
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Application of Rhodococcus jostii RHA1 glycolate oxidase as an efficient accessory enzyme for lignin conversion by bacterial Dyp peroxidase enzymes†
Awatif
Alruwaili
,
Goran M. M.
Rashid
and
Timothy D. H.
Bugg
 *
*
Department of Chemistry, University of Warwick, Coventry CV4 7AL, UK. E-mail: T.D.Bugg@warwick.ac.uk
First published on 11th April 2023
Abstract
Lignin oxidation by bacterial dye-decolorizing peroxidase enzymes requires hydrogen peroxide as a co-substrate, an unstable and corrosive oxidant. We have identified a glycolate oxidase enzyme from Rhodococcus jostii RHA1 that can couple effectively at pH 6.5 with DyP peroxidase enzymes from Agrobacterium sp. or Comamonas testosteroni to oxidise lignin substrates without addition of hydrogen peroxide. Rhodococcus jostii RHA1 glycolate oxidase (RjGlOx) has activity for oxidation of a range of α-ketoaldehyde and α-hydroxyacid substrates, and is also active for oxidation of hydroxymethylfurfural (HMF) to furandicarboxylic acid. The combination of RjGlOx with Agrobacterium sp. DyP or C. testosteroni DyP generated new and enhanced amounts of low molecular weight aromatic products from organosolv lignin substrates, and was able to generate high-value products from treatment of lignin residue from cellulosic biofuel production, and from a polymeric humin substrate.
Introduction
The conversion of aromatic biopolymer lignin to high-value products is a major unsolved problem in the lignocellulosic biorefinery concept, which is being studied around the world using a range of chemocatalytic and biocatalytic approaches.1 For biocatalytic lignin conversion, several classes of lignin-oxidising enzymes have been identified: from white-rot fungi, extracellular lignin peroxidase, manganese peroxidase and laccase enzymes have been identified for many years;2,3 in lignin-degrading soil bacteria, dye decolorizing peroxidases and multi-copper oxidase enzymes have also been identified in the last 10 years.4,5However, although peroxidase enzymes offer considerable potential for lignin bio-conversion, there are practical problems in achieving successful bioconversions: principally due to repolymerisation of free radical intermediates in lignin oxidation; and inactivation of peroxidase enzymes by their hydrogen peroxide substrate.4 Several fungal oxidase enzymes have been characterised that can generate extracellular hydrogen peroxide,6 of which aryl alcohol oxidase7 and glyoxal oxidase8 appear to be closely linked with lignin breakdown. Aryl alcohol oxidase from Pleurotus eryngii is able to oxidise benzylic alcohols and aliphatic unsaturated alcohols efficiently,9 and this oxidation has been shown to generate hydrogen peroxide extracellularly in Pleurotus eryngii.10 The crystal structure of this enzyme has been determined,11 which has been used to guide protein engineering studies.12 Glyoxal oxidase from Phanerochaete chrysosporium is able to oxidise aliphatic aldehydes, α-hydroxycarbonyl compounds and α-dicarbonyl compounds,13,14 and has been shown to support hydrogen peroxide generation extracellularly in P. chrysosporium.13 Two further glyoxal oxidase enzymes have been characterised from Pycnoporus cinnabarinus.15
We have recently identified a glycolate oxidase from lignin-degrading bacterium Rhodococcus jostii RHA1 as part of 4-hydroxybenzoylformate degradation gene cluster (gene ro02984, see Fig. 1) which is used to degrade aryl C2 fragments from lignin breakdown.16 Recombinant R. jostii glycolate oxidase is an FMN-dependent enzyme able to oxidise aromatic phenylglyoxal and mandelic acid substrates,16 and can catalyse successive oxidations of glycolaldehyde to glycolic acid, glyoxylic acid and oxalic acid.17R. jostii glycolate oxidase is not related to the fungal aryl alcohol oxidase or glyoxal oxidase families, but is a member of the vanillyl alcohol oxidase (VAO) superfamily that can oxidise 4-hydroxybenzyl alcohol substrates.18 Glycolate oxidase from spinach, which also utilises FMN, has been kinetically characterised,19 but bacterial glycolate oxidases have not been characterised biochemically. Since this enzyme generates hydrogen peroxide, we wished to investigate its substrate scope, and study whether it could act as an accessory enzyme for bacterial lignin degradation in vitro.
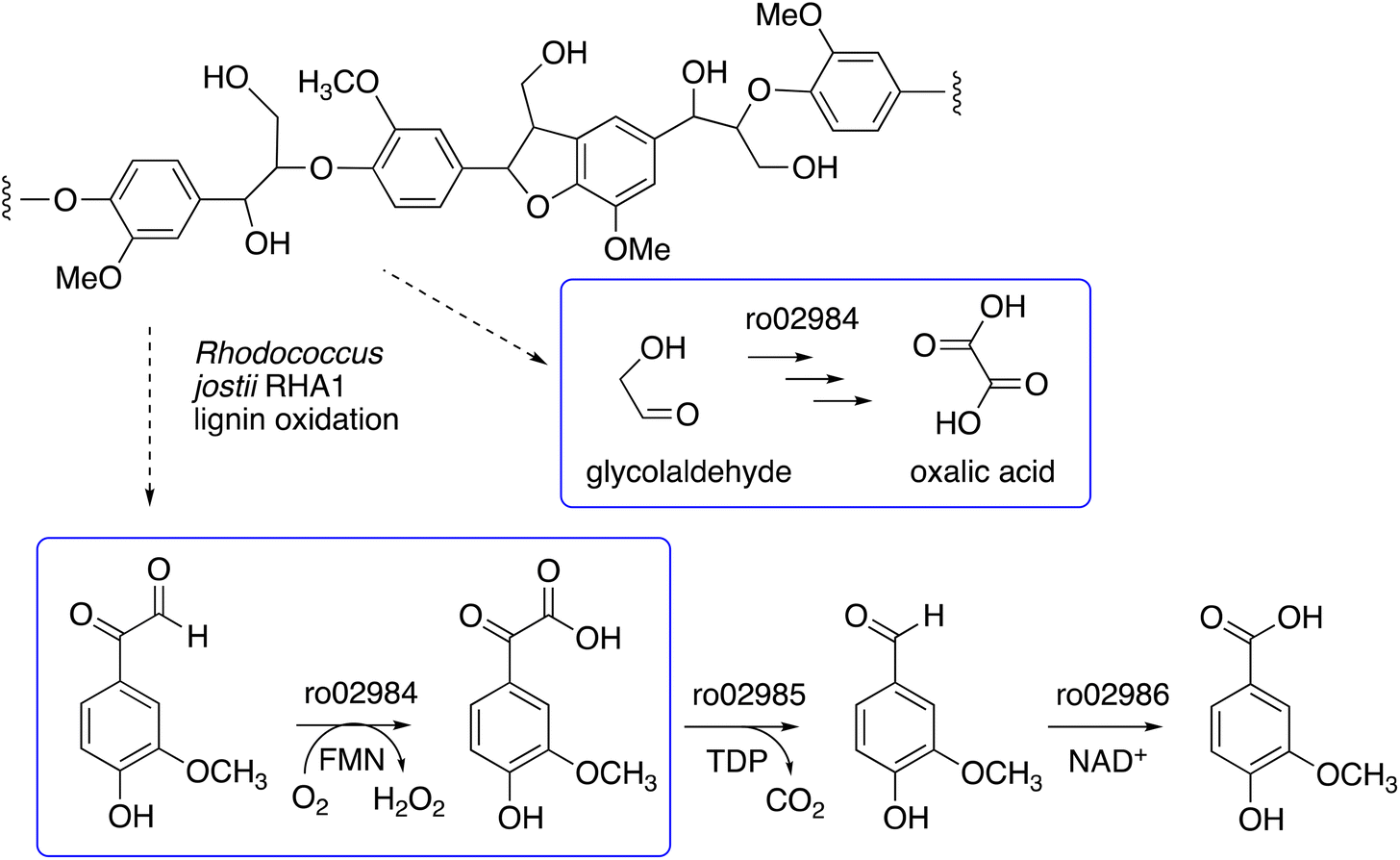 | ||
| Fig. 1 4-Hydroxybenzoylformate degradation pathway in Rhodococcus jostii RHA1, showing steps catalysed by glycolate oxidase (gene ro02984).16,17 | ||
Although fungal aryl alcohol oxidase and glyoxal oxidase have been shown to support lignin generation in vivo,9,12 there is only one report of their use in vitro in combination with a peroxidase, where P. eryngii AAO has been used in combination with fungal lignin peroxidase.20 Since fungal lignin peroxidases are challenging to overexpress in recombinant form, there are potential advantages for a bacterial oxidase that could be combined with bacterial Dyp-type peroxidase enzymes, which can oxidise lignin.4,5 A copper-dependent oxidase from Thermobifida fusca has been reported to modify lignin in sugarcane bagasse,21 while pyranose 2-oxidase from Kitasatospora aureofaciens has also been reported to couple effectively with manganese peroxidase.22
Use of an oxidase-peroxidase combination would allow biotransformation of lignin using dioxygen as oxidant, rather than hydrogen peroxide. As well as using dioxygen as a milder oxidant, this would also avoid inactivation of the peroxidase enzyme by hydrogen peroxide. Use of biocatalysis for lignin biotransformation avoids the use of organic solvents and transition metals for chemocatalytic transformation of lignin,1 and avoids the use of energy-intensive high pressure hydrogenation23 or pyrolysis for lignin depolymerisation.24
Here we report that R. jostii glycolate oxidase can be used as an effective accessory enzyme for bacterial DyP peroxidase-catalysed peroxide-free oxidation of lignin. We also describe the activity of R. jostii glycolate oxidase for oxidation of hydroxymethylfurfural (HMF), a common by-product found in biorefinery lignins, and we report its application to valorisation of two biorefinery streams. Bioconversion of biomass-derived polymeric lignin to small molecule products offers a green chemistry solution to the production of chemicals from petroleum.1
Experimental
Materials
Chemicals & biochemicals were purchased from Sigma-Aldrich, except as indicated. Standards for HMF bioconversion were gifts from Dr Andrew Carnell (University of Liverpool). Poplar ammonia organosolv lignin,25 poplar alkaline organosolv lignin,26 and eucalyptus organosolv lignin27 were prepared as previously described,25–27 and were structurally characterised in Lancefield et al.28 The hydrolysis lignin (HL, supplied through the Zelcor project by Prof. S. Baumberger, INRAE Versailles) was produced from wheat straw by steam explosion pretreatment, followed by enzymatic hydrolysis, and was received as a wet cake (∼50 wt% water). Before use, the sample was dried at room temperature for 4 days, then manually milled and dried under vacuum to obtain a fine powder. HL contained 55 wt% lignin and 39 wt% carbohydrates by Klason assay, including 33.9 wt% cellulose and 4.8 wt% hemicelluloses.29 Humins were supplied by Avantium N. V. and were produced in their pilot plant in Geleen, The Netherlands, by conversion of fructose and glucose. Structural characterisation of these humins has been reported.30Expression & purification of Rhodococcus jostii RHA1 glycolate oxidase (RjGlOx) from gene RHA1_ro02984 was described by Wei et al.16 Expression & purification of Agrobacterium sp. DyP peroxidase (AgroDyP) and Comamonas testosteroni DyP (CtDyP) was previously described by Rashid & Bugg.31
Assay methods for R. jostii glycolate oxidase
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 M−1 cm−1). All experiments were measured in triplicate, and substrate specificity was determined with the same assay. Michaelis–Menten plots with GraphPad Prism curve fitting software were used to calculate the kinetic parameters.
