
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective oxidation of biomass-derived carbohydrate monomers
Janvit
Teržan†
 *ab,
Anja
Sedminek†
b,
Žan
Lavrič
ac,
Miha
Grilc
ac,
Matej
Huš
acdf and
Blaž
Likozar
ae
*ab,
Anja
Sedminek†
b,
Žan
Lavrič
ac,
Miha
Grilc
ac,
Matej
Huš
acdf and
Blaž
Likozar
ae
aDepartment of Catalysis and Chemical Reaction Engineering, National Institute of Chemistry, Hajdrihova ulica 19, 1000 Ljubljana, Slovenia. E-mail: janvit.terzan@ki.si; Fax: +386 1 4760300; Tel: +386 1 4760 534
bDepartment for Materials Synthesis, Jožef Stefan Institute, Jamova cesta 39, 1000 Ljubljana, Slovenia
cUniversity of Nova Gorica, Vipavska cesta 13, Nova Gorica, 5000, Slovenia
dAssociation for Technical Culture of Slovenia (ZOTKS), Zaloška cesta 65, 1000 Ljubljana, Slovenia
eFaculty of Polymer Technology, Ozare 19, SI-2380 Slovenj Gradec, Slovenia
fInstitute for the Protection of Cultural Heritage of Slovenia, Poljanska cesta 40, 1000 Ljubljana, Slovenia
First published on 9th February 2023
Abstract
Current efforts in the decarbonisation and electrification of the chemical industry drive the interest in green production processes. We looked at the potential production processes for glucaric acid, which was recognized as one of the “highest value-added biomass-derived chemicals” by the US Department of Energy in 2004. Glucaric acid is a very interesting and important base chemical that can be converted into numerous end products, ranging from pharmaceuticals to bio-based polymers. Widespread use of glucaric acid would drastically reduce the carbon dioxide emissions and enable the renewable production of polymers, e.g. nylon 66. The current industrial nitric acid oxidation of glucose pushed the research towards the development of more sustainable oxidation practices, which can be classified into four broad categories: biocatalysis, heterogeneous catalysis, electrocatalysis, and photocatalysis. Since each approach requires a unique experimental setup, the comparison among them is often tedious. Enzymes require milder temperatures and pressures and provide high selectivity, but are fragile and thus unstable, compared with metallic catalysts, where selectivity is lower but productivity is usually higher. Electro- and photocatalytic processes are relatively new and promising techniques for glucose oxidation, harnessing electric potential and the power of photons, but currently suffer from low space-time yield. Furthermore, we used space-time yield as a robust comparison parameter, even though it is not as universal as, say, techno-economic analysis, which is almost impossible at this scale, due to the lack of data provided by each study. Therefore, we briefly review each approach, analyse the state of the art, and provide general guidelines for future research.
1. Introduction
In recent years, environmental awareness has become widespread among the public and the chemical industry. Environmental challenges, ranging from the climate crisis to ecosystem destruction, have necessitated green chemistry approaches that focus on reducing the consumption and production of environmentally harmful substances. Since glucose (Glc) and its derivatives are cheap and readily available raw materials, they have been traded as ideal sustainable feed-stocks for the chemical industry with few side effects. Although the bio-production of Glc can compete with food, the production of cellulose and other inedible glucose polymers and their subsequent de-polymerization offers a sustainable and environmentally friendly way to obtain glucose. The conversion of Glc to glucaric acid (GA) is an important first step in its utilisation. Therefore, the oxidation of Glc to GA would enable a new form of bio-based building block with many potential commercial applications. The extensive use of GA has led to its classification as one of the “highest value-added biomass chemicals” as defined by the US Department of Energy in 2004.As it stands, GA is used as a retardant for metallic mordants in textile dyeing,1 as a metal-sequestering agent2,3 or as a substitute for phosphates to inhibit corrosion in water treatment processes.4 It is also a good chelating agent for cations and therefore has great potential as a component of detergents and surfactants.3,5 As a chelating agent, GA can also be used to remove heavy metals from contaminated soils. It has been shown to remove more than 80% of Cu6 under optimized conditions. Subramanian and Madras7 have shown that the addition of GA can also dramatically increase the degradation of organic pollutants by chelating Fe. It can also help remove toxins from the body; supplements of its calcium salt have been shown to prevent tumour formation and produce tumour suppression in mice.8,9
A very interesting potential market for GA is its application in renewable polymers. For example, it could be used as an intermediate in the production of 2,5-furandicarboxylic acid (FDCA),10,11 which is a renewable substitute for polyethylene terephthalate (PET). In addition, GA can be used for the production of adipic acid.12,13 This is very important since the annual production of adipic acid exceeds 4 million metric tonnes, which is problematic, since there are almost 17 tonnes of CO2 released for every tonne of adipic acid produced.14 GA can also be polymerised directly. The latter leads to new nylons (polyhydroxypolyamides),5,15,16 which are desirable for several reasons. For example, activated carbohydrate monomers can be prepared from any aldose in a renewable manner. No protective step is required for synthesis and subsequent polymerization. Varying the composition of the copolymer results in significantly different properties such as solubility and melting temperature.17 As a polymer additive, it is used to improve the mechanical properties of industrial fibres such as polyacrylonitrile (PAN),18,19 which is an important precursor of carbon fibres. It is also used to reinforce recycled cotton for reuse in clothing. An emerging application is in wearable electronic textiles, where GA will play an important role as a renewable resource.20
Currently, most GA is produced by Glc oxidation with nitric acid.21–25 This process was developed and discussed by Kiliani26 as early as 1880. Some companies, such as Rivertop Renewables Inc.,27 have patented the nitric acid oxidation process. It provides reasonable selectivity, but is environmentally harmful due to NOx emissions and nitric acid discharge. For these reasons, and because of the high cost, GA is now mainly used as a pharmaceutical and nutraceutical chemical.28–33 Commercialization of various greener processes is under investigation, such as biocatalysis, electrocatalysis, photocatalysis, and heterogeneous catalysis (vide infra). A detailed review of the oxidation of glucose to gluconic acid and GA was written by Tsang et al.,34 with an emphasis on heterogeneous catalytic processes. Nevertheless, a substantial comparison of productivity between different state-of-the-art processes such as biocatalytic, heterocatalytic, photocatalytic and electrocatalytic processes seems to be lacking. Although patents describing these processes have already been filed, e.g., by Rennovia Inc.35,36 or Solvay,37 commercialization lags behind. Some patents38 describe the use of a mediator in the oxidation process that appears to increase selectivity for GA. To bolster the value of GA, thereby increasing its widespread utilization, alternative approaches for the production of GA are described in this review. Older research has also been included to better understand the chronological development of the alternative technologies and to have a better overview and more data for comparison, in terms of the space-time yield (STY) of each technology.
2. Biocatalysis
In the biocatalytic production route, we use enzymes that produce GA. The enzyme for the conversion of D-glucuronolactone to GA was purified and analysed almost 50 years ago.39 Moon et al. were the first to consider using a biological route to produce GA in an industrial process.40 They first analysed the mammalian route and later improved on this by using enzymes from different microorganisms (Saccharomyces cerevisiae, Pseudomonas syringae) and combining them in a recombinant Escherichia coli. They found that two factors prevented high productivity: the toxicity of the acids produced and the kinetics of the individual reaction steps (see Fig. 1 for the entire metabolic pathway). They concluded that the conversion of myo-inositol (MI) to D-glucuronic acid was the rate-limiting step. The authors41–43 tried several approaches to remove the bottlenecks and increase production. Using a modular scaffold to group the proteins required for GA production41,42 increased the titre fivefold compared with a 77-fold increase with mevalonate biosynthesis.42 This suggests that localising the production of MI to myo-inositol oxygenase (MIOX), thereby reducing mass transfer constraints, leads to a better titre. Based on the small increase, the authors assume that these constraints are not major factors in productivity.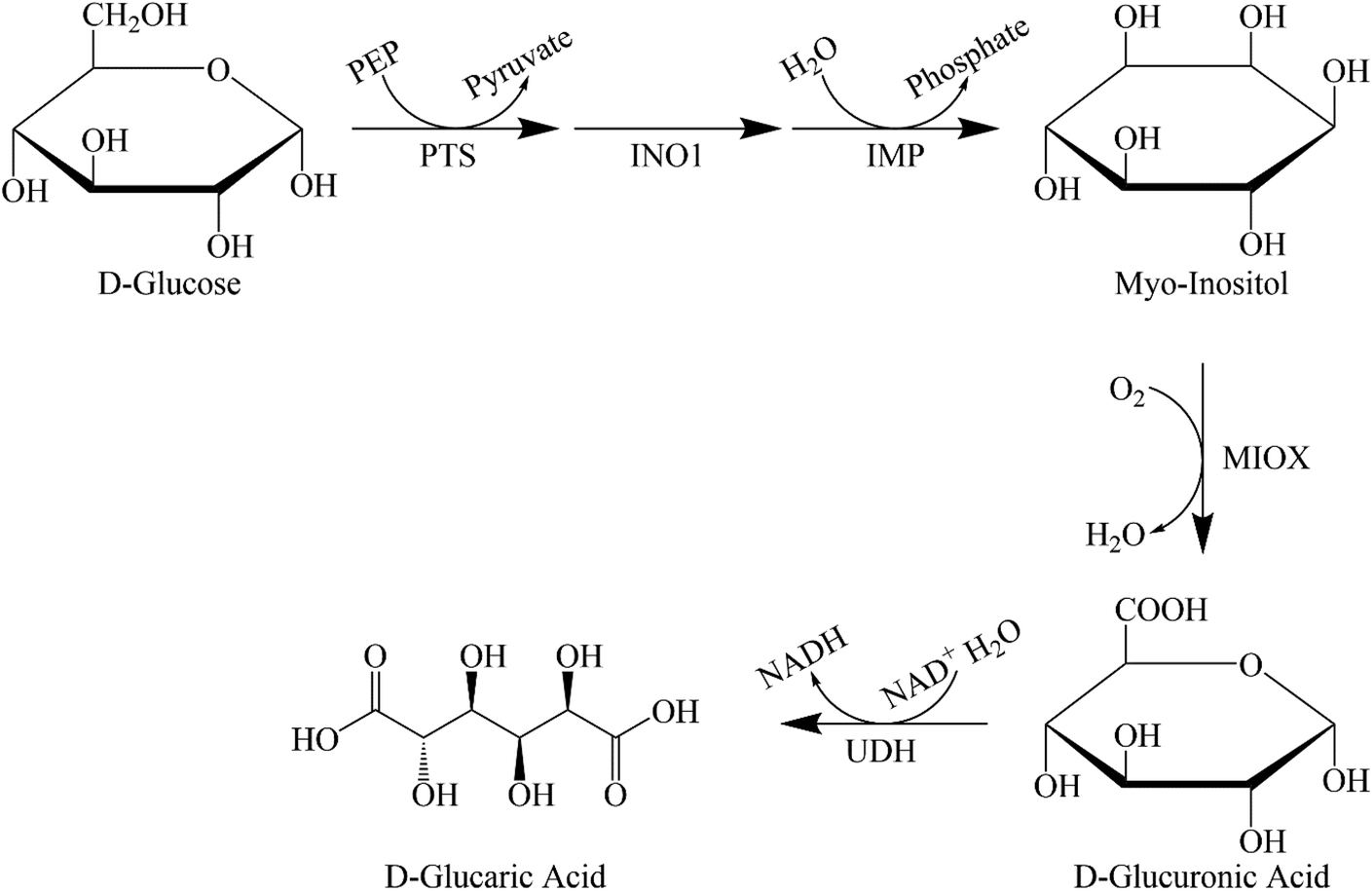 | ||
| Fig. 1 Schematic presentation of the synthetic pathway for the bio-based production of GA. PEP – phosphoenolpyruvate; PTS – phosphotransferase system; INO1 – myo-inositol-1-phosphate synthase; IMP – inositol phosphate-phosphatase; UDH – uronate dehydrogenase. | ||
The authors strived to further improve the yield by modifying MIOX with a small ubiquitin-related modifier (SUMO).43 The resulting enzyme improved the yield of GA from myo-inositol by 75%. However, fusion of the required enzyme with a maltose-binding protein and a green fluorescent protein rendered it completely inactive, demonstrating the importance of choosing the right fusion protein pair. Transport of myo-inositol into the cell was identified as another potential bottleneck for metabolic engineering. Expression of manX mRNA led to posttranscriptional upregulation of the expression of PtsG, a permease that functions as a transporter for myo-inositol. This approach improved the yield of D-glucaric acid by 65%.
In biocatalytic GA production an important parameter is what the organic carbon is utilized for. Brockman and Prather44 sought to improve the productivity of E. coli relative to GA while retaining the ability to direct the carbon flux toward cell growth. They achieved the goal of developing a host strain that can be switched between growth and production cycles by developing a strain in which phosphofructokinase-I (Pfk-I) is the only enzyme that utilizes glucose-6-phosphate (G6P) and fructose-6-phosphate (F6P). The expression of Pfk-I was modulated by an inducible promoter. Down-regulation of expression directed the carbon flux toward myo-inositol-1-phosphate synthase, which converts G6P to MI, thereby improving both the yield and titre of myo-inositol. Testing a similar strain for its ability to produce GA resulted in up to 42% improvement in yield.45
As with insulin, investigating other organisms, such as yeast, for the bio-production of organic molecules is important. Gupta et al.46 transferred the GA biosynthetic pathway to Saccharomyces cerevisiae because it tolerates low pH better. They found that deletion of the OPI1 gene (a negative regulator of phospholipid biosynthesis) significantly increased the yield of GA. The authors also discovered that the MIOX gene from Mus musculus produces an enzyme with a lower activity than the MIOX4 homolog from Arabidopsis thaliana. In the latter, they found an increase in the production of GA when the medium was enriched with myo-inositol. Thus the bottleneck is the availability of myo-inositol and not the low activity of MIOX, as is the case in Mus musculus. These findings are relevant only during the growth phase of the microbial biomass. During the production phase, myo-inositol accumulates, suggesting that the activity of MIOX could be further improved.
In the same manner as Gupta, Liu et al.47 investigated other organisms, analysed the native production pathway for GA in Pichia pastoris and discovered that native MIOX is present but no complete GA production pathway exists. The authors constructed a biosynthetic pathway using the mouse MIOX gene and the udh gene encoding uronate dehydrogenase (UDH) from Pseudomonas putida. They achieved a 9-fold increase in the yield of GA when the medium was enriched with myo-inositol. The lack of glucuronic acid enrichment suggests that MIOX activity is still the rate-limiting step. To further increase the production of GA, the authors fused the key enzymes MIOX and Udh end-to-end with protein linkers. This resulted in an almost 2-fold increase in the specific activities of MIOX, demonstrating the instability of MIOX as the reason for its low activity.
In 2018 Doong et al.48 considered previous research and developed a dynamic regulatory system to improve GA yield, focusing on the inherent instability of MIOX and its regulation. To delay the expression of MIOX, the repressor IpsA from Corynebacterium glutamicum was transduced into E. coli, which binds MI with high specificity. When its concentration increases above a certain threshold, the repressor dissociates from DNA and expresses MIOX (Fig. 2). This simple regulatory pathway increased the titre of GA by 2.5-fold. The authors also found that overexpression of IpsA can decrease the yield of GA. This biosensor MI was then combined with the previously reported regulation of PfK-I.44–46 They linked the downregulation of the latter to the build-up of N-acylhomoserine lactone (AHL). When intracellular AHL concentration increases, PfK-I expression is reduced, directing the carbon flux toward INO1. The combined use of all adjustable controls improved the total titre to nearly 2 g L−1.
