
Catalytic hydroconversion processes for upcycling plastic waste to fuels and chemicals
Junde
Wei
a,
Jieyi
Liu
a,
Weihao
Zeng
a,
Zichen
Dong
a,
Jingkuo
Song
a,
Sibao
Liu
 *a and
Guozhu
Liu
*abc
*a and
Guozhu
Liu
*abc
aKey Laboratory for Green Chemical Technology of Ministry of Education, School of Chemical Engineering and Technology, Tianjin, 300072, China. E-mail: liusibao@tju.edu.cn; gliu@tju.edu.cn
bHaihe Lab of Sustainable Chemical Transformations, Tianjin, 300072, PR China
cZhejiang Institute of Tianjin University, Ningbo, Zhejiang 315201, China
First published on 15th December 2022
Abstract
Plastic waste represents an environmental threat and a huge economic loss. Catalytic hydroconversion of plastic wastes can enable valorization of plastic wastes to diverse value-added products at high selectivity under mild reaction conditions, attracting significant attention. In this review, we focus on the recent advances in the catalytic hydroconversion processes for upcycling plastics wastes to fuels and valuable chemicals. The hydrocracking and hydrogenolysis of polyolefins as well as the hydrogenolysis and hydrodeoxygenation of heteroatom-containing plastics are summarized and exemplified. Besides, hydroconversion of plastic wastes by using in situ generated hydrogen is also described. We emphasize the recent progress in catalyst design and its performance. Further, we systematically discuss the functions of typical catalysts and the reaction mechanisms to gain insight into the hydroconversion of plastic wastes. In addition, the effect of some key factors, including macro- and microstructure of plastics, additives, and reaction conditions, on the catalytic performance is demonstrated as well. Given the existing contributions, a perspective is provided to address the challenges and opportunities in the field and to evaluate some potential solutions and avenues for future research. We hope that this review will inspire future research on the rational exploration of optimal catalysts with hydroconversion processes for chemical upcycling of plastic wastes to build a circular plastic economy.
1. Introduction
Synthetic plastics play an indispensable role in every aspect of daily life. Global plastic production reached 368 million tons in 2019 and is expected to increase to 589 million tons by 2050.1 Most of them are not biodegradable and less than 10% of the used plastics have been recycled, which cause them to accumulate in landfills and create serious environmental issues.2 Besides, plastic wastes represent a lost value of untapped carbon sources, and if utilized properly, could save 130 × 106 kJ per ton energy and up to 3.5 billion barrels of oil per year.3 Therefore, the proper end-of-life management of plastic wastes is urgent for a circular economy. Chemical recycling/upcycling of plastics is one of the promising ways.4–7 The chemical breakdown of “end-of-use” materials can be capable of reversing them to monomers/oligomers or generating new value-added products for a variety of industrial and commercial applications.8,9 There have been several techniques explored in this regard including gasification, pyrolysis, pyrolysis/hydrotreatment, hydrothermal liquefaction, solvolysis, and catalytic hydroconversion.10–17 Among them, catalytic hydroconversion can enable the conversion of plastic wastes to diverse products with high selectivity under mild reaction conditions, making it one of the most promising approaches towards a circular economy.18–20Although there are thousands of different plastics, approximately seven major categories of plastics are produced and consumed in large quantities: high-density polyethylene (HDPE), low-density polyethylene (LDPE), polypropylene (PP), polyvinyl chloride (PVC), polystyrene (PS), polyethylene terephthalate (PET), and other plastics (e.g. polycarbonates (PC), polylactic acid, polyamides (PA) and polyurethanes (PU)). All these plastics are linked with specific chemical bonds, including C–C, C–O and C–N bonds (Fig. 1). The hydroconversion reaction involves the use of H2 molecules to cleave C–C or C–heteroatom (C–O, C–N, etc.) bonds in the reacting polymers. The hydroconversion processes include hydrocracking, hydrogenolysis and hydrodeoxygenation. With these different routes, plastics can be efficiently converted to fuels and chemicals. In the case of conversion of polyolefins, hydrocracking and hydrogenolysis are the most reported processes to produce low molecular hydrocarbons that can be used as fuels, waxes, and lubricants.18,20 These reactions are usually carried out in a batch reactor with a supported metal catalyst in contact with melt-phase plastics in the absence of a solvent under mild reaction conditions. Bifunctional catalysts with metal and acid sites are used for hydrocracking, whereas only the metal sites catalyze the hydrogenolysis reaction. In the case of conversion of heteroatom containing plastics, hydrogenolysis and hydrodeoxygenation are the appealing ways.10 For instance, hydrogenolysis of polyesters, polycarbonates, polyamides and polyurethane can provide diacids, diols, amino alcohols and diamine monomers as well as phenols as products, while hydrodeoxygenation of polyesters, polycarbonates and polyethers can yield cyclic hydrocarbons and arenes. To achieve high yields of target products in high efficiency, the catalysts play an important role in realizing these processes. In recent years, there has been significant academic interest in exploring catalytic valorization of these plastic wastes through catalyst innovation.8,12,19,20
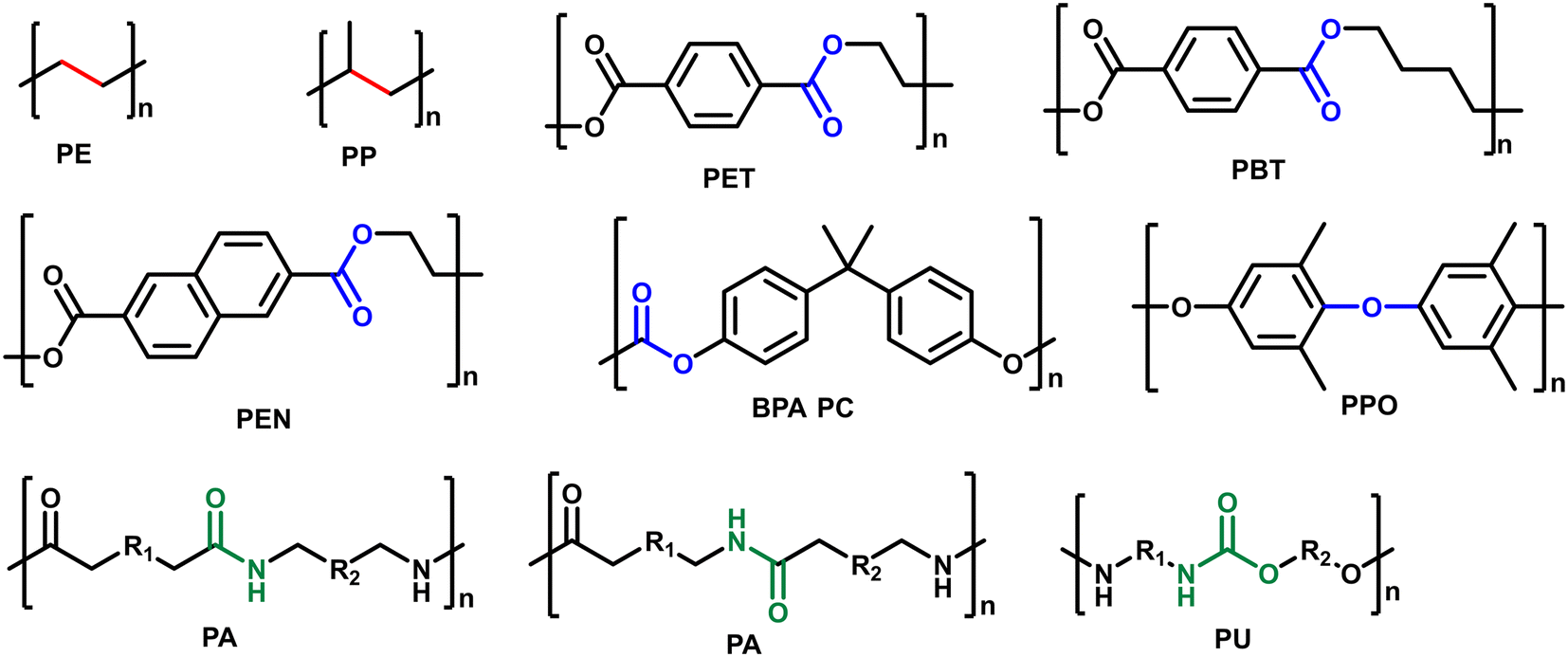 | ||
| Fig. 1 Different types of plastics for hydroconversion processes. | ||
Chemical conversion of plastic wastes to valuable fuels and chemicals is essential for the development of a sustainable world and catalysis is now playing significant role in achieving this goal. In recent years, over twenty excellent reviews have been published in this field.3–24 These review papers are broad in scope and aim to provide a brief description of each conversion strategy, rather than provide a comprehensive and systematic understanding of each method, including the hydroconversion process. Most of these published reviews focused on different strategic approaches for conversion of plastics including thermal pyrolysis, catalytic pyrolysis, hydrothermal liquefaction, hydroconversion, oxidation, photocatalysis, eletrocatalysis, solvolysis, biological catalysis, microwave- and plasma-assisted catalysis.3–24 In comparison, this review is a more focused review of only hydroconversion over a broad overview of various technologies and it will be beneficial to the catalysis and sustainability field. For example, Borkar et al. provided an excellent review on catalytic conversion of plastics and summarized several approaches, including hydroconversion, solvent treatments, and cross alkane metathesis.15 They summarized the hydroconversion of polyolefins by platinum, zirconium and ruthenium catalysts, but ignored the hydroconversion methods and non-noble metal catalysts used in polyolefin conversion as well as the impact of plastic structure and additives on the catalytic performance. However, in our review paper, we have emphasized different hydroconversion processes including hydrocracking, hydrogenolysis and hydroconversion with in situ generated hydrogen and related catalysts for the conversion of polyolefins. Besides, non-noble metal catalysts including Ni- and Co-based catalysts have also been highlighted. Additionally, the influence of several key factors, including macro- and microstructure of plastics, additives, reaction conditions, etc. on hydroconversion has also been well discussed. Furthermore, unlike the previous reviews on hydroconversion which focused on polyesters alone, our review has focused on multiple heteroatom containing plastics including polyesters, polycarbonates, polylactic acid, polyamides and polyurethanes. Overall, the purpose of this review is to offer a comprehensive elaboration of different hydroconversion technologies and comprehensive understanding on catalytic hydroconversion processes for upcycling plastic wastes, and to provide potentially assistance for the discovery of new opportunities. In this review, we attempt to highlight the most recent and impactful progress made in the catalytic hydroconversion processes for upcycling post-consumer plastics to fuels and chemicals. The plastic types are divided into polyolefins and heteroatom-containing plastics in this review. Hydrocracking and hydrogenolysis routes for conversion of polyolefins and hydrogenolysis and hydrodeoxygenation (HDO) approaches for deconstruction of polyesters, polycarbonates, polylactic acid, polyamides and polyurethane are systematically summarized. Besides, hydroconversion processes with in situ formed hydrogen were also discussed. It should be noted that the hydrogenolysis of polyolefins with non-noble metal catalysts, conversion with in situ generated hydrogen, and the hydroconversion of polyamides and polyurethanes are rarely reviewed in published review papers. The advances in catalyst design and their performance in every hydroconversion process for the conversion of polyolefins and heteroatom-containing plastics are highlighted. In addition, the reaction mechanisms and the function of typical catalysts in each case are discussed in detail to obtain improved insight into the upcycling of various plastic wastes. The influence of several key factors, including macro- and microstructure of plastics, additives, reaction conditions, etc. on hydroconversion of plastics is well discussed. In particular, the impact of plastic structure and additives on the catalytic performance was not included in previously published review papers. We hope that this review paper will encourage more and more researchers to participate in this fantastic field in the near future and inspire the rational exploration of catalysts with hydroconversion processes for upcycling of plastic wastes to build a circular plastic economy.
2. Catalytic hydroconversion processes for deconstruction of polyolefins
Polyolefins, mainly low-density polyethylene (LDPE), high-density PE (HDPE), polypropylene (PP) and polystyrene (PS), constitute the most abundant plastic waste streams. Catalytic conversion of polyolefin plastic wastes into fuels and useful chemicals over heterogeneous catalysts is one of the promising solutions. Owing to the rigidity of the C–C bonds in those polymers' backbone, a high-temperature energy-demanding process is required. Currently methods for polyolefin chemical upcycling involve catalytic or thermal pyrolysis, thermal pyrolysis followed by catalytic hydrotreatment, cross-alkane metathesis and direct hydroprocessing.5 The most widespread approach is catalytic pyrolysis with strong acidic materials such as zeolites. However, several issues including unselective manner, high operating temperatures (400–700 °C) and fast catalyst deactivation due to coking limit their practical applications.21 Although the catalytic cross-alkane metathesis method can be operated under mild reaction conditions (<200 °C), it is not efficient, which requires a second light alkane/alkene as a feedstock as well as a long reaction time (>72 h) to convert the plastics completely.25–27 Catalytic hydroprocessing of polyolefins, including hydrocracking over metal-acid bifunctional catalysts and hydrogenolysis over metal catalysts, is an attractive way to convert plastics into gaseous products, transportation fuels, waxes and lubricants efficiently.18,20 In addition, the metal catalysts used in hydrocracking can stabilize the unsaturated intermediates by hydrogenation, which eliminates the coking on the catalyst, to improve the catalyst stability. In this section, recent advances in the hydroprocessing of polyolefins will be discussed.182.1. Hydrocracking of polyolefins
Hydrocracking of polyolefins is an attractive pathway for chemical upcycling. Bifunctional catalysts combining metal catalysts and solid acids have been widely used for this process. The metal catalysts include Pt-,28–34 Pd-, Ni-35–37 and Co-36,37based catalysts, among which Pt-based catalysts are the most efficient ones. The solid acids used in the hydrocracking process contain zeolites,31–37 silica–alumina materials,35,37 acidic mixed oxides (WO3/ZrO2)29–31 and so on. The generally accepted mechanism for alkane hydrocracking relies on a synergism between acid sites and metal sites.38 The long-chain alkane firstly undergoes C–H activation on metal sites to form olefin intermediates by dehydrogenation. Then, these olefin intermediates diffuse to the Brønsted acid sites to generate carbocation active intermediates, followed by skeletal isomerization and β-scission (C–C cracking). Finally, the cracked and isomeric intermediates diffuse to the metal sites for hydrogenation to form short-chain alkanes. The metal–acid balance (MAB), intimacy between sites and site accessibility play a critical role in determining the reaction pathway and thus product distribution.In 2021, the Vlachos group demonstrated the Pt/WO3/ZrO2 catalyst for the hydrocracking of LDPE to valuable branched fuel- and lubricant-range alkanes in high yields at a low temperature of 250 °C and 3 MPa H2 for 1–24 h.29 They focused on the study of the mechanism for the hydrocracking of LDPE by using metal–acid balance (MAB) as a catalyst descriptor (Fig. 2). The systematic experiments and analysis indicated that the high MAB catalysts give a pseudo-equilibrium between the olefin and the associated paraffin over the metal sites while the acid-catalyzed reactions, including skeletal isomerization and subsequently β-scission, become slow, leading to heavy hydrocarbons and enhanced branching in the residual polymer. In contrast, over the low MAB catalysts, the rate-determining steps are the dehydrogenation and hydrogenation steps. The relatively fast reaction rates of isomerization and cracking over acid sites result in dominant lighter alkane products. These are quite similar to those in the hydrocracking of small alkanes. On the other hand, unlike the small alkanes' hydrocracking, the overall fraction of branched isomers is independent of the MAB. In addition, the composition of liquid isomers is far from thermodynamic equilibrium, owing to the competitive adsorption of the polymer over the liquid products and stereochemical hindrance of methines. Based on the above results, they proposed a new adhesive isomerization mechanism between the metal and Brønsted acid sites in parallel with slow polymer chain cracking. Very recently, they found that the additives (antioxidants) in the plastic significantly lower the hydrocracking activity of Pt/WO3/ZrO2 and thus change the product distribution.30 Both antioxidant concentration and chemical structure have complicated impact on the hydrocracking process by changing the effective MAB through interactions of phenols and/or other functional groups (e.g., acids, esters) with catalyst active sites. To further improve the activity of Pt/WO3/ZrO2 for the hydrocracking of polyolefins and even real plastic wastes, additional solid acids including zeolites, mesoporous materials, acidic mixture oxides as co-catalysts were introduced for conversion of LDPE under mild reaction conditions of 250 °C and 3 MPa H2.31 The same group found that the Pt/WO3/ZrO2 and HY zeolite mixture catalyst can almost catalyze hydrocracking of LDPE to liquid products with high yield (∼85%) within 2 h. Engineering solid acid types, acidity and porosity of the HY zeolite as well as reaction conditions is crucial for tuning the selectivity toward different alkane products. The combination between Pt metallic sites and zeolite acid sites is a prerequisite for an active catalyst and site separation is crucial for achieving high selectivity to valuable liquid products. More interestingly, the tandem catalyst mixture can be applicable to transform a variety of common plastic wastes, including LDPE, HDPE, PP, PS, everyday PE bags and bottles, and composite plastics to target fuels in high yield.
