
C60-β-cyclodextrin conjugates for enhanced nucleus delivery of doxorubicin†
Rohin
Biswas
 ,
Shilong
Yang
,
Ryan A.
Crichton
,
Patrick
Adly-Gendi
,
Tyler K.
Chen
,
William P.
Kopcha
,
Zheng
Shi
* and
Jianyuan
Zhang
*
,
Shilong
Yang
,
Ryan A.
Crichton
,
Patrick
Adly-Gendi
,
Tyler K.
Chen
,
William P.
Kopcha
,
Zheng
Shi
* and
Jianyuan
Zhang
*
Department of Chemistry and Chemical Biology, Rutgers University, Piscataway, NJ, USA. E-mail: zheng.shi@rutgers.edu; jy.zhang@rutgers.edu
First published on 28th February 2022
Abstract
We demonstrate the use of water-soluble C60-β-cyclodextrin conjugates to encapsulate and deliver doxorubicin to the cell nucleus. The behaviour of the fullerene aggregates inside cells is dictated by the functionalization of the C60 cage. While both the C60 conjugates are taken up by lysosomes upon cellular entry, only the one with a hydroxylated cage rapidly escaped the lysosome. The drug delivery system (DDS) with a hydroxylated C60 cage showed significantly enhanced doxorubicin delivery to the cell nucleus, whereas the DDS with a hydrophobic C60 cage was trapped in the lysosome for a longer time and showed significantly reduced doxorubicin delivery to the nucleus. This study opens new paths towards advanced fullerene-based DDSs for small molecule drugs.
 Jianyuan Zhang | Jianyuan Zhang received his BSc degree from Beijing Normal University in 2007 and conducted one year of research at the Institute of Chemistry, Chinese Academy of Sciences. He then moved to Virginia Tech to study fullerene and metallofullerene properties under the supervision of Harry Dorn, and obtained his PhD in December 2013. As a postdoctoral researcher, he subsequently performed research at the University of Washington (2014–15) and Massachusetts Institute of Technology (2015–17). In 2017, he joined the Department of Chemistry and Chemical Biology at Rutgers University – New Brunswick. His current research focuses on carbon nanomaterials and nanoparticles, with biomedical application of fullerenes and endohedral metallofullerenes being important themes of his work. |
Introduction
Doxorubicin is a small molecule therapeutic drug1 commonly used in the treatment of leukemia and various other types of cancers. During chemotherapy, doxorubicin enters the cancer cell nucleus and functions via interfering with the activity of topoisomerase II, thereby inhibiting DNA transcription.2 However, delivering native drug molecules into the nucleus is challenging due to intracellular barriers in reaching the target, low residence time inside the body and requirements of high drug dosage.3 To overcome these challenges, drug delivery systems (DDSs)4–6 such as hydrogels,7 polymers,8 peptides,9 nanoparticles,10 and carbon nanomaterials11,12 have been used.Fullerenes represent a small and precise molecular carbon nanomaterial with vast promise in biomedicine, used both for their intrinsic properties,13 and as a multivalent globular scaffold.14–18 The organic and molecular nature of fullerene derivatives provides high biocompatibility, well-defined structures, and tailorable metabolic pathways, making them a promising tool to develop DDSs compared to typical nanoparticles. When fullerenes are used in DDSs,19 their surface properties can be continuously tuned within a wide range of hydrophobicity, leading to tunable interactions with themselves (e.g., aggregation), with the drugs (e.g., hydrophobic interaction with doxorubicin),20,21 or with the biological system (e.g., enhancing cell membrane permeability).22–25 Additionally, fullerenes or their derivatives can be used as a secondary drug. A notable example is their ability to scavenge reactive oxygen species (ROS),26–31 thereby offering protection from side effects of chemotherapy drugs such as doxorubicin.32,33
Compared to hydrophobic interactions, host–guest chemistry is a more well-defined supramolecular approach34 that provides protection from the environment, and selective loading/release for the drugs.35–37 Cyclodextrins (CDs) are cost-efficient, eco-friendly and biocompatible supramolecular hosts that can hold doxorubicin in their hydrophobic cavity while their hydrophilic exterior imparts water-solubility.38,39 Although water-soluble fullerene–CD conjugates have been synthesized in various forms,40–42 their applications as DDSs are underexplored.
