
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNi single atoms on carbon nitride for visible-light-promoted full heterogeneous dual catalysis†
Minjoon
Kwak‡
a,
Jinsol
Bok‡
 ab,
Byoung-Hoon
Lee‡
ab,
Jongchan
Kim
a,
Youngran
Seo
a,
Sumin
Kim
a,
Hyunwoo
Choi
a,
Wonjae
Ko
ab,
Wytse
Hooch Antink
ab,
Chan Woo
Lee
ab,
Guk Hee
Yim
a,
Hyojin
Seung
ab,
Chansul
Park
ab,
Kug-Seung
Lee
c,
Dae-Hyeong
Kim
ab,
Taeghwan
Hyeon
*ab and
Dongwon
Yoo
*ab
ab,
Byoung-Hoon
Lee‡
ab,
Jongchan
Kim
a,
Youngran
Seo
a,
Sumin
Kim
a,
Hyunwoo
Choi
a,
Wonjae
Ko
ab,
Wytse
Hooch Antink
ab,
Chan Woo
Lee
ab,
Guk Hee
Yim
a,
Hyojin
Seung
ab,
Chansul
Park
ab,
Kug-Seung
Lee
c,
Dae-Hyeong
Kim
ab,
Taeghwan
Hyeon
*ab and
Dongwon
Yoo
*ab
aDepartment of Chemical and Biological Engineering, Institute of Chemical Processes, Seoul National University, Seoul 08826, Republic of Korea. E-mail: thyeon@snu.ac.kr; dwyoo@snu.ac.kr
bCenter for Nanoparticle Research, Institute for Basic Science (IBS), Seoul 08826, Republic of Korea
cPohang Accelerator Laboratory (PAL), Pohang University of Science and Technology (POSTECH), Pohang, Gyeongbuk 37673, Republic of Korea
First published on 20th June 2022
Abstract
Visible-light-driven organic transformations are of great interest in synthesizing valuable fine chemicals under mild conditions. The merger of heterogeneous photocatalysts and transition metal catalysts has recently drawn much attention due to its versatility for organic transformations. However, these semi-heterogenous systems suffered several drawbacks, such as transition metal agglomeration on the heterogeneous surface, hindering further applications. Here, we introduce heterogeneous single Ni atoms supported on carbon nitride (NiSAC/CN) for visible-light-driven C–N functionalization with a broad substrate scope. Compared to a semi-heterogeneous system, high activity and stability were observed due to metal–support interactions. Furthermore, through systematic experimental mechanistic studies, we demonstrate that the stabilized single Ni atoms on CN effectively change their redox states, leading to a complete photoredox cycle for C–N coupling.
Introduction
Heterogeneous photocatalytic reactions have received considerable attention for the greener synthesis of valuable fine chemicals and fuels from solar energy.1–3 In addition to the sole use of heterogeneous photocatalysts, dual catalysis (i.e., the merger of transition metal catalysis and photocatalysis) enables novel organic transformations, which are elusive under traditional methods.4–7 Although recent studies on semi-heterogeneous systems, exploiting metal salts together with a heterogeneous photocatalyst, showed some promise, several drawbacks, including the non-recyclability of metal catalysts and photocatalyst deactivation problems due to metal aggregation, remain.8,9Single-atom catalysts (SAC) are representative alternatives to complement the drawbacks of semi-heterogeneous dual catalysis as a bridge between heterogeneous and homogeneous systems.10–12 In SAC systems, various materials can be used to support single metal atoms, including carbon-based materials,13,14 oxides,15,16 and metals.17,18 Carbon nitride (CN), a metal-free 2D semiconductor, is one of the most promising supports for SACs because of its abundant anchoring sites for single metal atoms and low production cost. Moreover, CN has an appropriate band gap (about 2.7 eV) and band position to replace precious metal-based photocatalysts such as Ru and Ir complexes.1,19 Also, the polymeric ring structure makes carbon nitride highly stable under reaction conditions compared to photoactive complexes and organic dyes.20–22 Albeit a few photocatalytic organic transformations of C–O bond formation by SAC supported on CN have been reported,23–27 the range of organic reactions needs to be expanded for a broad application.
Herein, we demonstrate Ni-catalyzed photoredox C–N bond formation, which is an important moiety in natural and pharmaceutical products,28,29 using single Ni atoms supported on carbon nitride (NiSAC/CN). Notably, NiSAC/CN shows no aggregation of nickel species under blue light and a higher yield of the desired product compared to a semi-heterogeneous system. Intriguingly, we found that Ni single atoms on the heterogeneous surface of CN can dynamically change their oxidation state from the initial formation of Ni(0) to regulate the electron transfer process. We provide a picture of how the redox cycle of Ni atoms and photogenerated electrons and holes are interrelated to complete the heterogeneous photoredox process.
