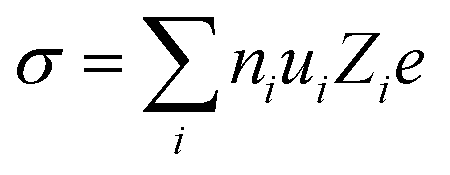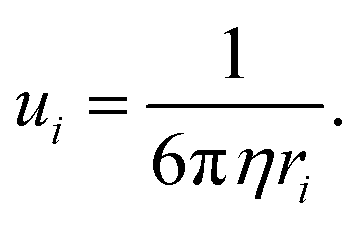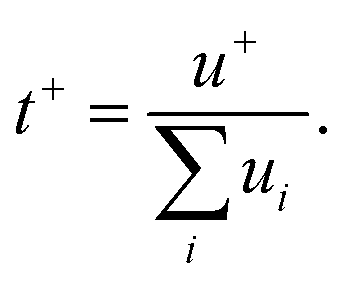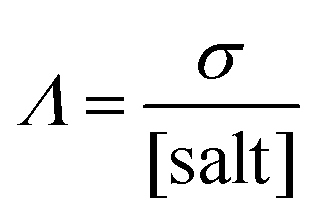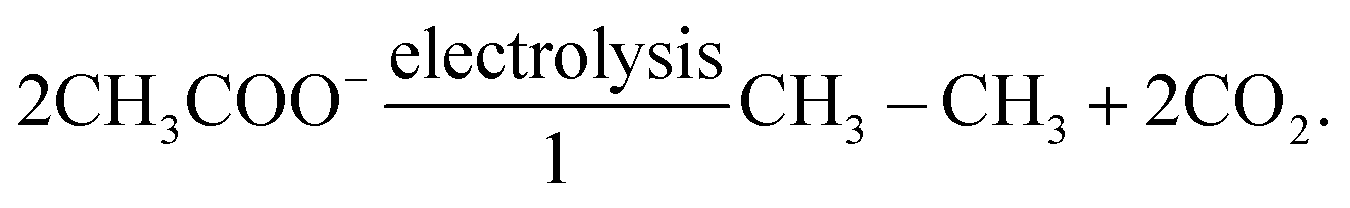DOI:
10.1039/D2EE03682G
(Perspective)
Energy Environ. Sci., 2023,
16, 1480-1501
A perspective on the role of anions in highly concentrated aqueous electrolytes
Received
14th November 2022
, Accepted 24th February 2023
First published on 27th February 2023
Abstract
Highly concentrated aqueous electrolytes enable a wider electrochemical stability window and, thus, higher energy batteries compared to conventional dilute aqueous solutions. Multiple properties of the electrolyte, e.g., ionic interactions, solvation structure, ion transport, tendency to hydrolyze, and capability to form a solid electrolyte interphase, distinctly change when the salt concentration is increased and highly depend on the salt anion. This work aims at reviewing, discussing and rationalizing the role of the salt anion in these physical and chemical properties in order to provide perspective guidelines for future developments.

Jin Han
| Jin Han received his MS degree from Southwest University under the supervision of Prof. Maowen Xu in 2017 and his PhD degree from Helmholtz-Institute Ulm (HIU) of Karlsruhe Institute for Technology (KIT) under the supervision of Prof. Stefano Passerini in 2021. After that, he continued postdoctoral research at HIU of KIT. His research interest is focused on the electrochemistry and materials for aqueous batteries. |

Alessandro Mariani
| Alessandro Mariani received his PhD in Physical Chemistry in 2016 at La Sapienza University of Rome (Italy). He then spent 2 years as a Postdoc at the European Synchrotron Radiation Facility (ESRF, Grenoble, France) on the ID02 TRUSWAXS beamline, serving as local contact for external users and focusing on the liquid–liquid phase transition of complex systems. In 2018, he moved to the Helmholtz Institute Ulm, where he started working on electrolytes for batteries. He is now co-responsible for the SAXS beamline at the italian synchrotron Elettra. He is an expert in the use of SAXS, Raman and NMR spectroscopies, and MD and DFT simulations. |

Stefano Passerini
| Stefano Passerini is Professor at the Chemistry Department of Sapienza University of Rome. Formerly professor at the Karlsruhe Institute of Technology, Helmholtz Institute Ulm (Germany) and the University of Muenster (Germany), he cofounded the MEET battery research center. His research focuses on the basic understanding and development of materials for high-energy batteries and supercapacitors, with the goal to create sustainable energy storage systems from environmentally friendly and available materials and processes. He is an internationally recognized pioneer in the field of ionic liquids and the development of alkali-ion batteries. |

Alberto Varzi
| Alberto Varzi studied Chemistry of Materials at University of Bologna (Italy) and received a PhD in 2013 from the Ulm University and the Center for Solar Energy and Hydrogen Research Baden-Württemberg (ZSW). After a postdoctoral period at MEET battery research center – University of Muenster, in 2014 he joined the Helmholtz-Institute Ulm (HIU) of the Karlsruhe Institute for Technology (KIT). Since 2021 he is Principal Investigator and leads the group “Electrochemistry of Materials and Interfaces”. His current research interests span from solid state to aqueous batteries, including beyond-Li systems. |
Broader context
Safety issues and high costs restrain the widespread application of conventional lithium-ion batteries (LIBs) for large-scale energy storage. The development of safe and affordable batteries is particularly important for stationary energy storage systems. Among the emerging battery chemistries, those employing an aqueous electrolyte are particularly promising in view of reducing material and manufacturing costs. However, the narrow electrochemical stability of water poses serious limits to the maximum output voltage of aqueous batteries. Recently, the introduction of highly concentrated aqueous electrolytes – the so-called “Water-in-salt” electrolytes (WiSE) – resulted in a wider electrochemical stability window of water and, in turn, allowed the development of higher energy batteries. Nevertheless, many aspects of these novel electrolyte systems remain unexplored. In fact, ionic interactions, solvation structure, ion transport, tendency to hydrolyze, and capability to form a solid electrolyte interphase, distinctly change compared to dilute solutions. These aspects are rarely discussed in detail, although advances in this field were generally summarized in specific reviews. This perspective aims at filling this gap and, in particular, at systematically discussing the crucial role of the salt anion.
|
1. Introduction
To reduce greenhouse gas emissions and mitigate climate change, abundant and readily available renewable energy sources, such as wind and solar energy, must be increasingly exploited.1 Indeed, diverse government policy-based incentives have facilitated the development of renewable energy harvesting technologies in the recent years.2 Nonetheless, these renewable energy sources are intrinsically intermittent and their availability depends on weather, diurnal cycle, seasons, and geographical location.3 Therefore, seeking an effective solutions to store large amount of energy from these intermittent resources is of pivotal importance.4,5 In the past decades, batteries have become one of the most popular energy storage technology for this purpose.6 In particular, lithium-ion batteries (LIBs) dominate the consumable electronics, house storage, and automotive market, where energy density and cycling ability are major priorities.7 Nevertheless, safety issues and high costs still hinder the widespread application of LIBs for large-scale energy storage.8,9 Therefore, the development of safe and affordable batteries is highly relevant for large-scale applications.
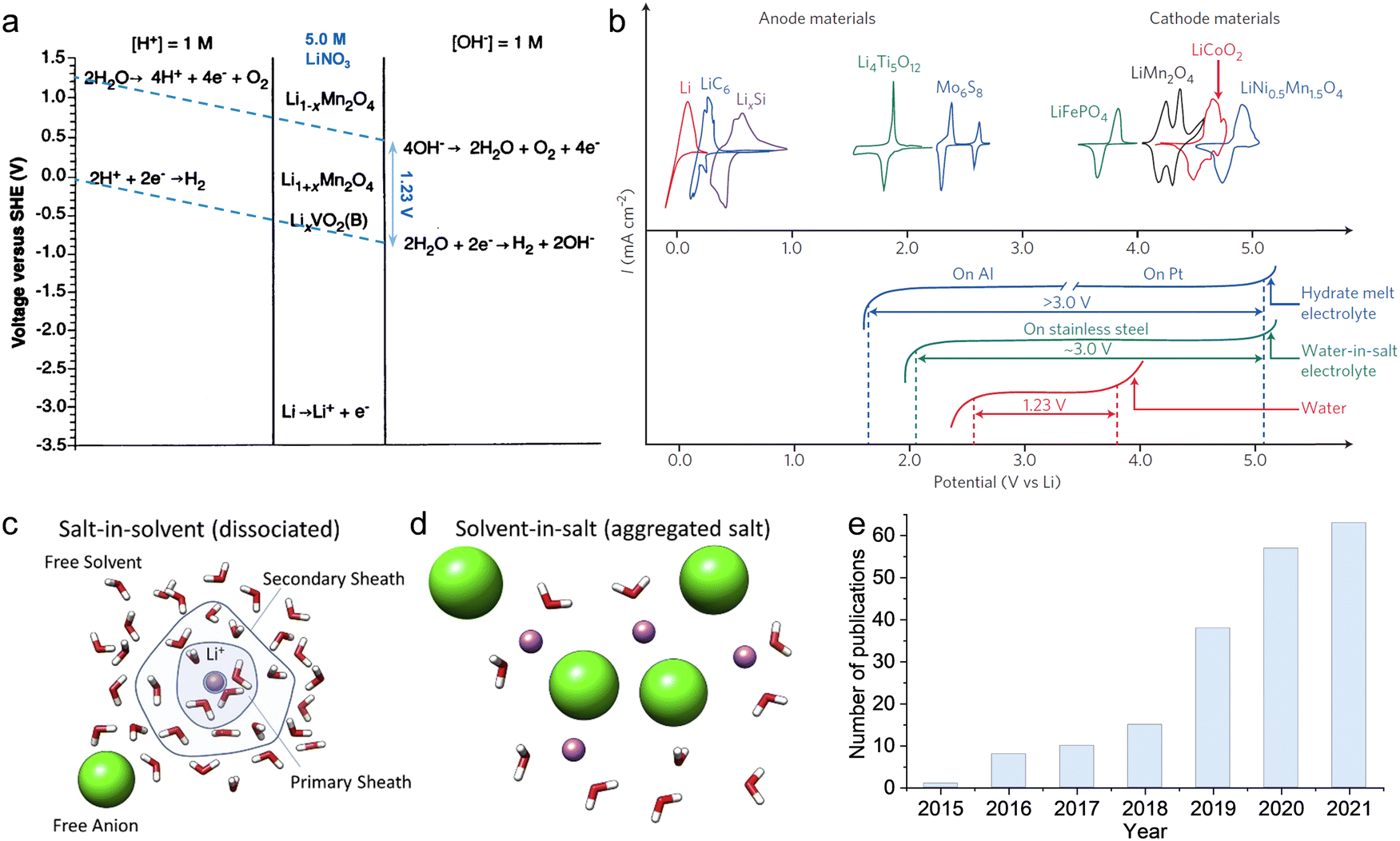
Since Dahn's group proposed the first VO2/LiMn2O4 aqueous lithium-ion battery (ALBs) employing 5 M LiNO3 electrolyte and displaying an average operating voltage of 1.5 V and an energy density of around 55 W h kg−1 in 1994 (see Fig. 1a),10 “rocking-chair” type batteries utilizing nonvolatile and nonflammable aqueous solutions as electrolyte have captured extensive attention of researchers.11 Nevertheless, “regular” dilute aqueous electrolytes suffer from narrow electrochemical stability window (ESW) (∼1.23 V), which restricts the output voltage of the battery and, in turn, its energy density.12 In fact, water decomposition easily take place when the electrodes approach the electrolyte's ESW limits, due to possible catalytic effects of the active materials and current collectors. The resulting oxygen evolution reaction (OER) and/or hydrogen evolution reaction (HER) cause, therefore, low Coulombic efficiency (CE), continuous electrolyte's consumption, battery swelling and capacity fading. These issues had stimulated researchers to further develop aqueous electrolytes with enlarged ESW.
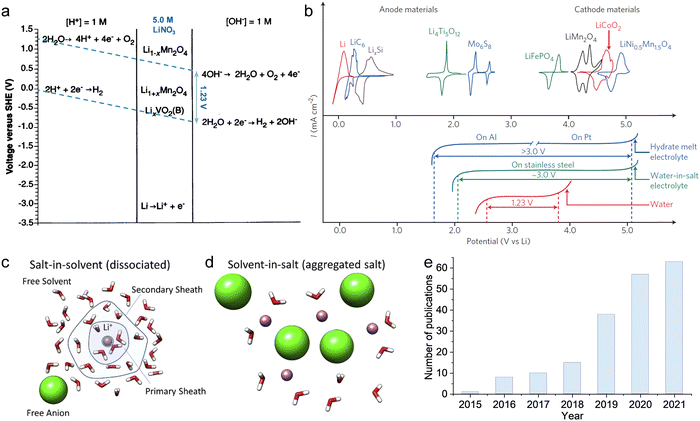 |
| | Fig. 1 (a) Potentials of the indicated reactions versus standard hydrogen electrode (SHE) in solutions where [Li+] = 1.0 M. Both acid and basic electrolytes with 1 M H+ or 1 M OH− are considered, respectively. Adapted and modified from ref. 10. (b) Comparison of ESW of multiple aqueous electrolytes and redox potentials of typical battery materials. The upper part displays the redox potentials of major anode and cathode materials: Li-metal, Mo6S8, Li4Ti5O12, LiMn2O4, LiFePO4, LiCoO2, and LiNi0.5 Mn 1.5O4; The bottom half shows that expanded ESW in super-concentrated electrolytes (21 m LiTFSI WiSE, and 27.8 m LiTFSI + LiBETI hydrate melt electrolytes) in comparison with the limited ESW of water at pH = 7 (1.23 V). Reproduced with permission.23 (c) Schematic representation of ion solvation sheath in “diluted” electrolytes evidencing the primary and the secondary solvation sheaths and the bulk free solvent. (d) Schematic representation of the solvation structure in a super-concentrated electrolyte, where the primary solvation sheath is disrupted by the lack of solvent molecules and the presence of anions in close proximity to the central cation. (c and d) Reproduced with permission.19 (e) Number of academic publications regarding “Water-in-salt” or “Hydrate-melt” concepts of aqueous batteries collected from Web of Science. | |
In 2015, Suo et al. proposed the first water-in-salt electrolyte (WiSE) by dissolving 21 m (mol kgsolvent−1) lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) into water, which enabled a wide ESW of about 3 V (see Fig. 1b).12 By definition, an electrolyte is defined as WiSE when the salt outnumbers the solvent by both weight and volume.12 This means that the number of free water molecules is drastically decreased as the concentration rises, while the amount of contact ion pairs (CIPs) and aggregates (AGGs) between cation and anion increases (see Fig. 1c and d).12 The extensive coordination between cation and anion generate a downward shift of anion's orbital levels, which make the reduction potential of the electrolyte being dominated by the ion's decomposition instead of water.13 Consequently, upon reduction, the anion is preferentially decomposed to form a solid electrolyte interphase (SEI) before the reduction of water takes place. The cathodic electrochemical stability is therefore enhanced by the ion-conductive and electron-insulating SEI.14 Simultaneously, the large majority of water molecules are coordinated to the cation via the lone pairs on the oxygen atom (being donated). As result, the oxidation potential of water is raised resulting into improved anodic stability.15 Moreover, anions gathered on the surface of the positive electrode would further prevent oxygen evolution.16 In this way, the ESW of aqueous electrolytes is effectively broadened, allowing for the development of batteries with high output voltage as well as high energy density.17 Since then, in fact, this concept has been further developed and extended to other types of highly concentrated aqueous electrolytes for ALBs, including aqueous sodium-ion batteries (ASBs), aqueous potassium-ion batteries (APBs), aqueous zinc-ion batteries (AZBs) and aqueous ammonium-ion batteries (AABs). Consequently, the number of academic publications regarding “Water-in-salt” or “Hydrate-melt” electrolyte concepts for aqueous batteries has rapidly grown in recent years (see Fig. 1d).17
Multiple reviews have summarized and discussed in general the advances in the field of aqueous batteries (ABs).16–22 However, neither reviews nor perspectives have comprehensively discussed the crucial role of the salt anion by systematically discussing ionic interactions, solvation structure, ion transport properties, hydrolysis and SEI. This review aims at filling this gap. However, before starting to discuss the above-mentioned properties, the state-of-the-art and most recent developments on aqueous batteries employing WiSEs are reviewed in Section 2.