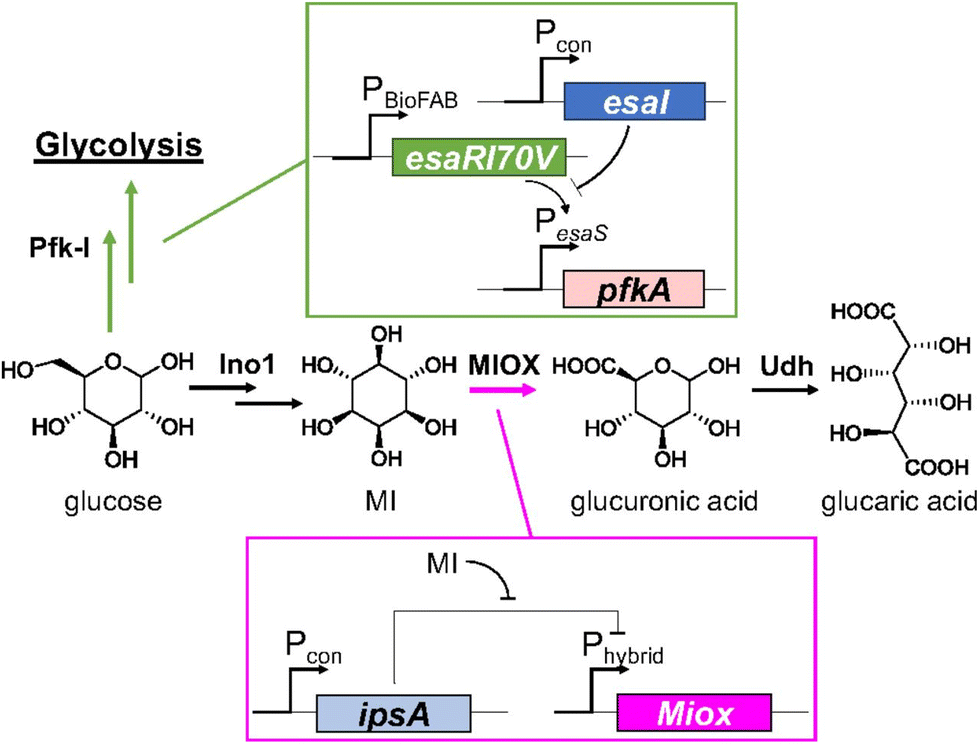 | ||
| Fig. 2 Schematic representation of layered dynamic regulation in the GA pathway. Reproduced with permission from ref. 48. Copyright 2018 Published under the PNAS license. | ||
Hou et al.49 combined the findings of Brockman and Doong and constructed a dynamic turn-off switch (dTFS) and turn-on switch (dTNS), which they used in the production of GA. They downregulated PfK-I during the production phase with the dTFS and upregulated myo-inositol-1-phosphate synthase with the dTNS. They achieved a slightly lower titre than Doong et al.48 but better productivity (0.0325 vs. 0.0275 g L−1 h−1). The authors believe that some of the problems identified in the past persist and reduce the final yield.
Since in most cases the instability of MIOX was cited as the issue for low productivity, Marques et al.50 studied more than 1500 MIOX variants and successfully expressed 35 productive variants in S. cerevisiae, demonstrating that similarity networks are a useful tool for identifying novel protein variants. Significant amounts of alcohol formed during the fermentation process reduce the overall yield of GA. A correlation was found between the length of the N-terminus and the GA titre, with the optimum being 40 amino acids. Other factors determining productivity include the fact that aspartate and lysine must occupy positions 88 and 257, respectively. In addition, mutations in the active site play an important role compared with M. musculus MIOX. The most efficient MIOX variants were from Flavobacterium johnsoniae and Talaromyces marneffei (1.85 and 1.76 g L−1, respectively). Marques et al. also noted the effects of MIOX stability on the GA yield. At the fermentation time of 10 days, a very high titre was obtained by Zhao et al.51
Su et al.52 followed a similar multistep approach as Marques to improve GA titres during fermentation with E. coli. Comparing the same MIOX variants as Gupta et al.,46 they obtained similar results (Fig. 3, GA1). High levels of active enzymes were achieved by including a T7 promoter in the plasmid used (Fig. 3, GA2). Removal of the lac repressor lacIq eliminated the need for the addition of IPTG (Fig. 3, GA3). The carbon flux was modulated by removing enzymes (uxaC and gudD) that catalyse further transformations of GA (Fig. 3, GA4 and 5). The titre was further improved by increasing the amount of available G6P by knocking out two genes (zwf and pgi) that either oxidize or isomerize it (Fig. 3, GA6–8). Because these modifications rendered the strain incapable of growing on medium with Glc as the sole carbon source, glycerol, L-arabinose and D-xylose were added. Ribosome-binding sites were optimized to achieve the optimal INO1/MIOX ratio of 1/4 (Fig. 3, GA9). All these changes decreased the intracellular NAD+/NADH ratio required for UDH to function. The nox gene from Lactobacillus pentosus should be ported to GA9 to improve cofactor regeneration. Finally, the agitation of the slurry was optimized to promote aerobic fermentation (Fig. 3, GA10).
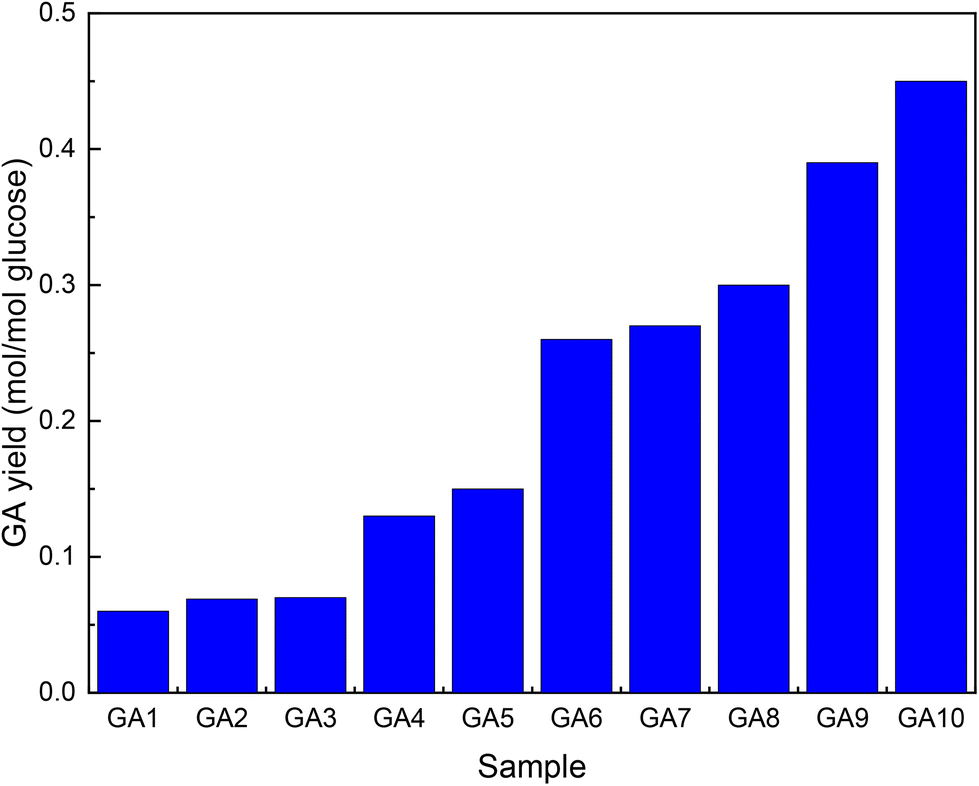 | ||
| Fig. 3 Increase in GA yield by metabolic engineering of E. coli. The graph was drawn from data reported by Su et al.52 GA1-unaltered enzyme, GA2-T7 promotor included in the plasmid, GA3-lacIq removed, GA4-uxaC additionally removed, GA5-gudD additionally removed, GA6–8-(zwf and pgi) additionally removed, GA9-ribosome binding sites optimized, and GA10-inclusion of the nox gene and improved agitation. | ||
Although fermentation studies with bacteria and fungi shows promising results, the space-time yield (STY) is rather low. Therefore, isolation of the enzymes present in the reaction pathway would enable a cell-free production of GA and also reduced the expensive upstream processing of the fermentation broth. Novel approaches include also the use of a different biomass source, xylan, the second most abundant biopolymer, which is an interesting GA source.53 In xylan, the ratio of 4-O-methyl D-glucaric acid ((4Me)-GA) to xylose can be as high as 1/3.54 To exploit this resource, Lee et al.55 synthesized so-called rosettasome enzyme assemblies that co-localized three different enzymes: xylanase, α-glucuronidase, and uronate dehydrogenase (UDH). They tested free and rosettasome-bound enzymes on branched Birchwood xylan, with the latter performing better than the former. The optimal ratio of the enzymes was 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1, proving that the localization effect of all three enzymes determines the activity. Complexation of the individual enzymes showed no improvement in productivity over the free enzyme.
:1:1, proving that the localization effect of all three enzymes determines the activity. Complexation of the individual enzymes showed no improvement in productivity over the free enzyme.
To further improve the extraction of GA from xylan, Li et al.56 investigated thermostable uronate dehydrogenase from Thermobispora bispora among several UDHs studied. It was most active at 60 °C and pH 7.0, but lost some activity after a His tag was added for purification purposes. Examination of the inhibition of the enzyme by a substrate showed that only glucuronic acid had some effect, whereas galacturonic acid did not. The inhibition could be avoided by continuous feed production. The use of Birchwood xylan as substrate and purified UDH in combination with xylanase and α-glucuronidase allowed a conversion rate of over 81%.
Recently, Vuong and Master57 succeeded in extracting 4-O-methyl-D-glucuronic acid from glucuronoxylan and converting it to (4Me)-GA in a yield of 62%. The remaining xylan was easily recovered and was enzymatically hydrolysable. The use of α-glucuronidase and gluco-oligosaccharide oxidase (GOOX) as catalysts showed no decrease in activity even at higher pH values, increased the solubility of the substrate, enabled the reactions with 6% glucuronoxylan, and prevented the formation of lactones from (4Me)-GA. Moreover, the α-glucuronidase used showed signs of inhibition by H2O2 (side product of oxidation by GOOX) in a sequential one-pot reaction, after pre-hydrolysis of glucuronoxylan for four hours.
Isolated enzyme systems were tested also on simpler substrates. Su et al.58 designed a seven-enzyme reaction cascade for the in vitro production of GA from sucrose, using sucrose phosphorylase (SP), phosphoglucomutase (PGM), myo-inositol 1-phosphate synthase, myo-inositol monophosphatase (IMPase), myo-inositol oxygenase (MIOX), uronate dehydrogenase (UDH), and NADH oxidase (NOX). As in many cases prior, MIOX was the rate-limiting enzyme due to its inherent instability. To rectify this, a fed-batch strategy of MIOX supplementation was employed. Since the increasing concentration of GA also lowers the pH, which has a detrimental effect on the catalyst activity, 10% NaOH was added after 36 h and 60 h of incubation. After 72 h, a 75% yield of GA was achieved.
Even though isolated enzyme systems reduce the need for product purification, there is still the issue of removing enzymes from the reaction mixture. To simplify this process, Petroll et al.59 immobilized the enzymes on zeolites using linkers and used glucose-1-phosphate (G1P) as substrate. Unfortunately, this had a detrimental effect on enzyme activity, which was minimal in some cases (36% for MIOX), and drastic in others (92% decrease for UDH). After immobilization, the activity decreased further, and for MIOX the activity decreased to 0%. When MIOX was added without immobilization, some conversion was observed. A two-pot reaction was required because the enzymes for the conversion of G1P to MI are thermostable (retain activity even at 85 °C), whereas NOX is not (maximum activity at 40 °C) (Fig. 4). It was also found that co-localization of the proteins gave better results than their separate immobilization. The optimized process using free enzymes resulted in an almost five-fold higher productivity than fermentation with whole cells. When the enzymes were immobilized, the increase in productivity was only about 2-fold, but the enzymes were stable enough to be recycled for multiple cycles.
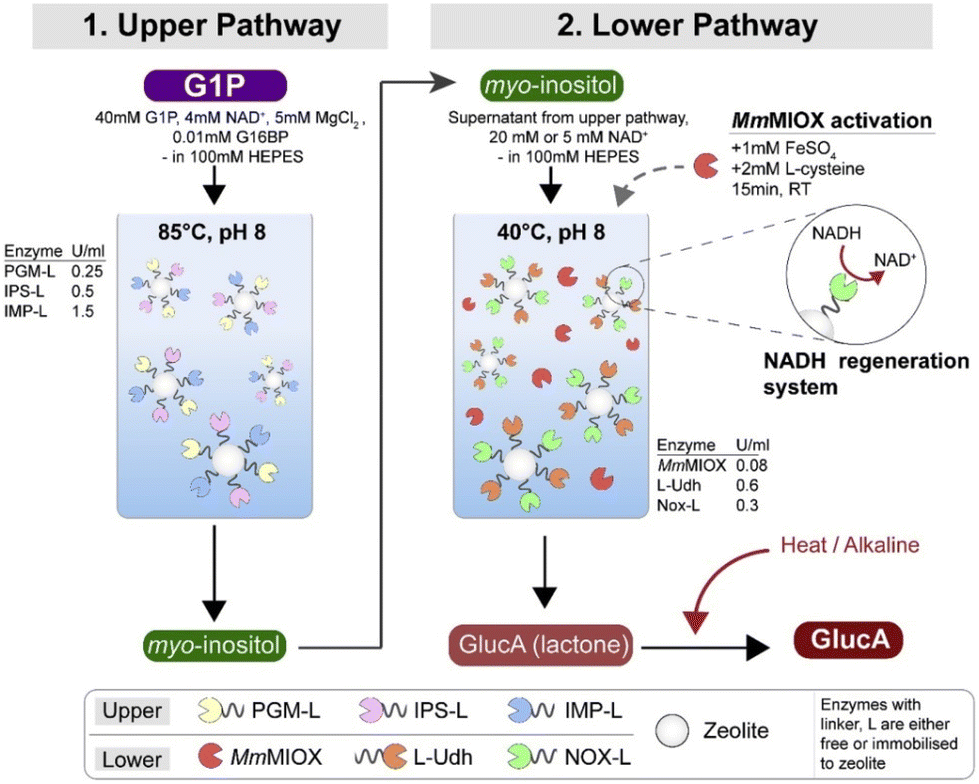 | ||
| Fig. 4 GA production through a two-batch reaction of the upper and lower pathways. Reproduced with permission from ref. 59. Copyright 2019, Elsevier. | ||
Table 1 summarizes the biotechnological experiments for the production of GA. The processes are not yet economically feasible due to low titres and yields. For example, comparing it with the production of citric acid, the minimum titre should be 29 g L−1 and productivity should reach 0.6 g L−1 h−1.60 An important thing to consider is the need for downstream processing. If industrial viability is the end goal, the potential removal of cells, co-factors, and other substrates must be considered. While several patents have been granted for the biotechnological production of GA,61–65 Kalion66 is the only company biotechnologically producing GA on a large scale.
| Ref. | Organism | Time [h] | T [°C] | Initial Glc [g L−1] | GA titre [g L−1] | STY [g (L h)−1] | Comment |
|---|---|---|---|---|---|---|---|
| a Myo-inositol. b MI titre. c Birchwood xylan substrate. d Glucuronic acid. e Sucrose. f Glucose 1,6-bisphosphate. | |||||||
| 40 | E. coli | 72 | 30 | 10 | 1.13 | 0.0157 | IPTG induction |
| 41 | E. coli | 48 | 30 | 10 | 2.50 | 0.0521 | IPTG and aTc induction |
| 42 | E. coli | 48 | 30 | 10 | 1.70 | 0.0354 | IPTG induction |
| 43 | E. coli | 72 | 30 | 10.8a | 4.85 | 0.0674 | IPTG induction |
| 44 | E. coli | 78 | 30 | 10 | 1.31b | 0.0168 | IPTG and aTc induction |
| 45 | E. coli | 72 | 30 | 10 | 1.56 | 0.0217 | IPTG and aTc induction |
| 46 | S. cerevisiae | 72 | 30 | 20 | 1.60 | 0.0222 | Supplemented with 60 mM myo-inositol |
| 47 | P. pastoris | 96 | 30 | 60 | 6.61 | 0.0689 | 13 g L−1 myo-inositol was added |
| 48 | E. coli | 72 | 30 | 10 | 1.98 | 0.0275 | IPTG induction |
| 49 | E. coli | 48 | 30 | 40 | 1.56 | 0.0325 | — |
| 50 | S. cerevisiae | 96 | 30 | 10a | 1.85 | 0.0193 | — |
| 51 | S. cerevisiae | 240 | 30 | 20 | 10.6 | 0.0442 | Supplemented with myo-inositol, 100 mM MgCl2, Glc was added at 24 and 48 h |
| 52 | E. coli | 72 | 30 | 10 | 5.35 | 0.0743 | — |
| 55 | Cell-free | 1.5 | 30 | 10c | 0.157 | 0.1047 | 50 nM of total enzyme; the product is (4Me)-glucaric acid |
| 56 | Cell-free | 1 | 50 | 0.194d | 0.179 | 0.1790 | 2 mM NAD+ was supplemented |
| 58 | Cell-free | 72 | 30 | 17.1e | 7.31 | 0.1016 | MIOX supplementation, pH modulation |
| 59 | Cell-free | 10 | 85/40 | 0.0034f | 1.70 | 0.17 | Two-step process |
A common denominator remains the instability of MIOX, which is addressed by using a modular scaffold,41 using a modifier such as SUMO,43 using protein linkers to fuse MIOX with UDH,47 developing a regulatory system to modulate MIOX synthesis,48,49 screening multiple variants from different organisms to find a more stable enzyme,46,47,50,51 or by site-directed mutagenesis.43,67,68 Other potential sources of GA or its derivatives are also worth investigating, such as xylan.55–57 Recently, cell-free enzyme systems have received increasing attention58,59 but deactivation of the enzyme by the substrate, cofactor, or product must be considered and mitigated. In some cases, cell-free systems may be more suitable for industrial applications because they can be immobilized and reused multiple times.59 To achieve industrial viability, multistep optimization must be performed, such as that of Su et al.52 with a 7.5-fold increase in GA production.