In this work, we report two versions of water-soluble C60-β-cyclodextrin conjugates as DDSs for doxorubicin for nucleus delivery. As shown in Fig. 1, the first C60-β-cyclodextrin conjugate is a direct conjugate of a fullerene derivative with β-cyclodextrin (FCD), and the second version is obtained via further functionalization of FCD with hydroxyl groups on the cage (hydroxyl-FCD, or hFCD for short). A non-fullerene control, poly(ethylene glycol) (PEG) – functionalized β-cyclodextrin was also investigated. Loading of doxorubicin to these DDSs yielded Dox@FCD, Dox@hFCD, and Dox@CD, respectively. Fluorescence imaging with HeLa cells showed that doxorubicin from Dox@CD randomly localized all over the cell; doxorubicin from Dox@FCD selectively localized near the nucleus; and most importantly, Dox@hFCD delivers doxorubicin directly into the nucleus. Mechanistic study revealed that the ability to take advantage of the nuclear pore complex (NPC) by reasonable-sized aggregates is a key reason for the enhanced nucleus targeting. Further microscopic characterization suggested that Dox@CD, Dox@FCD are trapped in lysosome for much longer time, while Dox@hFCD can efficiently escape lysosome, after endocytosis. Transmission electron microscope (TEM) images showed both fullerene derivatives FCD and hFCD could eventually enter the nucleus, which is consistent with a seminal study that demonstrated the nucleus uptake of a different fullerene derivative.43 Finally, the viability of cells was not affected by the DDSs themselves, while doxorubicin-loaded DDSs showed expected efficacy.
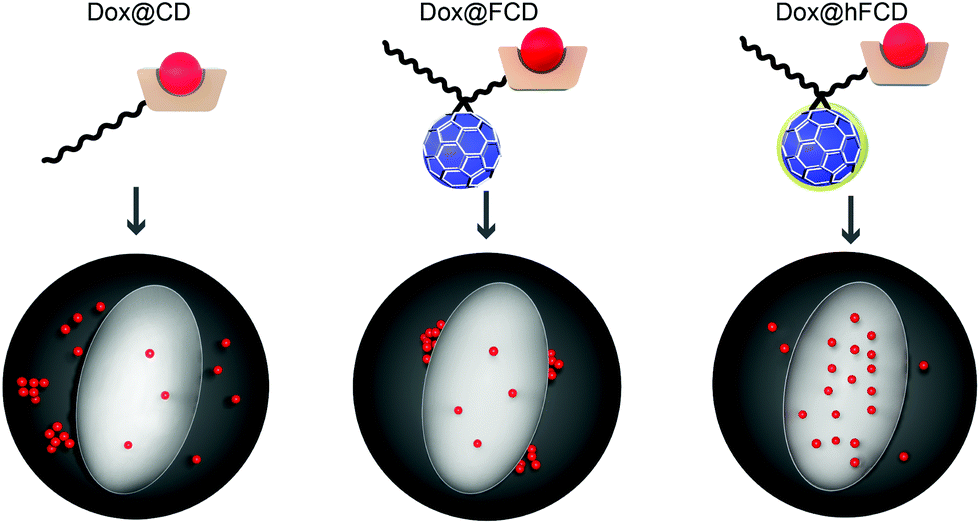 | ||
| Fig. 1 Schematic depiction of Dox@CD (PEG–β-CD conjugate), Dox@FCD (PEG–fullerene–β-CD conjugate), and Dox@hFCD (hydroxylated PEG–fullerene–β-CD conjugate) delivering doxorubicin in to cells. The dark grey sphere and light ellipse denote the cell and the nucleus, respectively. | ||
Results and discussion
