
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Parallel DNA circuits by autocatalytic strand displacement and nanopore readout†
Jinbo
Zhu
 *ab,
Jinglin
Kong
b,
Ulrich F.
Keyser
b and
Erkang
Wang
*a
*ab,
Jinglin
Kong
b,
Ulrich F.
Keyser
b and
Erkang
Wang
*a
aState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin 130022, P. R. China. E-mail: ekwang@ciac.ac.cn
bCavendish Laboratory, University of Cambridge, JJ Thompson Avenue, Cambridge CB3 0HE, UK. E-mail: jz421@cam.ac.uk
First published on 7th October 2022
Abstract
DNA nanotechnology provides a unique opportunity for molecular computation, with strand displacement reactions enabling controllable reorganization of nanostructures. Additional DNA strand exchange strategies with high selectivity for input will enable novel complex systems including biosensing applications. Herein, we propose an autocatalytic strand displacement (ACSD) circuit: initiated by DNA breathing and accelerated by a seesaw catalytic reaction, ACSD ensures that only the correct base sequence starts the catalytic cycle. Analogous to an electronic circuit with a variable resistor, two ACSD reactions with different rates are connected in parallel to mimic a parallel circuit containing branches with different resistances. Finally, we introduce a multiplexed nanopore sensing platform to report the output results of a parallel path selection system at the single-molecule level. By combining the ACSD strategy with fast and sensitive single-molecule nanopore readout, a new generation of DNA-based computing tools is established.
1. Introduction
DNA computing has developed over the last several decades, making valuable contributions to computer science and biomedicine,1,2 such as neural network-like computations reported recently.3,4 In a complex computing system, the information loaded on DNA needs to be processed and transmitted among different logic units and circuits. In early studies, enzymes were mainly used, which increased the complexity of both the design and operation of these circuits.5,6 The emergence of toehold-mediated strand displacement (TMSD) greatly simplified the development of DNA computing and nanotechnology.7–9 TMSD is an enzyme-free reaction to release the prehybridized incumbent DNA strand in the duplex upon the binding of the invader strand from the toehold end, which plays a vital role in TMSD, as its length dictates the kinetics of the reaction.10–12 This tunability has enabled the use of TMSD in DNA computing, molecular machines, biosensing, gene regulation, etc.13–16 Single base differences in the input strand can be detected by commonly used TMSD based on three-way branch migration. However, robust discrimination of a single base difference in the input strand is challenging due to inevitable crosstalk and leaks as the reaction can still proceed even with a single base mismatch.17,18The second important challenge for all complex DNA computing systems is the readout of multiple outputs at the same time, especially for parallel computing. While fluorescence is the main method to report on DNA computing systems,19,20 the cost of fluorescently labelled DNA is much higher than that of label-free assays and the overlap of the fluorescence wavelength limits the potential of multi-channel monitoring. Atomic force microscopy provides another solution for the readout of complex systems on surfaces, but is unsuitable for reactions in solution.21,22 One alternative readout system is based on single-molecule analysis using resistive-pulse sensing with solid-state nanopores.23 The nanopore technique provides a simple and label-free approach for nucleic acid detection and structural analysis.24–28 Our recently proposed solid state nanopore biosensing assay based on digitally encoded DNA nanostructures supplies an ideal label-free multiplexed sensing platform.29,30 Our multiplexed nanopore method is ideally suited to decode a parallel DNA computing system as recently suggested.31
Herein, we build an autocatalytic strand displacement (ACSD) circuit initiated by DNA breathing instead of toehold binding (Fig. 1a). Without toehold triggering, the ‘toeless’ strand displacement reaction32,33 is much slower than TMSD, while the selectivity for the trigger strand and adjustability for the DNA circuit design are greatly improved. We show the impact of a single base change in the invader strand on the kinetics of the strand displacement and demonstrate the enhanced sensitivity of the DNA circuit for single-base changes in the input strand in ACSD compared to that in TMSD. In contrast to the DNA breathing-initiated step at the beginning, the remaining parts of the circuit are connected by standard TMSD reactions. By adjusting the length of toehold ends and adding an external strand to control the intermediate reaction, we successfully imitate the variable resistor and switch inspired by standard electronic circuits in our DNA system, respectively. Furthermore, we show successful analysis of four parallel DNA circuits with our multiplexed nanopore sensing platform, which indicates the promising potential of the nanopore technique for the readout of complex DNA computing systems.
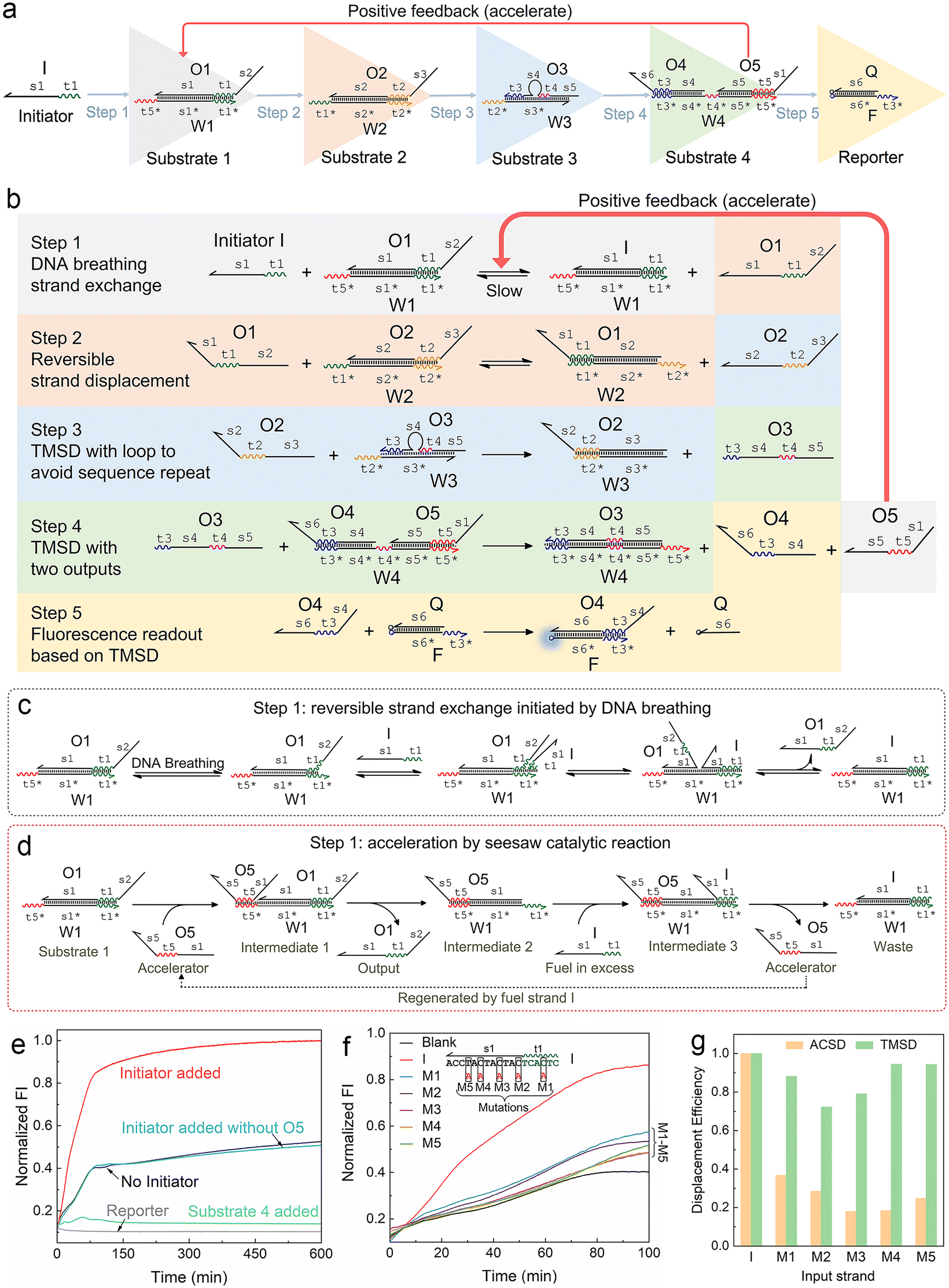 | ||
| Fig. 1 Mechanism of the autocatalytic strand displacement (ACSD)-based cascade DNA circuit. (a) Basic constitution of the ACSD circuit from the initial input strand I (Initiator) to the final fluorescent reporter (DNA complex F·Q). A series of YES gates are connected linearly to construct the ACSD circuit. Names of DNA strands are given in uppercase letters and domains in each strand are labelled in lowercase letters. (b) Reaction details of the whole ACSD circuit. (c) Details of the reaction in step 1 initiated by DNA breathing at the beginning before strand O5 is released. (d) Details of the reaction in step 1 accelerated by O5. (e) Kinetics measurements of the cascade DNA circuit under different conditions. Data of ‘No Initiator’ is from the mixture of DNA substrates in (a) without strand I. The blue curve shows another negative control, in which strand I is added but O5 is replaced by O5s (domain s1 is removed, Table S1†). The green curve shows the change in fluorescence when the reporter is mixed with substrate 4. (f) Kinetic measurements of the ACSD circuit triggered by different initiator strands. In groups of M1–5, strand I is replaced with its mutant strands M1–5, as shown above the curves, to investigate the effect of single base mutation on the ACSD circuit. No initiator is added in the blank. The fluorescence intensity (FI) is normalized by the FI of the ‘Initiator added’ group at 600 min. (g) Comparison between the ACSD circuit and the TMSD reaction for distinguishing single base mutation. The strand displacement efficiency (dE) is calculated based on the kinetics data at 100 min in (f) for the ACSD circuit and at 30 min in Fig. S3† for the TMSD reaction. | ||
2. Materials and methods
2.1 Design and preparation of DNA circuits
DNA sequences are designed by a modular strategy and listed in Table S1.† There were 6 nucleotides (nt) for most toehold ends with 50% GC content. For most strands, only three kinds of bases (GTA or CTA) were used for the sequence of a single strand to avoid the coexistence of G and C bases in the same strand and forming unwanted secondary structures. The sequences were validated on the NUPACK website to minimize the free energy. To prepare designed DNA structures for each substrate and reporter in the DNA circuit, single strands of each DNA complex were annealed together at a concentration of 2 μM in TM buffer solution (10 mM Tris-HCl, 10 mM MgCl2, pH 8.0) by heating at 88 °C for 5 min and cooling to 20 °C at a rate of 2 °C min−1.2.2 Fluorescence kinetics measurements
DNA substrates of the DNA circuit were mixed at room temperature (20 °C) in TM buffer solution. For a single circuit, the concentrations of the DNA complexes were 50 nM for the reporter, 50 nM for substrates 3 and 4, and 60 nM for substrates 1 and 2. A 100 nM strand initiator was added with gentle pipetting to trigger the reaction at the beginning of the fluorescence kinetics measurement. In a parallel competition between paths A and B, the same concentrations of DNA substrates were applied but only 50 nM strand I was added. 0.1 mL of mineral oil was added to the top of 0.8 mL of the solution to avoid evaporation during length measurement. The samples were monitored for 10 hours under 20 °C in a 10 mm path-length polystyrene semi-micro cuvette using a Cary Eclipse fluorescence spectrophotometer (Agilent Technologies, USA). The sample was excited at 495 nm for FAM and 535 nm for HEX, and the emission intensity was recorded at 520 nm for FAM and 556 nm for HEX.2.3 Synthesis of DNA carriers
The carrier synthesis follows our previous study.29 The carrier consists of one linear M13 scaffold and 190 short DNA staples. The 7228 nt DNA scaffold was linearized from M13mp18 ssDNA using a published protocol. Sequences of the 190 DNA staples can be found in our previous work.29 As shown in Fig. S4,† to construct the structures for barcodes on the carrier, staples 26–32, 40–46, 54–60, 68–74 and 82–88 were logically substituted by relevant dumbbell sequences (Table S1†) according to the barcode of the carrier. Staple 142 at the sensing site was substituted by RA, RB, RC or RD for carriers A, B, C or D, respectively. The staples need to be accurately mixed before preparation of the DNA carrier, and then the linear M13 scaffold was added into the solution (20 nM M13 scaffold, 60 nM staples, 120 nM dumbbell strands and 120 nM RX, X = A, B, C or D). The mixture was heated to 70 °C followed by a linear cooling ramp to 25 °C over 50 minutes. 1 μL of 4 μM DNA flower was added into 40 μL of DNA carrier solution (7WJa for carrier A, 7WJb for carrier B, 7WJc for carrier C and 7WJd for carrier D, ESI S1.2†) and incubated at room temperature for 2 h. Finally, the resulting solution was diluted with a washing buffer (10 mM Tris-HCl, 0.5 MgCl2, pH 8.0) to 500 μL and centrifuged with an Amicon Ultra 100 kDa filter to remove the excess DNA strands and 7WJ at 6000g for 10 min (repeated 3 times). About 35 μL of DNA carrier solution was obtained and quantified with a NanoDrop 2000 spectrophotometer.2.4 Strand displacement on DNA carriers
Carriers A, B, C and D were mixed and diluted with TM buffer to 1 nM for each carrier (4 nM for total carrier concentration). For the detection of strand Oa4 in Fig. 4c, 10 nM Oa4 was added into the carrier mixture solution (4 nM for total carrier concentration) and incubated at room temperature for 20 min. The same volume of 8 M LiCl was added after incubation and the sample was diluted with 4 M LiCl (in 1 × TE, pH 9.0) to 0.5 nM (total carrier concentration) for nanopore measurements.To detect the outputs of DNA circuits by nanopore sensing, DNA substrates of the 4 circuits were premixed in TM buffer solution at a concentration of 100 nM for substrates 3 and 4, 120 nM for substrates 1 and 2, and 500 nM for strand I. It was incubated at room temperature for 3 hours, and then 1 μL of the resulting solution was mixed with 4 μL of the carrier mixture (5 nM for total carrier concentration) to react for 20 min. 5 μL of 8 M LiCl was added after incubation and subsequently diluted with 30 μL of 4 M LiCl (in 1 × TE, pH 9.0) for nanopore measurement.
2.5 Nanopore measurements
The conical glass nanopore in the PDMS chip is fabricated and measured by following our previous study.29 Glass nanopores (14 ± 3 nm) were generated on a laser-heated pipette puller (P-2000, Sutter Instrument) by pulling quartz capillaries (outer diameter 0.5 mm and inner diameter 0.2 mm, Sutter Instrument). The resulting nanopores were fixed on a PDMS chip and filled with 4 M LiCl (in 1 × TE, pH 9.0). The DNA carrier mixture (carrier A–D) was incubated with DNA circuit solution in TM buffer at room temperature for 20 min and then diluted with 4 M LiCl to 0.5 nM (total carrier concentration) for measurement. Electrodes were placed at the two sides of the nanopore and the sample solution was added to the tip side of the glass nanopore. An Axon Axopatch 200B amplifier (Molecular Devices) connected to the electrodes, applied a voltage (600 mV) to drive the DNA through the nanopore. The current signal was recorded using an amplifier, filtered with an external Bessel filter (Frequency Devices) at 50 kHz and digitized at a 250 kHz sampling rate with a data card (PCI-6251, National Instruments). The introduction of nanopore data analysis can be found in the ESI S1.5.†3. Results and discussion
3.1 Design of the ACSD circuit
We firstly designed a DNA breathing initiated ACSD cascade circuit as shown in Fig. 1a: this can be simply seen as a series connection of five YES gates, and each gate is a DNA strand exchange (steps 1 and 2, reversible) or displacement reaction (steps 3, 4 and 5 irreversible) as shown in Fig. 1b. To identify different functional domains in each strand, toehold domains are named tx and represented by wavy lines, and binding domains are named sx and represented by straight lines. The asterisk after the domain name means a complementary sequence and all strand sequences are given in Table S1.†In step 1, there is no free toehold end in substrate 1 (DNA complex W1·O1) for initiator strand I to quickly trigger the reaction, and thus the DNA circuit starts from a slow reversible strand exchange initiated by DNA breathing32,33 as shown in Fig. 1c. A small amount of strand O1 output from substrate 1 will consequently interact with substrate 2 (complex W2·O2) to trigger a reversible seesaw strand exchange reaction and release strand O2 (Fig. 1b). step 3 is a classic TMSD reaction, where strand O3 will be replaced by O2 quickly as long as free O2 is present in solution. The loop in the middle of substrate 3 (complex W3·O3) is designed to avoid a sequence repeat. Rapid consumption of free O2 by substrate 3 will break the equilibrium of reactions in steps 2 and 1 and further promote the reactions to move forward. The released O3 interacts with substrate 4 in step 4 by binding with the middle toehold domain t4 to displace O4 and O5 simultaneously. Finally, O4 will be received by the reporter (complex F·Q) to yield a fluorescence signal, and strand O5 acts as positive feedback to accelerate the reaction in step 1. Therefore, the whole reaction gets accelerated with the accumulation of released O5 and the fluorescence intensity reaches the maximum much earlier (group ‘Initiator added’) than the control groups as shown in Fig. 1e. Gel electrophoresis was applied to investigate the reactions in the ACSD circuit. In Fig. S1,† the clear band of O4 is only observed when initiator I is added.
The acceleration mechanism of strand O5 in step 1 is based on a seesaw catalytic reaction. As illustrated in Fig. 1d, O5 interacts with substrate 1 to partly displace prehybridized O1 through the pre-reserved toehold end t5* on W1. When O1 is released from W1, the excess initiator I will work as fuel to exchange O5 on W1 and produce the waste (W1 I), and then the regenerated O5 can be reused in a cycle to catalyse the whole reaction. As the reaction progresses, the more O1 is output, the more O5 is fed back from step 4 to speed up the reaction in step 1. Thus, O5 plays a key role in guiding the reaction to completion. These prehybridized DNA complexes were not further purified after annealing, hence a small amount of unhybridized single strands, such as O5, O4, etc., may leak from the complexes and cause the background signal as observed from the blank group (‘No Initiator’) in Fig. 1e. Given the specific effect of O5 on the kinetics of the forward reaction in step 1, we infer that the strand leak, as the green curve shown in Fig. 1e, should be another factor to cooperate with strand I to initiate and accelerate the reaction. To demonstrate the significance of O5 in the ACSD circuit, we removed the domain s1 from O5 to generate the control strand O5s (Table S1†), which loses the ability to displace O1 in substrate 1. The resulting fluorescence signal (‘Initiator added without O5’ in Fig. 1e) is nearly the same as the blank, which indicates that the accelerator O5 is essential for the whole circuit to function properly. A similar result is gained by gel electrophoresis (Fig. S2†).
3.2 Detecting single base mutation
The ability of the ACSD circuit to detect single base mutation in the input strand is compared with the widely used TMSD reaction. As shown in Fig. 1f, initiator I and its mutant strands M1–M5 are applied to trigger the reactions in ACSD (Fig. 1a) and TMSD (Fig. S3†). For the ACSD circuit, the reaction is much slower, and hence we choose the time point of 100 min to determine the displacement efficiency (DE) for comparison (ESI S1.4†). At this point, the strand I-initiated ACSD reaction is nearly complete and the difference between I and its mutants is clear (Fig. 1f). For TMSD, the reaction quickly reaches the steady state even for the mutants (Fig. S3†), and we thus determine the DE at 30 min. As shown by comparison in Fig. 1g, a clear difference of the DEs with mutant inputs between ACSD and TMSD indicates that the ACSD circuit is more sensitive to single base mutation than TMSD due to the DNA breathing-based initiation mechanism of the ACSD circuit. The ability of the ACSD circuit to distinguish single base mutations within the input sequence renders it suitable for selective biosensing or constructing complex systems with multiple input strands and simultaneous computations.3.3 Modulating the circuit by adjusting step 2
Another feature of the ACSD circuit is that the reaction can be flexibly modulated by changing the design of substrate 2 in step 2, which is a reserved section that allows for easy adaptability of the circuit. For instance, by splitting W2 into two halves in step 2 (Fig. 2a), an external input strand Linker can be used to control the reaction, which is based on a four-way junction driven strand displacement reaction.34 Analogous to a switch within an electronic circuit, the strand displacement reaction only proceeds when the Linker is present in the solution (Fig. 2c). This stimuli-responsive feature of the ACSD circuit will be critical for the flexible control of DNA computers as well as biosensing applications based on DNA, such as using a DNA aptamer as the linker.35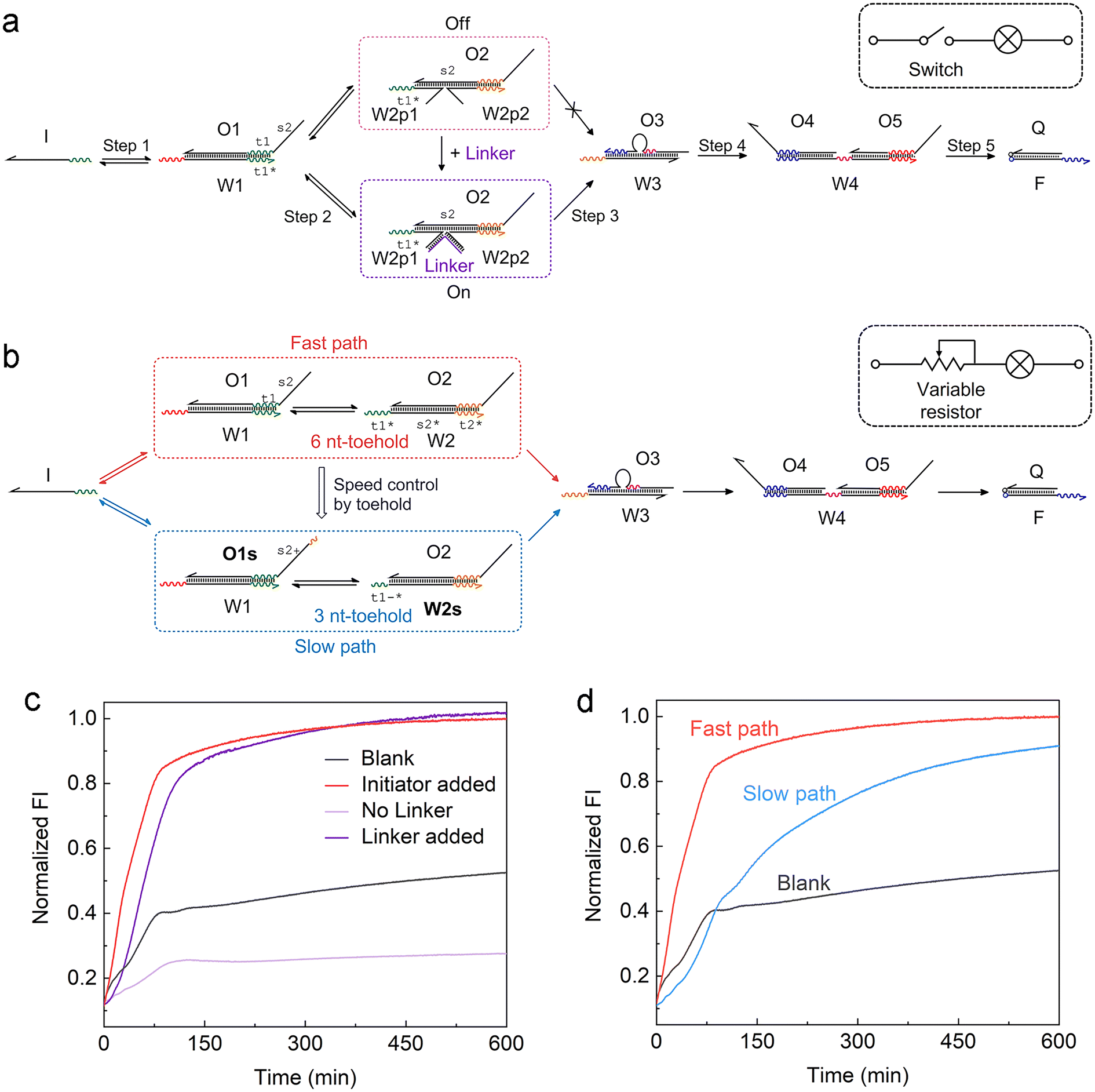 | ||
| Fig. 2 Adjusting the ACSD circuit to mimic the (a) switch and (b) variable resistor in an electronic circuit. (a) Scheme of the strand Linker controlled ACSD circuit. (b) Scheme of the kinetic modulation of the ACSD circuit by adjusting the toehold length. (c) Fluorescence kinetic result of the Linker controlled ACSD circuit. The groups ‘Blank’ and ‘Initiator added’ are the same data as shown in Fig. 1e. The strand Initiator was added in both the groups ‘No Linker’ and ‘Linker added’. (d) Kinetic measurements of the two paths in (b) with different rates. | ||
Furthermore, control over the reaction kinetics is realized by adjusting the toehold length on W2 as shown in Fig. 2b. The original design (fast path) is based on a toehold with 6 nucleotides (6 nt) on W2. We can slow down the reaction by shortening the toehold from 6 nt to 3 nt (W2s) and extend the 5′ end of O1 with 3 nt (O1s) to ensure that the strand exchange is still reversible. This operation is like tuning the variable resistor in an electronic circuit and for example, controlling the magnitude of current through a bulb as shown in the sketch in Fig. 2a and b. The kinetics result in Fig. 2d demonstrates that the reaction rate in the slow path dropped as expected but the whole reaction was still close to completion after 10 hours.
3.4 Parallel circuit imitated by ACSD
For the development of efficient computing systems, multiple ACSD circuits need to operate in parallel. As illustrated in Fig. 3a, two parallel ACSD circuits triggered by the same initiator strand I are designed. The main difference between them is the toehold length in step 2, which is adjusted to mimic two circuits with different resistance values. In common TMSD reactions, the effective rate constant would increase by a factor of about 103 when the toehold length increases from 3 nt to 6 nt (ref. 12) and is nearly irreversible. Herein, we adjust the design into two reversible seesaw reactions, so the dynamic distribution of strand I between the two paths is possible. The toeholds with different lengths are used to make the reaction rates of the two paths different. Since path B has a longer toehold (i.e., low resistance, Rb), it will be faster than path A. Two different fluorescent dyes are used to identify the report signals from different paths (HEX for path A and FAM for path B). The strand I randomly attempt to trigger the reactions in both circuits at the beginning, comparable to a case in a parallel circuit where the voltage across each branch path is the same. Due to the longer toehold end in the fast path B, more Ob5 will be released than Oa5 as the reactions progress to step 4 earlier. Then the Ob5 supplies more strand I to participate in the reactions in path B. Finally, more strand I will be involved in the reactions in path B and a high fluorescence output signal at 600 min was obtained from the FAM channel (green curve, Fig. 3c). Meanwhile, a low fluorescence signal was reported from the HEX channel (green curve, Fig. 3b) indicating only a small part of strand I was consumed in path A. In this case, strand I is like the current in a parallel circuit that splits into different branches based on the resistance value in each path, and then the bulbs showed different brightness (i.e., fluorescence intensity). As a positive control, DNA complex Wb1·Ob1 in step 1 of path B was removed to break it when only path A was activated, and it was the same for path A when only path B was activated. The FI ratio of the competition group to the positive control can indicate the relative extent of the reaction (RER, ESI S1.4†). 30.9% and 76.9% were obtained for the RERs of path A and path B, respectively, at 600 min in the competition group. Our results show that parallel DNA circuits can be constructed and the current distribution in parallel circuits can be imitated by the ACSD circuit.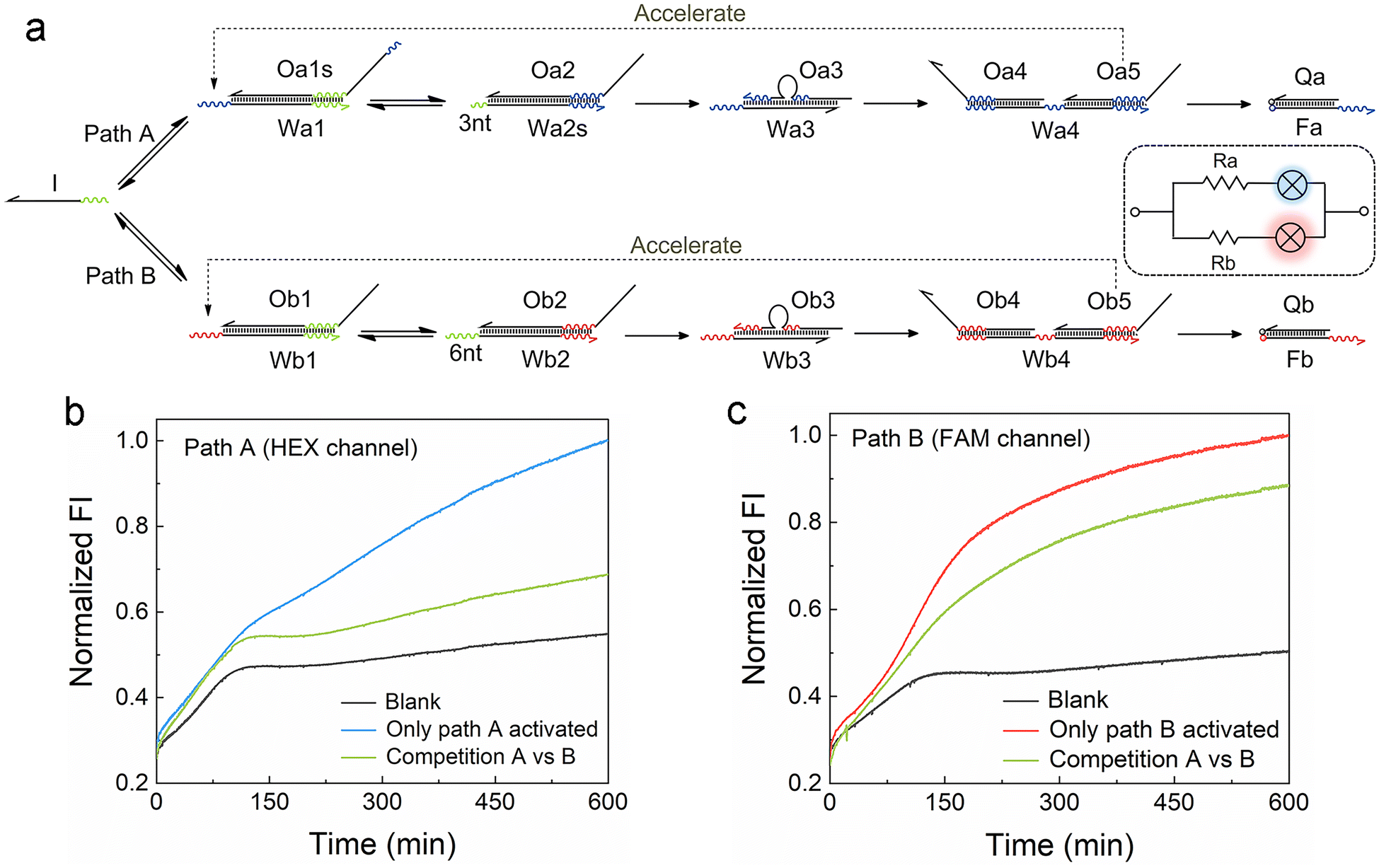 | ||
| Fig. 3 (a) Scheme of two ACSD circuits connected in parallel and triggered by the same initiator strand I. Fluorescent labels HEX and FAM are applied for paths A and B, respectively. (b and c) Kinetic measurements of the two branch paths of the parallel circuit to show the distribution of strand I in two paths. DNA substrates of the two paths were mixed with strand I in the competition group and without strand I in the blank. The complex of Wb1·Ob1 was absent when only path A was activated and the complex of Wa1·Oa1s was absent when only path B was activated. FI is normalized by the FI of the highest group at 600 min. | ||
3.5 Monitoring the multiple DNA circuits using a nanopore sensing platform
We now extend the competition model into a more complex path selection system with four parallel ACSD circuits (Fig. 4a). By changing the sequences of relative domains, the number of paths can be easily increased from one to four. The sequence of a binding domain in step 1 remained the same in all paths, so they all can be initiated by strand I. In specific path combinations, sections in the dashed boxes in Fig. 4a are removed to close the corresponding paths.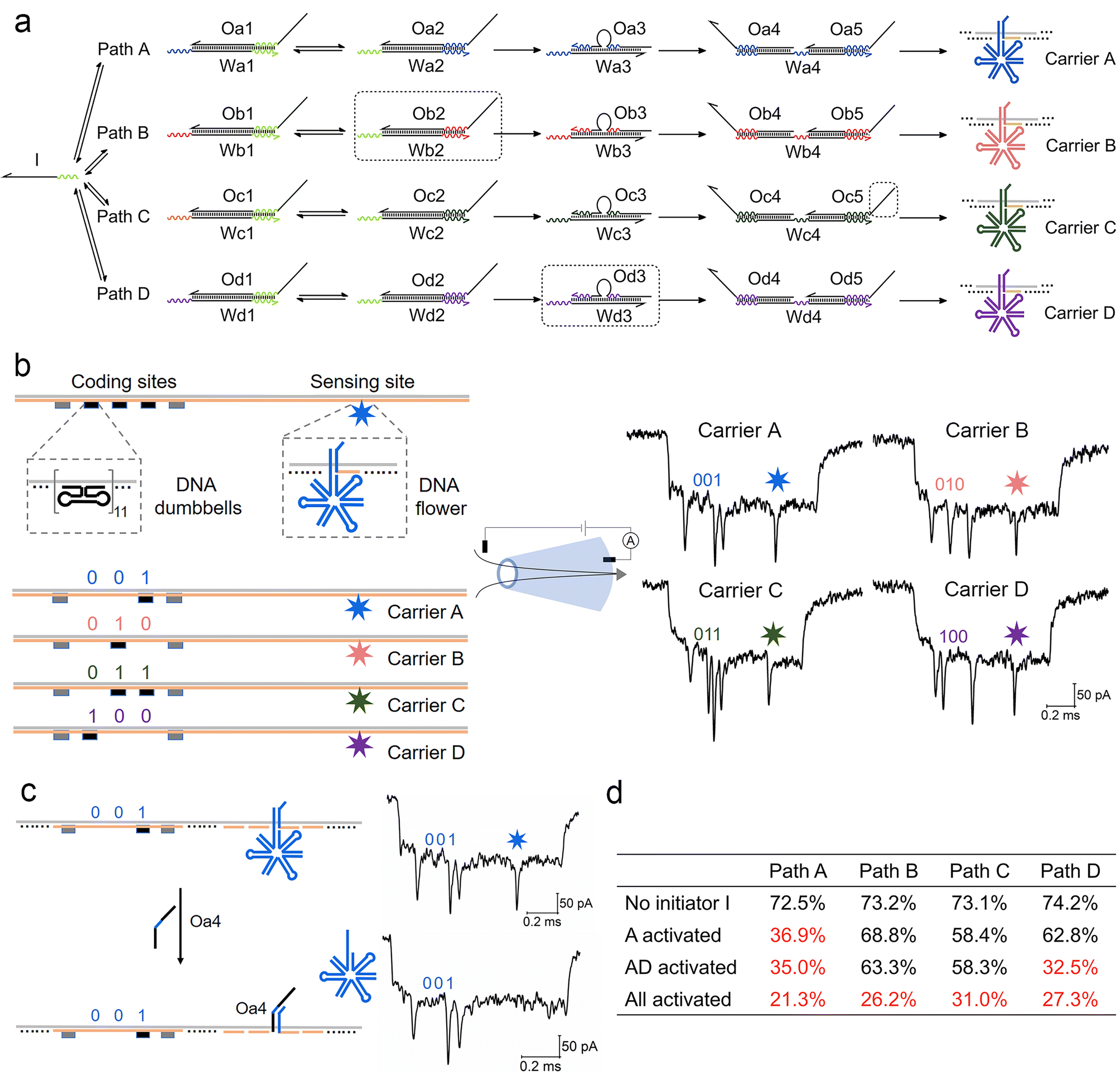 | ||
| Fig. 4 Measurement of a parallel ACSD circuit with four paths using nanopores. (a) Scheme of the parallel ACSD circuit with four paths triggered by the same initiator strand I. DNA carrier-based nanopore reporters (carrier A–D) are used to readout the four paths. (b) Design of the four DNA carriers (left) and their sample events (right) obtained from the glass nanopore (middle). The DNA barcode at the coding sites is applied to identify the carriers and a DNA flower (7-way junction) is used to report the strand displacement reaction at the sensing site. (c) Mechanism of carrier A for detecting the output strand Oa4 from path A in (a). Two events of carrier A before (top) and after (bottom) the addition of Oa4 are given on the right as an example. (d) Table of the occupied fractions at the sensing site of the four carriers under different path combinations. The data are from three repeated measurements and standard errors are given in Table S3.† Sections in the dashed box in (a) are removed from the mixture when the corresponding paths are closed. | ||
For multiplexed nanopore sensing, the other main change in the design of a parallel system with four branches is reporter units (Fig. 4a). To overcome the limitation of the wavelength overlap in fluorescence methodologies, we utilize a DNA carrier-based nanopore sensing platform to measure the final outputs of the four paths in parallel. The design of DNA carriers is given in Fig. 4b and Fig. S4.† The carrier is composed of a linear M13 strand and 190 short complementary strands (staples).29 DNA nanostructures can bind onto it by replacing the staples at specific sites. Five groups of DNA dumbbells (11 for each group) are used to construct a barcode system at the coding sites. The first and fifth groups of DNA dumbbells are set as reference structures, and the 3-bit binary codes between them are used to encode the carriers. At the other side of the carrier, a DNA flower (7-way junction, 7WJ) is bound by hybridizing with the overhang at the sensing site. Unlike the ordinary DNA junctions used in our recent work,36 a long strand with a small T-loop in the 7WJ can enhance the structural stability and current signal. Different toehold ends on the overhangs are reserved on relative carriers, carriers A–D, to receive output strands Oa4–Od4 from the DNA circuits, paths A–D, respectively by TMSD. Detection of Oa4 is given as an example in Fig. 4c.
Conical quartz nanopores (10–14 nm) are applied to analyse DNA structures bound to carriers.29 A negatively charged DNA carrier is driven through nanopores by electric force and causes the first level current drop (Fig. S5†). The DNA nanostructures on carriers cause the second level current drops as shown in Fig. 4b and Fig. S5,† whose detection times as measured from the start of each event correspond to the positions of nanostructures on the carriers. By decoding the barcode at the coding sites (ESI S1.5†), we can classify the translocation events into four groups corresponding to the four carriers. The occupied fraction (OF, percentage of the structure detected at the specific site) of the DNA flower at the sensing site for each carrier was counted to monitor the final output of the DNA circuit. As shown by the example in Fig. 4c, when the target output strand Oa4 was present, it displaced the DNA flower on carrier A mediated by the toehold end on the overhang. As a consequence, the frequency of the second level current drop at the sensing site decreased and a low OF was obtained for carrier A (Table S2†), because the short DNA double strand left on the carrier was insufficient to cause an obvious current change for nanopores with sizes above 10 nm.37,38 The first ten unfolded events of each carrier before and after the addition of strand Oa4 can be found in Fig. S6.†
As shown in Fig. 4d and Table S2,† we investigated four conditions of the parallel selection system. Firstly, when strand I was absent (blank), high OFs above 70% were obtained in all paths, which meant most DNA flowers remain on the carrier. Secondly, we closed paths B–D by removing the sections in the dashed boxes in Fig. 4a, hence only Path A was activated in the presence of strand I. A low OF below 40% was thus obtained for Path A indicating that strand I did choose this path and the final output Oa4 displaced most DNA flowers on carrier A. Thirdly, when paths A and D were both activated, the OF for path D also dropped to a low level, which demonstrated that the nanopore multiplexed sensing platform can sensitively detect the target output concentration change in a specific path. Finally, when all paths were activated, all four OFs decreased to less than 40%. To verify the nanopore results, fluorescence methods were also applied to investigate the change in paths A and B under the four conditions (Fig. S7†), consistent with the nanopore result. Overall, these experiments validate the DNA carrier-based nanopore sensing platform as a sensitive tool for measuring multiple output targets from these parallel systems.
4. Conclusions
In summary, we introduce a novel DNA breathing initiated DNA ACSD circuit. Unlike the ordinary TMSD, the initial strand displacement reaction is initiated by temporary instability in the DNA base pairs. The unique initiation mechanism of the ACSD circuit makes the reaction extremely sensitive to single base mutations at multiple sites in the target strand, which reduces crosstalk compared to the classic TMSD.Additionally, the ACSD circuit can be tuned to mimic specific electronic components. Firstly, step 2 was redesigned like a switch to be controlled by external stimuli. This feature is relevant for applications in biosensing and diagnosis. Secondly, toehold ends with different lengths were utilized in step 2 to adjust the reaction rates of the circuits. The possibility to connect ACSD circuits in parallel with different rates shows the equivalency to electronics. In addition, the initiator strand I interacts with the circuits according to their reaction rates, which is like the current distribution in branch paths with different resistance values in a parallel circuit.
Extending the path competition network, a four parallel path selection system is established and monitored with a nanopore multiplexed sensing platform. On the DNA carrier, the DNA dumbbells are used to encode the binary barcode to distinguish them, and the DNA flowers are used to detect the output strands from the circuits by calculating their OFs at the sensing site. Based on the circuit connection conditions, strand I can always correctly choose the activated paths and lead to the removal of DNA flowers on the relative carriers. Breaking the limitation of the wavelength overlap in the fluorescence assay, hundreds of carriers with different barcodes are possible to be simultaneously measured with the nanopore platform. The number of barcodes on a carrier can be extended via adding more coding sites on a carrier, increasing the number of digital codes on each coding site by using multi-level structures,30,36 increasing the coding sites by linking two carriers,37 or improving the resolution of the nanopore to read more bits in the same range.37 Multiplexed nanopore sensing and ACSD provide a promising approach for any parallel computing systems based on DNA.
Author contributions
J. Z, U. F. K and E. W conceived the project. J. Z designed and performed the experiments. J. Z and J. K analysed the data. All authors discussed the results and wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
J. Z and U. F. K are thankful for the support from an ERC Consolidator Grant (Designerpores No. 647144). EW is thankful for the support from the NSFC, Grant No. 21721003. We thank Filip Bošković for the discussion on the figures and Dr Casey Platnich for the critical reading of the manuscript.References
- D. Bhatia, C. Wunder and L. Johannes, ChemBioChem, 2021, 22, 763–778 CrossRef CAS PubMed.
- S. S. Wang and A. D. Ellington, Chem. Rev., 2019, 119, 6370–6383 CrossRef CAS PubMed.
- L. Qian, E. Winfree and J. Bruck, Nature, 2011, 475, 368–372 CrossRef CAS PubMed.
- K. M. Cherry and L. Qian, Nature, 2018, 559, 370–376 CrossRef CAS PubMed.
- J. Elbaz, O. Lioubashevski, F. Wang, F. Remacle, R. D. Levine and I. Willner, Nat. Nanotechnol., 2010, 5, 417–422 CrossRef CAS PubMed.
- Y. Benenson, T. Paz-Elizur, R. Adar, E. Keinan, Z. Livneh and E. Shapiro, Nature, 2001, 414, 430–434 CrossRef CAS PubMed.
- D. Y. Zhang and G. Seelig, Nat. Chem., 2011, 3, 103–113 CrossRef CAS PubMed.
- B. Yurke, A. J. Turberfield, A. P. Mills, F. C. Simmel and J. L. Neumann, Nature, 2000, 406, 605–608 CrossRef CAS PubMed.
- F. C. Simmel, B. Yurke and H. R. Singh, Chem. Rev., 2019, 119, 6326–6369 CrossRef CAS PubMed.
- J. Zhu, F. Bošković, B.-N. T. Nguyen, J. R. Nitschke and U. F. Keyser, Nano Lett., 2021, 21, 1368–1374 CrossRef CAS PubMed.
- A. J. Genot, D. Y. Zhang, J. Bath and A. J. Turberfield, J. Am. Chem. Soc., 2011, 133, 2177–2182 CrossRef CAS PubMed.
- N. Srinivas, T. E. Ouldridge, P. Šulc, J. M. Schaeffer, B. Yurke, A. A. Louis, J. P. K. Doye and E. Winfree, Nucleic Acids Res., 2013, 41, 10641–10658 CrossRef CAS PubMed.
- J. Zhu, L. Zhang, T. Li, S. Dong and E. Wang, Adv. Mater., 2013, 25, 2440–2444 CrossRef CAS PubMed.
- A. A. Green, P. A. Silver, J. J. Collins and P. Yin, Cell, 2014, 159, 925–939 CrossRef CAS PubMed.
- B. Li, A. D. Ellington and X. Chen, Nucleic Acids Res., 2011, 39, e110–e110 CrossRef CAS PubMed.
- Z. Zhang, D. Zeng, H. Ma, G. Feng, J. Hu, L. He, C. Li and C. Fan, Small, 2010, 6, 1854–1858 CrossRef CAS PubMed.
- N. E. C. Haley, T. E. Ouldridge, I. Mullor Ruiz, A. Geraldini, A. A. Louis, J. Bath and A. J. Turberfield, Nat. Commun., 2020, 11, 2562 CrossRef CAS PubMed.
- D. W. B. Broadwater, Jr. and H. D. Kim, Biophys. J., 2016, 110, 1476–1484 CrossRef PubMed.
- K. A. Schmidt, C. V. Henkel, G. Rozenberg and H. P. Spaink, in Fluorescence Spectroscopy, Imaging and Probes: New Tools in Chemical, Physical and Life Sciences, eds. R. Kraayenhof, A. J. W. Visser and H. C. Gerritsen, Springer-Verlag Berlin, Berlin, 2002, vol. 2, pp. 123–128 Search PubMed.
- M. You, G. Zhu, T. Chen, M. J. Donovan and W. Tan, J. Am. Chem. Soc., 2015, 137, 667–674 CrossRef CAS PubMed.
- A. J. Thubagere, W. Li, R. F. Johnson, Z. Chen, S. Doroudi, Y. L. Lee, G. Izatt, S. Wittman, N. Srinivas, D. Woods, E. Winfree and L. Qian, Science, 2017, 357, eaan6558 CrossRef PubMed.
- P. Petersen, G. Tikhomirov and L. Qian, Nat. Commun., 2018, 9, 5362 CrossRef CAS PubMed.
- L. Restrepo-Pérez, C. Joo and C. Dekker, Nat. Nanotechnol., 2018, 13, 786–796 CrossRef PubMed.
- R.-J. Yu, Y.-L. Ying, R. Gao and Y.-T. Long, Angew. Chem., Int. Ed., 2019, 58, 3706–3714 CrossRef CAS PubMed.
- M. Wanunu, T. Dadosh, V. Ray, J. Jin, L. McReynolds and M. Drndić, Nat. Nanotechnol., 2010, 5, 807–814 CrossRef CAS PubMed.
- K. Liu, C. Pan, A. Kuhn, A. P. Nievergelt, G. E. Fantner, O. Milenkovic and A. Radenovic, Nat. Commun., 2019, 10, 3 CrossRef CAS PubMed.
- K. Doroschak, K. Zhang, M. Queen, A. Mandyam, K. Strauss, L. Ceze and J. Nivala, Nat. Commun., 2020, 11, 5454 CrossRef CAS PubMed.
- Z. Zhu, R. Wu and B. Li, Chem. Sci., 2019, 10, 1953–1961 RSC.
- N. A. W. Bell and U. F. Keyser, Nat. Nanotechnol., 2016, 11, 645 CrossRef CAS PubMed.
- J. Kong, J. Zhu, K. Chen and U. F. Keyser, Adv. Funct. Mater., 2019, 29, 1807555 CrossRef.
- R. Kawano, Biotechnol. J., 2018, 13, 1800091 CrossRef PubMed.
- B. Wang, C. Thachuk, A. D. Ellington, E. Winfree and D. Soloveichik, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E12182–E12191 CrossRef CAS PubMed.
- L. P. Reynaldo, A. V. Vologodskii, B. P. Neri and V. I. Lyamichev, J. Mol. Biol., 2000, 297, 511–520 CrossRef CAS PubMed.
- J. Zhu, L. Zhang, S. Dong and E. Wang, ACS Nano, 2013, 7, 10211–10217 CrossRef CAS PubMed.
- J. Zhu, L. Zhang, Z. Zhou, S. Dong and E. Wang, Anal. Chem., 2014, 86, 312–316 CrossRef CAS PubMed.
- J. Zhu, N. Ermann, K. Chen and U. F. Keyser, Small, 2021, 17, 2100711 CrossRef CAS PubMed.
- K. Chen, J. Kong, J. Zhu, N. Ermann, P. Predki and U. F. Keyser, Nano Lett., 2019, 19, 1210–1215 CrossRef CAS PubMed.
- J. Kong, J. Zhu and U. F. Keyser, Chem. Commun., 2017, 53, 436–439 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, DNA sequences, and supporting data. See DOI: https://doi.org/10.1039/d2nr04048d |
| This journal is © The Royal Society of Chemistry 2022 |