
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical composition, coordination, and stability of Ca–organic associations in the presence of dissolving calcite†
Odeta
Qafoku
 *a,
Anil K.
Battu
a,
Tamas
Varga
*a,
Matthew A.
Marcus
b,
Brian
O'Callahan
a,
Qian
Zhao
*a,
Sebastian T.
Mergelsberg
c,
William R.
Kew
a,
John S.
Loring
c,
Nikolla P.
Qafoku
de and
Sarah I.
Leichty
a
*a,
Anil K.
Battu
a,
Tamas
Varga
*a,
Matthew A.
Marcus
b,
Brian
O'Callahan
a,
Qian
Zhao
*a,
Sebastian T.
Mergelsberg
c,
William R.
Kew
a,
John S.
Loring
c,
Nikolla P.
Qafoku
de and
Sarah I.
Leichty
a
aEnvironmental Molecular Sciences Laboratory, Pacific Northwest National Laboratory, 3335 Innovation Boulevard, Richland, WA 99352, USA. E-mail: odeta.qafoku@pnnl.gov; tamas.varga@pnnl.gov; qian.zhao@pnnl.gov; Tel: 1+(509) 371 6383 Tel: 1+(509) 371 6042 Tel: 1+(509) 371 6856
bAdvanced Light Source, Lawrence Berkeley National Laboratory, Berkeley, CA 94720, USA
cPacific Northwest National Laboratory, Energy and Environment Division, 900 Battelle Boulevard, Richland, WA 99352, USA
dPacific Northwest National Laboratory, Physical and Chemical Sciences Division, 3335 Innovation Boulevard, Richland, WA 99352, USA
eDepartment of Civil and Environmental Engineering, University of Washington, Seattle, WA 98195, USA
First published on 2nd May 2023
Abstract
Environmental biotic and abiotic factors and soil physical, mineralogical, and chemical properties control the chemical composition of soil organic matter (SOM). Particularly, soil mineralogy and the presence of multivalent cations affect SOM labile fraction composition, stability, and environmental persistence. The persistence of SOM in aridic or limestone deposit derived soils, i.e., calcareous soils, has been partially attributed to SOM stabilization through adsorption or inclusion into the calcite mineral structure. Recently, however, it was shown that Ca(aq) released during calcite dissolution formed aqueous Ca–organic associations with OM components, which were unbound to mineral surface sites. This study investigates the structure, composition, and coordination of these associations by characterizing lyophilized Ca–organic containing solutions with spectromicroscopy [Scanning Transmission X-ray Microscopy (STXM)] and with a nanoimaging chemical probe [Infrared scattering-Scanning Nearfield Optical Microscopy (IR s-SNOM)]. Chemical stability of Ca–organic associations is furthermore determined with pyrolysis mass spectrometry analysis. The results demonstrate that Ca–organic associations are formed in the presence of dissolving calcite and OM components relevant to soil chemistry, i.e., lignin and amino acids. This study further reveals a spatial homogeneity of solution-derived (bi) carbonate in Ca–organic associations indicating for the first time that an inorganic anion, such as (bi)carbonate, may be part of these associations. Most likely, Ca ions are bound to both the (bi)carbonate and the organic components. These Ca (bi)carbonate–organic associations seem to have greater chemical stability than the pristine organic mixtures and, possibly, a higher environmental stability and reduced mineralization rate.
Environmental significanceOrganic C persistence in soil environments is essential to decreasing natural CO2 emissions to the atmosphere. The interaction of organic matter (OM) with minerals controls the stability of OM labile fraction within soils and consequently organic C persistence. This study demonstrates that relevant OM components, lignin and amino acids, form Ca–organic associations in the presence of dissolving calcite, a common soil mineral. We demonstrate a greater chemical stability of Ca–organic associations compared to pristine organic components. Our results shed light on the composition and structure of Ca–organic associations and reveal inorganic anionic species, such as the (bi)carbonate, may play a role in the long-term stability of soil OM. Insights from our study are essential to predicting the stability of Ca–organic–inorganic associations and the environmental organic C lability. |
1. Introduction
Soil organic matter (SOM) represents the largest terrestrial carbon (C) reservoir at the global scale, estimated to be about three times greater than organic C content of the atmospheric or vegetation pools.1,2 SOM chemical composition and molecular structure is complex and diverse, ranging from organic acid monomers, low molecular weight aliphatic and aromatic compounds to complex mixtures of organic biopolymers.3 Characterization of SOM at the molecular level reveals occurrence of individual organic components and biochemical classes such as amino acids, lipids, lignin, carbohydrates but also inorganic cations, indicating that composition of SOM is influenced by environmental factors, soil minerals, and the physical and chemical nature of the soil matrix.4–8In particular, factors such as surface properties of soil minerals and the presence of multivalent cations in soil liquid phase significantly and uniquely affect not only the composition but also the stability and environmental persistence of the labile fraction of SOM.4,9 Soils that contain highly reactive minerals such as CaCO3, that can easily undergo dissolution, and/or amorphous Al or Fe oxyhydroxides with high sorption affinities, result in substantially higher content of SOM compared to soils low in these minerals.4,8,10,11 Of particular interest is the effect of multivalent cations (i.e., Ca) which build bridges among units of organic molecules and promote formation of (meta)stable complexes and linkages that may stabilize and protect organic compounds from decomposition.12–16 In addition, different forms of Ca, e.g., aqueous Ca [Ca(aq)] that is present in the soil liquid phase, exchangeable Ca [Ca(exch)] that is adsorbed on soil minerals' exchangeable sites, and structural Ca [Ca(str)] that is part of the soil minerals' structure, may interact with the soil organic matter (SOM) facilitating surface complexation and bonding, increasing SOM stability.10,15,17–20
Prior work has shown evidence of SOM accumulation in soils that contain reactive CaCO3, e.g., calcareous sand is found to contain approximately three times more SOM compared to silica sand21 and the presence of reactive CaCO3 or addition of Ca-salts showed an increase in SOM biostabilization.22–28 SOM-induced calcite (CaCO3) dissolution is an important source of Ca(aq),16,19 and Ca-mediated stabilization of SOM may occur through formation of Ca–organic associations,10,11,22–25 or through occlusion of organic biomolecules into calcite mineral voids.19,29
Recent work conducted by our research team, has demonstrated that interaction of CaCO3 with organic compounds representative of SOM classes (i.e., lipids, amino acids, lignin), induced calcite dissolution, releasing Ca ions into solution. Ca(aq) ions subsequently interacted with SOM components and formed complexes or Ca-cross linkages, which, surprisingly, were neither bound nor attached to the mineral Ca-surface sites. Theoretical modeling studies support the view that Ca(aq) binds strongly to dissolved organic species that contain carboxylic groups, and consequently through metal-carboxylation promote association of monomeric organic compounds into supramolecular colloidal aggregates.14,15,30 The formation of these large aggregates has not been evidenced when other metal cations (i.e., Mg, Na, or Cs) are present in solution.14,15,30 These computational molecular-scale studies, provide insights into structural organization of metal carboxylate complexes and associations, where high charge density Ca(aq) ions prefer to associate primarily with carboxylic organic groups in bidentate contact ion pairs, and less frequently in monodentate or in solvent separated ion pairs.30 However, to the best of our knowledge, experimental evidence of the structure and Ca coordination in the Ca–organic associations and their chemical stability has not yet been documented in the literature.
In this study we employed advanced analytical techniques to determine the interaction and association of Ca(aq) with SOM components from nano to macro scale. Specifically, we used Scanning Electron Microscopy (SEM), Transmission Electron Microscopy equipped with Energy Dispersive X-ray Spectroscopy (TEM/EDX), Scanning and Transmission X-ray Microscopy with Near-Edge X-ray Absorption Fine Structure/ (STXM/NEXAFS), and Infrared scattering-Scanning Nearfield Optical Microscopy (IR s-SNOM) to interrogate the functionality of organic compounds and Ca–organic local binding environment, aiming at understanding coordination of Ca(aq) ions and composition of functional groups bound to Ca metal center. One major objective of this research was to understand whether the presence of Ca influences the thermal decomposition of the Ca–organic associations, and to gather insights into their stability, with implications to the persistence of these associations in soil environment.