Synthesis and characterization of the DDSs and doxorubicin loaded DDSs
The synthetic scheme of FCD and hFCD is shown in Fig. 2. The monoderivative of C60 (2) was prepared via Bingel–Hirsch reaction, from malonic ester 1 that bears a PEG (750 g mol−1) and an alkynyl group. Compound 2 was then connected to a β-cyclodextrin-6-azide via the Cu(I)-catalyzed azide–alkyne cycloaddition44 click chemistry to yield the water-soluble C60-β-cyclodextrin conjugate, FCD (3). Compound 4, hFCD was prepared by further reacting FCD 3 with NaOH and H2O2, catalyzed by 18-crown-6 following the classical procedure of fullerenol synthesis, which typically randomly adds 20–30 hydroxyl groups on the cage.33,45,46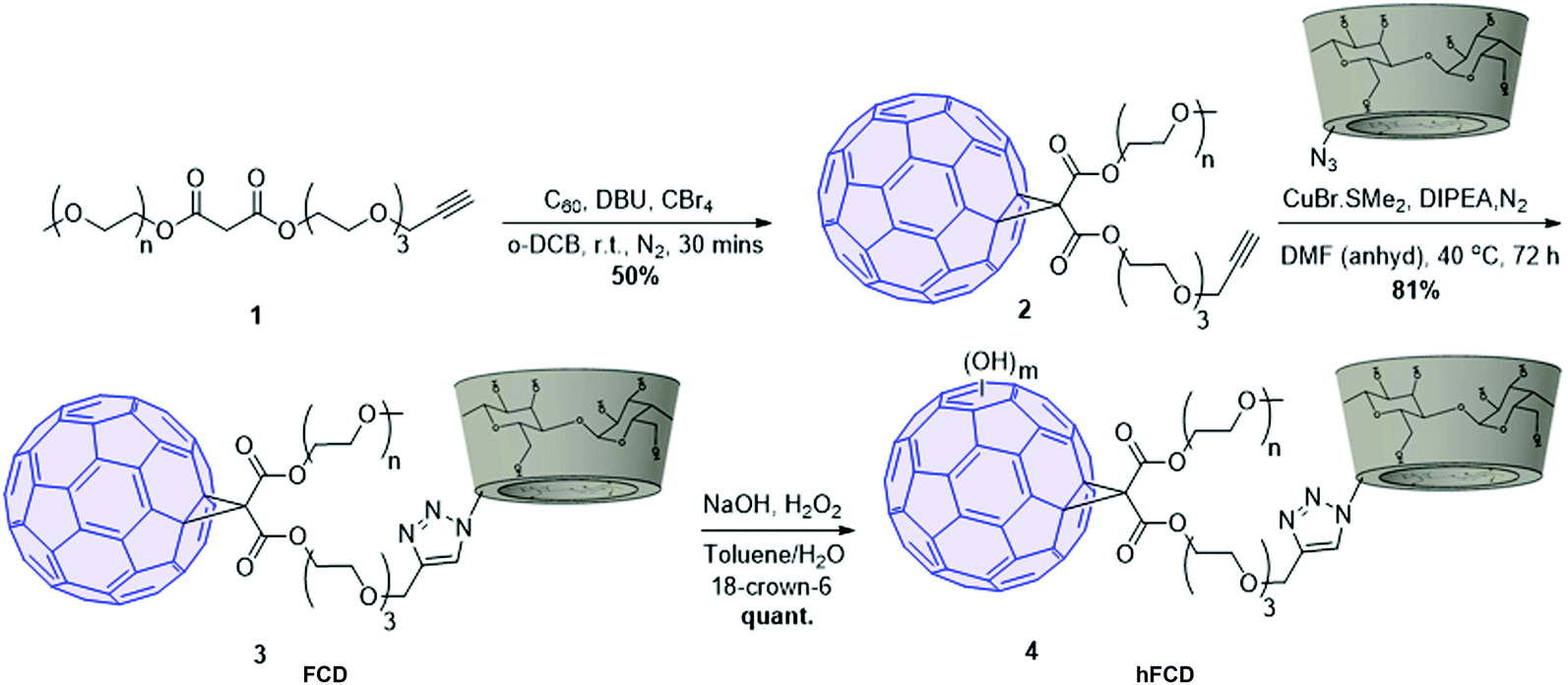 | ||
| Fig. 2 Synthetic scheme of C60-β-cyclodextrin conjugates FCD and hFCD. | ||
The key difference between FCD and hFCD is the former has a hydrophobic cage, while the latter has a hydroxyl-group covered cage with both hydrophobic and hydrophilic areas. Compounds FCD, hFCD and CD were characterized by NMR (Fig. 3a and S1–S10†) and UV-visible spectroscopy (Fig. 3b and S11†). In the proton NMR spectrum of FCD 3, the peak (a) at 8 ppm is from the triazole ring, confirming the successful conjugation via the click approach. The integration of peaks a–e showed a ratio of 1H/14H/7H/10H/3H, which is fully consistent with the molecular structure. After hydroxylation, the hydroxyl groups on the cage further broadened the peaks between 3–4 ppm (Fig. S8†), while the characteristic peaks from the triazole (7.9 ppm), CD (4.8 ppm) and methoxy group (3.2 ppm) remain in the ratio of 1H/7H/3H. UV-vis spectra of the FCD and hFCD before and after drug loading (Fig. 3b) showed that, upon hydroxylation, the extinction features of FCD as a fullerene monoderivative were lost as the pi-conjugated system is broken. A broad peak around 495 nm, corresponding to the doxorubicin absorption, emerged in the DDSs after drug loading.
