
Phase transition of metal–organic frameworks for the encapsulation of enzymes†
Zhen
Ren
a,
Weiqiang
Zhou
b,
Jiena
Weng
c,
Ziyue
Qin
a,
Liwei
Liu
d,
Ning
Ji
a,
Cheng
Chen
a,
Haohao
Shi
a,
Wenxiong
Shi
e,
Xinglong
Zhang
a,
Islam E.
Khalil
 a,
Bing
Zheng
a,
Jiansheng
Wu
a,
Weina
Zhang
*a and
Fengwei
Huo
*a
a,
Bing
Zheng
a,
Jiansheng
Wu
a,
Weina
Zhang
*a and
Fengwei
Huo
*a
aKey Laboratory of Flexible Electronics (KLOFE), Institute of Advanced Materials (IAM), Nanjing Tech University (Nanjing Tech), Nanjing 211816, China. E-mail: iamfwhuo@njtech.edu.cn; Tel: +8613814051684
bInstitute of Green Chemistry & Chemical Technology, School of Chemistry & Chemical Engineering, Jiangsu University, Zhenjiang 212013, China
cShaanxi Institute of Flexible Electronics (SIFE), Northwestern Polytechnical University (NPU), 127 West Youyi Road, Xi'an 710072, China
dCollege of Science, Northeastern University (NEU), 11 Wenhua Road, Shenyang 100819, China
eSeparation Membranes and Membrane Processes, School of Materials Science and Engineering, Tianjin Polytechnical University (TJPU), 399 Binshuixi Road, Tianjin 300387, China
First published on 10th May 2022
Abstract
Through encapsulating functional materials, metal–organic framework (MOF) composites show extraordinary potential in various fields due to the excellent synergistic effects between the host and guests. However, many attractive functional species, such as enzymes, could be easily damaged during the synthesis of MOF composites. Herein we report a new strategy, namely pressure-induced-stimulated-aging (PISA), in which crystalline MOFs were pressure-treated to induce partial disorder in MOF structures, followed by a recrystallization process to encapsulate enzymes into MOF crystals. The encapsulated enzymes were trapped in the overlap between MOF particles or on the surface of MOFs. Such reversible phase transition avoided high temperature, high ionic strength, strong acid/base conditions, etc., which was suitable for many types of enzymes. Different enzymes can not only be trapped into ZIF-8, but also into other stable MOFs such as Mg-MOF-74, UiO-66, UiO-66-NH2, MIL-53(Al) and MIL-53(Fe). After optimizing the experimental conditions, Glucose Oxidase (GOx)/UiO-66-WV exhibited similar activity compared to free enzymes. In addition, GOx/UiO-66-WV and GOx/MIL-53(Fe)-WV composites can retain more than 90% of the enzymatic activity after treatment with protease. This strategy will pave a new way for designing MOF composites and exploring further applications in various areas.
 Weina Zhang | Dr Weina Zhang is currently a Professor of the Institute of Advanced Materials, Nanjing Tech University. She received her B.S. and M.S. degrees from Xi'an University of Technology and Xi'an Jiaotong University in China in 2007 and 2011, respectively. She subsequently completed her PhD at Nanyang Technological University in Singapore in 2014. Then she joined the Institute of Advanced Materials, Nanjing Tech University as a researcher in 2015 and was promoted to professor in 2018. Her current research focuses on metal–organic framework materials in traditional catalysis, enzyme catalysis and microbial catalysis. |
Introduction
Metal–organic frameworks (MOFs) have shown potential applications in gas separation and storage due to their regular microporous structures.1,2 The rich and adjustable pore environment of MOFs has expanded their applications in catalysis,3 biomedicines,4,5 sensors,6–8etc. Additionally, through the combination of functional guest materials, MOF composites can obtain new properties that MOFs or guest materials do not have.9–11 In recent years, the encapsulation of functional materials into MOFs has become a sophisticated approach to obtain multifunctional MOF composites.12–14 For example, owing to the synergy between the catalytic capability of platinum nanoparticles (Pt NPs) and size-selectivity of the ZIF-8 shell, the hybrid Pt/ZIF-8 catalysts exhibited excellent size-selectivity for hydrogenation of olefins.15 The prepared MIL-101(Fe3+, Cr3+)@Pt@MIL-101(Fe3+, Cr3+) with “sandwich” structures encapsulated Pt NPs in MOF interlayers and showed increased selectivity for the hydroconversion of citronellal to citronellol, mainly due to the combination of the catalytic sites on Pt NPs and high selective adsorption of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bonds on metal sites from MOFs.16 A series of antitumoral and retroviral drugs enclosed into iron(III) based MOFs have shown potential applications in bio-imaging.17 These studies mentioned above showed an improvement in MOF properties after combination with functional materials.
O double bonds on metal sites from MOFs.16 A series of antitumoral and retroviral drugs enclosed into iron(III) based MOFs have shown potential applications in bio-imaging.17 These studies mentioned above showed an improvement in MOF properties after combination with functional materials.
Recently, enzyme@MOFs has gained extensive attention due to the attractive combination of stereoselective catalytic sites of enzymes and accessible porosity of MOFs.18–20 Traditional methods for preparing MOF composites usually involve high ionic strength and high temperature, and are prone to lose part or most of the enzyme activity during the synthesis of MOF composites.15,21–23 Ma's group used the infiltration encapsulation strategy to restrict enzymes in MOF structures.24 They focused on the loading of enzymes into mesoporous MOFs to boost the recyclability of enzymes. However, this strategy required the cage of MOFs to perfectly match the size of enzymes.18 To overcome this challenge, the biomimetic mineralization method25,26 and coprecipitation method27,28 have been explored to coat enzymes with MOF shells during the synthesis of MOF composites, which could encapsulate enzymes with different sizes. However, this strategy is only suitable for ZIF-based MOFs and usually involves a specific concentration of metal ions and organic ligands, which may negatively impact the biological activity of enzymes.29 Recently, a fast ball-milling strategy30 of encapsulating β-glucosidase, invertase, β-galactosidase and catalase into stable MOFs such as UiO-66-NH2 has been developed by Tsung's group. The synthesized composites maintained the enzyme activity after treatment with protease in an acidic environment. Undesirably, this method involved a trace of organic solvents and organic ligands, which may affect the activity of enzymes during the synthetic process. Although there are many reports about the fabrication of enzyme@MOFs composites, two main challenges which cannot be overlooked have been listed here. The first one is how to achieve a balance between high actively immobilized enzymes and various types of MOF substrates by a simple method. The second one is to understand the interactions between enzymes and MOFs after enzyme encapsulation, which is critical for the development of enzyme/MOF composites.
Previous research studies have shown that mechanical force can cause phase transformations of MOFs.31–34 If functional materials are not pressure sensitive, they might be trapped in MOF matrices during the phase transformation of MOFs without any damage.35 Herein, we propose a strategy to encapsulate enzymes into MOF structures through pressure-induced MOF phase transition and post-stimulated crystalline aging (Scheme 1), named pressure-induced-stimulated-aging (PISA). Enzymes were premixed with MOFs by applying a certain amount of mechanical pressure to transform MOFs into partially disordered MOFs. Then, by exposing them to water vapor, the enzymes/partially disordered MOFs could be converted to the enzyme/MOF composites. 5 min ball-milling was employed on MOFs to activate the surface of MOF particles.36,37 PISA has three advantages. Firstly, the overall encapsulation process occurred during the reversible phase transition of MOFs without involving any harsh conditions and the water vapor stimulated aging process is applicable to many sorts of enzymes. Secondly, nanoparticles were used to replace enzymes to explore the encapsulation mechanisms and figure out the possible position of enzymes encapsulated in MOFs. Combined with molecular dynamics simulations and control experiments, the interactions between enzymes and MOFs were clearer, which provided a research model for the regulation of the activity of enzymes. Thirdly, enzymes@MOFs composites prepared by PISA can preserve the activity of enzymes after treatment with protease, which may have potential application in bio-therapy, biological cascade reaction and biosensing.