000 M−1 cm−1). All experiments were measured in triplicate, and substrate specificity was determined with the same assay. Michaelis–Menten plots with GraphPad Prism curve fitting software were used to calculate the kinetic parameters.
The optimum temperature was determined using glyoxal as substrate at 30, 42, 55, 60, and 70 °C. The pH optimum was measured in 50 mM glycine HCl buffer (pH 2.2–2.8), sodium acetate (pH 3–5), MES (pH 5.5–6.7) HEPES (pH 6.8–8.2), and MOPS (pH 6.5–7.9) buffers.
The thermal stability of the enzymes was determined by incubating the proteins at 30, 42, 55, 60, and 70 °C for 15 min, 30 min, 1 h, and 2 h. The enzyme was cooled in ice before measuring the activity.
Conversion of 5-hydroxymethylfurfural
Conversions of 5 mg of lignin samples were carried out in a 1 mL total volume, containing 0.18 mg RjGIOx, 85 μg AgroDyp, 1 mM MnSO4, 0.1 mM FMN, 1 mM glycolic acid in 50 mM succinate buffer pH 6.5 at 30 °C. The mixture was left to incubate for 24 hours. A control incubation was set up without adding any enzymes. In order to perform LC/MS analysis, the material was acidified, and then extracted into ethyl acetate, dried in vacuo, and then reconstituted with 400 μL MeOH/water 1:1. A positive mode LC/MS analysis, with 270 nm monitoring, was used to examine the reaction components and compare them to authentic standards.
Biotransformation of 5 mg humin and futurol (dissolved in 200 μL DMSO) were carried out in a 1 mL total volume, containing 0.18 mg RjGlOx, 85 μg AgroDyp, 1 mM MnSO4, 0.1 mM FMN, 1 mM glycolic acid and 50 mM succinate buffer pH 6.5 at 30 °C. The mixture was left to incubate for 24 hours, and low molecular weight products extracted into ethyl acetate and examined by reverse phase C18 column by LC-MS.
Results
Biochemical characterisation of Rhodococcus jostii RHA1 GlOx
His6-tagged R. jostii glycolate oxidase was expressed and purified by immobilized metal affinity chromatography (see ESI Fig. S1†). The purified enzyme was yellow in colour, due to the FMN cofactor, with λmax values at 375 and 456 nm (see ESI Fig. S2†).The pH-rate profile of glycolate oxidase was determined via coupled enzyme assay, using recombinant P. fluorescens DyP1B peroxidase33 to couple the production of hydrogen peroxide with oxidation of 2,2′-azinobis (3-ethylbenz-thiazoline-6-sulfonic acid) (ABTS). The pH and temperature dependence of glycolate oxidase activity were measured with glyoxal as substrate, as shown in Fig. 2A. The enzyme showed optimal activity at pH 6.0, but showed >50% maximal activity in the range pH 4.5–6.5. The temperature dependence of the enzyme using glyoxal as substrate at pH 6.0 was measured at 30–70 °C, as shown in Fig. 2B. The enzyme retained activity at 30 °C or 42 °C, but lost activity upon incubation at 55 °C or above.
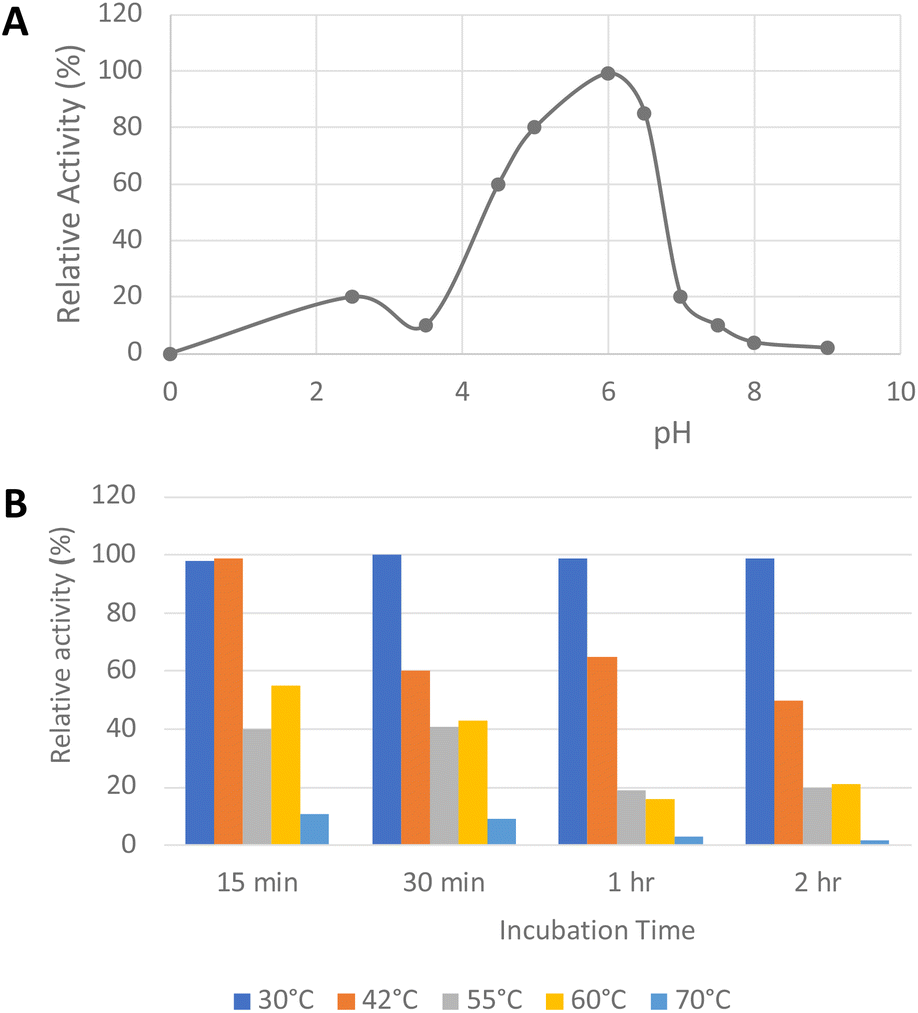 | ||
| Fig. 2 Activity of R. jostii glycolate oxidase versus pH (A) and temperature (B), using glyoxal as substrate, using coupled ABTS assay described in Experimental section. | ||
Several substrates were tested for oxidation by RjGlOx, using the Dyp1B/ABTS coupled enzyme assay. As well as glycolic acid, RjGlOx was able to oxidise α-hydroxy acids L-lactic acid, L-malic acid, and mandelic acid (see Fig. 3). It was also able to oxidise α-ketoaldehyde substrates glyoxal, methylglyoxal, phenylglyoxal, and 4-hydroxyphenylglyoxal. However, no activity was observed for benzaldehyde, hence the enzyme requires the presence of two adjacent functional groups in the substrate in order to show oxidation activity. Michaelis–Menten steady-state kinetic parameters were measured, as shown in Table 1 (kinetic plots shown in ESI Fig. S3†). The products obtained from RjGlOx conversion of each substrate were verified by HPLC analysis, as noted in Table 1 (see ESI Fig. S4†). Similar activities were observed for several α-dicarbonyl and α-hydroxycarboxylic acid substrates, hence this enzyme appears to show rather broad substrate specificity. In an earlier study of lignin metabolites observed in Rhodococcus jostii RHA1, we also observed that this enzyme can catalyse benzylic oxidation of 4-hydroxyphenylacetic acid to 4-hydroxymandelic acid and 4-hydroxybenzoylformic acid.17
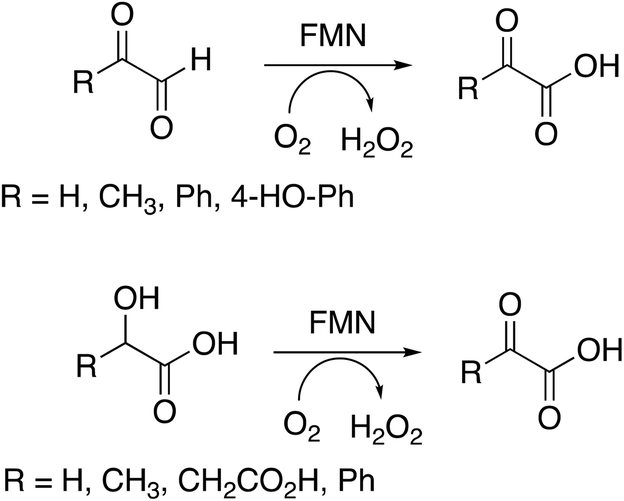 | ||
| Fig. 3 Substrates oxidised by R. jostii RHA1 glycolate oxidase. | ||
| Substrate | Product | Specific activity (μmol min−1 mg−1) | K M (mM) | k cat (s−1) | k cat/KM (M−1 s−1) |
|---|---|---|---|---|---|
| a Product identification by HPLC shown in ESI Fig. S4.† b Product identification shown previously.17 | |||||
| Glyoxal (R = H) | Oxalic acida | 0.45 | 1.92 | 0.44 | 230 |
| Methylglyoxal (R = CH3) | Pyruvic acida | 0.97 | 0.43 | 0.88 | 2050 |
| 4-Hydroxyphenylglyoxal | 4-Hydroxybenzoylformatea | 0.21 | 0.25 | 0.21 | 830 |
| Glycolaldehyde | Glycolic acidb | 0.49 | 0.69 | 0.48 | 700 |
