with the longest alkyl chain. This might be caused by the increased steric hindrance, which can hamper coordination to the aluminium complex, and/or by the observed lower solubility of the metal complex/PPNCl in 1,2-epoxydodecane compared to that in the other epoxides. With respect to selectivity, the opposite trend was observed: the longer the aliphatic chain, the higher the selectivity towards the polycarbonate (1,2-epoxyhexane < 1,2-epoxyoctane ≈ 1,2-epoxydecane < 1,2-epoxydodecane). The influence of the length of the aliphatic chain on the selectivity could be related to steric hindrance, which might hamper the intramolecular ring closure that leads to the cyclic carbonate; and/or to interaction with the methyl groups on the ligand of the Al complex, which might help to keep the carbonate intermediate in proximity to the Al centre and thus prevent the ring closure. Overall, the selectivity was found to be between 77% (for 1,2-epoxyhexane) and 95% (for 1,2-epoxydodecane), which is a remarkable increase when compared to the results achieved before at higher temperature. Under these optimised conditions, only a small percentage of ether linkages was found in the polymers prepared by reacting long-alkyl-chain epoxides with CO2 (11–21%, entries 2–5 Table 1). The molecular weight distributions of the prepared polycarbonates were determined by GPC (relative to polystyrene standards), see Table 2 (entries 1–4). The molecular weights were in the typical range observed for the copolymerisation of CO2 with epoxides with the chosen catalytic system and without special precautions to remove adventitious water,30 with a maximum Mw of around 10 kDa, a maximum Mn of around 7 kDa and dispersity values (Đ) ≤ 1.2, indicating a narrow distribution of polymer chain lengths. The thermal properties of the polycarbonates were investigated using DSC (Table 2, entries 1–4 and Fig. 3, Fig. S14–S18 in the ESI†). Notably, the anticipated effect of the long alkyl chains led to polymers with Tg (or Tm) values below 0 °C, implying that all these polycarbonates are low-viscosity fluids at room temperature. Within the set of long-alkyl-chain polycarbonates, the expected general trend of decreasing Tg value with the increasing length of the lateral chain was observed, from −29 °C for poly(1,2-hexene carbonate) up to −32 °C for poly(1,2-decene carbonate). On the other hand, poly(1,2-dodecene carbonate) exhibited a melting point (Tm) but no glass transition in the analysed temperature range (see Fig. 3 and Fig. S18 in the ESI†).27 The DSC peak was recorded at −16 °C, which is a higher temperature compared to the observed trend for the Tg values of the polycarbonates with shorter alkyl chains. This phenomenon might be related to the long pendant aliphatic chains of poly(1,2-dodecene carbonate), which might organise themselves in an ordered pattern leading to a certain degree of crystallinity in the polymer structure. In addition, since the polymer is a low-viscosity fluid at room temperature, these data suggest that 1,2-epoxydodecane-based polycarbonates display liquid crystal behaviour. This would be the first report of a liquid crystal CO2-based polycarbonate.32,37,38 However, further investigations that are beyond the scope of this paper would be required to ascertain the (liquid crystal) nature of poly(1,2-dodecene carbonate), for example by means of SAXS/WAXS. In comparison with the literature, the Tg values of poly(1,2-hexene carbonate) and poly(1,2-octene carbonate) are in fair agreement with previous reports,10,15,28 whereas poly(1,2-decene carbonate) and poly(1,2-dodecene carbonate) are reported here for the first time.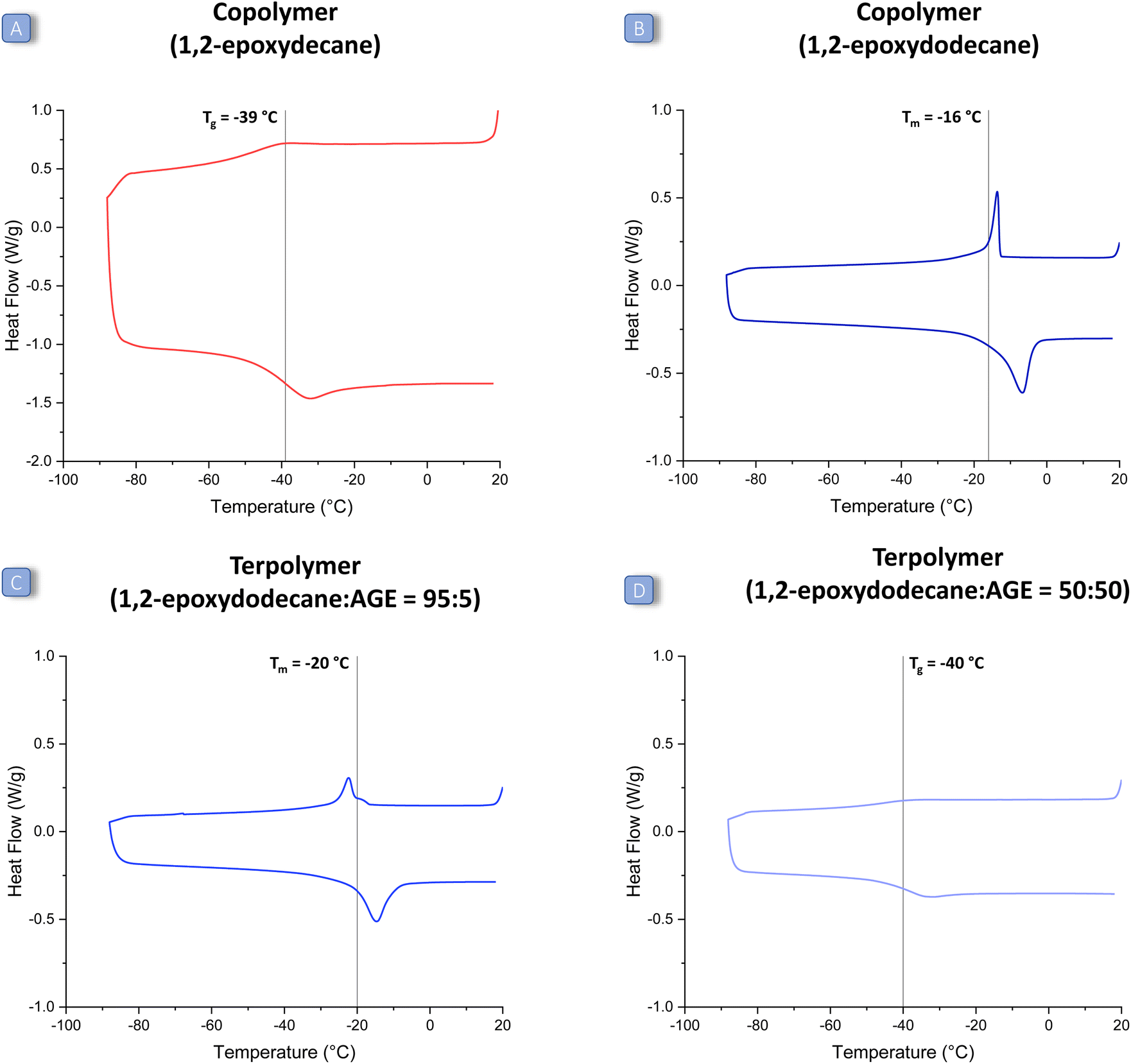 | ||
| Fig. 3 DSC curves of selected polycarbonates: (A) copolymer prepared from 1,2-epoxydecane and CO2; (B) copolymer prepared from 1,2-epoxydodecane and CO2; (C and D) terpolymers prepared from 1,2-epoxydodecane, allyl glycidyl ether (AGE) and CO2, with different ratios between the two epoxides (95:5 and 50:50). The Tg and Tm values were determined from the heating branch of the DSC curve, which is the bottom part of the curve. | ||
| Entry | Epoxide used to prepare the polycarbonate | T (°C) | Reaction time (h) | Conversiona (%) | Selectivity towards the polymerb (%) | Carbonate-to-ether linkages in the polymer c |
|---|---|---|---|---|---|---|
| Reaction conditions: 30 mmol epoxide, 3 mmol mesitylene (as internal standard), 0.5 mol% Al complex, 0.25 mol% PPNCl, 80 bar CO2, 45 or 60 °C.a Conversion of the epoxide calculated via1H NMR (see Fig. S3 and S5† for representative spectra).b Selectivity of the catalytic system towards the polycarbonate product, calculated via FTIR; the only other product was the cyclic carbonate (see Fig. S4 and S6† for representative spectra).c Determined by 1H NMR. | ||||||
| 1 | 1,2-Epoxyoctane | 60 | 18 | 97 | 46 | 75:25 |
| 2 | 1,2-Epoxyhexane | 45 | 24 | 98 | 77 | 89:11 |
| 3 | 1,2-Epoxyoctane | 45 | 24 | 83 | 85 | 87:13 |
| 4 | 1,2-Epoxydecane | 45 | 24 | 90 | 82 | 87:13 |
| 5 | 1,2-Epoxydodecane | 45 | 24 | 52 | 95 | 79:21 |
| Entry | Epoxide(s) used to prepare the polycarbonate | Epoxide ratio (mol/mol) | T g (°C) | T m (°C) | ΔH (J g−1) | M w (kDa) | M n (kDa) | Đ (Mw/Mn) |
|---|---|---|---|---|---|---|---|---|
| Reaction conditions: 30 mmol epoxide(s), 3 mmol mesitylene (as internal standard), 0.5 mol% Al complex, 0.25 mol% PPNCl, 80 bar CO2, 45 °C.a For these materials, Tm but no Tg was detected in the analysed temperature range. It is worth noting that for these polycarbonates the DSC analysis was carried out in a lower temperature range (from −90 to 20 °C) compared to the other polycarbonates (from −50 to 50 °C), as the Tg signal is expected to be observed at a lower temperature compared to the Tm signal.b Reaction conditions: 30 g of allyl glycidyl ether (AGE), 0.5 mol% Al complex, 0.25 mol% PPNCl, 80 bar CO2, 45 °C. | ||||||||
| 1 | 1,2-Epoxyhexane | — | −29 | 4.7 | 4.3 | 1.1 | ||
| 2 | 1,2-Epoxyoctane | — | −28 | 4.8 | 4.2 | 1.1 | ||