2. WiSE based on various anions
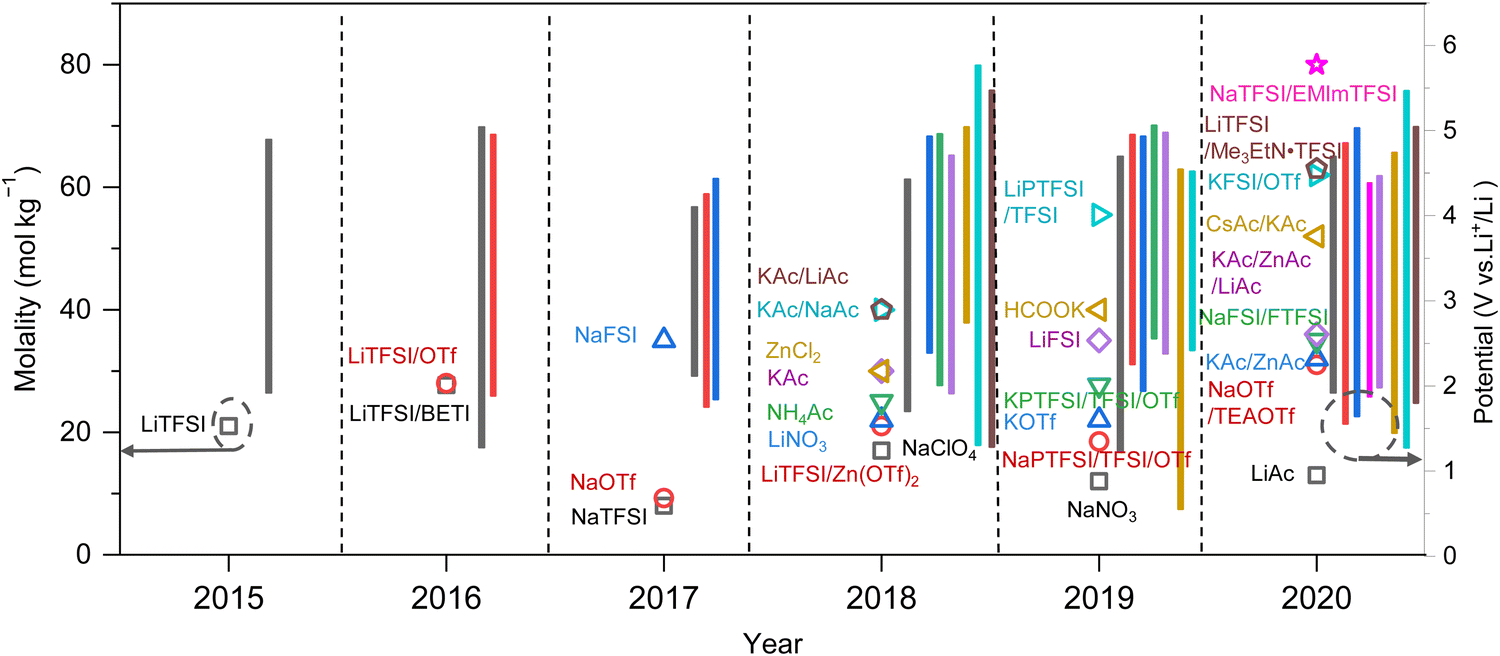
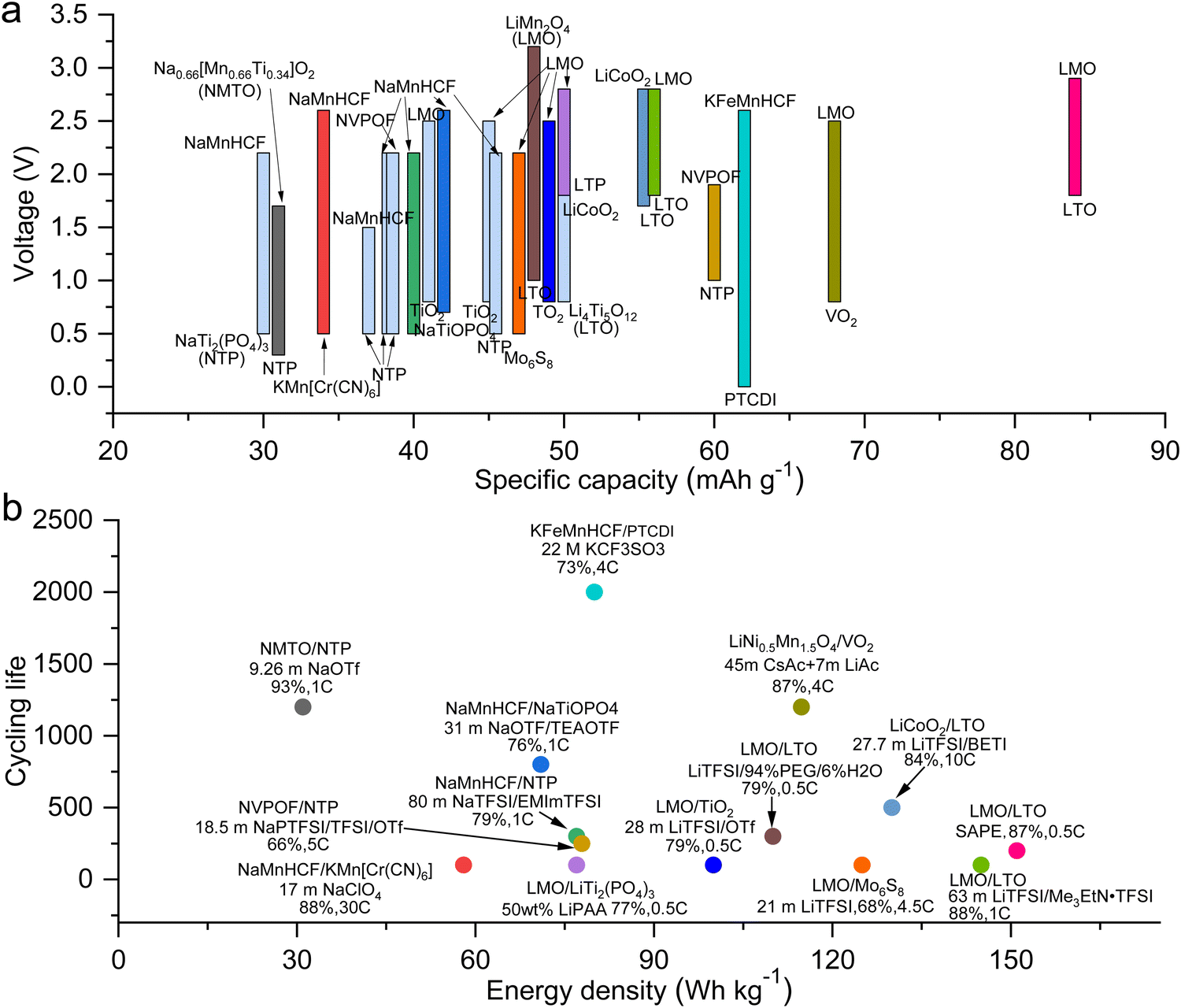
As previously mentioned, TFSI-based WiSEs have been the first and most investigated electrolyte systems. Despite the promising results obtained so far with these electrolytes, the presence of a perfluorinated anion rises concerns with regards to both cost and environmental impact. The cost aspect is a very important factor for large-scale energy storage applications. Thereby, alternative concentrated electrolytes employing fluorine-free salts whose anion are acetate, formate, perchlorate, nitrate, and chloride, are rapidly emerging (see Fig. 2). The most representative aqueous battery configurations employing these highly concentrated electrolytes are summarized in Fig. 3 in terms of operative voltage as function of specific capacity (see Fig. 3a) and cycle life as function of energy density (see Fig. 3b).
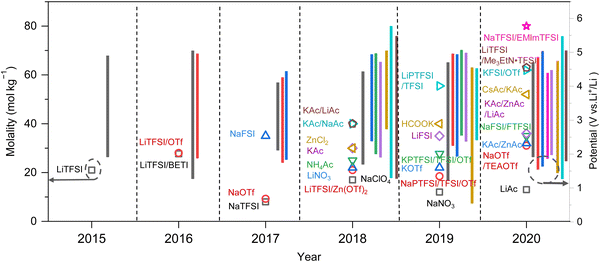 |
| | Fig. 2 Development of highly concentrated aqueous electrolytes in recent years: concentration (scattered points) and corresponding ESW (bars) of each electrolyte.12,24–51 | |
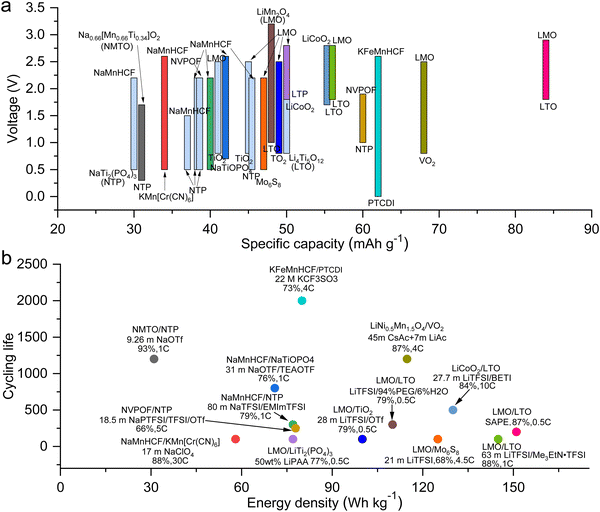 |
| | Fig. 3 Overview of the most representative aqueous batteries configurations employing highly concentrated electrolytes in terms of (a) operative voltage as function of specific capacity and (b) cycle life as function of energy density.12,24,27,29,30,36,39,42,44,45,48,50–52,61,62 | |
2.1 Fluorinated WiSE
The high solubility of LiTFSI was exploited by Wang's group to prepare a WiSE enabling Mo6S8//LiMn2O4 ABs with a high discharge voltage of 1.8 V and an energy density of 100 W h kg−1.12 After that, the 27.8 m Li(TFSI)0.7(BETI)0.3·2H2O electrolyte was proposed by Yamada's group, demonstrating an extremely wide ESW of 3.8V (∼1.25 to 5.05 V versus Li+/Li). The Li4Ti5O12//LiCoO2 and Li4Ti5O12//LiNi0.5Mn1.5O4 cells employing such electrolyte exhibited high operating voltage (∼2.3–3.1 V) and energy density (>130 W h kg−1).24 The room-temperature monohydrate melt of Li salts Li(PTFSI)0.6(TFSI)0.4·H2O 55.5 m was also reported by Yamada's group. By using an asymmetric imide anion featuring ultra-high solubility in water, a wide ESW of ∼5 V was claimed.42 Furthermore, a 63 m aqueous electrolyte (42 m LiTFSI + 21 m Me3EtN·TFSI) was shown to provide an ESW of 3.25 V, leading to the successful fabrication of a 2.5 V aqueous Li-ion battery (Li4Ti5O12//LiMn2O4) with an energy density of 145 W h kg−1.50 Lu's group employed the water-miscible polymer poly(ethylene glycol) to ease the concerns about cost and toxicity of fluorinated salts, and still develop a low-cost and eco-friendly aqueous electrolyte.52 In spite of the low salt concentration (2 m), such “molecular crowding” approach enabled a wide ESW of 3.2 V. The resulting Li4Ti5O12//LiMn2O4 cell delivered stable specific energy 75 W h kg−1 over 300 cycles. As demonstrated by online electrochemical mass spectroscopy, side reactions such as hydrogen and oxygen evolution were substantially suppressed compared to other concentrated electrolytes such as 21 m LiTFSI, and 32 m KAc + 8 m LiAc.52
Besides Li-based systems, a 9.26 m sodium trifluoromethanesulfonate (NaOTf) was reported for ASBs with an ESW of 2.5 V (1.7 to 4.2 V versus Na/Na+), which granted the NaTi2(PO4)3//Na0.66[Mn0.66Ti0.34]O2 cell with an energy density of 31 W h kg−1 and CE of 99.7% at 0.2 C for 350 cycles.53 Afterwards, several other types of WiSEs have been developed for ASBs, such as 35 m sodium bis(fluorosulfonyl)imide (NaFSI) with an ESW of 2.6 V (1.8 to 4.4 V versus Na/Na+),28 17 m sodium perchlorate (NaClO4) with an ESW of 2.7 V (1.7 to 4.4 V versus Na/Na+),54 and hybrid electrolytes with 25 m NaFSI + 10 m sodium (fluorosulfonyl)(trifluoromethanesulfonyl)imide (NaFTFSI)55 and 9 m NaOTf + 22 m tetraethylammonium trifluoromethanesulfonate (TEAOTf) with an ESW of 3.3 V.45 To further increase the concentration of the electrolyte, Kühnel's group employed a 1-ethyl-3-methylimidazolium (EMIm)-based ionic liquid to prepare a 80 m NaTFSI0.375EMImTFSI0.625 electrolyte featuring a small amount of water, which allowed the Ti2(PO4)3//Na2Mn[Fe(CN)6] cell to reach an energy density of 77 W h kg−1 and CE of >99.5% at 1C for 300 cycles.
Similar to ASBs, APBs are very appealing for large-scale energy storage due to large natural abundance and low cost of potassium-based materials.56 Besides, K+ ions have much weaker Lewis acidity than Li+ and Na+ due to the larger volume and, thus, lower charge density. This leads to smaller Stoke's radius of solvated K+ ions,57 which promotes high K+ mobility.58 The 22 M potassium trifluoromethanesulfonate (KOTf) electrolyte proposed by Hu's group was employed in KFeMnHCF)//PTCDI cells, which delivered a capacity of 63 mA h g−1 and an energy density of 80 W h kg−1 with 73% capacity retention over 2000 cycles at 4 C.39 Notably, this outstanding performance even exceeds most results reported on ASBs. Yamada's group also reported a 61.7 m electrolyte for APBs consisting of KFSI and KOTf.49 It was noted that the FSI− anion in this electrolyte did not suffer from hydrolysis. Moreover, this electrolyte showed a high ionic conductivity (12 mS cm−1 at 30 °C) and an ESW of 2.7 V (when determined with a Pt working electrode).49
AZBs have also recently captured intensive attention, mostly driven by the inherent merits of Zn, such as high theoretical capacity and low cost.59 However, the performance of the state-of-the-art aqueous Zn-based batteries are still far from being satisfactory, the main reason being Zn dendrite growth and water/oxygen-related side reactions leading to low CE.60 Notably, fluorinated and highly concentrated electrolytes have proven to improve the Zn stripping/plating efficiency. For instance, the 20 m LiTFSI + 1 m Zn(CF3SO3)2 electrolyte reported by Wang's group enabled dendrite-free Zn plating/stripping at nearly 100% CE and showed negligible tendency to evaporate.30 Zn-based batteries employing this electrolyte and either LiMn2O4 or O2 cathodes displayed energy density of 180 W h kg−1 with capacity retention of 80% for more than 4000 cycles and energy density of 300 W h kg−1 for more than 200 cycles, respectively.
2.2 Carboxylate-based WiSE
Carboxylate salts are organic compounds including a –C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)O− anionic group with the general formula M(RCOO)n, where M is a cation.63 They are classified based on the nature of the alkyl (R) chain, the most common being formates (R = H), acetates (R = CH3) and acrylates (R = CHCH2). In most cases, they are very soluble in water, which facilitates the achievement of WiSE regime.
O)O− anionic group with the general formula M(RCOO)n, where M is a cation.63 They are classified based on the nature of the alkyl (R) chain, the most common being formates (R = H), acetates (R = CH3) and acrylates (R = CHCH2). In most cases, they are very soluble in water, which facilitates the achievement of WiSE regime.
Formates.
Eco-friendly and cost-effective potassium formate (CHOOK) was employed to prepare 40 m HCOOK, in which the water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :salt mole ratio is 1.38:1. The electrolyte displayed an ESW of 4 V (from −2.5 to 1.5 V vs. Ag/AgCl) when glassy carbon was used as the working electrode. Notably, CHOOK-based electrolytes possess better cathodic stability and higher ionic conductivity than potassium acetate (KAc)-based electrolytes. A high specific capacitance of 321 F g−1 at 5 A g−1 and 121 F g−1 at 20 A g−1 were achieved by activated carbon-based positive electrodes in this electrolyte. Meanwhile, a reversible capacity of 15 mA h g−1 was delivered at 0.1 A g−1 for the KTi2(PO4)3 anode.41 Unfortunately, except for K, formate salts of other cations have a quite limited solubility in water.
:salt mole ratio is 1.38:1. The electrolyte displayed an ESW of 4 V (from −2.5 to 1.5 V vs. Ag/AgCl) when glassy carbon was used as the working electrode. Notably, CHOOK-based electrolytes possess better cathodic stability and higher ionic conductivity than potassium acetate (KAc)-based electrolytes. A high specific capacitance of 321 F g−1 at 5 A g−1 and 121 F g−1 at 20 A g−1 were achieved by activated carbon-based positive electrodes in this electrolyte. Meanwhile, a reversible capacity of 15 mA h g−1 was delivered at 0.1 A g−1 for the KTi2(PO4)3 anode.41 Unfortunately, except for K, formate salts of other cations have a quite limited solubility in water.
Acetates.
A super-concentrated potassium acetate (KAc) electrolyte with K:H2O molar ratio of 1:1.8 was reported by Li's group, demonstrating a wide ESW of around 2.5 V and enabling a AC//AC symmetric supercapacitor with an operating voltage of 2.0 V with 88% capacitance retention after 10000 cycles.64 Subsequently, 30 m KAc was reported by Ji's group to allow reversible insertion/deinsertion of K+ cations into KTi2(PO4)3 anode for APBs.33 The same electrolyte was also tested in combination with low-cost Prussian blue analogue (PBA) cathodes, but the instability of such an active material in the electrolyte resulted into poor cycling stability.61 This was attributed to the random distribution of vacancies in PBA, which can make the framework fragile and vulnerable to collapse.65,66 Moreover, the weak N-coordinated crystal field resulted in structural distortions when the N-coordinated metal is Co/Mn, and the lattice distortion may cause the cleavage of the M1–N![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C–Fe bridge and the dissolution of transition metal ions, especially in an aqueous electrolyte.67,68 To tackle this issue, 30 m KAc was gelified by adding a small amount of carboxymethyl cellulose (CMC), resulting into improved capacity retention and CE.69
C–Fe bridge and the dissolution of transition metal ions, especially in an aqueous electrolyte.67,68 To tackle this issue, 30 m KAc was gelified by adding a small amount of carboxymethyl cellulose (CMC), resulting into improved capacity retention and CE.69
Acetate-based WiSE were also studied for other types of ABs, such ALBs, ASBs and AZBs leveraging on the high solubility of KAc to reach the WiSE regime. For example, Cui's group developed an hybrid WiSE containing 32 m KAc and 8 m LiAc, which demonstrated an ESW of 3 V and enabled a TiO2/LiMn2O4 full ALBs with a high operational voltage of 2.5 V.36 Similarly, a 32 m KAc + 8 m sodium acetate (NaAc) solution was reported by Passerini and coll. and successfully employed in NaTi2(PO4)3//Na2MnFe(CN)6 ASBs.35 Considered the limited stability of hexacyanoferrate cathodes in this kind of acetate-based WiSE, symmetric full ASBs featuring NASICON-type Na2VTi(PO4)3/C as both the positive and the negative electrode were later reported, reaching a stable cycling performance with CE of 99.9% at 10C over 500 cycles.70
A hybrid WiSE consisting of 45 m cesium acetate (CsAc) and 7 m LiAc was proposed by Tao's group, which enabled VO2/LiNi0.5Mn1.5O4 ALBs with an energy density of 114.8 W h kg−1 at 1 C. Moreover, coin cells employing this chemistry delivered a promising capacity retention of 86.5% over 1200 cycles.48 Notably, the hybrid WiSE containing 1 m Zinc acetate (ZnAc2) and 31 m KAc proposed by Tao's group guaranteed a wide ESW of 3.4 V while the resulting Zn//MnO2 AZB displayed a discharge capacity of 304.6 mA h g−1.46 Subsequently, the 30 m KAc, 3 m LiAc, and 3 m ZnAc2 hybrid WiSE was developed by Passerini and coll., enabling outstanding zinc plating/stripping with an high average CE (99.6%) and long-term cycling stability. With this electrolyte, Zn//LiFePO4 and Zn//LiMn2O4 dual ion cells were realized, which delivered discharge capacity of 155 mA h g−1 and 121 mA h g−1 at 0.05 C, respectively.47
Finally, a novel 25 m ammonium acetate (NH4Ac) WiSE was proposed by Ji's group with a wide ESW of 2.95 V. This electrolyte was investigated with amorphous titanic acid (TiO1.85(OH)0.30·0.28H2O) and 1,4,5,8-naphthalenetetracarboxylic dianhydride-derived polyimide (PNTCDA) anodes displaying a reversible NH4+ insertion behavior.32,71
Acrylates.
The lithium polyacrylate (LiPAA) was prepared by neutralizing polyacrylic acid with dilute lithium hydroxide solution. The resulting leakless and dimensionally stable gel electrolyte containing 50 wt% LiPAA demonstrated a wide ESW. The TiO2//LiMn2O4 cells employing this electrolyte delivered a capacity of 59.2 mA h g−1 with an average output voltage of 2.1 V in the first cycle, corresponding to an energy density of 124.2 Wh kg−1.62 This novel electrolyte represents a further step towards truly sustainable and nontoxic ALBs with high energy density.