 | ||
| Fig. 3 Characterization data of the molecules; (a) 1H NMR of FCD 3. Characteristic proton signals from the glucopyranose units in β-cyclodextrin are labelled as (b–d). Signals from the PEG protons and the remaining protons from cyclodextrin overlap at around 3.5 ppm. Signal labelled as e represents the protons from the terminal methoxy group at the end of the PEG. (b) Normalized UV-vis spectra of the drug carriers and doxorubicin loaded DDSs. Inset: 10× the Y axis of the FCD spectra between 300–500 nm. DLS spectra of FCD and hFCD at (c) pH 7.4. (d) pH 5.5. TEM images showing aggregates of (e) FCD and (f) hFCD, aided with arrows (yellow). | ||
The loading of doxorubicin was optimized for all three systems to achieve a ∼1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio between the drug and the host (“full loading”). As seen in Table S1,† loading doxorubicin into CD was rather challenging, which required ∼10 eq. of the drug to achieve full loading, leading to poor encapsulation efficiency (EE). Dox@FCD and Dox@hFCD showed significantly higher EE with optimized loading using only slightly over 1 eq. of doxorubicin. Our results clearly show that the presence of fullerenes facilitates the encapsulation of doxorubicin, possibly because the cage could help break the doxorubicin aggregation.
:1 molar ratio between the drug and the host (“full loading”). As seen in Table S1,† loading doxorubicin into CD was rather challenging, which required ∼10 eq. of the drug to achieve full loading, leading to poor encapsulation efficiency (EE). Dox@FCD and Dox@hFCD showed significantly higher EE with optimized loading using only slightly over 1 eq. of doxorubicin. Our results clearly show that the presence of fullerenes facilitates the encapsulation of doxorubicin, possibly because the cage could help break the doxorubicin aggregation.
Dynamic light scattering (DLS) data revealed that the fullerene-based DDSs exist in large particles of 250–400 nm, which is expected for these fullerene derivatives (Fig. 3c). TEM images, Fig. 3 (e and f) also revealed that in fact FCD and hFCD exist as aggregates. The size of these DDSs was further studied at lower pH of 5.5 to mimic acidic cellular compartments (i.e., lysosome),47,48 and most tumor environments. Under acidic conditions, hFCD broke down to smaller particles of 50–80 nm, whereas the size of the FCD aggregates was not affected, indicating different aggregation mechanisms for FCD and hFCD (Fig. 3d). Like other amphiphilic molecules, FCD with a hydrophilic ligand and a hydrophobic cage would form aggregates that conceal the cage inside to minimize their interaction with water, which is further assisted by the strong π–π interaction among the fullerene cages.49,50 On the other hand, the aggregation of the hFCD is driven by intermolecular hydrogen-bond network between hydroxyl groups, which is well-established for fullerenols.51,52 Such aggregates can be broken up into smaller aggregates under certain conditions such as an acidic environment.