| 3 | 1,2-Epoxydecane | — | −39 | 8.7 | 7.3 | 1.2 | ||
| 4a | 1,2-Epoxydodecane | — | −16 | 14 | 4.8 | 4.0 | 1.2 | |
| 5 | AGE | — | −37 | 10.0 | 7.0 | 1.4 | ||
| 5bb | AGE | −35 | 7.4 | 6.4 | 1.1 | |||
| 6 | 1,2-Epoxyhexane + AGE | 95:05 |
−18 | 8.8 | 6.0 | 1.5 | ||
| 7 | 80:20 |
−14 | 9.8 | 8.4 | 1.2 | |||
| 8 | 50:50 |
−25 | 7.9 | 6.0 | 1.3 | |||
| 9 | 1,2-Epoxyoctane + AGE | 95:05 |
−33 | 10.2 | 8.8 | 1.2 | ||
| 10 | 80:20 |
−30 | 5.4 | 4.0 | 1.4 | |||
| 11 | 50:50 |
−40 | 2.1 | 1.6 | 1.3 | |||
| 12 | 1,2-Epoxydecane + AGE | 95:05 |
−33 | 4.2 | 3.3 | 1.3 | ||
| 13 | 80:20 |
−35 | 11.5 | 9.6 | 1.2 | |||
| 14 | 50:50 |
−35 | 7.6 | 6.0 | 1.3 | |||
| 15a | 1,2-Epoxydodecane + AGE | 95:05 |
−20 | 11 | 5.3 | 4.3 | 1.2 | |
| 16a | 80:20 |
−40 | 6 | 5.2 | 4.2 | 1.2 | ||
| 17 | 50:50 |
−40 | 5.3 | 4.4 | 1.2 | |||
| 18 | 1,2-Epoxydodecane + VCHO | 80:20 |
−23 | 6.2 | 5.2 | 1.2 | ||
2.2. Terpolymerisation of CO2 with long-alkyl-chain epoxides
After having proved the feasibility of the alternating copolymerisation of CO2 with long-alkyl-chain epoxides with the aluminium amino-tris(phenolate)/PPNCl catalytic system, our strategy to prepare CO2-based elastic rubbers entailed introducing a functional group that would allow cross-linking of the polycarbonates. For this purpose, we carried out a series of terpolymerisation reactions of CO2 with a long-alkyl-chain epoxide (1a–d in Fig. 2) and an epoxide with a terminal double bond (2a–b in Fig. 2).24,30 The presence of these pendant double bonds along the polycarbonate chain would then be used to cross-link the polymer chains using a peroxide (Fig. 1).39 As the monomer with an epoxide group and a pendant unsaturation, we initially selected allyl glycidyl ether (AGE, 2a), which is partially bio-based as it can be prepared from epichlorohydrin, which in turn can be synthesised from bio-based glycerol.40,41 With this compound, we investigated the effect of the ratio between the two epoxides used in the terpolymerisation (long-alkyl-chain epoxide:AGE = 95:5, 80:20 and 50:50) with each of the long-alkyl-chain epoxides 1a–d. The terpolymerisation experiments were run under the same reaction conditions as the optimised copolymerisation tests discussed above. The results were excellent, with conversion of both epoxides above 95% in most of the tests, regardless of the length of the chain of the aliphatic epoxides and their ratio relative to AGE (Table 3, entries 1–12). The optimal conversions were achieved when the long-alkyl-chain epoxides and AGE were used in an 80:20 ratio (Table 3, entries 2, 5, 8 and 11). Slightly lower conversions were achieved only when 1,2-epoxydodecane was terpolymerised with AGE in 95:5 and 50:50 ratios (Table 3, entries 10 and 12). The lower epoxide conversion when 1,2-epoxydodecane and AGE were employed in a 95:5 ratio is not unexpected considering the high concentration of 1,2-epoxydodecane and the fact that when this compound was copolymerised with CO2 a rather low conversion was observed (52%, see Table 1). On the other hand, further investigation would be required to rationalise the lower conversion obtained when using a 50:50 ratio between 1,2-epoxydodecane and AGE compared to the reaction with the 80:20 ratio. Notably, the selectivity towards the polycarbonate was very high (≥97%) in almost all cases and superior compared to the tests in the absence of AGE (see Table 1). This improved selectivity is ascribed to the use of AGE as monomer in the terpolymerisation reactions. This hypothesis stems from the results of the copolymerisation of AGE with CO2, which gave virtually full conversion (99%) of the epoxide with almost complete selectivity (99%) towards the polycarbonate (Table 3, entry 13). The copolymerisation of AGE and CO2 was also realised on a larger scale (30 g, entry 5b in Table 2 and Fig. S31†), achieving nearly full AGE conversion also in this case and without affecting the physical and chemical properties of the final polycarbonate. In order to explain the extremely high selectivity towards the polycarbonate product observed when AGE was reacted with CO2 (either alone or in a terpolymerisation), we hypothesised that the way in which this compound coordinates to the aluminium complex used as a catalyst may play a crucial role. As depicted in Fig. 4, the alkoxide intermediate obtained by ring-opening of AGE may interact with the aluminium centre through the oxygen of its ether group and/or with the aromatic ring of the ligand through its double bond functionality (π–π interaction). In order to establish if (and which of) these possible interactions play a role, we performed two different terpolymerisations with CO2 and 1,2-epoxydecane, but substituting AGE with a model epoxide bearing only one of the two functional groups in the AGE monomer that may interact with the complex. The first model epoxide (butyl glycidyl ether, BGE, 2c in Fig. 2) contains an ether group in the same position as in AGE but no unsaturation, whereas the second model compound (1,2-epoxy-5-hexene, EH, 2d in Fig. 2) has no ether group but a terminal double bond at approximately the same distance from the epoxide ring as in AGE. Both BGE and EH were less reactive than AGE (both towards the formation of the terpolymer and of the copolymer, see entries 13–17 in Table 3), which is ascribed to the presence of only one of the two functionalities characterising AGE. On the other hand, the selectivity of the reactions involving BGE (≥97%, entries 14 and 16 in Table 3) was similar to that with AGE and remarkably higher than the one with EH (80% for the terpolymer and 72% for the copolymer, entries 15 and 17 in Table 3). This difference suggests that the interaction between the oxygen of the ether group of AGE and the aluminium complex may be the factor leading to the observed high selectivity towards the polycarbonate. Future studies by means of density functional theory (DFT) calculations are advised for further investigating this hypothesis.