2. Materials and methods
Aqueous Ca–organic complexes, identified from our prior study,16 were investigated using microscopic and spectroscopic techniques, SEM, TEM/EDX, STXM/NEXAFS, and IR s-SNOM. Sample preparation procedures and characterization methods are as follows.2.1 Calcite and organic compounds
Samples were prepared by exposing synthetic calcite16 with particle size <3 μm for 24 h, to a four organic compounds mixture with various degrees of hydrophilicity and binding properties, at solid density of 30 g L−1 and at native pH of 8.45. The four organic compounds lauric acid, pentaglycine, trehalose, and lignin unit were selected to represent major biochemical classes found in natural SOM, respectively lipids, amino acids, carbohydrate, and lignin fragment (see ESI†). A stock solution prepared by mixing water-soluble portions of organic compounds was used for this study. After exposure of organic mixture to calcite followed by centrifugation and twice solid rinsing,16 suspensions (containing residual Ca(aq) and organic compounds) were drop cast on TEM grids or Si-substrates. Analyses conducted in this study, required lyophilization of solution droplets, through near instantaneous flash-and-freeze procedure (at −196 °C liquid N2). This technique is frequently used to preserve and accurately maintain elemental spatial distribution and integrity in biological and/or soil31 samples (see ESI† for more details on sample preparation). Lastly, solutions were analyzed with pyrolysis-gas chromatography/mass spectrometry (py-GC/MS) to study the stability of organic compounds before and after exposure to CaCO3.2.2 Characterization of reaction products
3. Results and discussion
3.1 Characterization of Ca–organic associations: electron microscopy results
Electron microscopy SEM/TEM/EDX interrogated the composition and elemental spatial distribution in the organic associations that formed in a representative lyophilized suspension (Fig. 1). The SEM and TEM characterization showed various morphological traits of the organic assemblages, e.g., predominately spherical-like and ring-like particles (representatives of ring-like and spherical particles are shown in Fig. 1 and S1†), which were arranged as aggregates and that we refer hereafter as “lacey aggregate” to distinguish them from calcite particles that were also present in the sample. In prior work we found that these morphological structures of organic aggregates were formed solely in the case when calcite was the reacted solid and they were not observed when other minerals, i.e., illite,16 Fe(oxy)hydroxides, or Al(OH)3 were exposed to same organic compound mixture.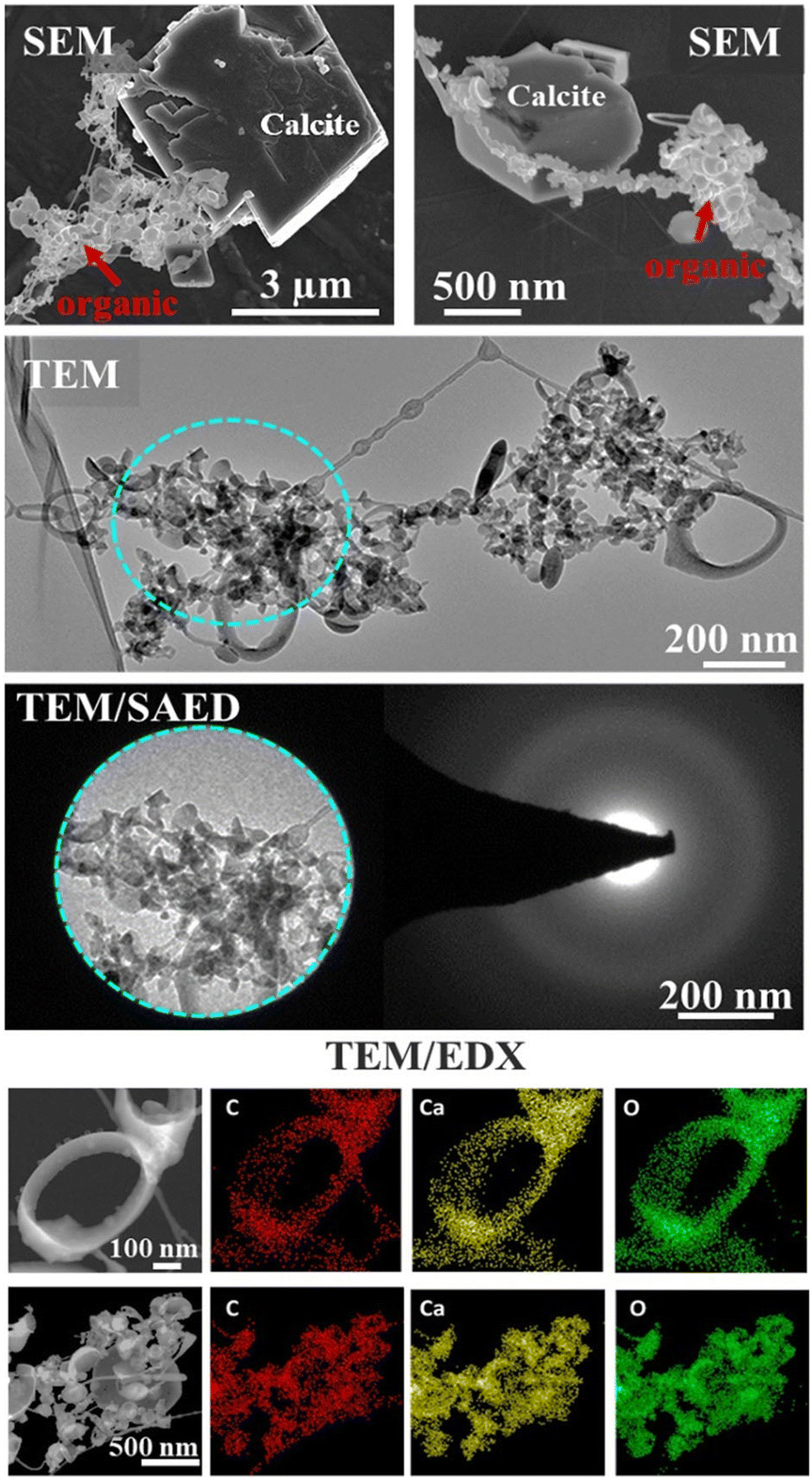 | ||
| Fig. 1 Electron microscopy images of a lyophilized sample, showing Ca–organic assemblages that formed after calcite was exposed to the organic mixture composed of lipids, amino acids, carbohydrate, and lignin. SEM micrographs show both calcite particles and organic assemblages. TEM images show only the regions of organics, which based on selected area electron diffraction analysis (SAED) analyses are amorphous and based on EDX analyses, are composed of Ca, C, and O, demonstrating a homogeneous spatial distribution. | ||
The chemical composition of the “lacey aggregate” determined through higher resolution TEM/EDX analysis (Fig. 1, bottom panels), revealed that particles within aggregates contain significant amount of Ca content in addition to the expected elements, carbon and oxygen that are part of the organic compounds. The EDX point analysis in addition showed that molar ratios on different particles ranged from 1 Ca: 3.8–5.8 C and from 1 Ca: 4.2–6.0 O (oxygen). The higher concentration was observed for ring-like particles. In comparison, the molar ratio of crystalline calcite with rhombohedral habit was calculated equal to 1 Ca: 1.4 C and 1Ca: 2.5 O (ref. 16) well below the measured values for the organic morphologies shown in Fig. 1.
Furthermore, the selected area electron diffraction analysis (SAED) reveals that (Ca, C)-containing particles are amorphous (Fig. 1), i.e., different from crystalline calcite.16 Based on composition, C content, morphological traits, and lack of crystallinity, it appears that the particles that compose “lacey aggregate” are Ca–organic associations that formed in solution. We hypothesized that particles with different morphologies might reveal different types of Ca–organic associations, i.e., associations with either amino acid, lignin, lipid, or a combination of them.
Formation of ring-like, globular, and chain-like structures has been previously observed during humic acid sorption into mica surface sites,35 or have been associated with changes in solution chemistry (i.e. pH, cations, and organic C concentration).36 For example, formation of ring-like morphologies was more present at acidic pH (i.e., 4–5).35 Yet, an earlier study36 indicated that ring-like, sheet, and chain-like morphologies were also present at alkaline pH (i.e., 8–12), particularly when concentration of multivalent cations (i.e., Ca, Cu, Fe) and organic C increased.
3.2 Characterization of Ca–organic associations: STXM/NEXAFS results
We employed STXM-NEXAFS on a representative lyophilized suspension to: i) collect data over C K-edge, energy range (280–320 eV), to characterize the functional groups of Ca–organic associations, and, ii) collect data over Ca L2,3-edge, energy range (340–360 eV), to gather insights at molecular scale on the local chemical environment and coordination of Ca in the organic associations (see Fig. S2† for the SEM image of the STXM/NEXAFS analyzed region). The STXM data (Fig. 2a and b and S3†) features grayscale optical density (OD) transmission images, which revealed the presence of two distinct regions that have varying sample thickness (based on Lambert–Beer's law).31,37 Region one (thinner, whiter, in the transmission images), was composed of aggregated particulates (∼30–100 nm in size), likely Ca–organic associations, that we referred to earlier as “lacey aggregate”, (Fig. 2a, image at 290.3 eV). Region two (thicker and darker) was composed of a larger-size single particle (∼2–3 μm), which most likely was the starting mineral, calcite. Hereafter, we refer to this region as “the particle”, (Fig. 2b, image at 301.3 eV; Fig. S2 and S3†). The C-K edge and Ca-L2,3 edge NEXAFS spectra results of these two distinct regions are discussed below.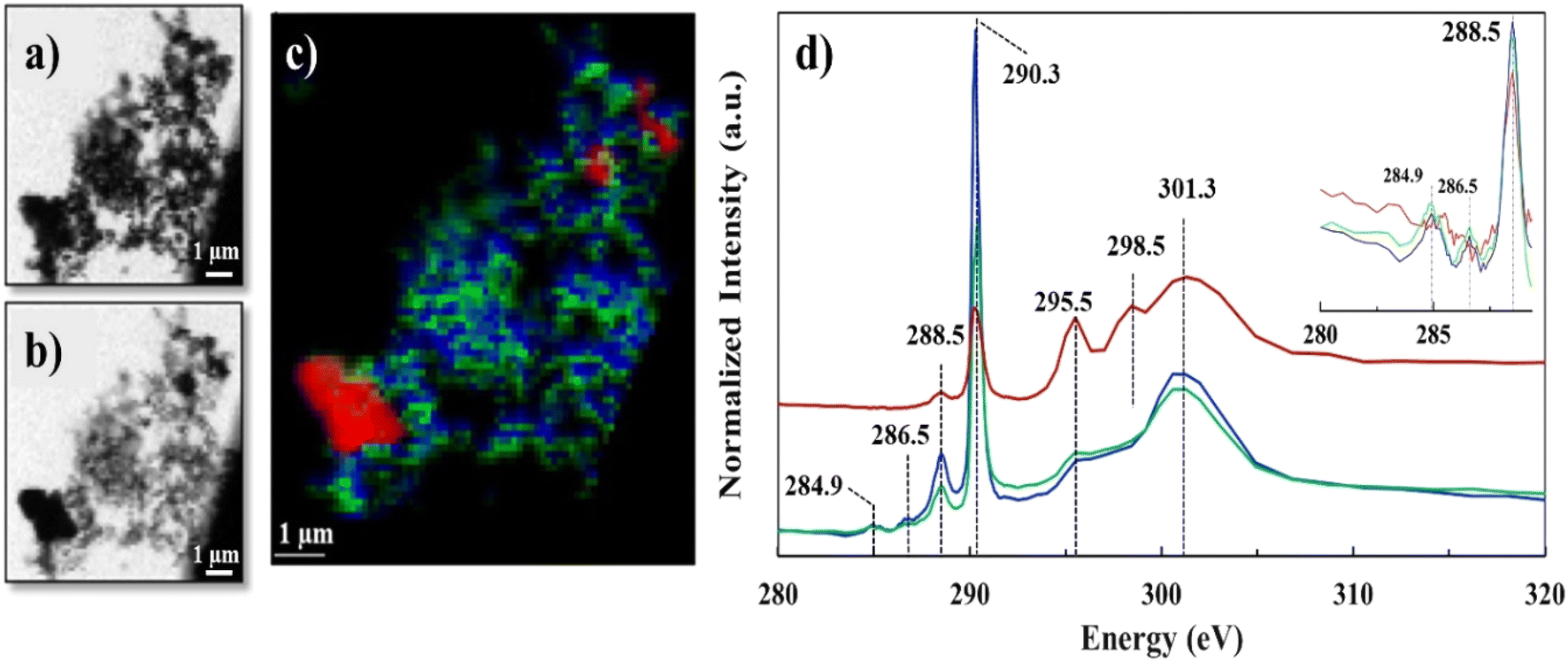 | ||
| Fig. 2 C K-edge STXM/NEXAFS data of lyophilized sample, a) grayscale transmission image recorded at 290.3 eV and b) at 301.3 eV; c) tricolor cluster map representing three distinct C regions, red (identified as calcite particle), green and blue (identified as Ca–organic association); d) averaged C K-edge NEXAFS spectra extracted from the three regions depicted in the tricolor image, using the same color coding. Spectra was normalized at the pre-edge at 284.9 and post-edge at 310 eV, similar to recent work.41 Vertical offset for red spectrum is applied. Insert shows spectra differences in the 284–289 eV region. | ||
The C K-edge NEXAFS spectra results shown in Fig. 2d, revealed significant resonance energy peaks at 290.3 eV and 295.5 eV that represent the 1s → π* electronic transition consistent with the carbonate functional group (RO(C*![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)OR) and peaks at 298.5 and 301.3 eV that arise from the structural features of calcite mineral39 (Table S2†) or other crystalline carbonate minerals.40 The sharp peaks at 290.3 eV and the broad peaks at 301.3 eV were common for all the three spectra components (red, blue and green); however, the peak positioned at 295.5 eV appeared only as a shoulder in the “lacey aggregate” represented by the blue and green spectra, but it was more resolved and more significant in “the particle” region, (red spectrum). We observed a broadening and a less intense carbonate peak at 290.3 eV for “the particle” compared to “the lacey aggregate”. Both, broadening and signal reduction of the carbonate signature peak, could have resulted from the saturation due to sample thickness effect, which is demonstrated by the higher OD value in the μ-thick region of “the particle”. Sample thickness has been previously recognized to broaden absorption of the major peaks and increase the intensity of the preceding minor peaks.42–44 However, it is generally accepted that sample thickness does not influence the spectra peak positions, which remain unchanged even at high absorption saturation.42–44 The comparison of peak positions reveals a resonant peak at 298.5 eV that is present only in the red spectrum of “the particle” and not in the green/blue spectra of “the lacey aggregate”. This peak, although slightly shifted in energy, has previously been assigned in literature as a secondary peak position for carbonate in calcite (Table S2†).40 Together, “the particle” spectrum peaks reproduce all C K-edge features of calcite structure.