Results and discussion
Synthesis and characterization of catalysts
NiSAC/CN was synthesized by the wet impregnation method followed by calcination in an Ar/H2 atmosphere based on a previous report.30 The X-ray diffraction (XRD) pattern of NiSAC/CN showed no additional peak other than the characteristic peaks of the CN structure, which are from the stacking structure at 27.5° and the in-plane repeating unit at 13.1°, indicating that the structure of CN is well preserved after the deposition of single Ni atoms (Fig. S1†). For comparison, Ni nanoparticles supported on CN (NiNP/CN) were synthesized by a light-assisted deposition method (Fig. S2†), and the XRD pattern of NiNP/CN shows the presence of Ni nanoparticles (Fig. S1†). No aggregated Ni-based species were observed in the transmission electron microscopy (TEM) image of NiSAC/CN, consistent with the XRD analysis (Fig. 1a and S1†). Energy dispersive X-ray spectroscopy (EDS) elemental mapping in scanning transmission electron microscopy (STEM) mode shows the abundance of C and N signals from CN and that the Ni atoms are uniformly dispersed on the CN surface (Fig. 1b). To visualize the Ni atoms, high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) was performed (Fig. 1c). The homogeneous distribution of Ni atoms was observed throughout the CN surface (the bright spots correspond to Ni atoms). Extended X-ray absorption fine structure spectroscopy (EXAFS) also confirms the homogeneous distribution of Ni atoms without agglomeration (Fig. 1d and S3†). The peaks observed at 1.66 Å correspond to nickel coordinated with nitrogen and the detailed local structure of NiSAC/CN was elucidated by curve-fitting analysis. The Ni content in NiSAC/CN was measured by inductively coupled plasma optical emission spectroscopy (ICP-OES) and was determined to be 0.53 wt%.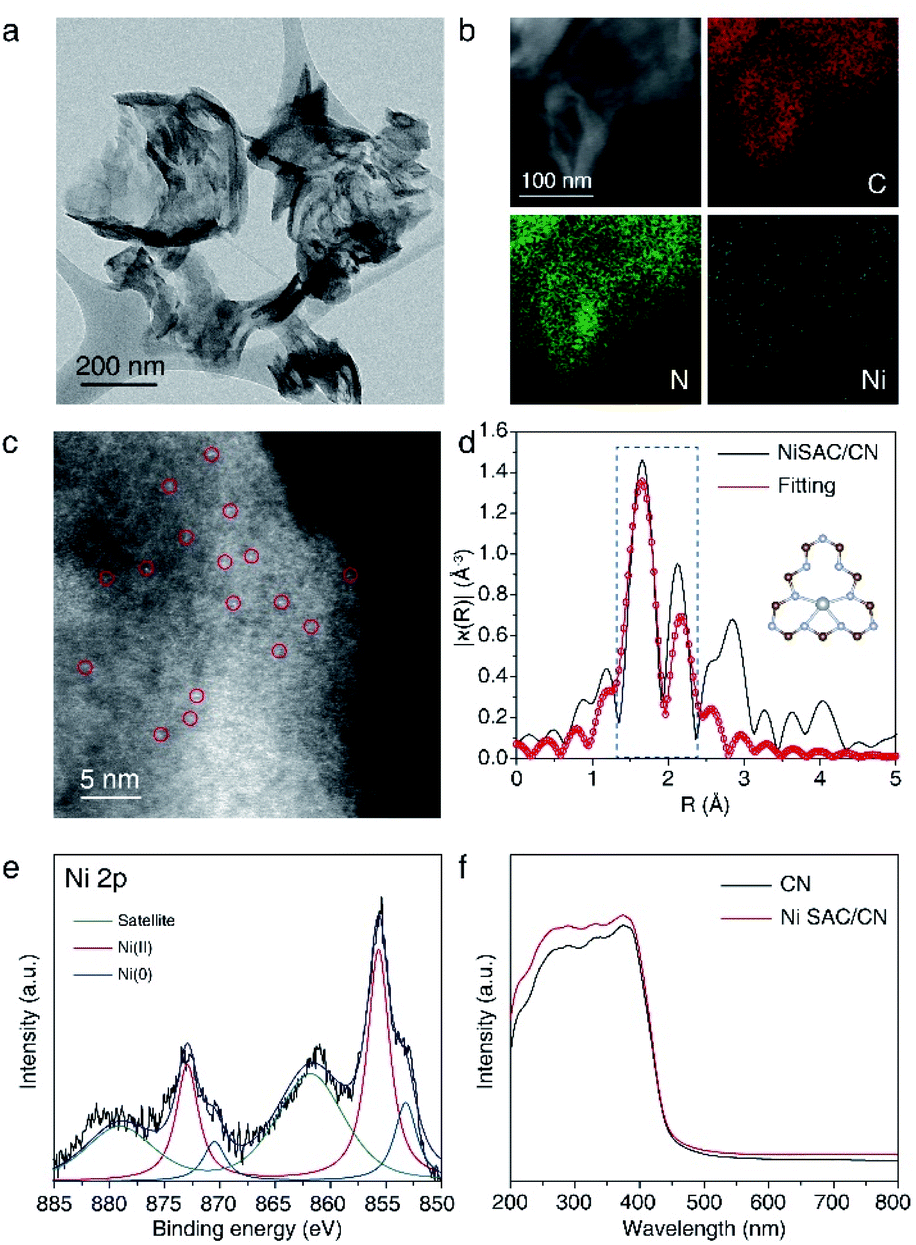 | ||
| Fig. 1 (a) Representative TEM image, (b) EDS mapping image, (c) HAADF-STEM image, (d) Fourier transform of Ni K-edge EXAFS spectra (silver: Ni, grey: N, brown: C), and (e) Ni 2p XPS spectra of NiSAC/CN. (f) UV-vis spectra of CN and NiSAC/CN. | ||
To verify the chemical states of the single Ni atoms on CN, X-ray absorption near edge structure (XANES) and X-ray photoelectron spectroscopy (XPS) studies were performed. In Ni K-edge XANES spectra (Fig. S4†), NiSAC/CN exhibited a higher edge position and white light intensity compared to Ni foil, implying the oxidized state of Ni species in NiSAC/CN. In the Ni 2p XPS spectra (Fig. 1e), three peaks were detected, attributed to Ni(0) species (853.2 eV), Ni(II) species (855.7 eV), and a satellite peak (861.8 eV), indicating that both oxidation states coexist. This implies that both oxidation states can be stabilized on the surface of CN, which provides the redox versatility necessary for photoredox coupling reactions. In the UV-vis spectra, CN and NiSAC/CN display similar spectra, implying that stabilization of the Ni atoms does not significantly modify the absorption properties of CN (Fig. 1f and S5†).
Photoredox C–N coupling
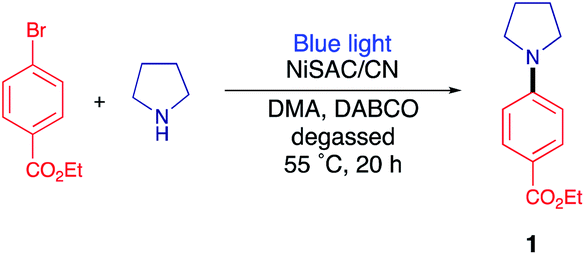