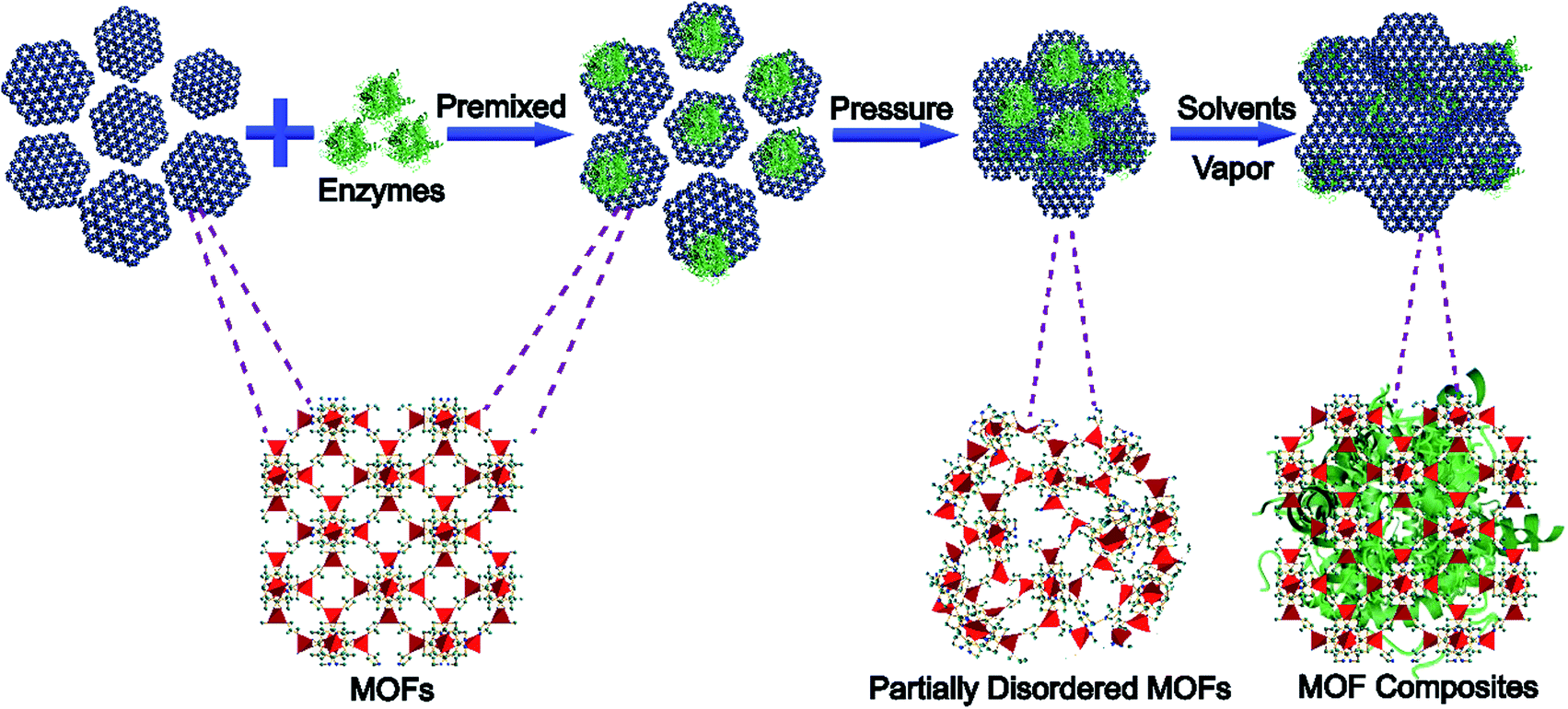 | ||
| Scheme 1 Schematic illustration of the pressure-induced-stimulated-aging (PISA) process of preparing MOF composites. | ||
Experimental section
Materials and measurements
Commercial ZIF-8 was purchased from Sigma-Aldrich (ACS grade). MIL-53(Al) and MIL-53(Fe) were purchased from HWRK. Glucose oxidase (GOx) from Aspergillus niger was bought from J&K Chemical. Peroxidase from horseradish (reagent grade) (HRP) was purchased from Solarbio. Cytochrome C (Cyt C) was purchased from Konoscience. Bradford solutions were purchased from Beyotime Biotechnology. Phosphate buffered saline (1×) was purchased from Shanghai Yuanye Biotechnology Co., Ltd. Glucose was purchased from Sinopharm Chemical Reagent Co., Ltd. 2,2′-azino-bis(3-ethyl benzothiazoline-6-sulfonic acid) diammonium salt (ABTS) was bought from MyBioScience. All the other reagents were purchased from Sigma-Aldrich and used without further purification.Fabrication of enzyme/MOF composites
Results and discussion
In order to demonstrate the concept of PISA, the processes of pressure-induced disorder32,38 and solvent-stimulated aging of MOFs36 were studied first. Different kinds of MOFs, such as ZIF-8, ZIF-67(Co), HKUST-1 and Mg-MOF-74, can be transformed into partially disordered MOFs by mechanical pressure or assisted by ball-milling. All the disordered MOFs can be recrystallized into crystalline structures stimulated by water vapor or organic solvents (Fig. S1–S14†). The study of this process served as a basis for enzyme encapsulation.As a proof of concept, the procedure of the PISA for pristine MOFs was demonstrated by ZIF-8 nanocrystals. First, 10 mg of activated ZIF-8 was ground and transferred into a 13 mm pellet die, and then a certain amount of mechanical pressure (0–1364 MPa) was applied. The effect of compression stress on the crystal structure has been systematically studied. The crystal structure, morphology and porosity of ZIF-8 were characterized by powder X-ray diffraction (PXRD), scanning electron microscopy (SEM) and Brunauer–Emmett–Teller (BET), respectively (Fig. 1a–d and S1–S5†). The characteristic diffraction peaks of ZIF-8 merged into a broad peak at 1364 MPa (Fig. 1a). The loss of crystallinity and porosity for pressed ZIF-8 has been determined by electron paramagnetic resonance (EPR)38 and BET, which was around 75% and 70%, separately (Fig. 1b and S5 and S6†). With increasing pressure, the packing density of the pressed ZIF-8 was also increased with few vacancies (Fig. 1c and d and S2†). Low compression loading only induced partial assembly of ZIF-8 NPs (Fig. 1c), while high compression loading resulted in the formation of tightly packed disordered ZIF-8 particles (Fig. 1d). All of this confirms that most of the ZIF-8 has been transformed into partially disordered ZIF-8 with compression loading of 1364 MPa.
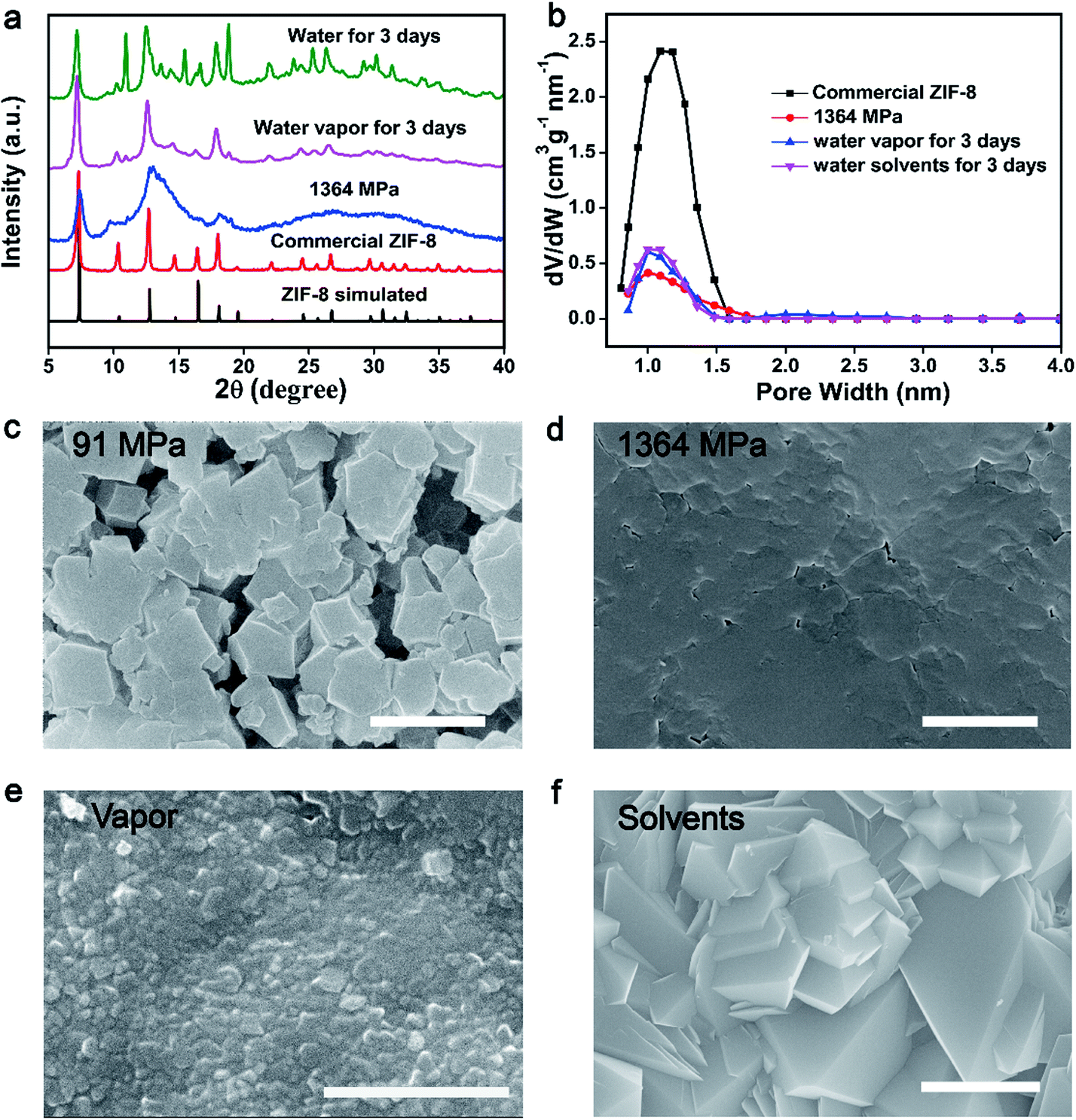 | ||
| Fig. 1 (a) PXRD patterns of simulated ZIF-8, commercial ZIF-8, and ZIF-8 under high pressure (1364 MPa), before and after recovery (water vapor or water at 25 °C for 3 days). (b) Pore volume analysis of commercial ZIF-8, and ZIF-8 under high pressure (1364 MPa), and after recovery (water vapor or water at 25 °C for 3 days). (c) SEM image of ZIF-8 at low pressure (91 MPa). (d) SEM image of ZIF-8 at high pressure (1364 MPa). (e) SEM image of disordered ZIF-8 recovered in water vapor. (f) SEM image of disordered ZIF-8 recovered in water (scale bars: 2 μm). | ||
Pressed ZIF-8 can be recrystallized in different solvents, such as water and methanol. Accordingly, water was chosen as the recovery solvent to gradually repair partially disordered MOFs. The prepared pressed ZIF-8 pellets (1364 MPa) were individually exposed to saturated water vapor or immersed in water for 3 days at 25 °C to recover the crystal structure. As expected, the crystallinity of ZIF-8 was reproduced as indicated by PXRD and SEM measurements. The characteristic and sharp Bragg peaks of the two samples centered at 12.7° and 18.07° appeared again after recovery, which could be ascribed to the (211) and (222) facets of ZIF-8 (Fig. 1a). Another four weak Bragg peaks centered at 14.7° (220), 16.5° (310), 24.5° (332) and 26.7° (510) also emerged. The Bragg peaks of both recovered ZIF-8 pellets were broader than but identical to those of the pristine ZIF-8 nanocrystals, indicating that partially disordered ZIF-8 was recrystallized under both treatments. Nevertheless, long time water treatment may induce the pressed ZIF-8 to transform into non-porous diamondoid (dia)-ZIF and ZIF-CO3-1 structures39–41 as reflected by PXRD (Fig. 1a and S7†), BET (Fig. 1b and S8†) and SEM (Fig. 1f and S9†). To further accelerate the conversion of disordered ZIF-8 into the crystalline one, 2-methyl imidazole/aqueous solvent was utilized because it could stabilize the structure of ZIF-8 and avoid further phase transition of ZIF-8 into other ZIF structures (Fig. S7 and S9†). In addition, methanol showed better performance in the recovery of disordered ZIF-8 structures (Fig. S7 and S10–S12†).