O)OR) and peaks at 298.5 and 301.3 eV that arise from the structural features of calcite mineral39 (Table S2†) or other crystalline carbonate minerals.40 The sharp peaks at 290.3 eV and the broad peaks at 301.3 eV were common for all the three spectra components (red, blue and green); however, the peak positioned at 295.5 eV appeared only as a shoulder in the “lacey aggregate” represented by the blue and green spectra, but it was more resolved and more significant in “the particle” region, (red spectrum). We observed a broadening and a less intense carbonate peak at 290.3 eV for “the particle” compared to “the lacey aggregate”. Both, broadening and signal reduction of the carbonate signature peak, could have resulted from the saturation due to sample thickness effect, which is demonstrated by the higher OD value in the μ-thick region of “the particle”. Sample thickness has been previously recognized to broaden absorption of the major peaks and increase the intensity of the preceding minor peaks.42–44 However, it is generally accepted that sample thickness does not influence the spectra peak positions, which remain unchanged even at high absorption saturation.42–44 The comparison of peak positions reveals a resonant peak at 298.5 eV that is present only in the red spectrum of “the particle” and not in the green/blue spectra of “the lacey aggregate”. This peak, although slightly shifted in energy, has previously been assigned in literature as a secondary peak position for carbonate in calcite (Table S2†).40 Together, “the particle” spectrum peaks reproduce all C K-edge features of calcite structure.
The missing resonant peak at 298.5 eV for the blue/green spectra of the “lacey aggregate” suggests that carbonate functional groups that are present in this region may have different local environment than in calcite. Furthermore, scatter plot analysis of C edge jump to CO3 peak intensity incident/transmitted count ratios (data not shown) in the “lacey aggregate” revealed a narrow distribution of carbonate ions. Consistent with the TEM/SAED characterization, which shows a uniform distribution of C in the “lacey aggregate”, the data suggests that carbonate ions may have a surrounding environment similar to other amorphous phases, and, as an example, similar to when carbonate is incorporated in amorphous calcium phosphates.45
In addition to the carbonate peak assignments, the low-intensity peak at ∼285 eV represented the 1s → π* electronic transition corresponding to aromatic or olefinic C (CC*–H of C), while the small peak at 286.5 eV represented the 1s → π* transition associated with aryl or vinyl-keto (CC–C*O) functional groups.39 These peak assignments, at 284.9 eV and at 286.5 eV were only present in the green and blue spectra that belong to “lacey aggregate” regions, but not in “the particle” (Fig. 1). Attempts to fit C K-edge NEXAFS spectra to the reference and measured lignin spectrum, showed ∼0.3 eV shifts in sample peak intensities and energy positions relative to the lignin standard. We hypothesize that such differences, may indicate a change in the coordination environment of lignin-based C as a result of complexation with Ca ions. Finally, the sharper peak at 288.5 eV that represented the 1s → π* transition associated with carboxyl (OR(C*O)C) groups was apparent in both “the particle” and “the lacey aggregate”, although it was more pronounced in the latter one (Fig. 1d insert and S4, Table S1†). It's possible that compounds with carboxylic functional groups (i.e., lauric acid) were adsorbed on “the particle”,16 yet the results indicate that the organics were significantly more abundant in the “lacey aggregate” region.
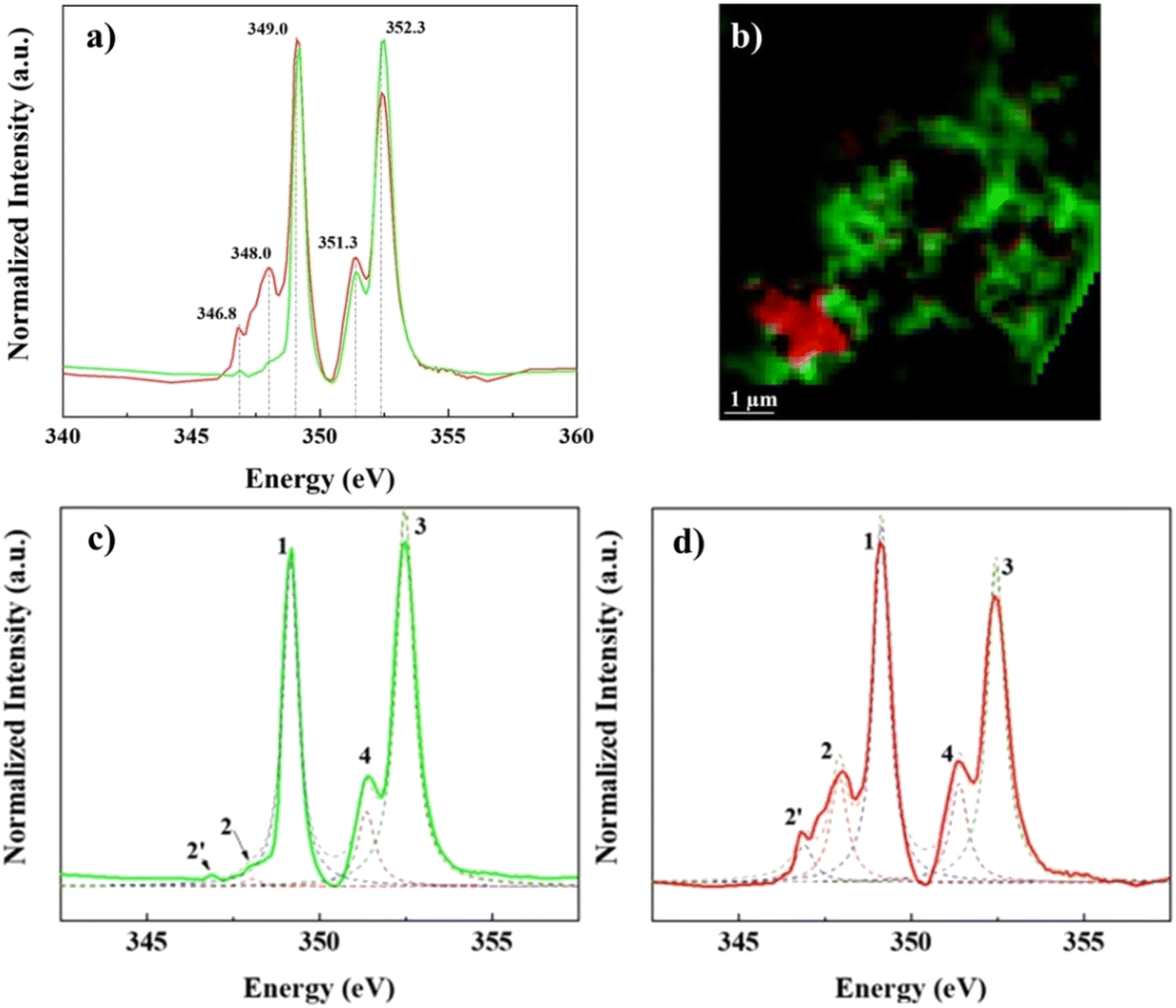 | ||
| Fig. 3 Ca L2,3-edge STXM/NEXAFS data collected in the same regions of the lyophilized sample as C-K edge, representing two distinct spectra components, red (identified as calcite particle) and green (identified as Ca–organic association). The two-color coded spectra in a) correspond to the bicolor cluster map in b). The bottom panels, show spectra least-squares fitting of the green Ca–organic association c) and red calcite particle d). | ||
In contrast, Ca L2,3-edge spectra of the “lacey aggregate” region indicates a different local coordination of Ca compared to that of calcite structure. Specifically, “lacey aggregate” spectrum lacked intensity in the expected calcite peak at 348 eV (Fig. 3d, see peak 2 assignment), implying a disordered or amorphous structure.50 Furthermore, the Ca L2,3-edge spectra fitting of this region showed a closer match to the aragonite reference spectra (Fig. S5†). This is a perplexing result, given the evidence from TEM/SAED that the “lacey aggregate” region was amorphous, and it lacked crystalline diffraction patterns expected for aragonite. Further NEXAFS fitting edge jumps showed a molar ratio of 1Ca![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.6C for the “lacey aggregate”. We note that recently NEXAFS data have been used to quantitatively determine molar ratios when standard materials are used to generate calibration curves.51 Although we did not use standard materials for calibration, the NEXAFS results appear in good agreement with the TEM/EDX results (Fig. 1). This collective evidence indicates that the “lacey aggregate” region has a much higher C content than what is expected for any of the CaCO3 polymorphs. In conclusion, the carbon K-edge and Ca L2,3-edge NEXAFS results combined indicate that, the “lacey aggregate” region has a higher degree of disorder compared to calcite, and that it is composed of Ca ions as well as inorganic and organic C components, that are homogeneously distributed.