As a model reaction, the C–N coupling of ethyl 4-bromobenzoate with pyrrolidine was conducted using NiSAC/CN as a heterogeneous catalyst under a blue LED light. When the reaction mixture was irradiated in dimethylacetamide (DMA) in the presence of diazabicyclo[2,2,2]octane (DABCO) as a base, desired product 1 was obtained in 40% yield after 20 h (Table 1, entry 1). To verify that the reaction proceeded by dual catalysis, control experiments were performed in the absence of light, carbon nitride, and nickel (Table 1, entries 2–4). No product was detected after 20 h, demonstrating that light, single Ni atoms, and carbon nitride are pivotal components for C–N coupling. Notably, a yield of 40% was obtained in the absence of the base (i.e., DABCO), suggesting that pyrrolidine served as both the base and substrate due to its basic properties (Table 1, entry 5). When the amount of pyrrolidine was increased from 3 eq. to 5 eq., the desired product was delivered in 96% yield. Furthermore, to determine which reactive species are involved in the reaction cycle, triethanolamine (TEOA), AgNO3, and (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) were added to the reaction as hole, electron, and radical scavengers, respectively (Table 1, entries 6–8). The addition of either scavenger led to no conversion of the starting material, implying that electrons, holes, and radicals are all engaged in the formation of the products. In addition, nickel nanoparticle/carbon nitride (NiNP/CN) was used instead of NiSAC/CN (Table 1, entry 9). Although NiNP/CN has a higher nickel content of 4.91 wt%, no desired product was detected. An even higher yield (79%) was obtained when DABCO was replaced with more basic quinuclidine (Table 1, entry 10). When a lower amount of catalyst was exploited, the yield decreased from 79% to 30%, suggesting that NiSAC/CN mediates the dual catalytic cycle in this reaction (Table S1†).|
|
||
|---|---|---|
| Entry | Deviation | Yield 1b (%) |
| a Reaction conditions: ethyl 4-bromobenzoate (0.1 mmol), pyrrolidine (0.3 mmol), DABCO (0.22 mmol), DMA (0.5 mL), NiSAC/CN (30 mg). b Determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as an internal standard. c Pyrrolidine (0.5 mmol) was added. d TEOA (excess, 0.1 mL) was added as a hole scavenger. e AgNO3 (0.1 mmol) was added as an electron scavenger. f TEMPO (0.1 mmol) was added as a radical scavenger. | ||
| 1 | None | 40 |
| 2 | No light | n.d. |
| 3 | NiCl2·6H2O | n.d. |
| 4 | Carbon nitride | n.d. |
| 5 | No DABCO | 40 (96c) |
| 6d | TEOA | n.d. |
| 7e | AgNO3 | n.d. |
| 8f | TEMPO | n.d. |
| 9 | NiNP/CN | n.d. |
| 10 | Quinuclidine | 79 |
Next, we investigated whether the anchoring of nickel atoms to carbon nitride affects photocatalytic activity. It was expected that the quick electron transfer from carbon nitride to nickel species on the heterogeneous surface contributes to the higher activity (Fig. 2a). In contrast, nickel complexes should diffuse near the carbon nitride surface to gain an electron from the photocatalyst in the semi-heterogeneous system (Fig. 2b). Thus, it was anticipated that inefficient charge transfer from carbon nitrides to free nickel is an obstacle to the reaction efficiency of the semi-heterogeneous system.31 