A significant surface morphology change was observed between the pressed and recrystallized ZIF-8 (Fig. 1c–f, S9, S11 and S12†). As compared to the disordered sample, polyhedral nanocrystals with varied sizes were found in both recovered samples. The surface area of pressed ZIF-8 and ZIF-8 aged in water or water vapor showed negligible differences (Fig. S8†). However, the pore volume of pressed ZIF-8 after water (water vapor) treatment increased from 0.42 cm3 g−1 nm−1 to 0.60 cm3 g−1 nm−1 (Fig. 1b). By treating pressed ZIF-8 with methanol solvent, around 76.3% of the porosity was recovered (Fig. S10†). The above experimental results jointly prove that the phase transition of disordered ZIF-8 can be triggered under the stimulation of different solvents.
In many reports, enzymes are relatively stable upon application of high mechanical pressure, which is also confirmed by our experiments. After applying the highest axial pressure for 5 min, the pellet machine can provide 5 mg GOx, and the enzyme activity remained almost the same as that of the free enzymes (Fig. S15†). Similar experimental results were also obtained for horseradish peroxidase (HRP) and cytochrome C (Cyt C) (Fig. S15†). The circular dichroism (CD) spectra of enzymes showed that the conformation of enzymes was maintained after the application of mechanical pressure (Fig. S16†). The results implied that such mechanical pressure had little impact on the activity of enzymes and may be a useful tool for the encapsulation of enzymes. Hence, in the process of PISA for the encapsulation of enzymes in MOFs, enzymes were mixed uniformly with MOFs. After compressing them together under a certain pressure, the enzymes were entrapped in the gaps between the MOF particles and the crystal structures of MOFs were destroyed partially due to mechanical pressure. These pressed composites were then recovered with water vapor at 25 °C, in which enzymes were trapped in the overlap of MOF particles or close to the surface of MOF particles during the dynamic coordination bond rearrangement and self-healing process of MOFs.
To further optimize and deeply understand the whole PISA encapsulation process, several key factors should be considered that may influence the encapsulation of enzymes into MOFs. Taking commercial ZIF-8 encapsulating GOx as an example, one factor should be the interactions between enzymes and MOFs. Commercial ZIF-8 (19.33 mV) was positively charged, while GOx was negatively charged (−10.91 mV). The enzyme and MOFs might be adsorbed together due to electrostatic interaction.29 While in the first step of PISA mixing process, the enzyme was fully adsorbed on the surface of MOFs and therefore favored the package of enzymes (Fig. S17†).
As we know, it is not easy for commercial ZIF-8 to disperse in water, which means the hydrophobic surface of commercial ZIF-8 will impede the adsorption of enzymes and lead to the low loading of enzymes. To enhance the interactions between enzymes and ZIF-8, ball-milling strategy was adopted to induce some defects on the surface of ZIF-8.37,42,43 After mixing 19 mg ZIF-8 that was premilled for 5 min with 1 mg GOx, the loading of GOx increased from 0 to 0.64% after washing with a large amount of water (the highest theoretical loading of GOx was 5%) (Fig. 2a). By pressing the mixers (grinding 19 mg ZIF-8 that was premilled for 5 min with 1 mg GOx) under 91 MPa and aging them into 1 mL water for 24 h, around 1.24% GOx was trapped in ZIF-8. In contrast, only 0.89% of GOx was loaded on ZIF-8 when the premilled process was not involved (Fig. 2a). The loadings of enzymes were determined by Bradford method (Fig. S18†). All of these prove that the 5 min milling (zeta potential for ZIF-8 premilled 5 min was 18.81 mV) can facilitate the interactions between ZIF-8 and GOx, thus enhancing the loading of enzymes in ZIF-8 by PISA strategy.