3. Heterogeneous catalysis
In heterogeneous catalysis, the reactants and the catalyst are in different phases, usually in liquid and solid form, respectively. Heterogeneous catalysis generally has a much less complex structure than enzymes and lower selectivity, but on the other hand allows the use of simple oxidants such as oxygen or even air. Competing reactions are not only possible but often non-negligible. For example, non-enzymatic Glc oxidation proceeds via several pathways and leads to different intermediates and products (Fig. 5), which usually prevents good selectivities at high conversions. Due to the simpler and cheaper precursors, heterogeneous catalysis remains popular.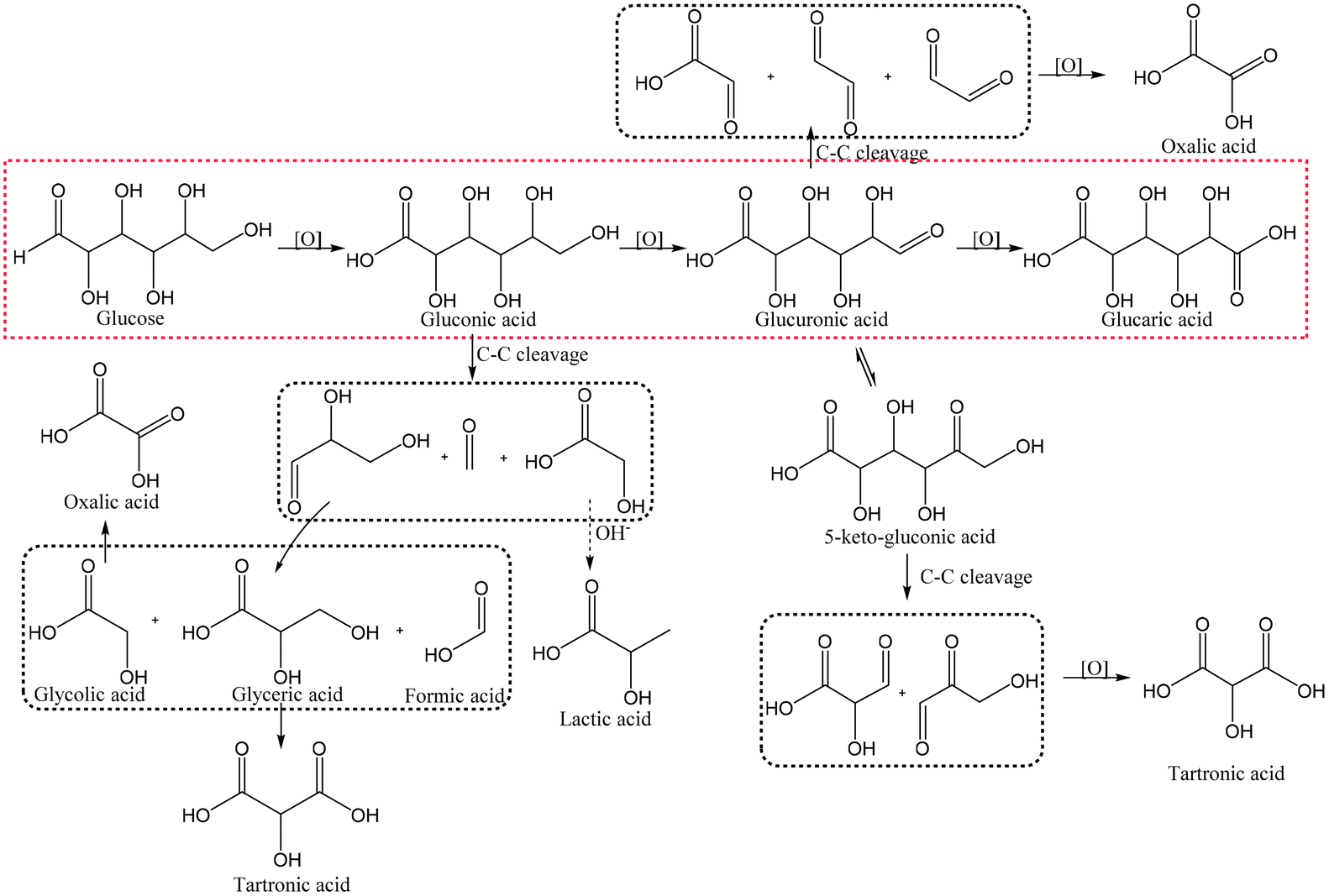 | ||
| Fig. 5 A schematic reaction pathway showing the possible products in Glc oxidation. The red dotted square represents the main pathway, whereas the black ones represent possible degradation products. | ||
The first patent describing the oxidation of Glc with air over platinum was filed as early as 1948 by Mehltretter,69 achieving the complete conversion of dextrose hydrate and a GA selectivity of 54%. While gluconic acid is readily produced de Wilt and van der Baan70 later found that the oxidation of Glc to ketogluconate is a detrimental reaction that reduces the overall yield of GA. In studying a catalyst with lower Pt loading (5 wt%), Dirkx et al.71 found that excessive amounts of oxygen reduced catalyst activity, which was attributed to the formation of strongly adsorbed oxygen (Pt–O) or the oxidation of Pt to PtO2.72 In both cases, Glc, gluconic acid, or nitrogen can reduce the amount of inhibitory species. While Glc oxidation is much faster than gluconic acid oxidation, both produce several by-products, including glycolic acid, tartaric acid, tartronic acid, oxalic acid, and others. The yield of GA was slightly higher (over 50%) when gluconic acid was oxidized.
Smits et al.73 focused on further improving the selectivity toward GA, by modifying the platinum catalyst used by Mehltretter with Pb, but 2-keto-D-gluconic acid was formed instead. In studying the deactivation of the Pt-based catalyst during Glc/gluconate oxidation, Dijkgraaf et al.74,75 confirmed that elevated O2 partial pressures and low gluconate concentrations accelerated the deactivation, while increasing the temperature slowed it down. It was found that sintering, irreversible adsorption of by-products, platinum oxidation, and the formation of Pt–O did not lead to deactivation. The authors developed a model of reversible but slow oxygen dissolution in the outer platinum layers.
Since Pt is immensely expensive, Besson et al.76 investigated how bismuth affects the PdBi/C catalyst in Glc oxidation. After a homogeneous deposition of bismuth on 1–2.5 nm Pd particles containing 4.7 wt% Pd (Bi/Pd atomic ratio of 0.1), the conversion increased from 82.6% to 99.6%. Unfortunately, the selectivity shifted from glucarate, which was already low (2.3%), to gluconate. The catalyst was stable after five cycles. Since the results on the Pd-based catalysts did not show any promise, the authors also77 modified Pt catalysts70,71 with either Bi or Au. Unfortunately, they observed no improvement in the yield of GA (97.2% gluconate conversion with 56.6% GA selectivity) although much more concentrated solutions (2 mol L−1) were used at lower gluconate-to-Pt ratios ([Glc]/[Pd] = 787). The addition of even minute amounts of Bi (Bi/Pt = 0.064) dramatically increased the selectivity for 2-ketogluconate, which was attributed to the formation of a complex between the Bi and the α-hydroxyl. Oxygen poisoning-induced deactivation is more pronounced for gluconate oxidation because Glc is a stronger reducing agent. The oxygen effect was further reduced by adding small amounts of Au to Pt, which decreased the oxophilicity (affinity for oxygen) of the surface.
Given that Pt-based materials exhibited good results, Rautiainen et al.78 investigated other noble metals. The authors prepared a Au/Al2O3 catalyst with 1.8 wt% Au and a dispersion of 43% (average Au particle size 2.4 nm) for the selective oxidation of uronic acids (glucuronic acid and galacturonic acid) to aldaric acid (glucaric and galactaric acid), which showed very high selectivity (>99%) to aldaric acids under optimal conditions. The conversion correlated with NaOH consumption, and an increase in pH led to a significant increase in the initial reaction rate, as did temperature. When Glc and glucuronic acid were used as precursors, TOFs of 10300 h−1 and 7920 h−1, respectively, were reported. However, reuse of the catalyst over several cycles resulted in coalescence and aggregation of the gold particles, leading to a decrease in activity.
Jin et al.,79 on the other hand, strived to improve the yield of Pt-based materials. They synthesized bimetallic PtCu/TiO2 catalysts for the oxidation of gluconate and found that the addition of Cu to Pt slightly decreased the size of the nanoparticles; the optimal Cu/Pt ratio was three. The use of sodium gluconate as a substrate caused the Cu-modified catalyst to lose some selectivity, but the conversion increased dramatically while the use of Glc increased both the selectivity for GA and the conversion. Overall, the addition of copper to Pt increased the yield of GA by more than 20-fold, but also decreased the selectivity due to the formation of tartronic acid and oxalic acid during the subsequent oxidation of GA. Recycling tests showed no decrease in catalyst activity and a small decrease in selectivity after three runs.
In heterogeneous catalysis, the support is as important as the active metal. The metal–support interactions can alter the selectivity/activity of the catalysts. The catalytic reaction can occur on the interface between the metal and the support. The latter can facilitate substrate adsorption, or product desorption. For this reason, Lee et al.80 investigated several supports for the Pt catalyst and were able to find a catalyst that had excellent yield in the oxidation of Glc to GA. Compared with Pt/SiO2 and Pt/Al2O3, Pt/C exhibited a 10-fold larger surface area and the highest activity, while its pore volume was 35–48% smaller. Evaluating different pH values, it was found that the oxidation of Glc in water at neutral pH leads to the best GA selectivity. Basic or acidic conditions increase the conversion but decrease the GA selectivity. The oxidation of Glc is also affected by the reaction temperature, O2 pressure, and Glc/Pt molar ratio. When the temperature range of 70 to 90 °C was studied, it was found that lower temperature favoured higher selectivity towards GA. The effect of the Glc/Pt ratio was insignificant, while an increase in the O2 partial pressure improved the conversion and selectivity, consistent with observations made by Dirkx et al.72 Under optimal conditions,80 yields of 74% were achieved. Furthermore, no significant deactivation of the catalyst was observed after five runs.
Jin et al.81 opted for a two-step approach, where they used a novel support and a bimetallic catalyst. They synthesized a TiO2-supported bimetallic PtPd catalyst in a one-pot reaction for the direct one-step oxidation of Glc to GA. The support was loaded with Pt and Pd metal contents in the range of 0.88–1.12 and 0.94–1.12 wt%, respectively. The average size of nanoparticles in the bimetallic Pt–Pd catalyst is 1.8–2.0-times larger than in monometallic Pt catalysts. Depending on the synthesis method, PtPd nanoparticles with alloy, core–shell, and cluster-in-cluster structures were formed on rutile or anatase TiO2 supports. Alloying the metals instead of core–shell or cluster-in-cluster formations maximized the selectivity to over 30%, while monometallic Pd/TiO2 had none and Pt/TiO2 had less than 10%. Reuse of the catalyst for three cycles showed no change in conversion or selectivity. Interestingly, the use of fructose as substrate impeded inhibition (no aldehyde group) but increased C–C cleavage (Fig. 6).
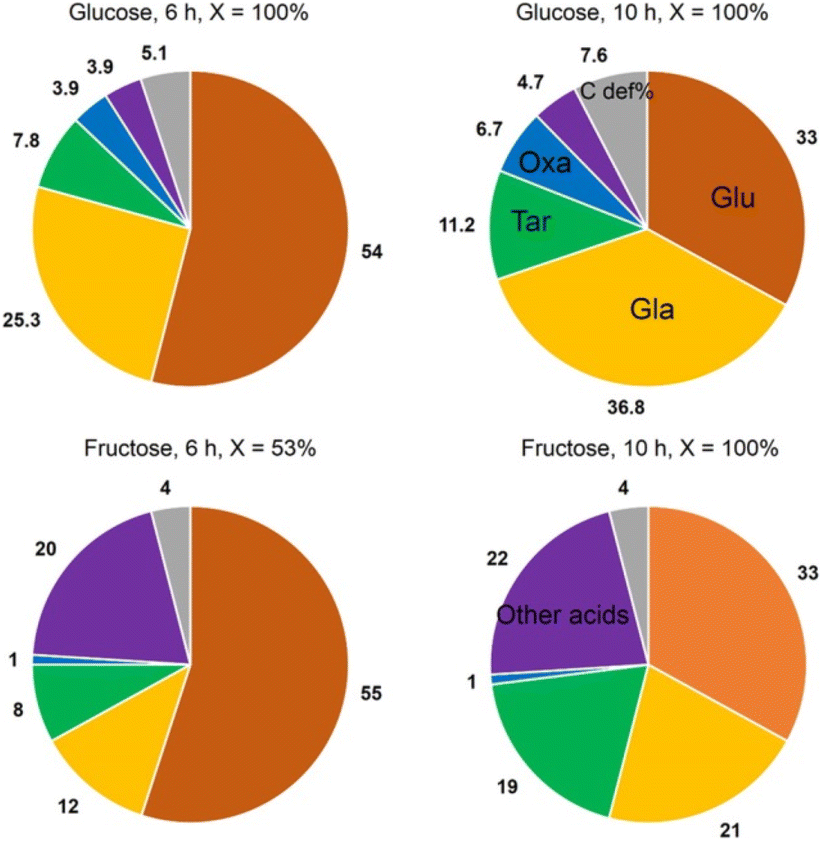 | ||
| Fig. 6 Graphic representation of the products formed in Glc and fructose oxidation. Glu, gluconic acid; GA, glucaric acid; Tar, tartronic acid; Oxa, oxalic acid. Other acids: glyceric, glycolic, lactic, and formic acid. Reproduced with permission from ref. 81. Copyright 2016, American Chemical Society. | ||
In studying the catalytic oxidation of gluconate or Glc to GA in an alkaline aqueous solution with a range of supported Pt and Au catalysts, Derrien et al.82 showed that Pt-based catalysts are superior to Au-based catalysts in terms of activity, but at the expense of lower selectivity. At an optimum pH of 9, bimetallic catalysts (AuPd) outperformed pure gold and trimetallic catalysts (AuPdBi). As other authors had noted, oxidation of Glc over Pt/C (3 wt% Pt) is very rapid with almost all Glc converted in the first hour without isomerization. After 24 hours, degradation products were detected in appreciable amounts (over 15% for tartrate). The maximum glucarate yield (54%) was reached after 10 hours. The use of bimetallic AuPd catalysts resulted in a lower maximum glucarate yield (37%) after 6 hours and greater further degradation after 24 hours (yield below 10%). An investigation of the effects of impurities (residues or by-products) from upstream hydrolysis on the reaction showed that furanaldehydes had little effect on the reaction rate, while phenol derivatives could significantly affect the reaction rate. For the Au-based catalysts,83 modified with either Pt or Pd, the Pd adatom results showed an increased rate of Glc oxidation but low GA yields. Among the different supports tested, Au–Pt on ZrO2 with 8 nm nanoparticles and some large agglomerates achieved a GA yield of 50%. Increasing the amount of catalyst by four times did not change the maximum yield of GA (56% compared with 50%), but shortened the required time by more than three times. The best yields of GA, which reached 50%, were obtained at 100 °C and a Au/Pt ratio of 1/1. After 6 cycles of Glc oxidation, the loss of efficiency was minimal, and long-term tests (200 hours TOS) confirmed the stability of the catalyst. As mentioned above,82 furan derivatives and phenolic fragments had a detrimental effect on reaction rates. This approach was patented by37 Rhodia Operations (a subsidiary of Solvay S.A.).
Similarly, to Derrien, Solmi et al.84 also investigated pure Pt/C catalysts. They achieved a somewhat smaller yield. They compared this benchmark material to a custom-synthesized carbon-supported bimetallic AuBi catalyst (metallic Au and Bi3+). The authors investigated the influence of particle distribution, Glc concentration, and Glc/metal/NaOH molar ratio. While GA does not form at higher Glc concentrations (10 to 20 wt%), lower concentrations (2.5 to 5 wt%) exhibit selectivity above 20%. Interestingly, in the absence of NaOH, GA does not form and the conversion of Glc is less than 20%. The optimum molar ratio of Glc/NaOH = 1/3 and Glc/metal = 500/1 leads to a conversion of 100% of Glc and GA selectivity of 30% even after 24 hours at 60 °C and 10 barg oxygen. Similar yields were observed with the commercial Pt/C catalyst under slightly different conditions (Fig. 7) except for the carbon balance, which is better when AuBi is used. Catalyst recycling showed a 50% decrease in selectivity of GA and a 20% decrease in carbon balance after 5 cycles, which was not caused by leaching but by organic matter deposition, nanoparticle agglomeration and sintering.