In line with our expectations, when carbon nitride and nickel species were separately introduced (i.e., semi-heterogeneous system), a lower yield of the desired product was observed compared to the reaction mediated by NiSAC/CN (Fig. 2c). Time-resolved PL spectra and steady-state photoluminescence (PL) spectra also corroborate the electron transfer from carbon nitride to single Ni atoms (Fig. S6†). When single Ni atoms were introduced, a decrease in PL intensity was observed due to the suppressed charge recombination. Moreover, a decrease in the PL lifetime (CN: 1.41 ns, NiSAC/CN: 1.07 ns) was observed due to the electron transfer from CN to single Ni atoms.
 | ||
| Fig. 2 The schematic picture of (a) NiSAC/CN and (b) semi-heterogeneous NiCl2·6H2O and carbon nitride (blue, gray balls, and yellow sheets represent Ni atoms, Ni aggregates, and carbon nitride). (c) Comparison of yields under NiSAC/CN or semi-heterogeneous NiCl2·6H2O and carbon nitride. (d) TEM image of carbon nitride after reaction under semi-heterogeneous system. * The indicated yields are the average of three replicate experiments. | ||
In addition, when g-CN and nickel precursors were separately introduced, the color of carbon nitride occasionally became gray (Fig. S7†). Material characterization studies, including TEM, STEM, and EDS elemental mapping, were conducted in the semi-heterogeneous system after reaction, and nickel agglomeration was observed on the surface (Fig. 2d). Severe aggregation of transition metal salts and subsequent deactivation of carbon nitride can reduce its versatility. Note that previous reports indicated that semi-heterogeneous systems for C–N coupling using NiCl2 salts and CN were not recyclable due to the formation of Ni particles under blue light.8
NiSAC/CN was analyzed after conducting the C–N coupling reactions using various characterization tools. TEM, STEM, and EDS elemental mapping images (Fig. S8†) show that the single Ni atoms do not aggregate into nanoparticles in contrast to a semi-heterogeneous system. Furthermore, the HAADF-STEM image (Fig. 3a) and EXAFS data (Fig. 3b) confirm that single Ni atoms maintain their single atomic form well after the reaction. The absence of metal aggregation, a major challenge of the single-atom catalyst due to high surface energy, can be attributed to the nitrogen-rich heterocycles of carbon nitride. Therefore, the surface free energy of a single atom can be minimized due to metal–support interactions.32,33 Additionally, in Ni 2p spectra (Fig. 3c), peak shift was negligible after the reaction, which was within about 0.1 eV, implying oxidation states of Ni atoms were well maintained. Also, the ratio of Ni(II)/Ni(0) remains almost identical (ca. 2.95). In this regard, NiSAC/CN maintains its activity after the light-driven C–N coupling reaction, and the reaction could be repeated three times with excellent yields (Fig. S9†), although 17% of Ni leaching was observed after the reaction. To determine whether the leached nickel participates in C–N coupling, a hot filtration test was conducted, and it was supported that the reaction proceeds in a heterogeneous manner (Table S2 and Fig. S10†).
