
New mechanistic insights into the role of water in the dehydration of ethanol into ethylene over ZSM-5 catalysts at low temperature†
L.
Ouayloul
ab,
M.
El Doukkali
 *bc,
M.
Jiao
de,
F.
Dumeignil
c and
I.
Agirrezabal-Telleria
*a
*bc,
M.
Jiao
de,
F.
Dumeignil
c and
I.
Agirrezabal-Telleria
*a
aDepartment of Chemical and Environmental Engineering, University of the Basque Country (UPV/EHU), Bilbao, Spain. E-mail: iker.aguirrezabal@ehu.eus
bDepartment of Chemistry, University of Sultan Moulay Slimane (USMS), Beni-Mellal, Morocco. E-mail: m.eldoukkali@usms.ma
cUniv. Lille, CNRS, Centrale Lille, Univ. Artois, UMR 8181, UCCS, Unité de Catalyse et Chimie du Solide, F-59000 Lille, France
dQingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, Shandong, China
eShandong Energy Institute, Qingdao 266101, Shandong, China
First published on 29th March 2023
Abstract
The low-temperature dehydration of bioethanol-to-ethylene is of great interest to reduce energy consumption and achieve high product purities in the biorefinery and olefin industry. Thermokinetic constraints, however, lead to low ethylene selectivity at low temperature. In this work, we integrate a new approach that combines a hierarchical acid H-form ZSM-5 (HZSM-5) with systematic catalytic testing to study how the physicochemical modification of the surface and intermediate catalytic species affect the ethanol-to-ethylene route at 225 °C. Four HZSM-5 zeolites were treated with OH species under basic conditions (OH−) or solely with H2O. Kinetic evidence coupled to 27Al-nuclear magnetic resonance, NH3-temperature-programmed desorption and N2 adsorption, as well as density-functional theory calculations, correlate ethylene selectivity with the appearance of new extra-framework Al(V) and Al(VI) species, acting as Lewis acid-sites. The adopted approach allows us to experimentally unveil the cooperative effect between Brønsted- and Lewis-acid sites that seem to play a key role in ethylene formation from ethanol at low-temperature via (i) a primary route via ethanol dimerization on neighboring Brønsted-acid sites to diethylether, which subsequently cracked on Lewis-acid sites to ethylene; (ii) a secondary route via the direct ethanol dehydration on Brønsted-acid sites. Theoretical calculations support the proposed catalytic cycle. These new insights shed light on the mechanism of ethanol-to-ethylene at low temperature, and on how the precise control over the strength of acid-sites and their population in HZSM-5 affects catalysis. This work progresses towards more active and stable catalysts, advancing into more mature low-temperature technologies for the dehydration of bioethanol into sustainable ethylene.
1. Introduction
In light of the current environmental issues, there is increasing interest in transforming excess bio-ethanol (ET) into value-added chemicals.1,2 The world production of ET is expected to increase from 125 to 138 metric million tons until 2026.3 In an effort to reduce CO2 emissions, the upscaling of ET conversion processes into valuable products such as bio-ethylene (ETY), bio-diethyl-ether (DEE), bio-acetaldehyde or bio-hydrogen can substantially enhance the environmental benefits. Among these compounds, ETY has broad industrial uses as a precursor for ethylene oxide, polyethylene or polyvinylchloride production.4 This makes the low-temperature bioethanol-to-ethylene route an environmentally friendly process at low energetic cost, most importantly because it avoids C–C bond cracking into CO2. Given the endothermic nature of the direct ET-to-ETY route (eqn (1)), the challenge at low temperature is to find new catalytic routes to ETY converting the intermediates arising from the exothermic reactions (i.e. DEE; eqn (2)). This suggests that catalytic proposals at low temperature will require new intermediate pathways beyond an optimal balance between the rates of the endo- and exothermic reactions described in eqn (1)–(4).| CH3CH2OH ↔ CH2 = CH2 + H2O ΔH298K = +44.9 kJ mol−1 | (1) |
| 2CH3CH2OH ↔ (CH3CH2)2O + H2O ΔH298K = −25.0 kJ mol−1 | (2) |
| CH3CH2OH ↔ CH3CHO + H2O ΔH298K = +68.7 kJ mol−1 | (3) |
| CH3CH2OH ↔ CH4 + CO + H2 ΔH298K = +49.0 kJ mol−1 | (4) |
The reported acid- or metal-based catalysts led to significant amounts of DEE at <250 °C, while the endothermic ETY production predominates at 300–500 °C. Dehydrogenation reactions to acetaldehyde (eqn (3)) also occur above 500 °C, while thermal decomposition or cracking of ET occurs at higher temperature to produce biogas or syngas mixtures (eqn (4)).3,5–8 ETY selectivity can be controlled via adequate catalyst surface features, reaction temperature, reactant space-velocity or H2O co-feeding. Few works achieved marked ETY selectivities below 225 °C, which is more interesting from energetic and environmental perspectives.9 Such mild conditions limit ET degradation into coke or aromatics formation, as observed for acid-catalysts such as HZSM-5 at temperatures around 350 °C.10,11 These latter conditions block catalyst micropores, making their broad use unfeasible. It is thus important to understand how to minimize the formation of coke and heavy compounds via new routes and provide new insights into how to maximize ETY rates from ET dehydration at low temperature.
Various solid catalysts such as transition metal oxides,12,13 heteropolyacids,14,15 zeolites16,17 and functionalized mesoporous silicas16,18 were reported for ET conversion. Suitable surface acidity and geometry are essential to promote ETY formation. γ-Al2O3 with remarkable Lewis acidity was extensively studied7,19 and commercialized for the SynDol® process above 500 °C.7 Its broad thermal stability and life cycle, however, remain a veritable challenge. Several improvements were explored for γ-Al2O3 by incorporating Fe oxides, SiO2 or MgO.14,20,21 Silico-alumino-phosphates (SAPOs) exhibited promising conversion and selectivity to light olefins (99.3% and 98.4%, respectively) at 450 °C, but acid sites still suffer from rapid deactivation.13,19,20 Mesoporous containers with acid-functions (i.e. Al-SBA-15 and Al-MCM-41) are catalytically more active.22,23 The expensive organic templating synthesis, however, limits their broad industrial application. In turn, zeolites such as Beta, Y, Mordenite, and Mobil Five (MFI) show promising ET-to-ETY yields.10,24–28 The modification of MFI-type zeolites with noble metals (Ru, Rh, Pd, Pt, etc.10), transition metals (Co, Ni, Cu, Fe, etc.10,29,30), rare-earth elements (Ce, La, etc.21,31,32), or non-metals (P, Ga, etc.10,16,21,29) significantly enhances ETY yields thanks to an increase in weak acidity. These catalysts, however, lead to fast deactivation rates. Among these zeolites, HZSM-5 zeolites possess a well-arranged 3D structure with attractive physiochemical properties such as high surface area, diffusivity, tunable acidity and good hydrothermal stability.25 Many mechanistic and kinetic studies attempted to elucidate their catalytic behavior for ethanol dehydration to ethylene.28,33–37 At temperatures slightly higher than 260 °C, HZSM-5 usually exhibits ETY selectivity and nearly complete ET conversion. However, such temperatures and narrow micropores (∼0.5 nm) facilitate pore-blocking.18,28 Several strategies are chosen to enhance ETY selectivity at low temperature: bottom-up synthesis (i.e. nanomaterials preparation and soft/hard templating17,38) and top-down modification (i.e. post-treatment dealumination or desilication11,28,39–42), which involve the formation of larger mesopores in ZSM-5 and a better control of their acid-site nature, strength and population. ZSM-5 nanosheets exhibit unique Brønsted acid-sites (BAS) over high external surfaces, which favor direct ETY formation.38 However, the acid ZSM-5 channels favor DEE formation.38 ZSM-5 channels at <100 nm greatly improved coke-resistance, featuring stable ETY selectivity at complete ET conversion.17 A dealumination treatment by steam can selectively remove Al from the MFI framework or transform Al(IV) in Al(VI), which allows controlling the strength/amount of Lewis acid sites (LAS) with respect to BAS.28,40,42 However, it usually modifies the MFI structure, particularly in Al-poor HZSM-5 structures, making them vulnerable under hydrothermal conditions (i.e. ethanol/H2O mixtures). In turn, a desilication treatment in ZSM-5 offers better control of enhanced mesopore formation and increase of weak acidity, improving the ET to ETY pathway without damaging the MFI framework.11,39,42 The hierarchization positively impacts the interconnectivity in HZSM-5, improving the internal diffusion of reactants/products through additional meso-channels. Even when works on HZSM-5 zeolites bring acceptable improvements in ETY selectivity and ET conversion and catalytic stability at a low temperature (<250 °C), a deeper understanding of these phenomena has still not been achieved. For example, the reported modifications are usually carried out in aqueous media.43,44 There is, however, no consensus on how water affects the surface characteristics of zeolites.37,45,46 To the best of our knowledge, a systematic study on the role of water as a function of ZSM-5 acidity is still not reported for the low-temperature ET-to-ETY route. Thus, these synthetic strategies are to be accompanied by a balanced strength and density of acid sites, as well as the elucidation of the role of water and the reaction temperature in catalysis.
The present study integrates a new approach that combines the modification of acid MFI zeolites with systematic kinetic analyses to provide new mechanistic insights into the ET-to-ETY route at 225 °C. We report how controlling the surface physiochemical properties of hierarchical HZSM-5 via OH-treatment and catalytic intermediates affects the ET-to-ETY catalysis underflow. While the usual zeolite modifications follow NaOH treatments to achieve different desilication degrees, here, we observe that marked catalytic consequences can also be achieved solely by treating with water, leading to the modification of the Al3+ environment. These modifications aim at unveiling and modulating the contribution of the OH content (as in water or in NaOH) on the number and strength of acid sites. The effect and role of such parameters at low temperature are scarce, especially as the origin of ETY yield enhancement is not clear.32 In an attempt to understand the effect of water during the alkali treatment, we also have water-treated parent HZSM-5 in the ex situ liquid- or in situ vapor-phase. Systematic analyses by NH3-TPD (temperature-programmed desorption), N2 sorption and 27Al-NMR (nuclear magnetic resonance) spectroscopy correlate ETY with the appearance of new extra-framework (EF) penta-coordinated Al(V) species emerging from water treatment. The obtained insights would help propose new catalytic pathways to ETY that explain the role of water in ethanol dehydration or as a co-feed, as it occurs in industrial bioethanol streams containing nearly 5% water.19,47 Such insights help propose a concerted Lewis- and Brønsted-acid catalytic contribution at low-temperature. This way, this work aims to provide new perspectives to progress into the design of low-temperature dehydration of bioethanol into ethylene.
2. Experimental procedures
2.1. Reactants and starting zeolite materials
Four types of NH4-form ZSM-5 zeolites (with SiO2/Al2O3 commercial molar ratios of 23, 50, 80, 280) were purchased from Zeolyst. The solids were transformed into H-form ZSM-5 by calcination under synthetic air with a ramp of 1 °C min−1 up to 550 °C for 5 h. The following chemicals are used: sodium hydroxide (NaOH, 99%) from Merck, ammonium nitrate (NH4NO3, 98.9%) from Acros Organics, NH3/He (5%) and N2 (99.9%) from Air Liquide, ethanol (C2H5OH, 99.8%) from PanReac, and in-house deionized water.2.2. Zeolite modification
The alkaline treatment of all H-form ZSM-5 zeolites was performed in an aqueous solution using NaOH solutions with two different concentrations (0.1 M, 1.0 M), two distinct temperatures (65 °C, 85 °C) and two treatment times (30 min, 60 min). In a typical experiment, about 3 g of the sample was vigorously stirred in the target NaOH solution at a given concentration, temperature and time. Subsequently, the alkaline solution was quenched by inserting the flask in ice-water. The solids were recovered by filtration and contacted with deionized H2O in subsequent steps. The as-obtained powder was treated under static air at 100 °C and converted into ammonium forms by three consecutive exchanges in 30 mL NH4NO3 (1.0 M). The solids were then transformed into H-form ZSM-5 by following air treatment in Section 2.1. Samples were named HZ(y)_x_OH, where y refers to the SiO2/Al2O3 molar ratio and x_OH refers to the initial NaOH molar concentration in solution.The modification by water in H-form ZSM-5 was similarly performed but in the absence of NaOH: treatment at 65 °C by aging the HZSM-5 powder under stirring in deionized water for 30 min. The samples named in-synthesis (flask solution) were filtered and treated overnight under static air at 100 °C and treated with synthetic air underflow at 1 °C min−1 and kept at 550 °C for 5 h. The samples named in-reactor were treated once the sample was placed in the reactor bed. These samples were treated under analogous conditions (65 °C) but under flowing water (0.04 mL min−1), and subsequently treated underflow of synthetic air at 100 °C for 5 h and at 1 °C min−1 until 550 °C for 5 h.