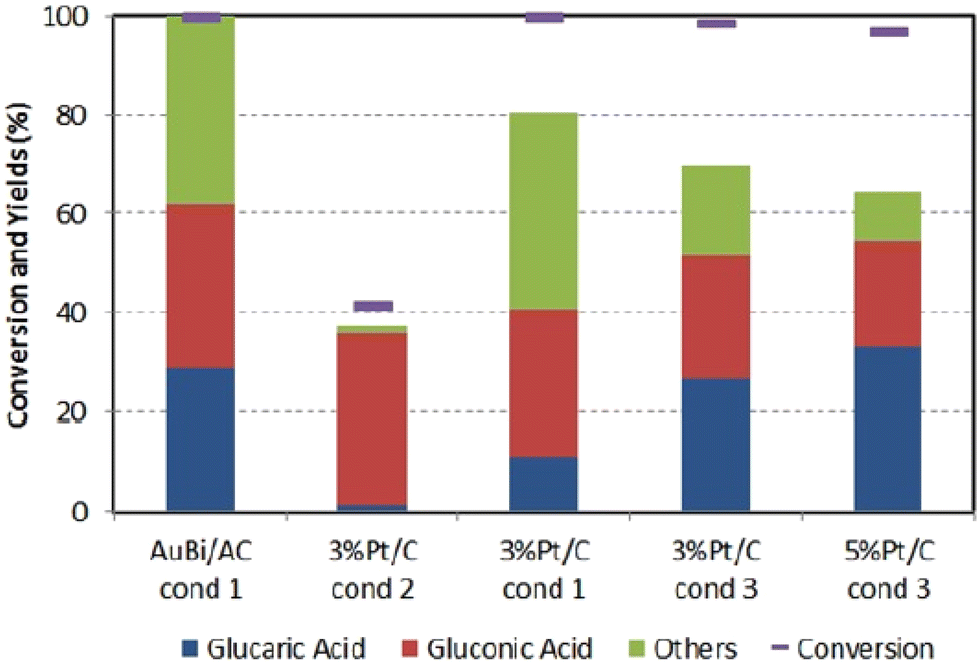 | ||
| Fig. 7 The figure represents a comparison of the synthesized catalyst AuBi/AC with two different commercial catalysts under various conditions. Condition 1: 5 wt% Glc in aqueous solution (15 mL water), Glc:metal:NaOH = 4.4:0.0088:13.2 mmol, T = 60 °C, O2 pressure 10 bar, reaction time 3 h. Condition 2: all parameters remain the same, except no base was added. Condition 3: 10 wt% Glc in aqueous solution (20 mL water), molar ratio Glc:metal:NaOH = 54:1:0, T = 80 °C, O2 pressure 13.8 bar, reaction time 3 h. Reproduced with permission from ref. 84. Copyright 2017 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim. | ||
In studying Glc oxidation, Shi et al.85 performed high-throughput screening, and tested several formulations of Pt-based mono- and bimetallic catalysts to determine the effects of support, synthesis method, secondary metals, and reducing agents. The use of TiO2 as the support and the application of the wet chemistry method yielded the best performing catalyst, which achieved a Glc conversion of 71% with a selectivity of GA over 19%, which was attributed to the strong interaction of the metal with the support. The optimised Pt–Cu catalyst had an alloyed structure and an average Pt–Cu particle size of 2.8 nm. The use of mono (Pt-) or bimetallic (Pt–Cu) catalysts showed no difference in the conversion, but the latter improved the selectivity. Interestingly, changing the Pt/Cu atomic ratio from 1/1 to 1/3 did not affect the yields of GA, but using a strong reducing agent (NaBH4) during the synthesis improved the activity. Performing the reaction in the temperature range between 80 and 105 °C showed an increase in Glc conversion with temperature, while the best GA selectivity was obtained at 90 °C. Higher temperatures or higher O2 partial pressures favoured C–C bond cleavage and decreased selectivity.
With the promising results reported by Derrien, Khawaji et al.86 prepared bimetallic AuxPty/Ti-NT catalysts by sol-immobilisation on Ti-NT with different atomic compositions. The average particle sizes ranged from 2.5 to 4 nm for monometallic Au/Ti-NT and 3.5 to 6.5 nm for the bimetallic catalysts. The best selectivity towards GA for the monometallic Au/T-NT and bimetallic Au56Pd44/Ti-NT catalysts was 18.5% and 13.8%, respectively. The selectivities reported were much lower than those observed by Derrien and at least 15 wt% of gold was required for higher conversion. Further increasing the Au content had a negligible effect on the activity but improved the selectivity. Contrary to the observations made by Derrien, the yield of GA correlated linearly with the Au content, as catalysts with low Au loading exhibited high selectivity (98.1%) for gluconic acid. However, the catalyst was not stable and deactivation due to metal leaching as a result of a lack of pH control was observed after six runs. A similar catalyst supported on Ce nanorods87 exhibited negligible selectivity for GA (below 2%).
To reduce the cost associated with the catalyst, Tsang et al.88 tested a series of low-cost Cu catalysts on a biochar support for base-free oxidation of glucose to glucuronic and gluconic acids under microwave irradiation. The maximum yields of 39.0% and 30.7% were obtained in 20 min reaction time at 160 °C. The results indicate the possibility of a two-step process in which Glc can first be oxidized to gluconic or glucuronic acid with a less expensive catalyst and then the products can be oxidized to GA with another catalyst that has higher selectivity for GA.
Recently, Deng et al.89 studied the entire pathway of the conversion of Glc to adipic acid, where GA is an intermediate. For this purpose, they first oxidized Glc over Pt/C catalysts with carbon in the form of commercial carbon nanotubes (CNTs). According to the known temporal evolution of the products, Glc is completely converted within 1 hour and the main product is gluconic acid. The optimal GA yield is achieved after 4 hours, while longer reaction times lead to further GA degradation. Temperature also plays an important role; below 60 °C gluconic acid predominates, above which cleavage of the C–C bond occurs. Interestingly, 2.5 nm Pt particles yield the most GA, while the yield decreases abruptly for particles larger than 3.5 nm. Increasing the initial pH to 12.9 also increases the yield of GA considerably (82%), as an alkaline medium inhibits the adsorption of acidic products on the CNT surface. Moreover, the degradation of GA is much slower due to the stabilization of GA in the glucarate form. A similar observation was made by Liu et al.90 who found that gluconic acid is also the main product for other dopants (Co and Mn).
To summarize, heterogeneous Glc oxidation generally occurs on Pt- and Au-based catalysts. Most research has focused on oxidation in a basic medium (NaOH or KOH). This is likely due to the accumulation of the negative charge on the surface of the catalysts, which facilitates desorption of the acidic products. In addition, this probably stabilizes GA in the salt form. If the end goal is the adoption of the developed process by the industry, base-free oxidation should be preferred. Formation of the salt adds additional, and potentially costly, steps to downstream processes such as adipic acid synthesis. The scientific community agrees that the oxidation of Glc proceeds much faster than the subsequent oxidation of gluconic acid to GA, which is the rate-limiting step.72,82 In most cases, bimetallic catalysts lead to much higher Glc yields than their monometallic counterparts, and the best selectivity is achieved when Pt and Au are present,76,83,86 although commendable results have also been obtained with Cu,79,85 Pd,81,82 or Bi,84 as shown in Table 2. The latter can be problematic, however, since the combination of Pd76 reduces the yield of GA. On the other hand, the combination of Bi with Pt77 may shift the selectivity towards 2-ketogluconate. Achieving a higher selectivity is essential as separation of the side-products, all acids, is not trivial. As with the formation of salts, this can add significant costs to down-stream processing. A higher temperature is required for Glc oxidation to proceed, but this also leads to overoxidation.85,89 A similar trend was observed with increasing oxygen pressure, with the glucarate yield following a Gaussian-like distribution as a function of pO2. The studies performed by Teržan et al. have shown that the main problem in the oxidation of Glc to GA seems to be oxygen activation (unpublished data), which is slow for catalysts favouring the formation of GA and too fast for catalysts exhibiting overoxidation.
| Ref. | Catalyst | Pressure/flow | Base | T [°C] | Time [h] | m catalyst [g] | Metal [wt%] | m substrate [g] | Glc conversion [%] | GA selectivity [%] | GA yield [%] | STY [gGA L−1 h−1] |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a O2. b Air. c Nominal value. d Total selectivity of organic acids. ♠Glucose; ♣gluconate; ◆glucuronic acid. | ||||||||||||
| 78 | Au/Al2O3 | 100 mL min−1a |
NaOH | 60 | 0.38 | 0.025 | 2 | 0.5 ♠ | 100 | 100 | 100.00 | 0.12 |
| 78 | Au/Al2O3 | 100 mL min−1a |
NaOH | 60 | 0.38 | 0.025 | 2 | 0.46 ◆ | 100 | 100d | 100d | 0.13 |
| 79 | Pt/TiO2 | 1 bara | NaOH | 60 | 6 | 0.1 | 0.52 | 3 ♣ | 70.8 | 38.1 | 26.97 | 5.19 |
| 79 | Cu/TiO2 | 1 bara | NaOH | 60 | 6 | 0.1 | 2c | 3 ♣ | 2.8 | 31.2 | 0.87 | 0.17 |
| 79 | PtCu/TiO2 | 1 bara | NaOH | 60 | 4 | 0.1 | 0.48–0.51 | 3 ♣ | 100 | 32.3 | 32.30 | 9.32 |
| 79 | PtCu/TiO2 | 1 bara | NaOH | 45 | 6 | 0.1 | 0.48–0.51 | 2.62 ♠ | 100 | 9.3 | 9.30 | 0.56 |
| 80 | Pt/C | 13.8 bara | — | 80 | 10 | 0.8 | 5 | 2 ♠ | 99 | 74 | 73.26 | 7.33 |
| 80 | Pt/SiO2 | 6.2 bara | — | 80 | 5 | 0.54 | 5 | 2 ♠ | N/A | N/A | N/A | 2.76 |
| 81 | Pd/TiO2 | 60 mL min−1a |
NaOH | 45 | 12 | 0.1 | 1.12 | 2.52 ♠ | 30 | 0 | 0.00 | 0.00 |
| 81 | Pt/TiO2 | 60 mL min−1a |
NaOH | 45 | 12 | 0.1 | 0.89 | 2.52 ♠ | 56 | 3.4 | 1.90 | 0.08 |
| 81 | PtPd/TiO2 | 60 mL min−1a |
NaOH | 45 | 10 | 0.1 | 0.94–0.95 | 2.52 ♠ | 100 | 31.1 | 31.10 | 1.57 |
| 81 and 82 | PtPd/TiO2 | 60 mL min−1a |
NaOH | 45 | 6 | 0.1 | 0.94–0.95 | 3.05 ♣ | 78.5 | 37.8 | 29.67 | 3.02 |
| 82 | Au–Pd/C | 0.5 L min−1b |
NaOH | 60 | 6 | 3.879 | 0.9–0.3 | 8.11 ♠ | 100 | 37 | 37.00 | 1.67 |
| 82 | Pt/C | 0.5 L min−1b |
NaOH | 60 | 8 | 3.106 | 3 | 13.51 ♠ | 100 | 54 | 54.00 | 3.04 |
| 83 | Au–Pt/ZrO2 | 50 barb |
— | 100 | 4 | 0.881 | 3.5–3.45 | 4.5 ♠ | 100 | 50 | 50.00 | 5.63 |
| 84 | Au/AC | 10 bara |
NaOH | 60 | 3 | 0.164 | 1 | 0.75 ♠ | 100 | 26 | 26.00 | 4.33 |
| 84 | AuBi/AC | 10 bara |
NaOH | 60 | 3 | 0.168 | 0.7–0.2 | 0.75 ♠ | 100 | 29.2 | 29.20 | 4.87 |
| 84 | Bi/AC | 10 bara |
NaOH | 60 | 3 | 0.174 | 1 | 0.75 ♠ | 79.17 | 2.1 | 1.66 | 0.28 |
| 85 | Pt–Cu/TiO2 | 15 bara |
NaOH | 90 | 12 | 0.4 | 5.08–3.30 | 1 ♠ | 92 | 60 | 55.20 | 2.30 |
| 86 | Pd/Ti-NT | 5 bara |
NaOH | 80 | 6 | 0.459 | 0.54 | 1.13 ♠ | 29.6 | 2.3 | 0.68 | 0.13 |
| 86 | Au/Ti-NT | 5 bara |
NaOH | 80 | 6 | 0.765 | 1.61 | 1.13 ♠ | 73 | 18.5 | 13.51 | 2.53 |
| 86 | Au56Pd44/Ti-NT | 5 bara |
NaOH | 80 | 6 | 0.542 | 1.27–0.54 | 1.13 ♠ | 59.7 | 13.8 | 8.24 | 1.55 |
| 86 | Au56Pd44/Ti-NT | 5 bara |
— | 80 | 6 | 0.542 | 1.27–0.54 | 1.13 ♠ | 74.4 | 6.1 | 4.54 | 0.85 |
| 89 | Pt/C | 10 bara | KOH | 60 | 4 | 0.1 | 5 | 0.0194 ♠ | 100 | 82 | 82.00 | 0.02 |
In general, promising results have been obtained in the heterogeneous oxidation of Glc to GA using Pt and Au catalysts, but a process with high selectivity and acceptable yields remains elusive. For the process to become truly industrially applicable, both conversion and selectivity should be improved.
4. Photocatalysis
Photocatalysis harnesses the effects of incident electromagnetic radiation from the visible or UV region of the spectrum. Photons with energies of a few eV are energetic enough to excite valence electrons and form electron–hole pairs. If the recombination rate is slow enough, they can diffuse to the surface and catalyse chemical reactions. In photocatalysis, the effect of the excited electrons goes far beyond simple energetic activation – i.e., the role of excited electrons is not just to provide the necessary activation energy that would otherwise be given off as heat energy but to change the reaction mechanism. Reactions occurring on the excited potential energy surface can follow other mechanisms and have lower activation barriers. TiO2 was originally used for water splitting, but it has been consistently touted as the photocatalyst of choice for a variety of chemical transformations because of its abundance, inexpensiveness, nontoxicity and performance. However, unmodified TiO2 is active in the UV region, so modifications are required to reduce its 3.2 eV band gap if it is to be used with visible light.More than a decade ago, Colmenares et al.91 investigated TiO2 for its ability to photocatalytically generate GA. Ultrasound-assisted synthesis, producing small crystallites with a large surface area and pure anatase with high crystallinity, was used to reduce the band gap to utilize visible light. The catalyst was compared with a similar catalyst prepared by a simple solvent reflux method and with the commercially available P25 from Degussa. Selectivity was highest at short irradiation times (15 minutes or less). The main products were gluconic acid, GA and some arabitol. The solvent used for the reaction was a mixture of acetonitrile (ACN) and water, the ratio of which significantly affected the conversion and selectivity. For all three catalysts, the conversion was highest in pure water, with zero selectivity toward organic acids/alcohols (only CO2 and H2O were formed). At a ratio of 1 (ACN/water = 50/50, v/v), the catalyst synthesized with ultrasound had the lowest conversion (11%) but the highest selectivity toward organics (71.2% glucaric + gluconic acid and arabitol). P25 and the catalyst prepared by the solvent reflux method exhibited the best selectivity (up to 32%) and the lowest conversion (up to 60.4%). The conditions used were a solvent ratio of 9 (ACN/water = 90/10, v/v), a Glc concentration of 2.8 mM in 150 mL of solvent, a catalyst concentration of 1 g L−1, and an irradiation time of 10 min (medium-pressure mercury lamp at 125 W).
This trend is probably due to an interplay between the zero-point charge of the catalyst and the stabilization effect of ACN. Supporting the active TiO2 phase on zeolites and fumed silica92 decreased the crystallite size (measured by XRD), to less than 8 nm or below the detection limit, respectively. The zeolite support exhibited slightly better conversion and selectivity (15.5% and 68.1%) than silica (14% and 64.8%). Supporting the active phase also shifted the selectivity as more gluconic acid but no arabitol was detected. Further doping of the zeolite-supported TiO2 with Crx+ decreased the activity of the catalyst but significantly increased the selectivity.93 Using a higher ACN/water ratio (ACN/water = 90/10, v/v) the cumulative selectivity for both acids reached 99.7% at 8% Glc conversion. Surprisingly, increasing the illumination time to 20 min resulted in a slightly higher conversion (10%) with a drastic decrease in cumulative selectivity (71%). Increasing the water content (ACN/water = 50/50, v/v) resulted in a further decrease in conversion (after 20 min) to 7%, but increased the selectivity to 87%. As before, the authors noted a shift towards gluconic acid when the water content was higher.
The promising results observed over TiO2 catalysts warranted further investigation of the support. To that end, Bellardita et al.94 used different types of TiO2 containing one or two polymorphs with different degrees of crystallinity and loaded them with Pt for photocatalytic Glc oxidation and hydrogen production (Fig. 8). The tests were performed under aerobic or anaerobic conditions with an initial Glc concentration of 1 mM and, depending on the sample used, a catalyst concentration of 0.3 to 0.8 g L−1, at room temperature and atmospheric pressure with a 125 W mercury lamp at medium pressure. Traces of GA were detected only in the photooxidation of Glc with pure commercial TiO2 from Merck and BDH under aerobic conditions. Both are pure anatase with a small surface area (about 10 m2 g−1) and moderate crystallinity. None of the platinum-modified catalysts produced GA. The Pt-BDH catalyst converted 100% (after 4.5 hours) of the Glc, but the main products were fructose (isomerization) and gluconic acid. Under anaerobic conditions, none of the synthesized catalysts produced any GA. In addition, they functionalized TiO2 with a commercial Keggin heteropolyacid H3PW12O40 (PW12) or its potassium salt K7PW11O39 (PW11).95 Under the same aerobic conditions, the highest GA selectivity was 1.5% with the PW12-modified commercial P25 (0.3 g L−1 catalyst concentration). The major products were fructose (55% selectivity) and gluconic acid (34% selectivity).