 | ||
| Fig. 3 (a) Representative HAADF-STEM image, (b) Ni K-edge EXAFS spectra, and (c) XPS spectra of NiSAC/CN after the model reaction. | ||
Mechanism study
We further conducted comprehensive experiments for elucidating the mechanism of reaction on the surface of NiSAC/CN. To gain insight into how redox-active Ni atoms mediate heterogeneous photoredox C–N coupling, we performed a mechanistic study through further experiments such as electron paramagnetic resonance (EPR), photoluminescence (PL), and fluorescence quenching studies. A quasi in situ EPR experiment was carried out to investigate whether the Ni atom possesses dynamic redox activity under the reaction conditions (Fig. 4a and S11†). Compared to the reaction mixture before the reaction, an enhanced signal at g = 2.0 originating from CN was observed after illuminating the reaction mixture. Additionally, another signal at g = 2.1, ascribed to Ni(0) on carbon nitride,30 was observed with increased intensity compared to that of the reaction mixture before the illumination. The results imply that photoexcited electrons are transferred to the single Ni atoms from CN. When the illumination stops after the reaction, the intensity of the signals originating from Ni(0) and CN returns to the original value, demonstrating the reversibility of the redox properties of the Ni atoms during the photoredox C–N coupling reaction.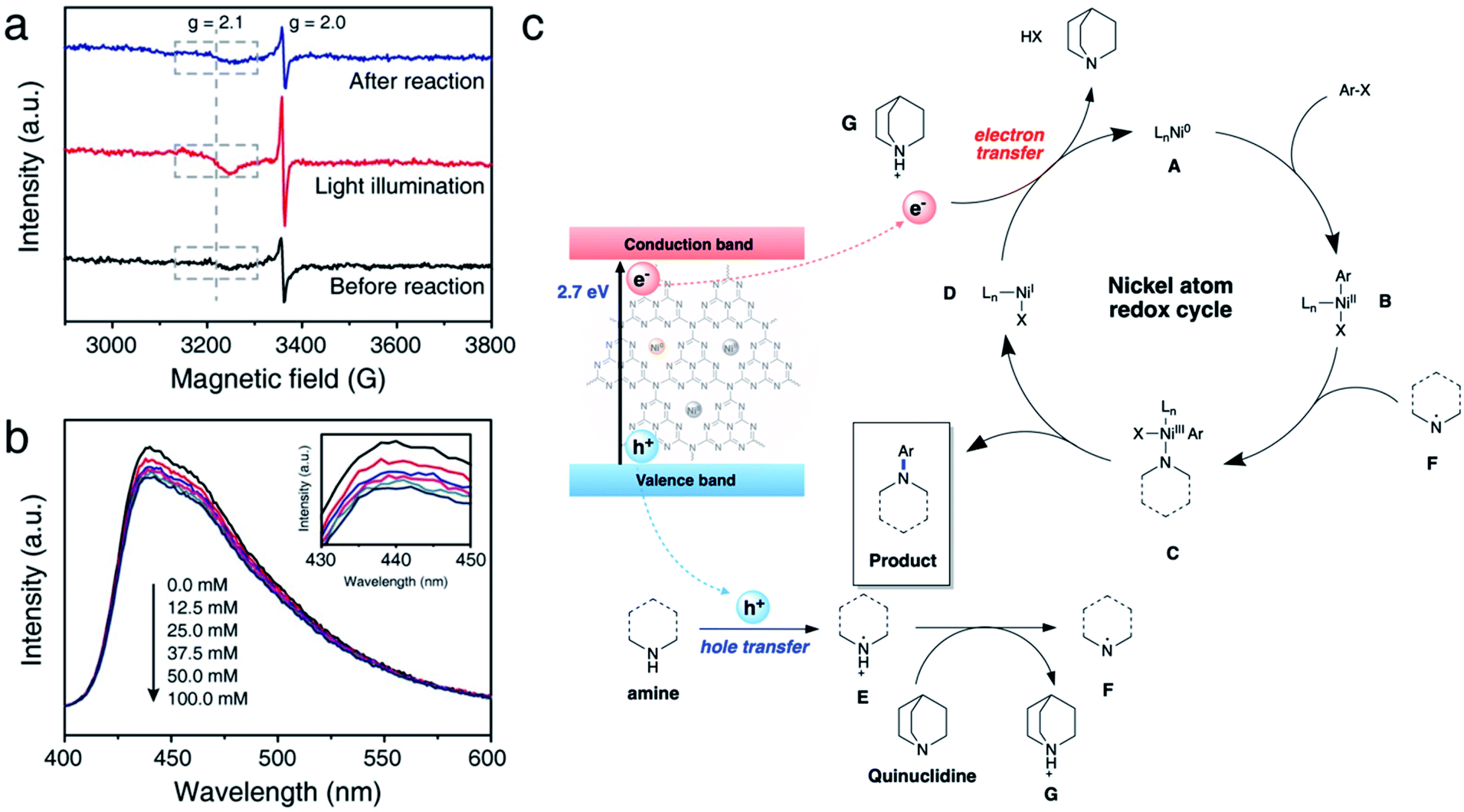 | ||
| Fig. 4 (a) Quasi in situ EPR spectra of the reaction mixture at 150 K under various conditions. (b) Emission quenching experiment using PL spectroscopy with various concentrations of pyrrolidine (inset: magnified spectra). (c) Proposed mechanism of photoredox C–N coupling on NiSAC/CN. | ||
Hence, it is concluded that Ni(0) species can play an essential role in mediating the reaction cycle. Recent reports on SAC-mediated photoredox organic reactions of C–O coupling proposed a catalytic cycle theoretically. For example, previous reports on C–O bond formation proposed a catalytic cycle via the Ni(I)/Ni(III) pathway, excluding Ni(0) formation.23,24 Through the EPR experiment, we clarify the sequential electron transfer process, demonstrating Ni(0) complex formation.