2.3. Catalyst characterization
The parent and treated HZSM-5 zeolites were examined by complimentary techniques to evaluate their elemental composition, texture, acidity and local chemical environment. Si and Al contents were determined by Inductively Coupled Plasma Optic Emission Spectroscopy (ICP-OES) using an OPTIMA 2000 OV apparatus (PerkinElmer). Prior to the analysis, about 50 mg of each sample was dissolved in an acid mixture (3 mL HNO3 and 2 mL HCl) at 180 °C (30 min) using a microwave digestor Ethos1 (Milestone). The textural properties in solids were evaluated by N2 physisorption at −196 °C, using an automated TriStar II plus apparatus (Micromeritics). Prior to each analysis, about 100 mg was degassed for 24 h under vacuum at 300 °C. The data were collected and treated using the MicroActive Software.The total acidity was evaluated by NH3-TPD using an Autochem II instrument (Micromeritics). Typically, 0.15 g of each zeolite was charged in a quartz cell and pre-treated in He (20 mL min−1) at 500 °C (10 °C min−1) for 1 h. After this, the sample was cooled to 100 °C in He and treated with 40 mL min−1 10% NH3/He for 2 h. Subsequently, the sample was treated for 2 h with 20 mL min−1 He at 100 °C to remove the weakly adsorbed NH3. The NH3-TPD profile was recorded by heating the sample up to 750 °C (10 °C min−1), while acid-sites were quantified using a TCD detector. A blank test was used as a reference, while the TCD signal was calibrated to the corresponding NH3 concentration range.
The coordination Al species in the framework of chosen HZSM-5 samples were analyzed by 27Al MAS-NMR using an Avance III 400 MHz NMR spectrometer (Bruker). 27Al nuclei were studied at resonance frequencies of 104.3 MHz by using a single-pulse π/2 excitation and a repetition every 0.5 s. Before the measurements, the samples were exposed in a desiccator to saturated vapors of Ca(NO3)2 solution at room temperature overnight. Hydrated powder of Al(NO3)3 was used as an external reference for the 27Al-NMR spectra. The 2D multiple-quantum MAS-NMR analyses were also performed over samples loaded with tri-methylphosphine oxide (TMPO) that was used as the probe molecule.
Fourier-transform infrared (FTIR) analysis was performed using a Nicolet iS50 spectrometer. The chosen samples were firstly calcined ex situ at 500 °C (5 h) and then compressed into a fine self-supporting disc (∼13.875 mg cm−2). Subsequently, they were introduced into the infrared cell and in situ heated at 350 °C and evacuated at 10−7 mbar overnight to remove moisture. After cooling under vacuum to room temperature, an FTIR spectrum was immediately recorded in the region of 4000–600 cm−1 using a resolution of 4 cm−1 and 128 scans. After background subtraction, the FTIR spectra were normalized for comparison.
2.4. Testing setup and catalytic evaluation
The catalytic performance in ET dehydration was evaluated at 225 °C using an automated plug-flow reactor set-up. The reaction temperature was controlled by a K-type thermocouple in contact with the catalyst bed. The total pressure was set to 1 bar by using 90 mL min−1 of pure N2 as the carrier gas. In a typical catalytic test, 0.12 g of the HZSM-5 sample was placed in the center of the reactor bed, and the sample was treated at 500 °C under 90 mL min−1 N2 for 2 h. The reactor was then cooled to the desired reaction temperature and the reactant (ET) was introduced using a HPLC pump at 0.04–0.23 mL min−1, obtaining a weight hourly space velocity (WHSV) of 17.9–89.1 gET gCAT−1 h−1. Analogous catalytic tests were performed with DEE as the reactant but fed through a N2 stream flow in a bubbler containing DEE at room temperature. In both cases, the reactor effluents were injected through heated lines into a gas-chromatograph (Agilent 6890, DB-5), equipped with FID and TCD detectors. The inlet and outlet C-balance was calculated after calibrating the reactant and product compounds using ET, H2O, DEE, N2 and ETY mixtures. The catalytic performance was determined as:- Reactant (i) conversion (Xi (%)):
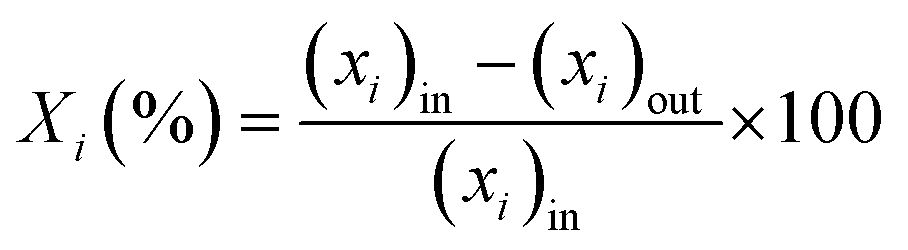 | (5) |
- Product (j) selectivity (Sj (%), C-based)
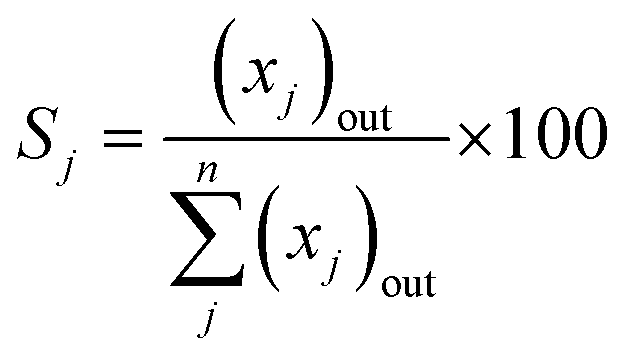 | (6) |
2.5. Calculation method
Density-functional theory (DFT) calculations were performed using the Vienna ab Initio Simulation Package (VASP) and plane wave basis sets.48–51 The electronic interactions were described using the projector–augmented wave (PAW) method52,53 with a plane-wave energy cut-off value of 600 eV. The exchange–correlation energies were calculated on the basis of the generalized gradient approximation (GGA) according to Perdew, Burke and Ernzerhof (PBE) functional.54 Brillouin zone sampling was restricted to the Γ-point. A maximum force convergence criterion of 0.02 eV Å−1 was used and each self-consistency loop was iterated until a convergence level of 10−6 eV was achieved. All atoms are free during the calculation. The adsorption energy of molecule A are calculated by:| ΔE = E(surf+molecule A) − E(surf) − E(molecule A) | (7) |
3. Results and discussion
3.1. Ethanol dehydration in parent and desilicated HZSM-5
| Catalyst | Treatment conditions | Elemental analysis | Textural properties | Surface acid-features (mmolacid gCAT−1) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| COH (M) | Temperature (°C) | Time (min) | SiO2/Al2O3 | S BET (m2 g−1) | V Total (cm3 g−1) | S μ (m2/g | V μ (cm3 g−1) | d μ (nm) | V meso (cm3 g−1) | d meso (nm) | WAS | SAS | WAS/SAS ratio | |
| CNaOH: molar concentration of OH from NaOH in solution; SiO2/Al2O3 molar ratio determined by ICP-OES; SBET: specific surface area estimated by the Brunauer–Emmett–Teller (BET) equation applied to the isotherm of N2 physisorption (P/P0 ∈ 0.10–0.3.0); VTotal: total pore volume estimated at single point P/P0 = 0.95; Sμ/Vμ: micropore surface/volume calculated by the “t-plot” method; dμ (nm): average pore diameter obtained by the Horvath–Kawazoe (HK) method; Vmeso: mesopore volume obtained by the Barrett–Joyner–Halenda (BJH) model applied to the desorption profile; dp: average pore diameter obtained by the BJH model. WAS and SAS; density of weak and strong acid sites measured from NH3-TPD profiles. | ||||||||||||||
| HZ(23) | — | — | — | 22.6 | 401 | 0.292 | 179 | 0.092 | 0.35 | 0.138 | 3.8/5.2 | 0.22 | 0.18 | 1.22 |
| HZ(23)_0.1OH | 0.1 | 65 | 30 | 20.4 | 383 | 0.257 | 217 | 0.105 | 0.33 | 0.110 | 3.8/7.3 | 0.24 | 0.18 | 1.33 |
| HZ(23)_H2O | 10−7 | 65 | 30 | 21.9 | 384 | 0.249 | 229 | 0.108 | 0.40 | 0.109 | 3.8/6.3 | 0.26 | 0.19 | 1.37 |
| HZ(23)_1.0OH | 1 | 85 | 60 | 13.3 | — | — | — | — | — | — | 0.02 | 0.01 | 2.00 | |
| HZ (50) | — | — | — | 48.8 | 405 | 0.297 | 198 | 0.103 | 0.38 | 0.152 | 3.7 | 0.10 | 0.08 | 1.25 |
| HZ(50)_ 0.1OH | 0.1 | 65 | 30 | 45.3 | 422 | 0.333 | 195 | 0.098 | 0.37 | 0.206 | 3.7–7.2 | 0.18 | 0.10 | 1.80 |
| HZ(50)_1.0OH | 1 | 85 | 60 | 24.4 | — | — | — | — | — | — | 0.11 | 0.10 | 1.10 | |
| HZ (80) | — | — | — | 72.2 | 428 | 0.284 | 209 | 0.105 | 0.39 | 0.117 | 3.8–5.7 | 0.07 | 0.05 | 1.40 |
| HZ(80)_0.1OH | 0.1 | 65 | 30 | 51.6 | — | — | — | — | — | — | — | 0.08 | 0.06 | 1.33 |
| HZ(80)_1.0OH | 1 | 85 | 60 | 42.4 | 484 | 0.611 | 169 | 0.087 | 0.38 | 0.611 | 3.8–18.6 | 0.17 | 0.07 | 2.43 |
| HZ(280) | — | — | — | 273.2 | — | — | — | — | — | — | — | 0.009 | 0.012 | 0.75 |
| HZ(280)_1.0OH | 0.1 | 85 | 60 | 253.2 | — | — | — | — | — | — | — | 0.01 | 0.02 | 0.50 |
The ratio between weak (WAS) and strong (SAS) acid-sites is derived from the integration of deconvoluted NH3-TPD profiles (Fig. S2†). Both parent and OH-treated samples contain two distinct peaks at 100–300 °C (assigned to WAS) and at 300–600 °C (assigned to SAS).58 Low OH− concentrations (0.1 M) are sufficient to duplicate the WAS content in HZ(23) and HZ(50) samples, while their SAS is essentially unaffected by such treatment. The number and strength of surface acid-species, namely Lewis – (L) and Brønsted – (B) acid sites, are caused by the presence of Al species in Si-frameworks.59 While B sites originate from the formation of H+ sites by charge compensation of Al3+ exchanged in Si4+ species, L sites originate from the electron sharing ability of Al species coordinated with OH species in distinct (IV, V) arrangements. The negligible changes in SAS contents for HZ(23)_0.1OH and HZ(50)_0.1OH can be attributed to the absence of new Si4+–Al3+ bonds leading to new H+ species. The WAS sites, present as L sites, however, arise as desilicated samples uncover Al species that were not accessible in parent zeolites. This leads to an increase in the formation of new Al(δ)–OH species with marked characterization and catalytic performance differences (as discussed later in Section 3.2.1).