After loading doxorubicin, the hydrodynamic size of Dox@FCD is substantially larger than that of the FCD (Fig. S12†), with the larger peak of the bimodal distribution into the micron regime, as expected from adding another hydrophobic molecule into an amphiphilic system. Given the molar ratio between doxorubicin and the FCD in Dox@FCD exceeded 1:1 (Table S1†), the significant increase in micelle size can be attributed to the doxorubicin molecules being carried through strong π–π interaction with the fullerene cage,21 in addition to the cavity of the CD. This interaction is absent in Dox@hFCD, as the aromatic surface of the C60 cage is covered by hydroxyl groups. With that said, the loading of doxorubicin also created bimodal distribution (Fig. S12†) under both neutral and acidic conditions, but with different peak positions and intensities. At lower pH, the smaller-sized peak was clearly more evident, showing that Dox@hFCD have different aggregate sizes in equilibrium,51 which can be shifted towards smaller size under acidic condition. This phenomenon suggests hydrogen-bond is still the major interaction in the Dox@hFCD system, which can be affected by [H+] concentration. The changes after drug loading can be ascribed to the new doxorubicin/CD supramolecular interactions altering the original CD-fullerenol hydrogen-bonds, while it is possible that hydrogen-bonding and static interaction between doxorubicin and fullerenol also played a role.53
Uptake of doxorubicin loaded DDSs in HeLa cells
We studied the cellular uptake of the doxorubicin loaded DDSs with microscopy, using DAPI fluorescence to label the nucleus, and the intrinsic fluorescence of doxorubicin to show the location of the drug molecules (Fig. 4). Native doxorubicin without DDS was first studied as a control (Fig. S13†), which provides a reference for the behavior of released drug molecules. First, the native doxorubicin control showed very poor drug uptake in the cells, i.e., significantly lower doxorubicin fluorescence in cells (Fig. S13 and S14†) compared to Fig. 4. Second, the signal pattern of the native drug was diffusive throughout the cell, without any subcellular enrichment, in contrast to the clustered signals from doxorubicin delivered via DDSs (Fig. 4 and S14†).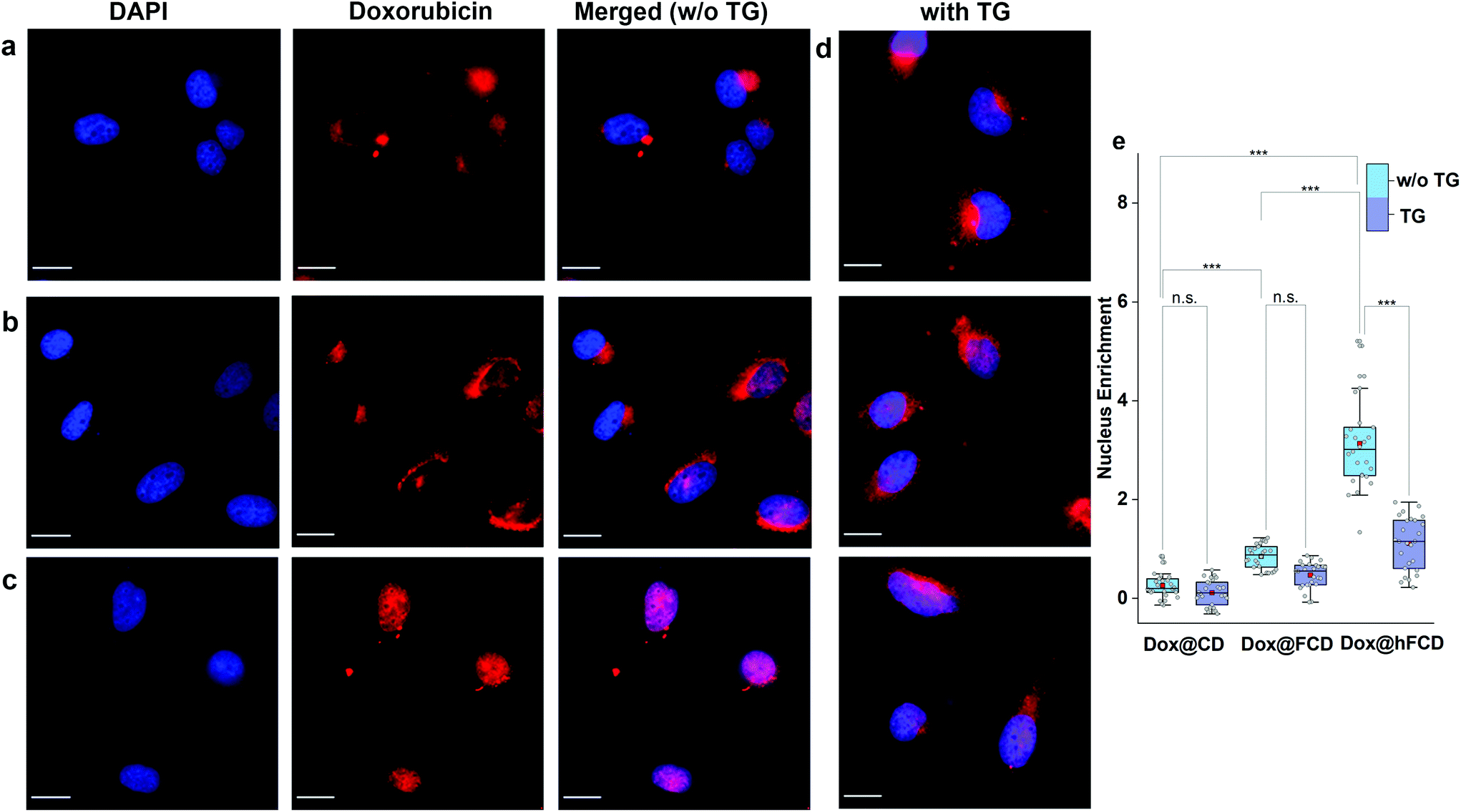 | ||
| Fig. 4 Microscope images of doxorubicin loaded DDSs in HeLa cells after incubation for 4 hours. Fluorescence image of DAPI, doxorubicin channels, and the merged image of the DAPI and doxorubicin fluorescence (magenta), are shown. Scale bars are 20 μm. (a) Dox@CD, (b) Dox@FCD, (c) Dox@hFCD. (d) HeLa cells treated with Thapsigargin (TG) for 45 minutes and then incubated with doxorubicin loaded DDSs while DAPI was used to stain the nucleus – Dox@CD (top), Dox@FCD (middle) and Dox@hFCD (bottom). (e) Box plots showing enrichment of Dox inside the nucleus w.r.t. the cytosol, without and with TG. The mid-lines represent the median and the red boxes show the mean. ***p ≤ 0.001, n.s. stands for not significant. | ||
Doxorubicin from all three DDSs showed strong intracellular fluorescence, i.e., enhanced uptake by HeLa cells compared to the native drug (Fig. S14†). However, Dox@CD showed very low signal intensity in the nuclei (Fig. 4a and S14†), suggesting Dox@CD is not a good option for doxorubicin delivery. Doxorubicin signals of the Dox@FCD system (Fig. 4b) were mainly observed around the nucleus (in lysosome, vide infra), although some were also from inside the nucleus. The perinucleus signal remained clustered, instead of being diffusive, suggesting that the drug remained in DDS particles. The cell uptake of the particles was likely via phagocytosis due to their large hydrodynamic size in the micron regime (Fig. S12†).54,55 However, such size is too large to penetrate through the nuclear pore. Due to the strong hydrophobic interaction between fullerene cage and doxorubicin,21 the departure of doxorubicin from FCD is slow, however some of the released doxorubicin from Dox@FCD explains the weak diffusive doxorubicin fluorescence overlap with the DAPI signal in the nucleus (Fig. 4b).
In striking contrast with previous two DDSs, the doxorubicin fluorescence signals from Dox@hFCD (Fig. 4c) were predominantly localized in the nuclei. The drastic difference can be explained by the special feature of the hFCD carrier. Under the acidic environment inside HeLa cells,47,48 the Dox@hFCD hydrogen-bond network is broken down to smaller size (Fig. S12†), which facilitates nucleus uptake.43 We hypothesized that the main mechanism for the nucleus entry is through the nucleus pore complex (NPC), so blocking the NPC could significantly reduce the nucleus uptake of doxorubicin for Dox@hFCD. To test this, we incubated HeLa cells with Thapsigargin (TG), a known inhibitor of the NPC which blocks the central and peripheral channels by the depletion of Ca2+.56 Dox@CD and Dox@FCD did not show any significant change in the fluorescence signal intensity in the nucleus; in contrast, Dox@hFCD showed a dramatic decrease of the overlap of DAPI and doxorubicin fluorescence (Fig. 4d).
We then determined the enrichment of doxorubicin signal in the nucleus in comparison to the cytosol to quantify the fluorescence data from microscopy, by using box plots as shown in Fig. 4e. The nucleus enrichment of doxorubicin w.r.t. cytosol of Dox@CD is (∼24%). Fullerene-based DDS Dox@FCD has a higher enrichment (∼83%) compared to that of Dox@CD, as it can slowly deliver doxorubicin to the nucleus. Finally, Dox@hFCD showed more than 300% enrichment in the nuclei. Both the increase in enrichment from Dox@CD to Dox@FCD, and from Dox@FCD to Dox@hFCD are statistically significant with p value ≤ 0.001 in our t-test. This suggests that Dox@hFCD is clearly the best candidate for nucleus delivery of doxorubicin. With the NPC blocker TG, the enrichment for Dox@CD and Dox@FCD both dropped (to 12% and 47%, respectively), but the decreases were statistically insignificant. It is likely that the NPC was not the pre-dominant pathway for nucleus uptake for these two systems. In contrast, the enrichment dropped from 300% to 110% for Dox@hFCD, a significant drop that clearly shows the strong involvement of NPC in the nucleus uptake mechanism of Dox@hFCD.
Sub-cellular localization of doxorubicin loaded DDSs and DDSs
Most nanoparticles upon cellular entry via endocytosis are taken up into endosomes, which mature into acidic lysosomes. To understand the influence of lysosomal environment on the DDSs, we tagged lysosome with Lysotracker Deep Red, and observed the fluorescence overlap with DAPI and doxorubicin signals (Fig. 5a–c), after incubation of the cells and the drug loaded DDSs for 1.5, 3 and 4 hours, respectively.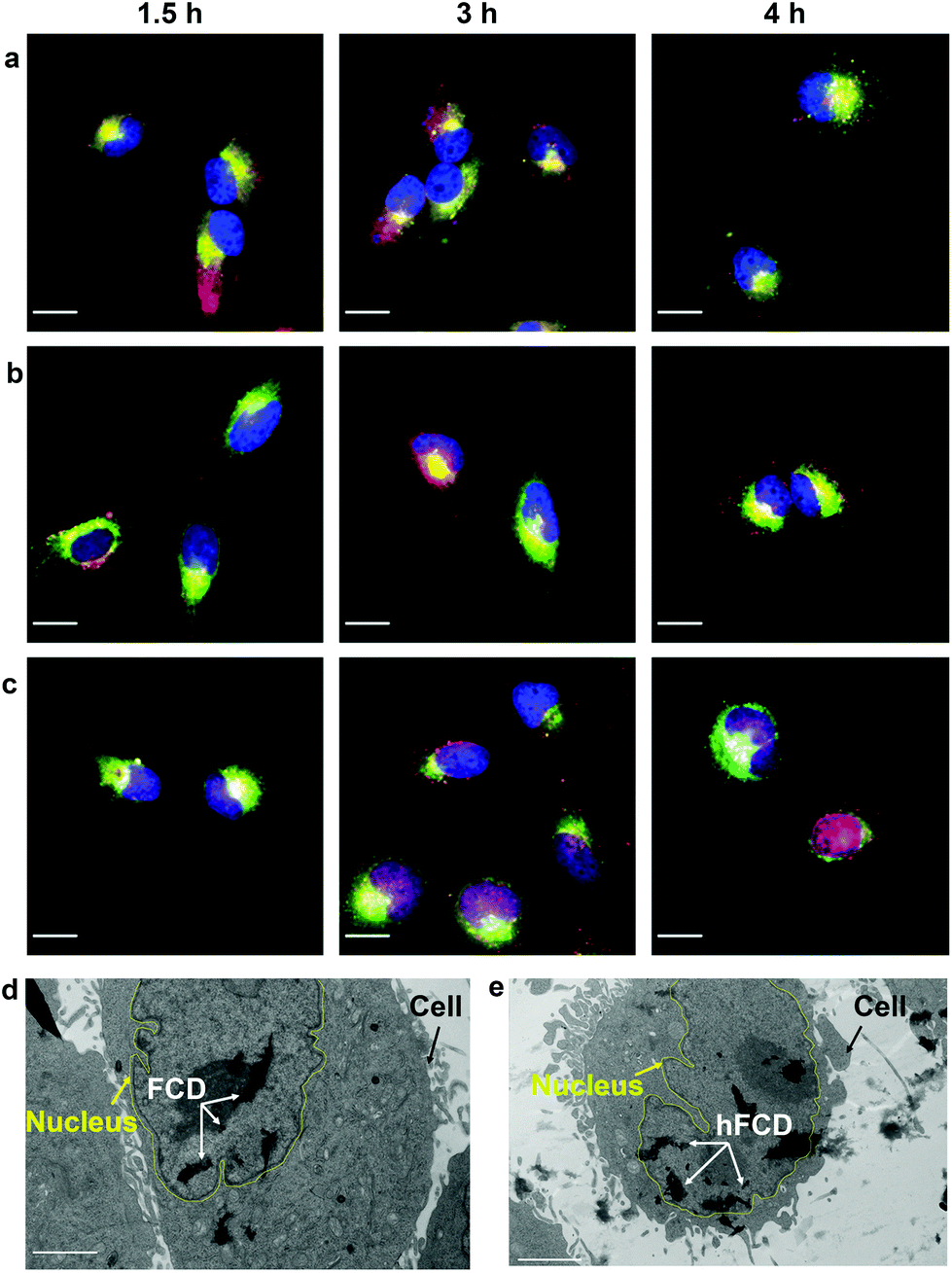 | ||
| Fig. 5 Microscope images showing merged fluorescence of DAPI (blue), Dox (red) and LysoTracker Deep Red (green) for (a) Dox@CD, (b) Dox@FCD, (c) Dox@hFCD. Scale bar is 20 μm. TEM images of HeLa cells after incubation with (d) FCD and (e) hFCD for 24 hours. Scale bar is 2 μm. Yellow lines are added to help the visualization of the nucleus membrane. | ||