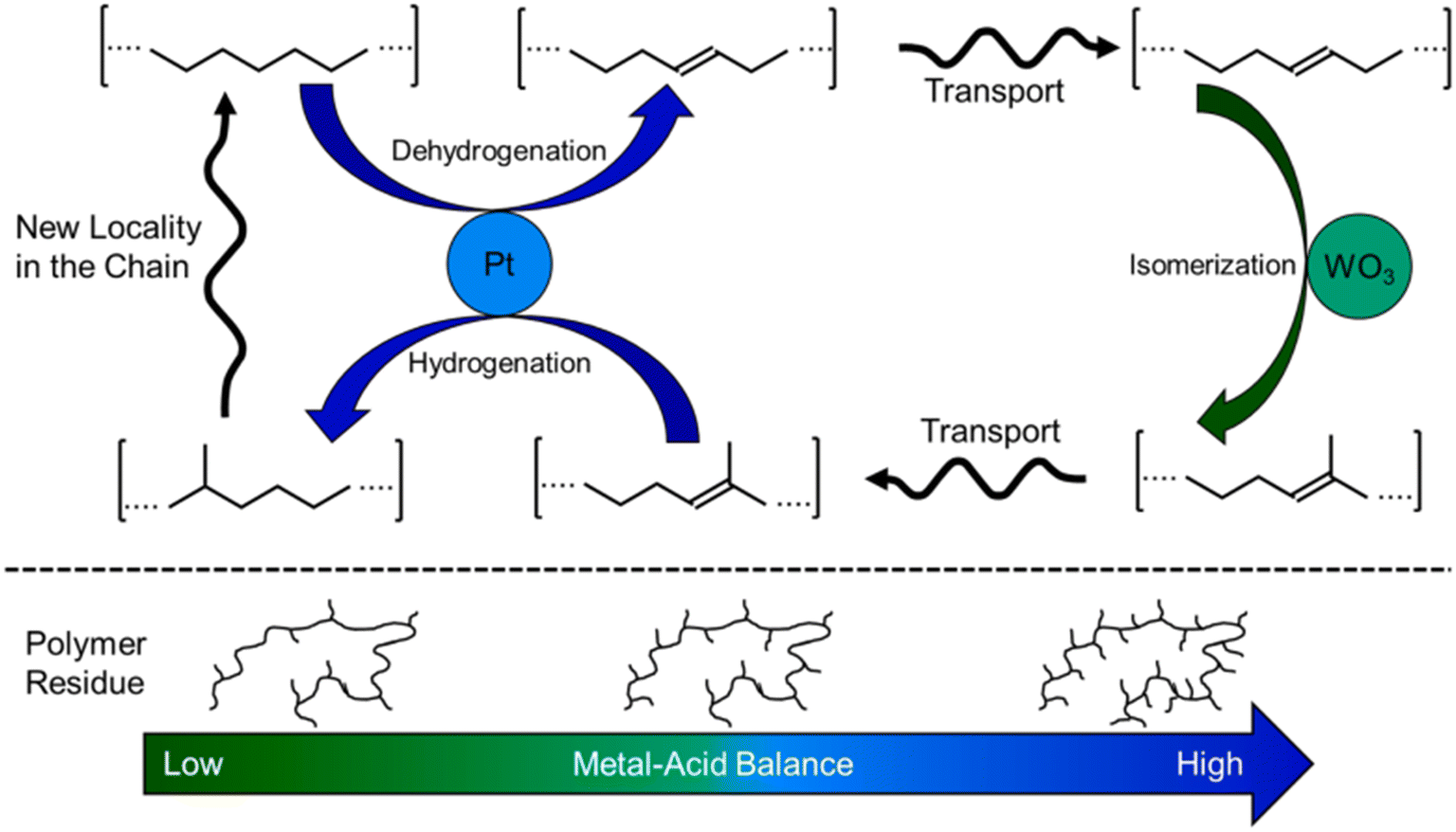 | ||
| Fig. 2 Proposed isomerization cycle in the hydrocracking of LDPE and the effect of the metal–acid balance on the structure of the residual polyethylene chains. Reproduced from ref. 29 with permission from Elsevier. | ||
Recently, Yury and his co-workers demonstrated Ru supported on FAU or BEA zeolites as active catalysts for the hydrocracking of polyethylene and polypropylene waste to liquid products with a yield up to 67%.39 Based on the ex situ and operando studies with model small hydrocarbons, they claimed that the dispersion of Ru metallic particles and the acidity properties rather than Ru reducibility play key roles in the conversion of polyolefins by hydrocracking.
In addition to Pt/zeolite and Ru/zeolite catalysts, cheap non-noble metal catalysts including Ni-, Co-, Ni–Mo, and Co–Mo supported on zeolites were also reported for the hydrocracking of POs.35–37 Compared to Pt/zeolite catalysts, non-noble metal catalysts showed lower activity, which need high reaction temperature and/or long reaction time to completely degrade polymers. Dyson and his coworkers reported Ni/HZSM-5 and Co/HZSM-5 catalysts for the hydrocracking of PE at 375 °C, 4.5 MPa H2 in 16 h.37 Ni/HZSM-5 is more active than Co/HZSM-5. It gives 87.1 ± 7.5% conversion of PE with 37.5% yield of C5–C20 alkanes. To overcome mass- and heat-transfer limitations in the solvent-free reactions, n-C16 alkane is used as a solvent. Consequently, Ni/HZSM-5 results in a near-quantitative conversion of 99.3 ± 0.6% along with 66.0% yield of C5–C20 liquid alkanes at 375 °C in a short reaction time (6 h). In addition, Ni sulfide supported on a hybrid support, a mixture of HZSM-5 and silica-alumina, possesses higher activity and could convert both HDPE and real plastic waste to gasoline-range products with high yield up to 64.8%, at 375 °C, H2 and 1 h.35 The metal sulfide-acid balance on the Ni/HSiAl catalyst could be the reason for its high hydrocracking and hydroisomerization activity. In addition, the catalyst is resistant to poisoning by nitrogen- and sulfur-containing compounds and can be regenerated simply by recalcination and re-sulfidation.
2.2. Hydrogenolysis of polyolefins with external hydrogen
In recent years, polyolefin hydrogenolysis over metal catalysts has received extensive attention. Pt- and Ru-based catalysts are predominantly used in the hydrogenolysis of polyolefins.40–47 The supports used are usually non-acidic materials, such as carbon materials,40,48–51 CeO2,52–54 TiO2,55,56 SrTiO3,47 ZrO2 (ref. 53) and so on. Unlike the hydrocracking reaction, the metal surface is the only active site for both C–H bond activation and C–C bond breaking in polyolefins, whereas no isomerization reaction occurs due to the lack of acidic sites and linear alkanes are formed as the main products from PE hydrogenolysis. In general, the reaction mechanism for polyolefin hydrogenolysis involves the following steps: (1) dissociative adsorption of the C–H bond in the polyolefin at the metal sites, (2) C–C bond breaking to form two alkyl adspecies, (3) hydrogenation of the alkyl adspecies and desorption.54,57,58 Both terminal C–C bond and internal C–C bond scissions occur on the metal sites and the regioselective internal C–C bond scission is the way to achieve high yields of valuable alkanes (>C5).52,57 In general, Ru-based catalysts are more active than Pt-based catalysts, which is due to the stronger metal–carbon bond strength. However, Ru-based catalysts produce high yields of low-value gases, predominantly methane. Therefore, achieving a balanced coverage between chemisorbed hydrogen (*H) and hydrocarbon species (*CxHy) is the route to obtain catalysts with high activity and selectivity towards valuable alkanes.Celik et al. showed that highly dispersed and uniformly distributed Pt nanoparticles (NPs) supported on SrTiO3 perovskite nanocuboids (Pt/SrTiO3) are very effective for the hydrogenolysis of PE and even a single-use plastic bag to high-quality lubricants and waxes with narrow distribution.47 The catalysts are prepared by the atomic layer deposition (ALD) method. The strong nanoparticle–support interaction, which is derived from cube-on-cube epitaxy for Pt NPs on the (100) facets of SrTiO3, ensures its high sintering resistance under the solvent-free, harsh reaction conditions (Fig. 3). The particle size, loading and edge to facet ratio can be controlled by varying the number of ALD cycles. It was found that Pt/SrTiO3 with 5 ALD cycles shows the best performance. Under the optimized reaction conditions of 300 °C, 1.2 MPa H2 and 96 h, this catalyst enables the conversion of different PEs and a single-use plastic bag to lubricants with a yield up to 99%. In addition, 99% yield of wax can be produced from HDPE by using the Pt/SrTiO3 catalyst, which is prepared by the strong electrostatic adsorption (SEA) method in a large scale.46 The obtained lubricant shows comparable property of wear scar volume to synthetic polyalphaolefin (PAO) based oils and far better than that of petroleum-based group III mineral oil.45 In addition, techno-economic analysis (TEA) showed that the production cost is in the range of $1.8–5.94 per gal of lubricant by using a mixture of colored and natural HDPE, which is economically profitable compared with the market price for group III and PAO based oils within the range of $6–10 per gal.44 Moreover, the life cycle assessment (LCA) indicated that emissions for plastic lubricants are in the range of 0.48–1.2 kgCO2 kgLub−1 with a 52% reduction relative to that for petroleum lubricants, and a 74% reduction relative to that for PAO lubricants.44 These calculations demonstrated that this hydrogenolysis process is a promising plastic upcycling technology with reduced CO2 emissions.44
 | ||
| Fig. 3 STEM images of Pt/SrTiO3 catalyst (left). 13C MAS (red) and CPMAS (black) spectra of 13C-enriched PE adsorbed on the SrTiO3 support (top), and Pt/SrTiO3 catalyst (right). Reproduced from ref. 47 with permission from American Chemical Society. | ||
The outstanding catalytic performance of Pt/SrTiO3 was rationalized by 13C magic-angle-spinning (MAS) solid-state nuclear magnetic resonance (13CMAS ssNMR) spectroscopy and DFT calculations.4713C MAS ssNMR for the 13C-enriched PE on SrTiO3 and Pt/SrTiO3 showed that the PE adsorbs on the support in an anti-conformer manner, whereas the mobile-polymer conformation is dramatically enhanced when Pt nanoparticles are induced (Fig. 3). In addition, DFT calculations for the adsorption of model n-alkanes (CnH2n+2, n = 4, 6, 8) on Pt(100) and Pt(111) surfaces as well as on the TiO2 double-layer terminated SrTiO3(001) were conducted. It showed that the binding energy for the interaction of n-hexane on the Pt(111) surface (−0.92 eV) is slightly lower than that on the Pt (100) surface (−0.86 eV), and much lower than that on the TiO2 double-layer terminated SrTiO3 surface (−0.36 eV) (Fig. 2). Furthermore, the binding energy of the n-alkane shows linear correlation with respect to the number of carbon atoms, which can be extended to larger alkanes. Both of the results suggested that Pt NPs strongly interacted with the large molecular polymer and prevented it from interacting with the SrTiO3 surface. Such preferential adsorption of a large molecular polymer chain onto the active Pt NP surface could be the reason for the selective hydrogenolysis behavior of Pt/SrTiO3.
The catalytic hydrogenolysis performance of the Pt/SrTiO3 catalyst is related to various macro- and microstructures of PEs and PPs, such as molecular weight, branch length and density and tacticity.43 It was found that the initial molecular weight of PEs has a moderate impact on the yield of the final liquid products (ranging from 55% for Mn 7600 Da to 67% for Mn 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 950 Da), but does not affect the product distribution. On the other hand, the length and density of branches in PEs are the main factors that determine the yields of liquid products (ranging from 67 wt% for Mn ∼50950 Da for linear low-density polyethylene (LLDPE) with C2 branches to 97 wt% for Mn ∼38850 Da for LLDPE with C6 branches). In the case of PP hydrogenolysis, the tacticity of PP with similar molecular weight has a profound effect on the molecular weight of the final product, while it has little effect on the conversion. Hydrogenolysis of isotactic polypropylene (iPP) yields Mn ∼250 Da with a wider distribution (Đ ∼1.4) compared to the narrower Mn ∼900 Da (Đ ∼ 1.0) and Mn ~750 Da (Đ ∼ 1.0) products from atactic (aPP) and syndiotactic (sPP) polypropylene, respectively. The stereochemistry of the methyl groups determines the shape and structure of the PP polymer in the melt, which in turn affects the interaction of the hydrocarbon chains with the catalyst surface and thus the number of C–C scission.
950 Da), but does not affect the product distribution. On the other hand, the length and density of branches in PEs are the main factors that determine the yields of liquid products (ranging from 67 wt% for Mn ∼50950 Da for linear low-density polyethylene (LLDPE) with C2 branches to 97 wt% for Mn ∼38850 Da for LLDPE with C6 branches). In the case of PP hydrogenolysis, the tacticity of PP with similar molecular weight has a profound effect on the molecular weight of the final product, while it has little effect on the conversion. Hydrogenolysis of isotactic polypropylene (iPP) yields Mn ∼250 Da with a wider distribution (Đ ∼1.4) compared to the narrower Mn ∼900 Da (Đ ∼ 1.0) and Mn ~750 Da (Đ ∼ 1.0) products from atactic (aPP) and syndiotactic (sPP) polypropylene, respectively. The stereochemistry of the methyl groups determines the shape and structure of the PP polymer in the melt, which in turn affects the interaction of the hydrocarbon chains with the catalyst surface and thus the number of C–C scission.
Perras and co-workers designed an artificial processive catalyst, mSiO2/Pt/SiO2, with a mesoporous shell/active site/core architecture, for HDPE hydrogenolysis to mimic the enzymatic deconstruction of macromolecules.42 During the catalytic process, the polymer chain penetrates into the pore and comes into contact with the surface of Pt NPs, where the C–C bond cleavage occurs to form smaller alkane fragments (Fig. 4). 13C MAS ssNMR spectroscopy revealed that the mesoporous silica shell plays a central role in providing the linear channel for hosting the head first adsorption of a polyethylene chain in the anti(zig-zag) conformation. In addition, the numerous cumulative dispersion interactions enable the binding of a long hydrocarbon polymer to be stronger than that of a small molecule, allowing a relatively efficient release. This unique interaction between the polyethylene chain and the pore facilitates the processive Pt-catalyzed hydrogenolysis of HDPE, resulting in a narrow distribution of short-chain alkanes. The studies on the Pt NP size effect and the pore size effect in identical mSiO2/Pt/SiO2 architectures indicated that the C–C bond cleavage activity is related to the Pt sites while the pore sizes of the mesoporous shell regulate the product distributions.41,42 This catalyst is applicable for catalyzing the conversion of isotactic polypropylene to a liquid product (79% yield) from C9 to C18. More interestingly, it can also efficiently transform environmentally polluted post-consumer bubble wrap into extracted wax products (28.8% yield), showing its strong resistance to common organic and inorganic impurities.
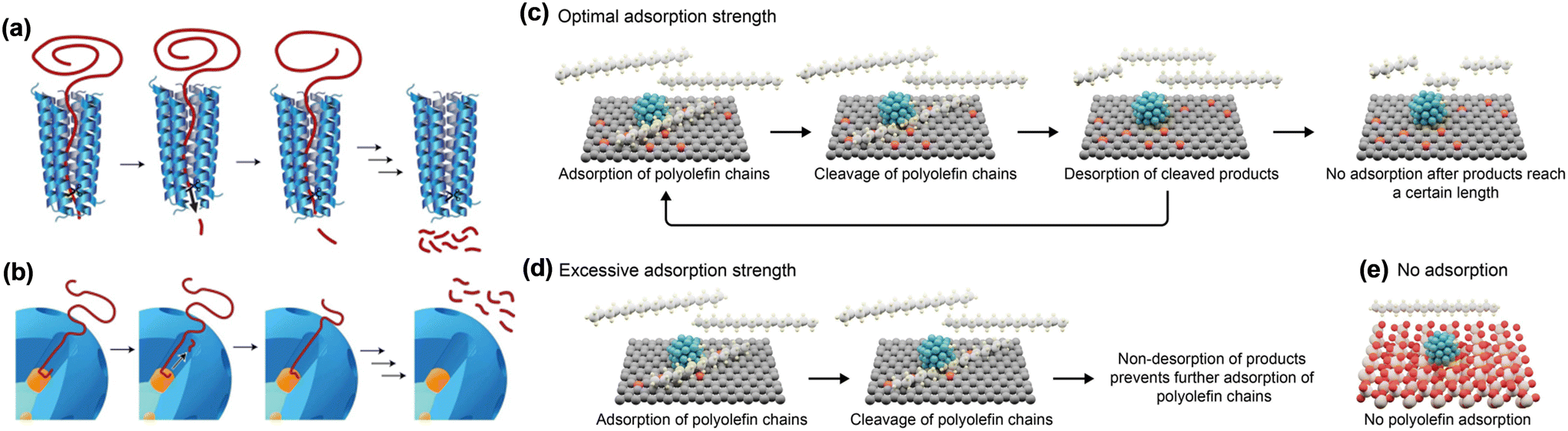 | ||
| Fig. 4 Processive deconstruction of macromolecules (a) over enzymes and (b) mSiO2/Pt/SiO2 catalyst. Adsorption/desorption processes on various carbon supports (c–e). Reproduced from ref. 42 and ref. 40 with permission from Springer Nature and Wiley, respectively. | ||
Besides the mesoporous silica shell, the carbon material is also reported to be an excellent carrier for the interaction with hydrocarbons.40 The binding strength of hydrocarbons is related to the relative oxygen concentration on the carbon surface. The higher the concentration, the weaker the binding strength. When it was loaded on Pt NP active sites for C–C bond breaking, the obtained carbon-supported platinum catalysts enable the efficient hydrogenolysis of PP into liquid hydrocarbons with high yields of motor oil (∼80%). The carbon with either excessive affinity (carbons with low oxygen concentration) or with no adsorption for hydrocarbons (silica or alumina) resulted in modest conversion into residues (Fig. 4). Pt NPs act as the active centers for the direct adsorption of C–C bonds, leading to the cleavage of PP chains. The reaction activity is correlated to the particle size and its rate increased with decreasing particle size, which is similar to that of the mSiO2/Pt/SiO2 catalyst for hydrogenolysis of HDPE. The catalyst is stable for four consecutive runs without loss of activity and selectivity to motor oils.
Yury and co-workers pioneered the application of the Ru/C catalyst for the hydrogenolysis of PEs and PP into liquid alkanes at 200–225 °C, 2–3 MPa H2 under solvent-free conditions.51 They screened several carbon supported metal nanoparticle catalysts for the hydrogenolysis of n-octadecane and the activity of the metal catalysts follows the order of Ru > Rh ≫ Ni/Pt. The balanced coverage between chemisorbed hydrogen (*H) and hydrocarbon species (*CxHy) on the Ru surface could be the reason for its superior activity to other metals. With the aid of the Ru/C catalyst, liquid n-alkanes with yield up to 45% can be obtained from PE under the reaction conditions of 200 °C, 2 MPa H2, 16 h and C5–C32 iso-alkanes with a yield of 68% can be achieved by hydrogenolysis of PP at 225 °C, 2 MPa H2, 16 h. In addition, this catalyst is also shown to be effective in converting realistic postconsumer plastic wastes including a LDPE plastic bottle, a PP centrifuge tube and a mixture of HDPE and PP to produce linear and branched liquid alkanes.50 After that, the Lin group and Szanyi group also demonstrated the feasibility of Ru/C as a highly active catalyst to convert PE and PP into liquid products.48,49 The conversion and the yield and composition of liquid products are significantly influenced by the reaction temperature, reaction time, hydrogen pressure, and solvent. As the reaction temperature and reaction time increase, the gaseous products, especially methane, become dominant. With the increase of hydrogen pressure, the conversions of PE show a volcano-shaped dependence, due to the competitive adsorption of hydrogen and hydrocarbon intermediates. In addition, the C–C bond cleavage position is also influenced by the hydrogen pressure with high hydrogen pressure facilitating internal C–C bond scissions. In the case of PE depolymerization in solvent, the solvation ability of PE and the interaction with solvent molecules are the key factors for degradation. Low solubility in polar solvents (subcritical water) gives rise to low conversion of HDPE. Among the selected non-polar hydrocarbon solvents, linear n-hexane is better for HDPE degradation than cyclic alkanes (methylcyclohexane and decalin). This can be rationalized by the low affinity of PE towards n-hexane solvent molecules, which causes the PE polymer to coil and pass through the solvent molecules to reach the catalyst surface, where it undergoes cracking reactions. As a result, 68% yield of jet-fuel range alkanes (C8–C16) can be obtained in the n-hexane solvent.