To examine how Ni(0) complex engages in the catalytic cycle, kinetic studies were performed for five para-substituted substrates (CF3, Cl, H, Me, and OMe), and the corresponding result was plotted based on the Hammett equation (Fig. S12 and S13†). The linear correlation with a slope value of 4.18 was obtained, demonstrating that the para-substituents on aryl halide influence the reaction rate of C–N coupling significantly. Furthermore, a positive correlation indicates that negative charge buildup occurs in the transition state of the catalytic cycle, and electron-withdrawing substituents accelerate the reactivity of aryl halides. Taken together with previous studies,24,27,34,35 we hypothesized that the photogenerated Ni(0) complexes participate in the subsequent oxidative addition process, and the oxidative addition step influences the overall reaction rate.
Next, we sought to investigate how pyrrolidine participates in the catalytic cycle. We hypothesized that pyrrolidine radicals are formed through the reaction with the photogenerated holes of carbon nitride.36–38 Therefore, to verify that pyrrolidine accepts photogenerated holes from carbon nitride, a steady-state emission quenching experiment was conducted (Fig. 4b). As the concentration of pyrrolidine increased from 0.0 mM to 100.0 mM, the emission intensity of carbon nitride decreased (Fig. S14†). These results confirm that pyrrolidine is capable of quenching photogenerated holes from carbon nitride. Through a control experiment in the presence of TEOA, it is corroborated that the photogenerated holes are involved in C–N bond functionalization (Table 1, entry 6). Furthermore, the pyrrolidine radical cation has a reduction potential of 0.85 V vs. SCE in acetonitrile, and the valence band of CN is located at 1.2 V vs. SCE.5,39 Consequently, the photogenerated holes from CN are sufficient to oxidize pyrrolidine, which is in good agreement with our results.
Encouraged by the results, we postulated a plausible dual catalytic pathway with synergistic interactions between single nickel atoms and carbon nitride to enable C–N coupling (Fig. 4c). Based on EPR experiments, electron transfer from CN to the single Ni atoms is demonstrated to be a prerequisite for C–N coupling. Correspondingly, Ni(0) species A is generated under light irradiation, and oxidative addition of aryl halides to Ni(0) results in the corresponding Ni(II) intermediate B. Based on kinetic studies, we postulated that an oxidative addition step influences the overall reaction rate, although complex mechanistic pathways cannot be ruled out. Simultaneously, CN quenches the photogenerated holes by pyrrolidine and forms pyrrolidine radical E, as demonstrated by the emission quenching experiment and redox properties. The resulting pyrrolidine radical E is relatively acidic and serves as a proton donor to quinuclidine, resulting in the formation of pyrrolidine radical F. Then, Ni(II) species B coordinates with the pyrrolidine radical to produce Ni(III) complex C. Reductive elimination of Ni(III) intermediate C leads to the formation of the desired product, and subsequent electron transfer from CN to Ni(I) complex D simultaneously regenerates Ni(0) species A and quinuclidine. In this cycle, quinuclidine is responsible for the generation of pyrrolidine radical F and the completion of the dual catalytic cycle. Therefore, the absence of a base or the use of another base can decelerate the conversion of the starting material.
Substrate scope
With the optimized conditions, C–N coupling was mediated with p-halobenzonitriles (Table 2). Coupling of 4-iodobenzonitrile was completed within 14 h under standard conditions. In addition to aryl iodide, aryl bromide was also effectively coupled. Furthermore, 4-chlorobenzonitrile, which is less reactive, produced the desired product in high yield at longer reaction times.

|
Then, we evaluated the scope of the C–N coupling reaction with aryl bromides containing an array of functional groups under standard conditions (Table 3). Aryl bromides with para-substituted electron-withdrawing groups, including an ester, nitrile, trifluoromethyl, or acetyl group, were tested, and the desired product was delivered with excellent to moderate coupling efficiency (1–4). The presence of the ortho-substituted cyano group was compatible (5). A meta-substituted compound (6) also delivered a corresponding product with a moderate yield (96% based on the recovered starting material). C–N coupling of 1,4-dibromobenzene gave a monoaminated product 7, providing sites for further functionalization. 1-Bromo-4-chlorobenzene underwent selective C–N coupling with a bromo substituent to produce product 8 with moderate yield (84% based on the recovered starting material). Selective amination of 1-bromo-4-chlorobenzene presumably occurs due to the weaker bond strength of C–Br than of C–Cl.

Then, C–N coupling with electron-rich aryl halides was conducted (9–12). Our catalytic system can also mediate transformation with aryl halides containing electron-donating groups. The relatively low yields can be explained by the sluggish oxidative addition process due to the presence of electron-donating groups. For heteroaryl bromides, which are common building blocks of bioactive compounds,29 the coupling product was also obtained, suggesting the possibility for pharmaceutical application (13–15).