:3.6C for the “lacey aggregate”. We note that recently NEXAFS data have been used to quantitatively determine molar ratios when standard materials are used to generate calibration curves.51 Although we did not use standard materials for calibration, the NEXAFS results appear in good agreement with the TEM/EDX results (Fig. 1). This collective evidence indicates that the “lacey aggregate” region has a much higher C content than what is expected for any of the CaCO3 polymorphs. In conclusion, the carbon K-edge and Ca L2,3-edge NEXAFS results combined indicate that, the “lacey aggregate” region has a higher degree of disorder compared to calcite, and that it is composed of Ca ions as well as inorganic and organic C components, that are homogeneously distributed.
3.3 Characterization of organic functionalities at nanoscale, from IR s-SNOM
The IR s-SNOM enables examination of organic particles at much higher spatial resolution than the spectromicroscopy and provides valuable insights about sample composition homogeneity at nanometer length scale.52,53 The results of the IR s-SNOM analysis were compared against measurements of the standard compounds and organic species, such as lauric acid, laurate (that is expected to form at the experimental pH), pentaglycine, trehalose, lignin, the mixture of four organics, and calcite; they were all recorded in the dry state with ATR-FTIR (Fig. S6 and S7†).The ATR-FTIR results indicated that the spectra of individual organic compounds (i.e., lauric acid, laurate, pentaglycine, trehalose, lignin, and calcite, Fig. S6†) were consistent with literature data, described in detail in the ESI.† The contribution of each of these organic compounds was also evident in the acquired ATR FTIR spectrum of the organic mixture, Fig. S6.† Specifically, lignin contribution was apparent by the peak at ∼1450 cm−1 (CH deformation) and peaks at ∼1500 cm−1 and ∼1600 cm−1 (aromatic vibrations). The contribution of laurate was recognized at ∼1564 cm−1 as shoulder feature, and that of pentaglycine at ∼1592 cm−1 as a weak peak based on least-squares fitting of spectral features (Fig. S6†). In the actual samples, which were obtained after organic mixture was exposed to calcite and the releases of (bi)carbonate anions, the contribution of (bi)carbonate to the IR spectra was also observed.
The IR s-SNOM spectra of calcite particle exposed to the mixture of organic compounds (Fig. 4) showed v3 carbonate asymmetric stretch mode as a broad peak slightly shifted at ∼1450 cm−1 (Fig. 4b blue circle and d blue line). The shifts could be due to the small particle size of calcite or could be due to presence of the organic compounds. The spectra collected at a second area (red circle, Fig. 4b, red line and d) showed a further shift and appearance of a double peak towards higher frequencies at 1480–1500 cm−1, which might correspond to the aromatic vibrations of lignin at ∼1500 cm−1 based on ATR FTIR results. Although the amorphous carbonate v3 response is slightly blue-shifted than that of calcite and may contribute to the peak at ∼1480 cm−1, contributions above 1500 cm−1 are more likely to arise from organic components. We note that on the IR s-SNOM image collected at 1515 cm−1, the likely lignin signal appeared to be more prominent around the outer edges of the calcite crystal (Fig. 4c). This could be an artifact due to topographic height differences between the particle and substrate. Yet, the results could also indicate that a portion of organic compounds gave rise to the IR spectrum consistent with the hypothesis that near-surface organic complexes can be present adjacent to the calcite particle.
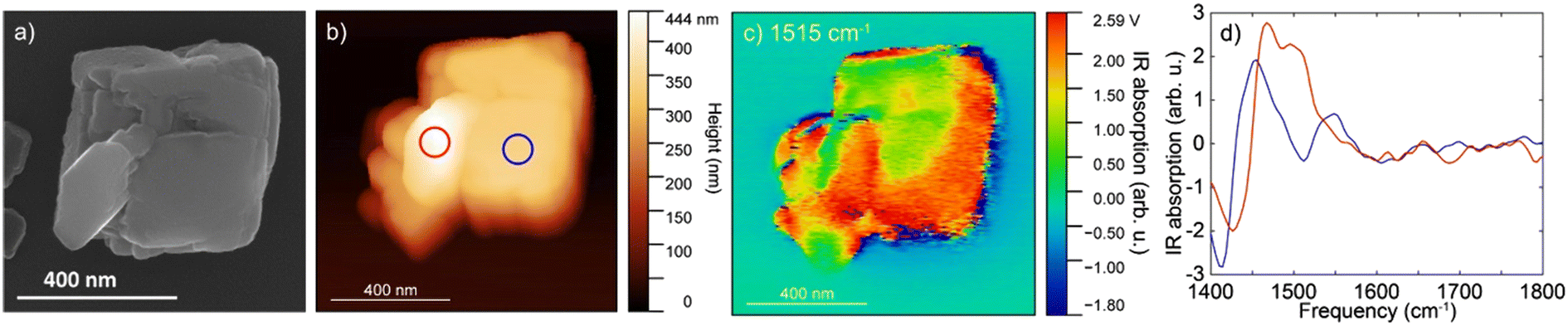 | ||
| Fig. 4 The morphology and IR s-SNOM spectroscopy of calcite crystal after being exposed to the mixture of the organic compounds: a) SEM image, b) AFM topography, and c) IR s-SNOM image at 1515 cm−1 of CaCO3 particle. d) Point spectra acquired at corresponding color-coded locations circled in b). | ||
The IR s-SNOM data were next collected on Ca–organic nano size particles that were mostly flat to the Si-substrate, spherical and ring-shaped structures identical to morphologies observed with SEM/TEM (Fig. 1). In general, the IR signal collected on these organic nanoparticles was relatively weak, without the necessary sharp features that could be assigned with certainty to frequencies of organic/inorganic functional groups. However, the IR broadband spectra collected on these particles revealed several broad features and peaks that were matched against the IR spectra of individual organic standards as well as against carbonate vibrational response (Fig. 5). We further applied PCA on several spectra, to aid our interpretation.
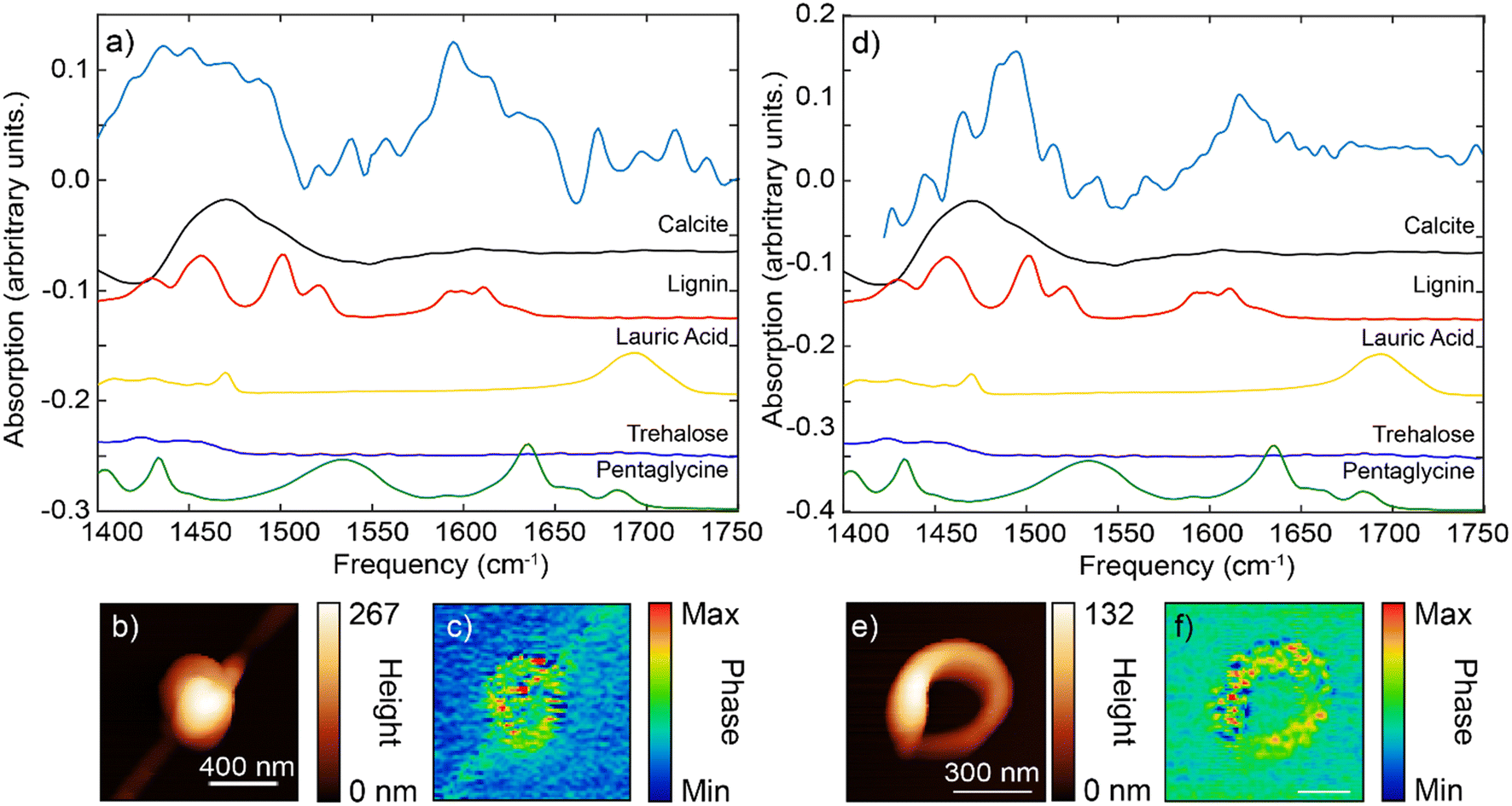 | ||
| Fig. 5 IR s-SNOM spectroscopy of Ca–organic morphologies: a) point spectra of a spherical particle revealing broad features at ∼1450 cm−1 and peaks at ∼1600 cm−1, matching carbonate stretching band and lignin vibrational signature, respectively, b and c) spherical particle AFM topography and IR s-SNOM image at 1600 cm−1, d) point spectra of ring-shaped particle showing peaks at ∼1500 cm−1 and ∼1600 cm−1, matching lignin spectra with a smaller contribution from carbonate band, e) and f) corresponding AFM height and IR s-SNOM image at 1600 cm−1. | ||
For the spherical particle, the main IR spectral features were located at the frequency range of 1400–1500 cm−1 and 1580–1620 cm−1 (Fig. 5a). The very broad feature at the ∼1450 cm−1 appeared to be more consistent with the carbonate dominant stretching bend (at 1400–1500 cm−1, Fig. 4d, S6 and S7†) with possible additional contributions from lignin (Fig. S6†). The peak at ∼1600 cm−1 matched more closely the aromatic stretch response of the lignin (given the absence of carbonate peaks in this region).