The Tomishige group firstly demonstrated that the Ru/CeO2 catalyst was active for the hydrogenolysis of various polyolefins, including LDPE, HDPE, PP and waste PEs, to produce value-added liquid fuels and waxes range alkanes (≥C5) with up to 90% yield under mild reaction conditions (200–240 °C, 2 MPa H2).54 The Ru/CeO2 catalyst with 5 wt% Ru loading shows superior performance to other CeO2 supported metal catalysts (Ru, Ir, Pt, Rh, Pd, Ni, Co, Cu), which can be ascribed to the lowest free-energy barrier of C–C bond rupture over the Ru surface. In addition, Ru/CeO2 exhibits much lower selectivity to gaseous products (C1–C4), compared with the degradation of PE over the Ru/C catalyst. This can be related to the basic support and the small Ru particle size (∼1 nm) which enable the internal C–C bond cleavage. Later, the same group discovered that the Ru/ZrO2 catalyst shows higher activity than Ru/CeO2.53 The structure–performance study showed a volcano correlation between the PE conversion and the Ru particle size (1–9 nm), and the highest PE conversion can be achieved at ∼2.5 nm Ru particle size. The selectivity towards gaseous products is low at medium to small Ru particle sizes (1–7 nm), but high at large Ru particles (>7 nm). In addition, the Tomishige group and Szanyi group both found that the intrinsic activity of Ru/CeO2 and Ru/ZrO2 in hydrogenolysis increases as the Ru particle size decreases in the high-loading range (≥0.5 wt%). In contrast, the Szanyi group disclosed the opposite trend in the low-loading range (≤0.25 wt%). Moreover, the Ru/CeO2 catalyst at low-loading (≤0.25 wt%) displays superior performance in conversion of PP and PE to high-loading (≥0.5 wt%) Ru/CeO2 catalysts.52 This abrupt changes in catalytic behavior can be attributed to the highly disordered, sub-nanometer and cationic Ru species in the low-loading Ru/CeO2 catalyst. Furthermore, this unique Ru structure with high coverage of adsorbed hydrogen favors the regioselective hydrogenolysis of internal C–C bonds, leading to low selectivity to gaseous products.
The challenge of obtaining high yields of liquid products with Ru catalysts is the suppression of methane formation. Mechanistically, the Vlachos group proposed that methane can be produced by a direct terminal C–C bond cleavage and a cascade of consecutive C–C bond scissions (Fig. 5).57,58 In the cascade cracking pathway, long-chain alkyls on the surface are initially formed through the alkane adsorption/dehydrogenation/C–C bond scission steps, and then are cleaved to produce several methane molecules in a cascade manner until they are saturated and desorb from the Ru surface. Methane is primary produced through the consecutive cracking pathway when desorption of long chain alkyls and hydrogenation are rate-determining. Thus, the hydrogen availability on Ru is crucial in controlling the hydrogenolysis selectivity to liquid products. A high intrinsic H coverage enhances liquid products. On the contrary, a low H coverage promotes a sequential cascade of C–C rupture to methane. With these in mind, they introduced a hydrogen storage species such as WOx, MoOx and VOx to modify the Ru/ZrO2 catalyst for hydrogenolysis of polyethylene. They found that the partially reduced WOx, MoOx or VOx domains can provide extra hydrogen storage by spillover and increase H coverage on Ru to promote the hydrogenation of surface alkyls, minimizing the cascade pathway for methane formation under H2-lean conditions. Furthermore, the methane suppression shows a volcano pattern with respect to the reducibility of hydrogen storage species and the catalysts with medium reducible doped oxides, such as WOx, MoOx and VOx, are the most effective. Consequently, higher yields of liquid products can be obtained over these catalysts. Furthermore, owing to the high H coverage on the Ru surface, the Ru-WZr catalyst shows much higher activity and can degrade polyethylene with a large molecule weight (Mw ∼ 76 kDa) in a short processing time (2 h).
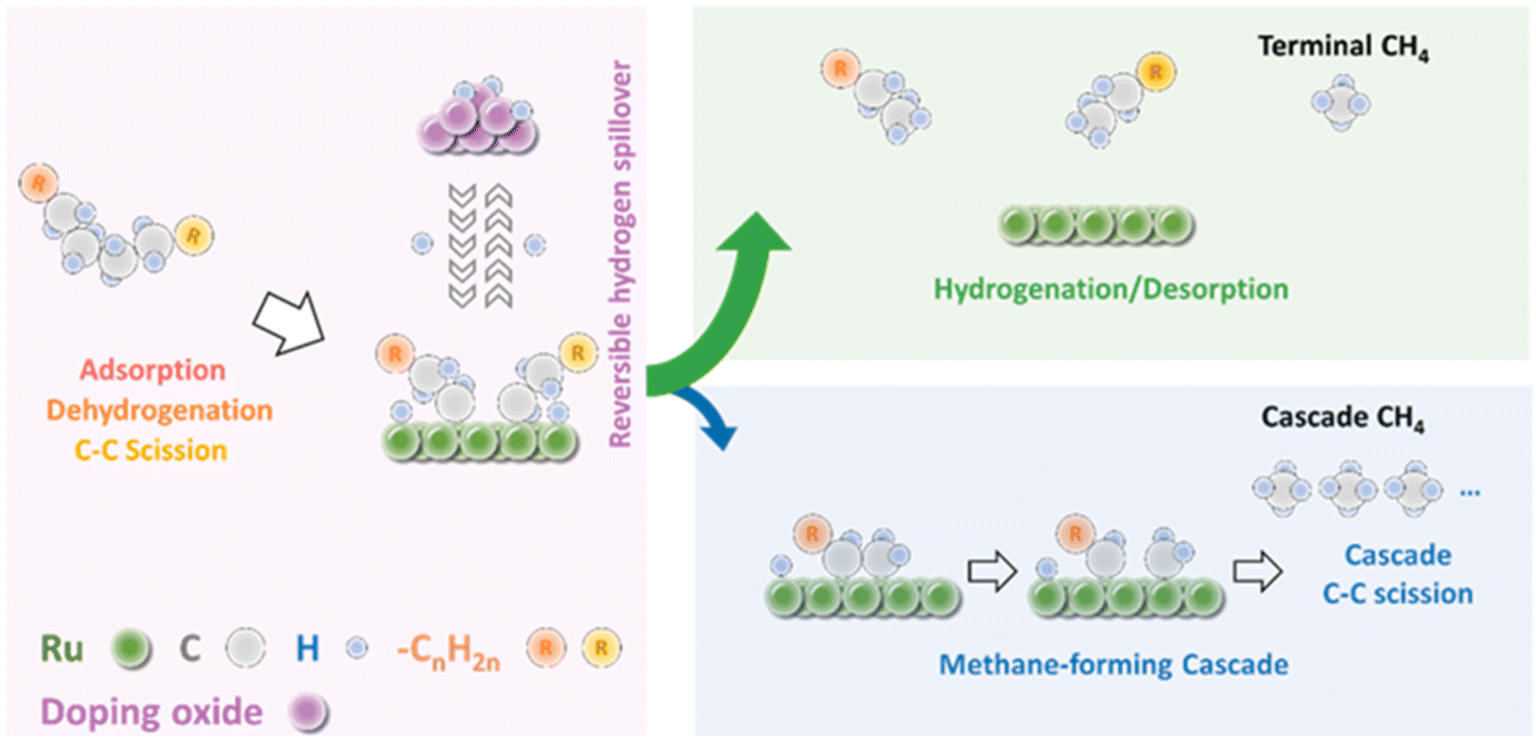 | ||
| Fig. 5 Schematic of the mechanism for LDPE hydrogenolysis. Reproduced from ref. 57 with permission from Elsevier. | ||
The products obtained over the above discussed Ru-based catalysts are mainly fuel-range hydrocarbons. Lubricants have higher value than fuels and the main components are C20–C60 iso-alkanes. However, the yields of lubricant-range hydrocarbons are quite low (<30%) by using the above discussed Ru-based catalysts. Recently, the Vlachos group demonstrated that a high yield of lubricant oils (66–80% depending on the plastic) with a narrow molecular weight distribution can be produced from PP hydrogenolysis by using an ruthenium deposited on titania (Ru/TiO2) catalyst under mild reaction conditions (250 °C, 3 MPa H2, 16 h).56 PP over Ru/TiO2 undergoes a dynamic adsorption/desorption and internal C–C bond scissions, lowering the molecular weight. Loss of polypropylene stereoregularity appears to be a prerequisite for hydrogenolysis. To further improve the catalyst activity, the same group tuned the catalyst's electronic properties by modifying the synthesis using ammonia.55 With the help of ammonia, the enhanced metal–support interactions in Ru/TiO2 boosted hydrogen spillover, reducing TiO2 more extensively, forming delocalized electrons, and increasing the positive charge of Ru. The positively charged Ruδ+ improves the hydrogen binding capacity for Ru particles by the formation of Ruδ+–H− ion pairs through the weak charge transfer from Ru to H. The higher hydrogen availability on Ru promotes the rate-determining hydrogenation and C–C bond scission, leading to degradation of PP with higher liquid yields (74% vs. 66%) in a shorter processing time (6 vs. 16 h).
In 1998, Dufaud and Basset reported that zirconium hydride supported on silica-alumina [(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiO)3ZrH] synthesized by surface organometallic chemistry converted LDPE and isotactic PP into alkanes via a hydrogenolysis reaction under very mild reaction conditions (<200 °C).60 The reaction mechanism follows σ-bond metathesis with first C–H activation and subsequently β-alkyl transfer and hydrogenolysis. The C–C cleavage mechanism is mainly based on the β-alkyl elimination, which is favored by the hydrogenolysis. DFT calculations indicated that β-methyl elimination favors methyl elimination, E-alkene products and primary Zr–C bond formation.64 In the case of hydrogenolysis of LDPE, the product can be tuned by adjusting the reaction time. For example, the dominant products are liquid C5–C17 medium chain alkanes after 5 hours of reaction while the main products become C1–C5 light alkanes after 62 hours of reaction. The conversion rate of PP is much slower than that of LDPE and the main products are C1–C5 light alkanes with a large portion of methane, due to the facile methyl elimination over the main chain cleavage, which does not significantly reduce the chain length. Consequently, a catalyst that favors β-alkyl elimination is the key to degrade PP and PE efficiently.
SiO)3ZrH] synthesized by surface organometallic chemistry converted LDPE and isotactic PP into alkanes via a hydrogenolysis reaction under very mild reaction conditions (<200 °C).60 The reaction mechanism follows σ-bond metathesis with first C–H activation and subsequently β-alkyl transfer and hydrogenolysis. The C–C cleavage mechanism is mainly based on the β-alkyl elimination, which is favored by the hydrogenolysis. DFT calculations indicated that β-methyl elimination favors methyl elimination, E-alkene products and primary Zr–C bond formation.64 In the case of hydrogenolysis of LDPE, the product can be tuned by adjusting the reaction time. For example, the dominant products are liquid C5–C17 medium chain alkanes after 5 hours of reaction while the main products become C1–C5 light alkanes after 62 hours of reaction. The conversion rate of PP is much slower than that of LDPE and the main products are C1–C5 light alkanes with a large portion of methane, due to the facile methyl elimination over the main chain cleavage, which does not significantly reduce the chain length. Consequently, a catalyst that favors β-alkyl elimination is the key to degrade PP and PE efficiently.
Recently, Wang and his coworkers demonstrated a Ni/SiO2 catalyst for the hydrogenolysis of PE into gasoline and diesel range hydrocarbons (C4–C22) with a yield of 81.2% at 280 °C, 3 MPa H2 for 8 h of reaction.61 Two different preparation methods including the impregnation method and deposition–precipitation method were investigated. Based on the characterization, the Ni/SiO2 catalyst prepared by the deposition–precipitation method possesses smaller nickel particles (∼0.87 nm) and strong metal–support interaction, due to the formation of nickel phyllosilicate, which renders its high hydrogenolysis activity and low selectivity to light alkanes. The Vlachos group also reported a Ni/SiO2 catalyst for the hydrogenolysis of various polyolefins including LDPE, PP and PS to liquid products (C6–C35) with yield up to 65 wt% under conditions of 300 °C and 3 MPa H2 for 2–12 h.65 However, the nickel particle size is quite larger in this case than that reported by Wang et al. Based on the product structure and distribution, they concluded that both the scissions of internal C–C bonds in the polymer and terminal C–C bonds in liquid products occurred on the metallic nickel surface. The stronger interaction between the polymers and nickel surface over liquid alkanes is critical for obtaining high yields of liquid products.
The Co/HZSM-5 catalyst was reported for the solvent-free hydrogenolysis of LDPE, PP, mixtures of LDPE and PP, and postconsumer PE into propane with a selectivity of 80 wt% in the gas phase at 280 °C, 4 MPa H2 for 20 h of reaction.62 The ZSM-5 zeolite support was found to be an important host for the stabilization of dispersed oxidic cobalt nanoparticles, as well as preventing further reduction to the metallic state which is the site for CH4 generation. In addition, the C2–C4 hydrocarbon gas-phase selectivity can be tuned by using zeolites with different framework topologies, such as FAU, MOR and BEA.
A metal oxide NbOPO4 catalyst was found to be active for the hydrogenolysis of PS into aromatic products with a yield of 51.9% at 280 °C, 0.5 MPa for 12 h of reaction.63 The main product is benzene, which suggests that this catalyst is effective for the cleavage of the Csp2–Csp3 bond. The in situ FTIR study and DFT calculations revealed that the cooperation between surface oxygen vacancies and strong Brønsted acid sites on the NbOPO4 catalyst enables the C–C scission and transformation of PS to arenes. The surface oxygen vacancies can dissociate hydrogen to hydride species via heterolytic and homolytic cleavages while the strong Brønsted acid sites activate the robust Csp2–Csp3 bond. The catalyst shows excellent stability and can be recycled three times with negligible changes in product yields and distribution.
2.3. Hydroprocessing of polyolefins without external hydrogen
External high pressure of H2 is essentially utilized for hydrocracking or hydrogenolysis of polyolefins to achieve high yields of liquid products and waxes. However, the introduction of highly pressurized H2 brings about the problems associated with safety concerns as well as high infrastructure cost, which poses an economic barrier for developing a sustainable plastic-upcycling industry. With this respect, the hydroprocessing of polyolefins to valuable hydrocarbon fuels and chemicals without the addition of external H2 is desirable. In fact, the polyolefins can serve as hydrogen donors to form in situ generated hydrogen through dehydrogenation and/or dehydroaromatization reactions of hydrocarbon chains. The self-generated hydrogen is compatible with hydrogenolysis of long chain polyolefins as well as hydrogenation of unsaturated alkenes to form saturated hydrocarbons.Zhang et al. demonstrated a tandem hydrogenolysis/aromatization strategy for upcycling of PE to high valuable long chain alkylaromatics and alkylnaphthenes (average ∼C30, dispersity Đ = 1.1) by using a simple heterogeneous catalyst, Pt/γ-Al2O3, in the absence of external hydrogen.66 In this strategy, Pt/γ-Al2O3 catalyzes the dehydroaromatization of long-chain polyolefins to generate H2, which are subsequently consumed by hydrogenolysis of PE chains and hydrogenation of aromatic rings (Fig. 6). Thus, a delicate balance is maintained between dehydroaromatization and hydrogenolysis to enable the tandem reaction with excellent performance. Control experiments with C30H62 showed a remarkably decreased amount of alkylaromatics and confirmed that PE with long-chain structure was a better feedstock for the tandem reaction system. Besides, the coupling of exothermic hydrogenolysis and endothermic aromatization reactions renders the overall transformation thermodynamically accessible, which allows the reaction to occur at low reaction temperature (280 °C). With this tandem catalytic conversion strategy, various grade PEs including low-molecular-weight PE, high-molecular-weight LDPE plastic bags and HDPE water-bottle caps were efficiently converted to liquid/wax products with dominant components of valuable long-chain alkylaromatics and alkylnaphthenes in high yields (55–80 wt%). These liquid products can be further transformed to various products, such as surfactants, lubricants, insulating oils and refrigeration fluids. However, the catalytic activity of the Pt/γ-Al2O3 catalyst decreases, due to the sintering of Pt nanoparticles during regeneration by calcination. Consequently, the stability of the Pt/γ-Al2O3 catalyst needs to be improved.
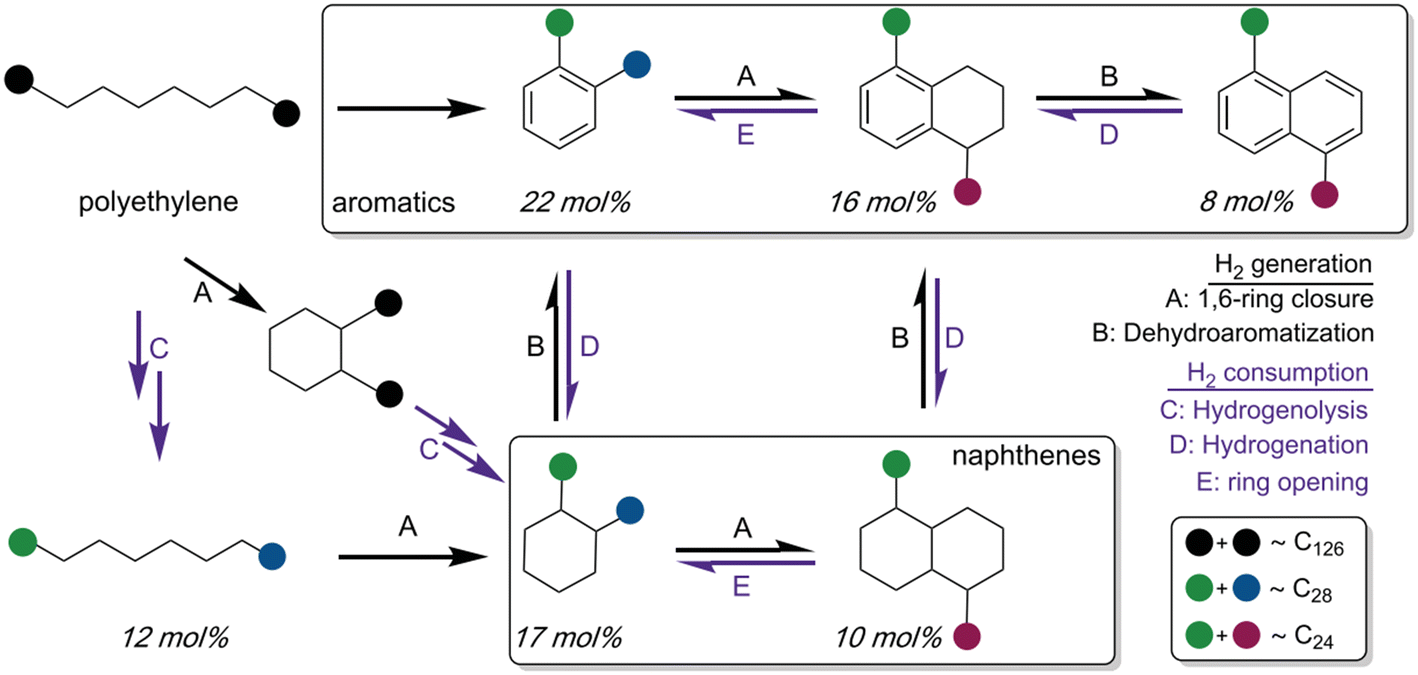 | ||
| Fig. 6 Proposed mechanism for tandem polyethylene hydrogenolysis/aromatization via dehydrocyclization. Reproduced from ref. 66 with permission from AAAS. | ||
Chen et al. developed a new bimetallic Pt–Fe/Al2O3 catalyst for the hydrogenolysis of PE to fuels without external H2. With a small amount of Fe species (Fe/Pt = 0.25), the Pt–Fe/Al2O3 catalyst shows the best performance in conversion of PE to fuel oils in a moderate yield of 55.6%.67 The oil contains mainly linear C7–C19 alkanes and alkenes with a small fraction of alkylaromatics. This is quite different from the case reported by Zhang, where long-chain alkylaromatics and alkylnaphthenes are the primary products in the oil. The reason could be ascribed to the higher acidity of Al2O3 and lower Pt loading (0.8 wt%) in the Pt–Fe/Al2O3 catalyst. Based on the product distribution, the in situ generated hydrogen is from both dehydrogenation and aromatization reactions. Systematic characterization showed that the Pt and Fe particles are evenly dispersed on Al2O3. The Pt nanoparticles play a key role in dehydrogenation of PE to generate in-situ H2 and hydrogenolysis of PE, but show mutual inhibition with the acid site of Al2O3 that contributes to PE cracking via a traditional carbocation chemistry. Modification of Fe on Pt/Al2O3 plays several roles in the catalysis: enhances the aromatization reaction, improves the hydrogenolysis activity of Pt by supplying active H during aromatic formation, and lowers Al2O3 acidity to eliminate gas production. The catalyst can be regenerated by combustion of the deposited residue and subsequent reduction.
Apart from hydrogenolysis of PE without external hydrogen over Pt-based catalysts, Pd-modified beta zeolite was also reported for the hydrocracking of LDPE into a paraffinic-rich hydrocarbon fuel with in situ generated hydrogen.68 The components of fuel oils are mainly C2–C5 and aromatic fractions, with high isoparaffin content and low olefinic compounds. As evidenced by FTIR operando studies, the induction of Pd not only promotes hydro/dehydrogenation and hydro-isomerization reactions, but also retards coke formation.
3. Catalytic hydroconversion processes for deconstruction of heteroatom-containing plastics
Heteroatom-containing plastics include polyesters, polycarbonates, polylactic acid, polyethers, polyamides, polyurethanes and so on, which are mainly constructed from monomers bridged by various C–O and C–N linkages. For example, polyethylene terephthalate (PET), polybutylene terephthalate (PBT), polyethylene naphthalate (PEN), poly(bisphenol A carbonate) (BPA PC), polyphenylene oxide (PPO), Nylon 6, Nylon 66 and so on are the common heteroatom-containing plastics, which are widely used in the food, clothing, automotive, electronics, and construction industries. Effectively recycling/upcycling them is urgent. The most common chemical processes for these heteroatom-containing plastics are glycolysis, methanolysis, aminolysis, ammonolysis, hydrolysis and so on.69–72 However, these processes are hindered by the requirement of a large amount of solvent/degradation agents and the formation of side products that are difficult to separate. On the other hand, hydrogenolysis and hydrodeoxygenation are appealing routes for upcycling of these heteroatom-containing plastics to achieve high yields of target products. Diacids, diols and phenols can be produced by C–O hydrogenolysis of polyesters and polycarbonates. Amino alcohols and diamines can be achieved through C–N hydrogenolysis of polyamides and polyurethanes. Besides these, cyclic alkanes and aromatics can be formed by hydrodeoxygenation with controlled aromatic ring saturation. To achieve high yields of target products, developing catalysts that enable selective C–O and C–N bond dissociation is the key. Various homogeneous and heterogeneous catalysts are employed and pretty high yields of various products can be obtained through hydroconversion processes.Compared to hydroconversion of polyolefins, higher selectivity to the target products can be obtained through hydroconversion of the heteroatom-containing plastics, which is due to the periodic C–O and C–N linkages. In addition, the reaction conditions used for this process are much milder, probably owing to the lower dissociation energies of C–O and C–N bonds.73 Solvents are often used for the hydroconversion of heteroatom-containing plastics, which is different from the hydroconversion of polyolefins, wherein no solvent is employed. The solvents include alcohols, ethers, water and alkanes depending on the catalyst, hydroconversion method and desired products. The solvent not only plays a role in dispersing the plastics, but also acts as a reagent to cleavage the C–O and C–N linkages. In addition, alcohol solvents have also been reported as hydrogen donors in hydroconversion without external hydrogen. In the mechanism for the hydrogenolysis of oxygen-containing plastics over heterogeneous catalysts including Ru/NbOx,73 MoOx-based catalyst74 and Hf(OTf)4 + Pd/C tandem catalysts,75 the synergistic catalysis of oxyphilic sites and metallic sites plays a significant role in C–O hydrogenolysis. The oxyphilic sites are responsible for adsorbing and activation of the C–O bond while the metal sites mainly play a role in the hydrogenation of unsaturated intermediates. This is different from the hydrogenolysis of polyolefins in which the metal is the only active site for C–C bond scission. Metal supported on solid acid catalysts and metal + solid acid mixture catalysts have been demonstrated to be effective for the HDO of plastic wastes. The catalyst systems include Ru/Nb2O5,76 Pt/C + Hβ,77 Rh/C + HUSY78 and so on. Mechanistically, the metal sites in these catalysts play a role in the hydrogenation of unsaturated intermediates, the oxyphilic oxide sites play a role in activating the C–O bonds, and the acid sites are responsible for the hydrolysis and dehydration of the oxygen-containing intermediates. In addition, the acid sites also could induce the C–C bond cleavage, which is not desirable in the HDO of oxygen-containing plastics. It is noted that the same catalyst systems including metal supported on solid acid catalysts and metal + solid acid mixture catalysts have also been widely used in the hydrocracking of polyolefins. However, the roles of each site are different, among which the metal sites are beneficial to the dehydrogenation of the polyolefin chain and hydrogenation of unsaturated olefinic intermediates, while the acid sites play the major role for C–C bond breaking and skeleton isomerization.
3.1. Hydrogenolysis processes for deconstruction of polyesters, polycarbonates, and polyethers
Hydrogenolysis of heteroatom-containing plastics is an attractive pathway for chemical conversion back to their initial monomers or even new monomers, which can be further repurposed for the production of known or new polymers. Homogeneous Ru and Mn complexes are often used for the hydrogenolysis of polyesters and polycarbonates to give diols and diphenols as the main products. Terephthalic acid and naphthalene dicarboxylic acid can be produced by using MoOx-based catalysts or Hf(OTf)4 + Pd/C tandem catalysts through hydrogenolysis of PET, PBT and PEN. In addition, mono-phenols can be obtained by using Ru/NbOx heterogeneous catalysts via hydrogenolysis of PPO and BPA PC plastics. The synergistic catalysis of oxyphilic sites and metallic sites in the heterogeneous catalysts plays a significant role in C–O hydrogenolysis. The oxyphilic sites play a major role in the activation of the C–O bond while the metal sites are mainly responsible for the hydrogenation of unsaturated intermediates.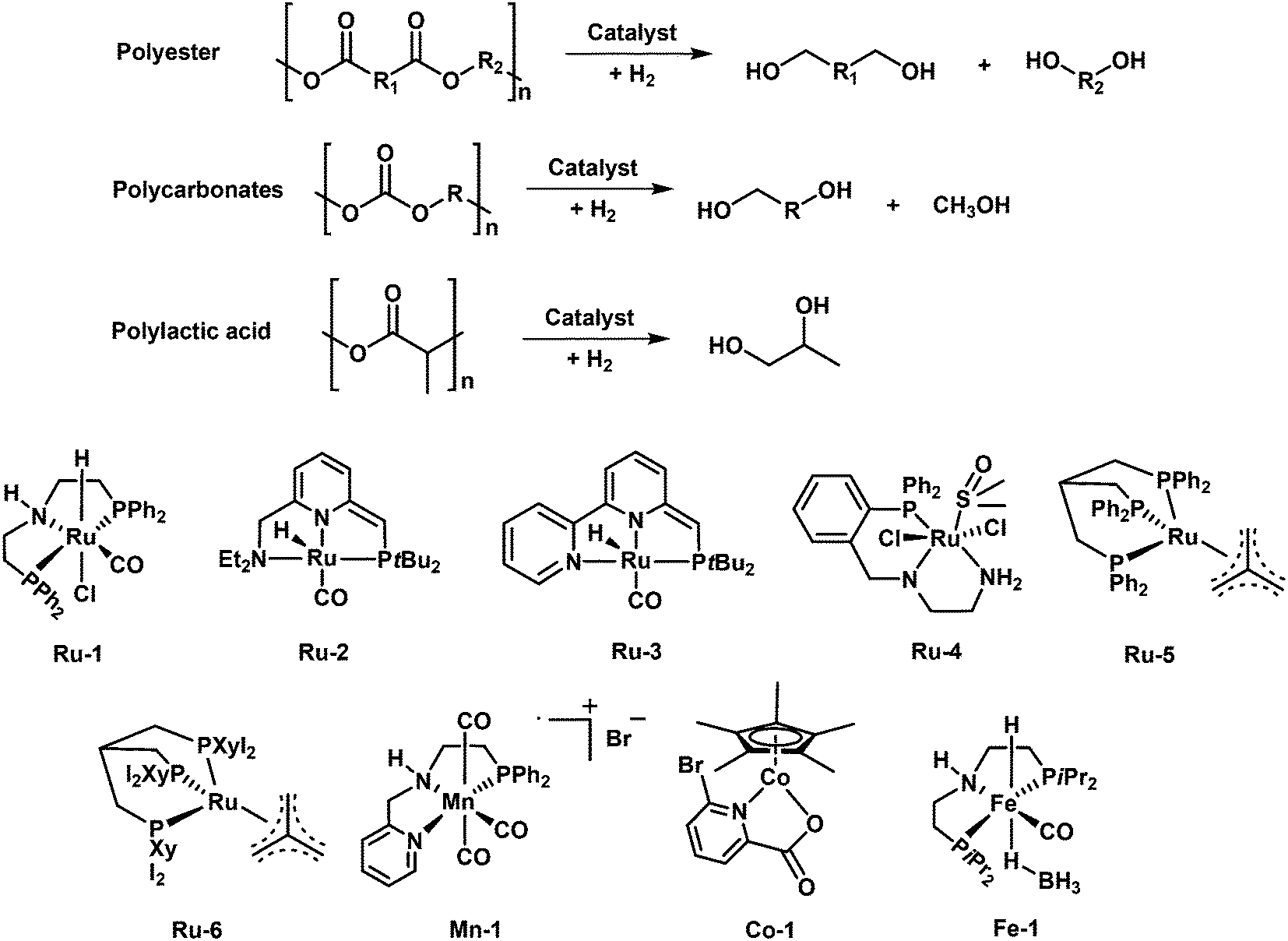 | ||
| Fig. 7 Hydrogenolysis of polyesters, polycarbonates and polylactic acid and the homogeneous catalysts. | ||
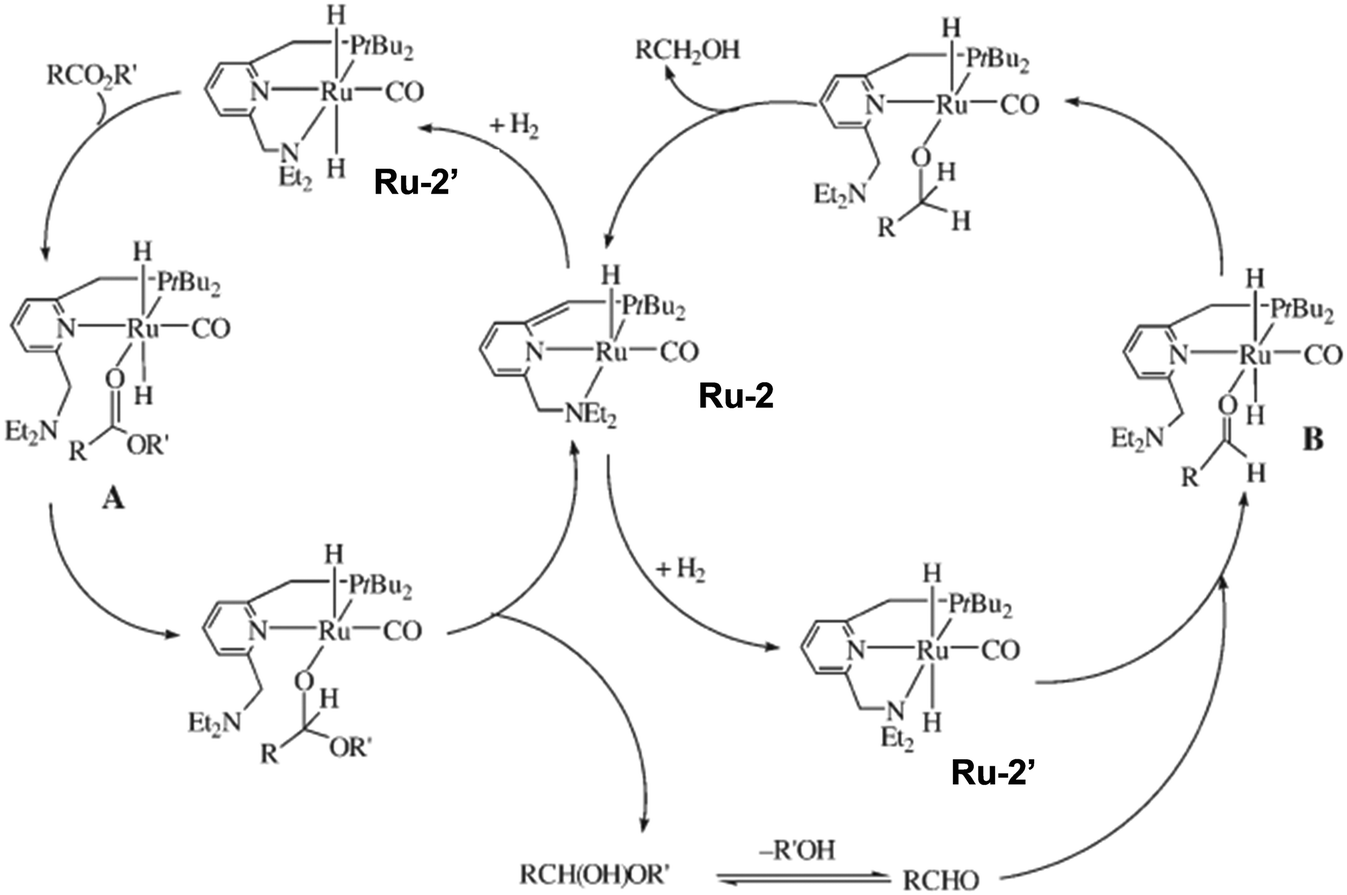 | ||
| Fig. 8 The mechanism for the hydrogenolysis of polyesters. Reproduced from ref. 77 with permission from Wiley. | ||
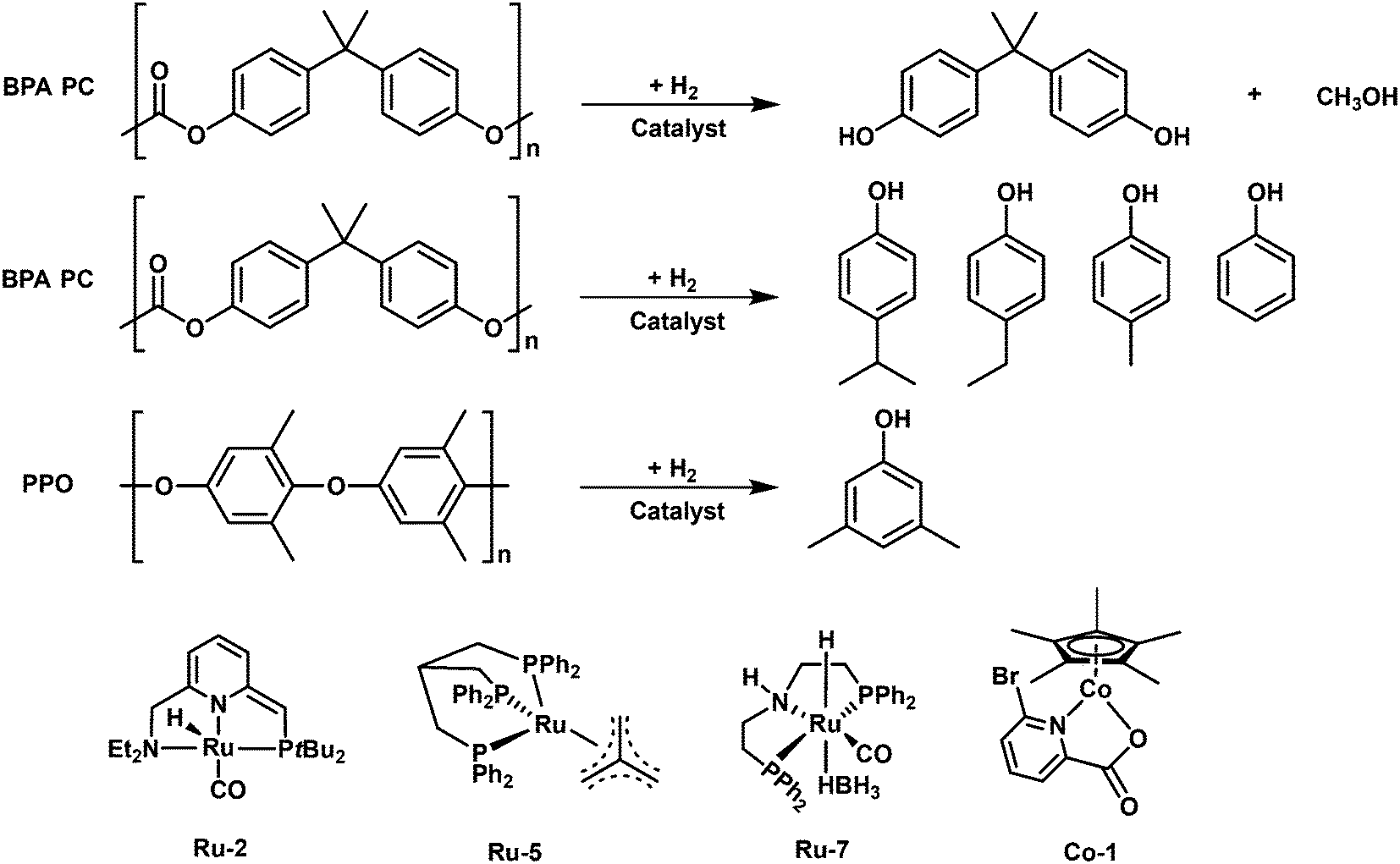
In 2018, Klankermeyer and coworkers demonstrated two Ru complexes with triphos ligands ([Ru(triphos)tmm] (Ru-5) and [Ru(triphos-xyl)tmm]) (Ru-6) and bis(trifluoromethanesulfnyl)imide (HNTf2) as a cocatalyst for the hydrogenolysis of various polyesters and BPA PC material (discussed below).85 The catalyst Ru-5 combined with HNTf2 can quantitatively catalyze the hydrogenolysis of PLA and polycaprolactone (PCL) into 1,2-propandiol and 1,6-hexanediol, respectively. However, the hydrogenolysis of PET and PBT is much more challenging, owing to the generation of a catalyst dimer accompanied with the release of free acid which boosted etherification side reactions. Further, the same group modified the catalyst by replacing the phenyl group with a xyl (3,5-dimethylphenyl) group on triphos ligands (Ru-6). As expected, the modified catalyst with the xyl group is able to realize high conversions of PET and PBT and high selectivity toward the respective diols. After further optimization, >99% of PET and PBT conversion and >99% selectivity for 1,4-benzenedimethanol and ethylene glycol (PET)/1,4-butanediol (PBT) are achieved. Remarkably, this catalyst is capable of dealing with a wide range of commercial polyester plastic wastes and even complex plastic mixtures, showing high tolerance of various polymer additives. In addition, this catalyst system enables separate hydrogenolysis of PLA and PET mixture to the corresponding diols by tuning the reaction temperatures. The scaling up of this approach is also feasible. They showed that hydrogenolysis of a completely coated PLA beverage cup and PET bottle flake in the presence of the PP screw cap and PE label could proceed even at >10 g scale. 11.2 g (93% isolated yield) of 1,2-propanediol and 8.13 g (87% isolated yield) of 1,4-benzenedimethanol and ethylene glycol can be easily isolated away from other additives and unconverted PP and PE present in the beverage cup and bottle.
The replacement of precious metal catalysts with earth-abundant base-metal substitutes is of high current interest, owing to the expensive noble-metal complexes. Based on the recent advances in manganese catalyzed hydrogenation of carbonyl compounds, the groups of El-Sepelgy and Rueping demonstrated the first example of a non-precious manganese homogenous catalytic system for hydrogenolysis of PPC.86 The utilization of the well-defined manganese catalyst Mn-1 for conversion of PPC results in 91% yield of 1,2-propanediol and 84% yield of methanol in the presence of a base (KOtBu) at 140 °C and 5 MPa H2 for 16 h. The free N-H group in the pincer ligand is crucial for achieving high yields of products. The deuterium labelling experiments and DFT analysis indicated that the catalytic cycle involves the heterolytic cleavage of three dihydrogen molecules by metal–ligand cooperation. In addition, a phosphine-free Cp*Co(III) complex (Co-1) was reported to catalyze the hydrogenolysis of PPC to 76% yield of 1,2-propanediol in the presence of a base (KOtBu) at 160 °C and 6 MPa H2 for 24 h.87
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and alkoxy O to form a well-precedented hexacoordinate Mo-dioxo complex. Such a six-membered transition state is crucial for C–O–β scission. This is also approved by the kinetic isotope effect study through hydrogenolysis kinetics of d4–1,2-ethanediol dibenzoate and 1,2-ethanediol dibenzoate. However, the hydrogen activation ability of C/MoO2 is low, which limits the hydrogenolysis step. To further improve the process efficiency, Cai et al. developed a new bimetallic CoMo@NC catalyst derived from Mo@ZIF-CoZn for selective solventless hydrogenolytic deconstruction of PET and PBT to 91% yields PTA within 10 h at 260 °C under 1 atmosphere of H2.88 The reaction pathway is the same as that over C/MoO2. The introduction of Co sites plays a critical role in efficient activation of hydrogen and the following hydrogenolysis. Thus, the catalytic activity was enhanced.
O and alkoxy O to form a well-precedented hexacoordinate Mo-dioxo complex. Such a six-membered transition state is crucial for C–O–β scission. This is also approved by the kinetic isotope effect study through hydrogenolysis kinetics of d4–1,2-ethanediol dibenzoate and 1,2-ethanediol dibenzoate. However, the hydrogen activation ability of C/MoO2 is low, which limits the hydrogenolysis step. To further improve the process efficiency, Cai et al. developed a new bimetallic CoMo@NC catalyst derived from Mo@ZIF-CoZn for selective solventless hydrogenolytic deconstruction of PET and PBT to 91% yields PTA within 10 h at 260 °C under 1 atmosphere of H2.88 The reaction pathway is the same as that over C/MoO2. The introduction of Co sites plays a critical role in efficient activation of hydrogen and the following hydrogenolysis. Thus, the catalytic activity was enhanced.
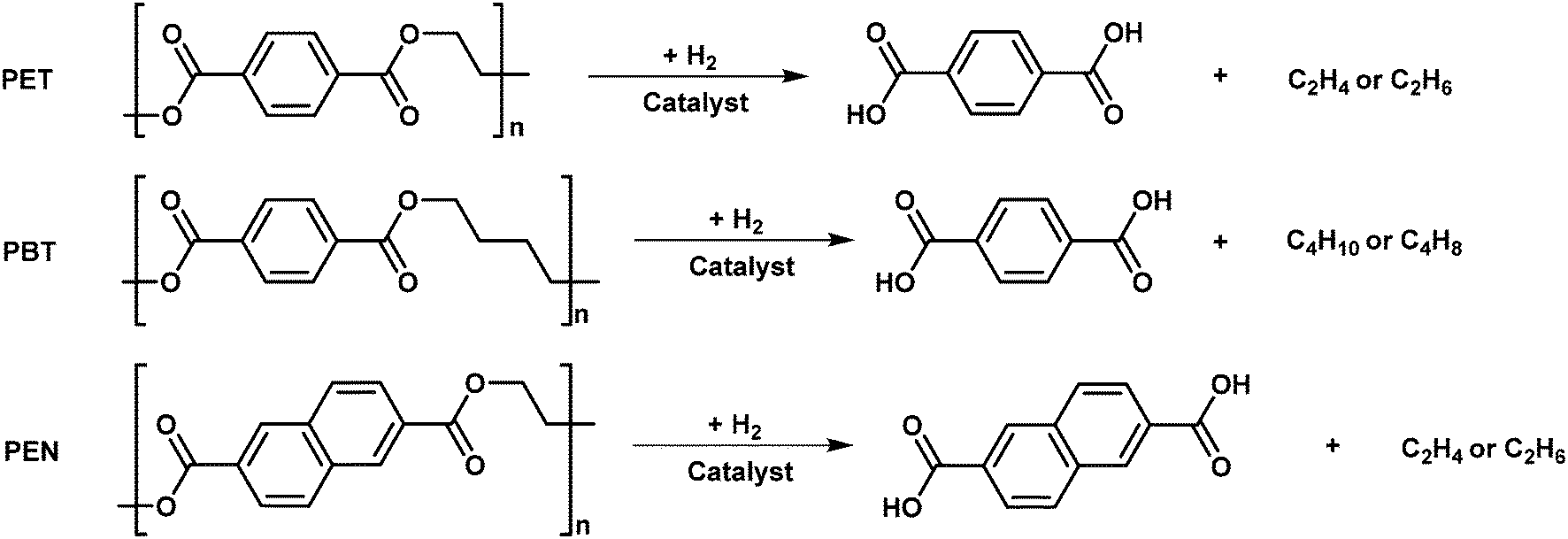 | ||
| Fig. 9 Hydrogenolysis of PET, PBT and PEN. | ||
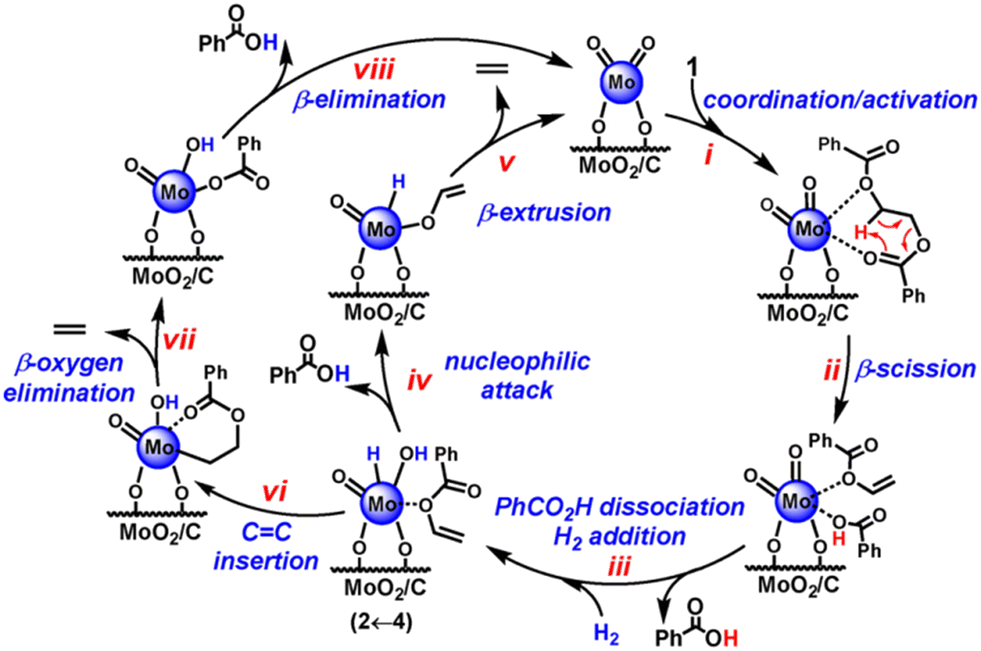 | ||
| Fig. 10 The mechanism for the hydrogenolysis of PET over the C/MoO2 catalyst. Reproduced from ref. 82 with permission from Wiley. | ||
The combination of a homogeneous metal triflate catalyst and a supported metal catalyst has been extensively used for the C–O hydrogenolysis of biomass derived oxygenates, including esters, furans and lignin.89–93 Inspired by this progress in biomass conversion, Marks' group employed a tandem catalytic system (Hf(OTf)4 + Pd/C) for the selective hydrogenolysis of polyester plastics, including PET, PBT and PEN in the absence of a solvent.75 This combo is capable of quantitative conversion of these polyester plastics into the corresponding monomers, PTA (PET and PBT) and naphthalene dicarboxylic acid (PEN), along with ethane (PET and PEN) and butane (PBT) under atmospheric H2. Experimental and DFT calculation studies with a model ester and diester suggest that the reaction pathway involves a tandem Hf(OTf)4-catalyzed retrohydroalkylation step to cleavage alkoxy C–O bonds and Pd/C-catalyzed olefin hydrogenation step to drive the overall reaction forward. This catalytic system is also effective for selective hydrogenolytic deconstruction of both commercial and real plastic wastes.
 | ||
| Fig. 11 Hydrogenolysis of polyesters, polycarbonates and polylactic acid and the homogeneous catalysts. | ||
The group of S. Enthaler applied a Milstein catalyst (Ru-2), for direct hydrogenolysis of BPA PC. High yield (>95%) of BPA can be achieved over 5.0 mol% Milstein catalyst with or without a base (KOtBu) at 140 °C and 4.5 MPa H2 for 24 h.94 However, the depolymerization of BPA PC is dependent on the amount of base at lower temperature and catalyst loading. The catalyst is also active for the conversion of DVD materials. 73% and 86% isolated yields of BPA can be obtained from the entire DVDs and cleaned DVDs without any additives, suggesting that the additives present in the DVDs affect the catalytic activity. To further improve the process efficiency, the same group employed another commercially available ruthenium complex (Ru-7) well known in CO2 hydrogenation, ruthenium-MACHO-BH, for the hydrogenolysis of BPA PC.95 This catalyst enables the conversion of the untreated DVDs and isolated PC from DVDs to 81% and 97% yields of BPA, respectively, in the absence of a base at 80 °C and 4.5 MPa H2 for 16 h. The loading amount of the catalyst sharply decreased to 0.5 mol%, highlighting a significant improvement over the Milstein catalyst. In 2020, Enthaler and coworkers employed an in situ generated Ru complex catalyst from the combination of precursors [RuClH(CO)(PPh3)3] and 2-(di-iso-propylphosphino)ethylamine with 1,8-diazabicyclo(5.4.0)undec-7-ene as a ligand for the hydrogenolysis of BAP PC.96 99% yield of BPA can be obtained by degradation of pristine BPA PC. However, the yields of BPA from DVDs and safety goggles (40% and 33%, respectively) are lower than those over previously discussed catalysts, suggesting the in situ formed catalyst may be seriously affected by the additives or impurities present in real PC materials.
To further lower the expense, a cheap non-noble Cp*Co(III) complex (Co-1) was applied by the group of Sundararaju for the hydrogenolysis of an entire CD with 66% isolated yield of BPA in the presence of a base (KOtBu) at 160 °C and 6 MPa H2 for 40 h.87
In the aspect of heterogeneous catalysis, Wang et al. reported an Ru/Nb2O5 catalyst with an ultralow loading of Ru (0.05 wt%) for the direct hydrogenolysis of PPO to 3,5-dimethyl phenol (3,5-DMP) with 63.3% yield at 280 °C and 2 MPa for 4 h.97 The catalyst is also effective for the conversion of common PPO, comprising PPO and PS components and 40.7% yield of 3,5-DMP along with 4.5% aromatics can be obtained. The catalyst after regeneration by reduction is quite stable that a constant yield of 3,5-DMP can be achieved after four consecutive runs. Although the calculated dissociation energy of the C(o)–O bond (305.20 kJ mol−1) is higher than that of the C(m)–O bond (290.39 kJ mol−1), Ru/Nb2O5 can be capable of selectively cleaving the C(o)–O bond to 3,5-DMP. The characterizations kinetics, in situ DRIFTS and control experiment studies demonstrated that the unique structure of Ru/Nb2O5 with small-size Ru particles, high oxyphilic NbOx species and Brønsted acid sites plays a crucial role in the selective C(o)–O hydrogenolysis of PPO into produce 3,5-DMP. Firstly, the small-size Ru has limited sites to adsorb the aromatic ring and supply H2 to remove of the phenolic hydroxyl group, leading to a high selectivity to phenolic products; secondly, the NbOx species with high oxygen affinity favors the adsorption-activation of C–O bonds; thirdly, the Brønsted acid sites are responsible for the stabilization the carbocation species via the δ–π hyperconjugation effect during C(o)–O cleavage, affording a remarkable selectivity to 3,5-DMP.
In the follow-up study, Wang and coworkers aimed to get mono phenol products via direct hydrogenolysis of PC. Unlike the hydrogenolysis of PPO to 3,5-DMP, conversion of PC to monocyclic phenolic compounds involves C–C bond cleavage and C–O bond preservation.73 However, in most cases, the dissociation energies of C–C bonds are much higher than those of C–O bonds. To overcome these challenges, they proposed a site-specific poisoning strategy for inhibiting C–O cleavage to achieve the selective hydrogenolysis of C–C bonds in PC plastics to produce monocyclic phenols. The same Ru/Nb2O5 catalyst, which is used in the hydrogenolysis of PPO, is applied. Methanol is used as a site-specific poisoning reagent. In situ DRIFTS and TPD-MS studies demonstrated that methanol not only dominantly occupies lower-coordinated NbOx species, which are the active sites for C–O bond activation, but also covers most of the Ru sites in a nonselective manner. This poisoning effect can inhibit the undesirable C–O bond cleavage. Moreover, with appropriate addition of methanol, the methanol poisoning effect does not reduce the activity for C–C bond cleavage, showing the reverse bond energy cleavage. Consequently, the Ru/Nb2O5 catalyst gives 83.9% yield of monocyclic phenols by hydrogenolysis of PC plastics and 28.9% yield of monocyclic oxygenates by direct conversion of phenolic resin at 270 °C and 0.6 MPa H2 in hexane + methanol mixture solvent with 10 wt% concentration of methanol. The catalyst can be recycled three times without loss of activity.
3.2. Hydrodeoxygenation of polyesters, polycarbonates and polyethers
The hydrodeoxygenation (HDO) reaction has been widely investigated in the past decade, in particular for upgrading biomass feedstocks into alkanes (Fig. 12).98–100 Various catalysts including metal supported on solid acid catalysts and metal + solid acid mixture catalysts have been demonstrated to be effective for the HDO of biomass-derived molecules. Given the similarity of oxygen-containing aromatic plastics to lignin, the catalysts used for HDO of biomass derived molecules could also be applicable for HDO of plastic wastes. Ru/Nb2O5, Pt/C + Hβ and Rh/C + HUSY catalysts have been reported for HDO of PET and BPA PC to arenes and cyclic alkanes. Mechanistically, the metal sites in these catalysts are responsible for the hydrogenation of unsaturated intermediates, the oxyphilic acidic oxide sites play a role in activating the C–O bonds, and the acid sites in oxides or zeolites play a role in the hydrolysis and dehydration of the oxygen-containing intermediates. In addition, the acid sites also could induce the C–C bond cleavage, which is not desirable in the HDO of oxygen-containing plastics.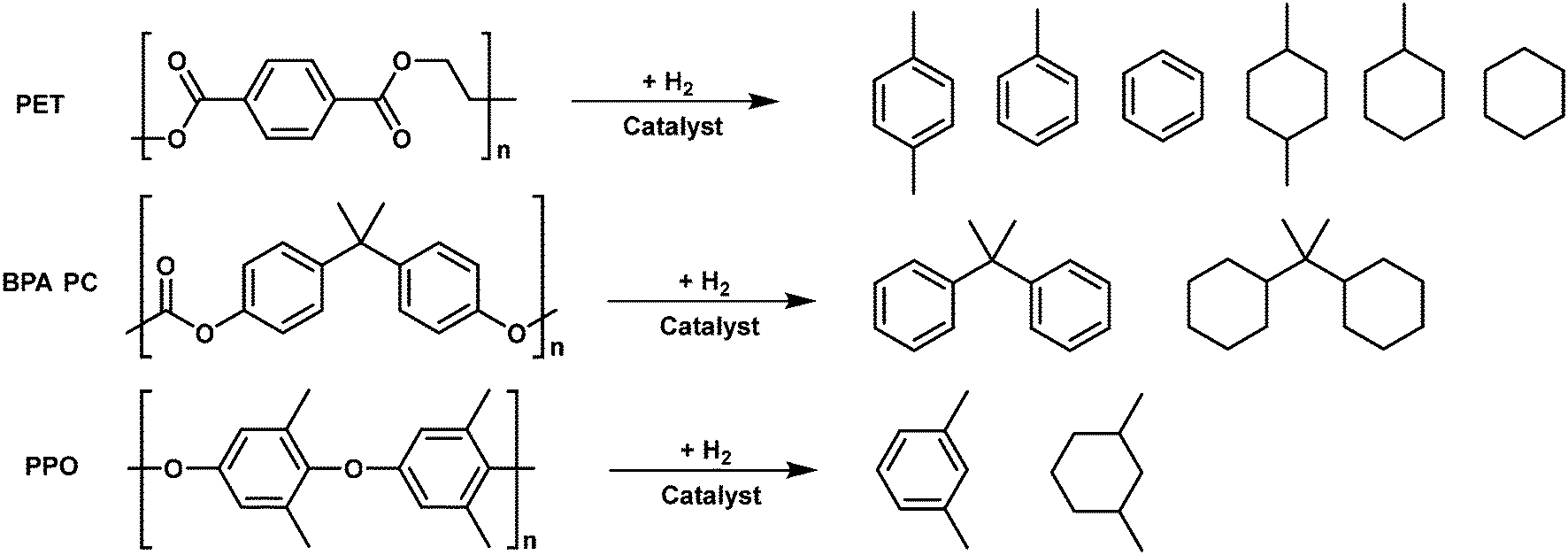 | ||
| Fig. 12 Hydrodeoxygenation of PET, BAP PC and PPO. | ||
Li et al. proposed a new process for the upcycling of PET waste to gasoline and jet-fuel range C7–C8 cycloalkanes and aromatics with a high overall yield (95%).101 This strategy consists of three steps: the methanolysis of PET waste to dimethyl terephthalate, hydrogenation of dimethyl terephthalate to dimethyl cyclohexane-1,4-dicarbonxylate (DMCD) over Pt/C catalysts and the subsequent HDO of DMCD to C7–C8 cycloalkanes and aromatic products. The bimetallic Ru–Cu/SiO2 catalyst was found to be effective for the HDO reaction, which can be attributed to the formation of smaller Ru–Cu alloy particles. The catalyst is quite stable and no evident deactivation is observed during 22 h of continuous reaction. The reaction pathway for the HDO of DMCD has been proposed. C8 cycloalkanes are produced by the initial hydrogenation of ester groups in DMCD followed by dehydration and hydrogenation steps, whereas C8 aromatics are formed through further dehydrogenation of C8 cycloalkanes. The reaction route for the production of C7 cycloalkanes and aromatics involves an additional decarbonylation step. However, this strategy is rather complicated, which is not beneficial for real application.
To fulfil the need of practical application, direct HDO of PET to cycloalkanes and arenes is more appealing. Recently, Wang et al. showed direct HDO of PET to value-added mono arenes in high yields (75–85%) by using a Ru/Nb2O5 catalyst in water or alkane solvent.76 This catalyst is also applicable for HDO of PPO to m-xylene and HDO of PC to benzene and cumin. In addition, the Coca-Cola bottle and commercial PC board can be effective converted to mono arenes over the same catalyst, proving its applicability to upgrade post-consumed plastic wastes. The FT-IR study and DFT calculations demonstrated that the excellent performance can be attributed to the multifunctional sites in the Ru/Nb2O5 catalyst: 1) the sub-nano Ru clusters on the Nb2O5 support prevent the adsorption of the benzene ring and its hydrogenation; 2) the oxyphilic NbOx species for C–O bond activation and Brønsted acid sites for C–C bond activation, respectively, making the production of monocyclic arenes feasible.
To further lower the production cost, a cheap Co/TiO2 catalyst was developed for the direct HDO of PET to produce xylene and toluene with a yield of 78.9% in octane under 3 MPa initial H2 pressure at 340 °C.102 The excellent performance can be attributed to the unique structural features including: 1) Co particles with weakly acidic CoOx species located the outer shell of the support, providing the accessible sites for direct interaction with large-molecule substrates PET; 2) the metal–acid bifunctional system beneficial for hydrodeoxygenation (HDO). However, the stability of this catalyst is not good, due to the formation of the CoTiO3 phase under harsh reaction conditions.
HDO of BPA PC plastic waste can provide propane-2,2-diyldicyclohexane, a C15 dicycloalkane product, which can be used as a high-density jet fuel to improve the volumetric heat values of current aviation fuels. For the production of the desired C15 dicycloalkane product, selective cleavage of C–O bonds and maintaining C–C bonds, especially the tertiary C–C bond that links with two benzene rings, is the key. Li et al. pioneered a two-step method for the production of high-density polycycloalkanes under mild reaction conditions. This route includes methanolysis of PC to bisphenol and subsequent HDO of formed bisphenol to C15 dicyclohexane over the Pt/C + Hβ catalytic system.103 The yield of the target bicyclohexane product is about 50%. A large amount of C–C cracking products (C7–C8 cycloalkanes) is formed, which might be due to the strong acidity of Hβ zeolite. To further simplify the process and improve the yield of bicyclohexane, the same group proposed an aqueous phase hydrodeoxygenation (APHDO) route for the direct catalytic conversion of PC to target C15 alkanes. Different combos of metal and solid acid catalysts were screened for the APHDO of PC. The mixture of Rh/C and H-USY was found to be active and stable for this process.78 High yields (94.9% and 86.9%) of C15 alkanes can be obtained by APHDO of pure PC pellets and a chopped DVD disk, respectively. Such excellent performance can be attributed to the ability of the Rh/C catalyst to catalyze the hydrogenation of carboxyl groups (or esters) and the high acid strength and amount of acid sites of H-USY zeolites. The reaction pathway study showed that the hydrolysis of PC to bisphenol A and direct C–O hydrogenolysis of PC to 4-benzylcycohexan-1-ol proceed in parallel for the depolymerization of PC. The H-USY zeolite and high-temperature water promote the hydrolysis of PC and dehydration of alcohols while the Rh/C catalyst plays a major role in hydrogenolysis and hydrogenation reactions.
3.3. Production of amino alcohols, diamines and diols by hydrogenolysis of polyamides and polyurethane
The hydrogenolysis of polyamides and polyurethanes can produce amino alcohols, or a mixture of diols and diamines or a mixture of diols, diamines and methanol, which can be used for the re-production of the same polymer. In this reaction, the selective hydrogenolysis of C–N in the amide and carbamate groups is the key. Several homogenous Ru-based, Ir-based and Mn-based catalyst were reported for this process and pretty high yields of target products can be achieved (Fig. 13).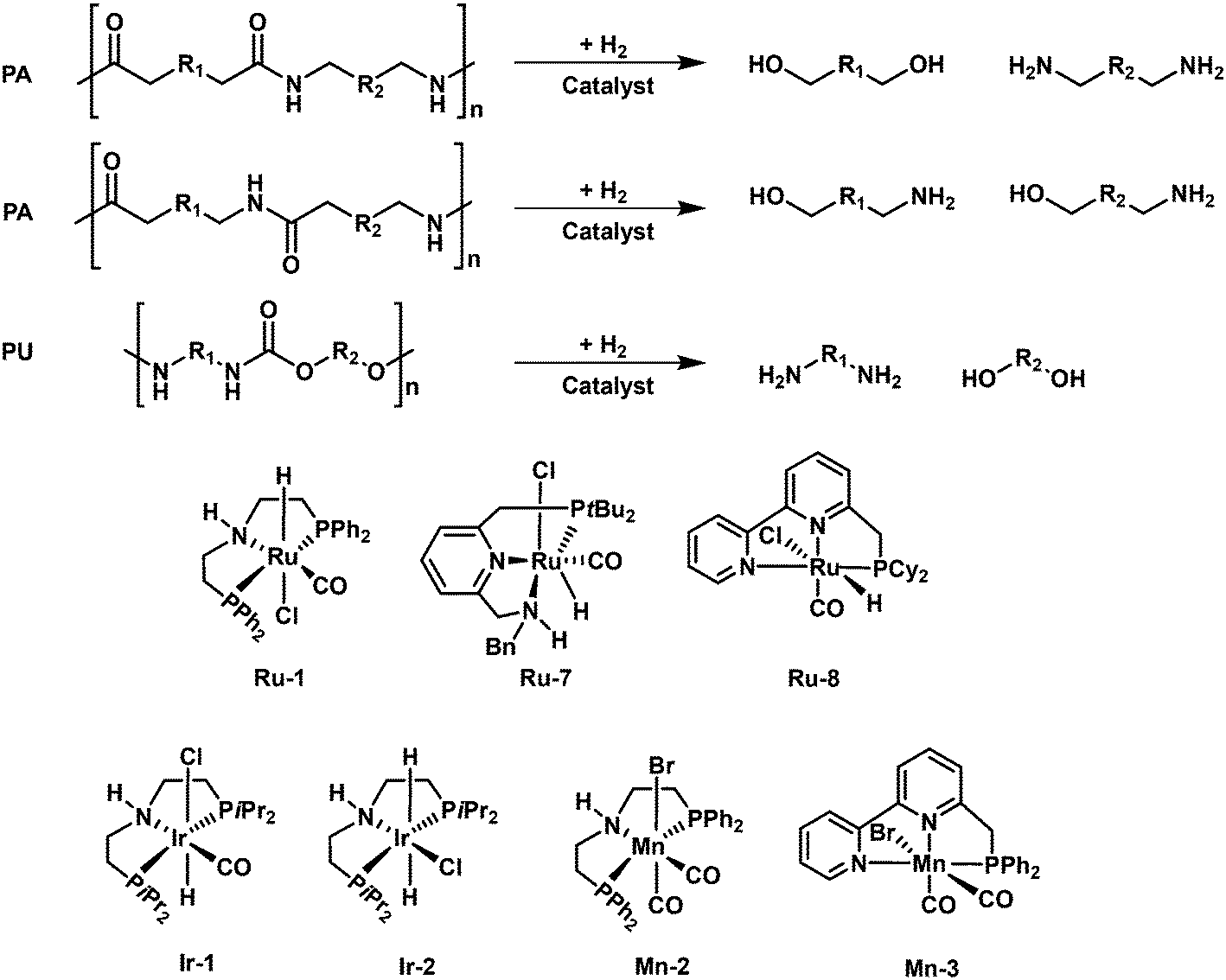 | ||
| Fig. 13 Hydrogenolysis of PA and PU and the homogeneous catalysts. | ||
Inspired by the recent progress on the hydrogenolysis of amides to alcohols and amines,104,105 the Milstein group firstly accomplished the first example of hydrogenolytic deconstruction of polyamides and polyurethanes by using a ruthenium pincer complex (Ru-8) as a versatile catalyst with the aid of KOtBu.106 Under the optimized reaction conditions (150 °C and 7 MPa H2) in DMSO solvent, several polyamides and polyurethanes can be converted. DMSO solvent is critical for the disruption of the hydrogen bond network of the polymers. However, DMSO solvent can deactivate the Ru catalyst and the catalyst shows limited turnover number (TON). A two-step approach, where first hydrogenation in DMSO solvent and subsequently hydrogenation in 1,4-dioxane solvent, is adopted to improve the yields of the target products. With this method, Nylon-6 (resins or powder) and nylon-12 (resins) are respectively converted to 6-amino-1-hexanol and 12-amnio-1-dodecanol in 24–55% yields, with the remaining oligomers (dimer-tetramer). Further, Nylon-66 and other synthesized poly(oligo)amides with the combination of aliphatic and aromatic parts can also undergo hydrogenolysis to produce the corresponding diols and diamines. The polymers containing aromatic parts are converted more easily. In addition, using complex 1 (2 mol%) and KOtBu (8 mol%), the polyurethane can be converted to afford diols, diamines and methanol in high yields. The reaction mechanism is quite similar to that of hydrogenolysis of esters. The metal–ligand cooperativity facilitated the catalytic cycle, where the hydrogenolysis of amide bonds takes place via an outer-sphere mechanism with hemiaminal as the intermediate. Further, the group of Schaub used an improved Ru pincer complex (Ru-9) for the hydrogenolysis of technical-grade nylon-66 and polyurethane materials to corresponding diamines and diols with high yields in THF solvent at 200 °C and 10 MPa H2.107 It should be noted that the high reaction temperature is critical for enhancing the solubility of polymers and thus accomplishment of high efficiency. In addition, Kristensen and Skrydstrup disclosed the use of a commercially available Ir-iPrMACHO complex (Ir-1) for the hydrogenolytic depolymerization of polyurethane materials.108 With the aid of K3PO4, the catalyst can be effective for the conversion of different commercial polyurethane materials, including foams, insulation materials, shoe soles, and inline skating wheels, back to aromatic diamines and polyols with high yields at 150–180 °C and 3 MPa H2 in iso-propanol solvent. What's more, Kumar and Luk demonstrated the first example of hydrogenolytic depolymerization of polyureas to diamines and methanol in moderate yields by using of a ruthenium (Ru-1) or an iridium (Ir-2) pincer complex at 140 °C and 5 MPa H2.109
The non-precious metal complexes, mainly Mn-based catalysts, were also reported to be effective for hydrogenolysis of polyamides and PU materials. Kristensen and Skrydstrup firstly revealed the possibility of a non-precious metal catalyst, Mn-iPrMACHO (Mn-2), for the hydrogenolysis of a commercial flexible PU foam.108 But the reaction conditions are harsher than those applied for the Ir-iPrMACHO complex (Ir-1). Further, the same group applied an improved Mn-PhMACHO catalyst for the efficient hydrogenolysis of PU molded foams, flexible foam, insulation, and end-of-life materials in high yields at 180 °C, 5 MPa H2 in the presence of 0.9 wt% KOH in iso-propanol solvent.110 Shortly after, Schaub and coworkers demonstrated that a manganese catalyst with a PNN-type ligand (Mn-3) is active for the hydrogenolysis of polyurethane materials with varying polyol and aromatic isocyanate compositions.111 In the presence of a Mn-PNN pincer complex at 200 °C and 6 MPa H2 in THF/toluene mixture solvents, various commercial polyurethane materials, including foams, packaging materials, chairs, and end-of-life mattresses can be converted back to aromatic diamines and polyols with high yields. The reaction pathway for the deconstruction of polyurethane involves the tandem C–O bond cleavage of the carbamate group to a polyol and a formate intermediate and following hydrogenation of the formed formate to amine and methanol.
3.4. Hydroconversion using in situ generated hydrogen
Wang's group pioneered the heterogeneous catalysis for H2-free PET conversion to BTX by utilizing the ethylene glycol fragments in PET structure to provide hydrogen via aqueous-phase reforming of ethylene glycol.112 To achieve this process, the key challenge is to obtain a multifunctional catalyst that can be capable of catalyzing three tandem steps including hydrolysis of PET, aqueous-phase reforming of ethylene glycol to hydrogen and subsequently C–O/C–C hydrogenolysis. Among the screened catalysts, both Ru/Nb2O5 and Ru/NiAl2O4 catalysts are effective for this process. H2 and CO2 are detected in the gaseous products, proving that the aqueous phase reforming of ethylene glycol reaction occurred in this process. The reforming reaction provides the hydrogen source, which can assist the following hydrogenolysis/hydrogenation reactions to obtain the desirable aromatic products. Ru/Nb2O5 gives excellent yield of BTX (91.3%) with 66.7% yields of alkyl aromatics (toluene and p-xylene) while Ru/NiAl2O3 showed 65% yield of BTX with benzene as the dominant product in water at 220 °C and 2 MPa N2. Remarkably, the Ru/Nb2O5 catalyst is applicable to convert a variety of waste PET including Coca-Cola bottles, polyester clothes and polyester film back to BTX with a yield above 81%. The reaction pathways for the conversion of PET into BTX include: (1) hydrolysis of PET to terephthalic acid and ethylene glycol; (2) the aqueous phase reforming of ethylene glycol to hydrogen; (3) parallel decarboxylation and hydrogenolysis reactions. Of them, the competitive decarboxylation and hydrogenolysis reactions are the rate-determining steps. The higher selectivity to alkyl aromatics over Ru/Nb2O5 and the dominant benzene product over Ru/NiAl2O3 demonstrate that Ru/Nb2O5 favors the C–O hydrogenolysis reaction, whereas the Ru/NiAl2O3 catalyst tends to decarboxylate. The characterization including XPS, H2-TPR, benzoic acid-adsorption DRIFTS and CO-adsorption DRIFTS showed that Ru0 is the active site for the decarboxylation reaction and Ru/NiAl2O4 shows more surface metallic Ru species than Ru/Nb2O5, which provides the high decarboxylation activity over Ru/NiAl2O4. On the other hand, the strong oxygen affinity of NbOx and the strong interaction between Ru and Nb2O5 with more Ruδ+ species contribute to the outstanding hydrogenolysis performance and weak decarboxylation ability of the Ru/Nb2O5 catalyst. The catalyst recycling experiments of Ru/Nb2O5 showed some activity lost in the second run, owing to the partial oxidation of metallic Ru during the posttreatment in the previous run. The activity can be recovered after the regeneration by reduction. However, high-cost Ru metal is utilized and the selectivity to p-xylene is rather low.To overcome above problems, Zhao et al. developed a cheap CuNa/SiO2 catalyst for a H2-free one-pot conversion of PET and PBT wastes into p-xylene as gasoline fuels and ethylene glycol as an antifreeze component in quantitative yield by using methanol as both a solvent and hydrogen donor.113 The catalyst is able to convert several common PET plastic wastes including Coca-Cola bottles, disposable lunch boxes, packaging bags, McDonald's drink caps and some polyester clothes and even beach sediment along the coastline of Phuket Island into PX with 100% yield under the same catalytic conditions. The catalyst is prepared by hydrothermal synthesis with appropriate addition of NaCl. The overall high activity could be attributed to the high Cu+/Cu0 ratio of the CuNa/SiO2 catalyst. The addition of NaCl during hydrothermal synthesis inhibits nucleation of layered copper silicate and results in the formation of granular copper silicate and thus the high Cu+/Cu0 ratio after reduction. Kinetic and in situ FTIR studies indicate that the conversion of PET back into PX and EG involves tandem PET methanolysis into DMT and EG and selective HDO of DMT to PX with the in situ generated hydrogen via methanol decomposition at 210 °C. The HDO of DMT intermediates to PX undergoes one-sided adsorption and subsequently HDO on the CuNa/SiO2 catalyst, based on the absence of 1,4-benzenedimethanol and the presence of 4-methylbenzyl alcohol and methyl p-toluate. The hydrogenation of methyl p-toluate to 4-methylbenzyl alcohol is the rate-determining step throughout the overall process. However, the catalyst suffers from deactivation, which was probably due to the sintering of Cu nanoparticles after recycling tests and reduction of Cu species. Further maintaining the state of Cu species is needed to improve the catalyst stability.
Li et al. demonstrated a one-pot transfer HDO of PC plastic waste into high-density cyclic hydrocarbons by using iso-propanol as both a solvent and hydrogen donor.77 The combination of RANEY® Ni and USY zeolite is effective for the one-pot two-step transfer HDO of PC plastic into C6–C15 cyclic hydrocarbons with a yield of ∼80% under the optimized reaction conditions. The catalytic mixture is also capable of catalyzing a chopped DVD disk and ∼75% yield of cyclic hydrocarbons can be obtained. The one-pot two-step transfer HDO process involves the degradation of PC in iso-propanol using RANEY® Ni as a catalyst at 190 °C for 1 h and subsequently HDO of the intermediates with introduction of USY zeolite as a co-catalyst at the same reaction temperature. A large amount of C6–C15 oxygenates (including cycloalcohols, diols and phenols) and a small number of cyclic hydrocarbons (including cycloalkanes and aromatics) are obtained over the RANEY® Ni catalyst. With addition of USY zeolite, the oxygenates are further converted into cyclic hydrocarbons. Mechanism studies showed that the transfer hydrogenation of the benzene ring group in aromatics (or phenol) to cycloalkane (or cyclohexanol) is facile over RANEY® Ni, while the transfer HDO of cyclohexanol to cycloalkane is difficult. The addition of USY zeolite catalyzes the dehydration of cyclohexanol to cyclohexene, which further undergoes transfer hydrogenation to cyclohexane over RANEY® Ni. Different solid acids including ZSM-5, USY and Al2O3 were investigated. Among them, USY shows the best promotion effect, which can be ascribed to its large pore size and suitably acidity. It is worth noting that the obtained C6–C15 cyclic hydrocarbon mixture shows better properties and density than those of RP-1 and RJ-4, whereas the volumetric heat value and freezing point are comparable to those of JP-10, which is appealing for the use of high-density aviation fuel.
Homogeneous molecular catalysts are also reported for the transfer hydrogenolysis of polyesters and polycarbonates to diols. Ethanol and iso-propanol are used as hydrogen donors. In 2018, the group of J. G. de Vries developed an iron pincer complex Fe-MACHO-BH (Fe-1) for the base-free transfer hydrogenolysis of Dynacol 7360, which is a polyester made from adipic acid and 1,6-hexanediol.114 By using ethanol as a hydrogen donor, 1,6-hexanediol is produced in 87% isolated yield through this process. In 2019, Werner and coworkers demonstrated the transfer hydrogenolysis of PPC by using a PNP-Fe complex Fe-2 as a catalyst and iso-propanol as a hydrogen source.115 Under optimized reaction conditions (140 °C, 30 h), 1,2-propanediol and methanol can be simultaneously obtained in 65% and 43% yields, respectively.
The transfer hydrogenolysis of polyurethanes is also feasible and this is firstly demonstrated by the Werner group.116 The authors revealed a manganese pincer complex catalyst (Mn-2) for the transfer hydrogenolysis of model, non-crosslinked polyurethanes to the corresponding polyols, amines, and methanol in moderate yields by using iso-propanol as a hydrogen donor. The reaction cycle is quite similar to that applying hydrogen. However, the recycling of real-life polyurethanes has not been addressed.
4. Conclusion and perspectives
Catalytic hydroconversion of plastic waste is an attractive approach to produce fuels and value-added chemicals for building a circular plastic economy. The catalytic hydroconversion mainly includes hydrocracking, hydrogenolysis and hydrodeoxygenation. A metal-based catalyst with high activity and selectively is the key to realizing these processes.Hydrocracking of polyolefins can produce fuel-range liquid hydrocarbons as the main products while hydrogenolysis of polyolefins can provide more valuable wax and lubricant-range hydrocarbons. The catalyst for hydrocracking is bifunctional, with metal and acid sites (e.g., Pt/WO3/ZrO2 and Pt/Zeolites), whereas the catalyst for hydrogenolysis has only metal sites (e.g., Pt and Ru). The internal C–C bond breaking in the polymer hydrocarbon chain is the way to achieve high yields of these high valuable hydrocarbons. Pt/Zeolite catalysts possess higher activity than Pt/WO3/ZrO2 catalysts in hydrocracking. The spatial distribution of Pt particles for hydrogenolysis is critical to obtain target products with high selectivity. Ru-based catalysts always outperform Pt-based catalysts for the hydrogenolysis of polyolefins. The issue for the Ru-based catalysts to obtain a high yield of valuable hydrocarbons is suppression of methane formation. Optimizing the reaction conditions especially hydrogen pressure, tuning the metal–support interaction and inducing hydrogen storage species have been explored to regulate the balance between chemisorbed hydrogen (*H and *CxHy) hydrocarbon species for improving the catalyst activity and inhibiting methane formation.
Homogenous Ru- and Mn- complexes are effective for the hydrogenolysis of polyesters, polycarbonates, polyamides and polyurethanes to diols, bisphenol, amino alcohols, and diamines by selective C–O bond and C–N bond cleavages. In addition, terephthalic acid and naphthalene dicarboxylic acid can be selectivity produced by C–O hydrogenolysis of polyesters over a carbon-supported single-site molybdenum-dioxo catalyst (C/MoO2) or a tandem catalytic system (Hf(OTf)4 + Pd/C) in the absence of a solvent. The obtained monomer products can be used for the reproduction of the polymers. Hydrodeoxygenation of polyesters, polycarbonates and polyethers can provide cyclic hydrocarbons and arenes as the main products. The bifunctional catalyst systems with metal and acid sites (Rh/C + USY) are effective for the production of cyclic hydrocarbons. The combination of metal with oxyphilic metal oxide catalysts (e.g., Ru/Nb2O5 and Co/TiO2) enables selective aromatic hydrocarbon production. Compared to the production of cyclic hydrocarbons, the formation of arene compounds is more challenging, due to hydrogenation of aryl groups, which easily occurs at the metal sites. Controlling appropriate Ru size and acidity of the Ru/Nb2O5 catalyst is the way to realize preferentially activation of C–O bonds instead of aryl groups and enable the selective production of arenes from PET. The low hydrogen pressure is also important for preventing from hydrogenation of aryl groups. This is also applicable to obtain high yields of 3,5-dimethyl phenol from PPO. By using methanol as a site-specific poisoning reagent, Ru/Nb2O5 can be capable of selective C–C bond cleavage while keeping C–O bonds inert, which result in monophenol as the main product from PC. Hence, tuning the properties of metal sites and oxyphilic sites is necessary to tune the product selectivity.
The catalytic hydroconversion process without external hydrogen as an emerging strategy has been applied to convert various plastic wastes into fuels and valuable chemicals to address the high-cost of hydrogen as well as safety concerns. The in situ generated hydrogen can be formed from hydrogen donors such as the polyolefin itself, methanol, and iso-propanol, through specific reactions, including dehydrogenation, dehydroaromatization and aqueous phase reforming. The self-generated hydrogen is compatible with hydrogenolysis or hydrodeoxygenation reactions to degrade plastic to valuable hydrocarbon products. These catalytic processes show promise in creating a pathway towards green chemistry.
Although great advances in hydroconversion of plastic waste have been obtained by using various catalysts, great efforts are still needed to develop more efficient new catalysts and to understand the mechanisms. Hydrocracking of polyolefins is becoming an important industrial process for the production of liquid fuels. Bifunctional catalysts combining metal catalysts and solid acids are very effective for this process, such as the Pt/zeolite catalyst. Rational modulation of the metal–acid balance (MAB), intimacy between sites, site location and site accessibility should be taken into the consideration for designing more powerful hydrocracking catalysts. Besides tuning the acidity properties of the solid acid materials, the introduction of mesopores in these materials (e.g. mesoporous zeolites) is also recommended to overcome the mass diffusion limitation due to the large molecular size of polymers. Based on the current progress in the development of metal-based catalysts for the hydroconversion of plastic waste, it can be found that noble metal based catalysts are dominantly employed and the loading amount is high (≥3 wt%). To lower the cost of expensive noble-metal hydroconversion catalysts, it is critical to develop many other efficient and durable catalysts using low loading noble metals or non-noble metals. In fact, the Ru/CeO2 catalyst with an ultra-low loading (≤0.25 wt%) has been developed by the Szanyi group and shows outstanding performance in the hydrogenolysis of PE and PP.52 Therefore, development of single-atom and subnanometric noble metal catalysts as well as alloy catalysts with low metal loading is encouraged for practical applications. Although some non-noble catalysts, such as Ni–SiO261 and Ni/HZSM-5,37 have been prepared for the hydrogenolysis and hydrocracking of polyolefins, the performances are still inferior to noble-metal catalysts. Based on the site requirements for C–C, C–O and C–N bond activation, selecting the optimal combination of non-noble metals, promoters, and supports, and carefully modulating the catalyst specific structure could be the key criteria for the development of non-noble metal-based hydroconversion catalysts with high efficiency. In addition, previous studies on the conversion of small hydrocarbons over non-noble metal catalysts could lend a hand for designing more efficient non-precious metal catalysts. For example, it was reported that the catalytic activity for C–C breaking with non-noble metals follows the order of Ni > Co > Fe in the hydrogenolysis of ethane117–120 and nickel metal is very selective for successive demethylations of alkanes to form methane due to the preferential adsorption with primary carbon.65,121 With this in mind, the suppression of methane formation and changing the hydrocarbon adsorption manner on the nickel metal surface would be the principle for designing highly efficient Ni-based catalysts for conversion of polyolefins. Moreover, these catalysts should be resistant to the presence of impurities, such as additives that are often introduced into plastics as plasticizers, antioxidants, flame retardants, or pigments. In addition, given the structural similarity to biomass polymers, the catalyst design principle that has been proved to be effective for biomass conversion can be transferred to plastic conversion, especially for heteroatom containing plastic wastes.22 Furthermore, it is highly desirable to design heterogeneous catalysts to replace homogeneous catalysts for hydroconversion of plastic waste, such as Ru- and Mn-complexes for the hydrogenolysis of polyesters, polycarbonates, polyamides and polyurethanes to diols, bisphenols, amino alcohols and diamines.85,104,106–111 Besides HDO, hydrodenitrogenation of polyamides and polyurethanes will be another promising way for upcycling, which has been not realized so far. We believe that the catalysis for hydrodenitrogenation in petroleum upgrading122 will provide a clue for this chemistry. What's more, the elucidation of catalyst active sites, interaction between the catalyst and plastic polymer, and reaction mechanisms is rather challenging, due to the complexity of plastic polymers. Although the model compounds by using small molecules can give some information, it cannot simply be transferred directly to plastic conversion, given the several orders of magnitude difference in molecular weight and differences in thermodynamics. Therefore, future studies with the aid of integrated product analysis, in situ and operando spectroscopy techniques, and theoretical calculations including DFT calculations and molecular simulations are necessary to identify the active sites and understand the reaction mechanisms. These will give insights into the rational design of highly efficient hydroconversion catalysts for upcycling plastic waste.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Natural Science Foundation of China (22208243) for financial support.References
- I. Tiseo, Global plastics industry - statistics & facts, 2021 Search PubMed.
- J. B. Lamb, B. L. Willis, E. A. Fiorenza, C. S. Couch, R. Howard, D. N. Rader, J. D. True, L. A. Kelly, A. Ahmad, J. Jompa and C. D. Harvell, Science, 2018, 359, 460–462 CrossRef CAS.
- J. M. Garcia and M. L. Robertson, Science, 2017, 358, 870–872 CrossRef CAS PubMed.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- K. Hu, Y. Yang, Y. Wang, X. Duan and S. Wang, Chem Catalysis, 2022, 2, 724–761 CrossRef.
- C. Jehanno, J. W. Alty, M. Roosen, S. De Meester, A. P. Dove, E. Y. X. Chen, F. A. Leibfarth and H. Sardon, Nature, 2022, 603, 803–814 CrossRef CAS.
- F. Zhang, F. Wang, X. Wei, Y. Yang, S. Xu, D. Deng and Y.-Z. Wang, J. Energy Chem., 2022, 69, 369–388 CrossRef CAS.
- Q. Hou, M. Zhen, H. Qian, Y. Nie, X. Bai, T. Xia, M. L. U. Rehman, Q. Li and M. Ju, Cell Rep. Phys. Sci., 2021, 2, 100514 CrossRef CAS.
- L. D. Ellis, N. A. Rorrer, K. P. Sullivan, M. Otto, J. E. McGeehan, Y. Román-Leshkov, N. Wierckx and G. T. Beckham, Nat. Catal., 2021, 4, 539–556 CrossRef CAS.
- S. C. Kosloski-Oh, Z. A. Wood, Y. Manjarrez, J. P. de los Rios and M. E. Fieser, Mater. Horiz., 2021, 8, 1084–1129 RSC.
- H. Chen, K. Wan, Y. Zhang and Y. Wang, ChemSusChem, 2021, 14, 4123–4136 CrossRef CAS PubMed.
- M. Chu, Y. Liu, X. Lou, Q. Zhang and J. Chen, ACS Catal., 2022, 12, 4659–4679 CrossRef CAS.
- P. S. Roy, G. Garnier, F. Allais and K. Saito, ChemSusChem, 2021, 14, 4007–4027 CrossRef CAS.
- M.-Q. Zhang, M. Wang, B. Sun, C. Hu, D. Xiao and D. Ma, Chem, 2022, 8, 2912–2923 CAS.
- S. S. Borkar, R. Helmer, F. Mahnaz, W. Majzoub, W. Mahmoud, M. M. Al-Rawashdeh and M. Shetty, Chem Catalysis, 2022, 2, 3320–3356 CrossRef.
- E. Barnard, J. J. Rubio Arias and W. Thielemans, Green Chem., 2021, 23, 3765–3789 RSC.
- J. Payne and M. D. Jones, ChemSusChem, 2021, 14, 4041–4070 CrossRef CAS PubMed.
- P. A. Kots, B. C. Vance and D. G. Vlachos, React. Chem. Eng., 2022, 7, 41–54 RSC.
- T. Tan, W. Wang, K. Zhang, Z. Zhan, W. Deng, Q. Zhang and Y. Wang, ChemSusChem, 2022, 15, e202200522 CAS.
- M. Chu, W. Tu, S. Yang, C. Zhang, Q. Li, Q. Zhang and J. Chen, SusMat, 2022, 2, 161–185 CrossRef.
- Y. Peng, Y. Wang, L. Ke, L. Dai, Q. Wu, K. Cobb, Y. Zeng, R. Zou, Y. Liu and R. Ruan, Energy Convers. Manage., 2022, 254, 115243 CrossRef CAS.
- K. Lee, Y. Jing, Y. Wang and N. Yan, Nat. Rev. Chem., 2022, 6, 635–652 CrossRef.
- Z. Dong, W. Chen, K. Xu, Y. Liu, J. Wu and F. Zhang, ACS Catal., 2022, 14882–14901, DOI:10.1021/acscatal.2c04915.
- J. Huang, A. Veksha, W. P. Chan, A. Giannis and G. Lisak, Renewable Sustainable Energy Rev., 2022, 154, 111866 CrossRef CAS.
- R. J. Conk, S. Hanna, J. X. Shi, J. Yang, N. R. Ciccia, L. Qi, B. J. Bloomer, S. Heuvel, T. Wills, J. Su, A. T. Bell and J. F. Hartwig, Science, 2022, 377, 1561–1566 CrossRef CAS PubMed.
- X. Jia, C. Qin, T. Friedberger, Z. Guan and Z. Huang, Sci. Adv., 2016, 2, e1501591 CrossRef.
- D. Kim, Z. R. Hinton, P. Bai, L. T. J. Korley, T. H. Epps and R. F. Lobo, Appl. Catal., B, 2022, 318, 121873 CrossRef CAS.
- K. R. Venkatesh, J. Hu, W. Wang, G. D. Holder, J. W. Tierney and I. Wender, Energy Fuels, 1996, 10, 1163–1170 CrossRef CAS.
- B. C. Vance, P. A. Kots, C. Wang, Z. R. Hinton, C. M. Quinn, T. H. Epps, L. T. J. Korley and D. G. Vlachos, Appl. Catal., B, 2021, 299, 120483 CrossRef CAS.
- Z. R. Hinton, P. A. Kots, M. Soukaseum, B. C. Vance, D. G. Vlachos, T. H. Epps and L. T. J. Korley, Green Chem., 2022, 24, 7332–7339 RSC.
- S. Liu, P. A. Kots, B. C. Vance, A. Danielson and D. G. Vlachos, Sci. Adv., 2021, 7, eabf8283 CrossRef CAS.
- A. Bin Jumah, V. Anbumuthu, A. A. Tedstone and A. A. Garforth, Ind. Eng. Chem. Res., 2019, 58, 20601–20609 CrossRef CAS.
- A. B. Jumah, A. A. Tedstone and A. A. Garforth, Microporous Mesoporous Mater., 2021, 315, 110912 CrossRef CAS.
- M. Sun, L. Zhu, W. Liu, X. Zhao, Y. Zhang, H. Luo, G. Miao, S. Li, S. Yin and L. Kong, Sustainable Energy Fuels, 2022, 6, 271–275 RSC.
- W. Ding, J. Liang and L. L. Anderson, Energy Fuels, 1997, 11, 1219–1224 CrossRef CAS.
- W. Sriningsih, M. G. Saerodji, W. Trisunaryanti, T. R. Armunanto and I. I. Falah, Procedia Environ. Sci., 2014, 20, 215–224 CrossRef CAS.
- W.-T. Lee, A. van Muyden, F. D. Bobbink, M. D. Mensi, J. R. Carullo and P. J. Dyson, Nat. Commun., 2022, 13, 4850 CrossRef CAS PubMed.
- W.-T. Lee, F. D. Bobbink, A. P. van Muyden, K.-H. Lin, C. Corminboeuf, R. R. Zamani and P. J. Dyson, Cell Rep. Phys. Sci., 2021, 2, 100332 CrossRef CAS.
- J. E. Rorrer, A. M. Ebrahim, Y. Questell-Santiago, J. Zhu, C. Troyano-Valls, A. S. Asundi, A. E. Brenner, S. R. Bare, C. J. Tassone, G. T. Beckham and Y. Román-Leshkov, ACS Catal., 2022, 12, 13969–13979 CrossRef CAS.
- S. D. Jaydev, A. J. Martín and J. Pérez-Ramírez, ChemSusChem, 2021, 14, 5179–5185 CrossRef CAS PubMed.
- X. Wu, A. Tennakoon, R. Yappert, M. Esveld, M. S. Ferrandon, R. A. Hackler, A. M. LaPointe, A. Heyden, M. Delferro, B. Peters, A. D. Sadow and W. Huang, J. Am. Chem. Soc., 2022, 144, 5323–5334 CrossRef CAS PubMed.
- A. Tennakoon, X. Wu, A. L. Paterson, S. Patnaik, Y. Pei, A. M. LaPointe, S. C. Ammal, R. A. Hackler, A. Heyden, I. I. Slowing, G. W. Coates, M. Delferro, B. Peters, W. Huang, A. D. Sadow and F. A. Perras, Nat. Catal., 2020, 3, 893–901 CrossRef CAS.
- R. A. Hackler, J. V. Lamb, I. L. Peczak, R. M. Kennedy, U. Kanbur, A. M. LaPointe, K. R. Poeppelmeier, A. D. Sadow and M. Delferro, Macromolecules, 2022, 55, 6801–6810 CrossRef CAS.
- V. Cappello, P. Sun, G. Zang, S. Kumar, R. Hackler, H. E. Delgado, A. Elgowainy, M. Delferro and T. Krause, Green Chem., 2022, 24, 6306–6318 RSC.
- R. A. Hackler, K. Vyavhare, R. M. Kennedy, G. Celik, U. Kanbur, P. J. Griffin, A. D. Sadow, G. Zang, A. Elgowainy, P. Sun, K. R. Poeppelmeier, A. Erdemir and M. Delferro, ChemSusChem, 2021, 14, 4181–4189 CrossRef CAS.
- I. L. Peczak, R. M. Kennedy, R. A. Hackler, R. Wang, Y. Shin, M. Delferro and K. R. Poeppelmeier, ACS Appl. Mater. Interfaces, 2021, 13, 58691–58700 CrossRef CAS.
- G. Celik, R. M. Kennedy, R. A. Hackler, M. Ferrandon, A. Tennakoon, S. Patnaik, A. M. LaPointe, S. C. Ammal, A. Heyden, F. A. Perras, M. Pruski, S. L. Scott, K. R. Poeppelmeier, A. D. Sadow and M. Delferro, ACS Cent. Sci., 2019, 5, 1795–1803 CrossRef CAS PubMed.
- C. Jia, S. Xie, W. Zhang, N. N. Intan, J. Sampath, J. Pfaendtner and H. Lin, Chem Catalysis, 2021, 1, 437–455 CrossRef.
- L. Chen, Y. Zhu, L. C. Meyer, L. V. Hale, T. T. Le, A. Karkamkar, J. A. Lercher, O. Y. Gutiérrez and J. Szanyi, React. Chem. Eng., 2022, 7, 844–854 RSC.
- J. E. Rorrer, C. Troyano-Valls, G. T. Beckham and Y. Román-Leshkov, ACS Sustainable Chem. Eng., 2021, 9, 11661–11666 CrossRef CAS.
- J. E. Rorrer, G. T. Beckham and Y. Román-Leshkov, JACS Au, 2021, 1, 8–12 CrossRef CAS PubMed.
- L. Chen, L. C. Meyer, L. Kovarik, D. Meira, X. I. Pereira-Hernandez, H. Shi, K. Khivantsev, O. Y. Gutiérrez and J. Szanyi, ACS Catal., 2022, 12, 4618–4627 CrossRef CAS.
- M. Tamura, S. Miyaoka, Y. Nakaji, M. Tanji, S. Kumagai, Y. Nakagawa, T. Yoshioka and K. Tomishige, Appl. Catal., B, 2022, 318, 121870 CrossRef CAS.
- Y. Nakaji, M. Tamura, S. Miyaoka, S. Kumagai, M. Tanji, Y. Nakagawa, T. Yoshioka and K. Tomishige, Appl. Catal., B, 2021, 285, 119805 CrossRef CAS.
- P. A. Kots, T. Xie, B. C. Vance, C. M. Quinn, M. D. de Mello, J. A. Boscoboinik, C. Wang, P. Kumar, E. A. Stach, N. S. Marinkovic, L. Ma, S. N. Ehrlich and D. G. Vlachos, Nat. Commun., 2022, 13, 5186 CrossRef CAS.
- P. A. Kots, S. Liu, B. C. Vance, C. Wang, J. D. Sheehan and D. G. Vlachos, ACS Catal., 2021, 11, 8104–8115 CrossRef CAS.
- C. Wang, K. Yu, B. Sheludko, T. Xie, P. A. Kots, B. C. Vance, P. Kumar, E. A. Stach, W. Zheng and D. G. Vlachos, Appl. Catal., B, 2022, 319, 121899 CrossRef CAS.
- C. Wang, T. Xie, P. A. Kots, B. C. Vance, K. Yu, P. Kumar, J. Fu, S. Liu, G. Tsilomelekis, E. A. Stach, W. Zheng and D. G. Vlachos, JACS Au, 2021, 1, 1422–1434 CrossRef CAS.
- S. Chen, X. Chang, G. Sun, T. Zhang, Y. Xu, Y. Wang, C. Pei and J. Gong, Chem. Soc. Rev., 2021, 50, 3315–3354 RSC.
- V. Dufaud and J.-M. Basset, Angew. Chem., Int. Ed., 1998, 37, 806–810 CrossRef CAS.
- Z. Zhao, Z. Li, X. Zhang, T. Li, Y. Li, X. Chen and K. Wang, Environ. Pollut., 2022, 313, 120154 CrossRef CAS.
- G. Zichittella, A. M. Ebrahim, J. Zhu, A. E. Brenner, G. Drake, G. T. Beckham, S. R. Bare, J. E. Rorrer and Y. Román-Leshkov, JACS Au, 2022, 2(10), 2259–2268 CrossRef CAS.
- H. Zhou, L. Chen, Y. Guo, X. Liu, X.-P. Wu, X.-Q. Gong and Y. Wang, ACS Catal., 2022, 12, 4806–4812 CrossRef CAS.
- A. Q. Kane, A. M. Esper, K. Searles, C. Ehm and A. S. Veige, Catal. Sci. Technol., 2021, 11, 6155–6162 RSC.
- B. C. Vance, P. A. Kots, C. Wang, J. E. Granite and D. G. Vlachos, Appl. Catal., B, 2023, 322, 122138 CrossRef CAS.
- F. Zhang, M. Zeng, R. D. Yappert, J. Sun, Y.-H. Lee, A. M. LaPointe, B. Peters, M. M. Abu-Omar and S. L. Scott, Science, 2020, 370, 437–441 CrossRef CAS.
- Z. Chen, L. Xu and X. Zhang, Chem. Eng. J., 2022, 446, 136213 CrossRef CAS.
- K. Pyra, K. A. Tarach, A. Śrębowata, I. Melián-Cabrera and K. Góra-Marek, Appl. Catal., B, 2020, 277, 119070 CrossRef CAS.
- P. Anggo Krisbiantoro, Y.-W. Chiao, W. Liao, J.-P. Sun, D. Tsutsumi, H. Yamamoto, Y. Kamiya and K. C.-W. Wu, Chem. Eng. J., 2022, 450, 137926 CrossRef CAS.
- A. M. Al-Sabagh, F. Z. Yehia, A.-M. M. F. Eissa, M. E. Moustafa, G. Eshaq, A.-R. M. Rabie and A. E. ElMetwally, Ind. Eng. Chem. Res., 2014, 53, 18443–18451 CrossRef CAS.
- F. R. Veregue, C. T. Pereira da Silva, M. P. Moisés, J. G. Meneguin, M. R. Guilherme, P. A. Arroyo, S. L. Favaro, E. Radovanovic, E. M. Girotto and A. W. Rinaldi, ACS Sustainable Chem. Eng., 2018, 6, 12017–12024 CrossRef CAS.
- J. G. Kim, Polym. Chem., 2020, 11, 4830–4849 RSC.
- Y. Jing, M. Shakouri, X. Liu, Y. Hu, Y. Guo and Y. Wang, ACS Catal., 2022, 12, 10690–10699 CrossRef CAS.
- Y. Kratish, J. Li, S. Liu, Y. Gao and T. J. Marks, Angew. Chem., Int. Ed., 2020, 59, 19857–19861 CrossRef CAS.
- Y. Kratish and T. J. Marks, Angew. Chem., Int. Ed., 2022, 61, e202112576 CAS.
- Y. Jing, Y. Wang, S. Furukawa, J. Xia, C. Sun, M. J. Hülsey, H. Wang, Y. Guo, X. Liu and N. Yan, Angew. Chem., Int. Ed., 2021, 60, 5527–5535 CrossRef CAS.
- L. Wang, F. Han, G. Li, M. Zheng, A. Wang, X. Wang, T. Zhang, Y. Cong and N. Li, Green Chem., 2021, 23, 912–919 RSC.
- L. Wang, G. Li, Y. Cong, A. Wang, X. Wang, T. Zhang and N. Li, Green Chem., 2021, 23, 3693–3699 RSC.
- Z. Han, L. Rong, J. Wu, L. Zhang, Z. Wang and K. Ding, Angew. Chem., Int. Ed., 2012, 51, 13041–13045 CrossRef CAS PubMed.
- E. M. Krall, T. W. Klein, R. J. Andersen, A. J. Nett, R. W. Glasgow, D. S. Reader, B. C. Dauphinais, S. P. Mc Ilrath, A. A. Fischer, M. J. Carney, D. J. Hudson and N. J. Robertson, Chem. Commun., 2014, 50, 4884–4887 RSC.
- E. Balaraman, C. Gunanathan, J. Zhang, L. J. W. Shimon and D. Milstein, Nat. Chem., 2011, 3, 609–614 CrossRef CAS PubMed.
- E. Balaraman, E. Fogler and D. Milstein, Chem. Commun., 2012, 48, 1111–1113 RSC.
- J. Zhang, G. Leitus, Y. Ben-David and D. Milstein, Angew. Chem., Int. Ed., 2006, 45, 1113–1115 CrossRef CAS PubMed.
- J. A. Fuentes, S. M. Smith, M. T. Scharbert, I. Carpenter, D. B. Cordes, A. M. Z. Slawin and M. L. Clarke, Chem. – Eur. J., 2015, 21, 10851–10860 CrossRef CAS.
- S. Westhues, J. Idel and J. Klankermayer, Sci. Adv., 2018, 4, eaat9669 CrossRef CAS PubMed.
- V. Zubar, Y. Lebedev, L. M. Azofra, L. Cavallo, O. El-Sepelgy and M. Rueping, Angew. Chem., Int. Ed., 2018, 57, 13439–13443 CrossRef CAS.
- P. Dahiya, M. K. Gangwar and B. Sundararaju, ChemCatChem, 2021, 13, 934–939 CrossRef CAS.
- P. Wu, G. Lu and C. Cai, Green Chem., 2021, 23, 8666–8672 RSC.
- T. L. Lohr, Z. Li, R. S. Assary, L. A. Curtiss and T. J. Marks, Energy Environ. Sci., 2016, 9, 550–564 RSC.
- T. L. Lohr, Z. Li and T. J. Marks, Acc. Chem. Res., 2016, 49, 824–834 CrossRef CAS.
- T. L. Lohr, Z. Li, R. S. Assary, L. A. Curtiss and T. J. Marks, ACS Catal., 2015, 5, 3675–3679 CrossRef CAS.
- Z. Li, R. S. Assary, A. C. Atesin, L. A. Curtiss and T. J. Marks, J. Am. Chem. Soc., 2014, 136, 104–107 CrossRef CAS.
- R. S. Assary, A. C. Atesin, Z. Li, L. A. Curtiss and T. J. Marks, ACS Catal., 2013, 3, 1908–1914 CrossRef CAS.
- C. Alberti, S. Eckelt and S. Enthaler, ChemistrySelect, 2019, 4, 12268–12271 CrossRef CAS.
- T.-O. Kindler, C. Alberti, J. Sundermeier and S. Enthaler, ChemistryOpen, 2019, 8, 1410–1412 CrossRef CAS.
- C. Alberti, J. Kessler, S. Eckelt, M. Hofmann, T.-O. Kindler, N. Santangelo, E. Fedorenko and S. Enthaler, ChemistrySelect, 2020, 5, 4231–4234 CrossRef CAS.
- B. Feng, Y. Jing, Y. Guo, X. Liu and Y. Wang, Green Chem., 2021, 23, 9640–9645 RSC.
- K. Tomishige, Y. Nakagawa and M. Tamura, Green Chem., 2017, 19, 2876–2924 RSC.
- S. Kim, E. E. Kwon, Y. T. Kim, S. Jung, H. J. Kim, G. W. Huber and J. Lee, Green Chem., 2019, 21, 3715–3743 RSC.
- Z.-H. Zhang, Z. Sun and T.-Q. Yuan, Trans. Tianjin Univ., 2022, 28, 89–111 CrossRef.
- H. Tang, N. Li, G. Li, A. Wang, Y. Cong, G. Xu, X. Wang and T. Zhang, Green Chem., 2019, 21, 2709–2719 RSC.
- S. Hongkailers, Y. Jing, Y. Wang, N. Hinchiranan and N. Yan, ChemSusChem, 2021, 14, 4330–4339 CrossRef CAS.
- H. Tang, Y. Hu, G. Li, A. Wang, G. Xu, C. Yu, X. Wang, T. Zhang and N. Li, Green Chem., 2019, 21, 3789–3795 RSC.
- S. Westhues, M. Meuresch and J. Klankermayer, Angew. Chem., Int. Ed., 2016, 55, 12841–12844 CrossRef CAS PubMed.
- E. Balaraman, B. Gnanaprakasam, L. J. W. Shimon and D. Milstein, J. Am. Chem. Soc., 2010, 132, 16756–16758 CrossRef CAS.
- A. Kumar, N. von Wolff, M. Rauch, Y.-Q. Zou, G. Shmul, Y. Ben-David, G. Leitus, L. Avram and D. Milstein, J. Am. Chem. Soc., 2020, 142, 14267–14275 CrossRef CAS.
- W. Zhou, P. Neumann, M. Al Batal, F. Rominger, A. S. K. Hashmi and T. Schaub, ChemSusChem, 2021, 14, 4176–4180 CrossRef CAS.
- L. Gausas, S. K. Kristensen, H. Sun, A. Ahrens, B. S. Donslund, A. T. Lindhardt and T. Skrydstrup, JACS Au, 2021, 1, 517–524 CrossRef CAS.
- A. Kumar and J. Luk, Eur. J. Org. Chem., 2021, 2021, 4546–4550 CrossRef CAS.
- L. Gausas, B. S. Donslund, S. K. Kristensen and T. Skrydstrup, ChemSusChem, 2022, 15, e202101705 CrossRef CAS.
- V. Zubar, A. T. Haedler, M. Schütte, A. S. K. Hashmi and T. Schaub, ChemSusChem, 2022, 15, e202101606 CrossRef CAS PubMed.
- S. Lu, Y. Jing, B. Feng, Y. Guo, X. Liu and Y. Wang, ChemSusChem, 2021, 14, 4242–4250 CrossRef CAS.
- Z. Gao, B. Ma, S. Chen, J. Tian and C. Zhao, Nat. Commun., 2022, 13, 3343 CrossRef CAS.
- R. A. Farrar-Tobar, B. Wozniak, A. Savini, S. Hinze, S. Tin and J. G. de Vries, Angew. Chem., Int. Ed., 2019, 58, 1129–1133 CrossRef CAS PubMed.
- X. Liu, J. G. de Vries and T. Werner, Green Chem., 2019, 21, 5248–5255 RSC.
- X. Liu and T. Werner, Chem. Sci., 2021, 12, 10590–10597 RSC.
- J. H. Sinfelt, in Advances in Catalysis, ed. D. D. Eley, H. Pines and P. B. Weisz, Academic Press, 1973, vol. 23, pp. 91–119 Search PubMed.
- C. J. Machiels and R. B. Anderson, J. Catal., 1979, 58, 260–267 CrossRef CAS.
- H. Matsumoto, Y. Saito and Y. Yoneda, J. Catal., 1971, 22, 182–192 CrossRef CAS.
- J. H. Sinfelt, W. F. Taylor and D. J. C. Yates, J. Phys. Chem., 1965, 69, 95–101 CrossRef CAS.
- G. Leclercq, S. Pietrzyk, M. Peyrovi and M. Karroua, J. Catal., 1986, 99, 1–11 CrossRef CAS.
- E. Furimsky and F. E. Massoth, Catal. Rev.: Sci. Eng., 2005, 47, 297–489 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2023 |