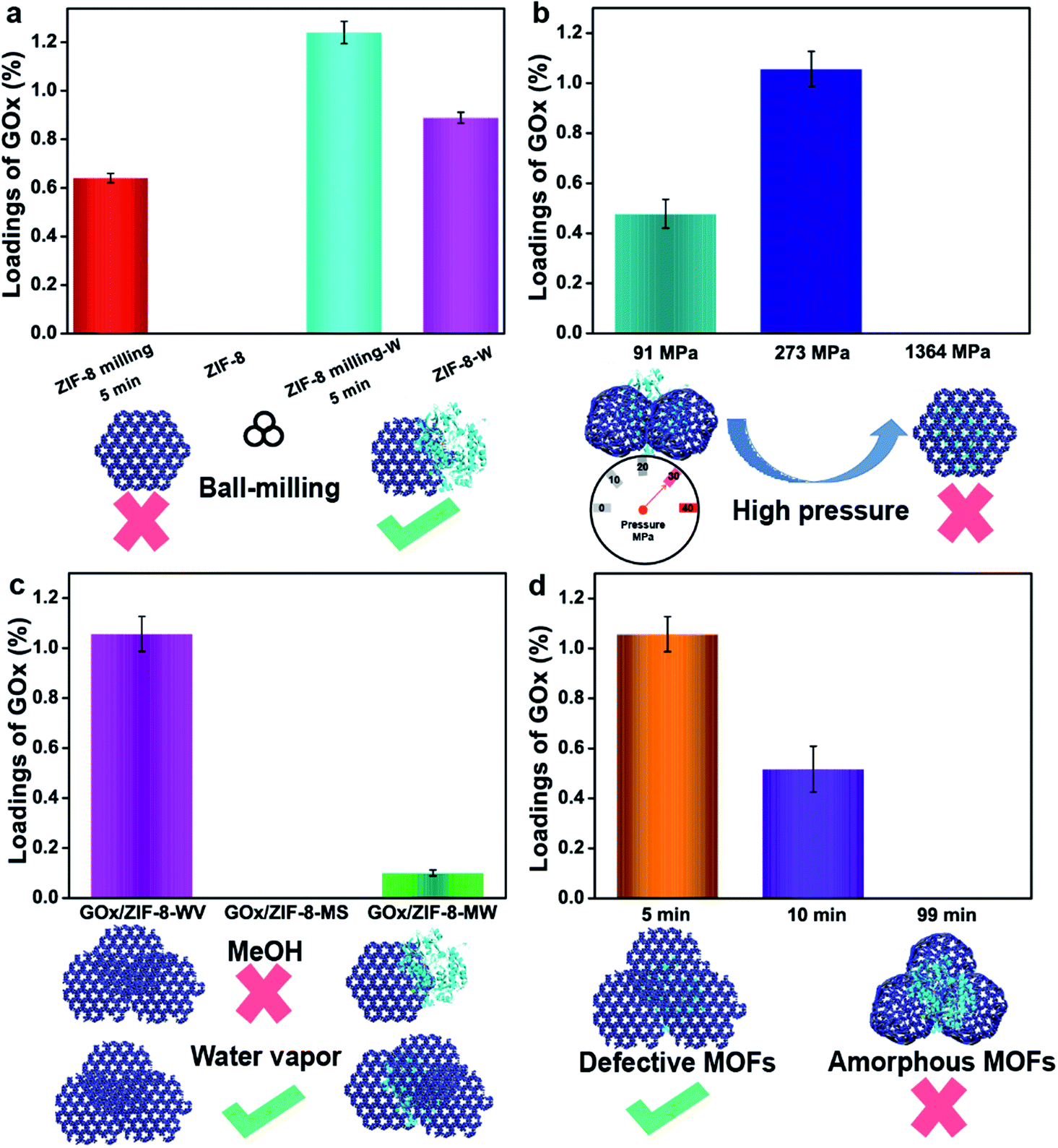 | ||
| Fig. 2 Exploration of the several factors influencing the encapsulation of enzymes. (a) Loadings of enzymes adsorbed on ZIF-8 with or without premilling treatment. The loadings of enzymes adsorbed on ZIF-8 with or without premilling through PISA after soaking in water for 24 h. (b) Loadings of enzymes in ZIF-8 through PISA with different pressures. (c) Loadings of enzymes in ZIF-8 through PISA with different stimulated solvents. (d) Loadings of enzymes in ZIF-8 through PISA with different premilling time. | ||
Secondly, mechanical pressure also plays a significant role in the process of encapsulation. Mechanical pressure provided higher internal stress, which accelerated the assembly and aging of ZIF-8 particles. To make our method milder, water vapor (WV) replaced water to finish the following experiments. After milling ZIF-8 for 5 min first, different pressure (91 MPa, 273 MPa and 1364 MPa) was introduced to analyze the effect of pressure on encapsulation. Under low pressure (91 MPa) or high pressure (1364 MPa), 20 mg of the mixture were recovered to obtain restored products, GOx/ZIF-8-WV. Unfortunately, the loading of enzymes was quite low as the high pressure blocked the solvents to infiltrate into the inner side of ZIF-8 pellets (0% for 1364 MPa) and the low pressure was not strong enough to assemble ZIF-8 particles and enzymes (0.47% for 91 MPa) (Fig. 2b). Through constant fine-tuning of pressure, we gladly found that the loading of enzymes was improved to around 1.05% at 273 MPa (Fig. 2b). This process unravels that only suitable pressure (not only suitable for encapsulation but also conductive to the penetration of solvent molecules) will benefit the encapsulation of enzymes.
Thirdly, stimulated solvents are also significant to the encapsulation of enzymes. Water or various organic solvents (methanol, DMF, etc.) have been used to synthesize MOFs44 and recover MOF crystal structures. During the encapsulation of enzymes, organic solvents and organic salts may negatively impact the activity of enzymes according to previous work.29,42–47 By contrast, water is mild and gentle for enzymes. In order to protect enzymes' active sites, water vapor recovery strategy was employed to recover the MOF crystal structures. After pressing the GOx and premilled ZIF-8 at 273 MPa, GOx/ZIF-8-X (X = WV, methanol solvents (MS), 2-methyl imidazole/water (MW)) was prepared. The loading of GOx in ZIF-8 recovered by water vapor was 1.05% (Fig. 2c), much higher than the GOx/ZIF-8 recovered under other conditions (0% for GOx/ZIF-8-MS and 0.1% for GOx/ZIF-MW). The superior loading was brought about by the infiltration of water molecules into ZIF-8 pellets and the acceleration of enzymes coordinating with unsaturated Zn clusters,37 which greatly enhanced the interactions between enzymes and ZIF-8. However, the methanol solvents and 2-methyl imidazole solution tended to accelerate the dissolution and regrowth of ZIF-8 particles themselves36 (Fig. S19†) instead of growing around enzymes, resulting in the unsatisfactory loadings. As a result, water vapor was supposed to be the best solvent to stimulate the aging of MOF particles for the enzyme encapsulation. Compared with traditional strategy, PISA strategy is quite mild for it avoids the use of organic solvents, large amounts of metal ions and organic ligands during enzyme encapsulation in MOFs.
The fourth important factor is the degree of disordered MOFs. Amorphous ZIF-8 (aZIF-8) was prepared by milling ZIF-8 under 30 Hz for 99 min. Through mixing aZIF-8 with GOx, pressing (273 MPa) and recovering it in water vapor, the loading of enzymes in ZIF-8 after aging in water vapor was 0% (Fig. 2d). By replacing aZIF-8 with ZIF-8 premilled for 10 min, the loading amount was improved to around 0.52%. The same data witnessed a further increase to around 1.05% when decreasing the milling time to 5 min (Fig. 2d), exhibiting 10.9% activity of free enzymes. This was probably caused by the mismatching of zeta potential between aZIF-8 (−1.42 mV) and GOx (−10.91 mV). As for ZIF-8 premilled for different lengths of time, ZIF-8 premilled for 5 min had a positive zeta potential (18.81 mV) while ZIF-8 premilled for 10 min had a negative zeta potential (−0.07 mV), which might improve the loading of enzymes encapsulated in ZIF-8. In addition, aMg-MOF-74 was also chosen to mix with GOx, and encapsulate it under pressure. After three days of water vapor treatment, the loading amount of GOx was about 2.05% with no enzymatic activity being observed (Fig. S20†). However, when using PISA, the loading of the encapsulated GOx was 3.66% and the activity of GOx was retained (6.9% activity of free enzymes) in GOx/Mg-MOF-74-WV. The comparison suggested that aMOFs were unfavorable for the encapsulation of certain enzymes (Fig. S20†). During the process of water vapor recovery, aMg-MOF-74 could release a large number of uncoordinated metal ions and ligands, resulting in enzyme inactivation. According to the above results, the process of using aMOFs and repairing them with water vapor is unfavorable for the encapsulation of GOx.
Based on the above study, the encapsulation of GOx in ZIF-8 can be improved by combining rapid ball milling with low pressure assisted water vapor stimulated recovery. The phase transition, and encapsulation process of GOx/ZIF-8-WV through the PISA strategy can be demonstrated by SEM, transmission electron microscopy (TEM), PXRD and laser confocal microscopy. The SEM images of pressed GOx/ZIF-8 showed the morphology of assembled ZIF-8 particles. Compared to the pressed GOx/ZIF-8, polyhedral nanocrystals with varied sizes were found in GOx/ZIF-8-WV samples, which indicated the aging of ZIF-8 particles (Fig. 3a–d). The crystal phase of GOx/ZIF-8-WV was changed as indicated by PXRD, which featured a mixed phase structure involving ∼94% phases of ZIF-CO3-1 and ∼6% ZIF-8 (ref. 39–41) (Fig. S19†). In order to prove the successful encapsulation of GOx, fluorescein isothiocyanate (FITC)-labeled GOx was prepared and found to evenly distribute in the ZIF-8 matrix after PISA treatment while the mixture of ZIF-8 particles with FITC-labeled GOx was observed to adsorb on the surface of ZIF-8 (Fig. 3h), which also corresponded to the results of the three-dimensional (3D) reconstruction confocal microscope graph (Fig. 3e–g). According to the above experimental results, GOx was encapsulated inside the crystalline ZIF-8 matrix through the PISA strategy.
 | ||
| Fig. 3 Characterization of GOx/ZIF-8-WV.54 (a) SEM image of pressed GOx/ZIF-8 (scale bar: 1 μm). (b) SEM image of GOx/ZIF-8-WV (scale bar: 1 μm). (c) TEM image of pressed GOx/ZIF-8 (scale bar: 100 nm). (d) TEM image of GOx/ZIF-8-WV (scale bar: 100 nm). (e) Laser confocal fluorescence microscopy imaging of the FITC-GOx/ZIF-8-WV (scale bar: 50 μm). (f) 3D reconstruction of laser confocal fluorescence microscopy imaging of the FITC-GOx/ZIF-8-WV. (g) Laser confocal fluorescence microscopy imaging of the FITC-GOx/ZIF-8-WV (scale bar: 5 μm). (h) Laser confocal fluorescence microscopy imaging of the FITC-GOx-on-ZIF-8 (scale bar: 10 μm). | ||
The PISA strategy can be used to encapsulate not only GOx but also other enzymes such as HRP and Cyt C in ZIF-8. Owing to the hydrophobic environment of ZIF-8, it may perturb the tertiary structure of enzymes and inhibit the transport of the substrate.48 During this study, we found that HRP/ZIF-8-WV exhibited 0.99% loading capacity and preserved 27.6% of the activity of the free enzymes when it was stored in a trace amount of aqueous solution (Fig. 4a). Compared with Cyt C, after the encapsulation by PISA, Cyt C/ZIF-8-WV had a high loading capacity of about 4.78%, and 79% of the free enzyme activity remained after being encapsulated into ZIF-8 (Fig. 4a). Partial crystals of HRP/ZIF-8-WV and Cyt C/ZIF-8-WV were transformed from ZIF-8 to ZIF-CO3-1 (Fig. S19†). The above experiments confirm that PISA is a general strategy for the loading of different enzymes into MOFs.
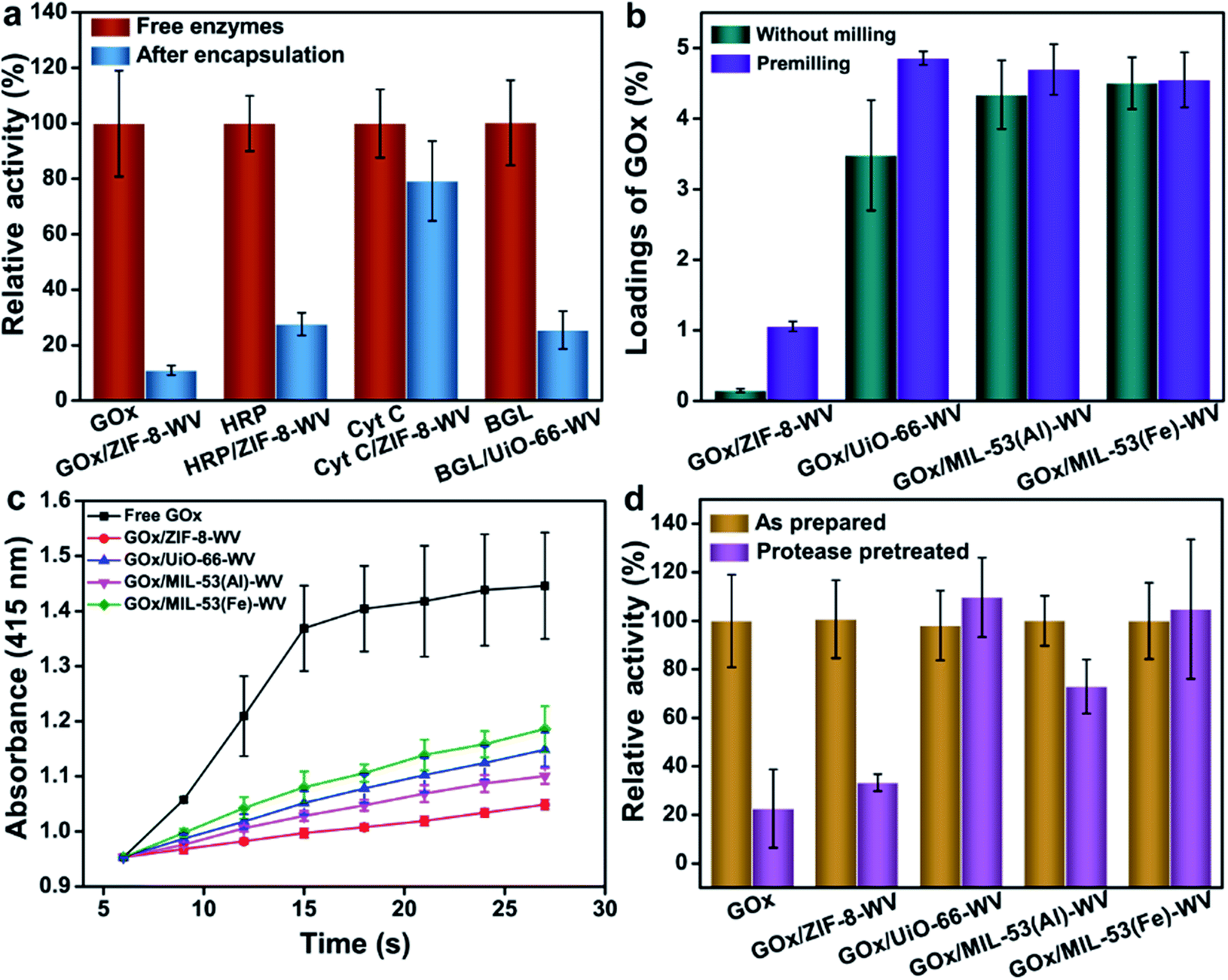 | ||
| Fig. 4 Universality of PISA for the encapsulation of enzymes. (a) Comparison of enzymatic activity between free enzymes, enzymes/ZIF-8-WV and BGL/UiO-66-WV. (b) Comparison of enzyme loadings in different MOFs. (c) Comparison of enzymatic activity between free GOx and GOx/MOFs-WV. (d) Biological activity of GOx and GOx/MOFs-WV before and after treatment with 1 mg mL−1 protease at 40 °C for 24 h. | ||
The PISA strategy can also be applied to other types of MOFs and realize the encapsulation and protection of enzymes. UiO-66, MIL-53(Al), and MIL-53(Fe) all have larger apertures compared to ZIF-8, which promise them to be desirable hosts for substrates' transportation. GOx was then encapsulated into UiO-66, MIL-53(Al) and MIL-53(Fe) by PISA and the loadings of enzymes were much higher than the loading of enzymes in ZIF-8 (Fig. 4b). For most MOFs, premilling treatment for 5 min boosted their loadings of enzymes while preserving their crystal structures (Fig. S21†). The loadings of enzymes in UiO-66, MIL-53(Al) and MIL-53(Fe) were around 4.86%, 4.7% and 4.55%, respectively (Fig. 4b). The crystal phases of UiO-66, MIL-53(Al) and MIL-53(Fe) changed a little due to crystal coarsening after the loading of enzymes, as reflected by the PXRD patterns and SEM images (Fig. S22 and S23†). It was found that the activity of MOFs loaded with GOx was lower than that of the free enzymes (22.7% and 26.9% that of the free enzymes for GOx/UiO-66-WV and GOx/MIL-53(Fe)-WV (Fig. 4c)). The activity difference between GOx/MOF composites may be ascribed to the effects of the metal-cluster in MOFs49 and MOF topologies. The Michaelis–Menten constants of GOx, GOx/UiO-66-WV and GOx/MIL-53(Fe)-WV were determined. Compared to the Km of free enzymes (1.72 mM), GOx/UiO-66-WV and GOx/MIL-53(Fe)-WV showed higher Km (11.82 mM and 5.99 mM), suggesting that the mass transfer was affected by the encapsulation (Fig. S24–S26†).
In order to further verify the universality and moderation of our strategy, β-glucosidase (BGL) has been encapsulated into UiO-66 by PISA. The results were analytically compared with those from the ball milling encapsulation strategy.30 The loading of BGL in UiO-66 was around 4.56% and the crystal structures were preserved well after the loading of enzymes (Fig. S22†). About 25.5% of the activity from BGL/UiO-66-WV was well maintained after encapsulation (Fig. 4a and S27†). While BGL was packaged by ball milling, it could only maintain about 5% (5 × 10−4 s−1 for BGL@UiO-66-NH2 and 1.0 × 10−2 s−1 for free BGL) of the enzymatic activity compared with that of the free enzyme. This proves that our method is milder and universal for enzyme encapsulation. To make our method more repeatable and efficient, several factors such as particle sizes of MOFs, pressure for the synthesis of enzyme/MOF composites, aging time, preparation process and electrostatic properties matching for the MOFs and enzymes have all been researched (Fig. S28–S32 and Movie S1†). The recovery time can be shortened to 24 h by adding 50 μL water to the pressed enzymes/MOFs (Fig. S32†), which can accelerate the aging process. It was surprising that GOx/UiO-66-WV exhibited 95% relative activity compared to native GOx at the same protein concentration when it was aged in a top covered mould to avoid coming into contact with condensed water (Fig. S32†). This means that the recovery condition is the most important for preparing enzyme/MOF composites with high activity. In addition, GOx/UiO-66-WV can still maintain ∼50% relative activity after 3 cycles which proved the stability of GOx/MOF composites prepared by PISA (Fig. S32†).