2.3 WiSE based on other anions: nitrate, perchlorate, and chloride
Besides the high solubility in water, nitrate salts possess appealing properties such as low-cost and eco-friendliness. A 22 m LiNO3 electrolyte with a wide ESW of 2.55 V was reported by Pan's group. Modelling results evidenced a unique local structure with a (Li+(H2O)2)n polymer-like aggregation network. The electrolyte also enabled reversible cycling of LiMn2O4 and LiNi1/3Mn1/3Co1/3O2 cathodes with high working potential.31 Subsequently, a 12 m NaNO3 WiSE was proposed by Cui's group, which exhibited an ESW of 2.56 V. The supercapacitor including this electrolyte displayed a specific capacitance of 32.68 F g−1 at 1 A g−1 with high operation voltage of 2.1 V and capacitance retention of 90% over 9000 cycles.37
Sodium perchlorate (NaClO4) was also proposed by Kim's group as salt for WiSE, owing to its high solubility in water (10 M or 17 m). The highly concentrated 17 m NaClO4 was employed in aqueous NaTi2(PO4)3–C//Na3V2O2x(PO4)2F3−2x/MWCNT cells.72 Later, Okada's group took full advantage of the wide ESW of this electrolyte (2.8 V) and constructed ASBs with output voltages higher than 2 V by coupling the sodium manganese hexacyanoferrate cathode with the potassium manganese hexacyanochromate anode. Additionally, a novel electrolyte was proposed by mixing the 17 m NaClO4 and 3 m ZnOTf solutions (volume ratio of 1:1), which could suppress HER and enable reversible and stable Zn plating/stripping (a high CE 99.96% and 1600 h long-term cycling).73
Zinc chloride (ZnCl2) also possesses an extremely high solubility in water leading to the formation of WiSEs as demonstrated by Ji's group.74 The ESW could be broadened from 1.6 to 2.3 V when the concentration of ZnCl2 was increased from 5 m to 30 m. As result of the increased concentration, the overpotential of Zn plating/stripping was increased and the HER onset potential was down-shifted. Overall, symmetric Zn‖Zn cells showed improved CE in 30 m ZnCl2.75 This electrolyte was then employed in Zn//Ca0.20V2O5·0.80H2O (CaVO) cells, that could deliver a high capacity of 496 mA h g−1 and a remarkable energy density of 206 W h kg−1. Interestingly, 30 m ZnCl2 substantially stabilized the structure of the electrode's active material as proven by XRD and FTIR.34 The same WiSE was later employed in rechargeable dual-ion batteries consisting of Zn3[Fe(CN)6]2 as cation-storage anode and ferrocene encapsulated in microporous carbon as the anion-storage cathode. The cell voltage could be increased by as much as 0.35 V compared to the corresponding dilute electrolyte (5 m).76 In addition, the concentrated electrolyte ZnCl2·2.33H2O (around 23.8 m) was reported by Xu's group, which offered a suite of properties similar to those of 30 m ZnCl2, endowing the resultant Zn–air cell with high energy density and good cycling stability.77
3. Interaction energy between water and ions
The interaction among water, cations and anions is particularly complex in WiSEs because of the intense interactions between cations and anions, which do not occur in dilute aqueous electrolytes where cations and anions are overwhelmingly solvated (and separated) by water. Moreover, the hydrophilic or hydrophobic properties of the anions can profoundly affect the physical and chemical properties of the electrolyte, e.g., solvation structure, transport property of anions, and viscosity. Especially for the electrolytes involving multiple cations, their charge density would also have some influence on the electrolyte physicochemical properties, e.g., solvation structure, transport of cations, viscosity, and degree of salt's dissociation. However, there is a lack of systematic studies on this aspect in WiSEs in the literature. To achieve a better understanding of the molecular interactions at an atomic level, we performed some DFT simulations. From the simulations we extracted the interaction energy for a variety of water–ion and cation–anion combinations (see Tables 1 and 2), and the charge density for selected ions (Table 1). The calculations were generally carried out by using Gaussian 1678 at the B3LYP/aug-cc-pvTZ level of theory, but the 6-311++G** basis set was used for systems containing potassium and calcium or for systems with a total number of electrons larger than 286. The starting guess geometries were hand-drawn using Avogadro, and then optimized in the gas phase using the int = ultrafine setting as implemented in Gaussian. The obtained geometries were tested to be real minima of the potential energy surfaces by calculating the vibrational spectra (as routinely done78). To obtain the interaction energy ΔGint298 between two species, we employed the following formula:| | | ΔGint298 = GAB298 − (GA298 + GB298) | (1) |
where GAB298 is the Gibbs free energy of the system comprised of the interacting species (A and B), and GA298 and GB298 are the Gibbs free energies of isolated A and B, respectively. The equation was adjusted to account for multiple ions in the case of multiple charged species. The interaction with water was calculated by simulating a single water molecule coordinated with a single ion. The contributions of thermal energy and zero-point energy (ZPE) were included in the calculated energy values to obtain the Gibbs free energies. The charge densities were calculated in two different ways for different ions. For the ions with a reasonably spherical geometry (e.g., all the considered cations, Cl−, ClO4−, and SO42−), the total net charge was divided by the ionic radius. For the ions where the charge is localized only on one portion of the structure (e.g., formate, acetate, OTf−, and TFSI−) or far from spherical (NO3−), the volume of the region with the highest charge localization was calculated. Then the volume was used to divide the charge present in that volume. To do so, an Atom-in-Molecule (AIM) Basin analysis was performed to derive the AIM atomic charges and volumes. Then, the volumes and charges of the relevant region of the ion were evaluated by summing the corresponding contributions, and the charge density was obtained by dividing the obtained quantities. The AIM analysis was performed using the Multiwfn software.79
Table 1 Volumes, charge densities and interaction energy with water for a variety of cations and anions
| Cation |
Volume (Å3) |
Charge density (e Å−3) |
Interaction energy with water (kJ mol−1) |
Anion |
Volume (Å3) |
Charge density (e Å−3) |
Interaction energy with water (kJ mol−1) |
| Li+ |
3.054 |
0.327 |
−115.8056 |
Formate |
54.028 |
0.016 |
−29.5973 |
| Na+ |
6.538 |
0.153 |
−71.1222 |
Acetate |
51.782 |
0.017 |
−25.6223 |
| K+ |
14.710 |
0.068 |
−78.5392 |
Cl− |
24.838 |
0.040 |
−32.4433 |
| NH4+ |
20.879 |
0.048 |
−45.4028 |
NO3− |
67.531 |
0.015 |
−17.8770 |
| Mg2+ |
2.664 |
0.751 |
−316.0577 |
ClO4− |
57.906 |
0.017 |
−7.0206 |
| Ca2+ |
6.206 |
0.322 |
−192.5017 |
OTf− |
69.396 |
0.010 |
−5.6921 |
| Zn2+ |
2.855 |
0.701 |
−405.3037 |
TFSI− |
110.663 |
0.005 |
5.6685 |
| Al3+ |
1.288 |
2.329 |
−827.7388 |
SO42− |
71.936 |
0.028 |
−34.7564 |
Table 2 Interaction energy between a variety of cations and anions. The interaction energy between NH4+ and ClO4− is not available because of the decomposition of NH4+ during the simulation. All the energies are expressed in kJ mol−1
|
|
Formate |
Acetate |
Cl− |
NO3− |
ClO4− |
OTf− |
TFSI− |
SO42− |
| Li+ |
−671.382 |
−684.404 |
−624.577 |
−618.896 |
−499.611 |
−562.269 |
−555.143 |
−1750.266 |
| Na+ |
−568.318 |
−575.819 |
−533.0316 |
−514.816 |
−410.985 |
−470.728 |
−451.833 |
−1526.993 |
| K+ |
−696.805 |
−703.778 |
−664.761 |
−649.1522 |
−567.725 |
−612.624 |
−589.913 |
−1784.337 |
| NH4+ |
−531.942 |
−537.833 |
−543.2973 |
−470.095 |
— |
−428.736 |
−392.207 |
−1450.919 |
| Mg2+ |
−2331.895 |
−2360.991 |
−2223.869 |
−2188.677 |
−2036.665 |
−2048.446 |
−2021.651 |
−2368.553 |
| Ca2+ |
−1961.474 |
−1998.527 |
−1836.739 |
−1825.069 |
−1609.539 |
−1710.760 |
−1687.345 |
−2088.356 |
| Zn2+ |
−2491.463 |
−2527.395 |
−2497.622 |
−2334.657 |
−2171.745 |
−2182.121 |
−2153.330 |
−2555.785 |
| Al3+ |
−5359.525 |
−5436.588 |
−5232.369 |
−5131.613 |
−4853.993 |
−4869.648 |
−4827.399 |
−11419.058 |
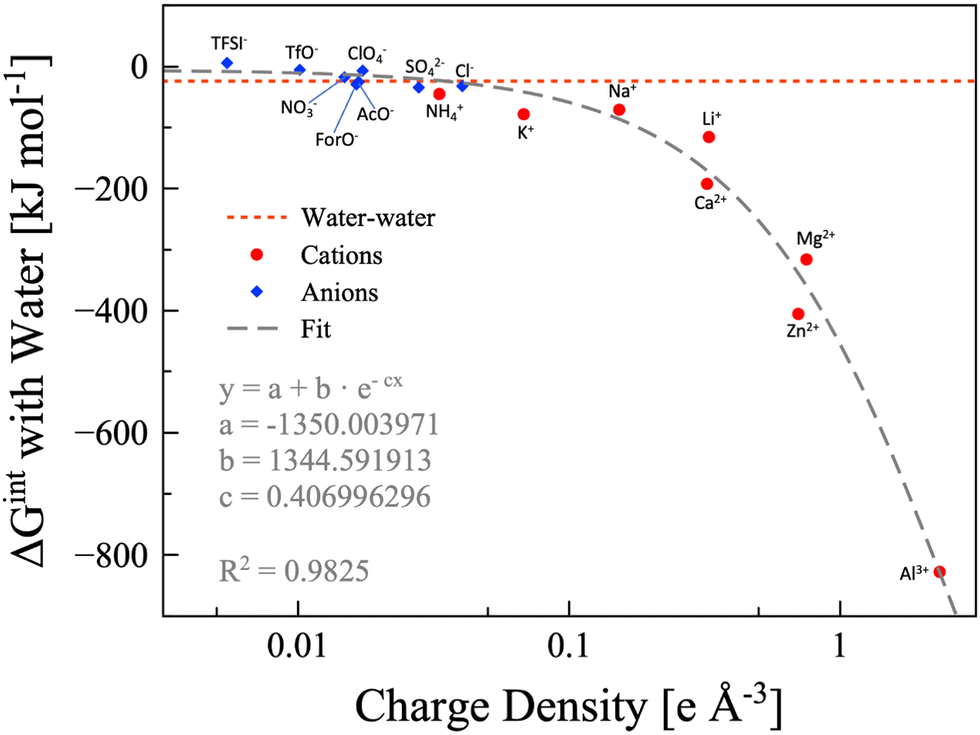
In the findings depicted in Fig. 4, it is clearly seen a well-defined dependency between hydration energy and ion charge density. Specifically, we found that the hydration energy exponentially increases with the ion charge density. On the basis of the hydration energy, hydrophilic or hydrophobic ions can be identified comparing the interaction energy between one ion and one water molecule with the interaction energy between two water molecules (−24.48 kJ mol−1, red dotted line in Fig. 4).69 We consider the ion to be hydrophobic if the interaction energy between the ion and one water molecule is more positive than −24.48kJ mol−1, otherwise it is hydrophilic. Using this metric, all the cations considered are hydrophilic, while some anions are hydrophobic, namely, TFSI− (5.67 kJ mol−1), OTf− (−5.69 kJ mol−1), ClO4− (−7.02 kJ mol−1), and NO3− (−17.89 kJ mol−1). Anyhow, it must be considered that the values are calculated for a single ion interacting with a single water molecule. If we consider that a single anion may interact with up to four water molecules (which is reasonable in a water-in-salt regime), NO3− can be safely considered hydrophilic and ClO4− is slightly hydrophilic. However, OTf− is slightly hydrophobic while TFSI− maintains its pronounced hydrophobicity. The hydrophobic or hydrophylic character of the anion will lead to two inherently different intermolecular interaction scenarios, and thus it would have a direct effect on the water signal in the Raman/IR spectra of WiSEs, which will be discussed in more details in a forthcoming paper. Here, the effect of the water affinity of the anion on Raman spectra is only briefly mentioned for the sake of discussion.
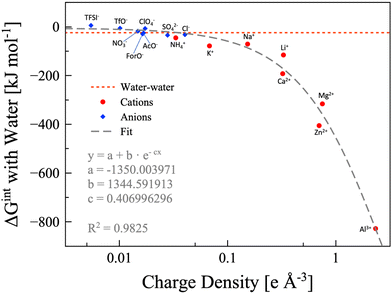 |
| | Fig. 4 Water-ion interaction energy as function of the ions charge density. Anions are depicted as blue diamonds, and cations as red circles. All the data are calculated at the B3LYP/aug-cc-pvTZ level of theory, except the potassium and calcium points, which are calculated at the B3LYP/6-311++G** level of theory. The dashed grey line is the exponential fitting, for which the equation and parameters are also shown. The water–water interaction energy is represented with the red dotted line. | |
In few words, the hydrophobic/hydrophilic character of anions has an extensive effect on the interaction between water, cations, and anions. The hydrophobic anions, e.g., TFSI−, hardly interact with water and drive the water molecules to the surrounding cations, which are generally much more hydrophilic. On the other hand, the hydrophilic anions, e.g., formate, acetate, and NO3−, could compete with cations in interacting with water. The complex interactions in the two kinds of WiSE (based on either hydrophilic or hydrophobic anions) are the reason behind the observations on the characteristic Raman band of water assigned to the O–H stretching (∼3000–3600 cm−1). In highly concentrated aqueous electrolytes consisting of hydrophobic anion salts, a sharp peak in the Raman spectra is found at 3565 cm−1,24,31,42 which indicates an unsaturated hydrogen bond state of water molecules. On the other hand, a much broader Raman band for O–H stretching vibrations still exists in highly concentrated aqueous electrolytes consisting of hydrophilic anion salts, which is attributed to the hydrogen bonding interactions between anions and water molecules.41,69 When preparing a WiSE, it is fundamental to take into account the interaction energy between water and the ions. In fact, the solubility of the considered salt strictly depends on the overall energy liberated by the dissociation and solvation processes. It is already evident from Raman spectroscopy how different anions have direct effects on the local structure of the electrolyte, with hydrophilic anions mimicking the hydrogen bond network of water (as witnessed by the broad O–H stretching band), while hydrophobic anions disrupt the network leading to unsaturated water molecules (asymmetric sharper band at ∼3565 cm−1). The capability of forming stable hydrogen bonds is a key point in the preparation of highly concentrated electrolytes, and chemical instinct could be misleading in this regard (TFSI− has eight hydrogen bond acceptors, while Ac− has only four, and yet the former is hydrophobic). Our calculation, though, did not account for entropy changes in the bulk system, since we only simulated single pairs. Nevertheless, we want to point at the necessity to include calorimetry measurements when characterizing highly concentrated aqueous solutions. The access to this kind of information can be readily used to design new systems based on the interaction energies. The DFT calculations, albeit extremely useful in rationalizing some observations, are not enough because of the size limitations. The interaction energies have direct effects on the solvation shells of all the species in the system, which have a pivotal role in the performances of this class of electrolytes.
4. Solvation shells structure
For the sake of brevity, the discussion here will be limited to one representative salt for each of the categories previously discussed, i.e., bis(fluoroalkylsulfonyl)imide, carboxylate, nitrate, perchlorate, and chloride. Specifically, we have selected LiTFSI, KAc, LiNO3, NaClO4, ZnCl2. Unfortunately, it is not possible to select a series of salts sharing the same cation because of the widely different solubilities (e.g., KAc > 3 kg L−1, LiAc about 0.45 kg L−1, LiTFSI about 6 kg L−1 and KTFSI only 0.01 kg L−1). Furthermore, these are among the most studied salts in literature. The first thing to mention is that the anions (TFSI−, Ac−, NO3−, ClO4−, Cl−) of these salts have different hydrophobic/hydrophilic character, while at the same time maintaining large solubility in water. Concretely, TFSI− is a hydrophobic anion (with a positive hydration energy, see Table 1) while Ac−, NO3−, Cl− and ClO4− are hydrophilic anions, which may bring far reaching impact on the interaction relationship between cations, anions, and water. The solvation structure of the representative 21 m LiTFSI,12 20 m KAc,69 22 m LiNO3,31 17 m NaClO4,82 31 m ZnCl281 aqueous solutions are briefly discussed based on the collected simulations results (Fig. 5 and 6) from literatures.