Next, we sought to explore the scope of amines. Secondary amines containing six-membered rings, including morpholine, piperidine, and 1-acetylpiperazine, were effective coupling partners (16–19). In addition to cyclic secondary amines, acyclic primary amine was investigated to produce product 20. Although primary amines are generally less operative to cross coupling due to their relatively low nucleophilicity,8 the desired product was delivered in 99% yield.
Conclusions
We successfully developed a totally heterogeneous NiSAC/CN photocatalytic system for C–N coupling. Additionally, we provide a unified picture of how the photogenerated electrons and holes on the CN surface control the redox cycle of single Ni atoms by regulating stepwise electron-transfer processes and radical generation to complete the heterogeneous photoredox reaction cycle. In light of mechanistic studies, including quasi in situ EPR and various spectroscopic experiments, we found that the sequential electron transfer process can be precisely controlled on the NiSAC/CN photocatalytic surface via the Ni(0)/Ni(II)/Ni(III)/Ni(I) pathway from the initial formation of Ni(0). Our work provides an important step toward understanding the photoredox process of SACs, implying the possibility of effective and sustainable synthesis of fine chemicals using heterogeneous photocatalysts consisting of earth-abundant components.Data availability
We have provided experimental data in ESI† and there are no additional experimental or computational data.Author contributions
T. H. and D. Y. directed the research and planned the experiments. M. K., J. K., Y. S., S. K., H. C., and G. H. Y. performed the photocatalytic experiments. J. B., B.-H. L., W. K., W. H. A., and C. W. L. synthesized and characterized the catalysts. H. S. and C. P. performed EPR and PL experiments under the direction of D.-H. K. K.-S. L. conducted X-ray absorption measurements. M. K., J. B., and B.-H. L. wrote the initial manuscript. All authors discussed the results and contributed to the final manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Synthesis and physicochemical property analysis of the nanomaterial samples were supported by the Research Center Program of the IBS (IBS-R006-D1) in Korea (T. H.). Photocatalytic experiments were supported by the National Research Foundation (NRF-2018M3A7B4071204 and NRF-2021R1A4A1032515) in Korea (D. Y.), and Research Resettlement Fund for new faculty of Seoul National University (D. Y.). X-ray absorption experiment at the Pohang Accelerator Laboratory (PAL) 8C beamline was supported by a National Research Foundation of Korea (NRF) grant funded by the Korea Government (2018M1A2A2061998) (K. L.).References
- A. Savateev, I. Ghosh, B. König and M. Antonietti, Angew. Chem., Int. Ed., 2018, 57, 15936–15947 CrossRef CAS PubMed.
- C. Bottecchia, N. Erdmann, P. M. Tijssen, L. G. Milroy, L. Brunsveld, V. Hessel and T. Noël, ChemSusChem, 2016, 9, 1781–1785 CrossRef CAS PubMed.
- Z. Chai, T.-T. Zeng, Q. Li, L.-Q. Lu, W.-J. Xiao and D. Xu, J. Am. Chem. Soc., 2016, 138, 10128–10131 CrossRef CAS.
- Y.-Y. Liu, D. Liang, L.-Q. Lu and W.-J. Xiao, Chem. Commun., 2019, 55, 4853–4856 RSC.
- I. Ghosh, J. Khamrai, A. Savateev, N. Shlapakov, M. Antonietti and B. König, Science, 2019, 365, 360–366 CrossRef CAS.
- Z. Zhang, Y. Xu, Q. Zhang, S. Fang, H. Sun, W. Ou and C. Su, Sci. Bull., 2022, 67, 71–78 CrossRef CAS.
- B. Pieber, J. A. Malik, C. Cavedon, S. Gisbertz, A. Savateev, D. Cruz, T. Heil, G. Zhang and P. H. Seeberger, Angew. Chem., Int. Ed., 2019, 58, 9575–9580 CrossRef CAS PubMed.
- S. Gisbertz, S. Reischauer and B. Pieber, Nat. Catal., 2020, 3, 611–620 CrossRef CAS.
- S. Das, K. Murugesan, G. J. Villegas Rodríguez, J. Kaur, J. P. Barham, A. Savateev, M. Antonietti and B. König, ACS Catal., 2021, 11, 1593–1603 CrossRef CAS.
- A. Wang, J. Li and T. Zhang, Nat. Rev. Chem., 2018, 2, 65–81 CrossRef CAS.
- S. Ji, Y. Chen, X. Wang, Z. Zhang, D. Wang and Y. Li, Chem. Rev., 2020, 120, 11900–11955 CrossRef CAS PubMed.
- Y. Chen, S. Ji, C. Chen, Q. Peng, D. Wang and Y. Li, Joule, 2018, 2, 1242–1264 CrossRef CAS.