To investigate whether MOF structures could prevent the enzymes from being attacked by enzyme inhibitors, free GOx and GOx/MOFs-WV were immersed in 1 mg mL−1 protease solution for 24 h at 40 °C, respectively. For most GOx/MOFs-WV, their enzyme activity slightly decreased after poisoning, probably due to the adsorption of a small number of enzymes on the surface. Meanwhile, most of them maintained more than 70% of the original activity except GOx/ZIF-8-WV (40% of the original activity after poisoning) (Fig. 4d). In particular, GOx/UiO-66-WV and GOx/MIL-53(Fe)-WV barely lost their activity after protease treatment (99% and 94%). For free enzymes, the activity they retained was only 22.7% of their original value. The results strongly evidence that the PISA strategy can not only maintain high activity of enzymes but also protect enzymes reliably.
In the process of enzyme encapsulation through PISA, we not only focus on the encapsulation of enzymes, but also pay more attention to the encapsulation mechanisms. How do MOFs encapsulate the enzymes in this system and realize the protective effect? Based on previous experiments, two packaging mechanisms have been predicted as below. The first assumption is that MOFs function as glue. Specifically, the glue here is the repair of the MOF dynamic coordination bond with no external substances (metal ions/organic solvents). The second one is that the existence of trace water is conducive to the dissociation and precipitation of local metal ions and organic ligands, especially at the defects, and tends to trap the enzyme more closely to the surface of MOF structures.50,51
To validate the two analytical predictions above, metal nanoparticles (MNPs) were used to replace enzymes to explore the encapsulation mechanisms through the technology of 3D TEM image reconstruction, cross-sectional images, control experiments and molecular dynamics simulations. Firstly, ∼13 nm-diameter Au NPs and ∼3 nm-diameter Pt NPs were used to replace enzymes and to be encapsulated into commercial ZIF-8 and UiO-66-NH2 through PISA, mainly because the enzymes were hardly observed from TEM. A series of 3D high angle annular dark-field scanning transmission electron microscope (HAADF-STEM) images of Au/ZIF-8-WV at consecutive tilt angles from −55° to 60° with a 1° tilt increment were taken (Fig. 5a–c and Movies S2 and S3†). As shown in the 3D reconstruction images, many Au NPs were embedded on ZIF-8 structures from the side direction of Au/ZIF-8-WV (Fig. 5d–f). This was consistent with the results of nanoparticle reconstruction alone (Fig. 5g–i). In addition, the merged Pt/UiO-66-NH2-WV samples were cut apart and Pt NPs inside were observed clearly by the change of contrast compared with Pt/UiO-66-NH2-WV samples (Fig. S33†). The 3D TEM image reconstruction and the Pt NPs exposed inside after cutting Pt/UiO-66-NH2-WV samples prove that the enzymes are trapped between the interlayer of MOF particles during the coalescence of MOF particles.
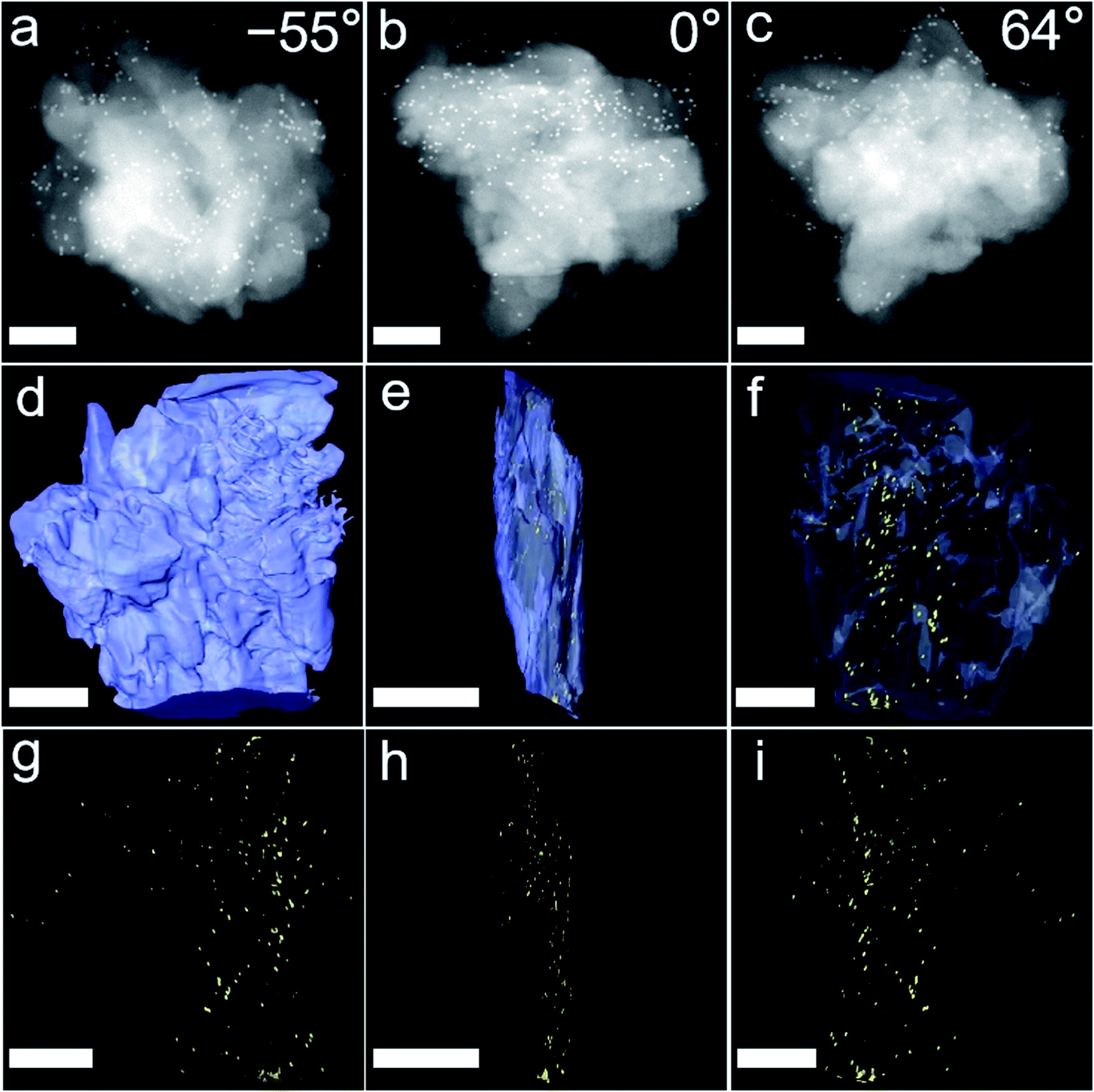 | ||
| Fig. 5 HAADF STEM and 3D reconstruction TEM images of 13 nm Au/ZIF-8-WV. (a–c) Representative HAADF-STEM images (−55°, 0° and 64°) captured from the video with a series of tilting angles for the Au/ZIF-8-WV sample taken with a 1° tilt increment step from −55° to 64°. The scale bar on the images is 250 nm. (d–f) Representative 3D reconstruction HRTEM images (0°, 90° and 180°) captured from the video for Au/ZIF-8-WV and (g–i) position of Au NPs; the scale bar in the images is 250 nm for (d), (f), (g), and (i), and 500 nm for (e) and (h). | ||
Moreover, partial enzymes may be encapsulated close to the surface of MOFs. To prove this prediction, control experiments have also been conducted. When mixing UiO-66 with GOx and aging them for 3 days, the GOx/UiO-66 showed 12.6% activity of free enzymes after being treated with protease (Fig. S34†). By mixing UiO-66 (premilled for 5 min) or UiO-66 (premilled for 10 min) with GOx and aging for 3 days, 50% of the enzymatic activity was retained (Fig. S34†) after being treated with protease, suggesting that defects were created on the MOF surface by ball milling and trapped enzymes during the dynamic coordination of MOFs. For all the samples, their crystal structures showed negligible change after the loading of GOx (Fig. S35†). Molecular dynamics simulations for the interactions between truncated MOFs and enzymes have been calculated, which proved that enzymes tended to adsorb on the defects of MOFs (Fig. S36 and S37, and Movies S4 and S5†). Based on the results above, low mechanical pressure combined with milling may create some defects on the surface of MOFs and lead to the encapsulation of enzymes close to the surface of MOFs. According to all of the results we obtained, enzymes should be trapped on the surface of MOFs or in the gaps between MOFs (Fig. 6).
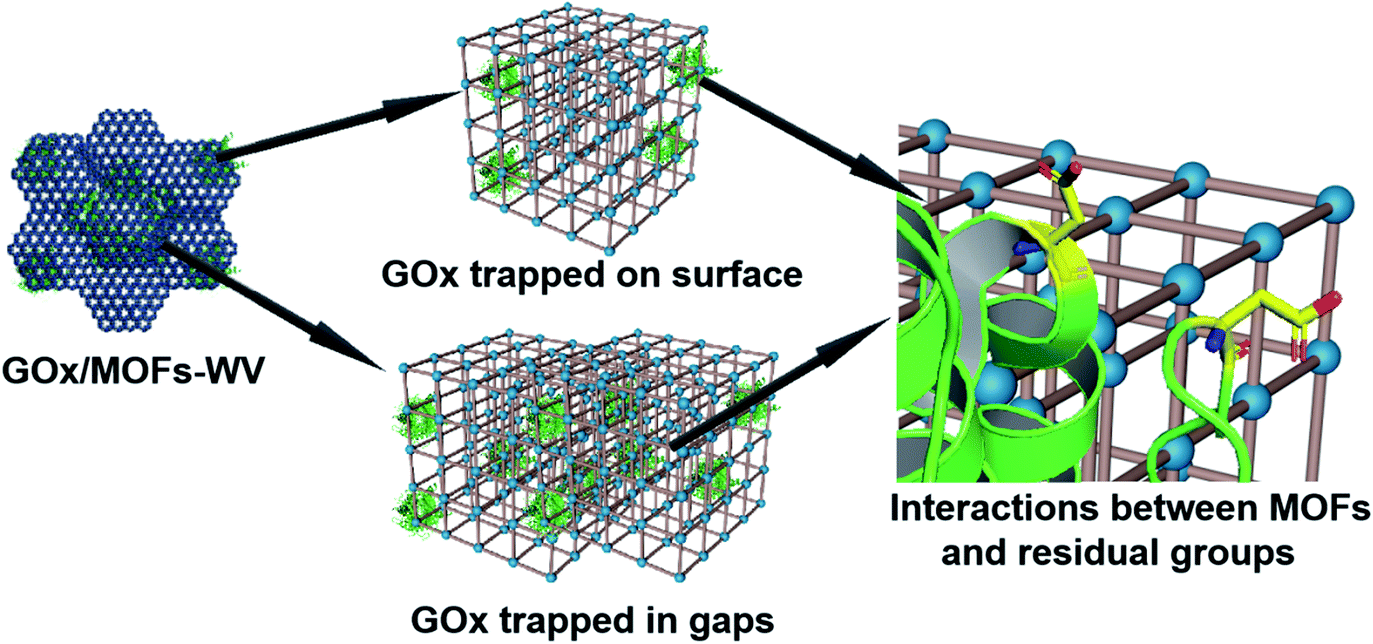 | ||
| Fig. 6 Schematic illustration of the possible position of GOx after being encapsulated into MOF structures and the interactions between MOFs and residual groups on GOx. | ||
Conclusions
In summary, we have developed a PISA strategy to encapsulate enzymes into MOF structures during the phase transition of MOFs by a mechanical pressure and water-vapor stimulated aging process. The PISA strategy is suitable for different enzymes (GOx, HRP, Cyt C and BGL) and can be extended to various MOFs (ZIF-8, UiO-66, UiO-66-NH2, Mg-MOF-74, MIL-53(Al) and MIL-53(Fe)). GOx/UiO-66-WV prepared under the best conditions showed 95% relative activity compared to free enzymes. The successfully synthesized GOx/MIL-53(Fe)-WV and GOx/UiO-66-WV composites retained more than 90% of the original activity after treatment with protease. Meanwhile, enzymes were trapped in the gaps between MOF particles or on the outer side of MOFs as proved by tilted HAADF-STEM, TEM images of sliced samples, control experiments and molecular dynamics simulations. This strategy serves as a good choice to design new MOF composites with enzymes. Moreover, other functional species such as luminescent molecules8,52 and drug molecules4,15,17 may also be accommodated into MOFs for preparing photonic MOF composites or dynamic drug delivery systems. The hosts are theoretically versatile to be other structures such as coordination networks53 and coordination compounds.54 We believe that the PISA strategy will be a potent toolbox for the synthesis of MOF composites without damaging the functionality of guest molecules and will also endow MOFs with new properties for a wide range of applications, such as biocatalysis, sensing, biotherapy and so on.Author contributions
Z. R., W. Q. Z. and J. N. W. designed and performed the experiments, analyzed the results and drafted the manuscript. Z. Y. Q. and L. W. L. assisted with catalytic experiments and manuscript revision. W. Q. Z., J. N. and C. C. were responsible for the PISA characterization process. H. H. S., X. L. Z., I. K., B. Z., and J. S. W. helped revise the manuscript. W. X. S. was responsible for molecular dynamics simulations. F. W. H. and W. N. Z. supervised the project, helped design the experiments and revised the manuscript. All authors contributed to the analysis of this paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Science Foundation for Distinguished Young Scholars (21625401), the National Natural Science Foundation of China (21727808, 21971114, and 22075139), and the Jiangsu Provincial Funds for Natural Science Foundation (BK20200090). We are thankful for the help from Prof. Matvey V. Fedin and Artem Poryvaev for calculating the ratios of disordered TEMPO@ZIF-8 under 1364 MPa by MATLAB.Notes and references
- Y. He, W. Zhou, G. Qian and B. Chen, Chem. Soc. Rev., 2014, 43, 5657–5678 RSC
.
- X. Zhao, Y. Wang, D.-S. Li, X. Bu and P. Feng, Adv. Mater., 2018, 30, 1705189 CrossRef PubMed
- Q. Yang, Q. Xu and H.-L. Jiang, Chem. Soc. Rev., 2017, 46, 4774–4808 RSC
- A. C. McKinlay, R. E. Morris, P. Horcajada, G. Férey, R. Gref, P. Couvreur and C. Serre, Angew. Chem., Int. Ed., 2010, 49, 6260–6266 CrossRef CAS PubMed
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed
- P.-L. Wang, L.-H. Xie, E. A. Joseph, J.-R. Li, X.-O. Su and H.-C. Zhou, Chem. Rev., 2019, 119, 10638–10690 CrossRef CAS PubMed
- Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed
- Y. Cui, J. Zhang, H. He and G. Qian, Chem. Soc. Rev., 2018, 47, 5740–5785 RSC
- L. Chen, R. Luque and Y. Li, Chem. Soc. Rev., 2017, 46, 4614–4630 RSC
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed
- T. Wang, L. Gao, J. Hou, S. J. Herou, J. T. Griffiths, W. Li, J. Dong, S. Gao, M.-M. Titirici and R. V. Kumar, Nat. Commun., 2019, 10, 1340 CrossRef PubMed
- A. Kirchon, L. Feng, H. F. Drake, E. A. Joseph and H.-C. Zhou, Chem. Soc. Rev., 2018, 47, 8611–8638 RSC
- P. Falcaro, A. J. Hill, K. M. Nairn, J. Jasieniak, J. I. Mardel, T. J. Bastow, S. C. Mayo, M. Gimona, D. Gomez, H. J. Whitfield, R. Riccò, A. Patelli, B. Marmiroli, H. Amenitsch, T. Colson, L. Villanova and D. Buso, Nat. Commun., 2011, 2, 237 CrossRef PubMed
- Y. Mao, J. Li, W. Cao, Y. Ying, P. Hu, Y. Liu, L. Sun, H. Wang, C. Jin and X. Peng, Nat. Commun., 2014, 5, 5532 CrossRef PubMed
- G. Lu, S. Li, Z. Guo, O. K. Farha, B. G. Hauser, X. Qi, Y. Wang, X. Wang, S. Han, X. Liu, J. S. DuChene, H. Zhang, Q. Zhang, X. Chen, J. Ma, S. C. J. Loo, W. D. Wei, Y. Yang, J. T. Hupp and F. Huo, Nat. Chem., 2012, 4, 310 CrossRef CAS PubMed
- M. Zhao, K. Yuan, Y. Wang, G. Li, J. Guo, L. Gu, W. Hu, H. Zhao and Z. Tang, Nature, 2016, 539, 76 CrossRef CAS PubMed
- P. Horcajada, T. Chalati, C. Serre, B. Gillet, C. Sebrie, T. Baati, J. F. Eubank, D. Heurtaux, P. Clayette and C. Kreuz, Nat. Mater., 2010, 9, 172 CrossRef CAS PubMed
- X. Lian, Y. Fang, E. Joseph, Q. Wang, J. Li, S. Banerjee, C. Lollar, X. Wang and H.-C. Zhou, Chem. Soc. Rev., 2017, 46, 3386–3401 RSC
- S. Huang, X. Kou, J. Shen, G. Chen and G. Ouyang, Angew. Chem., Int. Ed., 2020, 59, 8786–8798 CrossRef CAS PubMed