On the other hand, aggressive OH− treatments (1.0 M) lead to marked a desilication degree in HZ(23) and HZ(50) and drastically reduce the population of both surface acid types (Table 1). This is attributed to the partial dissolution of the zeolitic framework, leading to a disordered structure without the ability to form H+ species or yield accessible weaker Al(δ)–OH species. Such alkaline conditions, however, are necessary to observe significant changes in acid-site content for samples with a higher SiO2/Al2O3 ratio, as it occurs for HZ(80). In this case, HZ(80)_1.0OH enhances WAS by a factor 2.4 and SAS by a factor of 1.4 with respect to its parent HZ(80) sample. The HZ(280) sample barely shows any changes in the acid-site content (even at OH− contents up to 2.0 M), indicative of the large hydrothermal stability of such Si-rich samples even under very alkaline conditions.
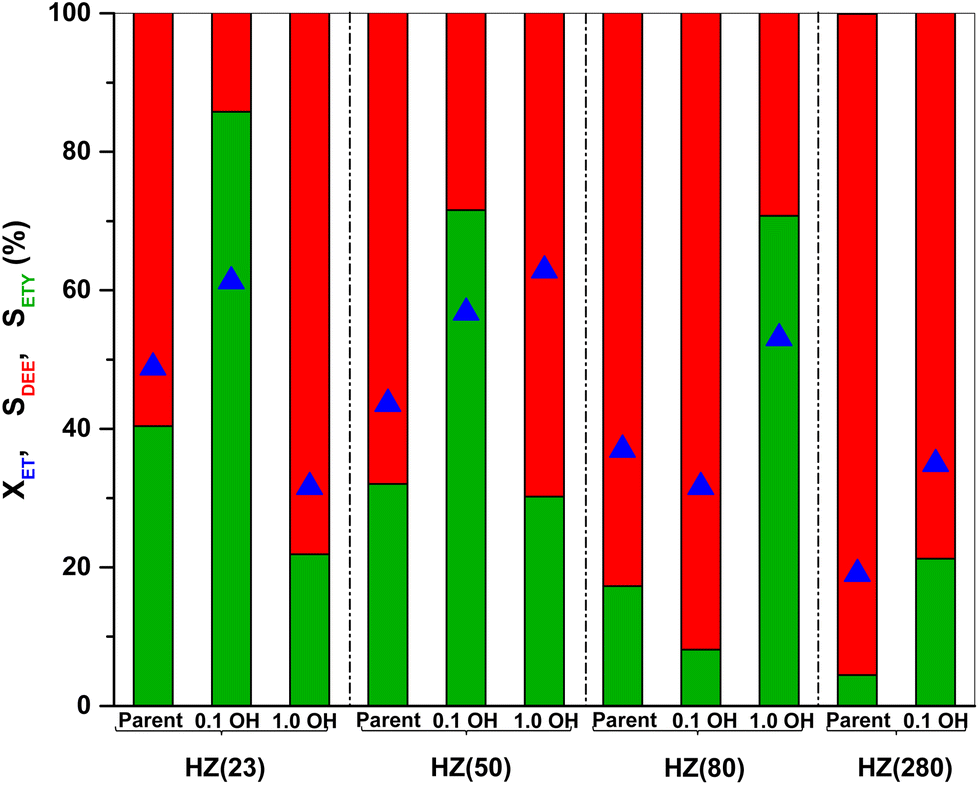 | ||
| Fig. 1 Conversion of ET (blue), selectivity of ETY (green) and of DEE (red) provided by parent HZSM-5 catalysts and by their counterparts treated in alkaline solution (values estimated at steady-state of reaction (5–6 h)). Test conditions: 225 °C, 1 bar, WHSV = 35.7 h−1. | ||
The SETY changes in Fig. 1 seem to be more affected by a variation in WAS content rather than in SAS. Fig. 2 shows SETY for parent and OH-treated samples, each at a similar XET value, as a function of the WAS content. The rigorous comparison at such XET values intends to neglect the kinetic contribution of the ET-derived product concentration in promoting secondary reactions at distinct XET, as shown in Section 3.2.3. Trends for the parent or OH-treated samples correlate with the WAS content (Fig. 2). At given WAS contents, however, all OH-treated samples show an increase of 20–40% in SETY with respect to parent samples. This indicates that WAS content cannot be used as the sole descriptor to explain the observed ETY selectivity differences (Fig. 2), suggesting that H-form ZSM-5 zeolites suffer a physicochemical modification from OH in solution that cannot be elucidated from NH3-sorption data. Given that OH-treated materials originate in different parent materials, with the consequent divergence in the degree of modification, but follow the same trend with the WAS content, the data in Fig. 2 indicate that OH-treatments have a larger influence (i.e. form more new active species) in samples with a higher Al content (HZ(23), HZ(50)). This indicates that such OH-contact predominantly leads to the modification of the Al environment. In contrast, if new active species (such as Si–OH derived from OH− in solution) were formed, samples with more abundant Si atoms (HZ(80)_1.0OH, HZ(280)_1.0OH) would have deviated from the trends shown in Fig. 2 (which is not the case). Observed catalytic consequences from OH-treatments provide the chance to propose new reaction pathways beyond the direct routes (eqn (1) and (2)), which could presumably be affected by the unexplored catalytic phenomena (new Al–OH species, diffusional effects, cooperative catalysis, etc.). In doing so, it is expected that the presence of H2O in synthesis or in ET dehydration may seemingly affect the catalytic surface and the behavior of zeolites towards ETY formation.
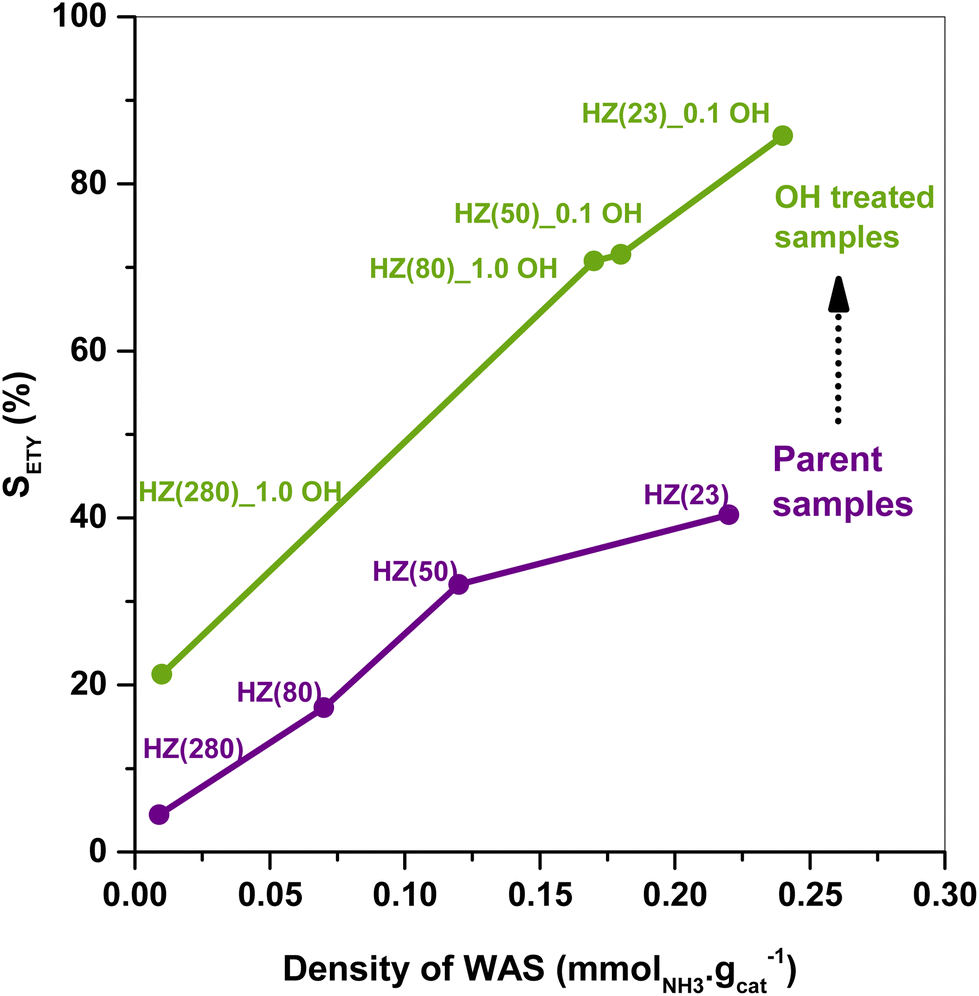 | ||
| Fig. 2 Correlation between the ETY selectivity (at similar ET conversion) and the density of WAS for parent (purple) and OH-treated (green) HZSM-5 catalysts tested at 225 °C, 1 bar, WHSV = 35.7 h−1. SETY data are obtained under stable catalytic behaviors. | ||
3.2. Effect of H2O treatment on HZSM-5 during ET dehydration
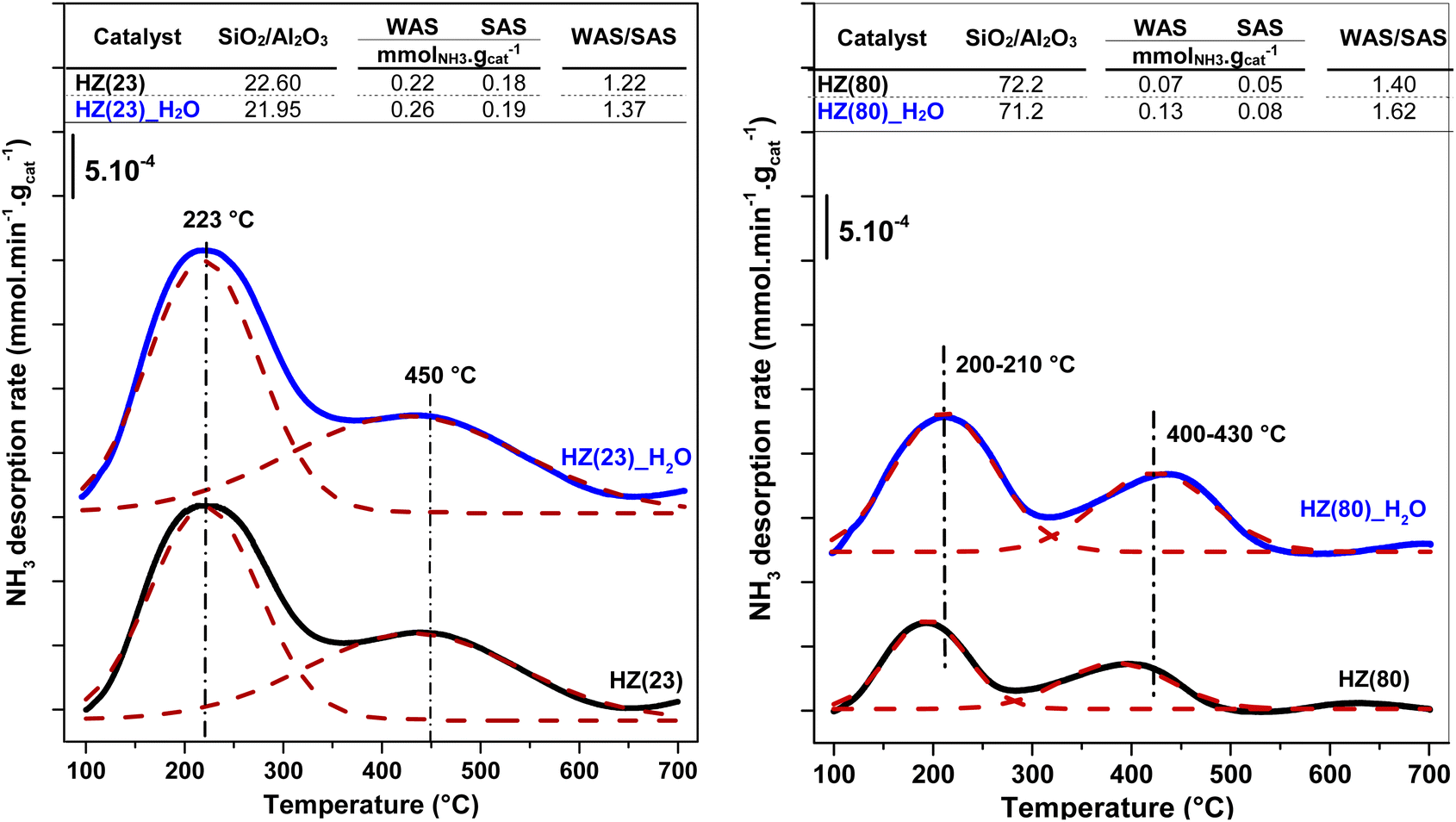 | ||
| Fig. 3 Elemental content and acidity changes of parent zeolites (black); HZ(23)-left and HZ(80)-right, compared their H2O-treated counterparts (blue). | ||
To unveil the consequences of H2O-treated Al-species in ET dehydration, solid-state 27Al MAS-NMR analyses were performed for HZ(23)_H2O and HZ(80)_ H2O zeolites and their parent counterparts. The 27Al MAS-NMR spectra (Fig. 4A and B) clearly indicate that parent HZ(23) and HZ(80) samples feature two signals: an intense peak centered at chemical shifts of 51 ppm and a small peak centered at −3.7 ppm. The first one corresponds to tetra-coordinated Al(IV) and the second one to hexa-coordinated Al(VI).42 Al(IV) coordination is mainly reported as a precursor for the formation of Si–OH–Al bridging generated via the substitution of Si4+ by Al3+ in the HZSM-5 framework, which leads to the appearance of B acid-sites. It is widely accepted that B in HZSM-5 are generated by H+ protons to compensate for the deficit of charges in the zeolite framework caused by Al(IV) species.60–62 The difference in the signal intensity between tetra-coordinated Al(IV) and hexa-coordinated Al(VI) is indicative of the B acid-site content in HZSM-5, which as expected is more pronounced for HZ(23). The treatment with H2O leads to both qualitative and quantitative changes in Al coordination (Fig. 4A–D). The spectra for HZ(23)_H2O become less intense but very broad and non-symmetrical, due to the overlapping of signals related with new Al(δ) species. The Gaussian deconvolution of this curve reveals the existence of additional extra-framework (EF) Al(IV), Al(V) and Al(VI) species. In situ FTIR spectra presented in Fig. 5 confirm a significant increase in the intensity of the bands at 3663 cm−1 for HZ(23)_H2O, which is mainly attributed to partially hydroxylated EF–Al(δ)–OH species acting as Lewis sites.60–62 The other bands (observed between 3740–3780 cm−1), which are generally attributed to the presence of different types of isolated silanol Si–OH groups, while the peak at 3611 cm−1 belongs to isolated bridged Si–O(H)–Al groups, remain practically in the same order of intensity. The apparition of new EF–Al(IV) and EF–Al(V) species might be caused by the interactions of bulk Al(IV) species with H2O and their transformation into Al(δ)–OH species after calcination, since a considerable decrease in the Al(IV) peak intensity is observed. These EF–Al(IV) species may also act as B sites on amorphous silica-alumina and zeolites,63 while EF–Al(V) and EF–Al(VI) originate from the hydroxylation of Al(IV) and Al(VI), respectively, leading to new surface L sites.64 In turn, HZ(80)_H2O features a substantially different tendency, as its spectra slightly widen at 51 ppm, whilst the shift at −3.1 ppm remains unchanged. Such negligible changes also occur due to the low Al content. The changes in the Al environment (Fig. 4A–D) may be related to an increase of WAS (NH3-TPD, Fig. 3), but based on the two distinct SETY trends observed in Fig. 2, not all the new Al(δ)–OH species seem to contribute to the appearance of new acid-sites, whilst they could have positive catalytic consequences during ETY formation. Based on the probability that higher Al contents lead to more abundant and detectable changes in the Al environment, HZ(23)_H2O is expected to show catalytic consequences during ET dehydration, as shown in the following.
 | ||
| Fig. 4 27Al MAS-NMR spectra of (A) parent HZ(23) and water-treated HZ(23); (B) HZ(80) and water-treated HZ(80). The (C) and (D) figures represent 27Al 3QMAS-NMR data for HZ(23) and HZ(23)_H2O respectively. | ||
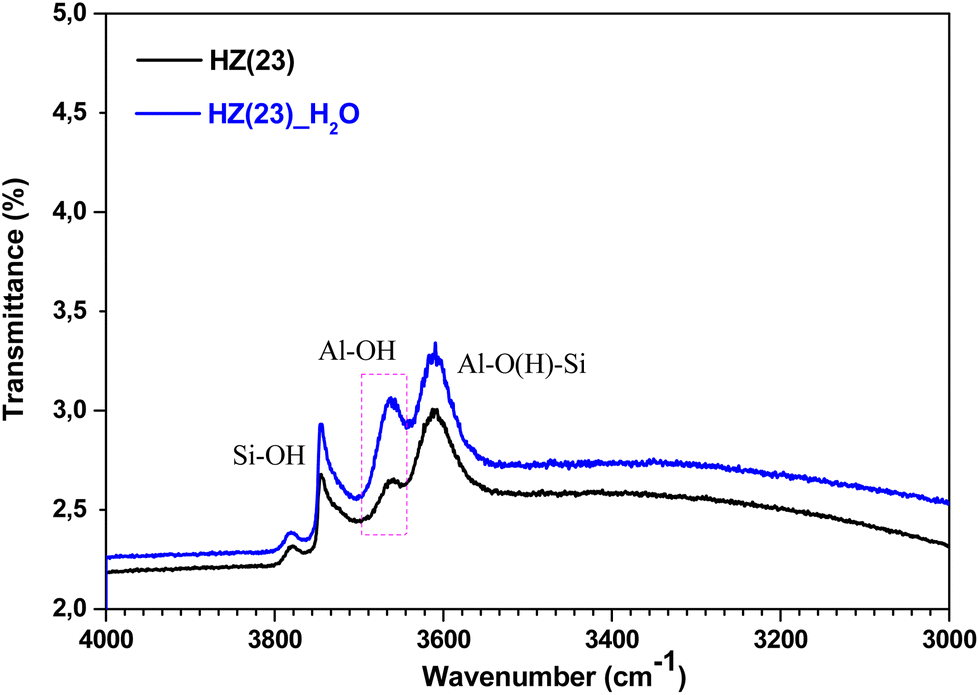 | ||
| Fig. 5 Normalized FTIR spectra of the OH-region for fresh parent HZ(23) and HZ(23)_H2O samples evacuated in situ at 350 °C (overnight) after ex situ calcination at 500 °C (5 h). | ||
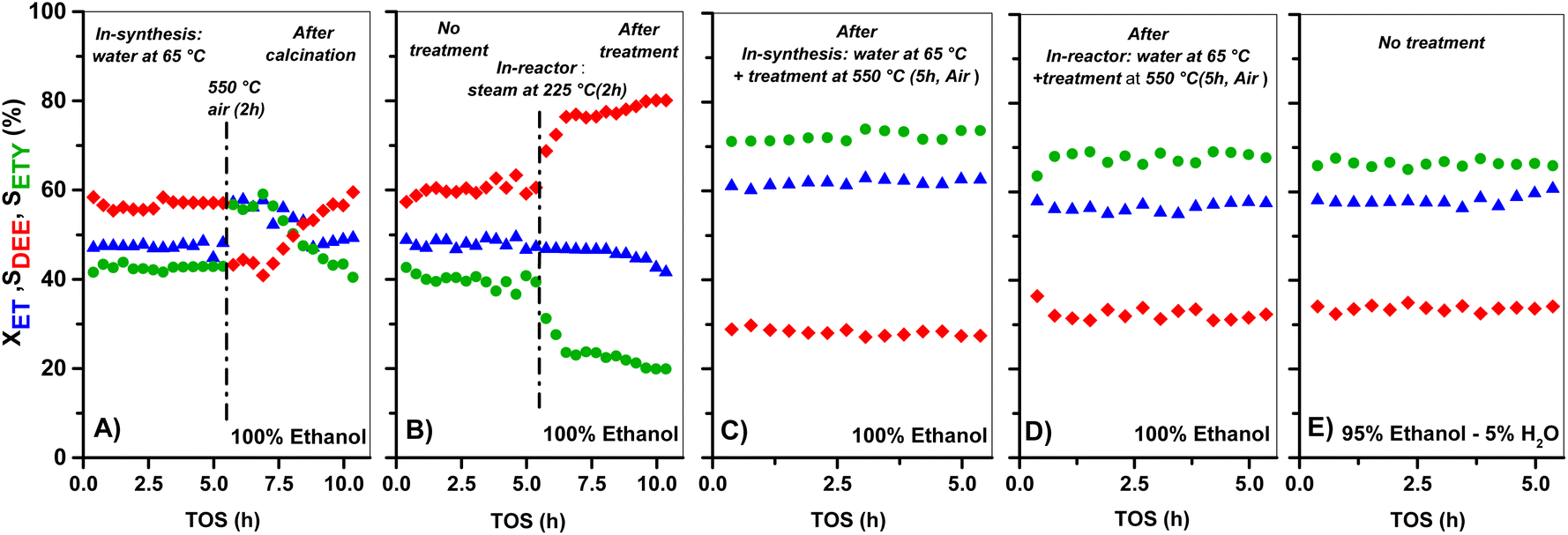 | ||
| Fig. 6 Evolution of ET conversion (XET), and selectivity to ETY (SETY) and DEE (SDEE) as a function of TOS for: (A) HZ(23) treated with water at 65 °C and in situ calcined; (B) HZ(23) treated in situ with steam at 225 °C; (C) HZ(23) treated with water at 65 °C and calcined ex situ, (D): HZ(23) treated with water at 65 °C and calcined in-reactor; (E) HZ(23) (only in H+-form) catalytic activity using 95%/5% ethanol/water as the feed. All data shown for 225 °C, 1 bar total pressure and WHSV = 35.7 h−1. | ||
The XET and product selectivity data in Fig. 6E, obtained by co-feeding ethanol with water at 225 °C, confirm the positive effect from water. Even when such water (gas-phase at 225 °C, Fig. 6E) is equivalent to the steam conditions shown in Fig. 6B, the minor water-concentration or the presence of ethanol species does not result in negative catalytic consequences towards ETY. In contrast, SETY increases from 40% (pure ethanol, Fig. 1) to 60% (5% water in ethanol, Fig. 6E) at similar XET, suggesting that co-fed water molecules may have a positive kinetic effect in inhibiting possible ethylene side-reactions, leading to higher SETY values. It is worth noting that ethanol dehydration conversions of around 50–60% (as shown in Fig. 1) already produce substantial amounts of water, but such conditions do not lead to similar catalytic consequences as observed in Fig. 6E, as discussed later. This indicates the relevance of the partial pressure of water in the feed,65,66 even when the adsorption of the ethanol reactant may differ from water (Section 3.3). Overall, the SETY data shown in Fig. 6 suggest catalytic benefits from water via catalyst modification or kinetic contributions, which justifies the use of nearly 95% ethanol feeding in ethanol-to-ethylene in industry.19 Such water effects, when compared to the catalytic consequences observed for solids treated with 0.1–1.0 M NaOH in synthesis (Fig. 1), corroborate the relevance of OH− species in solution for tuning HZSM-5 surface properties with low Si/Al contents. Some H2O dissociation may be favoring the apparition of new Al–OH sites, as suggested by 27Al-NMR (Fig. 4) and confirmed by FTIR analysis (Fig. 5). Such catalytic observations, however, are still inconclusive on the preferential routes to ETY (direct or indirect) that predominate for such H2O-treated samples. Thus, the control over H2O dissociation to OH− and H3O+ over BAS and/or LAS, and its role in metals cation modification is part of the ongoing research using wet ion-exchange and dry melt-infiltration of different acidic species (i.e. Al, Ce, La, P, Cs, etc.) into H-ZSM-5.
A similar water-treatment in HZ(80) yields analogous XET and SETY values (Fig. S3†) as for HZ(80)_0.1OH (Fig. 1), indicating that low OH concentrations in solution (∼10−7 M) are not sufficient to promote desilication with quantitative contribution to form ETY. This confirms that such high SiO2/Al2O3 samples require harsher OH-concentrations to open new porosity by reaching new Al sites and to generate significant changes in SETY (observed in Fig. 1, Fig. S1E and F;† discussed in section 3.2.3). These insights seem to be in good agreement with NH3-TPD (Fig. 3) and 27Al MAS-NMR (Fig. 4), indicating that OH-treatment modifies both acid features and Al-environment in samples containing a low Si/Al ratio (HZ(23)), with the apparition of new EF–Al(IV), EF–Al(V), and EF–Al(VI) species, and leads to detectable catalytic consequences (Fig. 6). Thus, the catalytic differences between OH-treated HZ(23) and HZ(80) do not seem to strictly relate to the nature or type of acid- or Al(δ)–OH sites, as they all originate in the same type of Si4+ or Al3+ species, but to their number on each catalyst surface. Among such sites, the contribution of water is mostly reflected in the considerable appearance of EF penta-coordinated Al(V) and EF hexa-coordinated Al(VI) species in HZ(23), which seem to be the origin of new L sites. The kinetic evidence between the parent and OH-treated samples allows an initial approximation to elucidate the origin of ETY from ET under studied reaction conditions, as discussed next.
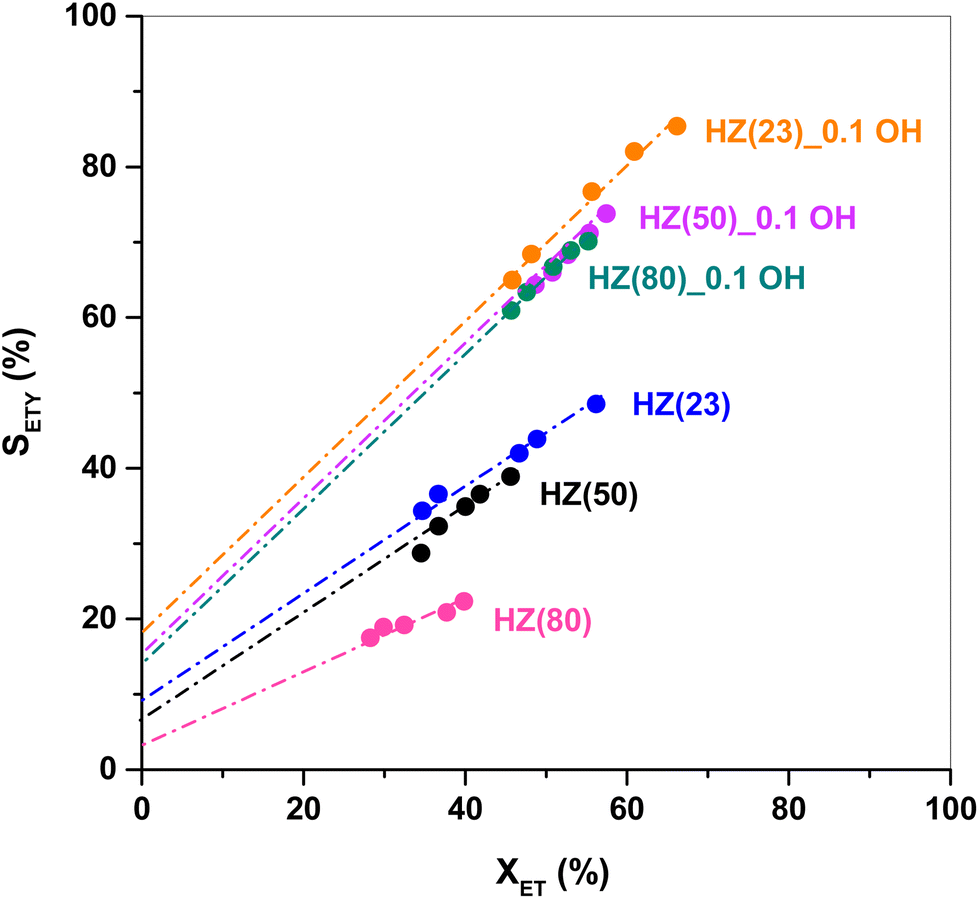 | ||
| Fig. 7 ETY selectivity (SETY) as a function of ET conversion (XET, changed with WHSV) over HZ(23), HZ(50), HZ(80), HZ(23)_0.1 OH, HZ(50)_0.1 OH and HZ(80)_0.1 OH. Test conditions: 225 °C, total pressure ∼1 bar, WHSV = 17.9–89.1 h−1. | ||
Fig. 8 shows the evolution of DEE conversion, and ETY and ET selectivity as a function of TOS for parent HZ(23) and HZ(80) and those OH-treated samples with detectable kinetic effects. In order to simplify the interpretation of data, even when H2O is detected as the product, the estimates in Fig. 7 neglect H2O in the selectivity term (eqn (6)). The nearly stoichiometric ETY and ET amounts for HZ(23) shown in Fig. 8A, as well as the absence of other dehydration products (in the GC), suggest that ETY and H2O form first via DEE cracking (eqn (8)) and subsequently via the macroscopic reverse of eqn (2) to give ET from DEE and H2O (eqn (9)). Both eqn (8) and (9) lead to eqn (10), which yields the observed stoichiometric ET and ETY ratio shown in Fig. 8A.
| (CH3CH2)2O ↔ 2CH2 = CH2 + H2O | (8) |
| (CH3CH2)2O + H2O ↔ 2CH3CH2OH | (9) |
| (CH3CH2)2O ↔ CH2 = CH2 + CH3CH2OH | (10) |
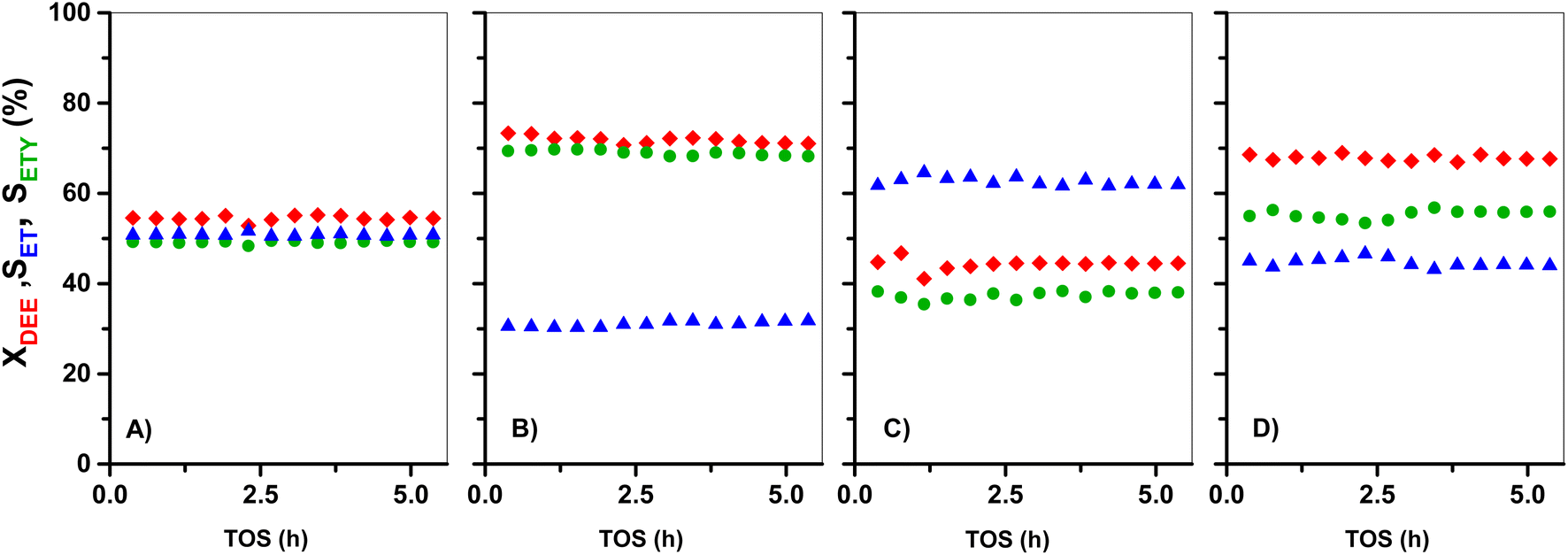 | ||
| Fig. 8 Conversion of pure DEE (XDEE, red) and selectivity of ethylene (SETY, green) and of ethanol (SET, blue) as a function of TOS for: (A) HZ(23), (B) HZ(23)_0.1OH, (C) HZ(80), and (D) HZ(80)_1.0OH catalysts. Test conditions: 225 °C, 1 bar and WHSV = 35.7 h−1. | ||
Similar conclusions are obtained for DEE reactions onto parent HZ(80) in Fig. 8C. The ETY and ET ratio, however, slightly deviate from expected values from eqn (10), which may originate from experimental errors. In contrast, the OH-treated in both HZ(23) and HZ(80) lead to a considerable increase of both XDEE and SETY. This reflects that the materials not only have a significantly bigger number of DEE cracking sites (emerging from OH treatment), but that these new sites also inhibit the formation of ET from DEE (eqn (10)). This leads to over-stoichiometric ETY/ET ratios, mostly evident for HZ(23)_0.1OH (Fig. 8B). These effects can only be attributed to the presence of Lewis-acid type Al(δ)–OH sites that inhibit the adsorption of H2O through repulsive interactions during DEE cracking into ET (eqn (9)), as discussed later from DFT inputs. This seems to be a clear confirmation of the predominance of DEE cracking reactions to give ETY when ET is used as the feed (Fig. 1, 2, 5 and 6). This becomes even more evident when such ET is co-fed with water (Fig. 6E). The proposal of a new indirect or secondary pathway to form ETY from ET seems thus related to the polarity of the molecules in the reaction mixture and the ability of the surface to interact. Such a hypothesis is assessed here through theoretical calculations based on the adsorption of such molecules on Al-derived species in HZSM-5 and the formation of kinetically-relevant transition states, as discussed below.
3.3 Analysis of the reaction mechanism
Further DFT calculations have been performed in an attempt to better understand the ET to ETY catalytic pathway over different surface acid species on HZSM-5. The structures of HZSM-5 (Fig. 9) were optimized according to the MFI's topology, where the atomic proportion of Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O:Al:H was set at 92:192:4:4, respectively (see the ESI for further structural and coordination details†).
:O:Al:H was set at 92:192:4:4, respectively (see the ESI for further structural and coordination details†).
 | ||
| Fig. 9 The optimized periodic structure of HZSM-5 (atomic ratio Si/Al ∼23, a = 20.615223 Å; b = 20.430119 Å and c = 13.716548 Å, α = 90.0248; β = 89.76483; γ = 90.05055). Yellow: Si; red: O; purple: Al; white: protons. | ||
According to previous reports67 there are four possible Al positions in the ZSM-5 structure; T1, T2, T3, and T4 (in blue, Fig. 9), which depend on the geometrical and elemental configuration. Among these four positions, which are usually altered under an aqueous medium,68 T2 was chosen as the representative position for evaluating the interaction with ET, DEE and ETY molecules under the possible presence of H2O. This choice is supported by the fact that T2 sites are located at the junction of three 10-membered rings, while T1 sites are located at the junction of only two 6-membered rings and one 10-membered rings. This makes the T2 position more relevant from a geometric perspective, and especially to assess the adsorption of molecules with different steric and polarization phenomena. The adsorption energy for individual molecules was calculated at the T2 position, by considering two distinct acid-sites: Brønsted (B) and Lewis (L). The effects from OH-treatment in HZSM-5 seem to be more noticeable in Al(δ)–OH species that relate to L sites, whilst OH or water-treatments are not expected to affect B sites as the resulting species may not be stable under the air-treatment (calcination) prior to catalysis.
As shown in the Fig. 9, the adsorption of ET on the B site involves lower energy (−0.8 eV) than for the rest of the molecules. This lowest energy would allow adsorbed ET to preferentially react through H–O bonding to be directly dehydrated into ETY or dimerized into DEE. A similar trend is observed for L sites, but with DEE molecules showing the lowest adsorption energy (−0.5 eV). This preferential adsorption of DEE over L sites is a critical step for its subsequent conversion into ETY (eqn (8)). The energies for H2O are nearly undetectable for B sites and 0.29 eV for L sites, suggesting that both species do not favor H2O adsorption. Thus, the DEE-to-ET route on L sites seems less favorable as it would require H2O species to adsorb on neighboring sites to form ET from DEE + H2O (eqn (9)). This indicates that adsorbed DEE species would preferentially form ETY and H2O (eqn (8)) on L species with a certain polar character, such as Al(δ)–OH species. This would indeed coincide with the non-stoichiometric ETY/ET ratio observed in Fig. 8B, where DEE is converted in higher quantities into ETY than in ET for OH-treated samples in comparison to parent samples. Such preferential catalysis on L sites is here confirmed by DFT calculations that determine the energy to form the kinetically-relevant transition states (TS) when DEE reacts into ETY and ET. Fig. 10B shows the reaction coordinate diagram for the ET → DEE → ETY route, which intends to compare the critical TS for the DEE → ETY step. The energy barrier for the TS on L sites is lower (0.99 eV) than that on B sites (1.24 eV), indicative that such ETY formation from DEE is favored on L sites featuring Al(δ)–OH-like species. This would still produce stoichiometric amounts of ETY and ET, but the data in Fig. 10B clearly indicate the prevalence of such DEE cracking reactions to form ETY on L sites in comparison to B sites.
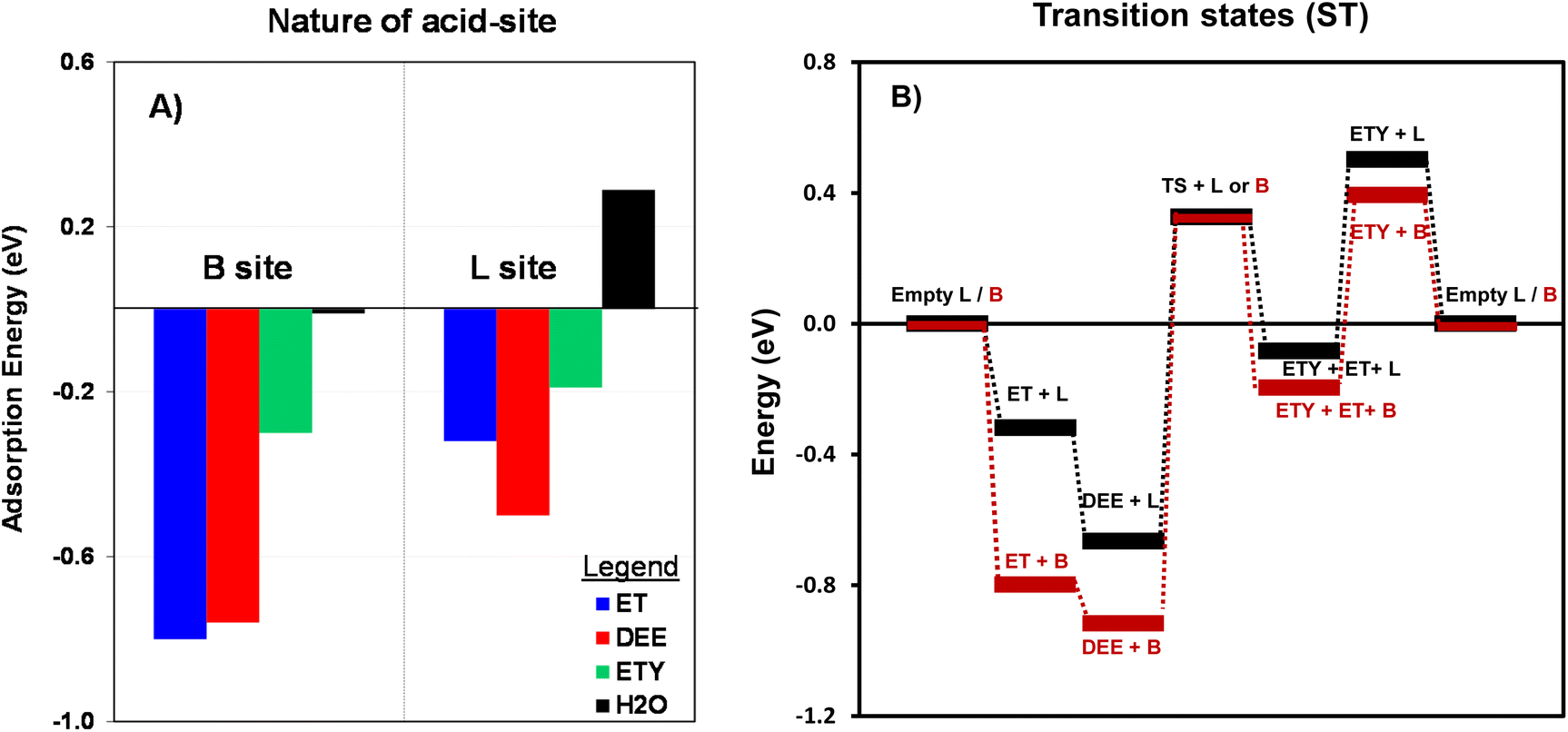 | ||
| Fig. 10 (A) The calculated adsorption energy (eV, eqn (8)) for ET, DEE, ETY and H2O on B sites and L sites for the T2 position (–Al–); and (B) Reaction coordinate diagram with DFT-derived energies normalized with respect to empty B or L sites at T2 positions. The TS for ET → DEE is not shown for simplicity. | ||
Based on the obtained experimental and theoretical outputs, we suggest a cooperative mechanism between B and L sites to produce ethylene from ethanol on HZSM-5 catalysts at low temperatures. This is shown via two reaction routes (Scheme 1):
(i) Primary: a sequential dimerization of two ET molecules catalyzed by species adjacent to B sites to form a DEE intermediate that is subsequently cracked on L sites to form ETY and H2O.
(ii) Secondary: a direct elimination of the hydroxyl group from ET to form ETY and H2O on B sites.
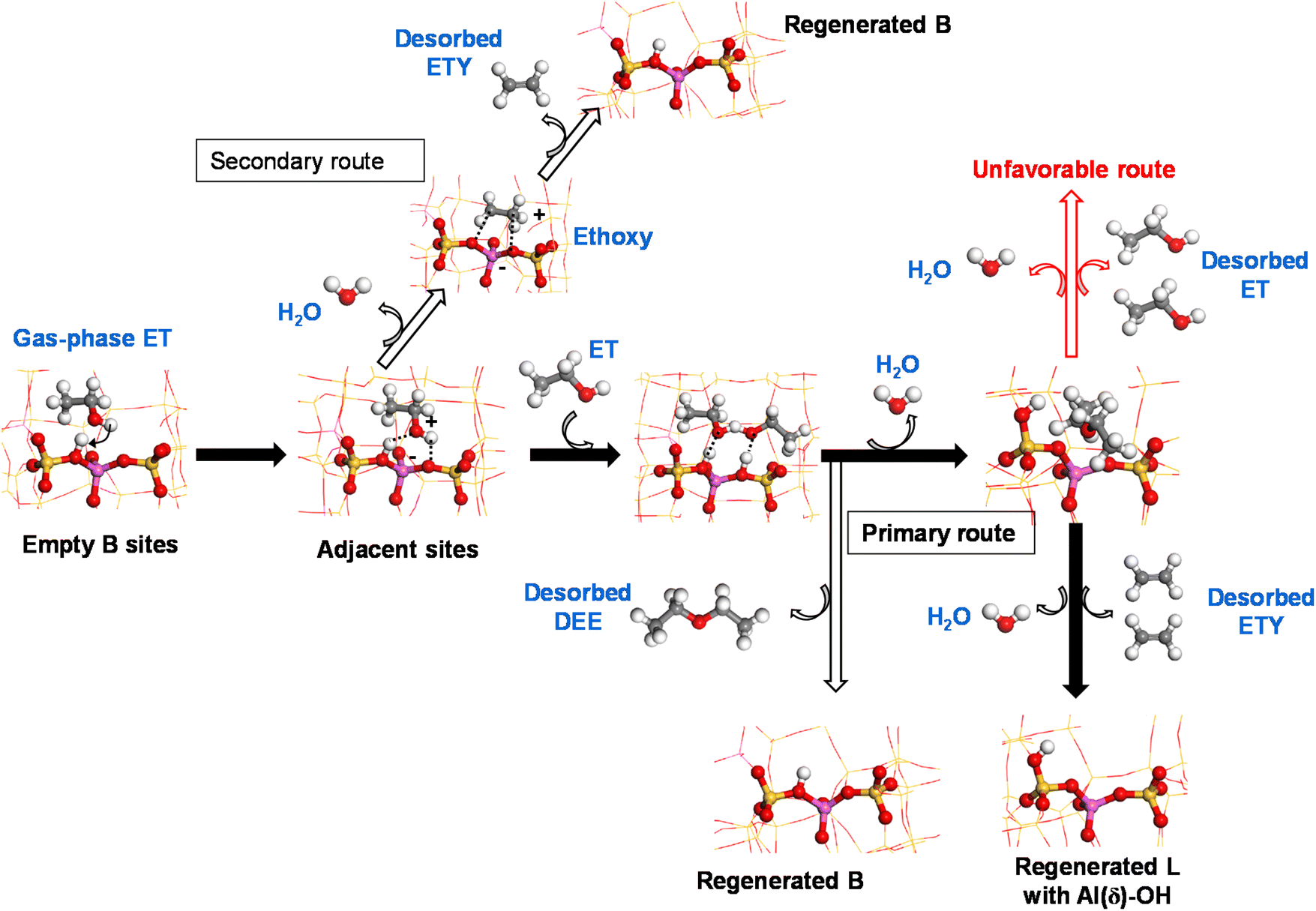 | ||
| Scheme 1 Identified routes for ET-to-ETY over HZSM-5 at low-temperature reaction via: (i) ET dimerization on neighboring sites on B species followed by DEE cracking on L (primary route), and (ii) direct elimination of the hydroxyl group of ET on B (secondary route). | ||
Following Scheme 1, ET is adsorbed over acid B sites (mainly Si–OH–Al) and dehydrated to form a surface-bound ethoxy intermediate after interacting with an adjacent OH group. This intermediate is thermodynamically unstable, leading to ETY by C–O cleavage and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) formation (secondary route) or immediately dimerized with another adsorbed ET on a neighboring similar site to form DEE (primary route). Previous DFT calculations38 reported that ethoxy species can be converted into ETY and DEE over external and internal –OH groups of the HZSM-5 framework, respectively. The abstraction of ethoxy intermediates by O to form ETY requires an activation energy of more than 41 kcal mol−1, while the formation of DEE at low temperature from dimeric ET requires only 25 kcal mol−1.69 This difference, which is mainly attributed to hydrophilic and acidic internal HZSM-5 channels,70 predominantly leads to DEE at low temperatures. These observations were confirmed by the experimental data in the present work (Fig. 7 and 8), revealing how DEE can re-adsorb on L sites (more probably in the form of EF–Al(V)) after contacting with H2O. Subsequently, two C–O bonds in DEE are decomposed by dehydration to form two ETY molecules in the gas-phase. This ET → DEE → ETY route is considered here as the primary route based on the experimental evidence using ET or DEE as reactants and HZSM-5 catalysts with different B (as SAS) and L (as WAS) contents, whilst the direct ET → ETY route seems to be less predominant on the studied system.
formation (secondary route) or immediately dimerized with another adsorbed ET on a neighboring similar site to form DEE (primary route). Previous DFT calculations38 reported that ethoxy species can be converted into ETY and DEE over external and internal –OH groups of the HZSM-5 framework, respectively. The abstraction of ethoxy intermediates by O to form ETY requires an activation energy of more than 41 kcal mol−1, while the formation of DEE at low temperature from dimeric ET requires only 25 kcal mol−1.69 This difference, which is mainly attributed to hydrophilic and acidic internal HZSM-5 channels,70 predominantly leads to DEE at low temperatures. These observations were confirmed by the experimental data in the present work (Fig. 7 and 8), revealing how DEE can re-adsorb on L sites (more probably in the form of EF–Al(V)) after contacting with H2O. Subsequently, two C–O bonds in DEE are decomposed by dehydration to form two ETY molecules in the gas-phase. This ET → DEE → ETY route is considered here as the primary route based on the experimental evidence using ET or DEE as reactants and HZSM-5 catalysts with different B (as SAS) and L (as WAS) contents, whilst the direct ET → ETY route seems to be less predominant on the studied system.
4. Conclusions
The present work has shown experimental and theoretical evidence of finding an optimal compromise between the surface content of L sites, mainly as EF–Al(V) species, and of B sites as Al–OH–Si species, for the catalytic conversion of ethanol into ethylene at low-temperature on HZSM-5 catalysts. The control over ethylene selectivity, for a given temperature, is predominantly affected by the Si and Al content on the parent sample and by the OH-treatment conditions (temperature and time) and the origin of such species (as from OH in an alkaline solution or water). Further calcination steps contribute to conditioning and stabilizing robust Al(δ)–OH bonds. Such treatment consequences are also evidenced when water is co-fed during the ET reaction, favoring ethylene formation. These experimental observations are confirmed with theoretical inputs, proposing a new catalytic route for the formation of ethylene from ethanol via diethylether cracking. In summary, this work provides compelling experimental and theoretical insights into the role of water in the production of ethylene from ethanol at low temperature, serving as the basis to set a relevant landmark towards the design of efficient catalysts and processes for the conversion of complex platform biomolecules into value-added compounds.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was carried out in the framework of academic collaboration between the University of Sultan Moulay Slimane (USMS, Morocco); the Sustainable Process Engineering (SUPREN, IT1554-22) group at the University of the Basque Country (UPV/EHU, Spain) and the “Unité de Catalyse et Chimie du Solide (UCCS)” at the university of Lille (ULille, France). The authors are grateful to the vice-chancellorship of research at UPV/EHU, the Moroccan Ministry of Higher Education, Scientific Research and Innovation for their respective fellowships (grant commitment 20-21-51) granted to L. Ouayloul, and the EU's H2020 program for funding the CatGTP project (MSCA agreement no. 101030636). The authors also thank Prof. P. L. Arias for hosting the PhD student in the SUPREN group. The authors would also like to thank Mrs J. C. Morin and J. Jezequel for their help in implementing In situ FTIR and N2 adsorption measurements at the laboratory of UCCS (France). The theoretical calculation part is supported by the Shandong provincial Natural Science Foundation (grant no. ZR2021QB157). We acknowledge the National Supercomputing Center in Shenzhen for providing the computational resources and materials studio (7.0 Visualizer).References
- C. Angelici, B. M. Weckhuysen and P. C. A. Bruijnincx, Chemocatalytic Conversion of Ethanol into Butadiene and Other Bulk Chemicals, ChemSusChem, 2013, 6(9), 1595–1614, DOI:10.1002/cssc.201300214.
- T. Takei, N. Iguchi and M. Haruta, Synthesis of Acetoaldehyde, Acetic Acid, and Others by the Dehydrogenation and Oxidation of Ethanol, Catal. Surv. Asia, 2011, 15(2), 80–88, DOI:10.1007/s10563-011-9112-1.
- M. El Doukkali, A. Iriondo and I. Gandarias, Enhanced Catalytic Upgrading of Glycerol into High Value-Added H2 and Propanediols: Recent Developments and Future Perspectives, Mol. Catal., 2020, 490, 110928, DOI:10.1016/j.mcat.2020.110928.
- E. Chaichana, N. Boonsinvarothai, N. Chitpong and B. Jongsomjit, Catalytic Dehydration of Ethanol to Ethylene and Diethyl Ether over Alumina Catalysts Containing Different Phases with Boron Modification, J. Porous Mater., 2019, 26(2), 599–610, DOI:10.1007/s10934-018-0663-7.
- T. K. Phung and G. Busca, Diethyl Ether Cracking and Ethanol Dehydration: Acid Catalysis and Reaction Paths, Chem. Eng. J., 2015, 272, 92–101, DOI:10.1016/j.cej.2015.03.008.
- G. Garbarino, R. Prasath Parameswari Vijayakumar, P. Riani, E. Finocchio and G. Busca, Ethanol and Diethyl Ether Catalytic Conversion over Commercial Alumina and Lanthanum-Doped Alumina: Reaction Paths, Catalyst Structure and Coking, Appl. Catal., B, 2018, 236, 490–500, DOI:10.1016/j.apcatb.2018.05.039.
- M. Zhang and Y. Yu, Dehydration of Ethanol to Ethylene, Ind. Eng. Chem. Res., 2013, 52(28), 9505–9514, DOI:10.1021/ie401157c.
- F. Epron, N. Bion, D. Duprez and C. Batiot-Dupeyrat. Steam Reforming of Alcohols from Biomass Conversion for H 2 Production, in Perovskites and Related Mixed Oxides; John Wiley & Sons, Ltd, 2016; pp 539–558. DOI:10.1002/9783527686605.ch24.
- V. Coupard, N. Touchais, S. Fleurier, H. G. Penas, P. D. Smedt, W. Vermeiren, C. Adam and D. Minoux. Process for Dehydration of Dilute Ethanol into Ethylene with Low Energy Consumption without Recycling of Water. US9079812B2, 2015. https://patents.google.com/patent/US9079812B2/en (accessed 2023-01-10).
- M. Inaba, K. Murata, M. Saito and I. Takahara, Ethanol Conversion to Aromatic Hydrocarbons over Several Zeolite Catalysts, React. Kinet. Catal. Lett., 2006, 88(1), 135–141, DOI:10.1007/s11144-006-0120-5.
- K. A. Tarach, J. Tekla, W. Makowski, U. Filek, K. Mlekodaj, V. Girman, M. Choi and K. Góra-Marek, Catalytic Dehydration of Ethanol over Hierarchical ZSM-5 Zeolites: Studies of Their Acidity and Porosity Properties, Catal. Sci. Technol., 2016, 6(10), 3568–3584, 10.1039/C5CY01866H.
- G. Chen, S. Li, F. Jiao and Q. Yuan, Catalytic Dehydration of Bioethanol to Ethylene over TiO2/γ-Al2O3 Catalysts in Microchannel Reactors, Catal. Today, 2007, 125(1), 111–119, DOI:10.1016/j.cattod.2007.01.071.
- X. Zhang, R. Wang, X. Yang and F. Zhang, Comparison of Four Catalysts in the Catalytic Dehydration of Ethanol to Ethylene, Microporous Mesoporous Mater., 2008, 116(1–3), 210–215, DOI:10.1016/j.micromeso.2008.04.004.
- V. V. Bokade and G. D. Yadav, Heteropolyacid Supported on Montmorillonite Catalyst for Dehydration of Dilute Bio-Ethanol, Appl. Clay Sci., 2011, 53(2), 263–271, DOI:10.1016/j.clay.2011.03.006.
- Y. Saito and H. Niiyama, Reaction Mechanism of Ethanol Dehydration on/in Heteropoly Compounds: Analysis of Transient Behavior Based on Pseudo-Liquid Catalysis Model, J. Catal., 1987, 106(2), 329–336, DOI:10.1016/0021-9517(87)90243-0.
- D. Zhang, R. Wang and X. Yang, Effect of P Content on the Catalytic Performance of P-Modified HZSM-5 Catalysts in Dehydration of Ethanol to Ethylene, Catal. Lett., 2008, 124(3), 384–391, DOI:10.1007/s10562-008-9481-x.
- J. Bi, X. Guo, M. Liu and X. Wang, High Effective Dehydration of Bio-Ethanol into Ethylene over Nanoscale HZSM-5 Zeolite Catalysts, Catal. Today, 2010, 149(1), 143–147, DOI:10.1016/j.cattod.2009.04.016.
- A. T. Aguayo, A. G. Gayubo, A. Atutxa, M. Olazar and J. Bilbao, Catalyst Deactivation by Coke in the Transformation of Aqueous Ethanol into Hydrocarbons. Kinetic Modeling and Acidity Deterioration of the Catalyst, Ind. Eng. Chem. Res., 2002, 41(17), 4216–4224, DOI:10.1021/ie020068i.
- D. Fan, D.-J. Dai and H.-S. Wu, Ethylene Formation by Catalytic Dehydration of Ethanol with Industrial Considerations, Materials, 2013, 6(1), 101–115, DOI:10.3390/ma6010101.
- Y. Chen, Y. Wu, L. Tao, B. Dai, M. Yang, Z. Chen and X. Zhu, Dehydration Reaction of Bio-Ethanol to Ethylene over Modified SAPO Catalysts, J. Ind. Eng. Chem., 2010, 16(5), 717–722, DOI:10.1016/j.jiec.2010.07.013.
- N. Zhan, Y. Hu, H. Li, D. Yu, Y. Han and H. Huang, Lanthanum–Phosphorous Modified HZSM-5 Catalysts in Dehydration of Ethanol to Ethylene: A Comparative Analysis, Catal. Commun., 2010, 11(7), 633–637, DOI:10.1016/j.catcom.2010.01.011.
- D. Varisli, T. Dogu and G. Dogu, Silicotungstic Acid Impregnated MCM-41-like Mesoporous Solid Acid Catalysts for Dehydration of Ethanol, Ind. Eng. Chem. Res., 2008, 47(12), 4071–4076, DOI:10.1021/ie800192t.
- A. Ciftci, N. A. Sezgi and T. Dogu, Nafion-Incorporated Silicate Structured Nanocomposite Mesoporous Catalysts for Dimethyl Ether Synthesis, Ind. Eng. Chem. Res., 2010, 49(15), 6753–6762, DOI:10.1021/ie9015667.
- T. Kamsuwan, P. Praserthdam and B. Jongsomjit, Diethyl Ether Production during Catalytic Dehydration of Ethanol over Ru- and Pt- Modified H-Beta Zeolite Catalysts, J. Oleo Sci., 2017, 66(2), 199–207, DOI:10.5650/jos.ess16108.
- G. Elordi, M. Olazar, G. Lopez, P. Castaño and J. Bilbao, Role of Pore Structure in the Deactivation of Zeolites (HZSM-5, Hβ and HY) by Coke in the Pyrolysis of Polyethylene in a Conical Spouted Bed Reactor, Appl. Catal., B, 2011, 102(1), 224–231, DOI:10.1016/j.apcatb.2010.12.002.
- S. Hamieh, C. Canaff, K. B. Tayeb, M. Tarighi, S. Maury, H. Vezin, Y. Pouilloux and L. Pinard, Methanol and Ethanol Conversion into Hydrocarbons over H-ZSM-5 Catalyst, Eur. Phys. J.: Spec. Top., 2015, 224(9), 1817–1830, DOI:10.1140/epjst/e2015-02501-1.
- P. Iadrat, K. Yomthong, C. Rodaum, P. Pornsetmetakul, A. Thivasasith, A. Prasertsab, X. Fan, T. Sooknoi and C. Wattanakit, Effects of Zeolite Frameworks and Hierarchical Structures on Catalytic Bioethanol Dehydration: In situ DRIFTS and DFT Studies, Fuel, 2023, 338, 127208, DOI:10.1016/j.fuel.2022.127208.
- C.-Y. Wu and H.-S. Wu, Ethylene Formation from Ethanol Dehydration Using ZSM-5 Catalyst, ACS Omega, 2017, 2(8), 4287–4296, DOI:10.1021/acsomega.7b00680.
- K. Van der Borght, V. V. Galvita and G. B. Marin, Ethanol to Higher Hydrocarbons over Ni, Ga, Fe-Modified ZSM-5: Effect of Metal Content, Appl. Catal., A, 2015, 492, 117–126, DOI:10.1016/j.apcata.2014.12.020.
- T. K. R. de Oliveira, M. Rosset and O. W. Perez-Lopez, Ethanol Dehydration to Diethyl Ether over Cu-Fe/ZSM-5 Catalysts, Catal. Commun., 2018, 104, 32–36, DOI:10.1016/j.catcom.2017.10.013.
- M. Ketkaew, S. Klinyod, K. Saenluang, C. Rodaum, A. Thivasasith, P. Kidkhunthod and C. Wattanakit, Fine-Tuning the Chemical State and Acidity of Ceria Incorporated in Hierarchical Zeolites for Ethanol Dehydration, Chem. Commun., 2020, 56(77), 11394–11397, 10.1039/D0CC04886K.
- J. Ouyang, F. Kong, G. Su, Y. Hu and Q. Song, Catalytic Conversion of Bio-Ethanol to Ethylene over La-Modified HZSM-5 Catalysts in a Bioreactor, Catal. Lett., 2009, 132(1–2), 64–74, DOI:10.1007/s10562-009-0047-3.
- R. Batchu, V. V. Galvita, K. Alexopoulos, T. S. Glazneva, H. Poelman, M.-F. Reyniers and G. B. Marin, Ethanol Dehydration Pathways in H-ZSM-5: Insights from Temporal Analysis of Products, Catal. Today, 2020, 355, 822–831, DOI:10.1016/j.cattod.2019.04.018.
- S. A. Kadam and M. V. Shamzhy, IR Operando Study of Ethanol Dehydration over MFI Zeolite, Catal. Today, 2018, 304, 51–57, DOI:10.1016/j.cattod.2017.09.020.
- S. Zeng, J. Li, N. Wang, W. Zhang, Y. Wei, Z. Liu and S. Xu, Investigation of Ethanol Conversion on H-ZSM-5 Zeolite by in Situ Solid-State NMR, Energy Fuels, 2021, 35, 12319–12328, DOI:10.1021/acs.energyfuels.1c02151.
- W. Choopun and S. Jitkarnka, Catalytic Activity and Stability of HZSM-5 Zeolite and Hierarchical Uniform Mesoporous MSU-SZSM-5 Material during Bio-Ethanol Dehydration, J. Cleaner Prod., 2016, 135, 368–378, DOI:10.1016/j.jclepro.2016.06.110.
- Z. Wang, L. A. O'Dell, X. Zeng, C. Liu, S. Zhao, W. Zhang, M. Gaborieau, Y. Jiang and J. Huang, Insight into Three–Coordinate Aluminum Species on Ethanol–to–Olefin Conversion over ZSM–5 Zeolites, Angew. Chem., Int. Ed., 2019, 58(50), 18061–18068, DOI:10.1002/anie.201910987.
- S. Shetsiri, A. Thivasasith, K. Saenluang, W. Wannapakdee, S. Salakhum, P. Wetchasat, S. Nokbin, J. Limtrakul and C. Wattanakit, Sustainable Production of Ethylene from Bioethanol over Hierarchical ZSM-5 Nanosheets, Sustainable Energy Fuels, 2018, 3(1), 115–126, 10.1039/C8SE00392K.
- Q. Sheng, S. Guo, K. Ling and L. Zhao, Catalytic Dehydration of Ethanol to Ethylene over Alkali-Treated HZSM-5 Zeolites, J. Braz. Chem. Soc., 2014, 25(8), 1365–1371, DOI:10.5935/0103-5053.20140118.
- M. Seifert, M. S. Marschall, T. Gille, C. Jonscher, O. Busse, S. Paasch, E. Brunner, W. Reschetilowski and J. J. Weigand, Ethanol to Aromatics on Modified H-ZSM-5 Part I: Interdependent Dealumination Actions, ChemCatChem, 2020, 12(24), 6301–6310, DOI:10.1002/cctc.202001344.
- Q. Sheng, K. Ling, Z. Li and L. Zhao, Effect of Steam Treatment on Catalytic Performance of HZSM-5 Catalyst for Ethanol Dehydration to Ethylene, Fuel Process. Technol., 2013, 110, 73–78, DOI:10.1016/j.fuproc.2012.11.004.
- H. Xin, X. Li, Y. Fang, X. Yi, W. Hu, Y. Chu, F. Zhang, A. Zheng, H. Zhang and X. Li, Catalytic Dehydration of Ethanol over Post-Treated ZSM-5 Zeolites, J. Catal., 2014, 312, 204–215, DOI:10.1016/j.jcat.2014.02.003.
- Y. Fang, F. Yang, X. He and X. Zhu, Dealumination and Desilication for Al-Rich HZSM-5 Zeolite via Steam-Alkaline Treatment and Its Application in Methanol Aromatization, Front. Chem. Sci. Eng., 2019, 13(3), 543–553, DOI:10.1007/s11705-018-1778-8.
- J. C. Groen, W. Zhu, S. Brouwer, S. J. Huynink, F. Kapteijn, J. A. Moulijn and J. Pérez-Ramírez, Direct Demonstration of Enhanced Diffusion in Mesoporous ZSM-5 Zeolite Obtained via Controlled Desilication, J. Am. Chem. Soc., 2007, 129(2), 355–360, DOI:10.1021/ja065737o.
- L. Zhang, K. Chen, B. Chen, J. L. White and D. E. Resasco, Factors That Determine Zeolite Stability in Hot Liquid Water, J. Am. Chem. Soc., 2015, 137(36), 11810–11819, DOI:10.1021/jacs.5b07398.
- D. E. Resasco, S. P. Crossley, B. Wang and J. L. White, Interaction of Water with Zeolites: A Review, Catal. Rev., 2021, 63(2), 302–362, DOI:10.1080/01614940.2021.1948301.
- P. G. Machado, A. Walter and M. Cunha, Bio-Based Propylene Production in a Sugarcane Biorefinery: A Techno-Economic Evaluation for Brazilian Conditions, Biofuels, Bioprod. Biorefin., 2016, 10(5), 623–633, DOI:10.1002/bbb.1674.
- G. Kresse and J. Hafner, Ab Initio Molecular Dynamics for Open-Shell Transition Metals, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48(17), 13115–13118, DOI:10.1103/PhysRevB.48.13115.
- G. Kresse and J. Hafner, Ab Initio Molecular-Dynamics Simulation of the Liquid-Metal–Amorphous-Semiconductor Transition in Germanium, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 49(20), 14251–14269, DOI:10.1103/PhysRevB.49.14251.
- G. Kresse and J. Furthmüller, Efficiency of Ab initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set, Comput. Mater. Sci., 1996, 6(1), 15–50, DOI:10.1016/0927-0256(96)00008-0.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54(16), 11169–11186, DOI:10.1103/PhysRevB.54.11169.
- P. E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1994, 50(24), 17953–17979, DOI:10.1103/PhysRevB.50.17953.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59(3), 1758–1775, DOI:10.1103/PhysRevB.59.1758.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77(18), 3865–3868, DOI:10.1103/PhysRevLett.77.3865.
- X. Zhu, L. L. Lobban, R. G. Mallinson and D. E. Resasco, Tailoring the Mesopore Structure of HZSM-5 to Control Product Distribution in the Conversion of Propanal, J. Catal., 2010, 271(1), 88–98, DOI:10.1016/j.jcat.2010.02.004.
- K. Sadowska, A. Wach, Z. Olejniczak, P. Kuśtrowski and J. Datka, Hierarchic Zeolites: Zeolite ZSM-5 Desilicated with NaOH and NaOH/Tetrabutylamine Hydroxide, Microporous Mesoporous Mater., 2013, 167, 82–88, DOI:10.1016/j.micromeso.2012.03.045.
- J. Vicente, A. G. Gayubo, J. Ereña, A. T. Aguayo, M. Olazar and J. Bilbao, Improving the DME Steam Reforming Catalyst by Alkaline Treatment of the HZSM-5 Zeolite, Appl. Catal., B, 2013, 130–131, 73–83, DOI:10.1016/j.apcatb.2012.10.019.
- W. Xia, A. Takahashi, I. Nakamura, H. Shimada and T. Fujitani, Study of Active Sites on the MFI Zeolite Catalysts for the Transformation of Ethanol into Propylene, J. Mol. Catal. A: Chem., 2010, 328(1), 114–118, DOI:10.1016/j.molcata.2010.06.008.
- J. Holzinger, P. Beato, L. F. Lundegaard and J. Skibsted, Distribution of Aluminum over the Tetrahedral Sites in ZSM-5 Zeolites and Their Evolution after Steam Treatment, J. Phys. Chem. C, 2018, 122(27), 15595–15613, DOI:10.1021/acs.jpcc.8b05277.
- S. Zhao, W. Yang, K. D. Kim, L. Wang, Z. Wang, R. Ryoo and J. Huang, Synergy of Extraframework Al 3+ Cations and Brønsted Acid Sites on Hierarchical ZSM-5 Zeolites for Butanol-to-Olefin Conversion, J. Phys. Chem. C, 2021, 125(21), 11665–11676, DOI:10.1021/acs.jpcc.1c02171.
- A. A. Gabrienko, I. G. Danilova, S. S. Arzumanov, L. V. Pirutko, D. Freude and A. G. Stepanov, Direct Measurement of Zeolite Brønsted Acidity by FTIR Spectroscopy: Solid-State 1H MAS NMR Approach for Reliable Determination of the Integrated Molar Absorption Coefficients, J. Phys. Chem. C, 2018, 122(44), 25386–25395, DOI:10.1021/acs.jpcc.8b07429.
- B.-T. L. Bleken, L. Mino, F. Giordanino, P. Beato, S. Svelle, K. P. Lillerud and S. Bordiga, Probing the Surface of Nanosheet H-ZSM-5 with FTIR Spectroscopy, Phys. Chem. Chem. Phys., 2013, 15(32), 13363–13370, 10.1039/C3CP51280K.
- Z. Wang, Y. Jiang, O. Lafon, J. Trébosc, K. Duk Kim, C. Stampfl, A. Baiker, J.-P. Amoureux and J. Huang, Brønsted Acid Sites Based on Penta-Coordinated Aluminum Species, Nat. Commun., 2016, 7(1), 13820, DOI:10.1038/ncomms13820.
- X. Yi, K. Liu, W. Chen, J. Li, S. Xu, C. Li, Y. Xiao, H. Liu, X. Guo, S.-B. Liu and A. Zheng, Origin and Structural Characteristics of Tri-Coordinated Extra-Framework Aluminum Species in Dealuminated Zeolites, J. Am. Chem. Soc., 2018, 140(34), 10764–10774, DOI:10.1021/jacs.8b04819.
- J. Lee, J. Szanyi and J. H. Kwak, Ethanol Dehydration on γ-Al2O3: Effects of Partial Pressure and Temperature, Mol. Catal., 2017, 434, 39–48, DOI:10.1016/j.mcat.2016.12.013.
- J. S. Bates, B. C. Bukowski, J. Greeley and R. Gounder, Structure and Solvation of Confined Water and Water–Ethanol Clusters within Microporous Brønsted Acids and Their Effects on Ethanol Dehydration Catalysis, Chem. Sci., 2020, 11(27), 7102–7122, 10.1039/D0SC02589E.
- D. Mei and J. A. Lercher, Mechanistic Insights into Aqueous Phase Propanol Dehydration in H-ZSM-5 Zeolite, AIChE J., 2017, 63(1), 172–184, DOI:10.1002/aic.15517.
- A. Vjunov, M. Wang, N. Govind, T. Huthwelker, H. Shi, D. Mei, J. L. Fulton and J. A. Lercher, Tracking the Chemical Transformations at the Brønsted Acid Site upon Water-Induced Deprotonation in a Zeolite Pore, Chem. Mater., 2017, 29(21), 9030–9042, DOI:10.1021/acs.chemmater.7b02133.
- S. Kim, D. J. Robichaud, G. T. Beckham, R. S. Paton and M. R. Nimlos, Ethanol Dehydration in HZSM-5 Studied by Density Functional Theory: Evidence for a Concerted Process, J. Phys. Chem. A, 2015, 119(15), 3604–3614, DOI:10.1021/jp513024z.
- F. Sannino, M. Pansini, A. Marocco, B. Bonelli, E. Garrone and S. Esposito, The Role of Outer Surface/Inner Bulk Brønsted Acidic Sites in the Adsorption of a Large Basic Molecule (Simazine) on H-Y Zeolite, Phys. Chem. Chem. Phys., 2015, 17(43), 28950–28957, 10.1039/C5CP05085E.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2gc04437d |
| This journal is © The Royal Society of Chemistry 2023 |