:05, 80:20 and 50:50)
| Entry | Long-alkyl-chain epoxide | Unsaturated epoxide | Ratioa (mol/mol) | Conversion of the two epoxidesb (%) | Selectivity towards the polymerc (%) | Ratio of carbonate-to-ether linkages in the polymerd |
|---|---|---|---|---|---|---|
| Reaction conditions: 30 mmol epoxides (long alkyl chain + unsaturated), 3 mmol mesitylene (as internal standard), 0.5 mol% Al complex, 0.25 mol% PPNCl, 80 bar CO2, 45 °C, 24 h; apart from * in which the reaction was carried out at 60 °C for 18 h. ◊ Not unsaturated.a In the synthesis mixture.b Conversion of the epoxide calculated via1H NMR (see Fig. S7, S9 and S11† for representative spectra).c Selectivity of the catalytic system towards the polycarbonate product, calculated via FTIR spectroscopy; the only other product was the cyclic carbonate (see Fig. S8, S10 and S12† for representative spectra).d Determined by 1H NMR. | ||||||
| 1 | 1,2-Epoxyhexane | AGE | 95:5 |
>99; >99 | 98 | 92:8 |
| 2 | 80:20 |
99; >99 | 97 | 95:5 |
||
| 3 | 50:50 |
98; 99 | 99 | 91:9 |
||
| 4 | 1,2-Epoxyoctane | AGE | 95:5 |
>99; >99 | 98 | 92:8 |
| 5 | 80:20 |
97; >99 | 97 | 90:10 |
||
| 6 | 50:50 |
95; 96 | 99 | 89:11 |
||
| 7 | 1,2-Epoxydecane | AGE | 95:5 |
97; >99 | 98 | 93:7 |
| 8 | 80:20 |
98; >99 | 99 | 94:6 |
||
| 9 | 50:50 |
99; 98 | 99 | 91:9 |
||
| 10 | 1,2-Epoxydodecane | AGE | 95:5 |
81; 93 | 97 | 91:9 |
| 11 | 80:20 |
98; >99 | 99 | 94:6 |
||
| 12 | 50:50 |
88; 90 | 99 | 85:15 |
||
| 13 | — | AGE | 0:100 |
99 | 99 | ND |
| 14 | 1,2-Epoxydecane | BGE◊ | 80:20 |
73; 60 | 98 | 94:6 |
| 15 | 1,2-Epoxydecane | EH | 80:20 |
88 (peaks overlap) | 80 | 90:10 |
| 16 | — | BGE◊ | 0:100 |
84 | 97 | ND |
| 17 | — | EH | 0:100 |
98 | 72 | 88:12 |
| 18 | 1,2-Epoxydodecane | VCHO | 80:20 |
76; >99 | 80 | 94:6 |
| 19* | 1,2-Epoxydodecane | VCHO | 80:20 |
99; >99 | 56 | 90:10 |
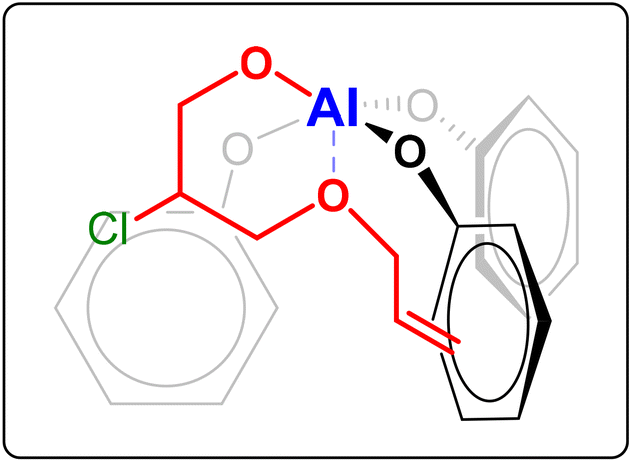 | ||
| Fig. 4 Possible coordination to the Al complex of the alkoxide intermediate formed by ring-opening of allyl glycidyl ether. The alkoxide intermediate is formed upon coordination of the epoxide to the metal centre and subsequent nucleophilic attack by the halide (Cl−), which can happen at both carbons of the epoxide ring.15 For the sake of clarity, in this figure only one of these two possibilities is shown, namely the one that would be more likely based on the electron withdrawing effect of the –OR group (R = allyl). | ||
After proving that the terpolymerisation is feasible with AGE and any of the long-alkyl-chain epoxides, we studied the possibility of using an alternative bio-based epoxide with a pendant unsaturation: 4-vinyl-1-cyclohexene 1,2-epoxide (VCHO, 2b), which can be completely obtained from renewable feedstock if prepared by the Diels–Alder dimerisation of bio-butadiene followed by epoxidation of the double bond with a green oxidant such as H2O2.10,42 The terpolymerisation was carried out with 1,2-epoxydodecane and VCHO in an 80:20 ratio and with the other conditions as in the tests with AGE (45 °C, 80 bar CO2). Although cyclohexene oxide and the related VCHO are well-known to be less reactive towards reaction with CO2 compared to linear, terminal epoxides (though they usually show a higher selectivity to form the polycarbonate over the competing cyclic carbonate),15,24 we were able to achieve excellent conversion of the two epoxides (76% and >99%, respectively) with good selectivity towards the polycarbonate (80%, entry 18 in Table 3). The fact that the selectivity is very similar to that obtained with EH, and lower compared to that with glycidyl ethers (Table 3), supports our hypothesis that the coordination of the O atom of the ether group to the Al complex promotes the selectivity towards the polycarbonate. We also attempted to carry out the terpolymerisation of CO2, 1,2-epoxydodecane and VCHO at a slightly higher temperature (60 °C, Table 3, entry 19), and this allowed reaching complete conversion of both epoxides, but at the cost of a lower selectivity (56%) towards the desired polycarbonate product. These tests with VCHO demonstrate the versatility of our terpolymerisation approach, which was proved to work with different long-alkyl-chain epoxides and with two very different bio-based epoxides with a terminal unsaturation, namely AGE and VCHO, achieving high conversions and selectivities at a mild reaction temperature (45 °C). In these terpolymerisations, the occurrence of ether linkages in the final polymer was in the range of 5–15%, which corresponds to a slightly higher degree of CO2 incorporation compared to the copolymerisation reactions (compare data in Tables 3 and 1). Characterisation by MALDI-TOF and 13C NMR was performed to investigate the structure and tacticity of the terpolymers. However, MALDI-TOF analysis was not successful, probably because the aliphatic side chains inhibit ionisation of the samples. The 13C NMR spectra showed the characteristic peaks of carbonate linkages (Fig. S13†), but the signals are rather complex due to the multiplicity of possible regiosequences (head-to-tail, head-to-head, tail-to-tail, for each of the possible combinations of the two epoxides, A–A, A–B and B–B), suggesting that the terpolymers are atactic. The thermal properties of the prepared terpolymers were determined by DSC (Table 2 and Fig. S19–S30†) and their molecular weights were estimated by GPC analysis (Table 2 and Fig. S32, 33† for examples of GPC curves). The molecular weights of the terpolymers were generally the highest when an 80:20 ratio between the long-alkyl-chain epoxide and AGE was employed (Table 2, entries 7, 10, 13, 16), i.e. under the conditions that also gave the highest degree of epoxide conversion (vide supra). For such polymers, the Mw and Mn values surpassed those of the corresponding copolymers prepared in the absence of AGE (Table 2, entries 1–4). This can probably be correlated to the relatively high Mw and Mn values obtained with the copolymer prepared by the reaction of CO2 with AGE (Table 2, entry 5). The terpolymers had a relatively narrow range of polymer chain lengths, as indicated by the relatively low dispersity values (Đ ≤ 1.5). The highest molecular weight was obtained with the terpolymer prepared from 1,2-epoxydodecane and AGE in 80 : 20 ratio (Mw = 11.5, Mn = 9.6, see Table 2, entry 13). Although these values indicate a relatively high molecular weight, future work might be aimed at increasing these values by working in a more controlled water-free environment to avoid chain-transfer reactions that limit the chain growth.15 This would be desirable because polymers used to produce technical rubbers have typical Mw values of 50 kDa or higher.34 All the prepared terpolymers can be defined as low-viscosity fluids at room temperature and accordingly they displayed glass transition temperatures below 0 °C (Table 2). A Tm but no Tg was observed in the terpolymers prepared with 95:05 and 80:20 ratios between 1,2-epoxydodecane and AGE, similarly to what was observed for the copolymer of 1,2-epoxydodecane and CO2 (Table 2, entries 4, 15 and 16), whereas for the terpolymer prepared with a 50:50 ratio, a Tg of −40 °C was observed (Table 2, entry 17). The fact that no Tg was detected for the terpolymers with high 1,2-epoxydodecane content in their backbone (80% and above) is probably due to the fact that their Tg signal is too weak or below the lower temperature limit of the DSC instrument (−90 °C). These results confirm the tendency of 1,2-epoxydodecane to lead to polycarbonates with liquid crystal behaviour, also in terpolymers in which this long-chain epoxide is the major epoxide monomer. Besides the determination of the Tg and Tm, the DSC curves also provide an insight into the structure of the terpolymers. More specifically, if the units derived from the two epoxides were randomly distributed in the prepared polycarbonate, the DSC would show a single Tg. In contrast, two Tg values would be observed if a block-copolymer was obtained.43 Since for each of the polymers prepared in this work a single Tg was found, we conclude that random alternating terpolymers were successfully synthesised. Importantly, the fact that all the prepared terpolymers have low Tg (or Tm) values makes them suitable candidates for preparing elastic rubbers by cross-linking of the pendant unsaturations.
2.3. Cross-linking of the polycarbonates
Once we successfully achieved the terpolymerisation between CO2, a long-chain epoxide and an additional epoxide bearing a terminal double bond, we cross-linked the polycarbonate chains by the reaction of these unsaturated groups using dicumyl peroxide (DCP) as the initiator39 to generate a three-dimensional polymeric network characterised by rubber elasticity (Fig. 1D).34,44 Peroxide-curing is generally the chosen method to synthesise cross-linked rubbers and DCP is a well-known curing agent in rubber applications34,45–48 as it is stable during elastomer processing and it degrades readily at the desired curing temperature.45–48 The terpolymers prepared with 1,2-epoxyoctane, 1,2-epoxydecane and 1,2-epoxydodecane as monomers were cross-linked in the presence of DCP in a compression mould. The curing was carried out at 170 °C, which is a high enough temperature to allow rapid cross-linking (Table S2†) and sufficiently low to avoid degradation of the polycarbonates (as assessed by TGA, see Fig. S34†). Even though peroxide cross-linking could theoretically lead to cross-linked polymers also in the absence of pendant terminal unsaturations,34,48,49 none of the terpolymers prepared with the lowest AGE content (5%) underwent cross-linking to a satisfactory degree under the employed reaction conditions, as proven by the fact that they were still low-viscosity fluids at the end of the test. This could be ascribed either to the steric hindrance of the aliphatic lateral chains, which could limit the accessibility of the pendant double bonds thus inhibiting their reaction with DCP and with each other, and/or to the insufficient incorporation of unsaturated groups.50,51 On the other hand, the terpolymers prepared with at least 20% of pendant terminal unsaturations were successfully cross-linked yielding solid elastic rubbers (Fig. 5 and Table 4). The synthesised elastic rubbers, which appeared as transparent, soft and flexible plates (∼1 mm thickness, Fig. 5), were able to undergo elastic deformation upon manual bending and to regain their initial shape and size when the strain was removed. However, the materials were also found to be breakable as a result of repeated handling, probably due to the low molecular weight of the parent terpolymers before undergoing cross-linking.33 The prepared elastic rubbers were then characterised to determine their degree of cross-linking by means of gel-content tests (Table 4),44,51 showing a rather similar high degree of cross-linking (≥77%). The highest gel content (89%) and thus the highest degree of cross-linking was achieved with the elastic rubber based on the terpolymer prepared from the reaction of CO2 with 1,2-epoxydecane and AGE (80:20), and this value was very similar to the gel content of the elastic rubber based on the copolymer of CO2 and AGE (Table 4), suggesting that the incomplete cross-linking was not caused by the long alkyl lateral chains that are present in the terpolymer. Instead, the observed incomplete degree of cross-linking can be ascribed to the presence of residual solvent after the addition of DCP to the terpolymer, which can be detrimental to the yield of the cross-linking reaction.49,52
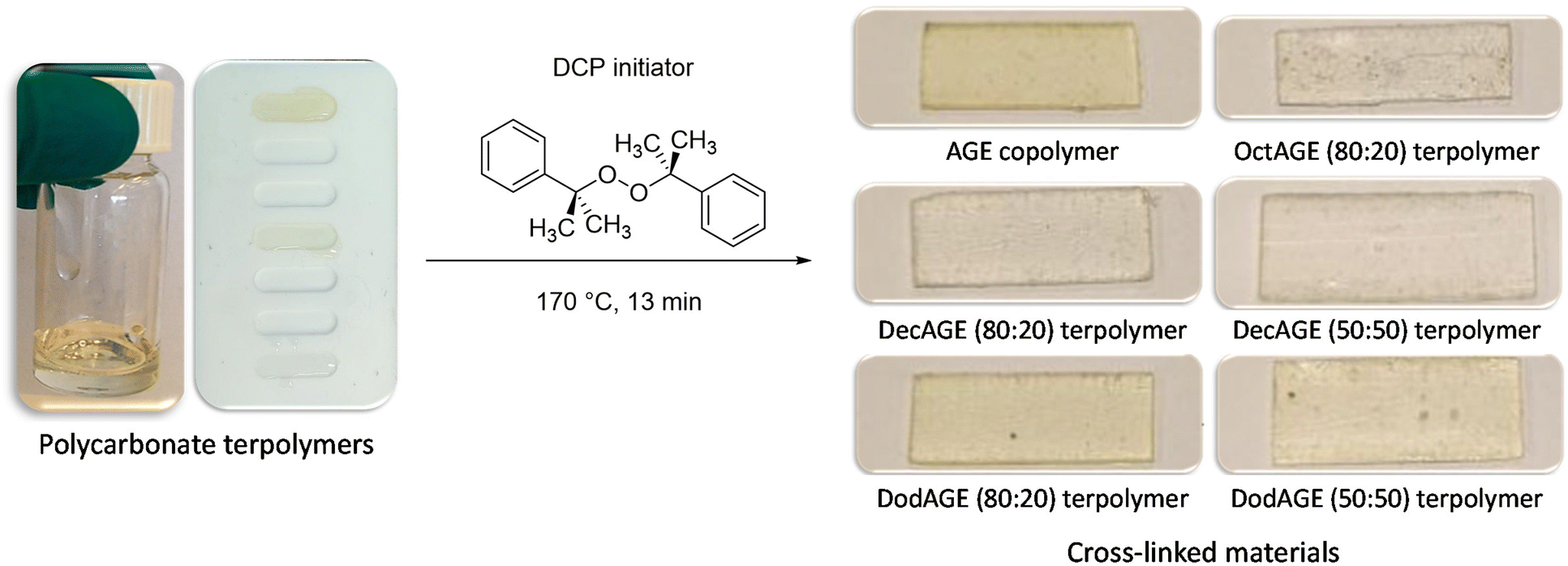 | ||
| Fig. 5 Conversion of polycarbonate terpolymers (low-viscosity fluids) into elastic rubbers (solid) through cross-linking with dicumyl peroxide (DCP) in a compression mould. The cross-linked AGE copolymer, which does not behave as an elastic rubber, is included for comparison purposes. The crossed-linked materials were obtained as plates (length: 2–3 cm, thickness: ∼1 mm). | ||
| Epoxide(s) used to prepare the polycarbonate | Epoxide ratio (mol/mol) | Gel content (wt%) | T g (°C) after cross-linking |
|---|---|---|---|
| a Dichloromethane was used as the solvent in the gel-content tests. | |||
| AGE | 00:100 |
88 | 59 |
| 1,2-Epoxyoctane + AGE | 80:20 |
77 | −14 |
| 1,2-Epoxydecane + AGE | 80:20 |
89 | −19 |
| 50:50 |
78 | ||
| 1,2-Epoxydodecane + AGE | 80:20 |
78 | −18 |
| 50:50 |
85 | ||
The behaviour as elastic rubbers of the most promising cross-linked polycarbonates (i.e. those based on AGE and 1,2-epoxydodecane, 1,2-epoxydecane and 1,2-epoxyoctane in an 80:20 ratio) was assessed by dynamic mechanical analysis (DMA). This allowed determination of the response of the synthesised cross-linked solid polymers to the application of stress (storage and loss modulus) and to a temperature increase. All the cross-linked polycarbonate terpolymers displayed a low Tg ranging from −19 to −14 °C and a clear rubbery plateau (corresponding to the state in which the material displays elastic properties) was reached well below room temperature (Fig. 6). These results indicate that the peroxide curing only slightly increased the overall Tg of the materials and that the final cured polycarbonates can be identified as elastic rubbers. On the other hand, the polycarbonate prepared by copolymerisation of AGE and CO2 displayed a remarkably higher Tg and the material did not show elastic behaviour (rubbery plateau) below 80 °C, meaning that the achieved material cannot be classified as elastic rubber and that the long-chain aliphatic epoxides are crucial to ensure elasticity to the cross-linked polycarbonates. The low Tg values of these cured terpolymers, which fall in a similar range to the low crystallinity hydrogenated acrylonitrile/butadiene rubbers (HBNR, −45/−10 °C), ethylene/vinyl acetate rubbers (EVM), and ethylene acrylic elastomers (AEM, −45/−10 °C),34 are even more interesting considering the relatively low Mw of the starting polycarbonates chains.
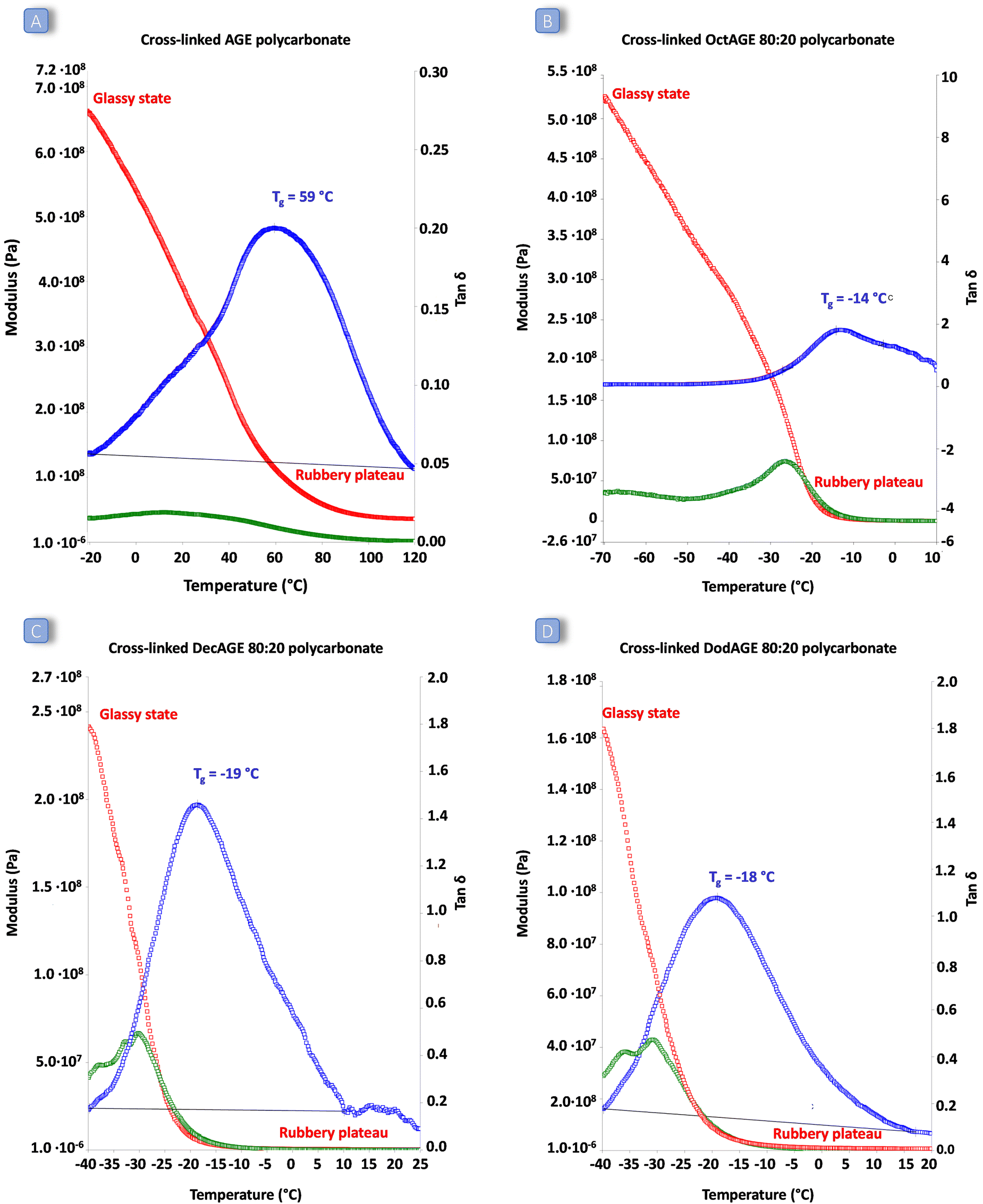 | ||
| Fig. 6 DMA graphs of: (A) solid obtained by cross-linking the AGE polycarbonate copolymer. (B) Solid obtained by cross-linking the 1,2-epoxyoctane:AGE = 80:20 polycarbonate terpolymer. (C) Solid obtained by cross-linking the 1,2-epoxydecane:AGE = 80:20 polycarbonate terpolymer. (D) Solid obtained by cross-linking the 1,2-epoxydodecane:AGE = 80:20 polycarbonate terpolymer. The storage modulus is shown in red, the loss modulus in green, and the tanδ in blue. | ||
Although there is room for improvement in the cross-linking procedure, for example by increasing the Mw of the polycarbonate scaffolds through chain elongation strategies (for instance by reaction with diisocyanates),53 by increasing the loading of peroxide relative to the terpolymer, and by tuning and optimising the peroxide curing operating conditions, these results represent a proof of principle of the possibility of preparing CO2-based elastic rubbers through the cross-linking of polycarbonate terpolymers.
2.4. Outlook
The novel polycarbonate-based elastic rubbers prepared by cross-linking of terpolymers of CO2, a long-alkyl-chain epoxide and AGE have several attractive features. Since they do not contain unsaturations in the polymer backbone, they are expected to exhibit resistance to heat, oxidation and ozonolysis that is superior to that of polydiene rubbers, such as natural rubber (NR), butadiene rubber (BR) and acrylonitrile/butadiene rubber (NBR), and similar to that of rubbers without unsaturations in the main chain, such as ethylene/propylene/diene terpolymers (EPDMs), fully hydrogenated nitrile/butadiene rubber (HNBR) and ethylene/vinyl acetate copolymers (EVMs).54 The polarity of these new polycarbonate elastic rubbers can be varied by adjusting the lengths of the alkyl side chains of the epoxide monomers, but will be in any case relatively high because of the presence of the carbonate linkages in the polymer backbone. The resistance to non-polar media, such as oil, fuel, grease and fat, is thus expected to be superior to that of non-polar rubbers, such as NR, BR and EPDM, and may be as good as that of polar rubbers, such as HNBR and EVM.54 Therefore, these new polycarbonate elastic rubbers could find potential applications in seals to be used in contact with oil, fuel and gas at relatively high temperatures as in (automotive) engines as well as in the oil and gas industry.54All the commercial rubbers mentioned above are based on unsaturated hydrocarbon monomers that are produced from crude oil with the obvious and major exception of NR. The polycarbonate elastic rubbers presented here are based on CO2, which is a renewable, cost-effective, and abundant C1 feedstock. The polycarbonate backbone of these new rubbers is expected to make these materials susceptible towards hydrolysis, which on the one hand is undesired in practical applications of rubber products, but on the other hand is beneficial in terms of recyclability and biodegradability. Covalently cross-linked rubbers are characterised by three-dimensional polymeric networks and, thus, are intractable and insoluble. As a result, such cross-linked rubber products are notoriously difficult to recycle into new, high-value rubber products. In addition, rubber waste discarded in the environment can persist for decades.55 In contrast, our polycarbonate-based rubbers can enable on-purpose hydrolysis for recycling or biodegradation of end-of-life rubber waste. In summary, these novel polycarbonate elastic rubbers are expected to combine excellent heat resistance with good oil resistance similar to high-value HNBR and EVM rubbers with the advantage of being more sustainable in terms of raw materials, recycling and biodegradability. One of the first steps to improve the practical performance of these polycarbonate-based rubbers is to increase their Mw, which should result in higher gel contents and thus enhanced elastic properties.
3. Conclusions
In this study, we introduced a novel class of polycarbonate-based elastic rubbers that were prepared from renewable CO2 and (partially) bio-based epoxides. First, we demonstrated that the copolymerisation of CO2 with long-alkyl-chain epoxides can be achieved selectively and at mild temperature (45 °C) using a binary homogeneous catalytic system consisting of an aluminium amino-tris(phenolate) complex and PPNCl. Moreover, these polymerisations can be scaled up, as demonstrated by the successful synthesis of the copolymer of CO2 and AGE at the 30 g scale. Next, we broadened the scope by including in the reaction mixture an epoxide with a pendant double bond, leading to the synthesis of terpolymers. These polycarbonates are characterised by long-alkyl-chains and by pendant unsaturations as the lateral groups along the polymer backbone. The presence of the long alkyl chains led to polymers with low glass transition temperatures (Tg < 0 °C) and thus low viscosities, whereas the pendant double bonds allowed the post-synthetic cross-linking of the terpolymer chains using a peroxide as the initiator. The combination of these two features was crucial to enable the formation of cross-linked solids displaying elastic rubber behaviour (as determined by DMA). Among these materials, the elastic rubber based on the terpolymer synthesised from 1,2-epoxydecane, AGE and CO2 (with an 80:20 ratio between the two epoxides) displayed the highest gel content (89%) and thus the highest degree of cross-linking, a Tg of −19 °C and a rubbery plateau at T < 0 °C. Additionally, this study highlighted some notable features of the epoxides used as substrates and of the polycarbonates obtained from their polymerisation with CO2. Among all the investigated copolymerisations and terpolymerisations, the reactions involving allyl glycidyl ether (AGE) as the substrate were found to be particularly effective, giving almost full epoxide conversion and nearly complete selectivity towards polycarbonates (>97%). Control tests with model compounds suggested that the excellent catalytic results with AGE originate from the oxygen atom present on the tail of this compound, which was hypothesised to lead to a favourable interaction with the metal centre of the catalytic complex. In the case of the CO2-based copolymers synthesised from 1,2-epoxydodecane and in the terpolymers prepared with at least 80% of this monomer, a melting point (Tm) was observed. This liquid crystal behaviour is a novel feature among CO2-based polycarbonates. In summary, in this work we investigated and demonstrated the copolymerisation and terpolymerisation of CO2 with epoxides with a long alkyl chain and epoxides with a pendant double bond. These novel polycarbonate terpolymers were successfully cross-linked to generate CO2-based elastic rubbers that are expected to display competitive and attractive properties compared to industrial fossil-based rubbers in terms of heat and oil resistance and with the advantage of being more sustainable. Future work will aim at optimising the chemical, thermal and mechanical properties of this new class of rubbers.
4. Experimental
4.1. Materials
For the synthesis of the amino-tris(phenolate) ligand, 2,4-dimethylphenol (98%, Sigma Aldrich), hexamethylenetetramine (HMTA, 99%, Sigma Aldrich) and formaldehyde (37 wt% in water, Sigma Aldrich) were used. For the synthesis of the aluminium complex, the previously synthesised amino-tris(phenolate) ligand and trimethylaluminium solution (2 M in heptane, Sigma Aldrich) were used. For the copolymerisation and terpolymerisation reactions, pressurised CO2 (≥99%, SOL Nederland B.V.), 1,2-epoxydodecane (90%, Sigma Aldrich), 1,2-epoxydecane (97%, Alfa Aesar), 1,2-epoxyoctane (95%, Sigma Aldrich), 1,2-epoxyhexane (97%, Sigma Aldrich), butyl glycidyl ether (BGE, 95%, Sigma Aldrich), 4-vinyl-1-cyclohexene 1,2-epoxide (VCHO, 97%, Sigma Aldrich), allyl glycidyl ether (AGE, 99%, Sigma Aldrich), and 1,2-epoxy-5-hexene (EH, 97%, Sigma Aldrich) were used. Mesitylene was used as the internal standard (98% Sigma Aldrich). Aluminium amino-tris(phenolate) and bis(triphenylphosphoranylidene)ammonium chloride (PPNCl, 97% Sigma Aldrich) were used as the binary catalytic system. For the cross-linking, dicumyl peroxide (DCP, 98%, Sigma Aldrich) was used. The solvents utilised in this work were diethyl ether, absolute methanol, n-hexane, dichloromethane, dry tetrahydrofuran (THF, dried over a series of columns), tetrahydrofuran, and deuterated chloroform (CDCl3, 99.8%, Sigma Aldrich).4.2. Synthesis of the amino-tris(phenolate) ligand
The methyl-substituted ligand was synthesised through a pseudo-Mannich reaction, following a previously reported procedure.56,57 2,4-Dimethylphenol (5.00 g, 40 mmol), HMTA (0.47 g, 3.3 mmol) and 37% aqueous formaldehyde solution (1.21 g, 14.2 mmol) were added to a glass round-bottom flask and stirred under reflux at 125 °C for 48 h. The reaction mixture was then quenched with chloroform (100 mL) and washed with water (3 × 100 mL each) in a two-liquid-phase separator funnel. The organic phase was collected, dried over magnesium sulphate, and separated from the solid by filtration. Chloroform was evaporated via rotary evaporation, yielding an orange–brown crude product. The pure ligand was isolated via crystallisation in a minimal amount of diethyl ether (a heat gun was used to increase the solubility of the compound in the solvent) followed by the addition of methanol (ratio roughly equal to 1:3 v/v) to give a yellow, crystalline product. The final product was separated from the solvent by filtration and dried in a vacuum oven for two days at 55 °C to remove any trace of solvent.
4.3. Synthesis of the aluminium amino-tris(phenolate) complex
The synthesis of the catalyst was carried out following a procedure from the literature.31 The synthesis was performed in a Schlenk line under nitrogen atmosphere, applying standard vacuum-line techniques. Before running the reaction, a three-necked round-bottom flask, a stopper, a Schlenk-line glass valve and a stirrer were placed overnight in a drying oven at 110 °C to remove any residual water. The amino-tris(phenolate) ligand was weighed (1.00 g, 1.92 mmol) in the reaction flask, which was then sealed with the stopper, the glass valve and a rubber cap (to enable further addition of reactants). Vacuum grease was used to ensure good adherence between the reaction flask and the stoppers. A nitrogen atmosphere and vacuum were then alternated three times to remove air from the reaction flask. This was done to avoid the reaction of AlMe3 with O2 from air, which may cause excessive heat development and possibly the risk of an explosion. Dry THF (20 mL) was then added to the reaction mixture using a (glass) syringe, followed by the dropwise addition of AlMe3 (1.19 mL, 2.38 mmol) to minimise heat generation (the reaction between AlMe3 and the ligand is extremely exothermic). The reaction mixture was allowed to react for 2 h, then it was concentrated by rotary evaporation. Finally, the formed Al complex was precipitated using a large excess of n-hexane. After filtration, the Al complex was dried in a vacuum oven for two days at 55 °C.4.4. Synthesis of the polycarbonates
The synthesis of the polycarbonates was performed following a well-established experimental procedure developed for the synthesis of other polycarbonates and cyclic carbonates.58,59 In this procedure, up to 10 tests can be carried out simultaneously in a high-throughput CO2 reactor unit manufactured by ILS-Integrated Lab Solutions GmbH (Fig. S2†). This CO2 workstation consists of two parts: a 10-reactors block, which allows performing 10 experiments at the same time in individually stirred batch reactors (84 mL volume each, 30 mm internal diameter); and a single, batch reactor of the same size, supplied with a borosilicate glass window to allow visualisation of the reaction mixture. The 10-reactors block and the single batch reactor can both be operated in a temperature range of 20–200 °C and a pressure range of 1–200 bar. In a typical experiment, 30 mmol of epoxide (as a single compound or as a mixture of two compounds in the case of the terpolymerisations), 3 mmol of mesitylene (internal standard), 0.15 mmol of aluminium amino-tris(phenolate) complex (0.5 mol% relative to the epoxide), and 0.075 mmol of PPNCl (0.25 mol% relative to the epoxide) were placed in a glass vial (46 mL volume, 30 mm external diameter) equipped with a magnetic stirring bar. The vial was then closed with a screw cap equipped with a PTFE-coated silicon septum. The septum was then pierced with two needles, allowing nitrogen and CO2 flux into and from the vial. The so-prepared vials were then placed into either the 10-reactors block or the single batch reactor of the high-throughput workstation, which was subsequently closed. The selected reactor unit was first purged three times with 5 bar N2 and then pressurised to 10 bar CO2. After 5–10 min, the reactor was depressurised to atmospheric pressure. After reaching atmospheric pressure and a 5–10 min pause, the selected unit was pressurised with 50 bar CO2. After reaching this pressure, the reactor unit was heated to the desired temperature (45 or 60 °C). After the desired temperature was reached, an extra 20–30 min was allowed to elapse to allow the Viton fluororubber O-rings sealing the reactors to adjust to the pressure and temperature. The reactor unit was then brought to 80 bar (supercritical CO2) and the stirring (900 rpm) was started. This was considered the time zero of the reaction (although it should be taken into account that the reaction had probably already proceeded slowly during the procedure above, which typically requires around 2 h). After the chosen reaction time (18 or 24 h), the magnetic stirring and the reactor heating were switched off and the water-cooling system was turned on to cool down the reactor unit. Upon reaching room temperature, the reactor unit was depressurised with an automated protocol and, after reaching atmospheric pressure, the reactor unit was opened and the vials were removed. The crude polymerisation products were first analysed by proton nuclear magnetic resonance (1H NMR) spectroscopy to calculate the conversion of the epoxide and by Fourier transform infrared (FTIR) spectroscopy to determine the selectivity towards the polycarbonates. Then, the products were purified by dissolution in a minimal amount of dichloromethane under vigorous stirring followed by precipitation in a large excess of methanol (roughly with a ratio 1:10 v/v). These steps were repeated three times, after which the sample purities were checked by 1H NMR spectroscopy. If cyclic carbonate peaks were still detected in the NMR spectra, additional purification steps were applied in order to fully remove the cyclic carbonates. Methanol was then used to wash the samples and remove low-molecular-weight polymer fractions. The molecular weight, decomposition temperature and Tg were determined by gel permeation chromatography (GPC), thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC), respectively. The chemical composition of the terpolymers was determined by carbon nuclear magnetic resonance (13C NMR) and matrix-assisted laser desorption/ionisation (MALDI-TOF) techniques. It is worth noting that the vast majority of these terpolymerisation tests were repeated in duplicate or triplicate and showed excellent reproducibility (Table S1†).
4.5. Large-scale synthesis of the copolymer of AGE and CO2
A batch Parr reactor (max T = 200 °C, max p = 200 bar) was filled with the reaction mixture (30 g of AGE, 0.25 mol% PPNCl, and 0.5 mol% Al ATP complex), sealed and pressurised to 50 bar. The reactor was heated up to the desired reaction temperature (45 °C) and topped up with CO2 until 80 bar was reached. The stirring was then activated (400 rpm) and the reaction was performed for 24 h. Once the reaction time was reached, the reactor was cooled down and depressurised, and the polymer characterised and purified following the procedure indicated in the previous paragraph.4.6. Synthesis and characterisation of the cross-linked polycarbonate rubbers
Dicumyl peroxide (DCP) cross-linking agent (1 wt% relative to the polymer) was added via a solution route to the purified polycarbonates. In this procedure, 1.00 g of the polymer was first dissolved in dichloromethane (minimal amount, ca. 1 mL). Next, DCP (0.010 g) was added to the mixture, and the solution was then stirred for 5–10 min. The solvent was subsequently removed overnight in a vacuum oven at 65 °C. The obtained solid sample was then cross-linked in a compression mould for 13 min at 170 °C and 50 bar (see Fig. S35† for the setup). The gel content of the obtained elastic rubber was determined as a measure of the degree of cross-linking. A pre-weighed amount of cross-linked polycarbonate (100–250 mg; W1) was allowed to swell and dissolve in 10 mL of dichloromethane for two days to remove the non-cross-linked chains. The swollen residue was then removed by filtration from the solution, dried overnight in a vacuum oven (40 °C) and finally weighed again (W2). The gel content60 was calculated as follows: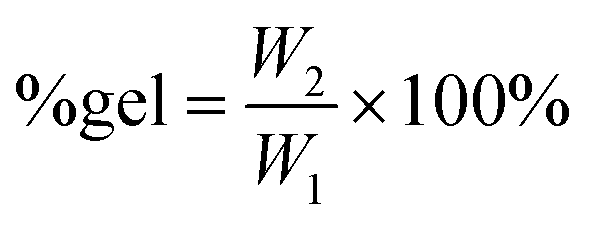
4.7. 1H and 13C NMR spectroscopy
1H NMR spectra were recorded using a Varian Oxford 300 MHz NMR spectrometer. Chemical shifts were measured relative to tetramethylsilane (TMS) as the internal reference to the solvent (CDCl3: 7.26 ppm). Samples were prepared by dissolving a small amount of compound (5–10 mg) in ca. 1 mL of CDCl3.13C NMR spectra were recorded using a Varian Oxford AS400 400 MHz NMR spectrometer. Chemical shifts were measured relative to TMS as the internal reference to the solvent (CDCl3: 77.33 ppm). Samples were prepared by dissolving ca. 40 mg of compound in 0.70 g of CDCl3.
The NMR spectra and data were analysed using the MestReNova software. 1H NMR spectra were obtained to characterise the ligand and the catalyst and to determine the conversion of the epoxides to the polymer products. 13C NMR spectroscopy was performed to investigate the chemical structure of the polycarbonate rubber terpolymers.
4.8. FTIR spectroscopy
FTIR spectra were recorded using a FTIR 100 Tracer Shimadzu spectrophotometer with a KBr beam splitter. Samples in the form of drops of the reaction mixtures were placed on the sample slot and analysed in the absorbance mode in the wavelength range from 4500 to 400 cm−1 with 64 scans. The data were elaborated with OriginLab software. FTIR spectroscopy was performed to calculate the selectivity of the reaction towards polycarbonates vs. cyclic carbonates by comparing the areas of the carbonyl peaks for the polycarbonates (at ca. 1750 cm−1) and the cyclic carbonates (at ca. 1800 cm−1).4.9. Gel-permeation chromatography (GPC)
The molecular weights (number average, Mn; and weight average, Mw) of the polycarbonates were determined by GPC on an Agilent Technology 1260 Infinity II apparatus equipped with a triple detector, consisting of a Viscotek Ralls detector, a Viscotek Viscometer model H502, and a Schambeck RI2912 refractive index detector. The analysis was carried out by utilising two PLgel 5 μm MIXED-C, 300 mm columns from Agilent Technologies at 35 °C. THF 99+% stabilised with butylated hydroxytoluene (BHT) was used as the eluent at a flow rate of 1.0 mL min−1. Data acquisition and calculations were performed using Viscotek OmniSec software version 5.0. Relative molecular weights were determined based on a calibration curve generated from narrow dispersity polystyrene standards (Agilent and Polymer Laboratories, Mw 0.645–3001 kg mol−1). Samples were prepared by dissolving a small amount of polycarbonate (3–5 mg) in THF (1 mL).4.10. Thermal gravimetric analysis (TGA)
Degradation temperature data were collected using a PerkinElmer TGA 4000 instrument. Samples were prepared by placing a small amount of sample (between 1 and 2 mg) inside a TGA cup. Samples were analysed under N2 with a temperature scan between 20 and 350 °C with a 10 °C min−1 heating rate. The results were analysed with the dedicated software of the instrument and the degradation temperature was calculated as the inflection point of the curve (Fig. S34†).4.11. Differential scanning calorimetry (DSC)
DSC analysis was performed on a PerkinElmer DSC Diamond instrument. DSC measurements are relative to an empty aluminium pan, used as the reference. The samples were prepared by placing 10–20 mg of the purified polymers in aluminium pans, which were sealed and then analysed using the following procedure. First, a preparation cycle going from −55/60 to 30/50 °C was run to erase the thermal history of the samples (not recorded), followed by the actual measurement with the same experimental settings, in the temperature range between −50 and 50 °C. In this way, the onset Tg or Tm of the purified polymers was determined.61 DSC data were analysed using OriginLab or via the instrument software. For the polymers that showed a Tm and not a Tg in the above-mentioned temperature range, additional tests were performed in a lower range (−90 to 20 °C) using a DSC 25 TA instrument. Samples were prepared as indicated above and the data were analysed with the dedicated instrument software.4.12. Dynamic mechanical analysis (DMA)
DMA was performed on a PerkinElmer DMA 8000 dynamic mechanical analyser having a 0–600 Hz dynamic range, and which can operate in a temperature range from 90 to 400 °C. The elastic rubber samples were analysed in the single cantilever mode following an isotherm phase at the initial temperature (−70 °C), followed by a temperature scan up to 50 °C (in the case of the terpolymer-based materials) or 150 °C (in the case of the AGE-based copolymer) with a 2 °C min−1 heating rate. The results were analysed using the PerkinElmer dedicated software. The Tg was calculated as the onset of the peak achieved from the tanδ curve (Fig. 6).
4.13. MALDI-TOF
Matrix-assisted laser desorption/ionisation time-of-flight (MALDI-TOF) mass spectrograms were acquired with a Voyager DE-Pro MALDI-TOF equipment from Applied Biosystems, using the recommended settings for polymer characterisation (linear positive mode and 2000–20000 m/z range). The samples were prepared as follows: 20–50 mg of the matrix (2,5-dihydroxybenzoic acid, DHB, Sigma Aldrich) was dissolved in 1 mL of THF and the polymer samples were also dissolved in THF (5 mg mL−1). The two solutions (matrix and polymer) were mixed in several ratios: 1:1, 3:1, 10:1, and 20:1 (v/v). Some samples were also analysed after adding a small quantity of sodium acetate as cationising agent to the matrix mixture (final matrix-to-polymer ratios of 1:1 and 3:1 v/v). One drop of the final mixture was then placed on a standard MALDI-TOF metal plate and left to dry at room temperature. MALDI-TOF analysis was aimed at determining the chemical structure of the terpolymers and at establishing whether the obtained polycarbonates were statistical terpolymers or mixtures of the two parent copolymers and whether the polymer chains were hydroxy-terminated.35
Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We acknowledge Marcel de Vries for technical support, and Léon Rohrbach, Dr Khaled Omar Sebakhy and Gert-Jan Boer for analytical support. Dr Arjen Kamphuis and Prof. Patrizio Raffa are acknowledged for useful discussions.References
- R. Lindsay, Climate Change: Atmospheric Carbon Dioxide, 2020. Published online at Climate.gov. Retrieved from https://climate.gov Search PubMed.
- P. N. Pearson and M. R. Palmer, Nature, 2000, 406, 695–699 CrossRef CAS PubMed.
- H. Ritchie and M. Roser, CO2 and Greenhouse Gas Emissions, 2017. Published online at OurWorldInData.org. Retrieved from: https://ourworldindata.org/co2-and-other-greenhouse-gas-emissions Search PubMed.
- L. Yang and H. Wang, ChemSusChem, 2014, 7, 962–998 CrossRef CAS PubMed.
- Q. Liu, L. Wu and R. Jackstell, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- M. K. Yu, I. Curcic, J. Gabriel and S. C. E. Tsang, ChemSusChem, 2008, 1(11), 893–899 CrossRef PubMed.
- G. Yuan, C. Qi, W. Wu and H. Jiang, Curr. Opin. Green Sustain. Chem., 2017, 3, 22–27 CrossRef.
- A. W. Kleij, M. North and A. Urakawa, ChemSusChem, 2017, 10(6), 1036–1038 CrossRef CAS PubMed.
- P. P. Pescarmona, Curr. Opin. Green Sustain. Chem., 2021, 100457 CrossRef CAS.
- A. J. Kamphuis, F. Picchioni and P. P. Pescarmona, Green Chem., 2019, 21(3), 406–448 RSC.
- W. Leitner, Acc. Chem. Res., 2002, 35, 746 CrossRef CAS PubMed.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS PubMed.
- Y. Li, X. Fang, K. Junge and M. Beller, Angew. Chem., 2013, 52(36), 9568–9571 CrossRef CAS PubMed.
- P. P. Pescarmona and M. Taherimehr, Catal. Sci. Technol., 2012, 2(11), 2169–2187 RSC.
- M. Taherimehr and P. P. Pescarmona, J. Appl. Polym. Sci., 2014, 131, 21 CrossRef.
- Y. Wang and D. J. Darensbourg, Coord. Chem. Rev., 2018, 372, 85–100 CrossRef CAS.
- S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255(13–14), 1460–1479 CrossRef CAS.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141 RSC.
- S. J. Poland and D. J. Darensbourg, Green Chem., 2017, 19(21), 4990–5011 RSC.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618 CrossRef CAS PubMed.
- S. Liu and X. Wang, Curr. Opin. Green Sustain. Chem., 2017, 3, 61–66 CrossRef.
- J. Langanke, A. Wolf, J. Hofmann, K. Böhm, M. A. Subhani, T. E. Müller and C. Gürtler, Green Chem., 2014, 16(4), 1865–1870 RSC.
- T. Stößer, C. Li, J. Unruangsri, P. K. Saini, R. J. Sablong, M. A. Meier and C. Koning, Polym. Chem., 2017, 8(39), 6099–6105 RSC.
- M. Taherimehr, J. P. C. Sertã, A. W. Kleij, C. J. Whiteoak and P. P. Pescarmona, ChemSusChem, 2015, 8(6), 1034–1042 CrossRef CAS PubMed.
- P. Wei, G. A. Bhat, C. E. Cipriani, H. Mohammad, K. Schoonover, E. B. Pentzer and D. J. Darensbourg, Angew. Chem., Int. Ed., 2022, 61(37), e202208355 CrossRef CAS PubMed.
- D. Kansal, R. Abouomar and M. Rabnawaz, J. Appl. Polym. Sci., 2021, 138(29), 50708 CrossRef CAS.
- R. J. Young and P. A. Lovell, Introduction to Polymers, CRC press, Boca Raton, 2011 Search PubMed.
- S. D. Thorat, P. J. Phillips, V. Semenov and A. Gakh, J. Appl. Polym. Sci., 2003, 89(5), 1163–1176 CrossRef CAS.
- F. W. Billmeyer, Textbook of Polymer Science, John Wiley & Sons, New York, 1984 Search PubMed.
- C. Martín and A. W. Kleij, Macromolecules, 2016, 49(17), 6285–6295 CrossRef.
- C. J. Whiteoak, N. Kielland, V. Laserna, F. Castro-Gómez, E. Martin, E. C. Escudero-Adán and A. W. Kleij, Chem. – Eur. J., 2014, 20(8), 2264–2275 CrossRef CAS PubMed.
- D. J. Darensbourg and Y. Wang, Polym. Chem., 2015, 6(10), 1768–1776 RSC.
- S. G. Allen and J. C. Bevington, Comprehensive polymer science supplement 2, Pergamon, Amsterdam, 1989, 533–569 Search PubMed.
- M. van Duin, C. Hassmann, E. Gandert and A. J. Rackstraw, Handbook of synthetic rubber, Arlanxeo GmbH, Dormagen, 2020 Search PubMed.
- M. Taherimehr, S. M. Al-Amsyar, C. J. Whiteoak, A. W. Kleij and P. P. Pescarmona, Green Chem., 2013, 15(11), 3083–3090 RSC.
- J. L. Kendall, D. A. Canelas, J. L. Young and J. M. DeSimone, Polymerizations in supercritical carbon dioxide, Chem. Rev., 1999, 99, 543–564 CrossRef CAS PubMed.
- F. Hessel and H. Finkelmann, Makromol. Chem., 1988, 189(10), 2275–2283 CrossRef CAS.
- W. Weissflog and D. Demus, Mol. Cryst. Liq. Cryst., 1985, 129(1–3), 235–243 CrossRef CAS.
- M. Van Duin, R. Orza, R. Peters and V. Chechik, Macromol. Symp., 2010, 291–292, 66–74 CrossRef CAS.
- E. A. Okhlopkova, L. A. Serafimov and A. V. Frolkova, Theor. Found. Chem. Eng., 2019, 53(5), 864–870 CrossRef CAS.
- C. M. Andrews, B. Dobinson, W. M. Rolfe and M. R. Thoseby, US Pat, US005429312A, 1995 Search PubMed.
- E. V. Makshina, M. Dusselier, W. Janssens, J. Degrève, P. A. Jacobs and B. F. Sels, Review of old chemistry and new catalytic advances in the on-purpose synthesis of butadiene, Chem. Soc. Rev., 2014, 43(22), 7917–7953 RSC.
- H. Daimon, H. Okitsu and J. Kumanotani, Polym. J., 1975, 7(4), 460–466 CrossRef CAS.
- L. M. Polgar, F. Fallani, J. Cuijpers, P. Raffa, A. A. Broekhuis, M. van Duin and F. Picchioni, Rev. Chem. Eng., 2018, 35(1), 45–72 CrossRef.
- J. Kruzelak, S. Hakosova, A. Kvasnicakova and I. Hudec, KGK, Kautsch. Gummi Kunstst., 2020, 73(10), 36–42 CAS.
- L. D. Loan, Rubber Chem. Technol., 1967, 40(1), 149–176 CrossRef CAS.
- J. Kruzelak, S. Hakosova, A. Kvasnicakova and I. Hudec, Adv. Ind. Eng. Polym. Res., 2020, 3(3), 120–128 Search PubMed.
- J. Lal and J. E. Mark, Elastomers and Rubber Elasticity, American Chemical Society, Washington D. C, 1982 Search PubMed.
- P. R. Dluzneski, Rubber Chem. Technol., 2001, 74(3), 451–492 CrossRef CAS.
- L. Gao, M. Huang, Q. Wu, X. Wan, X. Chen, X. Wei and J. Feng, Polymers, 2019, 11(9), 1467 CrossRef CAS PubMed.
- M. Huang, L. Gao, J. Feng, X. Huang, Z. Li, Z. Huang and L. Wang, ACS Omega, 2020, 5(28), 17808–17817 CrossRef CAS PubMed.
- H. S. Lee, J. H. Jeong, G. Hong, H. K. Cho, B. K. Baek, C. M. Koo and Y. W. Lee, Ind. Eng. Chem. Res., 2013, 52(20), 6633–6638 CrossRef CAS.
- C. M. Gomez, D. Gutierrez, M. Asensio, V. Costa and A. Nohales, J. Elastomers Plast., 2017, 49(1), 77–95 CrossRef CAS.
- I. Franta, Elastomers and Rubber Compounding Materials, Elsevier, Amsterdam, 1989 Search PubMed.
- J. E. Tamis, A. A. Koelmans, R. Dröge, N. H. Kaag, M. C. Keur, P. C. Tromp and R. H. Jongbloed, Microplas. Nanoplas., 2021, 1(1), 1–17 Search PubMed.
- M. Kol, M. Shamis, I. Goldberg, Z. Goldschmidt, S. Alfi and E. Hayut-Salant, Inorg. Chem. Commun., 2001, 4(4), 177–179 CrossRef CAS.
- S. Gendler, S. Segal, I. Goldberg, Z. Goldschmidt and M. Kol, Inorg. Chem., 2006, 45(12), 4783–4790 CrossRef CAS PubMed.
- Y. A. Alassmy and P. P. Pescarmona, ChemSusChem, 2019, 12(16), 3856–3863 CrossRef CAS PubMed.
- A. J. Kamphuis, F. Milocco, L. Koiter, P. P. Pescarmona and E. Otten, ChemSusChem, 2019, 12(15), 3635–3641 CrossRef CAS PubMed.
- E. M. Kampouris and A. G. Andreopoulos, Biomaterials, 1989, 10(3), 206–208 CrossRef CAS PubMed.
- J. He, W. Liu and Y.-X. Huang, PLoS One, 2016, 11, e0151454 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Scheme with the reaction mechanism, pictures of the used setups, NMR and FTIR spectra, DSC, GPC and TGA curves. See DOI: https://doi.org/10.1039/d3gc02374e |
| This journal is © The Royal Society of Chemistry 2023 |