For the ring-shaped particle, the overall IR spectrum similarly has two characteristic features, at 1450–1500 cm−1 region and at 1600–1640 cm−1 region (Fig. 5d and e). At the frequency range between 1450–1500 cm−1, several sharper peaks were evident. These peaks had more features and appeared to follow those of lignin standard spectrum. We note that the relative peak intensities of IR s-SNOM spectra can differ from those obtained using ATR FTIR spectroscopy with some peaks being absent due to the selection rules of IR s-SNOM.54,55 In addition, spectral shifts on the order of 5 cm−1 can be present in IR s-SNOM spectra due to dipole–dipole coupling between the tip and the sample.56 Compared to spherical particle spectrum, the ring-shaped particle spectrum showed a shift towards the higher frequencies. This shift is similar to our earlier observation that was interpreted as the lignin contribution on the IR spectrum of calcite particle (Fig. 4d, red line). Furthermore, the second feature/peak at ∼1600 cm−1 for the ring-shaped, similar to the spherical particle spectrum at this frequency, bear a resemblance to the features of the aromatic ring stretch mode of the lignin. Overall, our assessment of the IR spectra features for the ring-shaped morphology is that this structure had more contribution from the lignin compound, [see IR s-SNOM map at lignin functionalities 1600 cm−1 (Fig. 5f)]. However, the broader IR background between 1450–1500 cm−1 is similar to that observed in the IR spectra of calcite particle, indicating a contribution from carbonate stretching band although not as strong as in the spherical particle. Given the weak presence of pentaglycine, lauric acid (laurate) in the pristine organic mixture, their contribution in both particles were inconclusive (Fig. 5, S6†). Specifically, contribution of amide I and II (at ∼1630 cm−1 and ∼1540 cm−1) and the carbonyl group (at ∼1550 cm−1 and ∼1700 cm−1)] in the spectra was not clear, suggesting that that these organic species were either not present or had concentrations that were below the IR detection level.
Lastly, PCA conducted on several spherical and ring-shaped morphologies found no significant differences between their chemical composition, indicating that the contribution of organic/inorganic components to the IR spectra is rather similar (homogeneous) even at nanoscale.
3.4 Stability of Ca–organic association, from Py-GC/MS
Thermal stability of organic compounds via association with the Ca(aq) ions was studied using Py-GC/MS analysis. We compared the Py-GC/MS results between the pristine organic compound mixture and the same organic compound mixture after exposure to calcite. The evolved gas analysis (EGA) results revealed that pristine organic compounds mixture was pyrolyzed at 260 and 370 °C (Fig. 6a). The evolved gas peaks of pristine solutions aligned well with peaks of lignin, trehalose, and pentaglycine (although some overlap is observed for pentaglycine and trehalose). Meanwhile the contribution from lauric acid in the pristine OM compound mixture was not detectable due to its low concentration (limited by its solubility in water), compared to other compounds (Fig. 6a); however, as we describe later, lauric acid was identified in the pristine solutions during high-resolution GC/MS analysis. The EGA results revealed that organic compounds mixture exposed to calcite was pyrolyzed to higher temperatures 280, 370, and 415 °C (Fig. 6a). To understand the shift to the higher temperature we further conducted Py-GC/MS analysis and probed possible changes in the molecular composition of pyrolysis products. We identified several differences in peak assignment between pristine and solutions exposed to calcite, when we compared MS data collected at 260 °C with those at 280 °C, and data collected at 370 °C with those acquired at 415 °C (Fig. 6b). Despite the high complexity of spectra and challenges for accurate assignment of pyrolyzed organic compounds, we identified a number of new and unique compounds for the samples that were exposed to calcite, highlighted in Fig. 6b, and listed in the Tables S5 and S6.† The new compounds identified at 280 °C showed aromatic functionalities and appeared to be fragments of the lignin molecule, (Fig. 6b, see peaks 1, 2, 3, and 5; and Table S4†). Consistently, fragments of aromatic organic compounds were identified at the higher temperature of 415 °C (Fig. 6b, peak 1, 2, 3, 4; and Table S5†). These fragments were not present in the MS analysis of pristine organic compound mixture, based on data acquired both at 260 °C and 370 °C. Meanwhile, consistent with the finding that lauric acid is adsorbed to calcite,16 this compound was only identified in the pristine organic mixture (see peak 6 at 260 °C and peak 6 and 7 at 370 °C, (Fig. 6b). In general, our data suggest that the shift to higher temperatures observed during the EGA is most likely related to lignin changes in solution after its exposure to calcite. This finding also indicates an increase in thermal stability of organic compounds (specifically of the lignin compound) as a result of complexation with Ca(aq) ions in solution, that are released following calcite dissolution.16 Lastly, we note that we did not observe an increase in the EGA temperature when 1 mM CaCl2 was added to the pristine organic compound mixture, suggesting that (bi)carbonate ions, released in the aqueous phase during calcite dissolution, might also play an important role in increasing the stability of Ca–organic associations.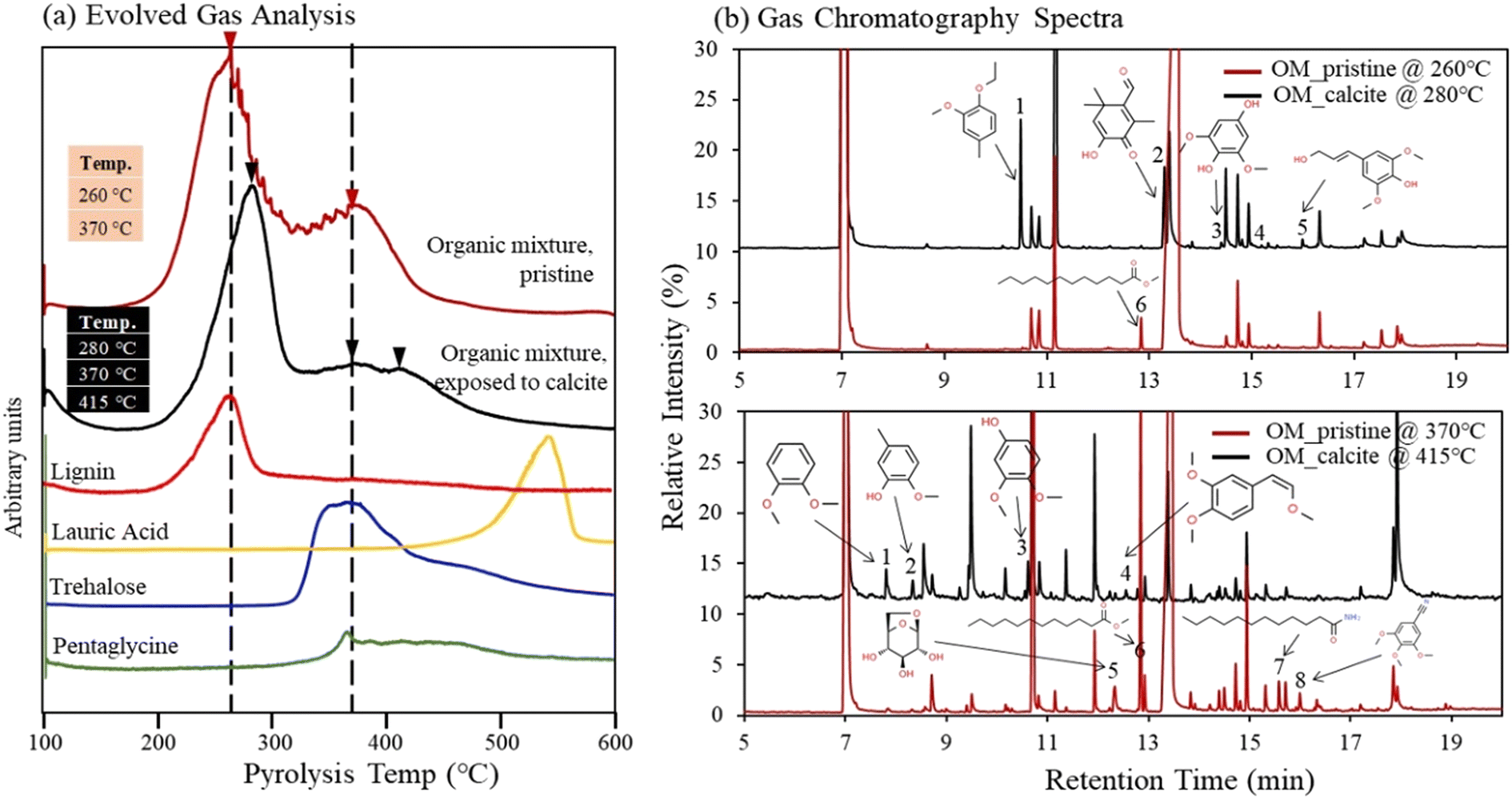 | ||
| Fig. 6 (a) EGA of pristine organic mixture, of organic mixture exposed to calcite, and of single organic components; (b) GC/MS spectra of pristine organic mixture and of organic mixture exposed to calcite. The pristine organic mixture pyrolyzed at 260 and 370 °C while organic mixture exposed to calcite, containing Ca–organic association pyrolyzed at 280 and 415 °C. | ||
4. Discussion
4.1 Ca–organic association
This study builds on recent work that revealed formation of Ca–organic associations from calcite exposure to SOM components, after organic ligand promoted calcite dissolution, and consequent to the release of Ca in the aqueous phase.16 The evidence presented therein suggested primarily aqueous Ca complexation with SOM species, lignin and amino acids. Even more importantly, the results of the previous study indicated that Ca–organic associations were not bound to calcite surface sites, although they exhibited reduced spatial mobilities.The objective of our follow up study was to reveal the chemical composition and Ca coordination in the complex Ca–organic associations. Experimental characterization and measurements at molecular scale of mixtures of classes of SOM bound to Ca ions, which then form Ca–organic associations, are challenging. Specifically, determining the chemical molecular identity and coordination environment of Ca bound to organic species requires a chemical aqueous probe (such as liquid microscopy/spectroscopy cells). In situ measurements of Ca–organic interactions of aqueous suspensions using Ca K-edge or L-edge X-ray scattering techniques, presented significant experimental challenges and were outside the scope of this study.
In the absence of the aqueous chemical probe, we used a powerful NEXAFS spectromicroscopy technique, combined with an IR s-SNOM fine scale nanoimaging chemical probe of lyophilized samples. The combined techniques provide molecular-level evidence of the Ca ions associated with organic moieties and specific evidence of the chemical nature of the organic functional groups of the Ca–organic associations. The Ca L-edge X-ray absorption spectra allowed interpretation of our results following a method previously developed to determine the Ca coordination environment for crystalline and poorly crystalline Ca containing materials formed during biomineralization processes.57 In few studies,48,49,57 Ca in calcite was identified in 6-fold coordination (CN = 6) by oxygen atoms while in the polymorph aragonite Ca was identified in the 9-fold coordination (CN ≅ 9) by oxygen atoms. The coordination number was used as an indicator to explain the differences between Ca L2,3-edge of aragonite from those of calcite and another CaCO3 polymorph, i.e., vaterite.
In our study, as expected, the spectral features of calcite particles showed a good match with the reference calcite energy positions provided in the study by DeVol et al., 2015.47 Meanwhile, the Ca–organic assemblages revealed spectral features that better matched the reference assigned positions of aragonite and demonstrated deviations from calcite and hydrated amorphous calcium carbonate features (Fig. S5†). Even more surprisingly, the spectra fit for Ca–organic association revealed a closer match to literature assigned peak positions for apatite.58 In this regard our observations appear to be consistent with a recent study, which investigated composition of organic C in soils in the presence of Ca ions.59 Similar to our findings Ca spectra signatures in the Chen et al., 2014 study are analogous to Ca phosphate compounds, bearing characteristics of a disordered or amorphous phase.50,59 Although the authors argue that these could be Ca–organic compounds, they also state that in soils it is possible that biomolecules, proteins, or extracellular polymeric substances could be closely associated with Ca phosphate phases. In our study however, phosphate bearing Ca minerals are an improbable occurrence based on the experimental conditions (i.e., phosphate was not present in the system). Taken together, these studies imply that Ca organic matter associations may exhibit features that are similar to those of amorphous Ca phosphates.
Prediction of local chemical environment and Ca coordination, based on peak energy differences for L3 compared to L2 component, is unattainable for Ca–organic associations in this study. This is due to missing of the minor signature peak at 348.0 eV in the L3-edge spectra (Fig. 3c and d and S5†), which is needed to calculate peak energy split ΔL3 (peak 2 – peak 1) of the L3 component and compare it to peak energy split ΔL2 (peak 4 – peak 3) of the L2 component.48 The differences in energy splits (Δ values) can be used to probe the local Ca structure.48–51,57 In addition to energy splits, spectral features in terms of the position and the number of smaller pre-edge peaks can also be used to make predictions for the Ca chemical environment.48,57 In this study we note the absence of two small peaks, expected to have about the same intensity, preceding (at ∼1 eV and ∼0.8 eV respectively) the calcite characteristic peak at 348.0 eV of the L3 component.48 The pattern that contains pre-edge small peaks with the same intensity has been determined to be characteristic of Ca compounds that have a CN = 6, i.e. calcite.48 Absence of all these features, in the Ca–organic association spectrum, together suggest that the Ca associated with organic compounds could have a chemical environment and site coordination that is similar to Ca in apatite and aragonite (see Fig. S5†), which both have Ca sites that satisfy CN > 6.43,48,49,51
In addition, both apatite and aragonite feature bidentate coordination of Ca, which is not observed in calcite. Bidentate Ca carbonate pairs are more commonly observed in solutions and in amorphous carbonates,60,61 for Ca salts of organic acids,61,62 and when Ca is coordinated within protein structures.63 Determining the type of coordination bonds of Ca ions requires an extensive molecular simulation approach,14 which was not within the scope of this study. However, indirect evidence collected here suggests that Ca atoms might be coordinated with both the organic compounds and carbonate and/or bicarbonate ions, which were both present in the solution of our experiments. Evidence in this study is provided by detailed microscopic analysis, TEM, and high-resolution nano-spectroscopic IR s-SNOM characterization. The combined results show uniform contribution of both organic and inorganic C in Ca rich structures (Fig. 1 and 5) and within detection of the analytical methods show no evidence of formation of organic or inorganic C clusters. According to a recent study, significant CO32−−CO32− clustering is absent when carbonate ions substitute into a disordered amorphous calcium phosphate phase.45 This agrees with the data in our study, which demonstrate spatial homogeneity of organic carbon (lignin primarily) and inorganic C species, [(bi)carbonate] at macro and nanoscale. This study for the first time suggests that an inorganic anion (i.e., (bi)carbonate) might be part of the Ca–organic associations, with inorganic and organic species in solution most likely to be both bound to Ca ions. We encourage further studies that employ solution spectroscopy probes, to provide direct evidence of (bi)carbonate presence as part of the Ca–organic associations and probe Ca chemical environment and coordination number.
Furthermore, consistent peak position shifts observed in NEXAFS spectra for organic compounds (in particular lignin), suggest that the corresponding deviations in the energy position can be related to the chemical environment of C atoms bonded to Ca neighboring atoms. This is in agreement with previous studies that have reported significant peak shifts at higher energy (from 288.4 to 288.6 eV) in the carboxylic group assignment when peptide molecules were bound to Ca ions.31,64 Similarly, shifts in the COO− spectral feature and changes in the relative peak intensities because of coordination with cation (as observed in our study) have been also reported elsewhere.65,66 Our findings in conjunction with these studies, highlight a critical need for detailed additional experimental or computational research on Ca or/and metal-containing organic model compounds to enable the accurate determination and quantification of Ca-induced or metal-induced shifts in energy position and peak assignments. The computational MD calculations are also necessary to deconvolute contributions of higher coordination numbers and Ca bidentate binding modes.
The evidence collected in this study demonstrates that calcite dissolution and release of Ca ions could have a significant role in promoting Ca–organic associations by neutralization of charged functional groups of the organic molecules promoting stabilization of organic biomolecules. Experimental measurements show that the organic compounds that were exposed to calcite have higher thermal stability based on the evidence that their decomposition (in the absence of O2 or their pyrolysis) occurs at higher temperatures. The decomposing products were most likely composed of aromatic functionalities indicating increased stabilities for lignin because of complexation with Ca ions in solution. Although indirect evidence presented in previous studies showed a decrease in organic matter mineralization after addition of Ca bearing salts, such as CaCO3 or CaSO4 in soils rich in organic matter,4,24,25 which could be attributed to the formation of Ca complexes with lignin – like compounds, future studies should determine whether the observed increase in thermal stabilization equates to protection of individual organic compounds, or generally SOM, from biologically driven decomposition.
5. Implications and future efforts
The use of soft X-ray spectromicroscopy and IR based nano-spectroscopy in this study has enabled the examination of chemical composition and Ca coordination of Ca–organic associations. In this process we discovered that inorganic anions [i.e. (bi)carbonate] are part of Ca–organic complexes. To our knowledge this is the first study that demonstrates the presence of inorganic anions in the Ca–organic associations. This finding suggests that, in addition to the bicarbonate, other anions that could be present in the soil solutions, could also be part of these inorganic/organic associations bridged to multivalent cations. Our study also shows that these Ca organic/inorganic complexes have a higher stability compared to pristine compounds. Although the higher stability could be due to stabilization of OM compounds through association with Ca ions, future studies should investigate the role of other inorganic anions in the stability of these associations.Apart from the above conclusion, spectromicroscopy-based results in our study show a plausible shift in the C energy spectra for organic compound classes when they are bound or bridged by cations compared to pristine compounds. The evidence here is for OM compounds bound to Ca ions; yet other multivalent cations, such Al or Zn, are known to form bridges between organic compounds pertaining to different SOM classes and could also affect the C spectra in ways that are currently unknown. The soft X-ray based spectromicroscopy has been frequently used in the last decade, for determining organic C speciation in soils based on energy assignments using reference pristine standards. Our findings underscore a need for characterization studies that accurately determine energy position of OM compounds when they are bound to Ca or other multivalent cations that are relevant in soils. Such characterization studies will provide more specific spectra assignments and, consequently a more accurate characterization of organic C species in soil environments.
Author contributions
Odeta Qafoku: investigation, data curation, management of the research, funding acquisition, writing the original draft, review & editing pre- and post-publication stages. Anil K. Battu: STXM/NEXAFS conducting analysis, visualization. Tamas Varga: STXM/NEXAFS section investigation, writing, reviewing, Matthew A. Marcus: STXM/NEXAFS section validation, data visualization, editing. Brian O'Callahan: IR s-SNOM section formal analysis, data visualization, writing. Qian Zhao: Py-GC/MS section investigation, writing, reviewing & editing. Sebastian T. Mergelsberg: STXM/NEXAFS section reviewing & editing. William R. Kew: formal analysis. John S. Loring: ATR FTIR conducting analysis. Nikolla P. Qafoku: conceptualization of research ideas, writing, review & editing pre- and post-publication stages. Sarah I. Leichty: providing analysis tools.Conflicts of interest
There are no conflicts to declare.Acknowledgements
(A portion of) This research was performed on a project award (https://doi.org/10.46936/intm.proj.2020.51672/60000251) from the Environmental Molecular Sciences Laboratory, a DOE Office of Science User Facility sponsored by the Biological and Environmental Research program under Contract No. DE-AC05-76RL01830. This research used resources of the Advanced Light Source, a U.S. DOE Office of Science User Facility under contract no. DE-AC02-05CH11231. We thank the anonymous reviewers for their careful reading of our manuscript and for their insightful suggestions and comments that improved the quality of our manuscript.References
- A. Fischlin, G. F. Midgley, J. T. Price, R. Leemans, B. Gopal, C. Turley, M. D. A. Rounsevell, O. P. Dube, J. Tarazona and A. A. Velichko, Ecosystems, their properties, goods, and services, in Climate Change 2007: Impacts, Adaptation and Vulnerability, ed. M. L. Parry, O. F. Canziani, J. P. Palutikof, P. J. van der Linden and C. E. Hanson, Cambridge University Press, Cambridge, UK, 2007, p. 211 Search PubMed.
- M. W. I. Schmidt, M. S. Torn, S. Abiven, T. Dittmar, G. Guggenberger, I. A. Janssens, M. Kleber, I. Kögel-Knabner, J. Lehmann, D. A. C. Manning, P. Nannipieri, D. P. Rasse, S. Weiner and S. E. Trumbore, Persistence of soil organic matter as an ecosystem property, Nature, 2011, 478, 49–56 CrossRef CAS PubMed.
- J. A. Baldock and P. N. Nelson, Soil organic matter, in Handbook of Soil Science, ed. M. Summer, CRC press, Boca Raton, FL, 2000, pp. 25–84 Search PubMed.
- J. A. Baldock and J. O. Skjemstad, Role of the soil matrix and minerals in protecting natural organic materials against biological attack, Org. Geochem., 2000, 31, 697–710 CrossRef CAS.
- A. J. Simpson, G. Song, E. Smith, B. Lam, E. H. Novotny and M. H. B. Hayes, Unraveling the structural components of soil humin by use of solution-state nuclear magnetic resonance spectroscopy, Environ. Sci. Technol., 2007, 41, 876–883 CrossRef CAS PubMed.
- H. Chen, R. C. Johnston, B. F. Mann, R. K. Chu, N. Tolic, J. M. Parks and B. Gu, Identification of mercury and dissolved organic matter complexes using ultrahigh resolution mass spectrometry, Environ. Sci. Technol. Lett., 2017, 4, 59–65 CrossRef CAS.
- A. Rivas-Ubach, Y. Liu, T. S. Bianchi, N. Tolic, C. Jansson and L. Pasa-Tolic, Moving beyond the van Krevelen diagram: A new stoichiometric approach for compound classification in organisms, Anal. Chem., 2018, 90, 6152–6160 CrossRef CAS PubMed.
- M. Kleber, I. C. Bourg, E. K. Coward, C. M. Hansel, S. C. B. Myneni and N. Nunan, Dynamic interactions at the mineral–organic matter interface, Nat. Rev. Earth Environ., 2021, 2, 402–421 CrossRef.
- M. S. Torn, C. W. Swanston, C. Castanha and S. E. Trumbore, Storage and turnover of organic matter in soil, in Biophysico-Chemical Processes Involving Natural Nonliving Organic Matter in Environmental Systems, ed. N. Senesi, B. Xing and P. M. Huang, Wiley, Hoboken, NJ, U.S., 2009, pp. 219–272 Search PubMed.
- M. C. Rowley, S. Grand and É. P. Verrecchia, Calcium-mediated stabilisation of soil organic carbon, Biogeochemistry, 2018, 137, 27–49 CrossRef CAS.
- T. D. Sowers, D. Adhikari, J. Wang, Y. Yang and D. L. Sparks, Spatial associations and chemical composition of organic carbon sequestered in Fe, Ca, and organic carbon ternary systems, Environ. Sci. Technol., 2018, 52, 6936–6944 CrossRef CAS PubMed.
- G. M. Bowers, H. E. Argersinger, U. V. Reddy, T. A. Johnson, B. Arey, M. Bowden and R. J. Kirkpatrick, Integrated molecular and microscopic scale insight into morphology and ion dynamics in Ca2+-mediated natural organic matter floccs, J. Phys. Chem. C, 2015, 119, 17773–17783 CrossRef CAS.
- J. Adusei-Gyamfi, B. Ouddane, L. Rietveld, J.-P. Cornard and J. Criquet, Natural organic matter-cations complexation and its impact on water treatment: A critical review, Water Res., 2019, 160, 130–147 CrossRef CAS PubMed.
- D. Devarajan, L. Liang, B. Gu, S. C. Brooks, J. M. Parks and J. C. Smith, Molecular dynamics simulation of the structures, dynamics, and aggregation of dissolved organic matter, Environ. Sci. Technol., 2020, 54, 13527–13537 CrossRef CAS PubMed.
- N. Loganathan, B. O. Ferguson, B. Arey, H. E. Argersinger and G. M. Bowers, A mechanistic exploration of natural organic matter aggregation and surface complexation in smectite mesopores, J. Phys. Chem. A, 2020, 124, 9832–9843 CrossRef CAS PubMed.
- O. Qafoku, A. Andersen, W. R. Kew, R. K. Kukkadapu, S. D. Burton, L. Kovarik, Q. Zhao, S. T. Mergelsberg, T. W. Wietsma and C. T. Resch, Selective interactions of soil organic matter compounds with calcite and the role of aqueous Ca, ACS Earth Space Chem., 2022, 6, 1674–1687 CrossRef CAS.
- R. Sutton and G. Sposito, Molecular simulation of humic substance Ca-montmorillonite complexes, Geochim. Cosmochim. Acta, 2006, 70, 3566–3581 CrossRef CAS.
- T. D. Sowers, J. W. Stuckey and D. L. Sparks, The synergistic effect of calcium on organic carbon sequestration to ferrihydrite, Geochem. Trans., 2018, 19, 4 CrossRef PubMed.
- J. Chi, W. Zhang, L. Wang and C. V. Putnis, Direct observations of the occlusion of soil organic matter within calcite, Environ. Sci. Technol., 2019, 53, 8097–8104 CrossRef CAS PubMed.
- I. Van Der Kellen, D. Derrien, J. Ghanbaja and M.-P. Turpault, Recent weathering promotes C storage inside large phyllosilicate particles in forest soil, Geochim. Cosmochim. Acta, 2022, 318, 328–351 CrossRef CAS.
- J. M. Oades, The retention of organic matter in soils, Biogeochemistry, 1988, 5, 35–70 CrossRef CAS.
- V. P. Sokoloff, Effect of neutral salts of sodium and calcium on carbon and nitrogen in soils, J. Agric. Res., 1938, 57, 201–216 CAS.
- P. Duchaufour, Dynamics of organic matter in soils of temperate regions: its action on pedogenesis, Geoderma, 1976, 15, 31–40 CrossRef.
- M. Muneer and J. M. Oades, The role of Ca-organic interactions in soil aggregate stability. I. Laboratory studies with glucose 14C, CaCO3 and CaSO4.2.H2O, Soil Res., 1989, 27, 389–399 CrossRef CAS.
- M. Muneer and J. M. Oades, The role of Ca-organic interactions in soil aggregate stability. II. Field studies with 14C-labeled straw, CaCO3 and CaSO4.2.H2O, Soil Res., 1989, 27, 401–409 CrossRef CAS.
- A. G. Kalinichev and R. J. Kirkpatrick, Molecular dynamics simulation of cationic complexation with natural organic matter, Eur. J. Soil Sci., 2007, 58, 909–917 CrossRef CAS.
- A. Majzik and E. Tombácz, Interaction between humic acid and montmorillonite in the presence of calcium ions II, Colloidal interactions: Charge state, dispersing and/or aggregation of particles in suspension, Org. Geochem., 2007, 38, 1330–1340 CrossRef CAS.
- E. Pihlap, M. Steffens and I. Kögel-Knabner, Initial soil aggregate formation and stabilisation in soils developed from calcareous loess, Geoderma, 2021, 385, 114854 CrossRef CAS.
- C. D. Grant, A. R. Dexter and J. M. Oades, Residual effects of additions of calcium compounds on soil structure and strength, Soil Tillage Res., 1992, 22, 283–297 CrossRef.
- E. Iskrenova-Tchoukova, A. G. Kalinichev and R. J. Kirkpatrick, Metal cation complexation with natural organic matter in aqueous solutions: molecular dynamics simulations and potentials of mean force, Langmuir, 2010, 26, 15909–15919 CrossRef CAS PubMed.
- J. Lehmann, D. Solomon, J. Brandes, H. Fleckenstein, C. Jacobson and J. Thieme, Synchrotron-based Near-Edge X-Ray Spectroscopy of natural organic matter in soils and sediments, in Biophysico-Chemical Processes Involving Natural Nonliving Organic Matter in Environmental Systems, ed. N. Senesi, B. Xing and P. M. Huang, Wiley, Hoboken, NJ, U.S., 2009, pp. 729–781 Search PubMed.
- T. Warwick, H. Ade, D. Kilcoyne, M. Kritscher, T. Tylisczcak, S. Fakra, A. Hitchcock, P. Hitchcock and H. Padmore, A new bend-magnet beamline for scanning transmission X-ray microscopy at the Advanced Light Source, J. Synchrotron Radiat., 2002, 9, 254–257 CrossRef CAS PubMed.