- W. Liang, P. Wied, F. Carraro, C. J. Sumby, B. Nidetzky, C.-K. Tsung, P. Falcaro and C. J. Doonan, Chem. Rev., 2021, 121, 1077–1129 CrossRef CAS PubMed
- J. Guan, Y. Hu, Y. Wang, H. Li, Z. Xu, T. Zhang, P. Wu, S. Zhang, G. Xiao, W. Ji, L. Li, M. Zhang, Y. Fan, L. Li, B. Zheng, W. Zhang, W. Huang and F. Huo, Adv. Mater., 2017, 29, 1606290 CrossRef PubMed
- J. V. Morabito, L.-Y. Chou, Z. Li, C. M. Manna, C. A. Petroff, R. J. Kyada, J. M. Palomba, J. A. Byers and C.-K. Tsung, J. Am. Chem. Soc., 2014, 136, 12540–12543 CrossRef CAS PubMed
- Z. Li, T. M. Rayder, L. Luo, J. A. Byers and C.-K. Tsung, J. Am. Chem. Soc., 2018, 140, 8082–8085 CrossRef CAS PubMed
- V. Lykourinou, Y. Chen, X.-S. Wang, L. Meng, T. Hoang, L.-J. Ming, R. L. Musselman and S. Ma, J. Am. Chem. Soc., 2011, 133, 10382–10385 CrossRef CAS PubMed
- K. Liang, R. Ricco, C. M. Doherty, M. J. Styles, S. Bell, N. Kirby, S. Mudie, D. Haylock, A. J. Hill, C. J. Doonan and P. Falcaro, Nat. Commun., 2015, 6, 7240 CrossRef CAS PubMed
- W.-H. Chen, M. Vázquez-González, A. Zoabi, R. Abu-Reziq and I. Willner, Nat. Catal., 2018, 1, 689–695 CrossRef CAS
- F. Lyu, Y. Zhang, R. N. Zare, J. Ge and Z. Liu, Nano Lett., 2014, 14, 5761–5765 CrossRef CAS PubMed
- X. Wu, J. Ge, C. Yang, M. Hou and Z. Liu, Chem. Commun., 2015, 51, 13408–13411 RSC
- G. Chen, X. Kou, S. Huang, L. Tong, Y. Shen, W. Zhu, F. Zhu and G. Ouyang, Angew. Chem., Int. Ed., 2020, 59, 2867–2874 CrossRef CAS PubMed
- T.-H. Wei, S.-H. Wu, Y.-D. Huang, W.-S. Lo, B. P. Williams, S.-Y. Chen, H.-C. Yang, Y.-S. Hsu, Z.-Y. Lin, X.-H. Chen, P.-E. Kuo, L.-Y. Chou, C.-K. Tsung and F.-K. Shieh, Nat. Commun., 2019, 10, 5002 CrossRef PubMed
- K. W. Chapman, G. J. Halder and P. J. Chupas, J. Am. Chem. Soc., 2009, 131, 17546–17547 CrossRef CAS PubMed
- K. W. Chapman, D. F. Sava, G. J. Halder, P. J. Chupas and T. M. Nenoff, J. Am. Chem. Soc., 2011, 133, 18583–18585 CrossRef CAS PubMed
- S. Yuan, X. Sun, J. Pang, C. Lollar, J.-S. Qin, Z. Perry, E. Joseph, X. Wang, Y. Fang, M. Bosch, D. Sun, D. Liu and H.-C. Zhou, Joule, 2017, 1, 806–815 CrossRef CAS
- T. Friščić, I. Halasz, P. J. Beldon, A. M. Belenguer, F. Adams, S. A. J. Kimber, V. Honkimäki and R. E. Dinnebier, Nat. Chem., 2013, 5, 66–73 CrossRef PubMed
- R. Monteagudo-Olivan, L. Paseta, G. Potier, P. López-Ram-de-Viu and J. Coronas, Eur. J. Inorg. Chem., 2019, 2019, 29–36 CrossRef CAS
- T. Panda, S. Horike, K. Hagi, N. Ogiwara, K. Kadota, T. Itakura, M. Tsujimoto and S. Kitagawa, Angew. Chem., Int. Ed., 2017, 56, 2413–2417 CrossRef CAS PubMed
- E. Shearier, P. Cheng, Z. Zhu, J. Bao, Y. H. Hu and F. Zhao, RSC Adv., 2016, 6, 4128–4135 RSC
- A. S. Poryvaev, D. M. Polyukhov and M. V. Fedin, ACS Appl. Mater. Interfaces, 2020, 12, 16655–16661 CrossRef CAS PubMed
- W. Liang, R. Ricco, N. K. Maddigan, R. P. Dickinson, H. Xu, Q. Li, C. J. Sumby, S. G. Bell, P. Falcaro and C. J. Doonan, Chem. Mater., 2018, 30, 1069–1077 CrossRef CAS
- F. Carraro, M. d. J. Velásquez-Hernández, E. Astria, W. Liang, L. Twight, C. Parise, M. Ge, Z. Huang, R. Ricco, X. Zou, L. Villanova, C. O. Kappe, C. Doonan and P. Falcaro, Chem. Sci., 2020, 11, 3397–3404 RSC
- W. Liang, H. Xu, F. Carraro, N. K. Maddigan, Q. Li, S. G. Bell, D. M. Huang, A. Tarzia, M. B. Solomon, H. Amenitsch, L. Vaccari, C. J. Sumby, P. Falcaro and C. J. Doonan, J. Am. Chem. Soc., 2019, 141, 2348–2355 CrossRef CAS PubMed
- T. D. Bennett, T. K. Todorova, E. F. Baxter, D. G. Reid, C. Gervais, B. Bueken, B. Van de Voorde, D. De Vos, D. A. Keen and C. Mellot-Draznieks, Phys. Chem. Chem. Phys., 2016, 18, 2192–2201 RSC
- B. Szczęśniak, S. Borysiuk, J. Choma and M. Jaroniec, Mater. Horizons, 2020, 7, 1457–1473 RSC
- S. Dai, F. Nouar, S. Zhang, A. Tissot and C. Serre, Angew. Chem., Int. Ed., 2021, 60, 4282–4288 CrossRef CAS PubMed
- T. T. Herskovits, B. Gadegbeku and H. Jaillet, J. Biol. Chem., 1970, 245, 2588–2598 CrossRef CAS PubMed
- S. Toba and K. M. Merz, J. Am. Chem. Soc., 1997, 119, 9939–9948 CrossRef CAS
- N. Vasileva and T. Godjevargova, Mater. Sci. Eng. C, 2005, 25, 17–21 CrossRef
- W. Liang, H. Xu, F. Carraro, N. K. Maddigan, Q. Li, S. G. Bell, D. M. Huang, A. Tarzia, M. B. Solomon, H. Amenitsch, L. Vaccari, C. J. Sumby, P. Falcaro and C. J. Doonan, J. Am. Chem. Soc., 2019, 141, 2348–2355 CrossRef CAS PubMed
- H. An, J. Song, T. Wang, N. Xiao, Z. Zhang, P. Cheng, S. Ma, H. Huang and Y. Chen, Angew. Chem., Int. Ed., 2020, 59, 16764–16769 CrossRef CAS PubMed
- C. Hu, Y. Bai, M. Hou, Y. Wang, L. Wang, X. Cao, C.-W. Chan, H. Sun, W. Li, J. Ge and K. Ren, Sci. Adv., 2020, 6, eaax5785 CrossRef CAS PubMed
- S. Gao, J. Hou, Z. Deng, T. Wang, S. Beyer, A. G. Buzanich, J. J. Richardson, A. Rawal, R. Seidel, M. Y. Zulkifli, W. Li, T. D. Bennett, A. K. Cheetham, K. Liang and V. Chen, Chem, 2019, 5, 1597–1608 CAS
- V. Glembockyte, M. Frenette, C. Mottillo, A. M. Durantini, J. Gostick, V. Štrukil, T. Friščić and G. Cosa, J. Am. Chem. Soc., 2018, 140, 16882–16887 CrossRef CAS PubMed
- N. Hosono and S. Kitagawa, Acc. Chem. Res., 2018, 51, 2437–2446 CrossRef CAS PubMed
- M. Jin, T. Sumitani, H. Sato, T. Seki and H. Ito, J. Am. Chem. Soc., 2018, 140, 2875–2879 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta02070j |
| This journal is © The Royal Society of Chemistry 2022 |