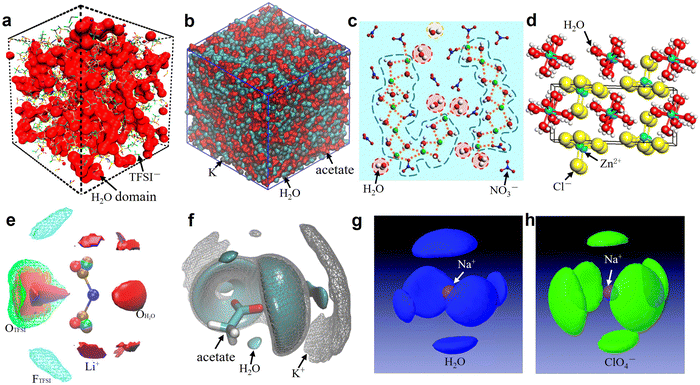 |
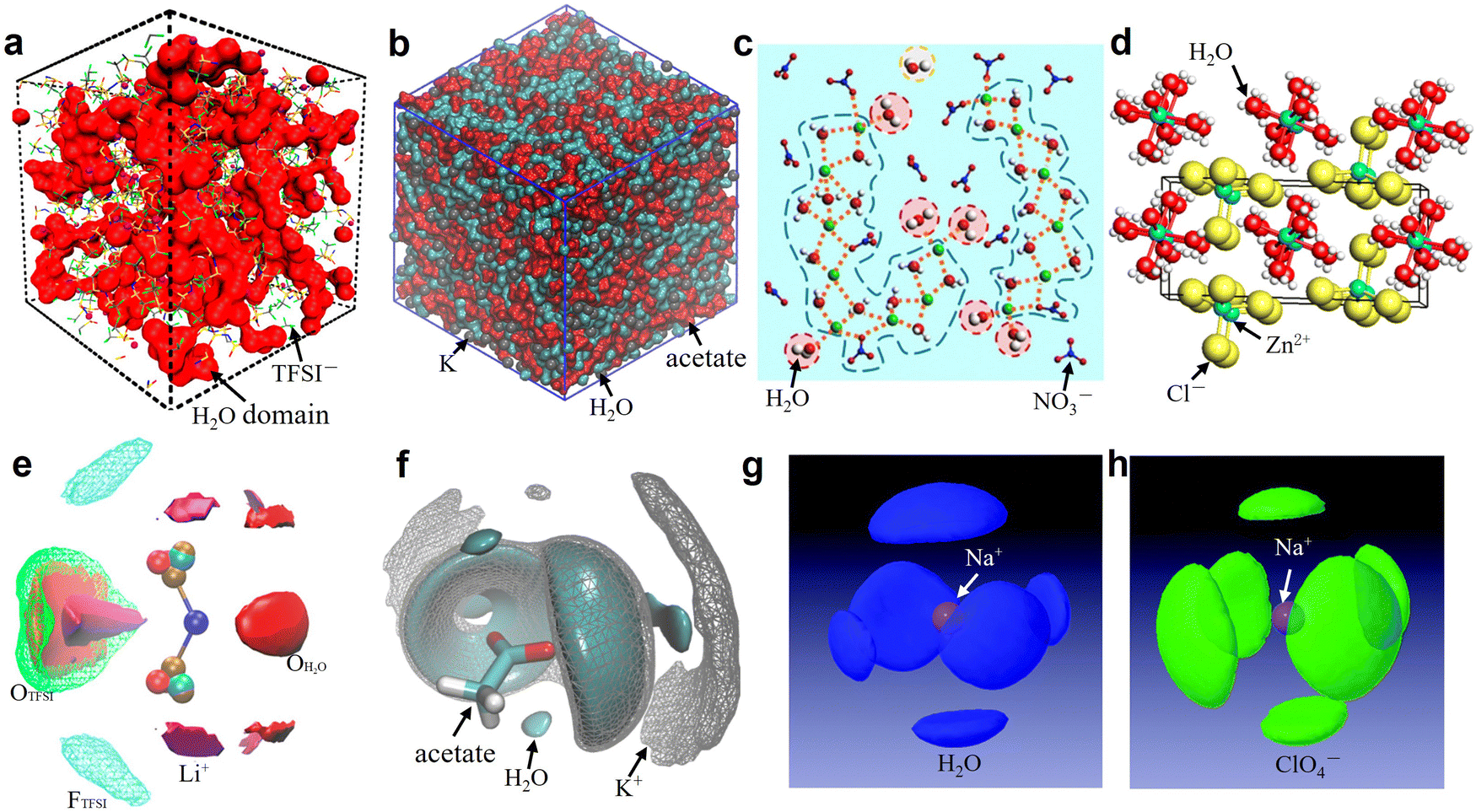
| | Fig. 5 (a) 3D snapshot showing an interconnected H2O domain in red and TFSI anions as wireframe from MD simulations of 21 m LiTFSI at 298 K. (a) Reproduced with permission.80 (b) Snapshots of the simulation boxes for 20 m KAc, in which water is represented in cyan, acetate in red, and potassium in gray. (b) Reproduced with permission.69 (c) MD simulated local structure evolution of LiNO3 aqueous solution with a concentration of 1:2 (LiNO3:H2O). (c) Reproduced with permission.31 (d) Crystal structure of zinc chloride trihydrate. (d) Reproduced with permission.81 (e) Isosurfaces of water oxygen (red solid, ρ/ρbulk = 5, ρ: local density; ρbulk bulk average density), oxygen of TFSI (green mesh, ρ/ρbulk = 2.5), fluorine of TFSI (cyan mesh, ρ/ρbulk = 2), and Li+ (purple solid, ρ/ρbulk = 12) for 21 m LiTFSI in H2O at 298 K. (e) Reproduced with permission.80 (f) 3D images of the surrounding of the acetate anion for 20 m KAc by selected spatial distribution functions. Water is represented in cyan, acetate in red, and potassium in gray. (f) Reproduced with permission.69 Spatial density functions of the neighboring (g) water molecules (blue lobes) and (h) ClO4− ions (green lobes) around Na+ ions in 17 m NaClO4 aqueous solution. The purple ball in the center represents a Na+ ion. (g and h) Reproduced with permission.82 | |
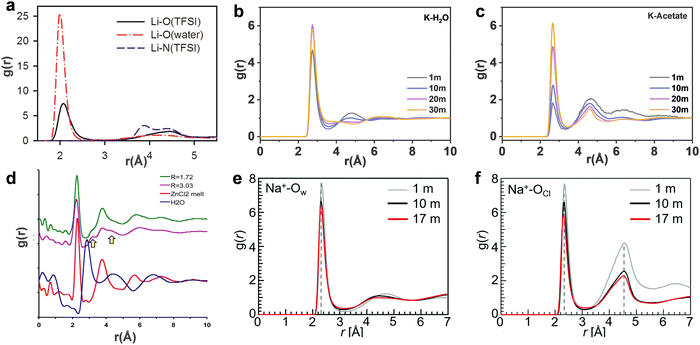 |
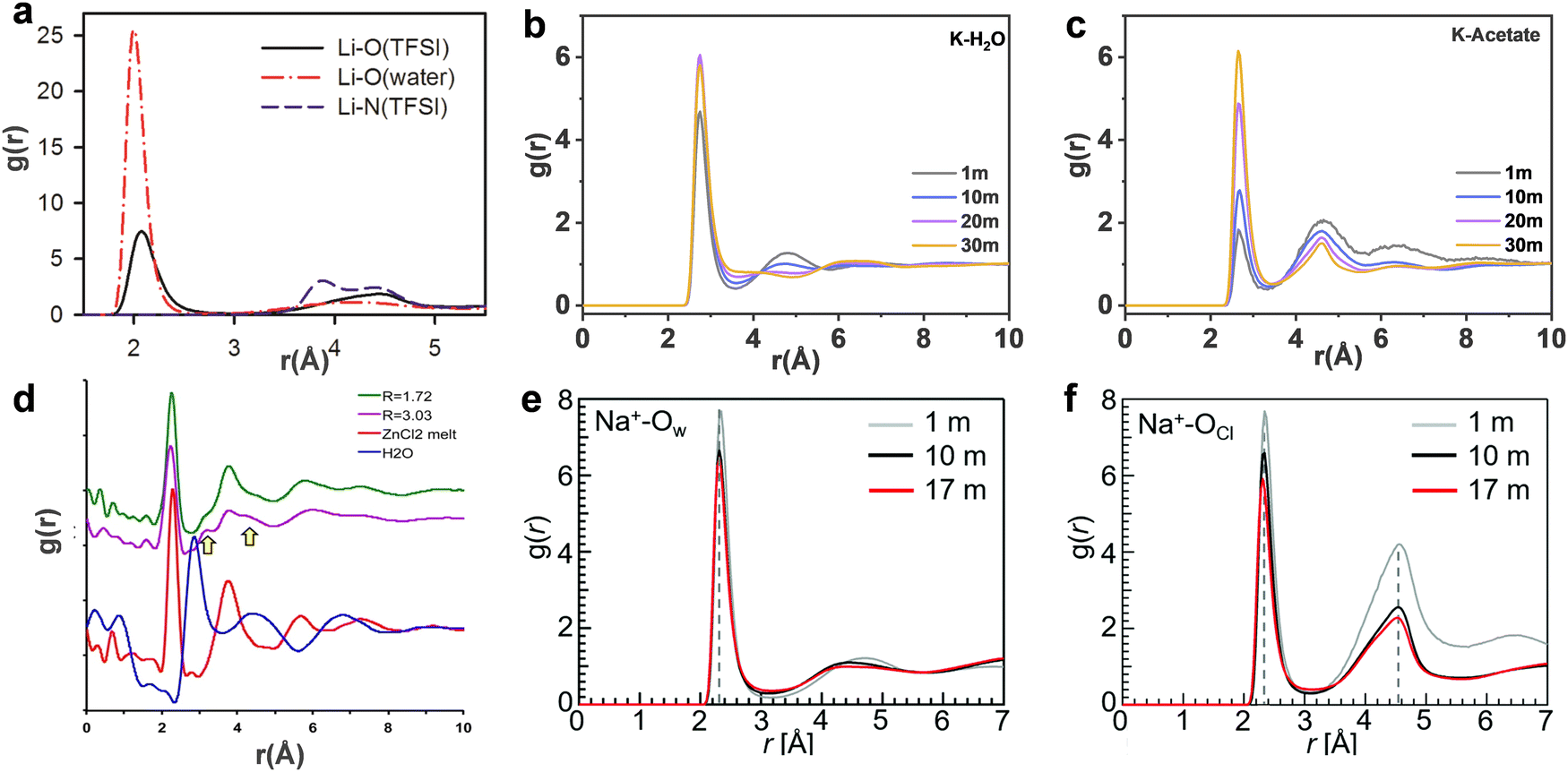
| | Fig. 6 (a) The Li-O and Li-N(TFSI) radial distribution functions (RDFs) from MD simulations of 21 m LiTFSI in water at 298 K. (a) Reproduced with permission.80 Centre of mass RDFs for (b) K-H2O and (c) K-acetate at 298 K in 1, 10, 20 m, 30 m KAc aqueous solutions, respectively. (b and c) Reproduced with permission.69 (d) Pair distribution function (PDF) plots of the X-ray scattering of the room temperature molten hydrates R = 3.03 (purple) (18.5 m ZnCl2), R = 1.72 (green) (31 m ZnCl2), water (blue), and molten anhydrous ZnCl2 at 320 °C (red). (d) Reproduced with permission.81 Pair correlation functions of (e) Na+-Ow and (f) Na+-OCl in 1, 10 and 17 m NaClO4 aqueous solutions. (e and f) Reproduced with permission.82 | |
4.1 Solvation structure in WiSEs based on TFSI−
In 21 m LiTFSI, contact ion pairs (CIP) and ionic aggregates (AGGs) dominate against solvent-separated ion pairs (SSIPs), whereas the opposite is observed in 1 m LiTFSI.12 As shown by the average coordination numbers extracted from the MD simulations of 21 m LiTFSI (Fig. 5a), each Li+ is coordinated by two oxygen atoms from TFSI− and two oxygen atoms from H2O in its first solvation sheath. Anyhow, average coordination numbers could sometimes be misleading, and a proper clusters population analysis is the best way to tackle solvation structures. In fact, at a closer look at the MD simulation it is possible to find two different populations of Li+ coordination states. The first, major, population (∼60%) consist of Li+ ions fully solvated by four TFSI oxygen atoms while the second (∼40%) is made of Li+ as SSIPs with no TFSI in their first coordination shell at all. Thus, a solvation disproportionation phenomenon is discovered for 21 m LiTFSI, with the Li+ ions tending to be solvated in two different ways rather than approaching the average of 1.75 oxygen atoms from TFSI and 2.25 from water.80 Looking at the Li+ solvation sheath by means of radial distribution functions (RDFs) (Fig. 6a), Li+ cation primarily binds to TFSI oxygen atoms rather than its nitrogen or fluorine atoms, and water competes for the direct contact. The hydrophobic –CF3 group of the anion approaches –CF3 groups of the other TFSI. The solvation disproportionation, induced by solvent-separated Li+ at high salt concentrations, generate the formation of a liquid structure with nano-heterogeneity. Although the anion movement is blocked by the TFSI-rich domains in the highly concentrated 21 m LiTFSI, a 3D percolating channel is build up by the Li+(H2O)4 domain, which enables fast Li+ transport with high transference number.80
4.2 Solvation structure in WiSEs based on Ac−
In 20 m KAc, the potassium cation is mainly interacting with the acetate anions, which progressively replaces water in comparison with the dilute solution. CIP and AGGs are the main components in the 20 m KAc system, similarly to the case of 21 m LiTFSI. A bicontinuous arrangement of polar and apolar channels is found in 20 m KAc (Fig. 5b). According to the 3D images representing the spatial distribution functions (Fig. 5f), the acetate anion is indeed surrounded by both potassium ions and water molecules. Water molecules compete with potassium ions for coordinating the carboxylic function of the acetate, but potassium appears to approach the anions closer than water. RDFs for potassium-water in a variety of different KAc concentrations (Fig. 6b) demonstrate that the quantity of water in the second solvation shell is gradually reduced with increased concentration (the peak at ∼5 Å vanishes at higher concentrations). Simultaneously, more and more potassium ions are coordinated by acetate as shown by RDFs for potassium-acetate (Fig. 6c), and thus CIP and AGGs are the dominating species in 20 m KAc.
4.3 Solvation structure in WiSEs based on NO3−
In the 22 m LiNO3 electrolyte, in which the salt:water mole ratio is around 1:2, most of the Li+(H2O)4 complex are condensed into (Li+(H2O)2)n accompanied with complete disappearance of hydrogen bonds between water molecules, and an extended linear chain of Li+ and water molecules is discovered (Fig. 5c). Such a structure of the (Li+(H2O)2)n chains, though, is conformationally flexible and allows Li+ ions mobility in the 22 m LiNO3 solution. The unique characteristic of the super-concentrated LiNO3 solution place it in a position in-between a crystal and a conventional solution, thus endowing the system with virtues from both the extremes.31
4.4 Solvation structure in WiSEs based on ClO4−
In the 17 m NaClO4 aqueous solution, water molecules and ClO4− anions hexacoordinate the Na+ cations. When comparing the Na+:H2O molar ratio (1:3.3) in 17 m NaClO4 with the average coordination number of Na+–Ow (2.8 water molecules for each Na+), it is possible to calculate an average of 0.5 free water molecules (i.e., not directly coordinated with the cation), indicating that most of the water molecules in 17 m NaClO4 aqueous solution participate in hydration of the Na+ cations (Fig. 5g).82 A percolating network structure is formed when cations and anions aggregate together (Fig. 5h), providing open spaces for water to form a water network.83 This self-segregating mechanism can find an explanation in the very mild hydrophilicity of the perchlorate anion (−7 kJ mol−1). In the 17 m NaClO4 system there are only 3.3 water moles for each mole of salt. Even if all the water molecules coordinate only the anion, the overall interaction energy (−23.17 kJ mol−1) would not be sufficient to break the water–water interaction (−24.48 kJ mol−1). Consequently, the ion and water networks are intricately intertwined with each other as described in previous literature.84,85 The lack of water molecules and the almost hydrophobic nature of ClO4− anions may generate the formation of the ion network in this kind of highly concentrated aqueous solution. As a result, the hydrogen bonds in the water structure suffer severe disruption, and the electrostatic interaction between ClO4− anions and Na+ cations is enhanced (Fig. 6e and f).83
4.5 Solvation structure in WiSEs based on Cl−
In the 31 m ZnCl2 system, [Zn(OH2)6][ZnCl4] is classified as a “zinc zincate” based on the solution of its single-crystal structure, in which the two types of ligands, water and chloride ion, are associated with two different zinc atoms in strong segregation (Fig. 5d).81 The O–H bonds are significantly activated to be hydrogen-bond donors by seizing the water molecules on the zinc ions (Fig. 6d). The interaction between water and zinc cations is so strong (∼−405 kJ mol−1) that causes the chemical breaking of water, forming hydroxide-zinc complexes. In accordance with the specific features of this hydrate melt, this aqueous solution could be described as a room temperature ionic liquid. Interestingly, anions of [ZnCl4]2− and [Zn(OH2)2Cl4]2− could serve as charge carriers.75 The incomplete solvation shells may lower the desolvation energy.76
4.6 Interplay between interaction energy and solvation structure
In summary, anions play also a critical role in the structure of WiSEs depending on their hydrophobic or hydrophilic character, leading to different solvation structures. In general, water molecules could interact with both cations and certain anions simultaneously, leading to rather limited water activity in hydrophilic-anion-based WiSEs. We have briefly discussed systems based on strongly hydrophobic (TFSI), mildly hydrophilic (ClO4−), and markedly hydrophilic (Ac−, NO3−, Cl−) anions, finding a very well-defined behavior pattern. Sorting the anions based on their overall interaction energy (i.e., the interaction energy with a single water molecule multiplied by the hydration number) – and setting apart chloride for the moment which deserve a special treatment – from the most hydrophobic to the most hydrophilic we have TFSI−, ClO4−, Ac−, and NO3−. In this order, we observe the structure passing from a situation where the anion mostly forms ionic clusters [Li(TFSI)n](n−1)− and is very weakly interacting with water, to a polymer-like NO3−–water–NO3− chain which negates hydrogen bonds between water molecules. In between we have the borderline hydrophylic ClO4−, forming aqueous channels like the ones in TFSI but with a less pronounced solvation disproportion, and the bicontinuous polar/apolar arrangement of Ac−, forming channel-like structures but with the anion strongly bound to water molecules and competing for hydration with the cation. Practically, what we observe following the hydrophilicity scale is a progressive homogenization of the system, displaying less and less clusters speciation as the hydrophilicity increases. Bringing this reasoning to its limit, we find the chloride-based system Zn(Cl)2, which combines two of the most hydrophilic ions, resulting de facto in the formation of a chemically homogeneous ionic liquid. It appears clear from the given examples how the anion profoundly tailors the solvation properties of this class of electrolytes, and thus one must consider the anion properties in order to achieve the desire properties.
5. Ion transport properties
Ionic transport in aqueous liquid electrolytes is accomplished by two processes: (i) solvation and dissociation of salts by the dipolar water molecules and (ii) migration of solvated ions through water.86 The ionic conductivity (σ) is extensively affected by the above mentioned two processes,86 being determined by the number of free ions ni and their ionic mobility ui as following:86| | 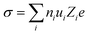 | (3) |
where Zi is the valence order of ionic species i, and e is the unit charge of electrons. It goes without saying that to improve the ion conductivity, the salt dissociation degree ni and/or the ionic mobility ui should be increased. In diluted electrolytes, the ionic mobility ui is determined by the solvation radius ri and the viscosity η of the medium according to the Stokes–Einstein relation:87| | 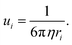 | (4) |
As mentioned before, the solvation structure, which is directly linked to the diffusing cations hydrodynamic radii, is profoundly affected by the anions. The charge carrier number (ni) and ionic mobility (ui) can be determined by the knowledge of the electrolyte's dielectric constant (ε) and viscosity (η).86 These latter characteristics are, in turn, heavily dependent on the salt content, i.e., the concentration of the solution.88 As known in the literature, the dielectric constant of water is 78.49 F m−1 at room temperature,89 while its viscosity is around 1 mPa s.90 However, these two parameters in WiSEs would dramatically vary with the salt concentration and the type of anions. So, it becomes crucial to characterize the ion transport properties in WiSEs.
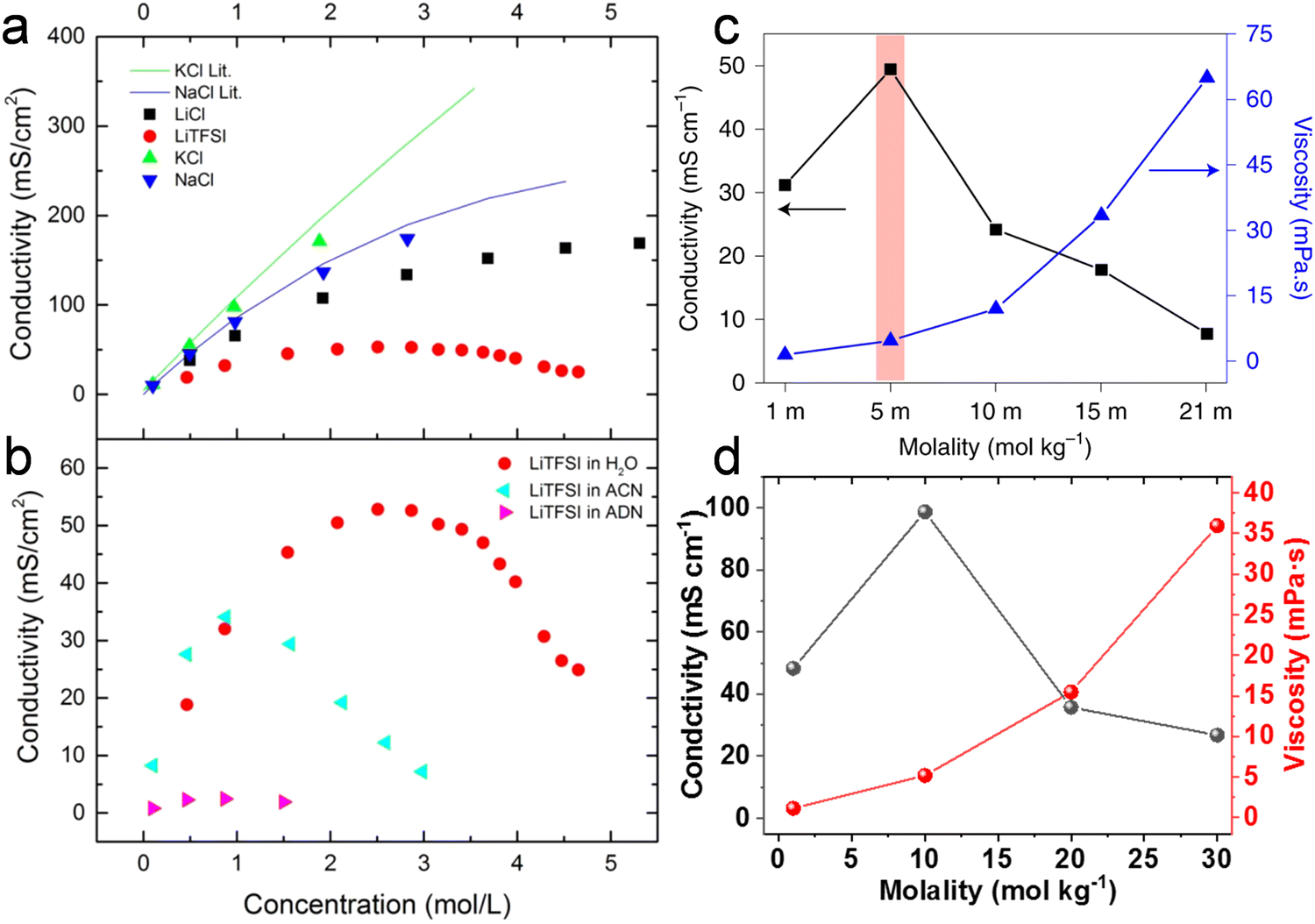
Abu-Lebdeh et al.91 studied the influence of salt concentration on ionic and molar ionic conductivity for a variety of non-aqueous and aqueous solutions (Fig. 7a and b). A typical Gaussian-like behavior is observed for most solutions, where the ion conductivity increases with the increased concentration until reaching a maximum at intermediate concentrations and then decreasing for higher salt contents up to saturation.91 The maximum ion conductivity (σ) is provided at a certain concentration, denoted as Cmax, which evidences a change in conduction mechanism.91 In very dilute conditions, the conductivity is governed by a vehicular mechanism. When the electrolyte concentration approaches Cmax, however, the hopping along the network structure also occurs via a cooperative mechanism involving the formation of stable ion pairs and ionic clusters in the electrolyte. When the concentration exceeds Cmax the vehicular mechanism drops down.91 Similar trends for the ion conductivity are observed in both nonaqueous and aqueous solutions when the salt concentration is increased. Of note, highly concentrated aqueous electrolytes follow the same ion transport mechanism as found in the nonaqueous solutions with a concentration beyond Cmax, which is a hopping process via the cooperative mechanism.93
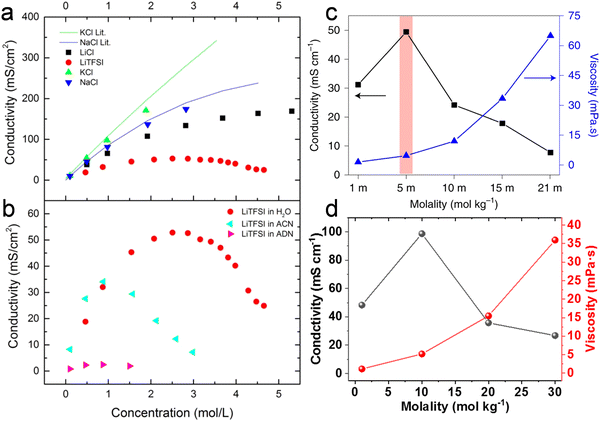 |
| | Fig. 7 (a and b) Ionic conductivity of several aqueous and non-aqueous solutions over a wide concentration range. (a and b) Reproduced with permission.91 (c) Viscosity and conductivity of LiTFSI aqueous solutions with varied concentration; 5 m shows a high conductivity and relatively low viscosity as indicated by the pink vertical line. (c) Reproduced with permission.92 (d) Ionic conductivity and viscosity of different concentrated KAc aqueous electrolytes. (d) Reproduced with permission.69 | |
The ionic conductivity of the LiTFSI aqueous electrolyte increases when the concentration is raised from 1 m to 5 m, while it gradually decreases beyond 5 m (Fig. 7c). Therefore, in this case Cmax is located near 5 m. On the other hand, the viscosity is continuously increased with rising concentration, as could be expected. Similar to what just said for LiTFSI, a Gaussian-like behavior was observed for the KAc aqueous electrolytes (Fig. 7d), where the Cmax is located near the 10 m concentration. By adding more salt after that threshold, the ion conductivity decreases while the viscosity increases. As mentioned before, the dissociation degree (i.e., charge carrier number) and the viscosity of the solutions govern the ion conductivity. In dilute solutions (i.e., for salt concentrations below Cmax), both the charge carriers and the viscosity increase with rising salt concentrations, but the charge carrier increase is predominant, resulting in an overall improvement of the ion conductivity. In highly concentrated solutions with limited free water (i.e., well above Cmax), however, plenty of ion pairs and ionic clusters exist and the salt dissociation degree sharply drops while the viscosity keeps rising, leading to a net decrease of the overall ionic conductivity.
It's worth noting that the drag force exerted on the anion by the surrounding water molecules is largely related to the hydrophilic/hydrophobic character of the former. For instance, TFSI− is hydrophobic while Ac− is hydrophilic, making the migration of hydrated Ac− more difficult than hydrated TFSI−. This would obviously affect the ratio of current transported by the cation, i.e., the cation transference number (t+). This latter is linked to the ionic mobility of each ionic species ui (cations and anions) as follows:86
| | 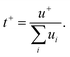 | (5) |
The hydrophilic/hydrophobic anions display different migration behavior which, in turn, affect the cation transference number.
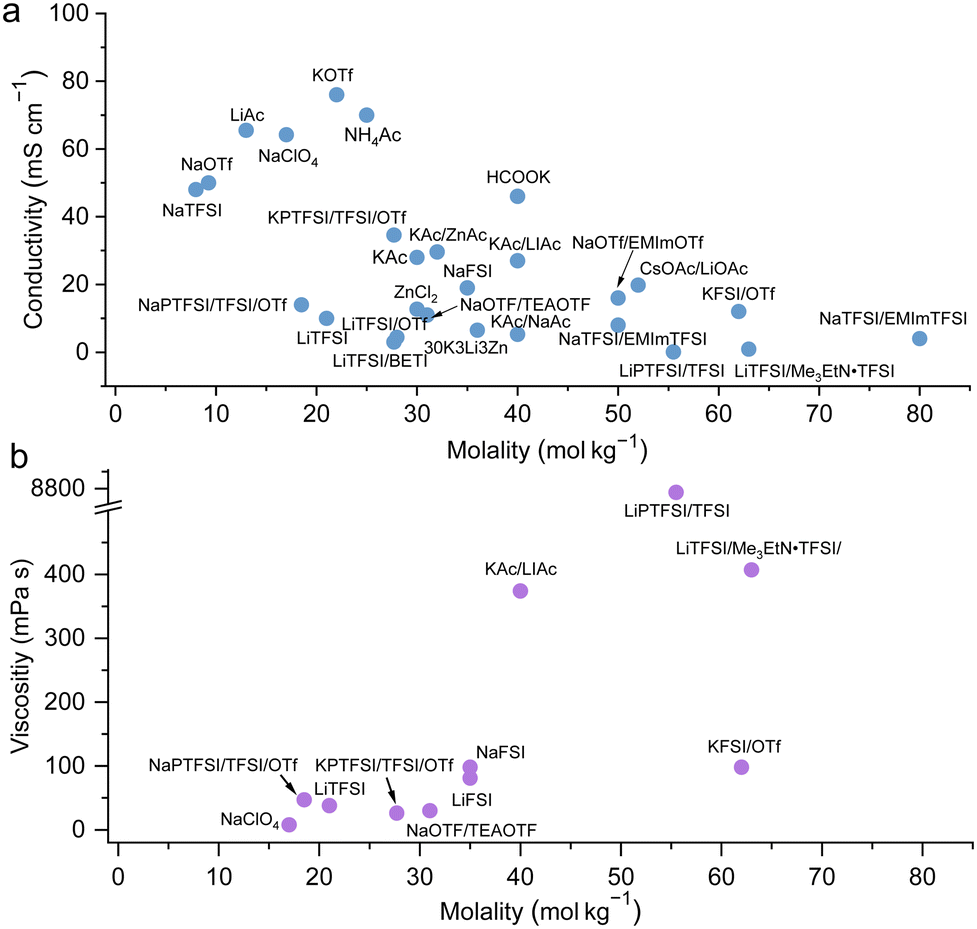
Collections of the ionic conductivity and the viscosity of a variety of electrolytes are shown in Fig. 8a and b, respectively. In general, the ionic conductivity of WiSEs falls while the viscosity rises when the salt concentration is increased. Nevertheless, it should be noted that the ionic conductivities of most WiSEs are still sufficient for battery applications. For instance, the 21 m LiTFSI system has conductivity of about 8 mS cm−1 at 25 °C, which is comparable to a typical non-aqueous electrolyte.12 The transference number of Li+ (t+) for the same system is 0.70 at 20 °C, which is much higher than that offered by typical non-aqueous electrolytes.80 The ion transport mechanism in this WiSEs was thoroughly investigated by Borodin et al.,80 who found that a nano-heterogeneous liquid structure was formed because of the solvation disproportionation of Li+ and TFSI− by H2O molecules. The Li+(H2O)4 were the dominant charge carriers in the solution with a vehicular motion mechanism. The TFSI anion was relatively immobilized in TFSI-rich domains, while fast Li+ transports with high transference number was guaranteed by the 3D percolating channels of Li+(H2O)4.80
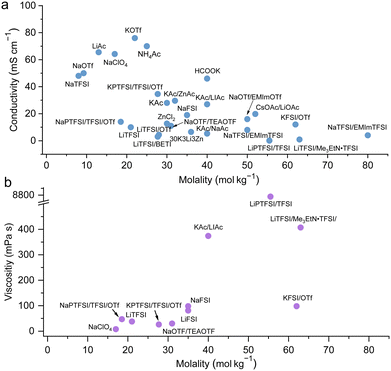 |
| | Fig. 8 Ion conductivity (a) and viscosity (b) for representative highly concentrated aqueous electrolytes, respectively.12,24–29,32–50 | |
With concentrations approaching water:salt ratios close to 2 or even lower, the ion transport mechanism changes to a hopping-type process in, e.g., Li(TFSI)0.7(BETI)0.3·2H2O hydrate melt,94 32 m KAc–8 m LiAc36 and 32 m KAc–8 m NaAc,61 among others. The 62 m KFSI/OTf possessed a relatively high conductivity (12 mS cm−1 at 30 °C) and a relatively low viscosity (98 mPa s) owing to the low Lewis acidity of K+, weakening its interaction with anions and water molecules.42
A more complete overview on the transport properties of the systems is achieved using the so-called Walden Plot introduced by Angell et al.96 In this representation, the viscosity (η) and the conductivity (σ) are directly related by the Walden Rule97
| | 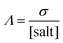 | (7) |
where
Λ is the molar conductivity defined as in
eqn (7),
α is the decoupling constant,
C is a temperature-dependent constant, and [salt] is the overall salt(s) concentration expressed in mol L
−1. The Walden plot is usually divided in three regions defined by two reference lines. The first line represents the “ideal” behavior,
i.e., a solution of
eqn (1) with
α = 1. Traditionally, this line is refereed as the “1 M KCl reference”. Although this definition has been widely debated,
98–100 it is useful to use this line as the bisector passing through the origin of the Walden plot. Electrolytes lying on this line have no closed (contact) ion pairs,
i.e., cations and anion may diffuse independently from each other (0% ion pairing). The second reference line is parallel to the first one, but it is shifted down one order of magnitude, thus assuming an ion pairing of 90%. Electrolytes lying between the two lines have “good ionicity” while above the first line or below the second line have “super ionicity” or “poor ionicity”, respectively. The ionicity is a measure of the amount of free (
i.e., non-paired) ions in the systems.
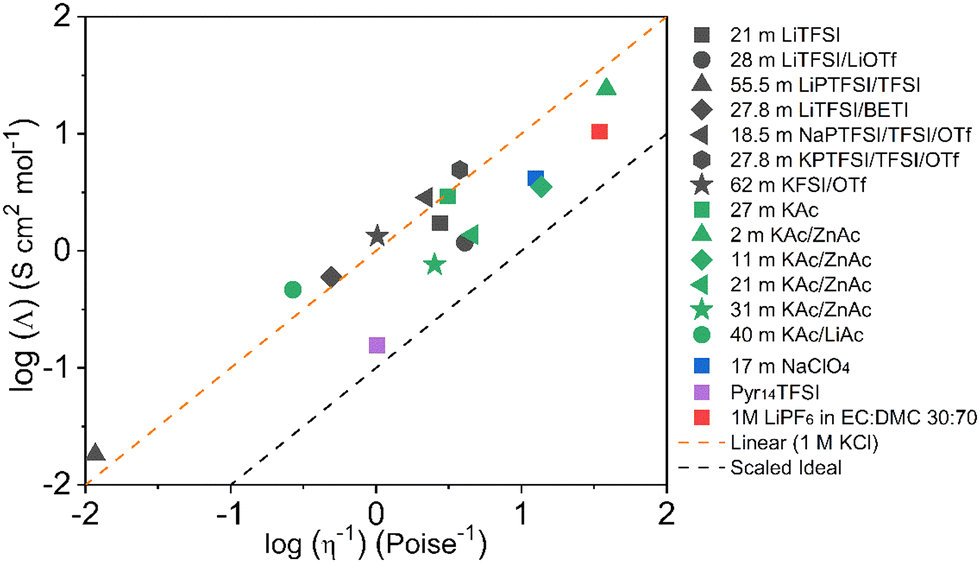
The Walden plot of a variety of highly concentrated aqueous electrolytes (Fig. 9 and Table 3) has been built using the molar ionic conductivity and fluidity values reported in the literature or measured in-house (2 m KAc/ZnAc (1 m KAc-1 m ZnAc), 11 m KAc/ZnAc (10 m KAc-1 m ZnAc), 21 m KAc/ZnAc (20 m KAc-1 m ZnAc), 31 m KAc/ZnAc (30 m KAc-1 m ZnAc)). It should be noted that some electrolytes, e.g., 18.5 m NaPTFSI/TFSI/OTf, 27.8 m LiBETFI/TFSI, 27.8 m KPTFSI/TFSI/OTf, 55 m LIPTFSI/TFSI, 62 m KFSI/OTf, 40 m KAc/LiAc, lie above the ideal KCl line, which classify them as superionic solutions.95
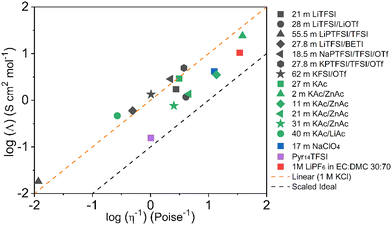 |
| | Fig. 9 Walden plot for a variety of highly concentrated aqueous electrolytes. | |
Table 3 Walden plot related parameters adapted from literatures and/or measured
| Salt |
m (mol kg−1) |
M (mol L−1) |
η
−1 (P−1) |
Λ (S cm2 mol−1) |
Log10η−1 |
Log10Λ |
| KAc |
27 |
10.727 |
3.125 |
2.927 |
0.495 |
0.466 |
| LiTFSI |
21 |
4.768 |
2.762 |
1.722 |
0.441 |
0.236 |
| LiTFSI/OTf |
28 |
5.534 |
4.082 |
1.175 |
0.611 |
0.070 |
| KLiAc |
40 |
11.396 |
0.267 |
0.465 |
−0.573 |
−0.332 |
| LiPTFSI/TFSI |
55.5 |
5.500 |
0.012 |
0.018 |
−1.932 |
−1.740 |
| LiBETFI/TFSI |
27.8 |
5.050 |
0.493 |
0.594 |
−0.307 |
−0.226 |
| NaPTFSI/TFSI/OTf |
18.5 |
4.912 |
2.230 |
2.850 |
0.348 |
0.455 |
| KPTFSI/TFSI/OTf |
27.7 |
7.033 |
3.780 |
4.920 |
0.577 |
0.692 |
| KFSI/OTf |
62 |
8.989 |
1.020 |
1.335 |
0.009 |
0.125 |
| 2KAc/ZnAc |
2 |
1.759 |
38.462 |
24.222 |
1.585 |
1.384 |
| 11KAc/ZnAc |
11 |
6.590 |
13.699 |
3.520 |
1.137 |
0.547 |
| 21KAc/ZnAc |
21 |
9.391 |
4.608 |
1.374 |
0.664 |
0.138 |
| 31KAc/ZnAc |
31 |
10.905 |
2.532 |
0.761 |
0.403 |
−0.119 |
| Pyr14TFSI |
10.13 |
14.179 |
1.013 |
0.155 |
0.006 |
−0.809 |
| LiPF6 in EC:DMC |
1.98 |
1.000 |
34.674 |
10.471 |
1.540 |
1.020 |
It is also important to notice that none of the electrolytes is poorly ionic. In fact, six show good ionicity (21 m LiTFSI, 17 m NaClO4, 28 m LiTFSI/OTf, 11 m KAc/ZnAc, 21 m KAc/ZnAc, 31 m KAc/ZnAc) and one lies almost exactly on the ideal line (27 m KAc). The systems with good ionicity behave like conventional aqueous salt solutions (e.g., 2 m KAc/ZnAc) or more common electrolytes (also represented in the plot for reference) like ionic liquids (e.g., Pyr14TFSI,) or carbonate-based ones (e.g., 1M LiPF6 in EC:DMC 30:70). In this regime, the ions are relatively free to diffuse even if some neutral ion-pairs hinder the conductivity and increase the viscosity. The charge transport is mostly vehicular, meaning that the conductivity is limited by the viscosity of the medium. On the other hand, for the super ionic systems the conductivity is decoupled from the viscosity, and the charge transport mechanism is, at least, partially structural.101,102 It is difficult to exclude a contribution from the Grotthuss effect of the water molecules, but it is more likely that an incipient gel-like structure favors the metal cations “jumping” (i.e., cooperative mechanism). The abundance of anions in the system may create a network in which the cations easily “slip” from one site to another.101,103,104 Moreover, the low water activity results in incomplete hydration shells, favoring the cations’ jumps because of their reduced effective radius. An honorable mention goes to KAc which lies almost exactly on the ideal line yet being the second most concentrated system (in mol L−1). In this system the water molecules are strongly bound to the acetate anion. Thus, extensive proton jumps could be expected considering the nature of the acetic acid. It is not trivial to estimate the amount of deprotonated water (pH is meaningless because of the very high salt concentration), but it is reasonable to expect a substantial presence of OH− anions which could help in the proton shuttling, thus boosting the conductivity despite the viscosity increase. By looking at the Walden plot, we observe that among the superionic systems, the only single-anion-based electrolytes are salts containing the hydrophilic Ac−. Differently, when the hydrophobic anions are used they have to be a mixture of different salts, often involving highly asymmetric structures. This highlights once more the differences induced by the nature of the anion, which in the case of marked hydrophilicity leads to more homogeneous systems resulting in a possible cooperative motion of ions, while for hydrophobic anions results in solvation disproportion facilitating the vehicular mechanism. Depending on the intended use of the electrolyte, one may take advantage of the two different regimes, finely tailoring the properties of the system by tuning the hydrophilicity of the anion. Albeit the Walden plot applied to WiSEs could be of great importance and could guide towards smarter design of this fairly new class of electrolytes, it is rare to find this kind of analysis in the literature.103,105,106 The widespread use of molality (mol kg−1) in the field is for sure useful, intuitive, and easy to use, but it could make difficult to perform important analysis like the Walden plot if not accompanied by density measurements (which are surprisingly rare in this field).
6. Reactions between anions and water
6.1 Hydrolysis in FSI−-based WiSE
Although the limited amount of water molecules presents in these electrolytes, hydrolysis can still take place affecting the concentration of H+/OH− and the stability of the electrolyte itself. For instance, electrolytes containing the FSI− anion may suffer from hydrolysis on account of the weaker S–F bond compared to the C–F in TFSI−.49 The hydrolysis may evolve as follows:40,49| | | [FSO2NSO2F]− + H2O → HF + [FSO2NSO3H]− | (8) |
Subsequently, the equilibria are established (A = Li+, Na+, or K+):| | | [FSO2NSO3H]− ⇌ H+ + [FSO2NSO3]2− | (11) |
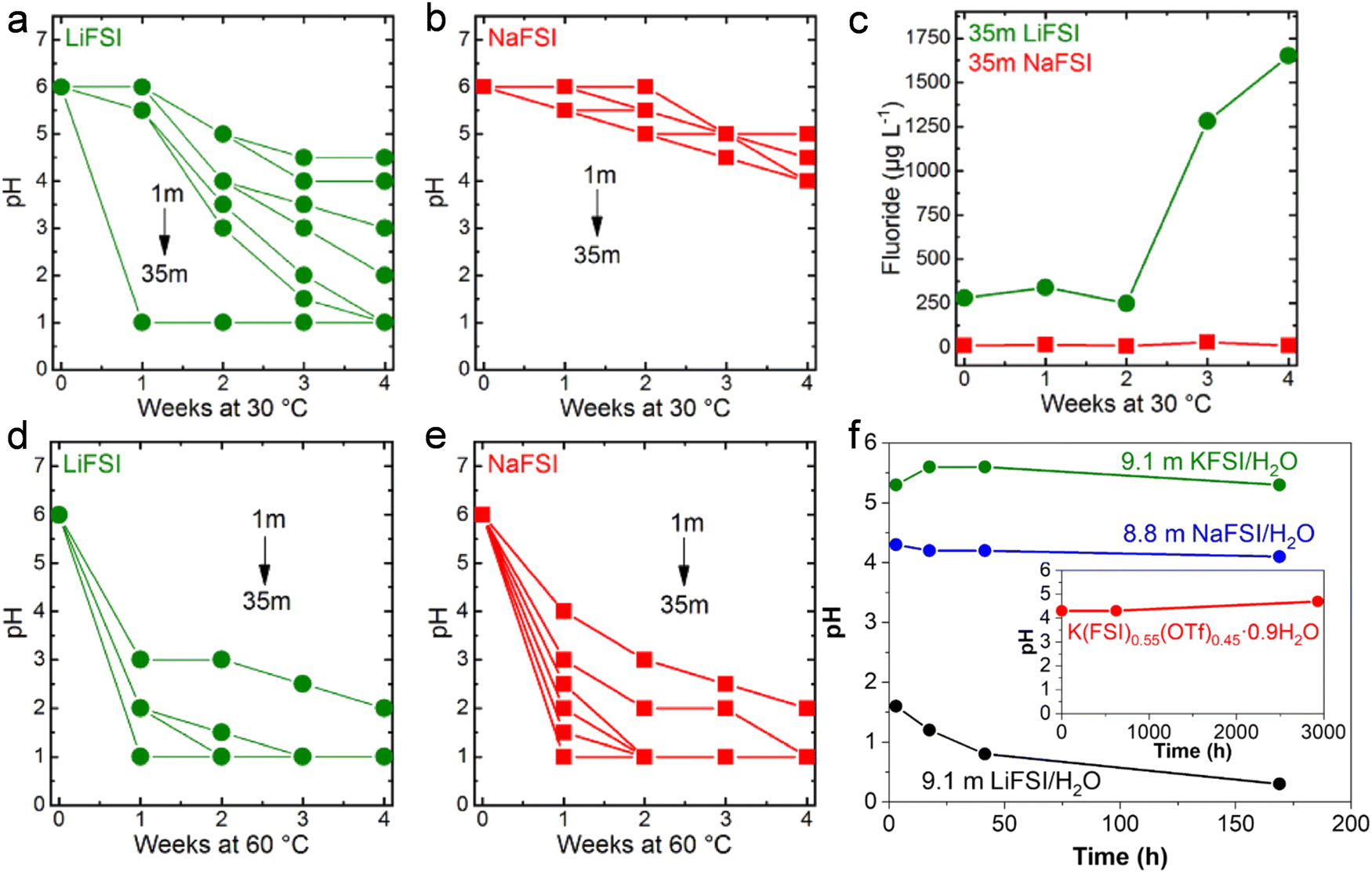
The pH of LiFSI (Fig. 10a) and NaFSI (Fig. 10b) aqueous solutions at different concentrations was studied by Kühnel et al. upon storage at 30 °C for 4 weeks.40 It was noted that the pH of highly concentrated LiFSI solutions drastically decreased to values of ≤1, while it was just slightly decreased to values comprised between 4 and 5 in case of NaFSI.40 In addition, the fluoride content in the 35 m LiFSI and 35 m NaFSI aqueous solutions was measured to better understand the rapid change in pH.40 After four weeks, the fluoride content in the LiFSI solution was 100 times higher than in the NaFSI sample.40 Moreover, a white precipitate, partially attributed to LiF (solubility in water 0.05 m at 30 °C), was observed in the LiFSI solution, indicating a much more pronounced hydrolysis of LiFSI.40 In contrast, all NaFSI solutions remained unchanged upon visual inspection during storage for four weeks. Nevertheless, the pH at 60 °C for all the LiFSI and NaFSI solutions severely dropped within the first week (Fig. 10d and e).40 In conclusion, FSI is highly prone to hydrolysis in both LiFSI and NaFSI inducing acidification of the solutions. However, NaFSI appears more stable possibly due to the different charge densities of Li+ and Na+40 and the lower solubility of LiF (0.05 m) compared to NaF (1 m).107 On the other hand, solutions based on LiTFSI, LiBETI, LiPTFSI and LiOTf did not evidence significant pH change over a period of 9 weeks, suggesting for a sufficient chemical stability of such salts to be employed as electrolytes for batteries.108 In addition, Yamada et al. reported the superior chemical stability of KFSI/H2O and K(FSI)0.55(OTf)0.45·0.9H2O compared to both LiFSI/H2O and NaFSI/H2O (Fig. 10f).49 Such a behavior was explained on the basis of the interaction between the cations A+ (Li+, Na+, K+) and FSI−. Specifically, the coordination between Lewis acidic A+ and the Lewis basic sulfonyl oxygen of FSI− decreases the electron density around the sulfonyl moiety and, thus, weaken the S–F bond leading to its cleavage upon attack of a water molecule as per in eqn (6).49 An additional role is played by the solubility of the respective fluoride. As reported by Reber et al.,40 the solubility of alkali metal fluoride (AF) in water is in the order of LiF < NaF ≪ KF. Therefore, LiF precipitates as a solid phase, leading equilibrium (8) to shift towards the products, i.e., the consumption of F− in solution.49 This would further trigger hydrolysis of the anions as shown in eqn (6). On the contrary, KF is soluble until relatively high concentrations, shifting the equilibrium (8) to the left and thus suppressing the hydrolysis in eqn (6).49
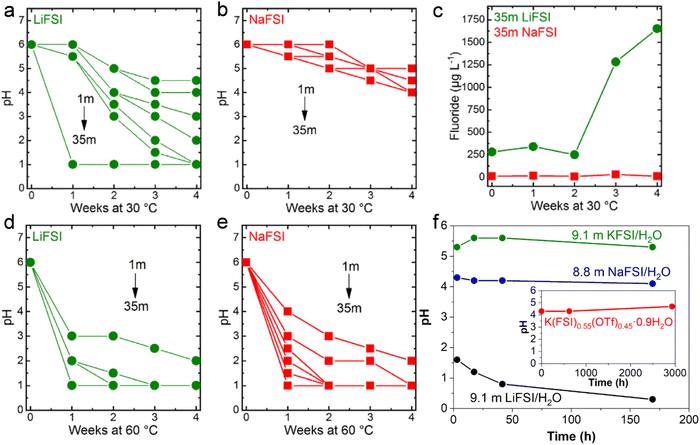 |
| | Fig. 10 Evolution of the pH in (a) LiFSI and (b) NaFSI aqueous solutions with different concentration stored at 30 °C. (c) Evolution of the fluoride content of 35 m LiFSI and 35 m NaFSI during storage at 30 °C. Evolution of the pH of (d) LiFSI and (e) NaFSI aqueous solutions with different concentration when stored at 60 °C. (a–e) Reproduced with permission.40 (f) Evolution of the pH for LiFSI/H2O, NaFSI/H2O, and KFSI/H2O solutions. In the inset the result for K(FSI)0.55(OTf)0.45·0.9H2O. (f) Reproduced with permission.49 | |
6.2 Acid base reaction in Ac−-based WiSEs
Apart from the hydrolysis of FSI− anion, being the conjugated basis of a weak acid, Ac− (pKb = 9.25) also suffers from protonation in aqueous solutions as it follows:109| | | CH3COO− + H2O ⇌ CH3COOH + OH−. | (12) |
This protonation of the Ac− anion results in LiAc, NaAc and KAc solutions become alkaline, thus affecting the ESW of the solutions as well as their compatibility with current collectors and active materials.61,69,110
6.3 Hydrolysis of cations in WiSEs
Besides the above-mentioned anions, some cations such as Zn2+, NH4+, also undergo hydrolysis in aqueous solutions. Specifically, hydrolysis equilibriums for Zn2+ (pKa = 10.98) and NH4+ (pKa = 9.40) are as following:109,111–113| | | pZn2+ + qH2O ⇌ Znp(OH)q(2p+q)+ + pH+ | (13) |
| | | NH4+ + H2O ⇌ NH3 + H3O+ | (14) |
Nevertheless, appropriate selection of anion and cation can also result into a neutral or near-neutral environment. For instance, Ji's group found that NH4Ac lead to near-neutral aqueous solutions at both dilute and high concentration,71,114 owing to the equilibrium between weak acid and weak base.109 In addition, as reported by Cui's group, a dual-salt solution consisting of 1 M Zn(Ac)2 and 4 M NaAc possessed a near-neutral environment (pH = 7.13), which enabled Zn stripping/plating with a nearly 95% CE and effectively restrained hydrogen evolution compared with the traditional “mild” electrolytes (pH < 5).109
6.4 Outlook on anion reactivity with water
The stability of the anion in aqueous environment is an important aspect to be considered when designing WiSE, since it can affect both operation and calendar life of the battery. The use of fluorinated salts prone to hydrolysis, e.g., LiFSI, is concerning also from an environmental point of view as F− ions are released. Meanwhile, reaction with water can induce changes in acidity/alkalinity of the solution, which may require adapting all cell components (not only active materials but also current collectors and casing) in order to avoid corrosion/dissolution issues.
7. Solid electrolyte interphase
The solid-electrolyte interphase (SEI) has been a key aspect of LIBs, since their first commercialization more than 30 years ago.115,116 In conventional organic electrolytes, the SEI is formed as results of the decomposition of salt and solvents on the electrodes’ surface when the potential exceeds the thermodynamic stability limits of the electrolyte. An effective SEI still holds the electrolyte nature, i.e., it avoids electron transport, but allows for ionic transport to support cell operation.117–119 Thus, the continuous electrolyte decomposition (and SEI growth) does not occur while the electrochemical reactions at the electrodes can still proceed.120,121 Although significant knowledge has been achieved regarding the chemistry,122 morphology123 and formation mechanism122 of SEI in non-aqueous electrolytes, the understanding about the interphase formed in aqueous electrolytes is still at an early stage. It should be noted that, unlike organic carbonates, the solvent (water) in this case does not decompose into solid products. This means that the SEI in aqueous electrolytes mostly arises from decomposition of the salt and, in particular, of the anion. Additionally, water can partially dissolve the typical components of an SEI formed in in nonaqueous electrolyte, e.g., Li2CO3, or LiF.124
7.1 SEI in fluorinated WiSEs
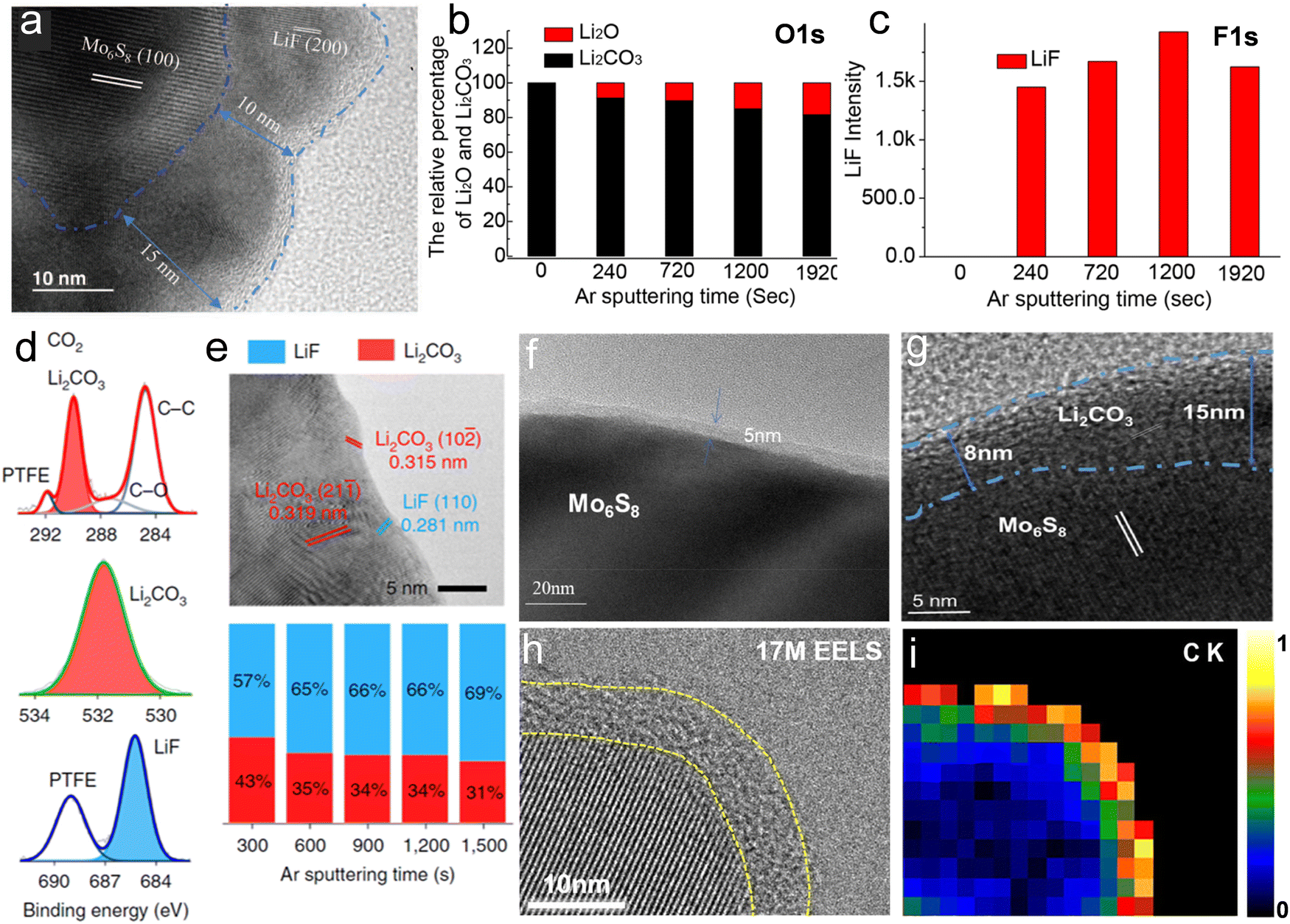
Suo et al. first reported the formation of a LiF-rich interphase on the surface of a Mo6S8 anode cycled in 21 m LiTFSI, as evidenced by TEM (Fig. 11a). The fully inorganic SEI functioned as an electron barrier preventing the reduction of water while allowing Li+ migration. A crystalline phase showing a 10 to 15 nm thickness was discovered on the cycled Mo6S8 anode, which according to its interplanar spacing could be identified as imperfect crystalline LiF.12 Similarly, a dense and robust NaF-based SEI was discovered by Suo et al. on a NaTi2(PO4)3 anode cycled in 9.26 m NaCF3SO3. To reveal other possible components other than LiF in the interphase generated in 21 m LiTFSI, Suo et al. performed XPS and collected the relative intensity of Li2CO3 and Li2O from the O1s spectra at various etching stages (Fig. 11b).124 Both Li2CO3 and Li2O were confirmed as components of the SEI. These findings were further verified when the full cell LiMn2O4/Mo6S8 was charged to 1.6 V.124 In addition, the same analysis performed on the F1s spectra (Fig. 11c) demonstrated that LiF resided in the inner region of SEI close to the electrode surface.124 The formation of LiF was attributed to the reduction of anion complexes (or clusters) at the potential of 2.9 V (vs. Li/Li+), while Li2O and Li2CO3 were produced by reduction of dissolved O2 and CO2 as shown in the following equations:124| | | 4Li+ + O2 + 4e− → 2Li2O E = 2.91 V | (15) |
| | | 2Li + 1/2O2 + CO2 + 2e− → Li2CO3 E = 3.82 V | (16) |
It should be noted that, given the non-negligible solubility of LiF, Li2O and Li2CO3, such an SEI is only stable in highly concentrated electrolytes with limited amount of free water.
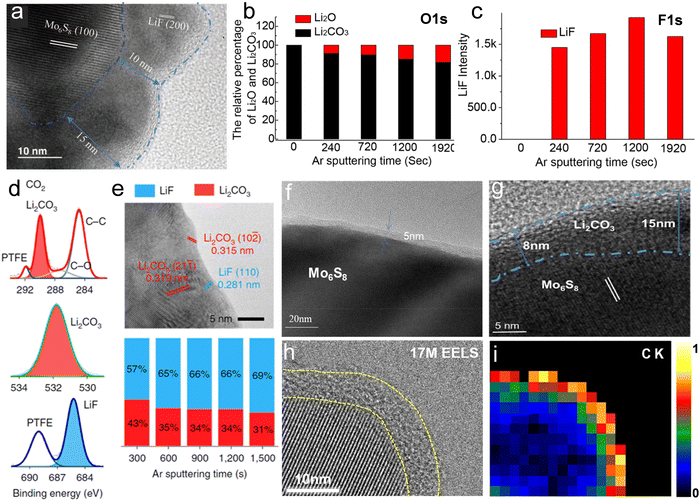 |
| | Fig. 11 (a) TEM images of cycled Mo6S8 electrode in 21 m LiTFSI. (a) Reproduced with permission.12 (b) Relative intensity of Li2O and Li2CO3 in O1s spectra collected by X-ray Photoelectron Spectroscopy (XPS) upon various etching times. (c) Relative intensity of LiF in F1s spectra collected by XPS upon various etching times.(b and c) Reproduced with permission.124 XPS spectra (d) and TEM image (e) relative to the characterization of the interphase on a cycled Mo6S8 electrode in 21 m LiTFSI saturated with CO2 in three-electrode cell. (d and e) Reproduced with permission.92 (f) TEM image of a Mo6S8 electrode after 10 cycles in saturated LiAc electrolyte. (g) TEM image of a Mo6S8 electrode after 10 cycles in 28 M LiCO2CF3. (f and g) Reproduced with permission.125 (h) High-resolution TEM images and corresponding elemental EELS maps of (i) carbon K-edge of the NaTi2(PO4)3 electrode surface after 20 cycles in 17 m NaClO4 electrolyte. (h and i) Reproduced with permission.126 | |
Despite the stable SEI formed in the 21 m LiTFSI system, this electrolyte suffers from multiple disadvantages such as high cost, high viscosity and limited ionic conductivity. For this reason, efforts are being done to develop SEIs that are stable also in lower concentration electrolytes. Suo et al., for example, introduced CO2 into 5 m LiTFSI (TFSI–CO2 complex) as an interphase formation additive.92 XPS (Fig. 11d) revealed that chemical components of the interphase were dominated by Li2CO3 and, to a lesser extent, LiF, whose corresponding lattice distances are marked by red and blue in the TEM image (Fig. 11e), respectively.92 Stepwise Ar+ sputtering and XPS analysis showed that Li2CO3 and LiF always coexist during etching.92 Tailoring the SEI composition via this CO2–TFSI interaction allowed stable cycling of aqueous LIBs with more sustainable low concentration LiTFSI electrolytes.92 By adopting a similar strategy, artificial introduction of a certain amount of CO2 was also reported by Zhao's group for the 1 M and 5 M LiNO3 electrolytes. The generated passivation film, mostly consisting of Li2CO3, broadened the ESW of the system.125
7.2 SEI in carboxylate-based WiSEs
As reported by Zhao's group, saturated LiAc electrolyte having a concentration more than 12 m,43 could induce the formation of an SEI mainly consisting of Li2CO3 on the Mo6S8 anode.125 The TEM image (Fig. 11f) of Mo6S8 electrode after 10 cycles evidenced that such layer possess a thickness of about 5 nm.125 The formation of a Li2CO3-dominated SEI results from both the protonation of acetate and the decarboxylation reaction of the carboxylate. The reaction mechanism for the decarboxylation can be described as follows:125Meanwhile, the electrochemical decarboxylation reaction could convert Ac− into CO2 as it follows:125| | 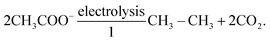 | (20) |
The decarboxylation reaction leads to CO2 production, which indeed favors the formation of Li2CO3. Notably, electrolytes were degassed by nitrogen for at least 1 h to remove dissolved oxygen as well as CO2.125 Zhao's group reported that 28 M LiCO2CF3 could also facilitate the production of CO2via decarboxylation to form a Li2CO3-rich SEI. The TEM image of a Mo6S8 electrode after 10 cycles in 28 M LiCO2CF3 showed a Li2CO3 layer twice as thick as that formed in the LiAc electrolyte (Fig. 11g).125 In addition, the trifluoromethyl groups in LiCO2CF3, possessing strong electron-withdrawing character, effectively improved the oxidative stability of the aqueous electrolyte.125
7.3 SEI in perchloric-based WiSEs
Kang's group reported that an SEI layer consisting of Na2CO3 and Na–O compounds was formed on a NaTi2(PO4)3 anode cycled in 17 m NaClO4, which guaranteed the excellent electrochemical storage stability of NaTi2(PO4)3/Na4Fe3(PO4)2(P2O7) full-cell.126 High-resolution TEM images of the NaTi2(PO4)3 anode clearly demonstrated a layer with thickness of around 5 nm (Fig. 11h).126 The corresponding elemental EELS maps showed carbon K-edge signals as elements comprising the SEI layer (Fig. 11i).126 In addition, XPS and Soft X-ray absorption spectroscopy (sXAS) spectra of the cycled NaTi2(PO4)3 electrode revealed the characteristic peak of Na2CO3 and NaOH.126 The SEI formation process was driven by the reduction of dissolved gases (O2 and CO2), and not by salt reduction. The specific formation process could be summarized as it follows:126–132| | | 2Na+ + O2 + e− → NaO2 E = 2.27 V (vs. Na/Na+) | (21) |
| | | 2Na+ + O2 + 2e− → Na2O2 E = 2.33 V (vs. Na/Na+) | (22) |
| | | 4Na+ + 3CO2 + 4e− → 2Na2CO3 + C E = 2.35 V (vs. Na/Na+) | (23) |
| | | NaO2 + 2H2O → Na+ + 4OH− | (24) |
| | | Na2O2 + 2H2O → 2Na+ + 4OH− | (25) |
Although the above reactions could take place in both 1 m and 17 m electrolytes, sodium carbonates and oxides are unstable in the more diluted solution due to the large amount of water.126 The highly concentrated 17 m electrolyte with limited free water guaranteed the durability of the carbonate and oxides although NaOH was formed in the surface layer by hydrolysis between the limited free water and the NaO2/Na2O2 compounds.126
7.4 Outlook on SEI formation
The SEI definitely plays an important role on the performance of aqueous batteries. Since water does not decompose into solid products, in WiSE the SEI formation mechanism is strictly linked to the decomposition of the anion. Specifically, fluorinated anions can lead to SEIs rich in fluorinated species (e.g., NaF and LiF). Decarboxylation reaction could take place in carboxylate-based WiSE, leading to CO2 evolution and formation of a carbonate-rich SEI. In case of WiSE featuring more stable anions, e.g., perchlorate, gases dissolved in the electrolyte (e.g., O2 and CO2) could promote the formation of an SEI anyway. Independently from the specific SEI composition, the main challenge is to form an SEI that is stable upon time and cycling and, ideally, not only at ultra-high salt concentration. To achieve that, new salts/additives capable of decomposing into highly insoluble compounds are needed.
8. Conclusions
The recent progress in highly concentrated aqueous electrolytes, so-called WiSE, based on a variety of anions has accelerated the development of high-energy aqueous batteries. The ionic interactions, solvation structure, transport properties, reactivity, and SEI formation of representative WiSEs based on various anions are here reviewed and discussed. It is found that the above-mentioned properties could be manipulated by selecting the appropriate anion. Among these properties, the SEI plays a crucial role in broadening the ESW of electrolytes and stabilizing the electrode materials, thus enabling high-energy aqueous batteries. However, the solubility of SEI components still remains an issue. Despite the numerous achievements obtained so far, highly concentrated electrolytes also still suffer from high viscosity and, depending on the salt, high cost.92
To benefit from the non-flammability of aqueous electrolytes and simultaneously circumvent the disadvantages of highly concentrated solutions, multiple strategies can be adopted. Of course, the development of new, low cost salts yielding WiSE with low viscosity, high ionic conductivity and large ESW would be very beneficial. Nevertheless, finding salts with sufficient solubility to achieve WiSE regime may be very challenging. In this sense, a reasonable approach would be to decouple the interphase formation from the high concentration, by identifying salts and/or additives capable of forming a stable SEI in moderate/low concentration electrolytes. Although WiSE are a wonderful playground for performing fundamental science, from a purely practical point view they still face serious limitations in terms of temperature operation range and self-discharge, as recently pointed out by Droguet et al.133 In view of this, a renewed interest in the classical “old” concept of salt-in-water electrolyte cannot be excluded in the future. This may likely happen in case the water splitting issue is effectively solved by other means such as, e.g., tuning electrode and SEI composition.
Conflicts of interest
There are no conflicts to declare.■■■■
References
- D. Gielen, F. Boshell, D. Saygin, M. D. Bazilian, N. Wagner and R. Gorini, The role of renewable energy in the global energy transformation, Energy Strategy Reviews, 2019, 24, 38–50 CrossRef.
- X. Yang, L. He, Y. Xia and Y. Chen, Effect of government subsidies on renewable energy investments: The threshold effect, Energy Policy, 2019, 132, 156–166 CrossRef.
- K. Engeland, M. Borga, J.-D. Creutin, B. François, M.-H. Ramos and J.-P. Vidal, Space-time variability of climate variables and intermittent renewable electricity production–A review, Renewable Sustainable Energy Rev., 2017, 79, 600–617 CrossRef.
- J. P. Barton and D. G. Infield, Energy storage and its use with intermittent renewable energy, IEEE Trans. Power Appar. Syst., 2004, 19(2), 441–448 Search PubMed.
- J. Han, A. Varzi and S. Passerini, The emergence of aqueous ammonium-ion batteries, Angew. Chem., Int. Ed., 2021, 61(14), e202115046 Search PubMed.
- Y. Gogotsi and P. Simon, True performance metrics in electrochemical energy storage, Science, 2011, 334(6058), 917–918 CrossRef CAS PubMed.
- Y. Ding, Z. P. Cano, A. Yu, J. Lu and Z. Chen, Automotive Li-ion batteries: current status and future perspectives, Electrochem. Energy Rev., 2019, 2(1), 1–28 CrossRef CAS.
- Y. Song, Y. Yang and Z. Hu, Present status and development trend of batteries for electric vehicles, Power System Technology, 2011, 35(4), 1–7 Search PubMed.
- M.-T. F. Rodrigues, G. Babu, H. Gullapalli, K. Kalaga, F. N. Sayed, K. Kato, J. Joyner and P. M. Ajayan, A materials perspective on Li-ion batteries at extreme temperatures, Nat. Energy, 2017, 2(8), 1–14 Search PubMed.
- W. Li, J. R. Dahn and D. S. Wainwright, Rechargeable Lithium Batteries with Aqueous Electrolytes, Science, 1994, 264, 1115–1118 CrossRef CAS PubMed.
- D. Chao, W. Zhou, F. Xie, C. Ye, H. Li, M. Jaroniec and S.-Z. Qiao, Roadmap for advanced aqueous batteries: From design of materials to applications, Sci. Adv., 2020, 6(21), eaba4098 CrossRef CAS PubMed.
- L. Suo, O. Borodin, T. Gao, M. Olguin, J. Ho, X. Fan, C. Luo, C. Wang and K. Xu, Water-in-salt electrolyte enables high-voltage aqueous lithium-ion chemistries, Science, 2015, 350(6263), 938–943 CrossRef CAS PubMed.
- Y. Yamada, K. Furukawa, K. Sodeyama, K. Kikuchi, M. Yaegashi, Y. Tateyama and A. Yamada, Unusual stability of acetonitrile-based superconcentrated electrolytes for fast-charging lithium-ion batteries, J. Am. Chem. Soc., 2014, 136(13), 5039–5046 CrossRef CAS PubMed.
- Y. Yamada, K. Usui, C. H. Chiang, K. Kikuchi, K. Furukawa and A. Yamada, General observation of lithium intercalation into graphite in ethylene-carbonate-free superconcentrated electrolytes, ACS Appl. Mater. Interfaces, 2014, 6(14), 10892–10899 CrossRef CAS PubMed.
- K. Yoshida, M. Nakamura, Y. Kazue, N. Tachikawa, S. Tsuzuki, S. Seki, K. Dokko and M. Watanabe, Oxidative-stability enhancement and charge transport mechanism in glyme–lithium salt equimolar complexes, J. Am. Chem. Soc., 2011, 133(33), 13121–13129 CrossRef CAS PubMed.
- M. Amiri and D. Bélanger, Physicochemical and Electrochemical Properties of Water-in-Salt Electrolytes, ChemSusChem, 2021, 14(12), 2487–2500 CrossRef CAS PubMed.
- T. Liang, R. Hou, Q. Dou, H. Zhang and X. Yan, The Applications of Water-in-Salt Electrolytes in Electrochemical Energy Storage Devices, Adv. Funct. Mater., 2020, 2006749 Search PubMed.
- H. Zhang, X. Liu, H. Li, I. Hasa and S. Passerini, Challenges and Strategies for High-Energy Aqueous Electrolyte Rechargeable Batteries, Angew. Chem., Int. Ed., 2021, 60(2), 598–616 CrossRef CAS PubMed.
- O. Borodin, J. Self, K. A. Persson, C. Wang and K. Xu, Uncharted waters: super-concentrated electrolytes, Joule, 2020, 4(1), 69–100 CrossRef CAS.
- D. Xiao, L. Zhang, Z. Li, H. Dou and X. Zhang, Design Strategies and Research Progress for Water-in-Salt Electrolytes, Energy Storage Mater., 2021, 44, 10–28 CrossRef.
- Y. Shen, B. Liu, X. Liu, J. Liu, J. Ding, C. Zhong and W. Hu, Water-in-salt electrolyte for safe and high-energy aqueous battery, Energy Storage Mater., 2021, 34, 461–474 CrossRef.
- T. Lv and L. Suo, Water-in-Salt Widens the Electrochemical Stability Window: Thermodynamic and Kinetic Factors, Curr. Opin. Electrochem., 2021, 100818 CrossRef CAS.
- K. Xu and C. Wang, Batteries: widening voltage windows, Nat. Energy, 2016, 1(10), 1–2 Search PubMed.
- Y. Yamada, K. Usui, K. Sodeyama, S. Ko, Y. Tateyama and A. Yamada, Hydrate-melt electrolytes for high-energy-density aqueous batteries, Nat. Energy, 2016, 1(10), 16129 CrossRef CAS.
- L. Suo, O. Borodin, W. Sun, X. Fan, C. Yang, F. Wang, T. Gao, Z. Ma, M. Schroeder, A. von Cresce, S. M. Russell, M. Armand, A. Angell, K. Xu and C. Wang, Advanced High-Voltage Aqueous Lithium-Ion Battery Enabled by “Water-in-Bisalt” Electrolyte, Angew. Chem., 2016, 55(25), 7136–7141 CrossRef CAS PubMed.
- D. Reber, R.-S. Kühnel and C. Battaglia, High-voltage aqueous supercapacitors based on NaTFSI, Sustainable Energy Fuels, 2017, 1(10), 2155–2161 RSC.
- L. Suo, O. Borodin, Y. Wang, X. Rong, W. Sun, X. Fan, S. Xu, M. A. Schroeder, A. V. Cresce and F. Wang, “Water-in-salt” electrolyte makes aqueous sodium-ion battery safe, green, and long-lasting, Adv. Energy Mater., 2017, 7(21), 1701189 CrossRef.
- R.-S. Kühnel, D. Reber and C. Battaglia, A High-Voltage Aqueous Electrolyte for Sodium-Ion Batteries, ACS Energy Lett., 2017, 2(9), 2005–2006 CrossRef.
- K. Nakamoto, R. Sakamoto, Y. Sawada, M. Ito and S. Okada, Over 2 V Aqueous Sodium-Ion Battery with Prussian Blue-Type Electrodes, Small Methods, 2019, 3(4), 1800220 CrossRef CAS.
- F. Wang, O. Borodin, T. Gao, X. Fan, W. Sun, F. Han, A. Faraone, J. A. Dura, K. Xu and C. Wang, Highly reversible zinc metal anode for aqueous batteries, Nat. Mater., 2018, 17(6), 543–549 CrossRef CAS PubMed.
- J. Zheng, G. Tan, P. Shan, T. Liu, J. Hu, Y. Feng, L. Yang, M. Zhang, Z. Chen and Y. Lin, Understanding thermodynamic and kinetic contributions in expanding the stability window of aqueous electrolytes, Chem, 2018, 4(12), 2872–2882 CAS.
- S. Qiu, Y. Xu, X. Li, S. K. Sandstrom, X. Wu and X. Ji, Reinforced potassium and ammonium storage of the polyimide anode in acetate-based water-in-salt electrolytes, Electrochem. Commun., 2021, 122, 106880 CrossRef CAS.
- D. P. Leonard, Z. Wei, G. Chen, F. Du and X. Ji, Water-in-Salt Electrolyte for Potassium-Ion Batteries, ACS Energy Lett., 2018, 3(2), 373–374 CrossRef CAS.
- L. Zhang, I. A. Rodríguez-Pérez, H. Jiang, C. Zhang, D. P. Leonard, Q. Guo, W. Wang, S. Han, L. Wang and X. Ji, ZnCl2 “Water-in-Salt” electrolyte transforms the performance of vanadium oxide as a Zn battery cathode, Adv. Funct. Mater., 2019, 29(30), 1902653 CrossRef.
- J. Han, H. Zhang, A. Varzi and S. Passerini, Fluorine-Free Water-in-Salt Electrolyte for Green and Low-Cost Aqueous Sodium-Ion Batteries, ChemSusChem, 2018, 11(21), 3704–3707 CrossRef CAS PubMed.
- M. R. Lukatskaya, J. I. Feldblyum, D. G. Mackanic, F. Lissel, D. L. Michels, Y. Cui and Z. Bao, Concentrated mixed cation acetate “water-in-salt” solutions as green and low-cost high voltage electrolytes for aqueous batteries, Energy Environ. Sci., 2018, 11, 2876–2883 RSC.
- J. Guo, Y. Ma, K. Zhao, Y. Wang, B. Yang, J. Cui and X. Yan, High-Performance and Ultra-Stable Aqueous Supercapacitors Based on a Green and Low-Cost Water-In-Salt Electrolyte, ChemElectroChem, 2019, 6(21), 5433–5438 CrossRef CAS.
- Q. Zheng, S. Miura, K. Miyazaki, S. Ko, E. Watanabe, M. Okoshi, C. P. Chou, Y. Nishimura, H. Nakai and T. Kamiya, Sodium-and Potassium-Hydrate Melts Containing Asymmetric Imide Anions for High-Voltage Aqueous Batteries, Angew. Chem., 2019, 131(40), 14340–14345 CrossRef.
- L. Jiang, Y. Lu, C. Zhao, L. Liu, J. Zhang, Q. Zhang, X. Shen, J. Zhao, X. Yu, H. Li, X. Huang, L. Chen and Y.-S. Hu, Building aqueous K-ion batteries for energy storage, Nat. Energy, 2019, 4, 495–503 CrossRef CAS.
- D. Reber, R. Figi, R.-S. Kuehnel and C. Battaglia, Stability of aqueous electrolytes based on LiFSI and NaFSI, Electrochim. Acta, 2019, 321, 134644 CrossRef CAS.
- T. Liu, L. Tang, H. Luo, S. Cheng and M. Liu, A promising water-in-salt electrolyte for aqueous based electrochemical energy storage cells with a wide potential window: highly concentrated HCOOK, Chem. Commun., 2019, 55(85), 12817–12820 RSC.
- S. Ko, Y. Yamada, K. Miyazaki, T. Shimada, E. Watanabe, Y. Tateyama, T. Kamiya, T. Honda, J. Akikusa and A. Yamada, Lithium-salt monohydrate melt: A stable electrolyte for aqueous lithium-ion batteries, Electrochem. Commun., 2019, 104, 106488 CrossRef CAS.
- S. Dong, Y. Wang, C. Chen, L. Shen and X. Zhang, Niobium Tungsten Oxide in a Green Water-in-Salt Electrolyte Enables Ultra-Stable Aqueous Lithium-Ion Capacitors, Nano-Micro Lett., 2020, 12(1), 1–11 CrossRef PubMed.
- D. Reber, R. Grissa, M. Becker, R. S. Kühnel and C. Battaglia, Anion Selection Criteria for Water-in-Salt Electrolytes, Adv. Energy Mater., 2021, 11(5), 2002913 CrossRef CAS.
- L. Jiang, L. Liu, J. Yue, Q. Zhang, A. Zhou, O. Borodin, L. Suo, H. Li, L. Chen and K. Xu, High-Voltage Aqueous Na-Ion Battery Enabled by Inert-Cation-Assisted Water-in-Salt Electrolyte, Adv. Mater., 2020, 32(2), 1904427 CrossRef CAS PubMed.
- S. Chen, R. Lan, J. Humphreys and S. Tao, Salt-concentrated acetate electrolytes for a high voltage aqueous Zn/MnO2 battery, Energy Storage Mater., 2020, 28, 205–215 CrossRef.
- J. Han, A. Mariani, A. Varzi and S. Passerini, Green and low-cost acetate-based electrolytes for the highly reversible zinc anode, J. Power Sources, 2021, 485, 229329 CrossRef CAS.
- S. Chen, R. Lan, J. Humphreys and S. Tao, Effect of cation size on alkali acetate-based ‘water-in-bisalt’electrolyte and its application in aqueous rechargeable lithium battery, Applied Materials Today, 2020, 20, 100728 CrossRef.
- S. Ko, Y. Yamada and A. Yamada, A 62 m K-ion aqueous electrolyte, Electrochem. Commun., 2020, 116, 106764 CrossRef CAS.
- L. Chen, J. Zhang, Q. Li, J. Vatamanu, X. Ji, T. P. Pollard, C. Cui, S. Hou, J. Chen, C. Yang, L. Ma, M. S. Ding, M. Garaga, S. Greenbaum, H.-S. Lee, O. Borodin, K. Xu and C. Wang, A 63 m Superconcentrated Aqueous Electrolyte for High-Energy Li-Ion Batteries, ACS Energy Lett., 2020, 5(3), 968–974 CrossRef CAS.
- J. Zhang, C. Cui, P.-F. Wang, Q. Li, L. Chen, F. Han, T. Jin, S. Liu, H. Choudhary and S. R. Raghavan, “Water-in-salt” polymer electrolyte for Li-ion batteries, Energy Environ. Sci., 2020, 13(9), 2878–2887 RSC.
- J. Xie, Z. Liang and Y. C. Lu, Molecular crowding electrolytes for high-voltage aqueous batteries, Nat. Mater., 2020, 19, 1006–1011 CrossRef CAS PubMed.
- L. Suo, O. Borodin, Y. Wang, X. Rong, W. Sun, X. Fan, S. Xu, M. A. Schroeder, A. V. Cresce, F. Wang, C. Yang, Y.-S. Hu, K. Xu and C. Wang, “Water-in-Salt” Electrolyte Makes Aqueous Sodium-Ion Battery Safe, Green, and Long-Lasting, Adv. Energy Mater., 2017, 7(21), 1701189 CrossRef.
- X. Bu, L. Su, Q. Dou, S. Lei and X. Yan, A low-cost “water-in-salt” electrolyte for a 2.3 V high-rate carbon-based supercapacitor, J. Mater. Chem. A, 2019, 7(13), 7541–7547 RSC.
- D. Reber, R.-S. Kühnel and C. Battaglia, Suppressing Crystallization of Water-in-Salt Electrolytes by Asymmetric Anions Enables Low-Temperature Operation of High-Voltage Aqueous Batteries, ACS Materials Letters, 2019, 1(1), 44–51 CrossRef CAS.
- J. Han, Y. Niu, S.-J. Bao, Y.-N. Yu, S.-Y. Lu and M. Xu, Nanocubic KTi 2 (PO 4) 3 electrodes for potassium-ion batteries, Chem. Commun., 2016, 52(78), 11661–11664 RSC.
- C. D. Wessells, S. V. Peddada, R. A. Huggins and Y. Cui, Nickel Hexacyanoferrate Nanoparticle Electrodes For Aqueous Sodium and Potassium Ion Batteries, Nano Lett., 2011, 11(12), 5421–5425 CrossRef CAS PubMed.
- K. Lei, F. Li, C. Mu, J. Wang, Q. Zhao, C. Chen and J. Chen, High K-storage performance based on the synergy of dipotassium terephthalate and ether-based electrolytes, Energy Environ. Sci., 2017, 10(2), 552–557 RSC.
- R. Trocoli and F. La Mantia, An aqueous zinc-ion battery based on copper hexacyanoferrate, ChemSusChem, 2015, 8(3), 481–485 CrossRef CAS PubMed.
- Y.-P. Deng, R. Liang, G. Jiang, Y. Jiang, A. Yu and Z. Chen, The Current State of Aqueous Zn-Based Rechargeable Batteries, ACS Energy Lett., 2020, 5(5), 1665–1675 CrossRef CAS.
- J. Han, H. Zhang, A. Varzi and S. Passerini, Fluorine-free water-in-salt electrolyte for green and low-cost aqueous sodium-ion batteries, ChemSusChem, 2018, 11(21), 3704–3707 CrossRef CAS PubMed.
- X. He, B. Yan, X. Zhang, Z. Liu, D. Bresser, J. Wang, R. Wang, X. Cao, Y. Su and H. Jia, Fluorine-free water-in-ionomer electrolytes for sustainable lithium-ion batteries, Nat. Commun., 2018, 9(1), 1–8 CrossRef CAS PubMed.
- R. G. Johnson and R. K. Ingham, The degradation of carboxylic acid salts by means of halogen-The Hunsdiecker reaction, Chem. Rev., 1956, 56(2), 219–269 CrossRef CAS.
- Z. Tian, W. Deng, X. Wang, C. Liu, C. Li, J. Chen, M. Xue, R. Li and F. Pan, Superconcentrated aqueous electrolyte to enhance energy density for advanced supercapacitors, Funct. Mater. Lett., 2017, 10(06), 1750081 CrossRef CAS.
- X. Wu, C. Wu, C. Wei, L. Hu, J. Qian, Y. Cao, X. Ai, J. Wang and H. Yang, Highly crystallized Na2CoFe (CN) 6 with suppressed lattice defects as superior cathode material for sodium-ion batteries, ACS Appl. Mater. Interfaces, 2016, 8(8), 5393–5399 CrossRef CAS PubMed.
- L. Wang, Y. Lu, J. Liu, M. Xu, J. Cheng, D. Zhang and J. B. Goodenough, A superior low-cost cathode for a Na-ion battery, Angew. Chem., 2013, 125(7), 2018–2021 CrossRef.
- M. Pasta, R. Y. Wang, R. Ruffo, R. Qiao, H.-W. Lee, B. Shyam, M. Guo, Y. Wang, L. A. Wray and W. Yang, Manganese–cobalt hexacyanoferrate cathodes for sodium-ion batteries, J. Mater. Chem. A, 2016, 4(11), 4211–4223 RSC.
- T. Shao, C. Li, C. Liu, W. Deng, W. Wang, M. Xue and R. Li, Electrolyte regulation enhances the stability of Prussian blue analogues in aqueous Na-ion storage, J. Mater. Chem. A, 2019, 7(4), 1749–1755 RSC.
- J. Han, A. Mariani, H. Zhang, M. Zarrabeitia, X. Gao, D. V. Carvalho, A. Varzi and S. Passerini, Gelified acetate-based water-in-salt electrolyte stabilizing hexacyanoferrate cathode for aqueous potassium-ion batteries, Energy Storage Mater., 2020, 30, 196–205 CrossRef.
- J. Han, M. Zarrabeitia, A. Mariani, Z. Jusys, M. Hekmatfar, H. Zhang, D. Geiger, U. Kaiser, R. J. Behm and A. Varzi, Halide-free water-in-salt electrolytes for stable aqueous sodium-ion batteries, Nano Energy, 2020, 77, 105176 CrossRef CAS.
- J. J. Holoubek, H. Jiang, D. Leonard, Y. Qi, G. C. Bustamante and X. Ji, Amorphous titanic acid electrode: its electrochemical storage of ammonium in a new water-in-salt electrolyte, Chem. Commun., 2018, 54(70), 9805–9808 RSC.
- P. R. Kumar, Y. H. Jung, C. H. Lim and D. K. Kim, Na3V2O2x (PO4)2F3−2x: a stable and high-voltage cathode material for aqueous sodium-ion batteries with high energy density, J. Mater. Chem. A, 2015, 3(12), 6271–6275 RSC.
- H. Ao, W. Zhu, M. Liu, W. Zhang, Z. Hou, X. Wu, Y. Zhu and Y. Qian, High-Voltage and Super-Stable Aqueous Sodium–Zinc Hybrid Ion Batteries Enabled by Double Solvation Structures in Concentrated Electrolyte, Small Methods, 2021, 2100418 CrossRef CAS PubMed.
- X. Ji, A Perspective of ZnCl2 Electrolytes: the Physical and Electrochemical Properties, eScience, 2021, 1(2), 99–107 CrossRef.
- C. Zhang, J. Holoubek, X. Wu, A. Daniyar, L. Zhu, C. Chen, D. P. Leonard, I. A. Rodríguez-Pérez, J.-X. Jiang and C. Fang, A ZnCl 2 water-in-salt electrolyte for a reversible Zn metal anode, Chem. Commun., 2018, 54(100), 14097–14099 RSC.
- X. Wu, Y. Xu, C. Zhang, D. P. Leonard, A. Markir, J. Lu and X. Ji, Reverse Dual-Ion Battery via a ZnCl2 Water-in-Salt Electrolyte, J. Am. Chem. Soc., 2019, 141(15), 6338–6344 CrossRef CAS PubMed.
- C. Y. Chen, K. Matsumoto, K. Kubota, R. Hagiwara and Q. Xu, A Room-Temperature Molten Hydrate Electrolyte for Rechargeable Zinc–Air Batteries, Adv. Energy Mater., 2019, 9(22), 1900196 CrossRef.
-
M. Frisch; G. Trucks; H. Schlegel; G. Scuseria; M. Robb; J. Cheeseman; G. Scalmani; V. Barone; G. Petersson and H. Nakatsuji, Gaussian 16, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef CAS PubMed.
- O. Borodin, L. Suo, M. Gobet, X. Ren, F. Wang, A. Faraone, J. Peng, M. Olguin, M. Schroeder and M. S. Ding, Liquid structure with nano-heterogeneity promotes cationic transport in concentrated electrolytes, ACS Nano, 2017, 11(10), 10462–10471 CrossRef CAS PubMed.
- R. J. Wilcox, B. P. Losey, J. C. Folmer, J. D. Martin, M. Zeller and R. Sommer, Crystalline and liquid structure of zinc chloride trihydrate: a unique ionic liquid, Inorg. Chem., 2015, 54(3), 1109–1119 CrossRef CAS PubMed.
- R. Sakamoto, M. Yamashita, K. Nakamoto, Y. Zhou, N. Yoshimoto, K. Fujii, T. Yamaguchi, A. Kitajou and S. Okada, Local structure of a highly concentrated NaClO 4 aqueous solution-type electrolyte for sodium ion batteries, Phys. Chem. Chem. Phys., 2020, 22(45), 26452–26458 RSC.
- J. Park, J. Lee and W. Kim, Water-in-Salt Electrolyte Enables Ultrafast Supercapacitors for AC Line Filtering, ACS Energy Lett., 2021, 6(2), 769–777 CrossRef CAS.
- S. Kim, H. Kim, J.-H. Choi and M. Cho, Ion aggregation in high salt solutions: Ion network versus ion cluster, J. Chem. Phys., 2014, 141(12), 124510 CrossRef PubMed.
- J.-H. Choi, H. R. Choi, J. Jeon and M. Cho, Ion aggregation in high salt solutions. VII. The effect of cations on the structures of ion aggregates and water hydrogen-bonding network, J. Chem. Phys., 2017, 147(15), 154107 CrossRef PubMed.
- K. Xu, Nonaqueous liquid electrolytes for lithium-based rechargeable batteries, Chem. Rev., 2004, 104(10), 4303–4418 CrossRef CAS PubMed.
- J. O. M. Bockris, A. K. Reddy and M. Gamboa-Aldeco, Electrodics, Modern Electrochemistry 2A: Fundamentals of Electrodics, 2000, 1035–1400 Search PubMed.
- N. Gavish and K. Promislow, Dependence of the dielectric constant of electrolyte solutions on ionic concentration: A microfield approach, Phys. Rev. E, 2016, 94(1), 012611 CrossRef PubMed.
- M. Uematsu and E. Frank, Static dielectric constant of water and steam, J. Phys. Chem. Ref. Data, 1980, 9(4), 1291–1306 CrossRef CAS.
- L. Korson, W. Drost-Hansen and F. J. Millero, Viscosity of water at various temperatures, J. Phys. Chem. Lett., 1969, 73(1), 34–39 CAS.
- C.-H. Yim, J. Tam, H. Soboleski and Y. Abu-Lebdeh, On the Correlation between Free Volume, Phase Diagram and Ionic Conductivity of Aqueous and Non-Aqueous Lithium Battery Electrolyte Solutions over a Wide Concentration Range, J. Electrochem. Soc., 2017, 164(6), A1002 CrossRef CAS.
- J. Yue, J. Zhang, Y. Tong, M. Chen, L. Liu, L. Jiang, T. Lv, Y.-S. Hu, H. Li and X. Huang, Aqueous interphase formed by CO2 brings electrolytes back to salt-in-water regime, Nat. Chem., 2021, 1–9 Search PubMed.
- J. Lim, K. Park, H. Lee, J. Kim, K. Kwak and M. Cho, Nanometric water channels in water-in-salt lithium ion battery electrolyte, J. Am. Chem. Soc., 2018, 140(46), 15661–15667 CrossRef CAS PubMed.
- K. Miyazaki, N. Takenaka, E. Watanabe, S. Iizuka, Y. Yamada, Y. Tateyama and A. Yamada, First-principles study on the peculiar water environment in a hydrate-melt electrolyte, J. Phys. Chem. Lett., 2019, 10(20), 6301–6305 CrossRef CAS PubMed.
- M. McLin and C. Angell, Contrasting conductance/viscosity relations in liquid states of vitreous and polymer solid electrolytes, J. Phys. Chem. Lett., 1988, 92(8), 2083–2086 CAS.
- C. A. Angell, N. Byrne and J.-P. Belieres, Parallel developments in aprotic and protic ionic liquids: physical chemistry and applications, Acc. Chem. Res., 2007, 40(11), 1228–1236 CrossRef CAS PubMed.
- D. R. MacFarlane, M. Forsyth, E. I. Izgorodina, A. P. Abbott, G. Annat and K. Fraser, On the concept of ionicity in ionic liquids, Phys. Chem. Chem. Phys., 2009, 11(25), 4962–4967 RSC.
- C. Schreiner, S. Zugmann, R. Hartl and H. J. Gores, Fractional Walden rule for ionic liquids: examples from recent measurements and a critique of the so-called ideal KCl line for the Walden plot, J. Chem. Eng. Data, 2010, 55(5), 1784–1788 CrossRef CAS.
- K. R. Harris, On the use of the Angell–Walden equation to determine the “Ionicity” of molten salts and ionic liquids, J. Phys. Chem. B, 2019, 123(32), 7014–7023 CrossRef CAS PubMed.
- A. Mariani, M. Bonomo, X. Gao, B. Centrella, A. Nucara, R. Buscaino, A. Barge, N. Barbero, L. Gontrani and S. Passerini, The unseen evidence of Reduced Ionicity: The elephant in (the) room temperature ionic liquids, J. Mol. Liq., 2021, 324, 115069 CrossRef CAS.
- P. Tan, J. Yue, Y. Yu, B. Liu, T. Liu, L. Zheng, L. He, X. Zhang, L. Suo and L. Hong, Solid-like nano-anion cluster constructs a free lithium-ion-conducting superfluid framework in a water-in-salt electrolyte, J. Phys. Chem. C, 2021, 125(22), 11838–11847 CrossRef CAS.
- M. Brinkkötter, A. Mariani, S. Jeong, S. Passerini and M. Schönhoff, Ionic Liquid in Li Salt Electrolyte: Modifying the Li+ Transport Mechanism by Coordination to an Asymmetric Anion, Adv. Energy Sustainability Res., 2021, 2(2), 2000078 CrossRef.
- Y. Yamada, J. Wang, S. Ko, E. Watanabe and A. Yamada, Advances and issues in developing salt-concentrated battery electrolytes, Nat. Energy, 2019, 4(4), 269–280 CrossRef CAS.
- G. Horwitz, C. R. Rodríguez, P. Y. Steinberg, G. Burton and H. R. Corti, Mobility-viscosity decoupling and cation transport in water-in-salt lithium electrolytes, Electrochim. Acta, 2020, 359, 136915 CrossRef CAS.
- M. P. Longinotti and H. R. Corti, Fractional Walden rule for electrolytes in supercooled disaccharide aqueous solutions, J. Phys. Chem. B, 2009, 113(16), 5500–5507 CrossRef CAS PubMed.
- K. R. Harris, Scaling the transport properties of molecular and ionic liquids, J. Mol. Liq., 2016, 222, 520–534 CrossRef CAS.
- J. H. Payne, The solubility of lithium and sodium fluorides, J. Am. Chem. Soc., 1937, 59(5), 947 CrossRef CAS.
- M. Becker, R.-S. Kühnel and C. Battaglia, Water-in-salt electrolytes for aqueous lithium-ion batteries with liquidus temperatures below− 10 °C, Chem. Commun., 2019, 55(80), 12032–12035 RSC.
- J. Wang, H. Qiu, Z. Zhao, Y. Zhang, J. Zhao, Y. Ma, J. Li, M. Xing, G. Li and G. Cui, Anti-corrosive Hybrid Electrolytes for Rechargeable Aqueous Zinc Batteries, Chem. Res. Chin. Univ., 2021, 37(2), 328–334 CrossRef CAS.
- J. Han, M. Zarrabeitia, A. Mariani, Z. Jusys, M. Hekmatfar, H. Zhang, D. Geiger, U. Kaiser, R. J. Behm, A. Varzi and S. Passerini, Halide-free water-in-salt electrolytes for stable aqueous sodium-ion batteries, Nano Energy, 2020, 77, 105176 CrossRef CAS.
- N. Demirkıran, A study on dissolution of ulexite in ammonium acetate solutions, Chem. Eng. J., 2008, 141(1–3), 180–186 CrossRef.
- K. Martinelle and L. Häggström, On the dissociation constant of ammonium: effects of using an incorrect pK a in calculations of the ammonia concentration in animal cell cultures, Biotechnol. Tech., 1997, 11(8), 549–551 CrossRef CAS.
- J. Yan, J. Wang, H. Liu, Z. Bakenov, D. Gosselink and P. Chen, Rechargeable hybrid aqueous batteries, J. Power Sources, 2012, 216, 222–226 CrossRef CAS.
- R. Williams and C. Lyman, A neutral buffered standard for hydrogen ion work and accurate titrations which can be prepared in one minute, J. Am. Chem. Soc., 1932, 54(5), 1911–1912 CrossRef CAS.
- K. Chayambuka, G. Mulder, D. L. Danilov and P. H. Notten, From Li-Ion Batteries toward Na-Ion Chemistries: Challenges and Opportunities, Adv. Energy Mater., 2020, 10(38), 2001310 CrossRef CAS.
- S. K. Heiskanen, J. Kim and B. L. Lucht, Generation and evolution of the solid electrolyte interphase of lithium-ion batteries, Joule, 2019, 3(10), 2322–2333 CrossRef CAS.
- M. Winter, The solid electrolyte interphase–the most important and the least understood solid electrolyte in rechargeable Li batteries, Z. Phys. Chem., 2009, 223(10–11), 1395–1406 CrossRef CAS.
- L. Wang, A. Menakath, F. Han, Y. Wang, P. Y. Zavalij, K. J. Gaskell, O. Borodin, D. Iuga, S. P. Brown and C. Wang, Identifying the components of the solid–electrolyte interphase in Li-ion batteries, Nat. Chem., 2019, 11(9), 789–796 CrossRef CAS PubMed.
- K. Xu, Electrolytes and interphases in Li-ion batteries and beyond, Chem. Rev., 2014, 114(23), 11503–11618 CrossRef CAS PubMed.
- E. Peled, The electrochemical behavior of alkali and alkaline earth metals in nonaqueous battery systems—the solid electrolyte interphase model, J. Electrochem. Soc., 1979, 126(12), 2047 CrossRef CAS.
- P. Verma, P. Maire and P. Novák, A review of the features and analyses of the solid electrolyte interphase in Li-ion batteries, Electrochim. Acta, 2010, 55(22), 6332–6341 CrossRef CAS.
- J. Besenhard, M. Winter, J. Yang and W. Biberacher, Filming mechanism of lithium-carbon anodes in organic and inorganic electrolytes, J. Power Sources, 1995, 54(2), 228–231 CrossRef CAS.
- M. Levi and D. Aurbach, Simultaneous measurements and modeling of the electrochemical impedance and the cyclic voltammetric characteristics of graphite electrodes doped with lithium, J. Phys. Chem. B, 1997, 101(23), 4630–4640 CrossRef CAS.
- L. Suo, D. Oh, Y. Lin, Z. Zhuo, O. Borodin, T. Gao, F. Wang, A. Kushima, Z. Wang, H. C. Kim, Y. Qi, W. Yang, F. Pan, J. Li, K. Xu and C. Wang, How Solid-Electrolyte Interphase Forms in Aqueous Electrolytes, J. Am. Chem. Soc., 2017, 139(51), 18670–18680 CrossRef CAS PubMed.
- S. Lin, H. Hua, J. Li, P. Zhang and J. Zhao, Lithium carboxylate: Effectively suppressing hydrogen evolution by self-introducing CO2 in aqueous electrolyte, J. Power Sources, 2020, 461, 228136 CrossRef CAS.
- M. H. Lee, S. J. Kim, D. Chang, J. Kim, S. Moon, K. Oh, K.-Y. Park, W. M. Seong, H. Park, G. Kwon, B. Lee and K. Kang, Toward a low-cost high-voltage sodium aqueous rechargeable battery, Materials Today, 2019, 29, 26–36 CrossRef CAS.
- P. Adelhelm, P. Hartmann, C. L. Bender, M. Busche, C. Eufinger and J. Janek, From lithium to sodium: cell chemistry of room temperature sodium–air and sodium–sulfur batteries, Beilstein J. Nanotechnol., 2015, 6(1), 1016–1055 CrossRef CAS PubMed.
- X. Hu, J. Sun, Z. Li, Q. Zhao, C. Chen and J. Chen, Rechargeable room-temperature Na–CO2 batteries, Angew. Chem., Int. Ed., 2016, 55(22), 6482–6486 CrossRef CAS PubMed.
- J. Kim, H. Park, B. Lee, W. M. Seong, H.-D. Lim, Y. Bae, H. Kim, W. K. Kim, K. H. Ryu and K. Kang, Dissolution and ionization of sodium superoxide in sodium–oxygen batteries, Nat. Commun., 2016, 7(1), 1–9 Search PubMed.
- X. Hu, Z. Li, Y. Zhao, J. Sun, Q. Zhao, J. Wang, Z. Tao and J. Chen, Quasi–solid state rechargeable Na-CO2 batteries with reduced graphene oxide Na anodes, Sci. Adv., 2017, 3(2), e1602396 CrossRef PubMed.
- H. Yadegari, Y. Li, M. N. Banis, X. Li, B. Wang, Q. Sun, R. Li, T.-K. Sham, X. Cui and X. Sun, On rechargeability and reaction kinetics of sodium–air batteries, Energy Environ. Sci., 2014, 7(11), 3747–3757 RSC.
- C. Liu, M. Carboni, W. R. Brant, R. Pan, J. Hedman, J. Zhu, T. R. Gustafsson and R. Younesi, On the Stability of NaO2 in Na–O2 Batteries, ACS Appl. Mater. Interfaces, 2018, 10(16), 13534–13541 CrossRef CAS PubMed.
- L. Droguet, A. Grimaud, O. Fontaine and J. M. Tarascon, Water-in-salt electrolyte (WiSE) for aqueous batteries: a long way to practicality, Advanced Energy Materials, 2020, 10(43), 2002440 CrossRef CAS.
Footnote |
| † Current address: ELETTRA Synchrotron of Trieste, 34012 Basovizza, Trieste, Italy. |
|
| This journal is © The Royal Society of Chemistry 2023 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
Stefano
Passerini
*ab,
Stefano
Passerini