- J. Zhang, Y. Zhao, C. Chen, Y.-C. Huang, C.-L. Dong, C.-J. Chen, R.-S. Liu, C. Wang, K. Yan and Y. Li, J. Am. Chem. Soc., 2019, 141, 20118–20126 CrossRef CAS PubMed.
- S. H. Lee, J. Kim, D. Y. Chung, J. M. Yoo, H. S. Lee, M. J. Kim, B. S. Mun, S. G. Kwon, Y.-E. Sung and T. Hyeon, J. Am. Chem. Soc., 2019, 141, 2035–2045 CrossRef CAS PubMed.
- J. Lin, A. Wang, B. Qiao, X. Liu, X. Yang, X. Wang, J. Liang, J. Li, J. Liu and T. Zhang, J. Am. Chem. Soc., 2013, 135, 15314–15317 CrossRef CAS PubMed.
- G. Ding, L. Hao, H. Xu, L. Wang, J. Chen, T. Li, X. Tu and Q. Zhang, Commun. Chem., 2020, 3, 1–8 CrossRef.
- C. J. Wrasman, A. R. Riscoe, H. Lee and M. Cargnello, ACS Catal., 2020, 10, 1716–1720 CrossRef CAS.
- P. N. Duchesne, Z. Li, C. P. Deming, V. Fung, X. Zhao, J. Yuan, T. Regier, A. Aldalbahi, Z. Almarhoon and S. Chen, Nat. Mater., 2018, 17, 1033–1039 CrossRef CAS PubMed.
- X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS PubMed.
- J. J. Devery III, J. J. Douglas, J. D. Nguyen, K. P. Cole, R. A. Flowers II and C. R. Stephenson, Chem. Sci., 2015, 6, 537–541 RSC.
- N. A. Romero and D. A. Nicewicz, Chem. Rev., 2016, 116, 10075–10166 CrossRef CAS PubMed.
- C. J. O'Brien, D. G. Droege, A. Y. Jiu, S. S. Gandhi, N. A. Paras, S. H. Olson and J. Conrad, J. Org. Chem., 2018, 83, 8926–8935 CrossRef.
- X. Zhao, C. Deng, D. Meng, H. Ji, C. Chen, W. Song and J. Zhao, ACS Catal., 2020, 10, 15178–15185 CrossRef CAS.
- A. Vijeta, C. Casadevall, S. Roy and E. Reisner, Angew. Chem., Int. Ed., 2021, 60, 8494–8499 CrossRef CAS PubMed.
- J. Wen, X. Yang, Z. Sun, J. Yang, P. Han, Q. Liu, H. Dong, M. Gu, L. Huang and H. Wang, Green Chem., 2020, 22, 230–237 RSC.
- J. Ma, F. Zhang, Y. Tan, S. Wang, H. Chen, L. Zheng, H. Liu and R. Li, ACS Appl. Mater. Interfaces, 2022, 14, 18383–18392 CrossRef CAS PubMed.
- A. Vijeta, C. Casadevall and E. Reisner, Angew. Chem., Int. Ed., 2022, 61, e202203176 CrossRef CAS PubMed.
- J. Bariwal and E. Van der Eycken, Chem. Soc. Rev., 2013, 42, 9283–9303 RSC.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- X. Jin, R. Wang, L. Zhang, R. Si, M. Shen, M. Wang, J. Tian and J. Shi, Angew. Chem., 2020, 132, 6894–6898 CrossRef.
- G. Neri, M. Forster, J. J. Walsh, C. Robertson, T. Whittles, P. Farràs and A. J. Cowan, Chem. Commun., 2016, 52, 14200–14203 RSC.
- Z. Chen, E. Vorobyeva, S. Mitchell, E. Fako, N. López, S. M. Collins, R. K. Leary, P. A. Midgley, R. Hauert and J. Pérez-Ramírez, Natl. Sci. Rev., 2018, 5, 642–652 CrossRef CAS.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS.
- H. Ren, G.-F. Li, B. Zhu, X.-D. Lv, L.-S. Yao, X.-L. Wang, Z.-M. Su and W. Guan, ACS Catal., 2019, 9, 3858–3865 CrossRef CAS.
- N. A. Till, L. Tian, Z. Dong, G. D. Scholes and D. W. MacMillan, J. Am. Chem. Soc., 2020, 142, 15830–15841 CrossRef CAS PubMed.
- M. Kudisch, C.-H. Lim, P. Thordarson and G. M. Miyake, J. Am. Chem. Soc., 2019, 141, 19479–19486 CrossRef CAS PubMed.
- T. Xiong and Q. Zhang, Chem. Soc. Rev., 2016, 45, 3069–3087 RSC.
- S. A. Morris, J. Wang and N. Zheng, Acc. Chem. Res., 2016, 49, 1957–1968 CrossRef CAS PubMed.
- M. Jonsson, D. D. Wayner and J. Lusztyk, J. Phys. Chem., 1996, 100, 17539–17543 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2sc02174a |
| ‡ M. K., J. B., and B.-H. L. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |