
Tunable gold nanorod/NAO conjugates for selective drug delivery in mitochondria-targeted cancer therapy†
Sergio
González-Rubio
ab,
Cástor
Salgado
c,
Vanesa
Manzaneda-González
a,
Mónica
Muñoz-Úbeda
ab,
Rubén
Ahijado-Guzmán
a,
Paolo
Natale
 ab,
Víctor G.
Almendro-Vedia
ab,
Elena
Junquera
a,
José Osío
Barcina
c,
Irene
Ferrer
bde,
Andrés
Guerrero-Martínez
*a,
Luis
Paz-Ares
*bdef and
Iván
López-Montero
*abg
ab,
Víctor G.
Almendro-Vedia
ab,
Elena
Junquera
a,
José Osío
Barcina
c,
Irene
Ferrer
bde,
Andrés
Guerrero-Martínez
*a,
Luis
Paz-Ares
*bdef and
Iván
López-Montero
*abg
aDepartamento Química Física, Universidad Complutense de Madrid, Avda. Complutense s/n, 28040 Madrid, Spain. E-mail: aguerrero@quim.ucm.es; ivanlopez@quim.ucm.es
bInstituto de Investigación Hospital Doce de Octubre (imas12), Avenida de Córdoba s/n, 28041 Madrid, Spain. E-mail: lpazares@ucm.es
cDepartamento Química Orgánica, Universidad Complutense de Madrid, Avda. Complutense s/n, 28040 Madrid, Spain
dCentro Nacional de Investigaciones Oncológicas (CNIO), Madrid, Spain
eCiberonc, Madrid, Spain
fDepartamento de Medicina, Universidad Complutense de Madrid, Avda. Complutense s/n, 28040 Madrid, Spain
gInstituto Pluridisciplinar, Ps. Juan XXIII 1, 28040 Madrid, Spain
First published on 17th May 2022
Abstract
Nonyl acridine orange (NAO) is a lipophilic and positively charged molecule widely used as a mitochondrial fluorescent probe. NAO is cytotoxic at micromolar concentration and might be potentially used as a mitochondria-targeted drug for cancer therapy. However, the use of NAO under in vivo conditions would be compromised by the unspecific interactions with off-target cells and negatively charged proteins present in the bloodstream. To tackle this limitation, we have synthesized NAO analogues carrying an imidazole group for their specific binding to nitrilotriacetic (NTA) functionalized gold nanorods (AuNRs). We demonstrate that AuNRs provide 104 binding sites and a controlled delivery under acidic conditions. Upon incubation with mouse embryonic fibroblasts, the endosomal acidic environment releases the NAO analogues from AuNRs, as visualized through the staining of the mitochondrial network. The addition of the monoclonal antibody Cetuximab to the conjugates enhanced their uptake within lung cancer cells and the conjugates were cytotoxic at subnanomolar concentrations (c50 ≈ 0.06 nM). Moreover, the specific interactions of Cetuximab with the epidermal growth factor receptor (EGFR) provided a specific targeting of EGFR-expressing lung cancer cells. After intravenous administration in patient-derived xenografts (PDX) mouse models, the conjugates reduced the progression of EGFR-positive tumors. Overall, the NAO-AuNRs provide a promising strategy to realize membrane mitochondria-targeted conjugates for lung cancer therapy.
1 Introduction
Traditional cancer treatments, such as surgery, chemotherapy and radiation therapy have given way in the recent years to targeted cancer therapy.1 This therapeutical approach is based on the administration of pharmacological drugs, mainly monoclonal antibodies, small molecule inhibitors or immunotoxins, that interfere with specific signaling proteins involved in tumor genesis and progression. Key targets are membrane receptor proteins that are overexpressed in malignant cells compared to the healthy counterparts. For instance, the epidermal growth factor receptor (EGFR) and human epidermal growth factor receptor 2 (HER2) are major biomarkers of lung cancer,2 being overexpressed in 41% and 2% of lung adenocarcinomas respectively.3,4 Current clinical treatments against lung cancer use Cetuximab and Trastuzumab monoclonal antibodies as EGFR and HER2 inhibitors respectively with relative success.3,5 It is known that cancer cells develop acquired resistance against such monoclonal antibodies,6–8 therefore, new approaches for cancer therapy able to bypass biochemical resistance are needed.Mitochondria are promising subcellular targets as they are key regulators of apoptosis.9 A new generation of antitumor agents aims to induce or facilitate mitochondrial fail, thus activating programmed cell death.10 Mitochondria-targeted molecules generally present common physicochemical properties, as learnt from the synthesis of mitochondrial fluorescent probes: they are positively charged and lipophilic. Indeed, mitochondria are membranous systems composed of two bilayers and their membrane potentials are the largest within the cell. This allows the accumulation of cationic molecules that usually monitor the polarization state of mitochondria. Interestingly, cytotoxic cationic molecules exhibit an unusual accumulation in carcinoma cells.11–14 The presence of a more negative membrane potential in mitochondria of cancer cells can explain the greater accumulation of those cationic species.15 Among the tens of mitochondrial tracer molecules, 10-nonyl-acridine (NAO) (Fig. 1) is a fluorescent compound widely used in cell microscopy. NAO is a lipophilic fluorescent molecule that stains the inner mitochondrial membranes (IMMs).16 However, incubation of eukaryotic cells with NAO at high concentrations induces cytotoxicity.16 A high accumulation of NAO in IMMs prevents cellular respiration that eventually produces the alteration of the mitochondria morphology altering the mitochondrial cristae into multi-lamellar stacked membranes and the final collapse of the organelle.16 In particular, NAO also inhibits the oxygen consumption, the activity of respiratory complexes and ATP synthesis.17 We have recently unveiled the supramolecular mechanism underlying such NAO cytotoxicity, which consists on the formation of IMM molecular zippers.18 NAO promotes strong adhesion of lipid membranes sustained by the formation of anti-parallel H-dimers of NAO molecules from apposing bilayers. The planar geometry of the acridine molecule promotes its molecular stacking that results from π–π interactions between stacked molecules. The NAO–NAO self-interactions induces a green-to-red emission shift that is mainly observed in high concentrated samples. The formation of IMM molecular zippers alter the ultrastructure of the mitochondrial cristae, leading to the final collapse of mitochondria and eventually inducing the apoptosis of the cell. In addition, enhanced cytotoxicty of NAO has been observed in human lung carcinoma H520 cells compared to healthy human lung primary fibroblasts.19 The hyperpolarized mitochondrial membrane potential of cancer cells15 has been hypothesized for the selective accumulation of NAO.
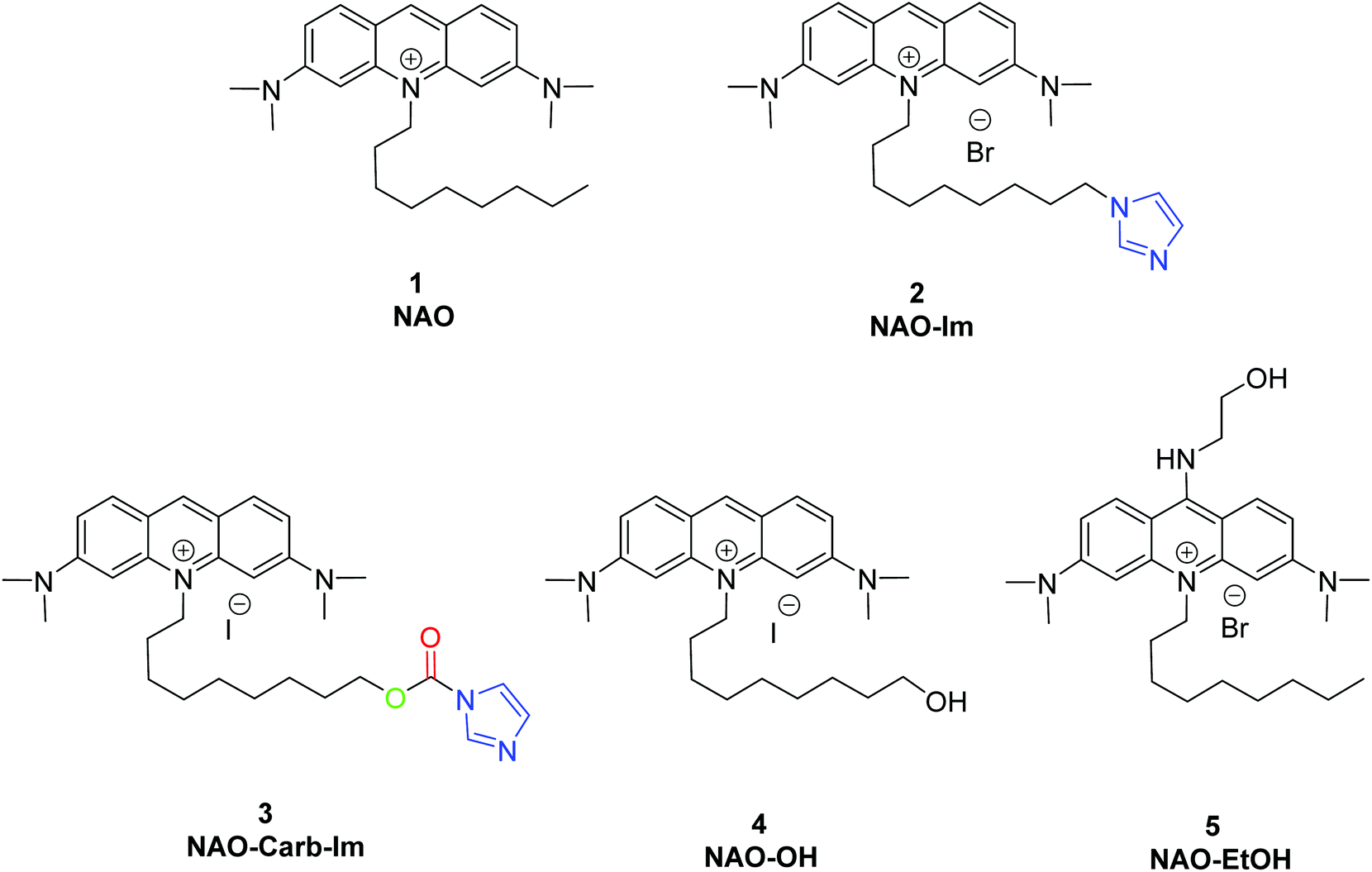 | ||
| Fig. 1 Synthesized and used compounds in this study. | ||
Here, we propose the combination of NAO's cytotoxicity with the ability of gold nanoparticles as efficient nanocarriers to engineer a highly specific anticancer agent able to trigger mitochondrial cell death exclusively in malignant cells. The use of gold nanoparticles for medical purposes has notably expanded in the last decade due to their remarkable optical properties and high in vivo stability.20 Compared to spherical nanoparticles with similar sizes, gold nanorods (AuNRs) have emerged as promising nanomaterials due to their larger absorption-to-scattering ratio and surface area available for the attachment of biofunctional molecules.21 The cellular response and high loading capacity of AuNRs make them particularly suitable for alternative cancer therapies, such as plasmonic photothermal therapy (PPTT)22 or the delivery of therapeutic molecules such as antibodies, peptides or nucleic acids.23
In the present study, we have built nano-conjugates composed of AuNRs and NAO derivatives as a novel approach for the treatment of lung cancer (Scheme 1). The AuNRs were functionalized with lipoic amide-nitrilotriacetic (NTA)-Co2+, which subsequently allowed their coating with the functional imidazole-containing molecules by exploiting the high affinity between NTA and imidazole. Then, by using in vivo functional switches that can be activated by temporally and spatially well-controlled biochemical signals, such as a pH drop provided by the lumen of endosomes or by enzymatic cleavage, NAO can be released within the cell and accumulate further within mitochondria to promote IMM adhesion and cell death. Moreover, the specific antibody Cetuximab, has been used to direct the nanovector cell internalization and target lung cancer cells through specific interactions.
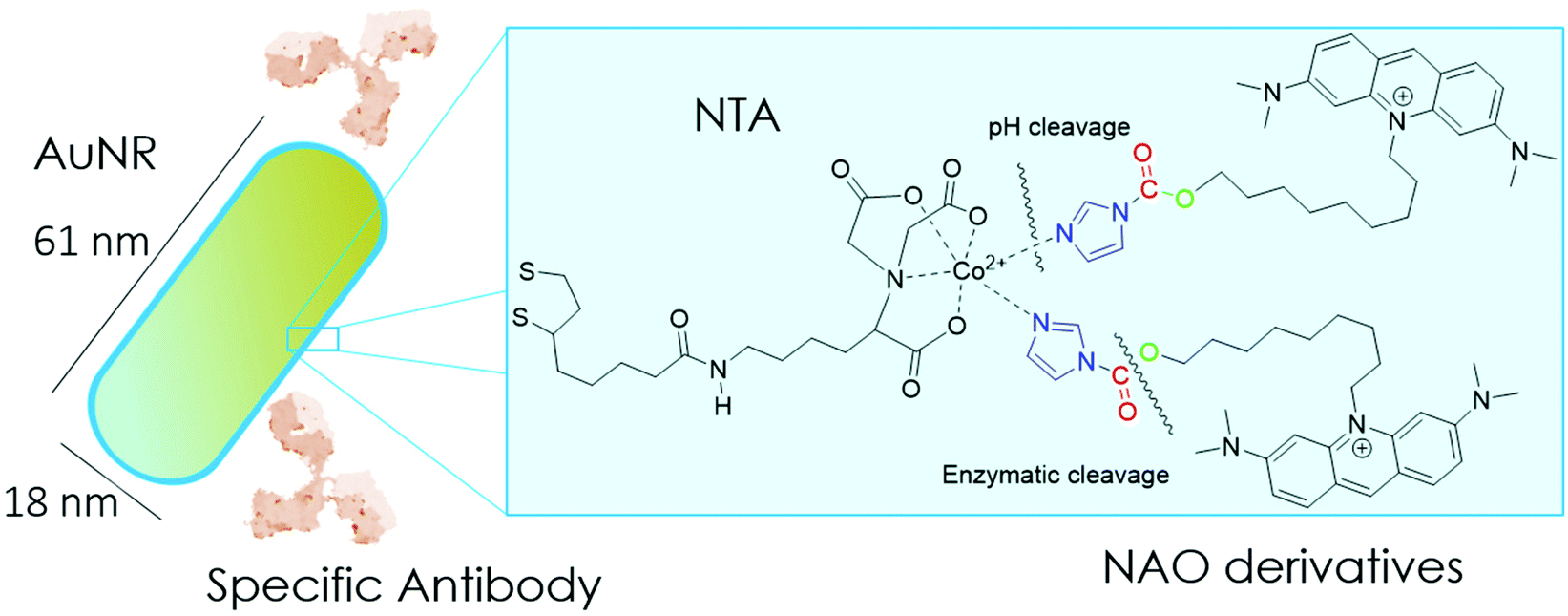 | ||
| Scheme 1 Cartoon of the mitochondrial membrane-targeted conjugate. The surface of gold nanorods is functionalized with NTA groups to specifically bind NAO derivatives carrying imidazole groups. NAO compounds can also have as a second cleavable carbamate linker. The conjugate is coated with the monoclonal antibody Cetuximab against the epidermal growth factor receptor EGFR. | ||
According to our strategy (Scheme 1), we have synthesized NAO analogues bearing imidazole groups and their corresponding degradation products after a pH drop or enzymatic cleavage. We then examined both the mitochondrial accumulation and cytotoxic effect in mouse embryonic fibroblasts (MEFs) upon incubation with the free molecules and with the AuNR conjugates. We further tested the conjugated nanovector on EGFR-negative and EGFR-positive lung cancer cell lines in vitro and on patient-derived xenografts (PDX) mouse models.24,25 The combined action of mitochondrial membrane-targeted compounds and AuNRs aims to activate the apoptosis by means of a mechanical action that compromise the ultrastructure of mitochondria. The findings of this study are discussed in terms of their potential application in cancer therapy.
2 Methods
2.1 Synthesis of NAO derivatives
See ESI† for details.2.2 Synthesis and characterization of gold nanorods (AuNRs) and conjugates
See ESI† for details.2.3 Cell cultures
NIH 3T3 mouse embryonic fibroblasts (MEF), purchased from ATCC, were cultured in Dulbecco Modified Eagle Medium (DMEM), 25 mM glucose (Gibco) supplemented with 10% fetal bovine serum (South Africa S1300; Biowest, Nuallé, France), penicilin and streptomycin (at a final concentration of 100 U mL−1 and 100 μg mL−1, respectively) and 1% of non-essential amino acids (all Gibco). The cells were grown in a humidified incubator (Forma Steri-Cycle Themofisher; 5% CO2) at 37 °C and maintained with a split ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 at 80% of confluence in T75 flasks (Nunc). PC-9 and H1944 cell lines, both purchased from ATCC, were cultured in RPMI Medium supplemented with 10% fetal bovine serum and penicillin/streptomycin. Cetuximab-ERBITUX (Merck KGaA, Merck Serono) was obtainied from Hospital 12 de Octubre (5 mg mL−1).
:10 at 80% of confluence in T75 flasks (Nunc). PC-9 and H1944 cell lines, both purchased from ATCC, were cultured in RPMI Medium supplemented with 10% fetal bovine serum and penicillin/streptomycin. Cetuximab-ERBITUX (Merck KGaA, Merck Serono) was obtainied from Hospital 12 de Octubre (5 mg mL−1).
2.4 Confocal laser scanning microscopy (CLSM)
Confocal microscopy images of cells were collected at 37 °C with a Nikon Ti-E inverted microscope equipped with a Nikon C2 confocal scanning confocal module, 488 and 561 nm continuous lasers, emission bandpass filters, and a Nikon Plan Apo 100 × NA 1.45 oil immersion objective. MEFs were seeded at 3 × 104 cells per cm2 in a four-chamber Lab-Tek® slide (Thermofisher) and incubated with complete DMEM for 24 h at 37 °C. Prior to confocal fluorescence imaging, cells were washed with PBS and then incubated with indicated NAO derivative and conjugate.2.5 Inductively coupled plasma mass spectrometry (ICP-MS)
To quantify the cellular uptake of conjugates we determined the number of nanoparticles per cell by ICP-MS. PC9 and H1944 cells were grown to a confluence of 60%. After incubation for 12 h with different conjugates at different concentrations (0.01 and 0.1 nM), cells were washed, digested and treated as described in.262.6 Cell viability assays
The inhibitory concentration (c50) was calculated by treating cells seeded in 96-well plates with a range of concentrations of each inhibitor under assay for 96 h. Cells were then fixed and stained with crystal violet (0.1% w/v). Then, crystal violet was diluted in 20% (v/v) acetic acid. Absorbance measured at 595 nm, which is correlated to the number of cells in each well, was quantified and analyzed as in.25 All experiments were performed in duplicate.2.7 Patient-derived xenografts (PDX) in mouse models
Tumor fragments from two PDX with a high (TP143) and low (TP60) expression levels of EGFR, respectively, were used.24,25 Tumor fragments were implanted in 5/6-week old nude female athymic nude mice (nu/nu; Envigo) in groups of 12 animals per PDX model (6 for each treatment to minimize interindividual variability). When the tumor size reached 180 mm3, the animals were randomly divided into one of the following groups: (a) control treated with the vehicle (physiological solution) and (b) mitochondrial membrane-targeting conjugate. The conjugate was administered intravenously in a volume of 0.2 mL. The treatment scheme was established by previous data in:27 4 mg kg−1 intravenously, 2 times a week for 3 consecutive weeks. The mice were monitored daily and weighted once a week to detect possible signs of stress during the treatment. The size of the tumor was measured with a caliber twice a week and the volume was estimated using the formula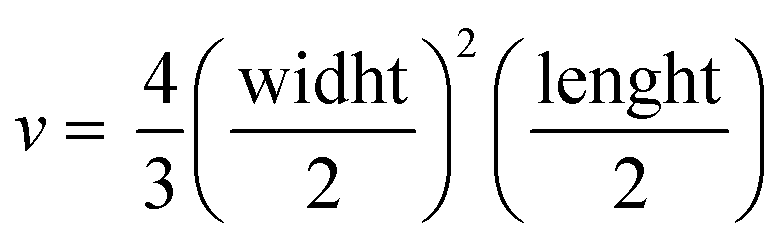 . The relative growth of the tumor from the initial volume is shown as a function of the number of doses. All animal procedures conformed to European Union Directive 86/609/EEC and Recommendation 2007/526/EC, enforced in Spanish law under RD 1201/2005. Animal protocols were approved by the CNIO Ethics Committee of Animal Experimentation. The experimental study protocols were additionally approved by local government (PROEX 138/18).
. The relative growth of the tumor from the initial volume is shown as a function of the number of doses. All animal procedures conformed to European Union Directive 86/609/EEC and Recommendation 2007/526/EC, enforced in Spanish law under RD 1201/2005. Animal protocols were approved by the CNIO Ethics Committee of Animal Experimentation. The experimental study protocols were additionally approved by local government (PROEX 138/18).
2.8 UV-vis spectroscopy
UV-Vis absorption using spectra were recorded at 20 °C (Genesis 10 spectrophotometer, Fisher scientific) with a spectral bandwidth of 1.0 nm and a scan rate of 200 nm min−1. All experiments were carried out using plastic cuvettes with a 1 cm optical path.2.9 Fluorescence spectroscopy
We determined the pH-dependent release of NAO-imidazole derivatives from conjugates by fluorescence spectroscopy after their incubation at different pH conditions. Briefly, 100 μL of imidazole-containing conjugates were washed twice by centrifugation (10000g × 10 minutes) in buffer (PBS buffer at pH 7.4 or Acetate buffer at pH 5.5). Then, the nanoparticles were resuspended in the corresponding buffer and incubated for 12 h. After centrifugation, the fluorescence spectra of supernatants were measured with an Aminco Bowman series 2 spectrofluoreometer.
2.10 Transmission electron microscopy (TEM)
TEM images were obtained on a JEOL JEM-1010 transmission electron microscope operating at an acceleration voltage of 80 kV (CNME, Spain). The cells were incubated with the AuNRs, washed twice with PBS buffer, and fixed with a solution containing 2% (w/v) glutaraldehyde in PBS. Then, the cells were stained with a mixture of 1% (w/v) osmium tetroxide and 1.5% (w/v) potassium cyanoferrate. After two washing cycles, the cells were gradually dehydrated in acetone. The samples were embedded in Epon, sectioned for analysis, and cut by ultramicrotomy for observation.3 Results
3.1 Synthesis of nonyl-acridine (NAO) derivatives
To coat AuNRs bearing NTA-Co2+ complexes with NAO, an imidazole moiety is required for specific binding (Scheme 1). A first approach consisted on the synthesis of the NAO-Im compound (2) (Fig. 1). This molecule bears an imidazole group at the end of the hydrophobic tail. However, the NAO-Im molecule presents an important limitation since, at the acidic intracellular pH, the imidazole moiety is protonated and the positively charged alkyl chain may modify the lipophilic properties of NAO, thus inhibiting its ability to penetrate the mitochondrial membranes. This inhibitory effect may also be enhanced by the bulkier size of the imidazole group. For this reason, we decided to introduce a carbamate functional group (–N-CO-O–, Scheme 1; NAO-Carb-Im (3), Fig. 1) as a second cleavable linker in our conjugate. Organic carbamates have been frequently used as pharmaceuticals in the form of drugs and prodrugs.28 Enzymatic cleavage of the carbamate linker will release the corresponding alcohol NAO-OH (4) (Fig. 1), a considerably less polar molecule than protonated and bulky compound 2. Moreover, a NAO-EtOH derivative (5) was synthesized as a tentative candidate to prevent NAO stacking within mitochondria. Note that, in the case of alcohol 5, the alkyl linker is placed at de C-9 position of the acridine moiety, resulting in a derivative with no polar groups in the alkyl chain. See the ESI† for experimental details and synthetic routes.3.2 Mitochondrial membrane staining of NAO derivatives
The use of NAO as a fluorescent probe for inner mitochondrial membranes (IMMs) relies on its cationic and lipophilic nature, allowing these molecules to spontaneously diffuse into membrane environments and accumulate within the mitochondria due to the negative membrane potential of IMMs. Structural modifications may thus influence the staining properties of NAO molecules. To determine whether the specific mitochondrial staining of the present NAO derivatives (2–5) was not compromised after functionalization, we incubated the different compounds with MEFs and imaged the cells by confocal laser scanning microscopy (CLSM). As in case of NAO,18 we found that its derivatives labelled progressively the mitochondria of single living cells when incubated at nM concentrations and that the mitochondrial network was unaltered upon staining after 24–48 h (Fig. 2). At higher concentrations, on the μM range, the cells experienced apoptosis, as shown by global morphological changes including blebbing and cell shrinkage (Fig. 2). As a result, the NAO analogues stained other membranous environments within the cell as the mitochondrial membrane potential is compromised during apoptosis. Also, at these high concentrations, the fluorescence emission of NAO derivatives shifted to red (Fig. 2). As in the case of NAO,18 the spectral shift of 2–5 may result from self-association into supramolecular stacks, partially promoted by the presence of negatively charged lipids, cardiolipin among them.29 Qualitatively, and compared to the NAO precursor, we found that compounds 2–4 present a lower capacity for IMM staining but retain a similar cytotoxic profile, whereas compound 5 requires higher concentrations for both membrane staining and apoptosis.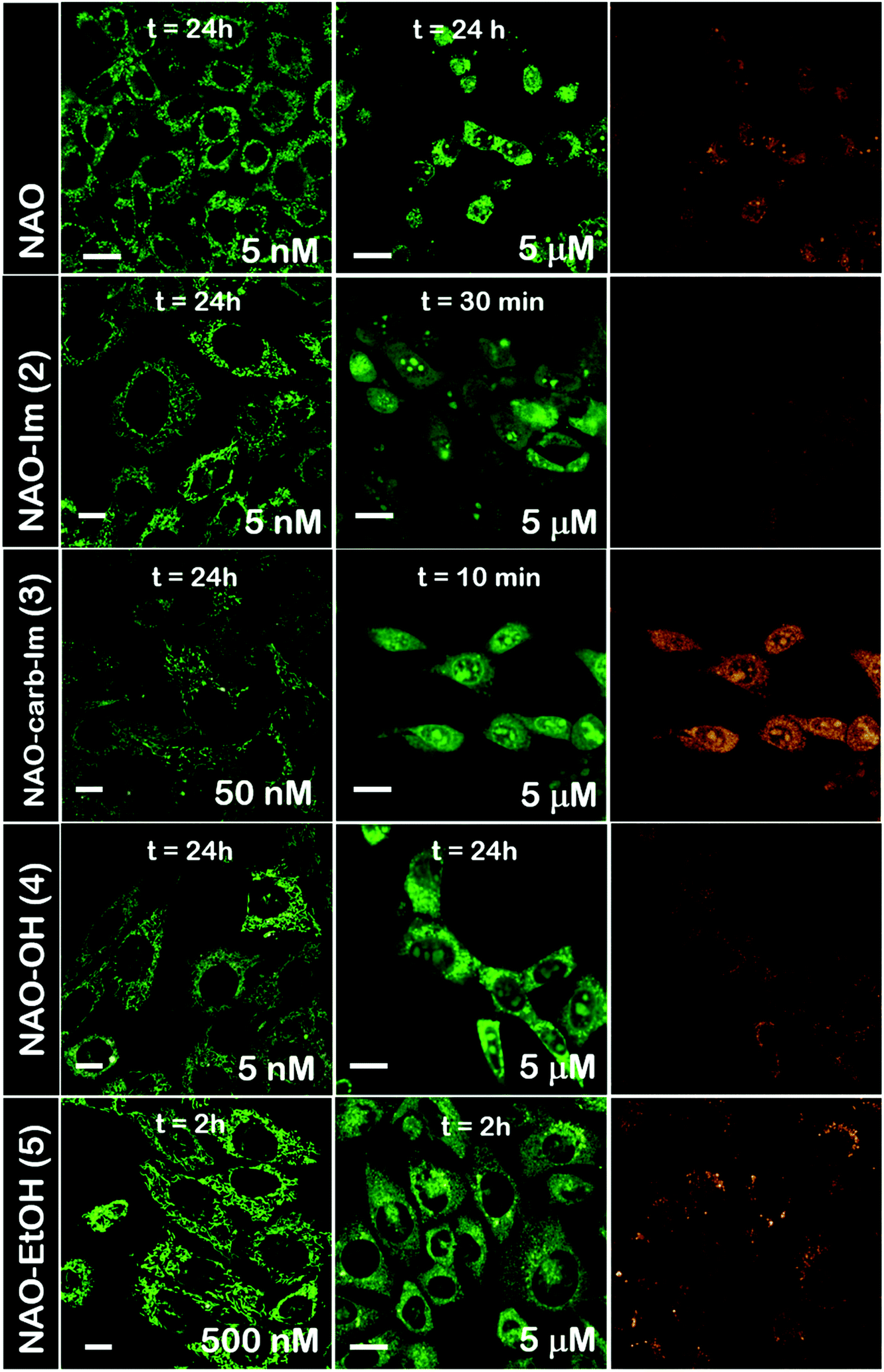 | ||
| Fig. 2 Cellular uptake of NAO derivatives within MEFs and mitochondrial stain. Confocal fluorescence micrographs (green channel, λexc = 488 nm; red channel, λexc = 561 nm) of MEFs upon incubation with NAO and different NAO derivatives (2–5). At low concentrations (in the submicromolar range), NAO derivatives stain the mitochondrial network and do not elicit cell death. At high concentrations (in the μM range), NAO derivatives induce cell death. Scale bars are 10 μm. | ||
3.3 Evaluation of the cellular uptake of NAO-loaded gold nanorods and the release of NAO analogues
Gold nanorods (AuNRs) were synthesized by a seed-assisted protocol.30 The seeds were prepared by the standard CTAB/NaBH4 procedure.31 The electron micrographs show that the bare AuNRs present an average length of 61 ± 4 nm and average diameter of 18 ± 2 nm. UV-VIS spectroscopy and ζ-potential measurements showed a localized surface plasmon resonance (LSPR) at 800 nm and a surface negatively charged −9.3 ± 0.9 mV (see Fig. S1 and S2 ESI†). AuNRs were then functionalized with both thiolated polyethylene glycol and lipoic acid. The final step consisted in the modification of the NRs surface with lipoic amide-NTA-Co2+ to obtain nanorods that were coated with functional NTA groups (Fig. S3†). Furthermore, before and after every biofunctionalization or bioconjugation step, the colloidal stability of the AuNRs was checked by measuring the absorption spectrum (UV-Vis-NIR region). All UV-Vis-NIR spectra showed that the LSPR band at 800 nm remained unaltered after each procedure (see ESI†) showing that the functionalized and conjugated AuNRs were stable in aqueous buffer during their synthesis and use.After the synthesis of NTA-functionalized AuNRs, the nanoparticles were incubated in excess with 2 and 3 to prepare 2-AuNRs and 3-AuNRs respectively (Fig. S4 and S5†). The addition of these NAO derivatives did not modify the LSPR of the AuNRs but altered the surface charge differently. Whereas 2 reduced the negative surface charge of Au-NRs (−8.4 ± 0.5 mV), the presence of 3 increased the negative surface charge up to −14.6 ± 0.5 mV. The cationic charge of the nitrogen heterocycle in 2 partially reduces the surface charge of the AuNRs. Although a straightforward explanation for the increased negative charge in 3 is not evident, the introduction of the carbamate group may contribute to this observation. The changes in the surface charge were taken as confirmation of the successful conjugation of imidazole derivatives on the surface of the AuNRs. Moreover, the extinction coefficient of 2 and 3 compounds allowed us to estimate the number of NAO derivatives loaded on each AuNR. From the UV-Vis spectra, an estimate of 104 NAO analogues per AuNR was calculated (see Fig. S6†). To assess the release of compounds 2 and 3 from AuNRs under acidic conditions, the NAO-loaded gold nanorods were incubated for 12 h at two different pH (7.4 and 5.5). The low pH mimicked the acidic medium found in the endocytic pathway. After centrifugation, the amount of released NAO derivative was then quantified and compared by fluorescence spectroscopy. Interestingly, both 2- and 3-AuNRs showed a much higher release of NAO compounds into the aqueous solution at low pH than at physiological conditions (Fig. 3A). The controlled release of compounds 2 and 3 by a pH drop is demonstrated.
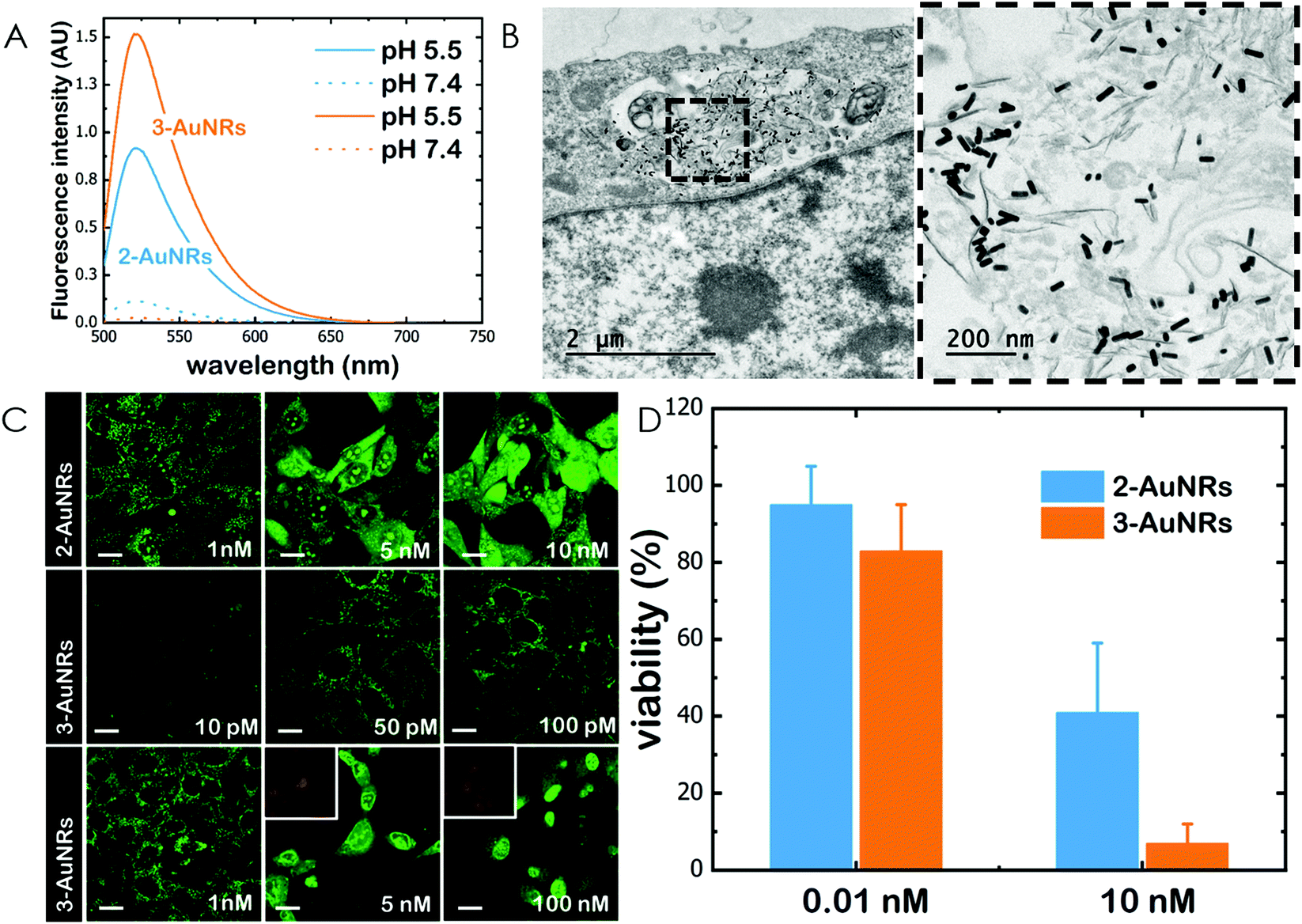 | ||
| Fig. 3 Cellular uptake of AuNRs and conjugates within MEFs. (A) Fluorescence emission spectra of 2-AuNRs and 3-AuNRs supernatnats after incubation for 12 h at different pH conditions. (B) TEM micrograph of a single cell loaded with AuNRs. The right panel is an image at higher magnification. (C) Confocal fluorescence micrographs (green channel, λexc = 488 nm; red channel, λexc = 561 nm) of MEFs upon incubation with 2-AuNRs and 3-AuNRs. At low concentrations (in the subnanomolar range), NAO conjugates stain the mitochondrial network and do not elicit cell death. At high concentrations (in the nM range), NAO conjugates induce cell death. Scale bars are 10 μm. (D) Cell viability of MEF cells upon incubation with several doses (0.01 and 10 nM) of 2-AuNRs and 3-AuNRs. | ||
MEFs were then incubated with 2-AuNRs and 3-AuNRs to evaluate the release of 2, 3 and 4 compounds to the cytosolic medium through the endosomal acidic environment. Gold nanoparticles are known to enter the cell through the endocytic pathway (see Fig. 3B) and released again to the extracellular medium32 unless active molecules, usually pH-sensitive peptides, are present and promote endosomal escape.33,34 The release of NAO derivatives in living MEFs cultures was visualized and traced again by CLSM (Fig. 3C). Similar to previous experiments with the free NAO species, the mitochondrial network of MEFs was labelled with both 2- and 3-AuNRs. Surprisingly, cell staining occurred at subnanomolar concentration of 2- and 3-AuNRs and cell death was now induced at nM concentrations of the conjugates. As the cytotoxic concentration of free NAO is ≈10 μM,19 this suggests that each AuNR is coated with ≈104 NTA groups, which allow the binding of 2–3 imidazole moieties, in agreement with the estimate obtained by UV-vis spectroscopy (Fig. S6†). As both 2-AuNRs and 3-AuNRs release their NAO analogue by a pH drop, the mitochondrial staining at low concentration was likely produced by the free NAO analogues released within the endosomes and diffusing across the cytosol of MEFs. At high concentrations, a closer assessment of fluorescence micrographs (Fig. 3C) showed that the cells did not exhibit a clear collapse when incubated with 2-AuNRs whereas the treatment with 3-AuNRs promoted the apoptotic hallmarks of blebbing and cell shrinkage. To quantitatively compare the cytotoxicity of 2-AuNRs and 3-AuNRs in MEFs, cells were exposed for 24 h to two different amounts of NAO-loaded gold nanorods (0.01 and 10 nM). After incubation, cell viability was determined (Fig. 3D). As expected, the viability of MEFs was not compromised in the presence of 0.01 nM of NAO-loaded gold nanorods (cell viability >80%) but it was severely affected at high concentration (10 nM). The survival rate of MEFs exhibited approximately 40% in the presence of 2-AuNRs whereas 3-AuNRs induced around 100% of cell mortality. As both 2-AuNRs and 3-AuNRs have a similar pH-dependent release of the corresponding NAO analogue, this suggests that the additional cleavable linker from the carbamate group is able to enhance the efficiency of 3-AuNRs in promoting cell apoptosis. Taking into account these results as well as the straightforward preparation of 3, we centred our attention in this derivative for the design of the final conjugate, which included the presence of the monoclonal antibody Cetuximab.
3.4 In vitro cytotoxicity of Cmab-3-AuNRs in EGFR cancer cells
The epidermal growth factor receptor (EGFR) is overexpressed in numerous lung adenocarcinomas. EGFR is therefore an essential target in lung cancer therapy. Specific monoclonal antibodies against EGFR, such as Cetuximab (Cmab), inhibit the EGFR signalling pathway that leads to tumour progression. The unspecific binding of Cmab to gold nanoparticles provides them with an enhanced cytotoxicity.27 However, it is well known that EGFR-overexpressing cancer cells acquire Cmab resistance.7,8 Here we employed Cmab as a selective targeting strategy to specifically deliver 3-AuNRs into cells showing high expression levels of EGFR. For this, Cmab molecules were unspecifically bound to 3-AuNRs upon incubation with antibodies in large excess. After washing, we could estimate by means of Coomassie blue staining gel electrophoresis that the surface coating was ≈30 Cmab molecule per AuNR (see Fig. S7 and S8† for details). The addition of Cmab did not change significantly the surface charge of 3-AuNRs (See ESI†).Then, CLSM showed that Cmab-3-AuNRs were effectively internalized into the cells after incubation with MEFs (Fig. 4A). Although the mitochondrial network was labelled within the first hours at nM concentration, the presence of Cmab shifted the concentration at which apoptotic morphological changes were observed. To gain a deeper insight into the selective uptake and cytotoxicity of the different conjugates we incubated AuNRs, 3-AuNRs and Cmab-3-AuNRs at two different non-cytotoxic concentrations (0.01 and 0.1 nM) with lung cancer cells with high (PC9) and low (H1944) expression levels of EGFR. The internalization of different conjugates was determined by ICP-MS (Fig. 4B). As expected, the number of internalized nanoparticles per cell was higher at higher nanoconjugate concentrations. Moreover, the PC9 cells systematically internalized more nanoconjugates than H1944 cells in all conditions. The presence of compound 3 affected differently the uptake of 3-AuNRs depending on the particle concentration and the type of cell and a clear correlation was not found though. However, the presence of Cmab enhanced ≈2-fold the uptake of nanoparticles in both PC9 and H1944 cells in comparison with the uptake of bare AuNRs and 3-AuNRs, suggesting a selective internalization of Cmab-3-AuNRs by the presence of EGFR in both cancer cells. As PC9 and H1944 cells have different expression levels of EGFR, a small amount of EGFR copies seems to be enough for the selective uptake of Cmab-containing nanoparticles.
 | ||
| Fig. 4 In vitro cytotoxicity of Cmab-3-AuNRs in cells. (A) Confocal fluorescence micrographs (green channel, λexc = 488 nm) of MEFs in the presence of different concentrations of Cmab-3-AuNRs. At low concentration (below 1 nM) the mitochondrial network is stained and cytotoxicity is produced at higher concentrations (above 1 nM). Scale bars are 10 μm. (B) Cellular uptake of AuNRs, 3-AuNRs and Cmab-3-AuNRs in PC9 (EGFR+) and H1944 (EGFR−) cells as measured by ICP-MS. (C) Cell viability of PC9 (EGFR+) and H1944 (EGFR−) cells upon incubation with several doses of AuNRs, 3-AuNRs and Cmab-3-AuNRs. Dose–response curves are shown and fitted with the Hill model (see main text for details). (D) Cell viability of PC9 (EGFR+) and H1944 (EGFR−) cells upon incubation with several doses of Cmab. | ||
Then, lung cancer cells were incubated with increasing amounts (pM–10 nM) of AuNRs, 3-AuNRs and Cmab-3-AuNRs and the cell viability was determined (Fig. 4C). The experimental data were adequately fitted by a classical sigmoidal curve that is obtained from the Hill equation:35
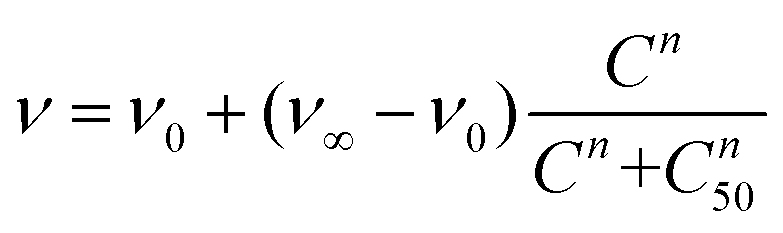 | (1) |
The incubation of PC9 and H1944 cells with 3-AuNRs reduced the c50 to 0.03 nM and 0.1 nM, respectively. Again, this observation suggests that AuNRs are extensively coated with 3 species, as the c50 of free NAO is 5–6 orders of magnitude larger than that of 3-AuNRs. Remarkably, the Hill coefficient also decreased to values closer to 1 (n ≈ 1.6 and n ≈ 1.2 for PC9 and H1944 respectively). A likely interpretation for this observation is the release of 3 compounds during the cellular uptake of 3-AuNRs, in agreement with the rapid labeling of the mitochondrial network within the first hours of incubation of MEFs with 3-AuNRs (see Fig. 3). As a result, the dose-response curve captures a synergetic effect where the non-cooperative cytotoxic effect of NAO is observed at lower concentrations than the c50 of AuNRs.
Cmab-3-AuNRs were also applied to PC9 and H1944 cells (Fig. 4C). The viability curve of PC9 cells displayed a combined trend resulting from the AuNRs and 3-AuNRs curves. The Hill coefficient was closer to n ≈ 2 but the c50 value felt in the subnanomolar range (c50 ≈ 0.06 nM). However, the viability curve of Cmab-3-AuNRs on H1944 cells exhibited an overlapping tendency with that obtained for AuNRs. The toxicity of Cmab-3-AuNRs was therefore compromised by the presence of the monoclonal antibody. To assess the effect of Cmab alone and isolate the effect of each component of the delivery nanosystem, we performed the cell viability assay for Cmab in both PC9 and H1944 cells. In agreement with previous reports,36,37 Cmab showed no growth inhibitory effect in PC9 and H1944 cell lines up to a concentration of 1 μM (Fig. 4D), despite of the difference in the EGFR expression level between cell lines (c50 = 5.85 μM and 14 μM, for PC9 and H1944 cells, respectively). As the average binding of Cmab is 30 antibody per 3-AuNR nanoparticle, we can discard any cytotoxic effect coming from Cmab as the c50 of 3-AuNRs c50 = 0.03 nM and 0.1 nM, for PC9 and H1944 cells, respectively. The slight differences in c50 of 3-AuNRs and Cmab-3-AuNRs found in PC9 cells suggests that the unspecific binding of Cmab may hamper the release of 3 molecules upon cellular uptake. Interestingly, Cmab seems to stimulate the cell growth of H1944 cells in the non-cytotoxic submicromolar range. The presence of Cmab might counterbalance the cytotoxic effect of 3 molecules on H1944 cells and the cytotoxic mechanism of the whole conjugate would correspond to that observed for bare AuNRs, where the viability curve is characterized by high values of the Hill coefficient. This hypothesis is supported by the absence of apoptotic morphological changes upon incubation of MEFs with Cmab-3-AuNRs (Fig. 4A). The nanosystem behaves differently at the cytotoxic concentration range (0.01–1 nM) depending on the EGFR expression level. The cytotoxicity of Cmab-3-AuNRs in mainly caused by the AuNRs in low expressing EGFR H1944 cells (c50 ≈ 0.3 nM), whereas the action of 3 molecules within high expressing EGFR PC9 cells reduces the c50 of AuNRs one order of magnitude.
3.5 In vivo therapy on PDX models
Finally, the antitumor effect of Cmab-3-AuNRs was evaluated using tumour fragments from two PDX models with a high (TP143) and low (TP60) expression levels of EGFR.24,25 After subcutaneous implant in nude female mice, the tumours reached a size of 200 mm3 and the treatment was applied (see Methods). Cmab-3-AuNRs were administrated intravenously and the relative change of the tumour size was monitored to test whether the presence of the monoclonal antibody had a selective impact on tumour progression (Fig. 5). In the absence of treatment, both PDX models exhibited a similar linear growth with a progression rate of ≈30%. Intravenous injection of Cmab-3-AuNRs reduced significantly the growth of TP143 tumours, which grew at a lower progression rate (≈15%) compared to the control. However, Cmab-3-AuNRs increased the progression rate (≈50%) of the T60 subcutaneous tumours. The dissimilar output of Cmab-3-AuNRs in TP143 and TP60 tumours can be refereed to the behaviour of the nanosystem found in PC9 and H1944 cells (Fig. 4). As no antitumor effect was found, as visualized by a decrease of the tumour size, the stimulated growth of TP60 tumours at this particular dose is in agreement with the increased cell cycle progression of H1944 cells promoted by Cmab in the non-cytotoxic submicromolar range. Also, the reduced progression rate of TP143 tumours is in agreement with the action of Cmab-3-AuNR in EGFR expressing cells, without reaching cytotoxic concentrations able to reduce the volume of the tumours tough. Overall, these results suggest that the presence of Cmab in 3-AuNRs exploits the selective targeting and action of the conjugate in in vivo tumour models.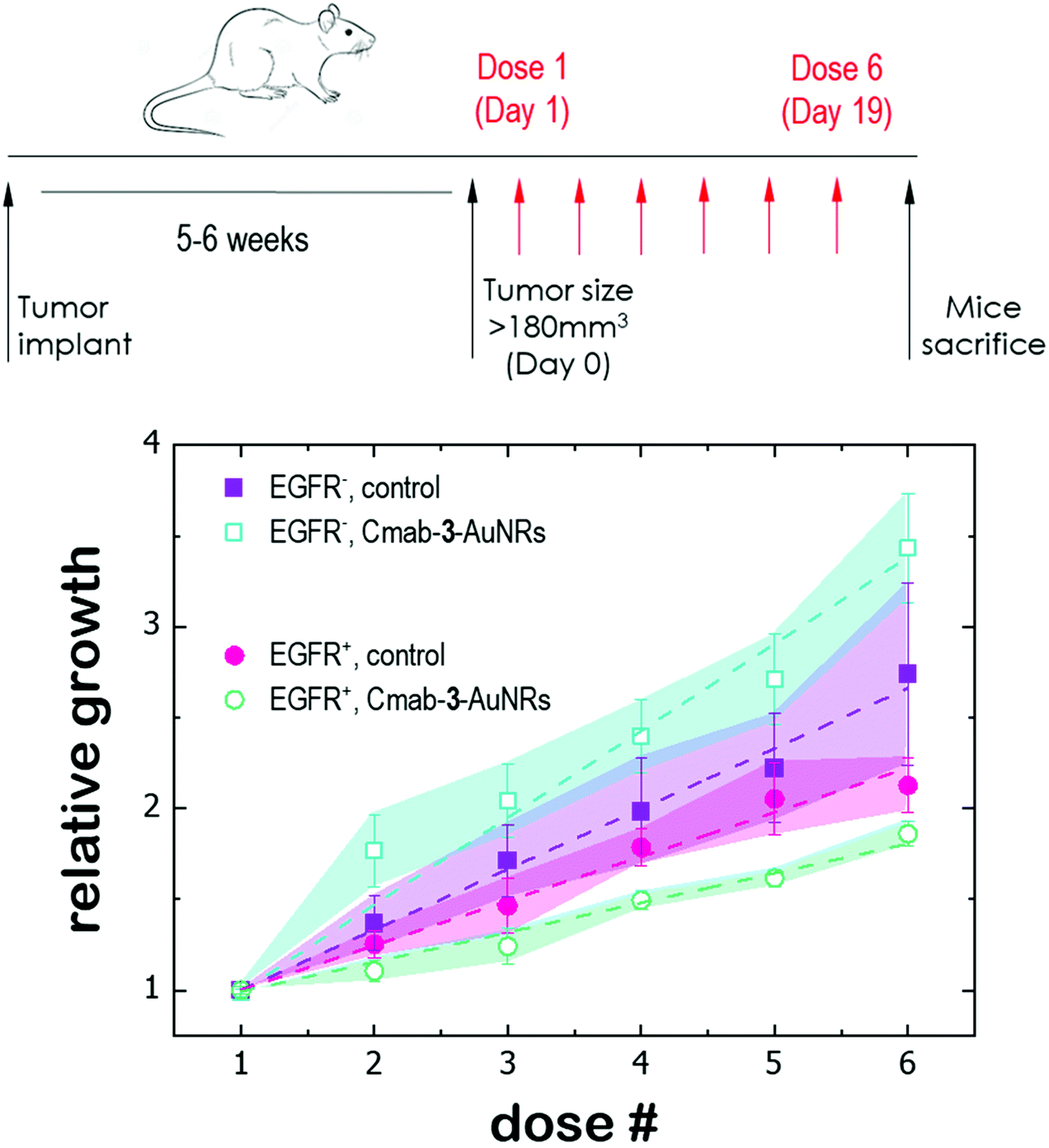 | ||
| Fig. 5 Antitumor effect of Cmab-3-AuNRs on subcutaneous PDX models with a high (EGFR+) and low (EGFR−) expression levels of EGFR respectively. PDX in nude female mice were intravenous treated with PBS (control) and Cmab-3-AuNRs (4 μg kg−1, 2 times a week for 3 weeks, red arrows) and tumor volumes were then monitored up to day 19. | ||
4 Discussion
The multiple essential functions of mitochondria make this important organelle an attractive target in cancer therapy. Mitochondria produce the biochemical energy in the form of adenosine triphosphate (ATP) through various metabolic pathways: oxidative phosphorylation (OXPHOS), glycolysis and the tricarboxylic acid (TCA) cycle. Moreover, mitochondria regulate the redox homeostasis and the metabolic signalling. Importantly, mitochondria are also the key controllers of the apoptotic pathways.38 Hacking any of these processes has become a promising strategy for cancer therapy, ultimately aiming at inducing mitochondrial failure and damage in malignant cells. The vast knowledge available of mitochondrial biochemistry enables the use of specific inhibitors molecules such as tamoxifen, 2-deoxyglucose, metformin, dichloroacetate, capsaicin and triphenylamine to target the electron transport chain and the OXPHOS function, glycolysis, the TCA cycle, reactive oxygen species (ROS) homeostasis and the apoptotic pathways, respectively.39A challenge in terms of mitochondrial-targeted drugs is the difficulty to cross the multiple barriers that represent the plasma membrane and the double membrane of the organelle itself. To overcome this problem, the selective delivery and accumulation of drugs into mitochondria can be rationalized through the design and synthesis of conjugates using lipophilic molecules bearing a delocalized positive charge, which allows them to cross membranous environments and remain within the negatively charged mitochondrial matrix.15 A first approach consists on bioconjugating the drug with peptides. Szeto-Schiller (SS) peptides40 and mitochondria-penetrating peptides (MPPs)41 combine highly hydrophobic residues such as cyclohexylalanine (Fx) or phenylalanine with positively charged amino acids such as arginine (R) or lysine (K). The alternate location of those residues provides the required degree of delocalized positive charge and hydrophobicity. A second strategy is the formation of prodrugs through combination of the active principle with small cationic and lipophilic transporters. Here the possibilities are manifold. The canonical example is the triphenyl phosphonium cation (TPP). TPP bears a delocalized positive charge over three phenyl groups, which also provides hydrophobicity to the molecule.42 Pyridinium-based molecules, rhodamine-based carriers and guadinine derivatives, have shown intrinsic mitochondria-penetrating capacity.43–45 In some cases, as that of Rhodamine 123 or NAO here, the carrier itself has been evaluated for cancer treatment without any other conjugated molecule.19,45 Other delocalized lipophilic cations (DLC) have been reviewed in the literature.46,47
The success of prodrugs lies on the efficient release of the drug within the cell. Stability and responsiveness are key features for optimal linkage of drugs to the molecular carriers. Usual cleavage linkers are covalent bonds such as amide, hydrazine, disulfide, ester, and ether bonds that are degraded by peptidase proteins, aqueous media, glutathione, acidic environments, or under strong acidic or basic conditions, respectively. Although mitochondria-targeted peptides and DLCs have shown specific mitochondrial targeting and delivery in in vitro experiments, two major drawbacks have been identified for their in vivo administration. The delocalized positive charge and lipophilicity of the conjugates result first in unspecific interaction with negatively charged proteins and secondly in molecular uptake by healthy cells. As a result, the absence of selectivity and the fast clearance of the conjugates hamper their use under in vivo conditions and represent the first barriers to their evaluation in clinical trials.48 To overcome these issues, the integration of mitochondrial-targeted drugs in larger nanocarriers with tuneable size, surface charge and multiple binding sites has been introduced as a new strategy to prevent off-target effects and blood clearance, as well as enhance the water solubility and in situ release of the active agents through stimulus responsiveness. Multiple options are available to implement this approach including lipoplexes,49 micelles,50 liposomes,51 polymersomes52 and metallic nanoparticles.53
Here, we coated the surface of AuNRs with NAO derivatives. NAO is a DLC whose cytotoxic mechanism does not rely on a specific mitochondrial protein but rather on the damages to the ultrastructure of mitochondrial membranes through the formation of NAO dimers from adhering membranes.18 The control of cell proliferation by means of physical perturbation renders our approach less prone to acquired drug resistance. The specific binding of NAO to AuNRs required the chemical modification at the rim of its aliphatic tail by introducing imidazole and carbamate groups (see compounds 2, 3 and 4 in Fig. 1). This allowed us to explore the efficiency of cleavable linkers based on the NTA-imidazole coordination bonds and carbamate bonds. Interestingly, the free 2–4 compounds were found to accumulate within mitochondria and triggered the apoptotic programs upon incubation at high μM concentration (Fig. 2). Although the red shift of the fluorescence intensity indicated that the cytotoxic mechanism remained unaltered after chemical modification, a fluorescence lifetime imaging microscopy (FLIM) analysis would confirm the formation of H-aggregates of NAO derivatives.18,19
The conjugation of compounds 2 and 3 with AuNRs throught the imidazole-NTA coordination bond was shown to be reversible at low pH conditions, a required step for a controlled released within cells using the acid environment of the endocytic pathway (Fig. 3). As AuNRs are not able to escape from endosomes and lysosomes, the specific staining of the mitochondrial network after incubation with 2- and 3-AuNRs showed that compounds 2 and 3 were released upon the degradation of the NTA-imidazole bond through the pH drop found in endosomal vesicles. Moreover, the higher cytotoxicity of 3-AuNRs compared to 2-AuNRs in MEFs suggests an additional cleavage of the carbamate bond in compound 3 that might result in an enhanced activity through the release of compound 4. Carbamates are generally enzymatically more stable than the corresponding esters and more prone to hydrolysis than amides in base catalysis reactions. As physiological environments are characterized mainly by acid/neutral pHs, the use of carbamate bonds in drug delivery is limited to prodrugs designed as substrates of specific enzymes.
The unspecific addition of Cetuximab to the conjugate did not compromise the cytotoxic character of 3-AuNRs as shown by the confocal fluorescence micrographs that evidenced also the release of the acridine compounds within MEFs (Fig. 4A). The specific targeting of Cmab was then assessed in EGFR-positive and EGFR-negative cancer cells. The enhanced cellular uptake of Cmab-3-AuNRs in EGFR-positive cells showed a selective interaction of Cmab with overexpressed EGFR membrane receptors (Fig. 4B). The cytotoxicity of different constituents of Cmab-3-AuNRs in cancer EGFR-expressing cancer cells was evaluated based on cell viability properties (Fig. 4C). The presence of compound 3 in 3-AuNRs reduced the c50 of AuNRs was by a factor ≈5, but also the dose-dependent response followed a different trend. In general, gold nanoparticles are believed to present low cytotoxic effects, as they are internalized by cells via a mechanism involving endocytosis and sent off the cell by exocytosis.32 However, it has been reported that gold nanoparticles exhibit potential cytotoxicity through induction of autophagy and oxidative stress.54 Here, a severe accumulation of AuNRs in cells was found to produce cytotoxicity at the nM range with a sigmoidal curve characterized by a Hill coefficient close to n ≈ 2, whereas this value decreased to n ≈ 1 for the conjugate 3-AuNRs, an akin value to that obtained for free NAO.19 Taken all these observations together, we suggest that compound 3 is transported by the AuNRs and released upon cellular uptake, so the cytotoxic effect is caused by the cargo molecules at lower concentrations than the c50 corresponding to bare AuNRs. Also, the large shift of c50 from the micromolar range of free NAO19 to the subnanomolar range of 3-AuNRs allows us to affirm that a large number of molecules (≈104, see estimation on Fig. S6†) are coating each AuNR.
However, the efficient action of NAO derivatives was weakened by the presence of the antibody molecules, as the cytotoxicity curve for Cmab-3-AuNRs tend to reach the curve of bare AuNRs (Fig. 4C). As Cmab did not affect the cell viability of EGFR-positive cells, the direct effect on the cell viability seems to be related to the capacity of NAO analogues to reach the cell. However, the cell internalization of conjugates is improved by the presence of Cmab (Fig. 4B), so the antibody seemed to hinder the release of the NAO derivative. This effect was more pronounced in EGFR-negative cancer cells in combination with the stimulated cell cycle progression of those cancer cells upon incubation with Cmab alone (Fig. 4D). In addition, the inert action of Cmab on the cell viability of both cancer cells up to the micromolar range allows to discard any cytotoxic effect of the antibody for the conjugates.
The dissimilar effect of Cmab on EGFR-expressing cancer cells might ground the experimental observations found in in vivo experiments (Fig. 5). First, the apparent increase of the progression rate of EGFR-negative tumours after Cmab-3-AuNRs administration may result from a stimulated cell cycle progression of EGFR-negative cells by Cmab. Second, the small but significant reduction of the progression rate of EGFR-positive tumours might be due to the selective targeting and uptake of Cmab-3-AuNRs within EGFR-positive cells. However, the current conjugate was not able to completely inhibit the tumour growth. The interaction between Cmab-3-AuNRs and the bloodstream proteins may mediate its rapid elimination through the reticuloendothelial system.55 A better selectivity and accumulation of NAO derivatives in tumours might be achieved with a more versatile platform. Conjugation chemistry to biomolecules is simple and well developed for gold nanoparticles,53 so that a controlled binding of Cmab to the conjugate may be achieved with other linker bonds using spacers with different lengths to prevent the unspecific interaction between the Cmab and NAO species. Also, an additional polymer coating of poly ethylene glycol (PEG), which would enhance the conjugate furtiveness, is conceivable as it will allow for wide and sustained biodistribution after intravenous administration.56
The use of mitochondrial membrane-targeted drugs with conjugated-loaded nanoparticles as promising antitumor agents57 may represent a more selective and effective option for cancer therapy. We have shown that conjugates can be internalized within cells through the endocytic pathway, accumulate into the mitochondria and release the active cargo inducing cell death. Although a better controlled binding is required, the presence of monoclonal antibodies offers an additional level of specificity provided by the selective uptake of conjugates in EGFR-positive cancer cells and the stimulated cell cycle progression of EGFR-negative cells. Further, the system has proven to be safe in murine PDX models of lung cancer after intravenous injection. However, the administrated dose was not efficient for tumour regression but the growth rate was partially reduced in EGFR-positive tumours. Higher doses may be needed for optimal treatment. Gold nanoparticles are chemically inert in the blood stream and the release of NAO derivatives was observed during cellular uptake; therefore, mitochondrial membrane-targeted anticancer conjugates might exhibit fewer side effects upon increasing the dosage. Also, the intravenous injection through the tail vein often results in the rapid accumulation of drugs mainly in lung, liver and spleen after 24 h of dose administration. A better efficiency might result with intratumoral administration due to the closer proximity of drugs with cancer cells and the absence of other conditions such as furtiveness and biodistribution or protein corona effects, among others.
Albeit unexplored here, additional advantages might be considered of the NAO/AuNRs conjugate. Both gold nanoparticles and NAO molecules may serve as well as self-contrast agents and allow for their tracing in in vitro or in vivo experiments by dark field microscopy and two photon fluorescence microscopy, respectively. Finally, our approach is easily transferable to a broad variety of cancers by using specific ligands targeting different cells.
5 Conclusions
In this study, mitochondrial membrane-targeting anticancer conjugates were prepared by loading the mitochondrial fluorescent dye NAO and the monoclonal antibody Cmab into a drug delivery system based on AuNRs bearing NTA groups. The binding of NAO molecules required the chemical modification of the probe with imidazole and carbamate groups at the aliphatic tail, whereas Cmab molecules coated the AuNR surface unspecifically. The conjugates were able to deliver and release NAO products into the mitochondria upon uptake by lung cancer cells, producing cell death after accumulation in mitochondria. The high loading capacity of AuNRs (≈104 NAO molecules per AuNR) shifted the c50 of free NAO from the micromolar range into the subnanomolar range for the conjugates. The mitochondrial membrane-targeting conjugates were also administered intravenously to murine PDX models of lung cancer. The growth rate was partially reduced in EGFR-positive tumours but further optimization is required to improve the outcome in terms of tumour regression.Abbreviations
| AuNRs | Gold nanorods |
| ATP | Adenosine triphosphate |
| CLSM | Confocal laser scanning microscopy |
| Cmab | Cetuximab |
| CTAB | Cetyl trimethyl ammonium bromide |
| DLC | Delocalized lipophilic cations |
| DMEM | Dulbecco modified eagle medium |
| EGFR | Epidermal growth factor receptor |
| FLIM | Fluorescence lifetime imaging microscopy |
| HER2 | Epidermal growth factor receptor 2 |
| IMM | Inner mitochondrial membranes |
| LSPR | Localized surface plasmon resonance |
| MEFs | Mouse embryonic fibroblasts |
| MPPs | Mitochondrial penetrating peptides |
| MTT | Methylthiazoletetrazolium |
| NAO | 10-Nonyl-acridine orange |
| NAO-Im | Compound 2 |
| NAO-Carb-Im | Compound 3 |
| NAO-OH | Compound 4 |
| NAO-EtOH | Compound 5 |
| NTA | Nitrilotriacetic |
| PDX | Patient-derived xenografts |
| PPTT | Plasmonic photothermal therapy |
| OXPHOS | Oxidative phosphorylation |
| ROS | Reactive oxygen species |
| SS | Szeto-Schiller |
| TCA | Tricarboxylic acid |
| TPP | Triphenyl phosphonium |
| UV-VIS | Ultraviolet – visible |
Author contributions
IL-M, JOB, AG-M and LP-A designed research. SGR, CS, VM-G, MM-U, RA-G, VA-V and IF performed research. SGR, MM-U, IF and IL-M analysed data. PN, EJ, AG-M and IL-M wrote the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ERC Starting Grant MITOCHON (ERC-StG-2013-338133) and ERC Proof of Concept mitozippers (ERC-PoC-2017-780440). I. L.-M. also acknowledges the financial support from the Spanish Ministry of Science, Innovation and Universities through the grant PGC2018-097903-B-I00 the TECNOLOGÍAS 2018 program funded by the Regional Government of Madrid (Grant S2018/BAA-4403 SINOXPHOS-CM and REACT ANTICIPA-UCM) with FEDER and FSE funds. A. G.-M. and E. J. acknowledge the financial support from Spanish Ministry of Science, Innovation and Universities (MICIU) (Grant number RTI2018-095844-B-100). A. G.-M. also acknowledges the Regional Governent of Madrid (Grant number P2018/NMT-4389 and REACT ANTICIPA-UCM).References
- T. A. Baudino, Curr. Drug Discovery Technol., 2015, 12, 3–20 CrossRef CAS PubMed.
- F. R. Hirsch, M. Varella-Garcia and F. Cappuzzo, Oncogene, 2009, 28, S32–S37 CrossRef CAS PubMed.
- A. Al Olayan, H. Al Hussaini and A. R. Jazieh, J. Infect. Public Health, 2012, 5(Suppl. 1), S50–S60 CrossRef PubMed.
- E. K. Kim, K. A. Kim, C. Y. Lee and H. S. Shim, PLoS One, 2017, 12, e0171280 CrossRef PubMed.
- C. J. Langer, P. Stephenson, A. Thor, M. Vangel and D. H. Johnson, J. Clin. Oncol., 2004, 22, 1180–1187 CrossRef CAS PubMed.
- T. M. Brand, M. Iida and D. L. Wheeler, Cancer Biol. Ther., 2011, 11, 777–792 CrossRef CAS PubMed.
- D. Westover, J. Zugazagoitia, B. C. Cho, C. M. Lovly and L. Paz-Ares, Ann. Oncol., 2018, 29, I10–I19 CrossRef CAS PubMed.
- S. Shimoyama, Mol. Clin. Oncol., 2014, 2, 175–181 CrossRef CAS PubMed.
- C. Wang and R. J. Youle, Annu. Rev. Genet., 2009, 43, 95–118 CrossRef CAS PubMed.
- S. Fulda, L. Galluzzi and G. Kroemer, Nat. Rev. Drug Discovery, 2010, 9, 447–464 CrossRef CAS PubMed.
- I. C. Summerhayes, T. J. Lampidis, S. D. Bernal, J. J. Nadakavukaren, K. K. Nadakavukaren, E. L. Shepherd and L. B. Chen, Proc. Natl. Acad. Sci., India, Sect. B, 1982, 79, 5292–5296 CAS.
- K. K. Nadakavukaren, J. J. Nadakavukaren and L. B. Chen, Cancer Res., 1985, 45, 6093–6099 CAS.
- J. S. Modica-Napolitano and J. R. Aprille, Adv. Drug Delivery Rev., 2001, 49, 63–70 CrossRef CAS PubMed.
- V. R. Fantin, M. J. Berardi, L. Scorrano, S. J. Korsmeyer and P. Leder, Cancer Cell, 2002, 2, 29–42 CrossRef CAS PubMed.
- L. D. Zorova, V. A. Popkov, E. Y. Plotnikov, D. N. Silachev, I. B. Pevzner, S. S. Jankauskas, V. A. Babenko, S. D. Zorov, A. V. Balakireva, M. Juhaszova, S. J. Sollott and D. B. Zorov, Anal. Biochem., 2018, 552, 50–59 CrossRef CAS PubMed.
- M. Septinus, T. Berthold, A. Naujok and H. W. Zimmermann, Histochemistry, 1985, 82, 51–66 CAS.
- A. Maftah, J. M. Petit and R. Julien, FEBS Lett., 1990, 260, 236–240 CrossRef CAS PubMed.
- V. G. Almendro-Vedia, C. Garcia, R. Ahijado-Guzman, D. de la Fuente-Herreruela, M. Munoz-Ubeda, P. Natale, M. H. Vinas, R. Q. Albuquerque, A. Guerrero-Martinez, F. Monroy, M. P. Lillo and I. Lopez-Montero, BBA, Biochim. Biophys. Acta, Gen. Subj., 2018, 1862, 2824–2834 CrossRef CAS PubMed.
- S. G. Rubio, N. M. Pastor, C. Garcia, V. G. Almendro-Vedia, I. Ferrer, P. Natale, L. Paz-Ares, M. P. Lillo and I. Lopez-Montero, Front. Oncol., 2018, 8, 514 CrossRef PubMed.
- D. A. Giljohann, D. S. Seferos, W. L. Daniel, M. D. Massich, P. C. Patel and C. A. Mirkin, Angew. Chem., Int. Ed., 2010, 49, 3280–3294 CrossRef CAS PubMed.
- J. Perez-Juste, I. Pastoriza-Santos, L. M. Liz-Marzan and P. Mulvaney, Coord. Chem. Rev., 2005, 249, 1870–1901 CrossRef CAS.
- R. Ahijado-Guzman, G. Gonzalez-Rubio, J. G. Izquierdo, L. Banares, I. Lopez-Montero, A. Calzado-Martin, M. Calleja, G. Tardajos and A. Guerrero-Martinez, ACS Omega, 2016, 1, 388–395 CrossRef CAS PubMed.
- X. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Nanomedicine, 2007, 2, 681–693 CrossRef CAS PubMed.
- S. Molina-Pinelo, R. Melendez, R. Suarez, L. Garcia, L. Ojeda, N. Moreno, M. V. Guijarro, L. Paz-Ares and I. Ferrer, Cancer Res., 2017, 77, 4801 Search PubMed.
- A. Quintanal-Villalonga, S. Molina-Pinelo, P. Yague, A. Marrugal, L. Ojeda-Marquez, R. Suarez, S. Ponce-Aix, A. B. Enguita, A. Carnero, I. Ferrer and L. Paz-Ares, Lung Cancer, 2019, 131, 112–121 CrossRef PubMed.
- S. Lopez-Sanz, N. R. Farinas, R. S. Vargas, R. Martin-Doimeadios and A. Rios, Talanta, 2017, 164, 451–457 CrossRef CAS PubMed.
- Y. C. Qian, M. T. Qiu, Q. Q. Wu, Y. Y. Tian, Y. Zhang, N. Gu, S. Y. Li, L. Xu and R. Yin, Sci. Rep., 2014, 4, 7490 CrossRef CAS PubMed.
- A. K. Ghosh and M. Brindisi, J. Med. Chem., 2015, 58, 2895–2940 CrossRef CAS PubMed.
- P. M. Oliver, J. A. Crooks, M. Leidl, E. J. Yoon, A. Saghatelian and D. B. Weibel, J. Bacteriol., 2014, 196, 3386–3398 CrossRef PubMed.
- G. Gonzalez-Rubio, V. Kumar, P. Llombart, P. Diaz-Nunez, E. Bladt, T. Altantzis, S. Bals, O. Pena-Rodriguez, E. G. Noya, L. G. MacDowell, A. Guerrero-Martinez and L. M. Liz-Marzan, ACS Nano, 2019, 13, 4424–4435 CrossRef CAS PubMed.
- N. R. Jana, L. Gearheart and C. J. Murphy, Adv. Mater., 2001, 13, 1389–1393 CrossRef CAS.
- R. Ahijado-Guzman, N. Sanchez-Arribas, M. Martinez-Negro, G. Gonzalez-Rubio, M. Santiago-Varela, M. Pardo, A. Pineiro, I. Lopez-Montero, E. Junquera and A. Guerrero-Martinez, Nanomaterials, 2020, 10, 590 CrossRef CAS PubMed.
- I. Plaza-Ga, V. Manzaneda-Gonzalez, M. Kisovec, V. Almendro-Vedia, M. Munoz-Ubeda, G. Anderluh, A. Guerrero-Martinez, P. Natale and I. L. Montero, J. Nanobiotechnol., 2019, 17, 1–10 CrossRef CAS PubMed.
- D. de la Fuente-Herreruela, A. K. Monnappa, M. Munoz-Ubeda, A. Morallon-Pina, E. Enciso, L. Sanchez, F. Giusti, P. Natale and I. Lopez-Montero, J. Nanobiotechnol., 2019, 17, 1–14 CrossRef CAS PubMed.
- R. L. Prentice, Biometrics, 1976, 32, 761–768 CrossRef CAS PubMed.
- T. Mukohara, J. A. Engelman, N. H. Hanna, B. Y. Yeap, S. Kobayashi, N. Lindeman, B. Halmos, J. Pearlberg, Z. Tsuchihashi, L. C. Cantley, D. G. Tenen, B. E. Johnson and P. A. Janne, J. Natl. Cancer Inst., 2005, 97, 1185–1194 CrossRef CAS PubMed.
- H. Kimura, K. Sakai, T. Arao, T. Shimoyama, T. Tamura and K. Nishio, Cancer Sci., 2007, 98, 1275–1280 CrossRef CAS PubMed.
- E. Murphy, H. Ardehali, R. S. Balaban, F. DiLisa, G. W. Dorn 2nd, R. N. Kitsis, K. Otsu, P. Ping, R. Rizzuto, M. N. Sack, D. Wallace, R. J. Youle, American Heart Association Council on Basic Cardiovascular Sciences, Council on Clinical Cardiology and Council on Functional Genomics and Translational Biology, Circ. Res., 2016, 118, 1960–1991 CrossRef CAS PubMed.
- L. F. Dong and J. Neuzil, Cancer Commun., 2019, 39, 63 CrossRef PubMed.
- D. Liu, F. Y. Jin, G. F. Shu, X. L. Xu, J. Qi, X. Q. Kang, H. Yu, K. J. Lu, S. P. Jiang, F. Han, J. You, Y. Z. Du and J. O. Ji, Biomaterials, 2019, 211, 57–67 CrossRef CAS PubMed.
- K. L. Horton, K. M. Stewart, S. B. Fonseca, Q. Guo and S. O. Kelley, Chem. Biol., 2008, 15, 375–382 CrossRef CAS PubMed.
- J. Zielonka, J. Joseph, A. Sikora, M. Hardy, O. Ouari, J. Vasquez-Vivar, G. Cheng, M. Lopez and B. Kalyanaraman, Chem. Rev., 2017, 117, 10043–10120 CrossRef CAS PubMed.
- Y. N. Antonenko, A. V. Avetisyan, D. A. Cherepanov, D. A. Knorre, G. A. Korshunova, O. V. Markova, S. M. Ojovan, I. V. Perevoshchikova, A. V. Pustovidko, T. I. Rokitskaya, I. I. Severina, R. A. Simonyan, E. A. Smirnova, A. A. Sobko, N. V. Sumbatyan, F. F. Severin and V. P. Skulachev, J. Biol. Chem., 2011, 286, 17831–17840 CrossRef CAS PubMed.
- J. Wang, X. Y. Fan, L. Y. Yang, H. He, R. Huang, F. L. Jiang and Y. Liu, MedChemComm, 2016, 7, 2016–2019 RSC.
- T. J. Lampidis, Y. Hasin, M. J. Weiss and L. B. Chen, Biomed. Pharmacother., 1985, 39, 220–226 CAS.
- G. Battogtokh, Y. S. Choi, D. S. Kang, S. J. Park, M. S. Shim, K. M. Huh, Y. Y. Cho, J. Y. Lee, H. S. Lee and H. C. Kang, Acta Pharm. Sin. B, 2018, 8, 862–880 CrossRef PubMed.
- M. T. Jeena, S. Kim, S. Jin and J. H. Ryu, Cancers, 2020, 12, 4 CrossRef CAS PubMed.
- L. Dong, V. Gopalan, O. Holland and J. Neuzil, Int. J. Mol. Sci., 2020, 21, 7941 CrossRef CAS PubMed.
- M. Munoz-Ubeda, A. Tolosa-Diaz, S. Bhattacharya, E. Junquera, E. Aicart, P. Natale and I. Lopez-Montero, Mol. Pharmaceutics, 2019, 16, 4787–4796 CrossRef CAS PubMed.
- Y. A. Tan, Y. Zhu, Y. Zhao, L. J. Wen, T. T. Meng, X. Liu, X. Q. Yang, S. H. Dai, H. Yuan and F. Q. Hu, Biomaterials, 2018, 154, 169–181 CrossRef CAS PubMed.
- Y. Yu, Z. H. Wang, L. Zhang, H. J. Yao, Y. Zhang, R. J. Li, R. J. Ju, X. X. Wang, J. Zhou, N. Li and W. L. Lu, Biomaterials, 2012, 33, 1808–1820 CrossRef CAS PubMed.
- Z. Khatun, Y. S. Choi, Y. G. Kim, K. Yoon, M. Nurunnabi, L. Li, E. Lee, H. C. Kang and K. M. Huh, Biomacromolecules, 2017, 18, 1074–1085 CrossRef CAS PubMed.
- C. Fernandez, G. Gonzalez-Rubio, J. Langer, G. Tardajos, L. M. Liz-Marzan, R. Giraldo and A. Guerrero-Martinez, Angew. Chem., Int. Ed., 2016, 55, 11237–11241 CrossRef CAS PubMed.
- J. J. Li, D. Hartono, C. N. Ong, B. H. Bay and L. Y. L. Yung, Biomaterials, 2010, 31, 5996–6003 CrossRef CAS PubMed.
- P. Opanasopit, M. Nishikawa and M. Hashida, Crit. Rev. Ther. Drug, 2002, 19, 191–233 CrossRef CAS PubMed.
- G. D. Zhang, Z. Yang, W. Lu, R. Zhang, Q. Huang, M. Tian, L. Li, D. Liang and C. Li, Biomaterials, 2009, 30, 1928–1936 CrossRef CAS PubMed.
- K. G. Roth, I. Mambetsariev, P. Kulkarni and R. Salgia, Trends Mol. Med., 2020, 26, 119–134 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2nr02353a |
| This journal is © The Royal Society of Chemistry 2022 |