| Glycolic acid (R = H) | Glyoxylic acidb | 0.78 | 0.49 | 0.77 | 1560 |
| L-Lactic acid (R = CH3) | Pyruvic acida | 0.48 | 0.24 | 0.47 | 1970 |
| Malic acid (R = CH2CO2H) | Oxaloacetic acida | 0.29 | 0.29 | 0.60 | 2060 |
| Mandelic acid (R = Ph) | Benzoylformic acida | 1.92 | 0.50 | 1.90 | 3800 |
| Glyoxylic acid | Oxalic acidb | 0.55 | 0.64 | 0.62 | 970 |
Activity of RjGlOx for oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid
There is considerable interest in the oxidation of 5-hydroxymethylfurfural (HMF) to 2,5-furandicarboxylic acid (FDCA), since FDCA can be used to generate polyester bioplastics.34 Enzyme-catalysed oxidation of HMF to FDCA has been reported by HMF oxidase from Methylovorus sp. MP688,35,36 and a combination of oxidase PaoABC with glucose oxidase variants,37 however, both systems show modest rates of turnover to FDCA. A further oxidase from Moesziomyces antarcticus has been reported to convert HMF to FFCA, and a further unspecific peroxygenase used to convert FFCA to FDCA.38 Since R. jostii glycolate oxidase shows activity for oxidation of bifunctional aldehyde substrates, we tested HMF as a substrate for RjGlOx-catalysed oxidation. We observed 20–30% conversion on a small scale of 2 mM HMF to 5-hydroxymethyl-2-furancarboxylic acid (HMFCA) and FDCA (see Fig. 4A), indicating that the aldehyde sidechain is oxidised first to a carboxylic acid, followed by oxidation of the hydroxymethyl group. On a 200 mg scale, conversion of 16 mM HMF with addition of 9 mg RjGlOx in 50 mM phosphate buffer pH 7.5 gave FDCA as the major product peak in 45–50% yield, but with residual HMF substrate, and some HMFCA (see Fig. 4B). Addition of 40 μg bovine liver catalase on a small-scale conversion of 2 mM HMF was observed to improve the bioconversion of HMF (see ESI Fig. S5†), but under these conditions the major product was HMFCA, with a 23% conversion to FDCA. R. jostii glycolate oxidase is therefore a new biocatalyst for HMF oxidation, which although it has lower catalytic efficiencies than some reported oxidases,37,38 can oxidise HMF to FDCA in a single enzyme biotransformation, and can be expressed efficiently in Escherichia coli.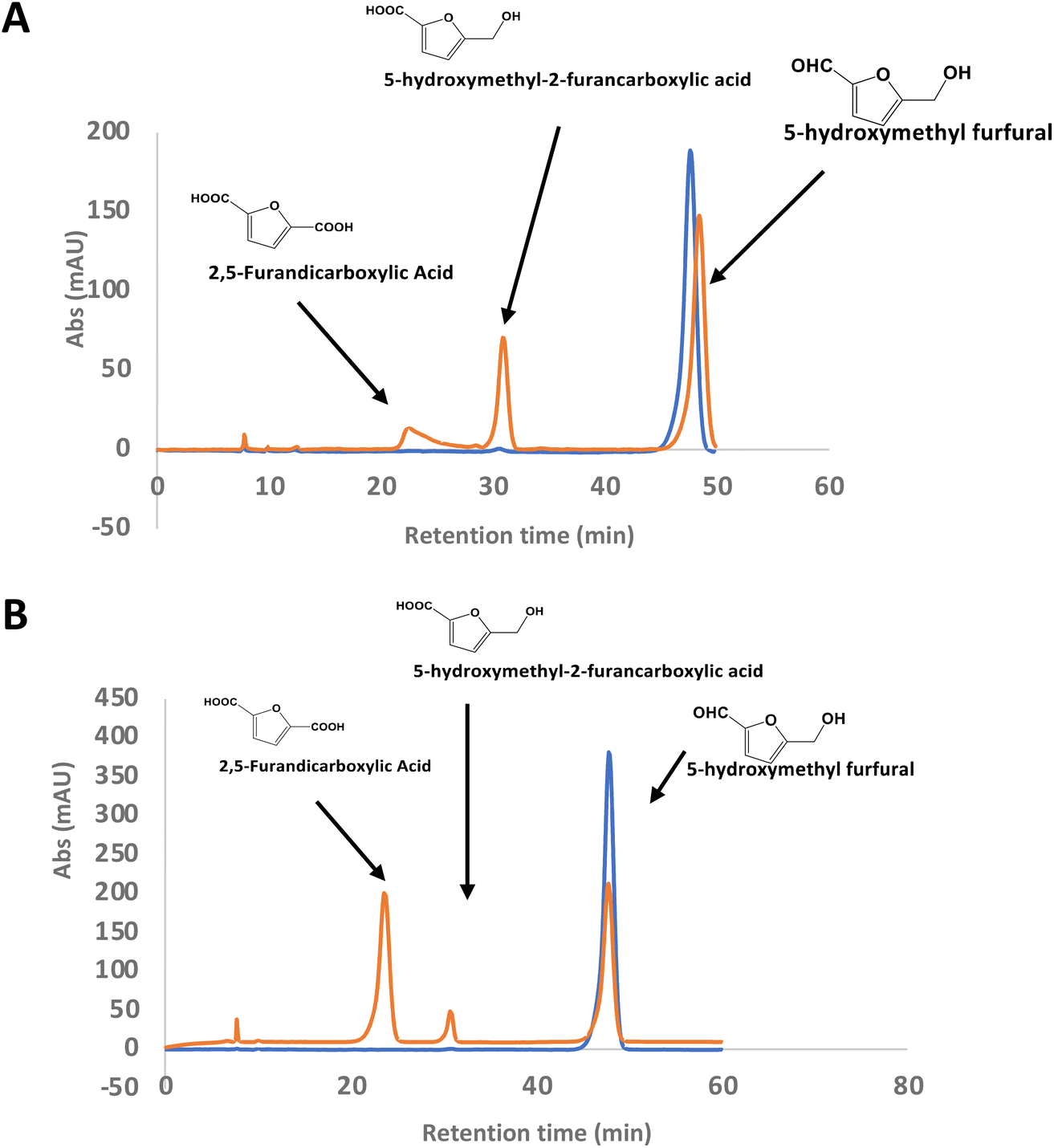 | ||
| Fig. 4 Conversion of HMF to FDCA by R. jostii RHA1 glycolate oxidase. (A) Small scale (1 ml) conversion of 2 mM HMF by 90 μg Rj GlOx; (B) large scale (100 ml) conversion of 200 mg (16 mM) HMF by 9 mg Rj GlOx. Orange, reaction mixture containing enzyme; blue, control reaction lacking enzyme. All bioconversions incubated in 50 mM phosphate buffer pH 7.5 for 24 h at 30 °C. | ||
Coupling of RjGlOx with AgroDyp and CtDyP
Since RjGlOx generates hydrogen peroxide, it could potentially act as an accessory enzyme for peroxidase-catalysed oxidation of lignin. We have previously identified bacterial dye-decolorizing peroxidases DypB from Rhodococcus jostii RHA139 and Dyp1B from Pseudomonas fluorescens pf-533 with activity for oxidation of polymeric lignin, however, in common with most peroxidases, these enzymes are most active at pH 4–5. We have recently identified two further bacterial dye decolorizing peroxidases from Agrobacterium sp. and Comamonas testosteroni that retain significant activity at pH 6–7.31 Comparing their pH-rate profiles with R. jostii glycolate oxidase (see ESI Fig. S6†), we found that all three enzymes retained >80% maximum activity at pH 6.5. Therefore we tested RjGlOx in combination with AgroDyP or CtDyP at pH 6.5, using different polymeric lignins.We first tested for the formation of low molecular weight products from polymeric lignin, using two published colorimetric assays, using 2,4-dinitrophenylhydrazine to derivatise ketone and aldehyde products, and the Folin–Ciocalteu assay to measure low molecular weight phenol products,31 as shown in Fig. 5. Four different samples of polymeric were used, that were characterised in a previous study:28 poplar alkaline and ammonia organosolv lignin, eucalyptus organosolv lignin, and a miscanthus ionic liquid lignin. Activity of Agrobacterium DyP in the presence of 1 mM hydrogen peroxide was compared with AgroDyP and RjGlOx in the presence of 1 mM glycolic acid. As shown in Fig. 5A, using the DNP assay, the conversion with AgroDyP/RjGlOx in the absence of hydrogen peroxide in fact generated more low molecular weight products than AgroDyP/H2O2: it showed 30% higher activity after 30 min, and remained active over 24 h, whereas in the presence of hydrogen peroxide, AgroDyP lost activity after 3 h. Using the FCA assay, for AgroDyP/H2O2, product formation after 24 h was reduced, consistent with some repolymerisation of phenolic products, but this was not observed for the peroxide-free conversion, suggesting that the coupled RjGlOx conversion may also reduce the degree of repolymerisation. Similar trends were observed using other polymeric lignin substrates (see ESI Fig. S7 and S8†).
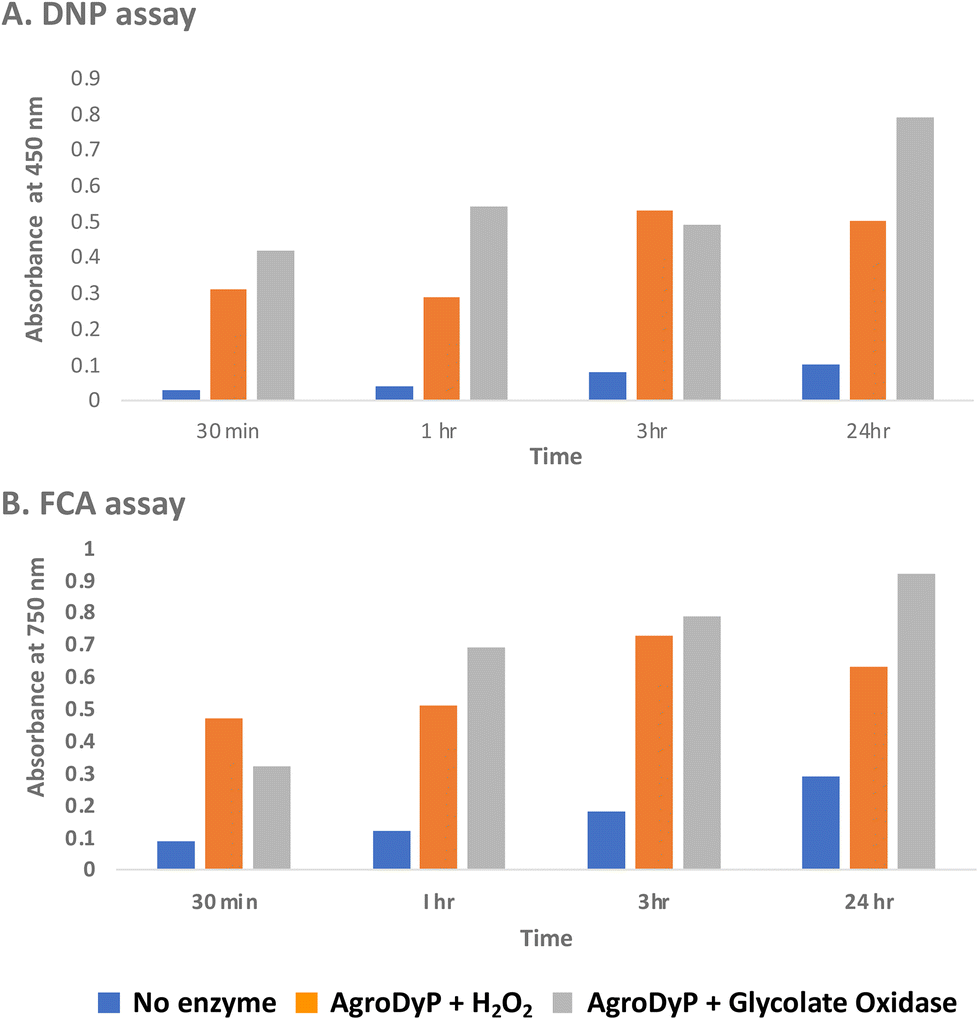 | ||
| Fig. 5 Formation of low molecular weight ketone/aldehyde products via 2,4-dinitrophenylhydrazine assay (A) and phenolic products via Folin–Ciocalteu assay (B) from conversion of poplar alkaline organosolv lignin by Agrobacterium DyP/1 mM hydrogen peroxide (in orange) vs. Agrobacterium DyP/R. jostii glycolate oxidase/1 mM glycolic acid (in grey). Assay data for conversion of other polymeric lignins are shown in ESI.† | ||
In order to investigate whether the product distribution had changed in the presence of R. jostii glycolate oxidase, the low molecular weight products were extracted and analysed by LC-MS, using poplar ammonia organosolv lignin, poplar alkaline organosolv lignin, and eucalyptus organosolv lignin, which had been shown previously to generate low molecular weight products using Pseudomonas fluorescens Dyp1B.28 Analysis of transformations with AgroDyp or CtDyP in the presence or absence of RjGlOx showed the formation of new product peaks, and enhanced peak heights for existing peaks, in the presence of RjGlOx (see Fig. 6), indicating that the combined DyP/GlOx bioconversion generates increased product yield and some new reaction products, compared with DyP alone. Although there are few published studies on lignin bioconversion with enzyme combinations, we observed previously the formation of new products from lignin bioconversion with DyP peroxidases and other accessory enzymes.31
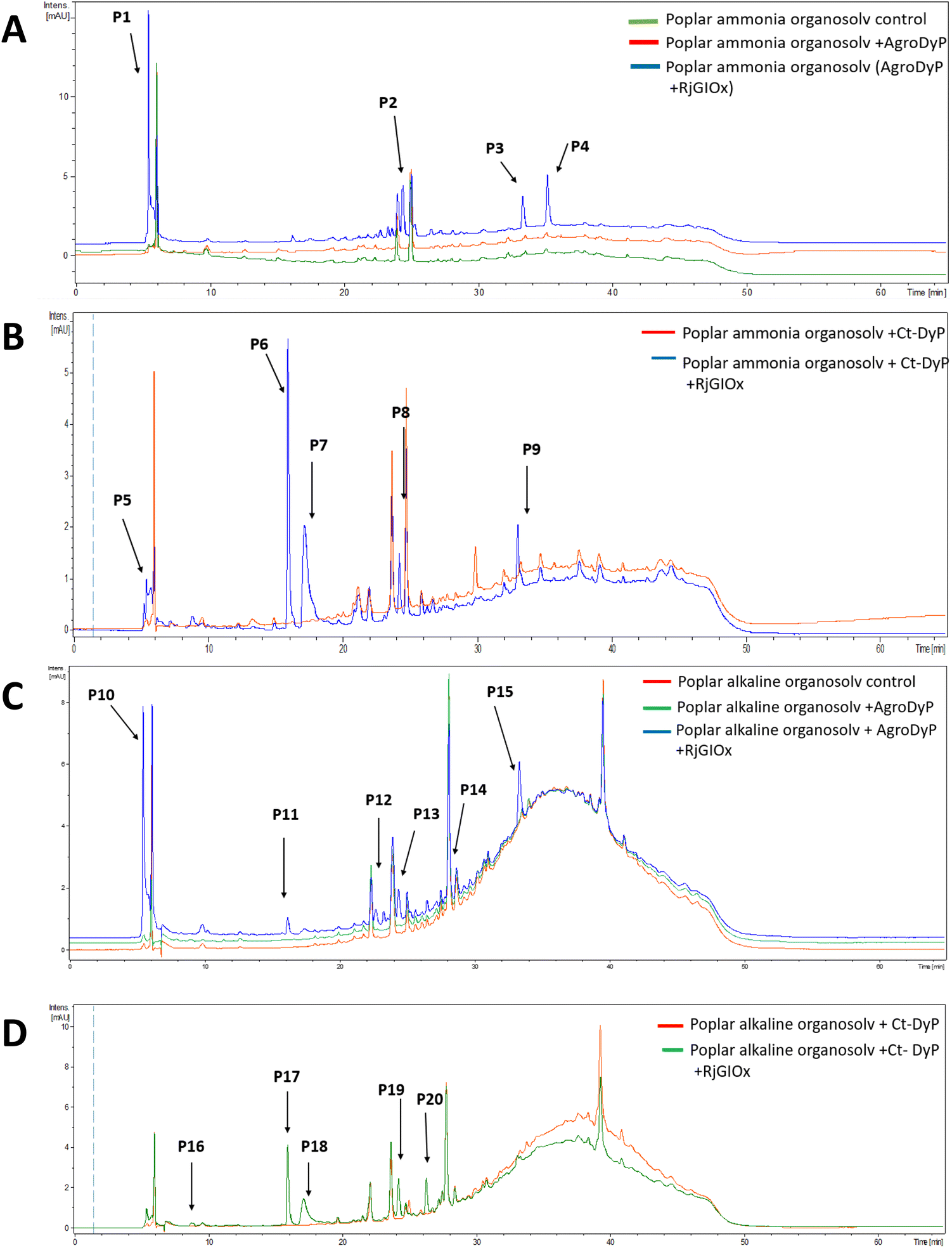 | ||
| Fig. 6 Total ion chromatograms for product formation from combined DyP/GlOx transformations of polymeric lignin substrates. Bioconversion of poplar ammonia organosolv lignin by AgroDyp/RjGlOx (A) and CtDyp/RjGlOx (B), and conversion of poplar alkaline organosolv lignin by AgroDyp/RjGlOx (C) and CtDyp/RjGlOx (D), are compared with control incubations containing DyP peroxidase alone, or no enzyme. Peaks that show increased intensity or are absent without RjGlOx are indicated, and they are identified in Table 2. | ||
| Poplar ammonia organosolv lignin | Poplar alkaline organosolv lignin | |||||
|---|---|---|---|---|---|---|
| RT (min) | m/z | AgroDyp/RjGlOx | CtDyp/RjGlOx | AgroDyp/RjGlOx | CtDyp/RjGlOx | Product identity |
| 5.2 | 228 | P5 | ND | |||
| 5.5 | 99 | P1 | P10 | Glyoxylic acid | ||
| 8.7 | 157 | P16 | 2-Methoxy-6-hydroxy-1,4-hydroquinone | |||
| 16.0 | 125 | P6 | P11 | P17 | Guaiacol | |
| 17.2 | 153 | P7 | P18 | Vanillin | ||
| 22.0 | ND | P12 | ND | |||
| 24.3 | 155 | P8 | P13 | P19 | Protocatechuic acid | |
| 24.5 | 169 | P2 | Vanillic acid | |||
| 26.3 | 183 | P20 | Syringaldehyde | |||
| 29.0 | 146 | P14 | ND | |||
| 33.1 | 243 | P3 | P9 | P15 | Syringyl 1-keto-2,3-dihydroxypropane | |
| 35.2 | 582 | P4 | Unidentified oligomer | |||
As shown in Table 1, a number of products were identified by LC-MS analysis. Many of these products have been observed in previous lignin bioconversions,28,31 and their formation will be discussed further in the Discussion section. Products guaiacol and 2-methoxy-6-hydroxy-1,4-hydroquinone arise from aryl-Cα bond cleavage, whereas vanillin, syringaldehyde, vanillic acid and protocatechuic acid arise from Cα–Cβ bond cleavage. Ct-DyP showed a stronger preference to generate guaiacol and vanillin products. A syringyl monomer containing an oxidised keto-diol sidechain was observed in the presence of RjGlOx, which could have arisen from sidechain oxidation by glycolate oxidase. Glyoxylic acid was also observed as a product, which can be generated by RjGlOx-catalysed oxidation of glycolaldehyde.17 Further LC-MS data for transformation of eucalyptus organosolv lignin is shown in ESI (Fig. S12†), in which several of the same products were also generated, and the case of CtDyP/RjGlOx, syringic acid was also observed as a product.
Application to biorefinery streams
In the lignocellulose-based biorefinery concept, pretreatment of lignocellulosic biomass improves for availability of cellulosic sugars for saccharification, however, a practical problem with pretreatment is the formation of fermentation inhibitors and enzyme inhibitors,40 notably aldehydes such as 5-hydroxymethylfuran (HMF) and furfural,41 which limits the utilisation of both cellulose and lignin streams. Since RjGlOx shows activity for oxidation of HMF and other aldehydes, we investigated whether the combination of DyP peroxidases and RjGlOx would improve the generation of high-value chemicals from biorefinery streams arising from lignocellulose pretreatment.A combination of AgroDyP and RjGlOx was applied to bioconversion of two biorefinery streams: hydrolysis lignin (HL) produced after pretreatment and enzymatic saccharification of wheat straw for cellulosic bioethanol production;29 and humins, polymeric materials derived from polymerisation of HMF.30 In the case of HL, 4 new peaks were observed in the presence of the AgroDyP/RjGlOx combination, as shown in Fig. 7. Peak F3 was identified as syringaldehyde (m/z 183 MH+), by comparison with an authentic standard, and has been observed in previous lignin bioconversions.28 Peak F4 (33.0 min, m/z 243) was also observed in lignin bioconversions above (peak P3 in Table 1), and is proposed to be syringyl 2,3-dihydroxypropan-1-one, a monomer containing a keto-diol C3 sidechain. A syringyl monomer containing a C3 triol sidechain has been observed in a previous lignin bioconversion,28 and a keto-diol sidechain has been observed in a lignin dimer product arising from lignocellulose oxidase by Dyp1B.33 Peak F2 (24.2 min, m/z 227) is proposed to be syringyl 3-hydroxypropan-1-one, for which a related guaiacyl product has been observed previously in a DyP peroxidase biotransformation.28 Peak F1 (5.2 min, m/z 228) is an unknown product.
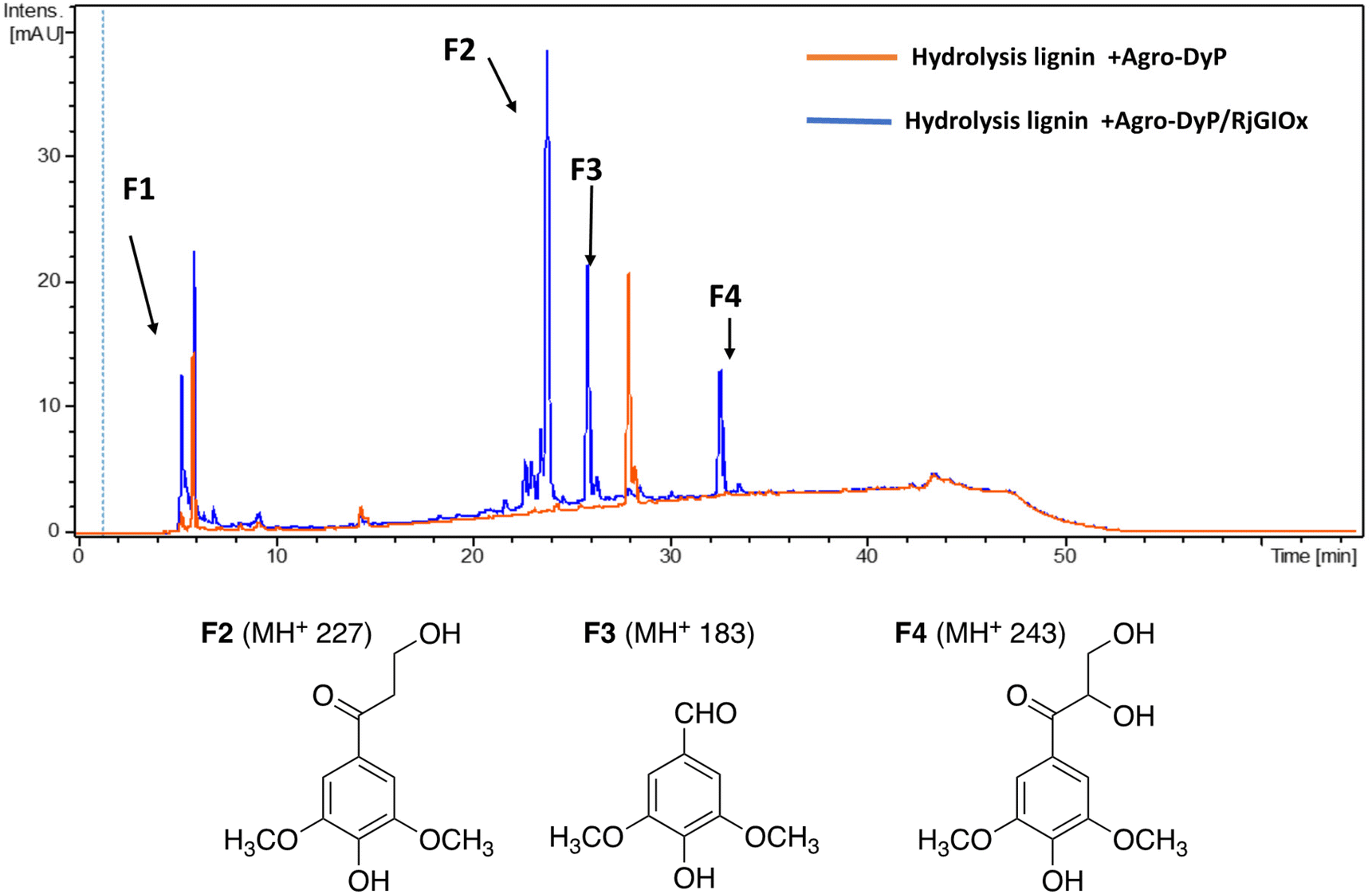 | ||
| Fig. 7 Total ion chromatogram of products from bioconversion of hydrolysis lignin by AgroDyP and RjGlOx, and chemical structures of identified products. | ||
In the case of Avantium humins, 7 new peaks were observed with the AgroDyp/RjGlOx combination, as shown in Fig. 8. Peak H2 was identified as 5-hydroxymethylfurfural (HMF), which was also observed in much smaller amounts (<10%) in a control incubation lacking enzyme. 5-Hydroxymethylfurfural (peak H2) and 5-hydroxymethyl-2-furancarboxylic acid (HMFCA, peak H3) were produced as major products, while further compounds related to HMF were identified by comparison with authentic standards, namely 2,5-furandicarboxylic acid (FDCA, peak H1) and 5-formyl-2-furancarboxylic acid (FFCA, peak H7). Based on the proposed structure for polymeric humins,30 and the observed activities of RjGlOx, we have proposed feasible hypothetical dimeric and trimeric structures in Fig. 7 for peaks H6 (25.5 min, m/z 259) and H4 (21.8 min, m/z 323), while peak H5 (24.0 min, m/z 457) appears to be a tetrameric furanic compound.
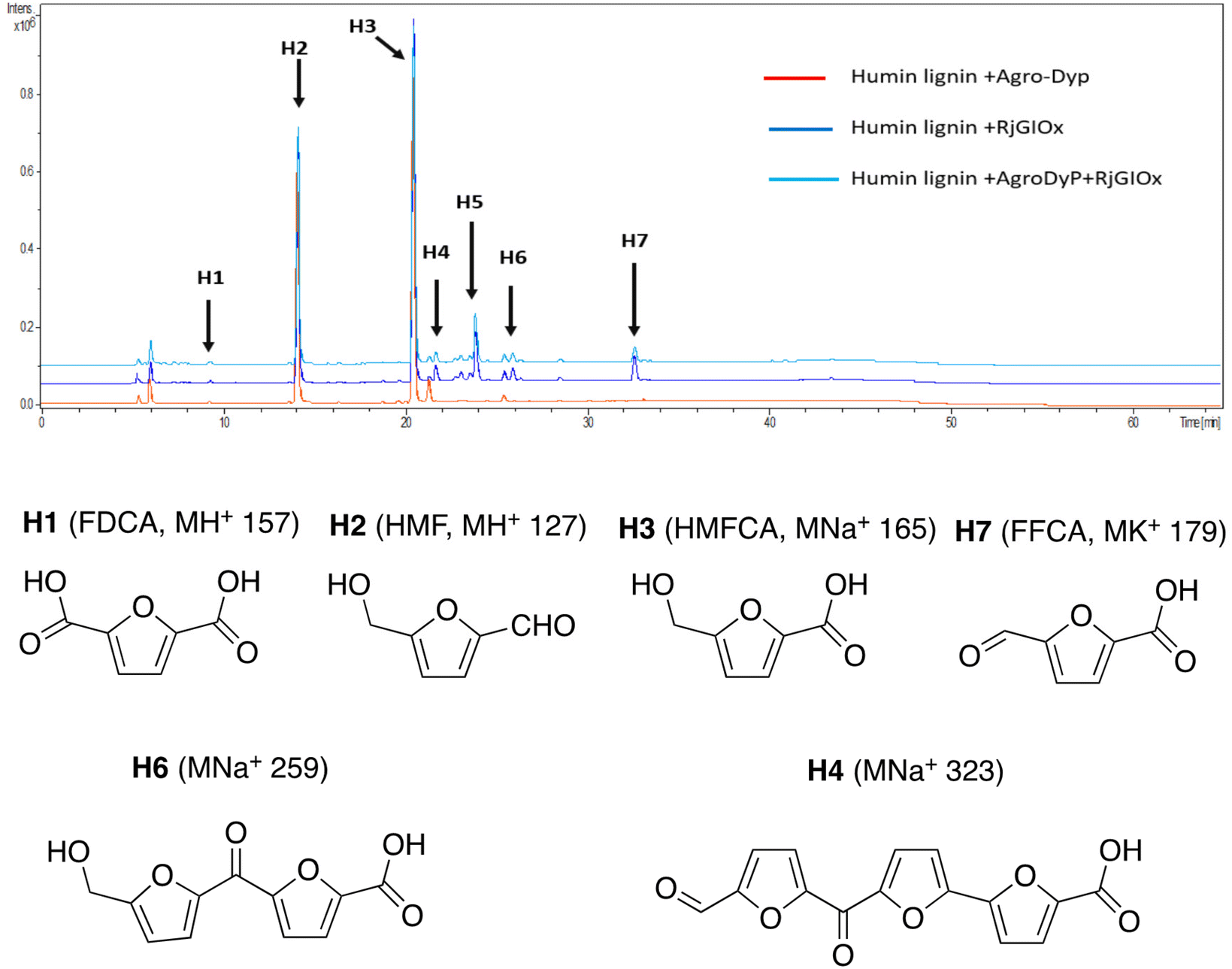 | ||
| Fig. 8 Total ion chromatogram of products from bioconversion of Avantium humins by AgroDyP and RjGlOx. Structures for H1–H3 and H7 confirmed by co-elution with authentic standards, structures for H4 and H6 based on predicted humin structure.30 | ||
Discussion
Rhodococcus jostii RHA1 glycolate oxidase is shown to be a valuable accessory enzyme for degradation of polymeric lignin in vitro. Although first identified as an oxidase for generation of aromatic benzoylformate pathway intermediates,16 we show that it has a broad substrate scope for α-ketoaldehyde and α-ketoacid substrates, and for oxidation of HMF to 2,5-FDCA. The kinetic parameters measured in Table 1 indicate that highest kcat and kcat/KM were observed for mandelic acid, but for dicarbonyl substrates, methylglyoxal has a higher kcat and kcat/KM than 4-hydroxyphenylglyoxal. α-Substituted aldehydes such as glycolaldehyde and methylglyoxal are known by-products of biomass conversion,42 therefore, this enzyme could oxidise these aliphatic aldehydes during lignin oxidation, and we have already shown that this enzyme oxidises glycolaldehyde to oxalic acid during lignin breakdown.17 The R. jostii RHA1 glycolate oxidase gene ro02984 contains no signal sequence, therefore it is unlikely that this enzyme is used to generate hydrogen peroxide extracellularly, however, it could participate in the oxidation of lignin fragments intracellularly. We note that overexpression of Pseudomonas putida KT2440 glycolate oxidase genes glcDEF has been recently reported to permit growth on ethylene glycol, catalysing the removal of toxic intermediates.43 On the basis of the experimental data, there does not seem to be one preferred substrate for R. jostii glycolate oxidase, we therefore suspect that it has a dual role in the cell (as shown in Fig. 1), for oxidation of aryl-C2 substrates to be processed by the 4-hydroxybenzoylformate pathway,16 but also to oxidise glycolaldehyde and methylglyoxal generated from lignin degradation, which would otherwise be toxic to the cell.The enzyme requires two functional groups to be present for oxidation to take place, which may be a consequence of the enzyme mechanism. We have previously observed conversion of 4-hydroxyphenylacetic acid to 4-hydroxybenzoylformate, via hydroxylation of benzylic position.17 The likely mechanism for this reaction involves hydrogen atom abstraction (or deprotonation followed by 1-electron transfer) in the benzylic position, via a radical intermediate. If a similar hydrogen atom abstraction mechanism applied to aldehyde and α-hydroxy-acid oxidation, as shown in Fig. 9, then this would rationalise why two functional groups are needed, since the radical intermediate would be stabilised by adjacent carboxylic acid (or ketone) group. In the case of HMF oxidation, a radical intermediate would be stabilised by the adjacent furan ring.
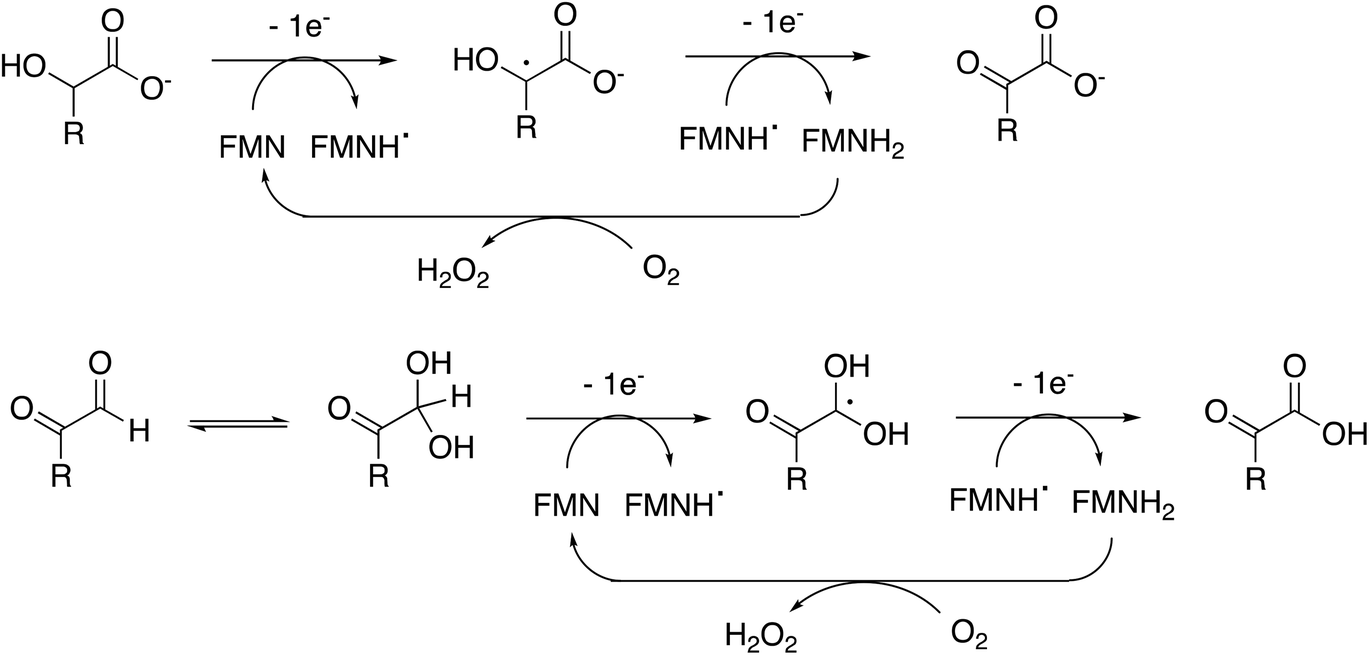 | ||
| Fig. 9 Proposed catalytic mechanism for RjGlOx-catalysed oxidation of α-hydroxy-carboxylic acid and α-dicarbonyl substrates. R can be either an aromatic or aliphatic (H, –CH3, –CH2CO2H) group. | ||
RjGlOx co-operates effectively with DyP peroxidases AgroDyP and CtDyp in vitro, since they retain activity at pH 6.5, to enable a bioconversion driven by dioxygen as an oxidant, rather than hydrogen peroxide. This is a milder, more sustainable process than using a peroxidase with added hydrogen peroxide, which also leads to less enzyme inactivation (shown in data in Fig. 5). Although some hydrogen peroxide will be generated during the biotransformation by RjGlOx, it will likely be sub-millimolar concentrations, since KM for hydrogen peroxide by DyP peroxidases is in the range 50–70 μM,33 and kcat values for DyPs are in the range 10–20 s−1,33 compared with 0.5–2 s−1 for RjGlOx (see Table 1). Since inactivation of DyPs occurs in the presence of 1–2 mM hydrogen peroxide,39 it is logical that controlled release of hydrogen peroxide should be beneficial.
The formation of higher product yields from polymeric lignin oxidation (see Fig. 7–9) is most likely due to a more effective DyP oxidation, since there is less enzyme inactivation, but there is a possibility that RjGlOx might also prevent repolymerisation of radical products, via further 1-electron oxidation, or 1-electron reduction by a flavin semiquinone intermediate. We have shown previously that combination of DyP peroxidases with other accessory enzymes that prevent lignin repolymerisation can lead to enhanced yield of products from polymeric lignin substrates.31 The formation of alternative products may be aided by RjGlOx-catalysed oxidation reactions: two new products (ketodiol P3/9/15 and glyoxylic acid P1/10) contain carbonyl groups that could arise from GlOx-catalysed oxidation. The formation of the various reaction products is rationalised in Fig. 10: Cα–Cβ oxidative cleavage is precedented for DyP peroxidases;33,39 the formation of ketodiol P3/9/15 and guaiacol P6/11/17 seems most likely via a benzylic ketone intermediate; and ketoalcohol product F2 could perhaps be formed via elimination of the β-O-aryl substituent via a benzylic radical, which might be catalysed by RjGlOx on an oligomeric lignin fragment. The formation of a reaction product containing an oxidised ketodiol sidechain has also been observed previously from lignocellulose, by P. fluorescens Dyp1B.33
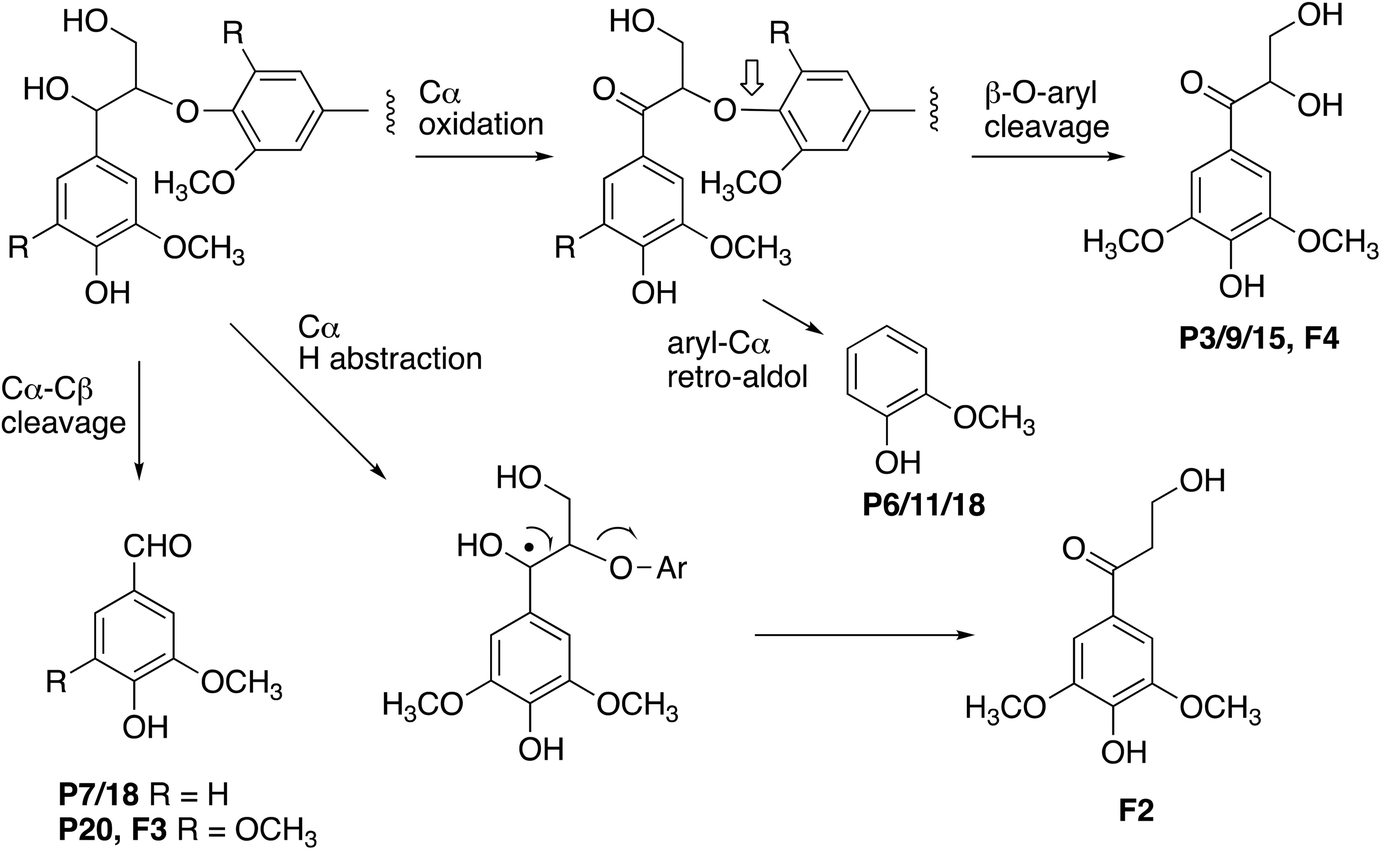 | ||
| Fig. 10 Formation of products from polymeric lignin. R = H (guaiacyl G lignin unit) or OCH3 (syringyl S lignin unit). Further oxidation of vanillin (P7/18) generates the corresponding carboxylic acid, vanillic acid (P2). Poplar organosolv lignin contains both S and G units (with proportion of S > G). | ||
One interesting point is that for the combined DyP/GlOx conversion of polymeric lignin, each enzyme provides the substrate for the other, since GlOx provides hydrogen peroxide for DyP, but also DyP generates oxidised small molecule products, such as aldehydes, from lignin breakdown. This is seen in the formation of ketodiol product P3/9/15 from organosolv lignin. Hence there is a kind of symbiotic relationship between these two enzymes.
The observation that the RjGlOx/DyP combination is effective for oxidation of biorefinery streams which contain high levels of aldehydes could lead to new applications for generation of high value chemicals from a lignocellulosic biorefinery. Reaction products F4 from HL and H4 and H6 from bioconversion of humins each contain ketone groups which could arise from GlOx oxidation. The low background in the case of HL implies that GlOx actively participates in product release, whereas in the case of humins, some products are released by GlOx alone (see Fig. 8). Although we do not know the precise mechanism for oxidation of humins, it is possible that GlOx could catalyse oxidation of an ether-linked unit found in humins, as shown in Fig. 11, leading to the formation of an aldehyde sidechain, which could be further oxidised to a carboxylic acid, and the methylene unit also oxidised to a ketone. Hence we see new products and new mechanisms through the use of enzyme combinations, which offer new possibilities for oxidation of polymeric lignin/humins in vitro.31
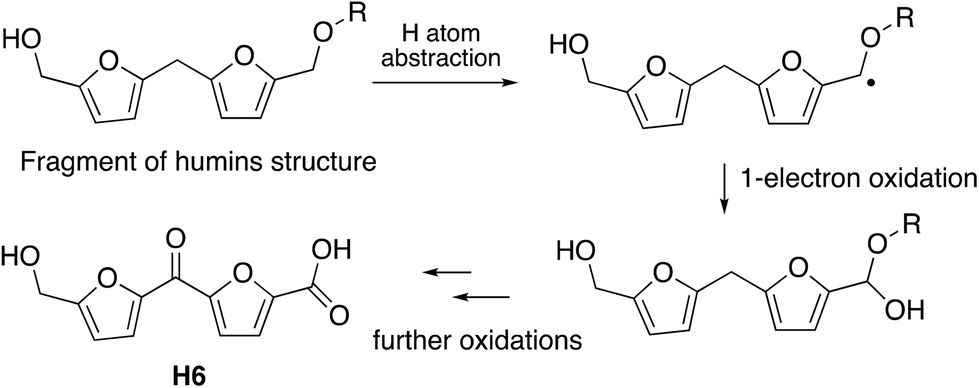 | ||
| Fig. 11 Possible mechanism for generation of product H6 from oxidation of humins via combined RjGlOx/DyP biotransformation. A similar mechanism can be drawn for the formation of product H4. | ||
Author contributions
The research was carried out by AA and GMMR. The project was supervised by TDHB. The funding for the work was obtained by TDHB and AA. The first draft of the manuscript was written by TDHB and AA.Conflicts of interest
The authors declare no conflict of interest associated with this article.Acknowledgements
This research was supported by a PhD studentship (to A. A.) funded by Northern Border University, Saudi Arabia, and by BBSRC research grant BB/T010622/1. We thank Dr Andrew Carnell (University of Liverpool) for the gift of authentic standards for HMF oxidation. Hydrolysis lignin and Avantium humins were supplied through the EU Zelcor project, funded from the Bio-Based Industries Joint Undertaking under the European Union Horizon 2020 Research and Innovation Programme under Grant Agreement 720303. We would like to thank Dr Annelie Jongerius (Avantium) for provision of samples of humins, and Prof Stéphanie Baumberger (UMR IJPB, AgroParisTech-INRAE-Université Paris Saclay, France) for supplying the hydrolysis lignin sample.References
- J. Zakzeski, P. C. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- A. T. Martínez, M. Speranza, F. J. Ruiz-Duenas, P. Ferreira, S. Camarero, F. Guillen, M. J. Martínez, A. Gutierrez and J. C. del Rio, Int. Microbiol., 2005, 8, 195–204 Search PubMed.
- D. W. S. Wong, Appl. Biochem. Biotechnol., 2009, 157, 174–209 CrossRef CAS PubMed.
- T. D. H. Bugg and R. Rahmanpour, Curr. Opin. Chem. Biol., 2015, 29, 10–17 CrossRef CAS PubMed.
- T. D. H. Bugg, J. J. Williamson and G. M. M. Rashid, Curr. Opin. Chem. Biol., 2020, 55, 26–33 CrossRef CAS PubMed.
- B. Bissaro, A. Várnai, A. K. Røhr and V. G. H. Eijsink, Microbiol. Mol. Biol. Rev., 2018, 82, e00029-18 CrossRef PubMed.
- A. Hernández-Ortega, P. Ferreira and A. T. Martínez, Appl. Microbiol. Biotechnol., 2012, 93, 1395–1410 CrossRef PubMed.
- M. Daou and C. B. Faulds, World J. Microbiol. Biotechnol., 2017, 33, 87 CrossRef PubMed.
- F. Guillén, A. T. Martínez and M. J. Martínez, Eur. J. Biochem., 1992, 209, 603–611 CrossRef PubMed.
- F. Guillén, A. T. Martínez, M. J. Martínez and C. S. Evans, Appl. Microbiol. Biotechnol., 1994, 41, 465–470 Search PubMed.
- I. S. Fernández, F. J. Ruíz-Dueñas, E. Santiliana, P. Ferreira, M. J. Martínez, A. T. Martínez and A. Romero, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2009, 65, 1196–1205 CrossRef PubMed.
- J. Viña-Gonzalez, D. Jimenez-Lalana, F. Sancho, A. Serrano, A. T. Martínez, V. Guallar and M. Alcalde, Adv. Synth. Catal., 2019, 361, 2514–2525 Search PubMed.
- P. J. Kersten and T. K. Kirk, J. Bacteriol., 1987, 169, 2195–2201 CrossRef CAS PubMed.
- P. J. Kersten, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 2936–2940 CrossRef CAS PubMed.
- M. Daou, F. Piumi, D. Cullen, E. Record and C. B. Faulds, Appl. Environ. Microbiol., 2016, 82, 4867–4875 CrossRef CAS PubMed.
- Z. Wei, R. C. Wilkinson, G. M. M. Rashid, D. Brown, V. Fülöp and T. D. H. Bugg, Biochemistry, 2019, 58, 5281–5293 CrossRef CAS PubMed.
- A. Alruwaili, G. M. M. Rashid, V. Sodré, J. Mason, Z. Rehman, A. Menaketh, S. P. Brown and T. D. H. Bugg, RSC Chem. Biol., 2023, 4, 47–55 RSC.
- T. A. Ewing, G. Gygli, M. W. Fraaije and W. J. H. van Berkel, Enzymes, 2020, 47, 87–116 CrossRef CAS PubMed.
- P. Macheroux, V. Massey, D. J. Thiele and M. Volokita, Biochemistry, 1991, 30, 4612–4619 CrossRef CAS PubMed.
- E. Liu, F. Segato, R. A. Prade and M. R. Wilkins, Bioresour. Technol., 2021, 338, 125564 CrossRef CAS PubMed.
- C. C. Lee, H. S. Chen, C. H. Yang, S. P. Wang, J. H. Wu and M. Meng, Process Biochem., 2016, 51, 1486–1495 CrossRef.
- P. Herzog, L. Sützi, B. Eisenhut, D. Maresh, D. Haltrich, C. Obinger and C. K. Peterbauer, Appl. Environ. Microbiol., 2019, 85, e00390-19 CrossRef PubMed.
- T. Renders, G. Van den Bossche, T. Vangeel, K. Van Aelst and B. Sels, Curr. Opin. Biotechnol., 2019, 56, 193–201 CrossRef CAS PubMed.
- W.-J. Liu, H. Jiang and H.-Q. Yu, Green Chem., 2015, 17, 4888–4907 RSC.
- F. P. Bouxin, S. D. Jackson and M. C. Jarvis, Bioresour. Technol., 2014, 162, 236–242 CrossRef CAS PubMed.
- F. P. Bouxin, A. McVeigh, F. Tran, N. J. Westwood, M. C. Jarvis and S. D. Jackson, Green Chem., 2015, 17, 1235–1242 RSC.
- R. P. Castillo, J. Baeza, J. Rubilar, A. Rivera and J. Freer, Appl. Biochem. Biotechnol., 2012, 168, 2028–2042 CrossRef CAS PubMed.
- C. S. Lancefield, G. M. M. Rashid, F. Bouxin, A. Wasak, W.-C. Tu, J. Hallett, S. Zein, J. Rodríguez, S. D. Jackson, N. J. Westwood and T. D. H. Bugg, ACS Sustainable Chem. Eng., 2016, 4, 6921–6930 CrossRef CAS.
- G. N. Rivière, F. Pion, M. Farooq, M. H. Sipponen, H. Koivula, T. Jayabalan, P. Pandard, G. Marlair, X. Liao, S. Baumberger and M. österberg, Sustainable Mater. Technol., 2021, 28, e00269 CrossRef.
- I. van Zandvoort, E. J. Koers, M. Weingarth, P. C. A. Bruijnincx, M. Baldus and B. M. Weckhuysen, Green Chem., 2015, 17, 4383–4392 RSC.
- G. M. M. Rashid and T. D. H. Bugg, Catal.: Sci. Technol., 2021, 11, 3568–3577 RSC.
- T. V. Votyakova and I. J. Reynolds, Arch. Biochem. Biophys., 2004, 431, 138–144 CrossRef CAS PubMed.
- R. Rahmanpour and T. D. H. Bugg, Arch. Biochem. Biophys., 2015, 574, 93–98 CrossRef CAS PubMed.
- E. de Jong, H. A. Visser, A. S. Dias, C. Harvey and G.-J. M. Gruter, Polymers, 2022, 14, 943 CrossRef CAS PubMed.
- W. P. Dijkman and M. W. Fraaije, Appl. Environ. Microbiol., 2014, 80, 1082–1090 CrossRef PubMed.
- W. P. Dijkman, D. E. Groothuis and M. W. Fraaije, Angew. Chem., Int. Ed., 2014, 53, 6515–6518 CrossRef CAS PubMed.
- S. M. McKenna, P. Mines, P. Law, K. Kovacs-Schreiner, W. R. Birmingham, N. J. Turner, S. Leimkühler and A. J. Carnell, Green Chem., 2017, 19, 4660–4665 RSC.
- A. Lappe, N. Jankowski, A. Albrecht and K. Koschorreck, Appl. Microbiol. Biotechnol., 2021, 105, 8313–8327 CrossRef CAS PubMed.
- M. Ahmad, J. N. Roberts, E. M. Hardiman, R. Singh, L. D. Eltis and T. D. H. Bugg, Biochemistry, 2011, 50, 5096–5107 CrossRef CAS PubMed.
- L. J. Jönsson and C. Martin, Bioresour. Technol., 2016, 199, 103–112 CrossRef PubMed.
- M. Ask, M. Bettiga, V. Mapelli and L. Olsson, Biotechnol. Biofuels, 2013, 6, 22 CrossRef CAS PubMed.
- L. N. Jayakody, T. L. Turner, E. J. Yun, I. I. Kong, J.-J. Liu and Y.-S. Jin, Appl. Microbiol. Biotechnol., 2018, 102, 8121–8133 CrossRef CAS PubMed.
- M. A. Franden, L. N. Jayakody, W.-J. Li, N. J. Wagner, N. S. Cleveland, W. E. Michener, B. Hauer, L. M. Blank, N. Wierckx, J. Klebensberger and G. T. Beckham, Metab. Eng., 2018, 48, 197–207 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3gc00475a |
| This journal is © The Royal Society of Chemistry 2023 |