- A. L. D. Kilcoyne, T. Tyliszczak, W. F. Steele, S. Fakra, P. Hitchcock, K. Franck, E. Anderson, B. Harteneck, E. G. Rightor and G. E. Mitchell, Interferometer-controlled scanning transmission X-ray microscopes at the Advanced Light Source, Synchrotron Radiat., 2003, 10, 125–136 CrossRef CAS PubMed.
- H. A. Bechtel, S. C. Johnson, O. Khatib, E. A. Muller and M. B. Raschke, Synchrotron infrared nano-spectroscopy and-imaging, Surf. Sci. Rep., 2020, 75, 100493 CrossRef CAS.
- C. Colombo, G. Palumbo, R. Angelico, H. G. Cho, O. Francioso, A. Ertani and S. Nardi, Spontaneous aggregation of humic acid observed with AFM at different pH, Chemosphere, 2015, 138, 821–828 CrossRef CAS PubMed.
- S. C. B. Myneni, J. T. Brown, G. A. Martinez and W. Meyer-Ilse, Imaging of humic substance macromolecular structures in water and soils, Science, 1999, 286, 1335–1337 CrossRef CAS PubMed.
- R. C. Moffet, T. Henn, A. Laskin and M. K. Gilles, Automated chemical analysis of internally mixed aerosol particles using X-ray spectromicroscopy at the carbon K-edge, Anal. Chem., 2010, 82, 7906–7914 CrossRef CAS PubMed.
- R. Mak, M. Lerotic, H. Fleckenstein, S. Vogt, S. M. Wild, S. Leyffer, Y. Sheynkin and C. Jacobsen, Non-negative matrix analysis for effective feature extraction in X-ray spectromicroscopy, Faraday Discuss., 2014, 171, 357–371 RSC.
- G. D. Cody, H. Ade, C. M. O. D. Alexander, T. Araki, A. Butterworth, H. Fleckenstein, G. Flynn, M. K. Gilles, C. Jacobsen, A. L. D. Kilcoyne, K. Messenger, S. A. Sandford, T. Tyliszczak, A. J. Westphal and S. Wirick, Quantitative organic and light-element analysis of comet 81P/Wild 2 particles using C-, N-, and O-μ-XANES, Meteorit. Planet. Sci., 2008, 43, 353–365 CrossRef CAS.
- J. A. Brandes, S. Wirick and C. Jacobsen, Carbon K-edge spectra of carbonate minerals, J. Synchrotron Radiat., 2010, 17, 676–682 CrossRef CAS PubMed.
- M. A. Marcus, Data analysis in spectroscopic STXM, J. Electron Spectrosc. Relat. Phenom., 2023, 264, 147310 CrossRef CAS.
- S. Hanhan, A. M. Smith, M. Obst and A. P. Hitchcock, Optimization of analysis of soft X-ray spectromicroscopy at the Ca 2p edge, J. Electron Spectrosc. Relat. Phenom., 2009, 173, 44–49 CrossRef CAS.
- G. Geng, R. J. Myers, A. L. D. Kilcoyne, J. Ha and P. J. M. Monteiro, Ca L2,3-edge X-ray absorption fine structure of tricalcium aluminate, gypsum, and calcium (sulfo)aluminate hydrates, Am. Mineral., 2017, 102, 900–908 CrossRef.
- G. Geng, J. Li, Y. Yu, D. A. Shapiro, D. A. L. Kilcoyne and P. J. M. Monteiro, Nanometer-Resolved spectroscopic study reveals the conversion mechanism of CaO·Al2O3·10H2O to 2CaO·Al2O3·8H2O and 3CaO·Al2O3·6H2O at an elevated temperature, Cryst. Growth Des., 2017, 17, 4246–4253 CrossRef CAS.
- O. F. Yasar, W.-C. Liao, B. Stevensson and M. Edén, Structural role and spatial distribution of carbonate ions in amorphous calcium phosphate, J. Phys. Chem. C, 2021, 125, 4675–4693 CrossRef CAS.
- R. T. DeVol, R. A. Metzler, L. Kabalah-Amitai, B. Pokroy, Y. Politi, A. Gal, L. Addadi, S. Weiner, A. Fernandez-Martinez and R. Demichelis, Oxygen spectroscopy and polarization-dependent imaging contrast (PIC)-mapping of calcium carbonate minerals and biominerals, J. Phys. Chem. B, 2014, 118, 8449–8457 CrossRef CAS PubMed.
- R. T. DeVol, C.-Y. Sun, M. A. Marcus, S. N. Coppersmith, S. C. B. Myneni and P. U. P. A. Gilbert, Nanoscale transforming mineral phases in fresh nacre, J. Am. Chem. Soc., 2015, 137, 13325–13333 CrossRef CAS PubMed.
- S. J. Naftel, T. K. Sham, Y. M. Yiu and B. W. Yates, Calcium L-edge XANES study of some calcium compounds, J. Synchrotron Rad., 2001, 8, 255–257 CrossRef CAS PubMed.
- M. E. Fleet and X. Liu, Calcium L2,3-edge XANES of carbonates, carbonate apatite, and oldhamite (CaS), Am. Mineral., 2009, 94, 1235–1241 CrossRef CAS.
- Y. L. Politi, R. A. Metzler, M. Abrecht, B. Gilbert, F. H. Wilt, I. Sagi, L. Addadi, S. Weiner and P. U. P. A. Gilbert, Transformation mechanism of amorphous calcium carbonate into calcite in the sea urchin larval spicule, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(45), 17362–17366 CrossRef CAS PubMed.
- J. Cosmidis, K. Benzerara, N. Nassif, T. Tyliszczak and F. Bourdelle, Characterization of Ca-phosphate biological materials by scanning transmission X-ray microscopy (STXM) at the Ca L2,3-, P L2,3- and C K-edges, Acta Biomater., 2015, 12, 260–269 CrossRef CAS PubMed.
- B. T. O'Callahan, K. T. Crampton, I. V. Novikova, T. Jian, C.-L. Chen, J. E. Evans, M. B. Raschke, P. Z. El-Khoury and A. S. Lea, Imaging nanoscale heterogeneity in ultrathin biomimetic and biological crystals, J. Phys. Chem. C, 2018, 122, 24891–24895 CrossRef.
- D. Kim and V. H. Grassian, Attenuated total reflection-fourier transform infrared and atomic force microscopy-infrared spectroscopic investigation of Suwannee River fulvic acid and its interactions with α-FeOOH, ACS Earth Space Chem., 2021, 6, 81–89 CrossRef.
- E. A. Muller, B. Pollard, H. A. Bechtel, P. van Blerkom and M. B. Raschke, Infrared vibrational nano-crystallographyand nano-imaging, Sci. Adv., 2016, 2(2), e1601006 CrossRef PubMed.
- H. A. Bechtel, S. C. Johnson, O. Khatib, E. A. Muller and M. B. Raschke, Synchrotron infrared nano-spectroscopy and -imaging, Surf. Sci. Rep., 2020, 75, 100493 CrossRef CAS.
- A. Cvitkovic, N. Ocelic and R. Hillenbrand, Analytical model for quantitative prediction of material contrasts in scattering-type near-field optical microscopy, Opt. Express, 2007, 15, 8550–8565 CrossRef CAS PubMed.
- K. Benzerara, T. H. Yoon, T. Tyliszczak, B. Constantz, A. M. Spormann and G. E. Brown Jr, Scanning transmission X-ray microscopy study of microbial calcification, Geobiology, 2004, 2, 249–259 CrossRef.
- C. A. Stifler, N. K. Wittig, M. Sassi, C.-Y. Sun, M. A. Marcus, H. Birkedal, E. Beniash, K. M. Rosso and P. U. P. A. Gilbert, X-ray linear dichroism in apatite, J. Am. Chem. Soc., 2018, 140, 11698–11704 CrossRef CAS PubMed.
- C. Chen, J. J. Dynes, J. Wang, C. Karunakaran and D. L. Sparks, Soft X-ray spectromicroscopy study of mineral-organic matter associations in pasture soil clay fractions, Environ. Sci. Technol., 2014, 48(12), 6678–6686 CrossRef CAS PubMed.
- F. Yang, Y.-S. Liu, X. Feng, K. Qian, L. C. Kao, Y. Ha, N. T. Hahn, T. J. Seguin, M. Tsige and W. Yang, Probing calcium solvation by XAS, MD and DFT calculations, RSC Adv., 2020, 10, 27315–27321 RSC.
- M. P. Prange, S. T. Mergelsberg and S. N. Kerisit, Ab Initio molecular dynamics simulations of amorphous calcium carbonate: interpretation of pair distribution function and X-ray absorption spectroscopy data, Cryst. Growth Des., 2021, 21, 2212–2221 CrossRef CAS.
- S. Bette, G. Eggert, S. Emmerling, M. Etter, T. Schleid and R. E. Dinnebier, Crystal structure, polymorphism, and anisotropic thermal expansion of α-Ca (CH3COO)2, Cryst. Growth Des., 2020, 20, 5346–5355 CrossRef CAS.
- V. Martin-Diaconescu, M. Gennari, B. Gerey, E. Tsui, J. Kanady, R. Tran, J. Pécaut, D. Maganas, V. Krewald and E. Gouré, Ca K-edge XAS as a probe of calcium centers in complex systems, Inorg. Chem., 2015, 54, 1283–1292 CrossRef CAS PubMed.
- G. De Stasio, M. A. Schmitt and S. H. Gellman, Spectromicroscopy at the organic-inorganic interface in biominerals, Am. J. Sci., 2005, 305, 673–686 CrossRef CAS.
- M. Plaschke, J. Rothe, M. Altmaier, M. A. Denecke and T. Fanghänel, Near edge X-ray absorption fine structure (NEXAFS) of model compounds for the humic acid/actinide ion interaction, J. Electron Spectrosc. Relat. Phenom., 2005, 148, 151–157 CrossRef CAS.
- M. Plaschke, J. Rothe, M. K. Armbruster, M. A. Denecke, A. Naber and H. Geckeis, Humic acid metal cation interaction studied by spectromicroscopy techniques in combination with quantum chemical calculations, J. Synchrotron Radiat., 2010, 17, 158–165 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2en01143c |
| This journal is © The Royal Society of Chemistry 2023 |