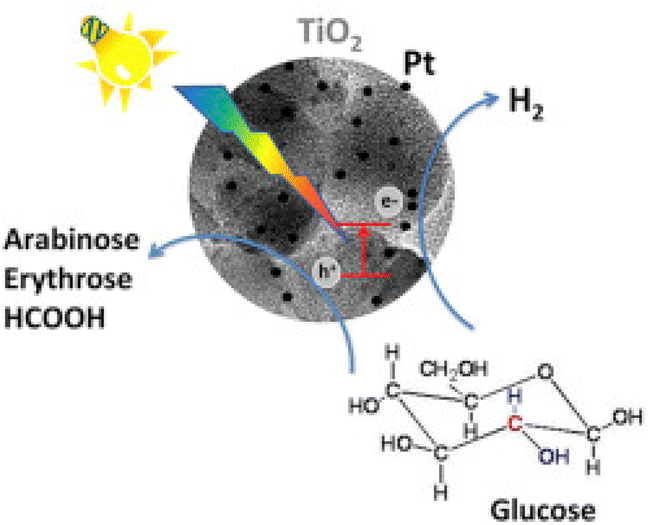 | ||
| Fig. 8 Graphical illustration of the photoconversion of Glc in the presence of Pt-loaded TiO2 catalysts. Reproduced with permission from ref. 94. Copyright 2016, Elsevier. | ||
A common photoactive substitute for TiO2 is ZnO with a comparable band gap of 3.37 eV. Cheng et al.96 therefore modified ZnO with cobalt thioporphyrazine (CoPz(SBu)8). Tests were performed under aerobic and anaerobic conditions with a Glc concentration of 8 mM and a catalyst concentration of 0.4 g L−1, while air or nitrogen was injected at 20 mL min−1. A xenon lamp was used to simulate sunlight (light intensity 1.5 W cm−2). While the performance of the catalysts was negligible under N2 and without illumination, the CoPzS8 modification improved the conversion more than fourfold (∼78%) compared with pure ZnO. The optimum loading of CoPzS8 was 0.5 wt%, with lower (0.25%) and higher (1%) loading reducing the conversion. Selectivity to arabinose and formic acid also decreased significantly (by more than 50%), while more gluconic acid was formed (∼10-fold increase). However, after 5 hours of reaction time, only traces of GA were formed. Reuse of the catalyst for three cycles decreased the conversion by 20%, but the selectivity for gluconic acid and GA increased slightly. Using different radical scavengers, O2˙−, ˙OH, 1O2 and h+ were identified as the major reactive species.
Also substituting the TiO2 support, Zhang et al.97 modified SnO2 with iron thioporphyrazine (FePz(SBu)8) in a manner similar to Cheng et al.96 The band-gap is somewhat larger than TiO2 at 3.6 eV, but the authors assumed the modification can shift it to the visible light range. They used slightly different conditions than Cheng, where the Glc concentration was reduced to 1 mM and the air flow was increased to 400 mL min−1, while the catalyst concentration remained the same (0.4 g L−1). At the slightly higher light intensity (2 W cm−2), the optimum loading of FePz(SBu)8 was 0.1%, which gave the GA a selectivity of 12.9% with a Glc conversion of 34.2% after 3 h. The main product was still gluconic acid (32.9% selectivity). At high Glc concentration, the conversion and GA selectivity decreased, while the addition of base increased the conversion but prevented the formation of GA. Radical scavengers showed that h+ is the dominant active species in this photocatalytic process, while for the production of GA, h+, O2˙− and 1O2 appear to be the required active species. Reuse of the catalyst for 4 cycles reduced the Glc conversion by more than 15% and GA selectivity by more than 40%.
Also, from Deng's group, like Cheng and Zhang, Chen et al.98 used a thioporphyrazine. The authors used iron thioporphyrazine (FePz(SBu)8) on a zeolite, HZSM-5, and studied the photooxidation of Glc by H2O2 under a Xe lamp (1.7 W cm−2) with a UV cut-off filter (420 nm) with a catalyst concentration of 1 g L−1 and a Glc/H2O2 ratio of 1/3.3 at a Glc concentration of 3 mM. The highest selectivity for both acids (gluconic acid and GA) was achieved at an FePz(SBu)8 loading of 1 wt% at the expense of lower conversion (below 7%). The optimum loading in terms of yield was determined to be 5 wt%, with a GA selectivity of 13.1% at a Glc conversion of 35.8%, although the major product was still gluconic acid (31.9%). Increasing the illumination wavelength to 500 nm further decreased the conversion (14.3%) and GA selectivity (11.1%). Further increasing the wavelength (650 nm) decreased the conversion even more (7.9%) but increased the GA selectivity (21.9%). Radical scavengers showed that O2˙− and ˙OH are the dominant oxidation species. It is believed that the radicals can be stabilized in the pores of the zeolite, which improves their longevity and, consequently, their oxidation capacity. Reuse of the catalyst for 5 cycles reduced the Glc conversion to 26% and the GA selectivity to less than 8%.
While Deng's group was observing interesting results using a single modifier, they wanted to see if a second modifier with induce a significant change in selectivity/activity. Yin et al.99 used the original TiO2 support, and modified it with phosphotungstic acid (HPW)/CoPz (cobalt tetra(2-hydroxymethyl-1,4-dithiin)porphyrazine) modifiers. They tested the composites for Glc oxidation with air (without aeration) under a Xe lamp (1.7 W cm−2) at a catalyst concentration of 1 g L−1 and a Glc concentration of 5 mM. Under optimal conditions, the selectivity to GA was 16.9% and the Glc conversion was 22.2%. The addition of 29% HPW significantly increased the selectivity, while the addition of CoPz also increased the conversion. Selectivity for GA showed a local maximum at a Glc concentration of 5 mM. Using radical scavengers, the authors concluded that 1O2 was a crucial radical species in this reaction. After five cycles, the catalyst retained high Glc conversion (over 16%) with moderate GA selectivity (about 16%).
In recent years another promising photocatalytic support has emerged, g-C3N4. With a somewhat smaller band-gap than TiO2 (under 3 eV), it can harvest a part of visible light. To that end, Zhang et al.100 successfully prepared a hybrid material of g-C3N4 and CoPz (cobalt tetra(2-hydroxymethyl-1,4-dithiin)porphyrazine), in which a thin layer of CoPz was deposited on the surface of g-C3N4 nanosheets, improving the visible light response and the photocatalytic efficiency (Fig. 9). With H2O2 and a Xe lamp (2 W cm−2), a catalyst concentration of 0.67 g L−1 and a Glc concentration of 1 mM, Glc was successfully oxidised. Without the catalyst, a Glc conversion of 91.3% was achieved, but with poor selectivity toward GA (about 10%). The use of the catalyst improved the GA selectivity to 20%, but at the expense of lower conversion (52.1%). Increasing CoPz improved conversion but decreased GA selectivity; the optimal level is 0.5 wt%. In the absence of the catalyst, O2˙−, ˙OH, and 1O2 dominated the oxidation, while the catalysed reaction was controlled by O2˙− and 1O2. Recycling tests confirmed that the catalyst did not lose significant activity (44.6% conversion) or selectivity (18.6% GA selectivity) after five cycles. Testing of a CoPz-modified SnO2 engineered to have more oxygen vacancies101 gave similar results (Table 3).
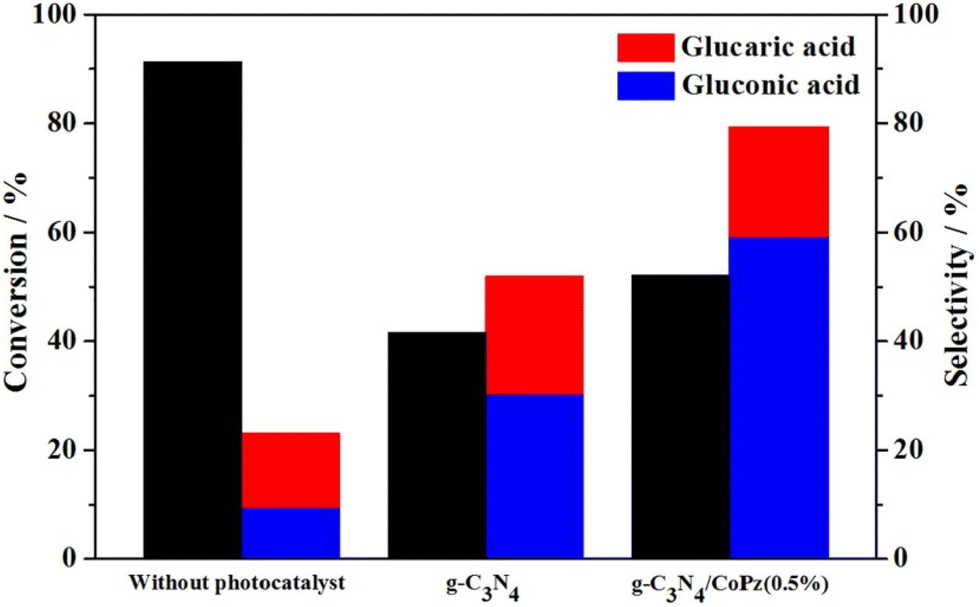 | ||
| Fig. 9 Glc photocatalytic oxidation under three different conditions: without using a catalyst, in the presence of g-C3N4, and in the presence of a composite g-C3N4/CoPz(0.5%). Reproduced with permission from ref. 100. Copyright 2020, Elsevier. | ||
| Ref. | Catalyst | T [°C] | Irradiation time [h] | m catalyst [g] | m Glc [g] | Glc conversion [%] | GA selectivity [%] | GA yield [%] | STY [gGA L−1 h−1] |
|---|---|---|---|---|---|---|---|---|---|
| a Total selectivity for organic compounds; N/A not applicable. | |||||||||
| 91 | TiO2 | 30 | 0.17 | 0.15 | 0.0757 | 11.0 | 71.3a | N/A | 0.2369 |
| 92 | TiO2/SiO2 | 30 | 0.17 | 0.15 | 0.0757 | 14.0 | 64.8a | N/A | 0.2740 |
| 92 | TiO2/zeolite | 30 | 0.17 | 0.15 | 0.0757 | 15.5 | 68.1a | N/A | 0.3188 |
| 93 | Cr-TiO2/zeolite | 30 | 0.33 | 0.15 | 0.0757 | 7.0 | 87a | N/A | 0.0931 |
| 93 | Cr-TiO2/zeolite | 30 | 0.33 | 0.15 | 0.0757 | 10.0 | 71a | N/A | 0.1085 |
| 94 | Pt-TiO2 | 25 | 7.00 | N/A | N/A | 100 | 0 | 0 | N/A |
| 95 | PW12-P25 solv | 25 | 6.00 | N/A | N/A | 85.0 | 1.5 | 1.28 | N/A |
| 96 | ZnO/CoPzS8 | 25 | 5.00 | 0.02 | 0.0721 | 78.0 | <0.01 | N/A | N/A |
| 97 | SnO2/FePz(Sbu)8 | 25 | 3.00 | 0.02 | 0.0090 | 34.2 | 12.9 | 4.41 | 0.0026 |
| 97 | SnO2/FePz(Sbu)8 | 25 | 3.00 | 0.03 | 0.0090 | 11.1 | 1 | 0.111 | 0.0001 |
| 98 | ZSM-5/FePz(Sbu)8 | 25 | 4.00 | 0.03 | 0.0162 | 35.8 | 13.1 | 4.69 | 0.0063 |
| 99 | TiO2/HPW/CoPz | 25 | 3.00 | 0.05 | 0.0450 | 22.2 | 16.9 | 3.75 | 0.0113 |
| 100 | g-C3N4/CoPz | 25 | 0.33 | 0.02 | 0.0054 | 52.1 | 20 | 10.4 | 0.0569 |
| 101 | SnO2-OVs/CoPz | 25 | 3.00 | 0.02 | 0.0108 | 43.6 | 28.5 | 12.4 | 0.0149 |
| 102 | Ce6@BNCN | 25 | 2.00 | 0.01 | 0.0054 | 62.3 | 12 | 7.48 | 0.0067 |
Bai et al.102 went one step further and synthesized a completely metal-free photocatalyst based on graphitic carbon nitride (g-C3N4), modified with NaBH4 and chlorin e6. When hydrogen peroxide was used as the oxidant without a catalyst, almost 90% of the glucose was converted in 2 h. The conversion of Glc by catalyst was not observed. The catalyst drastically suppressed the conversion (less than 15%) but significantly increased the selectivity (from 32% to 95%) towards value-added products (arabinose, gluconic acid, and GA). However, selectivity toward GA was rather low, peaking at 12% when a Glc conversion of 62% was achieved. The authors performed quantum chemical calculations at the density functional theory level to determine the oxidation mechanism (Fig. 10), suggesting that the predominant pathway of Glc oxidation is via gluconic rather than glucuronic acid. It should be noted that the authors performed DFT calculations at the basic level and neglected electron excitation. With radical scavengers, the authors were able to noticeably shift the selectivity of the catalytic reaction, but the activity decreased significantly (up to 85%).
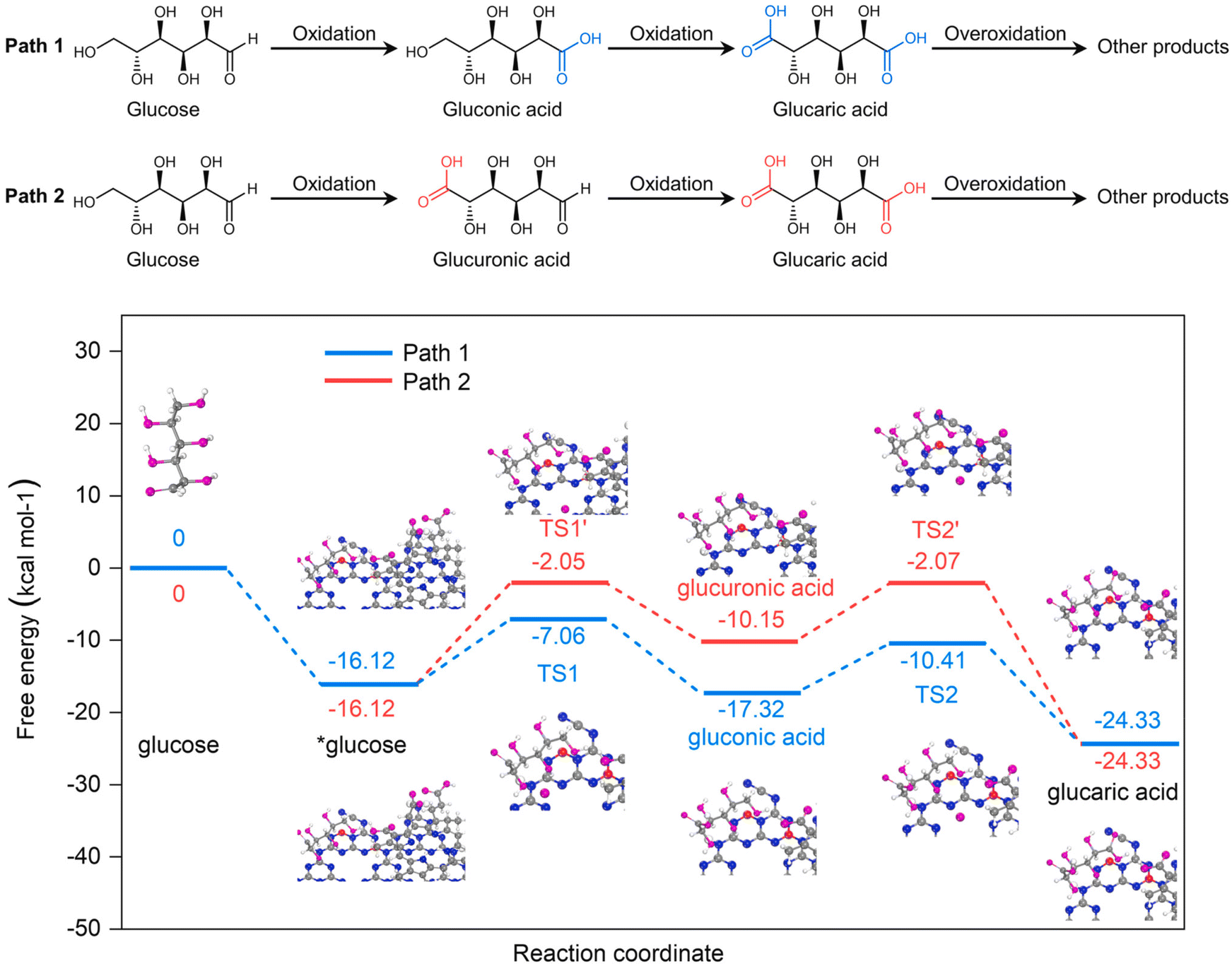 | ||
| Fig. 10 At the top of the figure, glucose oxidation pathways over Ce6@BNCN proposed by Bai et al. are depicted. At the bottom, quantum chemical calculations for the proposed pathways are shown. Reproduced with permission from ref. 102. Copyright 2021, Elsevier. | ||
In summary (Table 3), the most studied photocatalyst, TiO2, can produce GA without any modification but under UV light.91 Dispersion on a support and/or additional modification with active metals dramatically increases the GA selectivity.92–94,99 Several other supports have been tested, such as ZnO,96 SnO2,97 HZSM-5,98 and g-C3N4,100,102 and although considerable GA selectivity (20%100) can be achieved, the activity and conversion still need improvement.
Several factors determine the mode of action of a photocatalyst. A quick and easy indicator of the latter is the use of quenching agents.96–100 By selectively quenching the reactive species, one can determine which species is the major oxidant of Glc. Subsequently, the species that facilitate the deep oxidation of Glc to short chain acids or even CO2 can also be determined. Care should be taken, however, since there are several reaction steps in Glc oxidation all the way to GA. With the use of quenching agents several reactions using intermediates (gluconic acid) as the reactants have to be performed. It is unlikely that all reactive species are equally represented in each reaction step. Three parameters determine how the catalyst reacts with incident light and which radicals are formed: the band gap, the recombination rate of holes and electrons, and the absolute positions of the valence and conduction bands. The actual reaction pathway also depends on the reaction mechanism on the excited potential surface. To fully understand the function of the synthesized catalysts, various experimental methods can be used, such as XPS and UV/vis-DR. Researchers are often eager to shift the catalyst function to the visible region, which would improve the use of sunlight. Since a mere decrease in bandgap can result in a suboptimal position of either the conduction or the valence band or both, as well as a change from direct to indirect bandgap and vice versa, such changes do not necessarily improve the activity of the catalyst. In general, photocatalysts have been shown to reduce conversions, which are exceptionally high but completely nonselective at vacancies with pure oxidants, and to increase selectivity. When catalysts are used, too high a concentration of Glc hinders the selectivity, as also does excessive catalyst loading. Catalyst leaching is also a common problem in photocatalysis, limiting catalyst reusability, and should be considered in all research studies.
5. Electrocatalysis
In electrocatalysis, reactions can be fine-tuned by using the electrical potential at the electrodes to selectively favour the desired reactions. Although common in oxidation and reduction reactions, electrocatalysis is usually limited to smaller molecules than carbohydrate derivatives. Nevertheless, there have been several attempts to use it for the oxidation of Glc to GA, including one patent (Fig. 11).103,104 | ||
| Fig. 11 Schematic representation of the production of fuels and value-added chemicals by electrocatalysis. Reproduced with permission from ref. 104. Copyright © 2017, American Association for the Advancement of Science. | ||
The electrocatalytic oxidation (EO) of Glc to GA was studied 30 years ago,105,106 when Kokoh et al. tested pure Au and Pt electrodes, as well as Pt electrodes modified with either Pb or Bi, in acidic, neutral, and basic media.105 They used a two-chamber glass cell with a total volume of 120 mL and HClO4 or KH2PO4/NaOH as acidic or neutral electrolytes, respectively. The authors used a double- or triple-pulse potential program with a low current density of about 0.22 mA cm−2, with a maximum of 6.3 mA cm−2, using a gold electrode in a neutral medium. At 1.1 V rhe−1 on a pure Pt electrode in acidic medium, a negligible amount of GA was formed and only about 3% of 200 mM Glc was oxidized after 35 h. Modification of the electrode with Ti did not increase the yield, while modification with a Pb adatom at 0.65 V rhe−1 produced small amounts of GA (3.5% of Glc oxidized and 0.2 mA GA detected). Reducing the initial Glc concentration to 100 mM increased the conversion to 13%. In neutral medium over a Pt electrode and at 0.62 V rhe−1, no GA was produced after 19 hours despite a 7% oxidation of Glc with an initial concentration of 50.6 mM.
Electrolysis on gold electrodes at 0.95 V rhe−1 in a 200 mM Glc solution gave no GA and only 1% of the Glc was oxidized. In a neutral medium at 0.87 V rhe−1, 14% of a 50.2 mM Glc solution was oxidized after 20 h and the final GA concentration was 0.4 mM (5% selectivity). Other oxidation products such as gluconic, glucuronic, glyoxylic, oxalic, formic, tartaric, and other acids were also detected. In all cases, however, gluconic acid was the main product.
In a basic medium (0.1 M NaOH), only minute amounts of GA formed at 0.6 V rhe−1 on pure gold and Ti-modified gold electrodes when a lower Glc concentration (10 mM)106 was used. Modification with Bi slightly increases the yield of GA, while Pb significantly improves the yield. Increasing the potential to 0.9 V rhe−1 caused the yield of GA to increase to 33.3% after 24 hours but the concentrations of other oxidation products also increased. At a pure Pt electrode in a basic medium at 0.8 V rhe−1, about 80% of the Glc was oxidized after 21 hours, with a meagre 5% yield of GA. Modification of this electrode with Pd significantly increased the activity, and at 0.7 V rhe−1, all Glc was oxidized after 4 hours. However, while the yield of GA increased slightly, the major products were 2-keto-D-gluconic acid, oxalic acid and formic acid.
Basically, Glc can be oxidized much more effectively in basic media. The first step of oxidation is always the formation of gluconic acid via dehydrogenation of the anomeric carbon atom. The selectivity can be significantly affected by varying the electrode and modifying adatoms.
As with the processes described in the sections before, overoxidation is an omnipresent problem. To determine its extent in electrocatalysis, Bonfatti et al.107 studied the complete mineralization of Glc in a single-body electrochemical cell with a volume of 200 mL, 1 M H2SO4, and modified titanium plate electrodes. Using 10 g L−1 Glc, they found that modification of the plates with Pt decreased the activity the most, regardless of the temperature (25 and 57 °C) and current density (10–90 mA cm−2). While modification with pure Pt slightly improved the activity and a combined modification with SnO2 and Pt performed slightly better, only the electrodes modified with PbO2 showed reasonable production rates. At 57 °C, Glc mineralization was complete after 13 hours even at lower current densities (30 mA cm−2), with minimal leaching of Pb. Significant oxidation activity toward GA, which was even higher than that of Glc, was the result of decarboxylation of the terminal carbon as the activity increased in the following order: aldose < aldonic acid < aldaric acid.
To prevent overoxidation, a mediator molecule can be added to the reaction cell. This facilitates the formation of softer oxidizing species. Koga and Taniguchi108 oxidized Glc in the presence of 2 mM TEMPO as a mediator (2,2,6,6-tetramethyl-1-piperidinyloxy). In a 50 mL reactor with a glassy carbon electrode, electrolysis was complete within 20 minutes when using an alkaline medium (1 M NaOH). The optimum potential for the potential-controlled electrolysis of Glc was found to be +0.6 V (Ag/AgCl). At high Glc concentrations ([Glc]/[TEMPO] = 50), gluconate formed predominantly, while at lower concentrations ([Glc]/[TEMPO] = 1), more GA was formed. At even lower concentrations the selectivity shifted towards oxalic acid. While oxidation to gluconate is a two-electron reaction, 4 additional electrons are transferred when glucarate is formed. To oxidize Glc to oxalic acid, 18 electrons must be transferred. The reactions occur in sequence. First, Glc is oxidized to gluconate and it can then be further oxidized to glucarate.
Habrioux et al.109 used a multidisciplinary approach, where the biocatalytic route was combined with electrocatalysis. The authors designed and tested a membraneless Glc/O2 biofuel cell in which the cathode was impregnated with laccase immobilized with 2,2′-azinobis (3-ethylbenzothiazoline-6-sulfonate) diammonium salt (ABTS2−), and the carbon anode was impregnated with Au–Pt nanoparticles. In a neutral medium (0.2 M phosphate buffer) with a Glc concentration of 10 mM, the maximum power output of the cell was over 70 μW and the only by-products were gluconic acid and GA. This represents a promising opportunity to harness energy while producing value-added chemicals. A similar fuel cell was studied by Li et al.,110 which they constructed from a 4-acetamido-2,2,6,6-tetramethyl piperidine-1-oxyl (ACT) based anode and an air-breathing Pt–C based cathode. However, a high electrode potential and the slow rate of the electrode reaction limit the feasibility of such a cell.
In electrocatalysis, another issue manifested and was first observed by Ibert et al.,111,112 who studied the TEMPO-mediated electrooxidation of Glc using a graphite felt anode and a stainless-steel cathode and investigated different combinations of reactants and different pH values. As in other work, a strong correlation was observed between the ratio of Glc and TEMPO and the GA yields (0.02 mol Glc and 0.0005 mol TEMPO performed best with 600 mA at 0.8 V per SCE). The activity was also strongly dependent on the pH of the solution, with the optimum being 11.8. An unexpected product, later identified as maribersonic acid, was formed via a benzylic rearrangement.112
Bin et al.113 also used a multidisciplinary approach, where they also investigated the engineering aspect of catalysis, as well as the material design. They prepared a porous MnO2/Ti electrode with 10 g L−1 Na2SO4 as the electrolyte (Fig. 12), and the average diameter of the MnO2 nanoparticles was about 23 nm. The Glc conversion and GA selectivity increased with MnO2 loading up to 4.98 wt%, which is correlated with the charge transfer resistance (CTR) shown in the Nyquist diagrams. The CTR decreased with increasing loading up to the optimum loading, where the trend reversed to a higher CTR. Glc conversion was only weakly dependent on pH value but selectivity showed a clear maximum at pH 7 at 30 °C. Because the current density was fixed at 3 mA cm−2, an increase in the Glc concentration from 25 to 126 mM resulted in a decrease in conversion and selectivity toward GA. Lower temperatures adversely affected conversion but increased GA selectivity. At higher current densities (5–6 mA cm−2), an increase in selectivity toward GA was observed, but current efficiency drops precipitously above 3 mA cm−2. At a loading of 4.98 wt%, a Glc concentration of 50.5 mM, a temperature of 30 °C, a residence time of 19 min, a current density of 4 mA cm−2, and a pH of 7, a Glc conversion of 99% and a combined selectivity of more than 99% for gluconic acid and GA were observed.
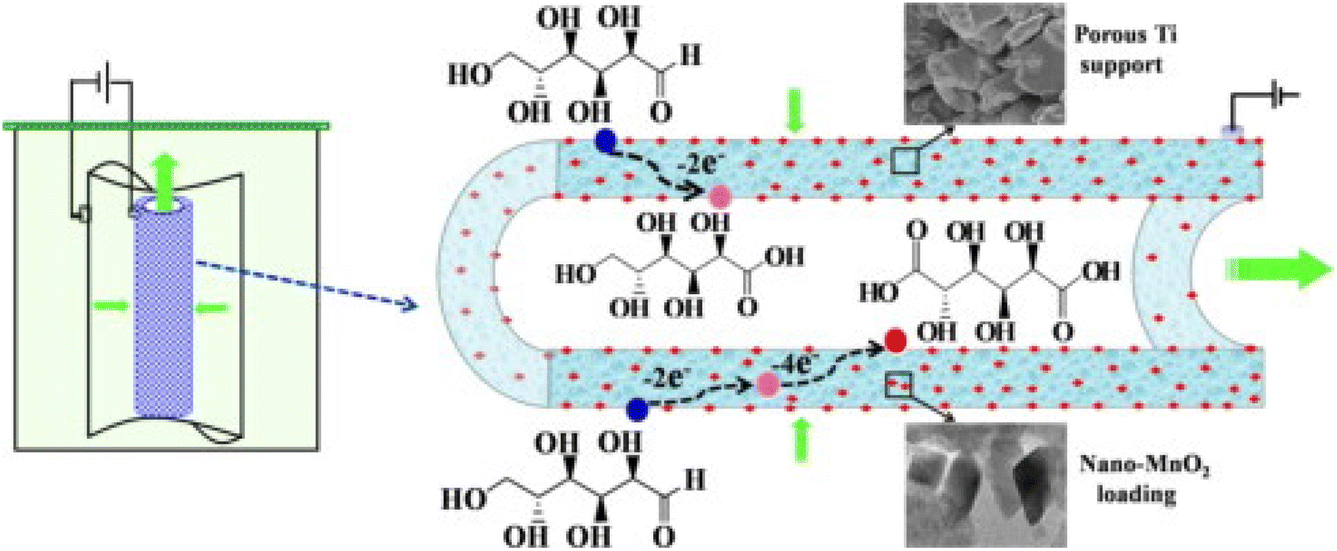 | ||
| Fig. 12 Schematic representation of the nano-MnO2-loading tubular porous titanium electrode used by Bin et al. Reproduced with permission from ref. 113. Copyright 2014 Elsevier. | ||
With the whole world slowly shifting to, at least to some extent, a hydrogen-based economy, Zhao et al.114 had a brilliant idea and combined the oxidation reaction at the anode with the hydrogen evolution reaction (HER) at the cathode but used sodium gluconate (SG) instead of Glc. For the anode and cathode, they supported a Ni-based metal–organic framework (MOF) on nickel foam with benzene-1,3,5-tricarboxylic acid (H3BTC) as an organic linker in the MOF. With this setup, the yield of hydrogen was increased threefold compared with the same setup without SG, outperforming the oxygen evolution reaction. The researchers also found that a cell potential as low as 1 V rhe−1 could be used. In an 8 M KOH solution, the electrode outperformed even a Pt electrode with the lowest Tafel slope (46.6 mV dec−1) and the highest exchange current density (8.15 × 10−3 mA cm−2). After 20 hours at 1 V rhe−1 in 8 M KOH, the conversion of SG was over 99% and the selectivity toward GA was nearly 98% with a faradaic efficiency of 100% and a stable current density of 50 mA cm−2. Liu et al.115 noticeably improved on the work done by Zhao and used modified nickel foam electrodes and successfully oxidized over 90% of 10 mM Glc with a GA yield of over 85% at 1.3 V rhe−1. With NiFeOx-modified nickel foam as the anode and NiFeNx-modified nickel foam as the cathode, H2 was produced simultaneously with high efficiency. The catalysts were reused for 5 cycles with negligible loss of activity. The authors confirmed that the oxidation of Glc on the anode was more efficient than OER on the anode, as already reported by Zhao et al.114
The faradaic efficiency toward GA and gluconic acid production was 87%. Long-term tests (24 h) in 1 M KOH and 0.5 M Glc showed a Glc conversion of 21.3% and a yield of 11.6% and 4.7% of GA and gluconic acid, respectively, with a decrease in current density of less than 4% (from 101.2 to 97.8 mA cm−2). A similar observation was made by Zhang et al.116
In studying Glc oxidation on pure Cu, Pt, and Au electrodes in an alkaline medium, Moggia et al.117 found that the oxide layer formed on the copper electrode was active in Glc oxidation. A peak in the voltammogram of the Cu electrode, which could be attributed to the formation of soluble products from the CuO species, confirmed this. The performance of these catalysts was evaluated by long-term electrocatalysis (65 h) with 0.04 M Glc in 0.1 M NaOH. Since more than 46% of Glc is isomerized to fructose at 20 °C, the reactions were carried out at 5 °C. They used a potential program in which the oxidation potential (0.7–1.8 V rhe−1) was applied for 30 seconds, followed by a desorption step for 1 s at 2.4 V rhe−1 and an adsorption step for 1 s at 0 V rhe−1. Copper was the most selective catalyst for GA with a selectivity of 38% at 0.84 V rhe−1, but with a low activity that could be somewhat improved by increasing the potential (1.8 V rhe−1) at the expense of selectivity. The Pt electrode also exhibited some selectivity for GA (12.6%) at lower potentials (0.7 V rhe−1), but mainly gluconic acid was formed (68%). Gold outperformed the other two catalysts in terms of GA yield, despite a lower selectivity for GA (13.5%), at 1.34 V rhe−1.
Electrocatalysis is an efficient method to produce GA, but fine-tuning the setup (electrodes, media, cell potential …) is difficult. As we have already seen (Table 4), even the same type of electrode can have drastically different selectivity depending on the pH, as shown by Kokoh et al.105,106 Since deep oxidation of Glc is also a problem,107 a two-step process was proposed by Moggia et al.118 However, the process still needs to be improved because of the low gluconic acid conversion and electrode poisoning. A persistent problem, throughout the final three processes described, is that even softer oxidizing species can oxidize glucose to gluconic acid, while they are mostly insufficient for further oxidation to glucaric acid. The use of a mediator that is oxidized and in turn oxidizes Glc has already been discussed38 and successfully attempted.108,111,112 The use of TEMPO allows good GA selectivity provided that a suitable Glc/TEMPO ratio is used. So far, GA yields have been too low to justify the addition of a mediator. An interesting concept is the combination of Glc oxidation with electricity generation,109 for which several types of Glc fuel cell has already been patented.119 When hydrogen is also produced, the combined reaction efficiency is higher than that of conventional water splitting. Glc oxidation can be coupled with the hydrogen evolution reaction.115,120 Researchers have found that the latter can improve the energy efficiency of the HER. Electrode leaching is also possible117 and should be investigated in any case. Finally, GA yield can be improved by clever design of the cell, as shown by Bin et al.113 who used a porous MnO2/Ti electrode.
| Reference | Current density [mA cm−2] | Work potential | Catalyst material | Initial Glc [mol L−1] | Time [h] | Reaction volume [mL] | Glc conversion [%] | GA selectivity [%] | GA yield [%] | STY [gGA L−1 h−1] |
|---|---|---|---|---|---|---|---|---|---|---|
| N/A not applicable.a Gluconate. | ||||||||||
| 105 | 0.22 | 0.55 V rhe−1 | Pt | 0.200 | 30 | 60 | 3.5 | 3.0 | 0.1 | 0.0013 |
| 106 | N/A | 0.9 V rhe−1 | Au–Pb | 0.010 | 24 | 60 | 90.0 | 37.0 | 33.3 | 0.0250 |
| 107 | 30 | N/A | PbO2 | 0.056 | 13 | 200 | 100.0 | N/A | N/A | N/A |
| 108 | N/A | 0.6 V/(Ag/AgCl) | GCE | 0.002 | 0.33 | 50 | 100.0 | 60.0 | 60.0 | 0.6551 |
| 109 | N/A | N/A | AuPt | 0.010 | N/A | 20 | 100.0 | N/A | N/A | N/A |
| 110 | N/A | N/A | N/A | 0.050 | 72 | 10 | N/A | N/A | N/A | N/A |
| 111 | 600 | N/A | N/A | 0.1a | 20 | 330 | 100.0 | 85.0 | 85.0 | 0.7657 |
| 113 | 3 | N/A | MnO2 | 0.051 | 0 | 400 | 99.0 | 45.0 | 44.6 | 0.1642 |
| 116 | 50 | 1 V rhe−1 | Ni3(BTC)2/NiF | 1a | 20 | N/A | 100.0 | 98.0 | 98.0 | N/A |
| 117 | N/A | 1.34 V rhe−1 | Au | 0.040 | 65 | N/A | 100.0 | 13.5 | 13.5 | N/A |
| 115 | 100 | 1.3 V rhe−1 | Ni–Fe | 0.500 | 24 | 100 | 21.3 | 54.5 | 11.6 | 0.4357 |
6. Discussion
The various Glc oxidation processes discussed in this review are fundamentally different and have drastically different process parameters, making them difficult to compare. A useful, nearly universal parameter for determining process utility, particularly the potential for upscaling, is the STY. Where the necessary data have been published, we have calculated it, defined as grams GA produced per hour per unit volume required (Fig. 13). This parameter is a suitable indicator for evaluating the industrial profitability of the process. If dilute reactant concentrations and consequently large volumes or long reaction times are required, the feasibility of the process decreases. We should note at this point, that in many of the processes described, the STY would increase noticeably if smaller volumes are used. In many cases, this could be straightforwardly achieved by rational engineering.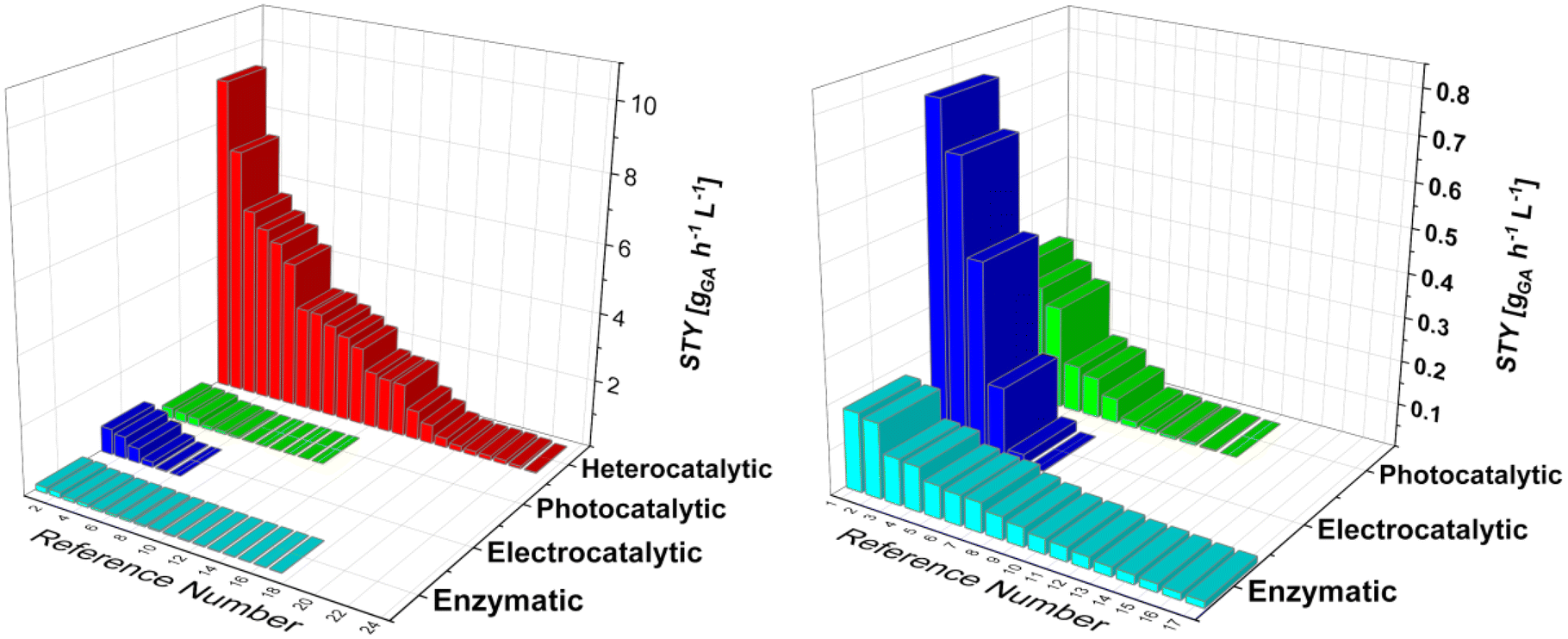 | ||
| Fig. 13 The left bar graph illustrates the STY of all the processes encompassed in this review. On the right, the heterogeneous catalytic processes were omitted for clarity. The calculated STYs were sorted by size to further improve clarity. The reference numbers can be correlated with actual references and data with the help of Table 5. | ||
| Reference number in Fig. 13 | Reference in article | |||
|---|---|---|---|---|
| Enzymatic | Electrocatalytic | Photocatalytic | Heterocatalytic | |
| 1 | 56 | 111 | 92 | 78 |
| 2 | 59 | 108 | 92 | 79 |
| 3 | 55 | 115 | 91 | 80 |
| 4 | 58 | 113 | 93 | 83 |
| 5 | 52 | 106 | 93 | 79 |
| 6 | 47 | 105 | 100 | 84 |
| 7 | 43 | 101 | 84 | |
| 8 | 41 | 99 | 82 | |
| 9 | 51 | 102 | 81 and 82 | |
| 10 | 42 | 98 | 80 | |
| 11 | 49 | 97 | 86 | |
| 12 | 48 | 97 | 85 | |
| 13 | 46 | 82 | ||
| 14 | 45 | 81 | ||
| 15 | 50 | 86 | ||
| 16 | 44 | 86 | ||
| 17 | 40 | 79 | ||
| 18 | 84 | |||
| 19 | 79 | |||
| 20 | 78 | |||
| 21 | 86 | |||
| 22 | 81 | |||
| 23 | 89 | |||
| 24 | 81 | |||
At first glance, heterogeneous catalysis appears to be clearly superior to other types of catalysis. However, the caveat here is that the two best performing heterogeneous processes used gluconate as the substrate, which converts faster than Glc. On the other hand, enzymatic GA production is compromised by long reaction times. Electrocatalysis and photocatalysis require very dilute concentrations, which lowers the STY.
The STY, however, is not the only relevant parameter in determining the profitability of a process. The cost of reactants and possible co-factors must be considered, as well as downstream processing. Product purification (separation) can account for a large portion of the operating costs. The solvents required also affect the environmental impact of the process. Therefore, the selectivity of the reaction is crucial, as high selectivity simplifies the purification of GA, as Choi et al. aptly noted.121 In most cases, the selectivity obtained in heterogeneous catalytic processes is quite low (less than 50%), but this value is somewhat higher when the catalyst is supported by Pt (about 80%). Several acids, produced as by-products of the reaction, affect the final downstream process.
In photocatalysis, the development of catalysts is hampered by the difficulty of performing in situ studies in the liquid phase. The relevance of reaction factors such as intermediates, the restructuring of the catalyst and its oxidation state, etc., is difficult to determine, which hinders targeted catalyst development. Therefore, it is a particular challenge to accurately define and control the reactive oxygen species, which in turn determine the reaction selectivity. This can be seen in Table 3, where quite low selectivity was determined in photocatalytic experiments for the production of GA. In many cases, catalyst synthesis is difficult, even at the laboratory scale, and scale-up is rarely investigated. This is in no small part because there is no practical way to efficiently illuminate tons of photocatalysts.122
High selectivity can be achieved with electrocatalytic glucose oxidation, which in turn simplifies product separation. The disadvantage of electrocatalytic processes is the instability of the catalytic electrodes, questionable faradaic efficiency (FE), current density and cell voltage, while the advantage is that oxidation takes place at the anode, while hydrogen gas can be generated at the cathode. This brings significant economic advantages. The price of electricity (and its source) is still an important factor. It has been reported123 that for CO2 reduction to CO, the current density should be higher than 300 mA cm−2, the FE should be higher than 80%, the cell voltage should be lower than 1.8 V, and the stability should be over 80000 hours to make the CO economically viable. Liu et al.115 calculated that with their electrocatalytic method, the minimum selling price for GA should be $9.32 per kg. According to their calculations, this reduces the cost by more than 45% compared with conventional chemical Glc oxidation ($17.04).
The selectivity of electrocatalytic systems can only be achieved with biological systems, which, however, have other disadvantages. The extremely low space-time yields can be improved by using isolated and immobilized enzymes, which eliminate the need for batch operation as in fermentation. These typically allow STYs one to two orders of magnitude higher yields than fermentation. An additional advantage is easier removal of the enzymes, which simplifies downstream processing. The use of enzymes from hyperthermophilic organisms allows operation at elevated temperatures, further shortening reaction times and thus increasing the STY. An important parameter to consider is the price of co-factors, (co-)substrates and other components that need to be added to the medium (IPTG, MIOX…). While the stability of the catalyst plays a crucial role for the feasibility of any catalytic process, the stability of the enzymes and their price are even more crucial for the profitability.
To evaluate the environmental impact of these processes, several factors must be considered. First, catalyst stability is important because the use of short-lived and non-recyclable catalysts is wasteful. If low stability is due to leaching, the impact of metals (enzymes) must also be considered. Another important factor is the cost of producing the catalytic materials, whether they are based on enzymes or metals (oxides). While heterogeneous catalysis offers high STYs, the best catalysts require critical raw materials and/or noble metals (Pt, Au, and Pd). In addition, most of the heterogeneous catalysis processes discussed in this article require pH control with NaOH or KOH, which is environmentally problematic. Additionally, base addition increases the production costs, which offsets the feasibility of upscaling of the process. Finding a suitable Cu- or Bi-based catalyst would sufficiently reduce the cost.
It is possible to combine the oxidation of Glc with the production of hydrogen, which can offset some costs associated with the process. This is particularly important in electrocatalysis, where the price of the electrolyte, its toxicity, and its transformation during the process can greatly alter the environmental impact and financial design.
Most processes require mild reaction conditions, with the exception of heterogeneous catalysis, which requires high pressures. A thorough life-cycle analysis (LCA) is beyond the scope of this review, but Liu et al.115 have shown that both electrocatalytic and heterogeneous catalysis are viable options, if chemical oxidants are avoided. In short, in addition to energy requirements, the energy source and other externalities must be considered for each process. For example, while electrocatalysis and photocatalysis require only a modest amount of energy, the cost of biocatalytic processes is even lower, although they involve higher costs due to the use of genetically modified organisms.
As of 2022, heterogeneous catalysis is the most viable production route for GA, provided adequate downstream processing is available and purity is not the limiting factor. If higher purity is required, electrocatalytic and bio-enzymatic processes can be used. Photocatalysis remains limited to laboratory experiments due to practical difficulties in upscaling despite its low energy consumption.
7. Conclusion and future outlook
As shown here, there are several emerging technologies (bio-enzymatic, heterogeneous catalysis, photocatalysis, electrocatalysis) for the environmentally friendly production of GA. Due to their fundamentally different backgrounds, they suffer from various drawbacks that are being intensively researched. Recommendations and the main challenges related to these shortcomings are evaluated and critically discussed in this review. Broad comparisons are difficult because published research results vary widely with respect to the parameters presented. Preferably, catalyst selectivity, conversion, productivity (per mass and per time), and stability should be reported, along with space-time yields and other transferable quantities. In a general, catalysts should be compared with available data.To date, most work has been done in batch reactor systems, which are inherently difficult to scale up. Trickle bed or plug flow systems would greatly facilitate the scalability and allow better techno-economic analyses. With modern software, such as Aspen Plus, entire production chains can be analysed for feasibility. The accuracy of the proposed models differs from actual values, but still allows researchers to evaluate their catalysts on an industrial scale.
There are several properties that determine the viability of a catalyst, such as activity, selectivity, stability, price, and complexity. Some of these properties are not easy to assess which can drastically affect the feasibility of the process. Little research has been done on potential impurities, such as hemicellulose, present in the starting stream. Purification in an industrial setting can quickly and significantly increase the price of the proposed process. Finding a renewable pathway for the production of GA is essential for the transition to a carbon-neutral society, and Glc remains a suitable feedstock.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was funded by the Slovenian Research Agency (ARRS), research core funding P2-0152 and P2-0089. M. H. was funded by the fundamental research projects J1-3020 and J4-3085 from the ARRS. M. G. was funded by the fundamental research project N2-0242 from the ARRS. Infrastructure funding I0-0039 from the ARRS is gratefully acknowledged.References
- C. L. Mehltretter and C. E. Rist, J. Agric. Food Chem., 1953, 1, 779–783 CrossRef CAS.
- C. L. Mehltretter, B. H. Alexander and C. E. Rist, Ind. Eng. Chem., 1953, 45, 2782–2784 CrossRef CAS.
- T. N. Smith and R. Shirley, 9347024B2, 2016.
- T. N. Smith, D. E. Kiely and K. Kramer-Presta, 9404188B2, 2016.
- T. Werpy and G. Petersen, Top Value Added Chemicals from Biomass: Volume I – Results of Screening for Potential Candidates from Sugars and Synthesis Gas, Golden, CO (United States), 2004 Search PubMed.
- K. Fischer and H.-P. Bipp, Water, Air, Soil Pollut., 2002, 138, 271–288 CrossRef CAS.
- G. Subramanian and G. Madras, Water Res., 2016, 104, 168–177 CrossRef CAS PubMed.
- J. Singh and K. P. Gupta, Biomed. Environ. Sci., 2003, 16, 9–16 Search PubMed.
- J. Singh and K. P. Gupta, J. Environ. Pathol., Toxicol. Oncol., 2007, 26, 63–73 CrossRef CAS PubMed.
- N. van Strien, S. Rautiainen, M. Asikainen, D. A. Thomas, J. Linnekoski, K. Niemelä and A. Harlin, Green Chem., 2020, 22, 8271–8277 RSC.
- S. Li, W. Deng, S. Wang, P. Wang, D. An, Y. Li, Q. Zhang and Y. Wang, ChemSusChem, 2018, 11, 1995–2028 CrossRef CAS PubMed.
- S. Gyergyek, A. Kocjan, M. Grilc, B. Likozar, B. Hočevar and D. Makovec, Green Chem., 2020, 22, 5978–5983 RSC.
- J. Rios, J. Lebeau, T. Yang, S. Li and M. D. Lynch, Green Chem., 2021, 23, 3172–3190 RSC.
- M. L. R. Liman and M. T. Islam, J. Mater. Chem. A, 2022, 10, 2697–2735 RSC.
- F. H. Isikgor and C. R. Becer, Polym. Chem., 2015, 6, 4497–4559 RSC.
- D. E. Kiely, L. Chen and T. H. Lin, J. Am. Chem. Soc., 1994, 116, 571–578 CrossRef CAS.
- D. E. Kiely, L. Chen and T.-H. Lin, Polymers from Agricultural Coproducts, 1994, vol. 575, pp. 149–158 Search PubMed.
- C. Lu and E. Ford, Macromol. Mater. Eng., 2018, 303, 1700523 CrossRef.
- M. C. Biswas, B. Bush and E. Ford, Carbohydr. Polym., 2020, 245, 116510 CrossRef CAS PubMed.
- M. L. R. Liman and M. T. Islam, J. Mater. Chem. A, 2022, 10, 2697–2735 RSC.
- C. L. Mehltretter, 2436659A, 1948.
- D. E. Kiely, A. Carter and D. P. Shrout, 5599977A, 1997.
- D. E. Kiely and G. Ponder, 6049004A, 2000.
- D. E. Kiely and K. R. Hash, 2008/021054A3, 2008.
- D. E. Kiely and K. R. Sr, 7692041B2, 2010.
- H. Kiliani, Justus Liebigs Ann. Chem., 1880, 205, 145–190 CrossRef.
- S. Donen and K. Jensen, 2014/0275622A1, 2014.
- C. Dwivedi, W. J. Heck, A. A. Downie, S. Larroya and T. E. Webb, Biochem. Med. Metab. Biol., 1990, 43, 83–92 CrossRef CAS PubMed.
- Z. Walaszek, J. Szemraj, M. Narog, A. K. Adams, J. Kilgore, U. Sherman and M. Hanausek, Cancer Detect. Prev., 1997, 21, 178–190 CAS.
- A. S. Heerdt, C. W. Young and P. I. Borgen, Isr. J. Med. Sci., 1995, 31, 101–105 CAS.
- J. K. Selkirk, G. M. Cohen and M. C. MacLeod, Arch. Toxicol., 1980, 171–178 CAS.
- N. Yoshimi, Z. Walaszek, H. Mori, M. Hanausek, J. Szemraj and T. J. Slaga, Int. J. Oncol., 2000, 16, 43–51 CAS.
- C. Dwivedi, A. A. Downie and T. E. Webb, J. Environ. Pathol., Toxicol. Oncol., 1989, 9, 253–259 CAS.
- Q. Zhang, Z. Wan, I. K. M. Yu and D. C. W. Tsang, J. Cleaner Prod., 2021, 312, 127745 CrossRef CAS.
- T. R. Boussie, E. L. Dias, Z. M. Fresco, V. J. Murphy, J. Shoemaker, R. Archer and H. Jiang, 2010/144862A2, 2010.
- G. M. Diamond, E. L. Dias, R. H. Archer, V. J. Murphy and T. R. Boussie, 2016/054065A2, 2016.
- N. Potrzebowska, M. Besson, C. Pinel, N. Perret, E. Derrien and P. Marion, 2020/001840A1, 2020.
- N. Merbouh, J. M. Bobbitt and C. Brueckner, 6498269B1, 2000.
- P. G. Tonkes and C. A. Marsh, Aust. J. Biol. Sci., 1973, 26, 839 CrossRef CAS PubMed.
- T. S. Moon, S.-H. Yoon, A. M. Lanza, J. D. Roy-Mayhew and K. L. J. Prather, Appl. Environ. Microbiol., 2009, 75, 589–595 CrossRef CAS PubMed.
- T. S. Moon, J. E. Dueber, E. Shiue and K. L. J. Prather, Metab. Eng., 2010, 12, 298–305 CrossRef CAS PubMed.
- J. E. Dueber, G. C. Wu, G. R. Malmirchegini, T. S. Moon, C. J. Petzold, A. v. Ullal, K. L. J. Prather and J. D. Keasling, Nat. Biotechnol., 2009, 27, 753–759 CrossRef CAS PubMed.
- E. Shiue and K. L. J. Prather, Metab. Eng., 2014, 22, 22–31 CrossRef CAS PubMed.
- I. M. Brockman and K. L. J. Prather, Metab. Eng., 2015, 28, 104–113 CrossRef CAS PubMed.
- I. M. B. Reizman, A. R. Stenger, C. R. Reisch, A. Gupta, N. C. Connors and K. L. J. Prather, Metab. Eng. Commun., 2015, 2, 109–116 CrossRef PubMed.
- A. Gupta, M. A. Hicks, S. P. Manchester and K. L. J. Prather, Biotechnol. J., 2016, 11, 1201–1208 Search PubMed.
- Y. Liu, X. Gong, C. Wang, G. Du, J. Chen and Z. Kang, Enzyme Microb. Technol., 2016, 91, 8–16 CrossRef CAS PubMed.
- S. J. Doong, A. Gupta and K. L. J. Prather, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, 2964–2969 CrossRef CAS PubMed.
- J. Hou, C. Gao, L. Guo, J. Nielsen, Q. Ding, W. Tang, G. Hu, X. Chen and L. Liu, Metab. Eng., 2020, 61, 47–57 CrossRef CAS PubMed.
- W. L. Marques, L. A. Anderson, L. Sandoval, M. A. Hicks and K. L. J. Prather, Enzyme Microb. Technol., 2020, 140, 109623 CrossRef CAS PubMed.
- Y. Zhao, J. Li, R. Su, Y. Liu, J. Wang and Y. Deng, J. Biotechnol., 2021, 332, 61–71 CrossRef CAS PubMed.
- H.-H. Su, F. Peng, X.-Y. Ou, Y.-J. Zeng, M.-H. Zong and W.-Y. Lou, N. Biotechnol., 2020, 59, 51–58 CrossRef CAS PubMed.
- P. Yadav, J. Clin. Diagn. Res., 2015, 9, ZE21–ZE25 CAS.
- F. M. Gírio, C. Fonseca, F. Carvalheiro, L. C. Duarte, S. Marques and R. Bogel-Łukasik, Bioresour. Technol., 2010, 101, 4775–4800 CrossRef PubMed.
- C. C. Lee, R. E. Kibblewhite, C. D. Paavola, W. J. Orts and K. Wagschal, Mol. Biotechnol., 2016, 58, 489–496 CrossRef CAS PubMed.
- Y. Li, Y. Xue, Z. Cao, T. Zhou and F. Alnadari, World J. Microbiol. Biotechnol., 2018, 34, 102 CrossRef PubMed.
- T. v. Vuong and E. R. Master, Biotechnol. Biofuels, 2020, 13, 51 CrossRef CAS PubMed.
- H. Su, Z. Guo, X. Wu, P. Xu, N. Li, M. Zong and W. Lou, ChemSusChem, 2019, 12, 2278–2285 CrossRef CAS PubMed.
- K. Petroll, A. Care, P. L. Bergquist and A. Sunna, Metab. Eng., 2020, 57, 162–173 CrossRef CAS PubMed.
- M. Sauer, D. Porro, D. Mattanovich and P. Branduardi, Trends Biotechnol., 2008, 26, 100–108 CrossRef CAS PubMed.
- W. A. Schroeder, P. M. Hicks, S. C. Mcfarlan and T. W. Abraham, 2004/0185562A1, 2008.
- T. Ito, H. Tadokoro, H. Masaki, K. Mikuni, H. Murakami, T. Kiso and T. Kiryu, 2013/183610A1, 2013.
- T. S. Moon, S.-H. Yoon and L. J. P. Kristala, 8835147B2, 2014.
- K. Konishi and S. Imazu, 9506091B2, 2016.
- I. M. Brockman, L. J. Prather Kristala and A. Gupta, 20170130210A1, 2017.
- Kalion, https://www.kalioninc.com/products (accessed: 15.12.2021).
- H.-H. Su, F. Peng, P. Xu, X.-L. Wu, M.-H. Zong, J.-G. Yang and W.-Y. Lou, Bioresour. Bioprocess., 2019, 6, 36 CrossRef.
- A. Murugan, R. Prathiviraj, D. Mothay and P. Chellapandi, Int. J. Biol. Macromol., 2019, 140, 1214–1225 CrossRef CAS PubMed.
- C. L. Mehltretter and B. H. Alexander, 2472168A, 1949.
- H. G. J. de Wilt and H. S. van der Baan, Ind. Eng. Chem. Prod. Res. Dev., 1972, 11, 374–378 CrossRef CAS.
- J. M. H. Dirkx, H. S. van der Baan and J. M. A. J. J. van den Broek, Carbohydr. Res., 1977, 59, 63–72 CrossRef CAS.
- J. M. H. Dirkx and H. S. van der Baan, J. Catal., 1981, 67, 14–20 CrossRef CAS.
- P. C. C. Smits, B. F. M. Kuster, K. van der Wiele and S. van der Baan, Appl. Catal., 1987, 33, 83–96 CrossRef CAS.
- P. J. M. Dijkgraaf, M. J. M. Rijk, J. Meuldijk and K. van der Wiele, J. Catal., 1988, 112, 329–336 CrossRef CAS.
- P. J. M. Dijkgraaf, B. F. M. Küster, H. A. M. Duisters and K. van der Wiele, J. Catal., 1988, 112, 337–344 CrossRef CAS.
- M. Besson, F. Lahmer, P. Gallezot, P. Fuertes and G. Fleche, J. Catal., 1995, 152, 116–121 CrossRef CAS.
- M. Besson, G. Flèche, P. Fuertes, P. Gallezot and F. Lahmer, Recl. Trav. Chim. Pays-Bas, 1996, 115, 217–221 CrossRef CAS.
- S. Rautiainen, P. Lehtinen, J. Chen, M. Vehkamäki, K. Niemelä, M. Leskelä and T. Repo, RSC Adv., 2015, 5, 19502–19507 RSC.
- X. Jin, M. Zhao, J. Shen, W. Yan, L. He, P. S. Thapa, S. Ren, B. Subramaniam and R. v. Chaudhari, J. Catal., 2015, 330, 323–329 CrossRef CAS.
- J. Lee, B. Saha and D. G. Vlachos, Green Chem., 2016, 18, 3815–3822 RSC.
- X. Jin, M. Zhao, M. Vora, J. Shen, C. Zeng, W. Yan, P. S. Thapa, B. Subramaniam and R. v. Chaudhari, Ind. Eng. Chem. Res., 2016, 55, 2932–2945 CrossRef CAS.
- E. Derrien, P. Marion, C. Pinel and M. Besson, Org. Process Res. Dev., 2016, 20, 1265–1275 CrossRef CAS.
- E. Derrien, M. Mounguengui-Diallo, N. Perret, P. Marion, C. Pinel and M. Besson, Ind. Eng. Chem. Res., 2017, 56, 13175–13189 CrossRef CAS.
- S. Solmi, C. Morreale, F. Ospitali, S. Agnoli and F. Cavani, ChemCatChem, 2017, 9, 2797–2806 CrossRef CAS.
- H. Shi, P. S. Thapa, B. Subramaniam and R. v. Chaudhari, Org. Process Res. Dev., 2018, 22, 1653–1662 CrossRef CAS.
- M. Khawaji, Y. Zhang, M. Loh, I. Graça, E. Ware and D. Chadwick, Appl. Catal., B, 2019, 256, 117799 CrossRef CAS.
- M. Khawaji, I. Graça, E. Ware and D. Chadwick, Catal. Today, 2021, 365, 257–264 CrossRef CAS.
- Q. Zhang, S. Xu, Y. Cao, R. Ruan, J. H. Clark, C. Hu and D. C. W. Tsang, Green Chem., 2022, 24, 6657–6670 RSC.
- Y. Wang, W. Deng, L. Yan, B. Wang, Q. Zhang, H. Song, S. Wang and Q. Zhang, Angew. Chem., Int. Ed., 2021, 60, 4712–4719 CrossRef PubMed.
- M. Liu, X. Jin, G. Zhang, Q. Xia, L. Lai, J. Wang, W. Zhang, Y. Sun, J. Ding, H. Yan and C. Yang, ACS Catal., 2020, 10, 10932–10945 CrossRef CAS.
- J. C. Colmenares, A. Magdziarz and A. Bielejewska, Bioresour. Technol., 2011, 102, 11254–11257 CrossRef CAS PubMed.
- J. C. Colmenares and A. Magdziarz, J. Mol. Catal. A: Chem., 2013, 366, 156–162 CrossRef CAS.
- J. C. Colmenares, A. Magdziarz, K. Kurzydlowski, J. Grzonka, O. Chernyayeva and D. Lisovytskiy, Appl. Catal., B, 2013, 134–135, 136–144 CrossRef CAS.
- M. Bellardita, E. I. García-López, G. Marcì and L. Palmisano, Int. J. Hydrogen Energy, 2016, 41, 5934–5947 CrossRef CAS.
- M. Bellardita, E. I. García-López, G. Marcì, B. Megna, F. R. Pomilla and L. Palmisano, RSC Adv., 2015, 5, 59037–59047 RSC.
- M. Cheng, Q. Zhang, C. Yang, B. Zhang and K. Deng, Catal. Sci. Technol., 2019, 9, 6909–6919 RSC.
- Q. Zhang, Y. Ge, C. Yang, B. Zhang and K. Deng, Green Chem., 2019, 21, 5019–5029 RSC.
- R. Chen, C. Yang, Q. Zhang, B. Zhang and K. Deng, J. Catal., 2019, 374, 297–305 CrossRef CAS.
- J. Yin, Q. Zhang, C. Yang, B. Zhang and K. Deng, Catal. Sci. Technol., 2020, 10, 2231–2241 RSC.
- Q. Zhang, X. Xiang, Y. Ge, C. Yang, B. Zhang and K. Deng, J. Catal., 2020, 388, 11–19 CrossRef CAS.
- Q. Zhang, C. Yang, B. Zhang and K. Deng, ACS Sustainable Chem. Eng., 2021, 9, 2057–2066 CrossRef CAS.
- X. Bai, Q. Hou, H. Qian, Y. Nie, T. Xia, R. Lai, G. Yu, M. Laiq Ur Rehman, H. Xie and M. Ju, Appl. Catal., B, 2022, 303, 120895 CrossRef CAS.
- L. Zhao and Z. Wang, 109675639A, 2019.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, 1–1 CrossRef PubMed.
- K. B. Kokoh, J.-M. Léger, B. Beden and C. Lamy, Electrochim. Acta, 1992, 37, 1333–1342 CrossRef CAS.
- K. B. Kokoh, J.-M. Léger, B. Beden, H. Huser and C. Lamy, Electrochim. Acta, 1992, 37, 1909–1918 CrossRef CAS.
- F. Bonfatti, S. Ferro, F. Lavezzo, M. Malacarne, G. Lodi and A. de Battisti, J. Electrochem. Soc., 1999, 146, 2175–2179 CrossRef CAS.
- T. Koga and I. Taniguchi, Electrochemistry, 2004, 72, 858–860 CrossRef CAS.
- A. Habrioux, K. Servat, B. Kokoh and N. Alonso-Vante, ECS Trans., 2019, 6, 9–17 CrossRef.
- G. Li, Y. Wang, F. Yu, Y. Lei and Z. Hu, Chem. Commun., 2021, 57, 4051–4054 RSC.
- M. Ibert, P. Fuertès, N. Merbouh, C. Fiol-Petit, C. Feasson and F. Marsais, Electrochim. Acta, 2010, 55, 3589–3594 CrossRef CAS.
- M. Ibert, P. Fuertès, N. Merbouh, C. Feasson and F. Marsais, Carbohydr. Res., 2011, 346, 512–518 CrossRef CAS PubMed.
- D. Bin, H. Wang, J. Li, H. Wang, Z. Yin, J. Kang, B. He and Z. Li, Electrochim. Acta, 2014, 130, 170–178 CrossRef CAS.
- L. Zhao, X. Kuang, X. Sun, Y. Zhang and Q. Wei, J. Electrochem. Soc., 2019, 166, H534–H540 CrossRef CAS.
- W.-J. Liu, Z. Xu, D. Zhao, X.-Q. Pan, H.-C. Li, X. Hu, Z.-Y. Fan, W.-K. Wang, G.-H. Zhao, S. Jin, G. W. Huber and H.-Q. Yu, Nat. Commun., 2020, 11, 265 CrossRef CAS PubMed.
- Y. Zhang, Y. Qiu, Z. Ma, Y. Wang, Y. Zhang, Y. Ying, Y. Jiang, Y. Zhu and S. Liu, J. Mater. Chem. A, 2021, 9, 10893–10908 RSC.
- G. Moggia, T. Kenis, N. Daems and T. Breugelmans, ChemElectroChem, 2020, 7, 86–95 CrossRef CAS.
- G. Moggia, J. Schalck, N. Daems and T. Breugelmans, Electrochim. Acta, 2021, 374, 137852 CrossRef CAS.
- P. Simons and L. M. Jennifer, 2019/164839A1, 2019.
- L. Zhao, X. Kuang, X. Sun, Y. Zhang and Q. Wei, J. Electrochem. Soc., 2019, 166, H534–H540 CrossRef CAS.
- H. Choi, N. E. Soland, B. L. Buss, N. C. Honeycutt, E. G. Tomashek, S. J. Haugen, K. J. Ramirez, J. Miscall, E. C. D. Tan, T. N. Smith, P. O. Saboe and E. M. Karp, Green Chem., 2022, 24, 1350–1361 RSC.
- L. Xiong and J. Tang, Adv. Energy Mater., 2021, 11, 2003216 CrossRef CAS.
- S. Jin, Z. Hao, K. Zhang, Z. Yan and J. Chen, Angew. Chem., Int. Ed., 2021, 60, 20627–20648 CrossRef CAS PubMed.
Footnote |
| † These authors are equally contributed to this work. |
| This journal is © The Royal Society of Chemistry 2023 |