All three systems were taken up by lysosomes as deduced by the fluorescence overlap of Dox and Lysotracker Deep Red after 1.5 hours (Fig. 5a–c). Dox@CD remained trapped in the lysosomes even after 4 hours, and no nucleus entry was observed, consistent with Fig. 4a. For Dox@FCD, doxorubicin signals gradually moved away from lysosome in the 4-hour period, suggesting that some doxorubicin from Dox@FCD can escape the lysosomes. Correspondingly, minor nuclear entry of doxorubicin was observed (more evident in Fig. 4b). Initially, Dox@hFCD was also captured by the lysosomes but majority of the DDS was shown to escape the lysosomes in 3 hours, and almost completely in 4 hours. Meanwhile, very high levels of overlap between the doxorubicin and DAPI signals at 4 hours (both Fig. 4c and 5c) suggests efficient nucleus uptake.
In light of this, to investigate the stability of the fullerene DDS aggregates in lysosome, we dissolved Dox@FCD and Dox@hFCD in artificial lysosomal fluid (ALF) and investigated the absorbance (Fig. S15†) and aggregate sizes (Fig. S16†) after 24 h. ALF did not cause appreciable change in the UV-vis spectra, suggesting the inherent molecular stability. On the other hand, FCD formed larger aggregates, and hFCD formed smaller aggregates in the acidic ALF, compared to neutral buffer solutions (Fig. 3c), because of the difference in the mechanism of aggregation (π–π interaction vs. hydrogen bond). This trend is consistent with the DLS result in our acidic buffer (Fig. 3d) although the exact sizes are different due to the more complex chemical environment in the ALF. Overall, the fullerene derivative aggregates are stable in the ALF, while the smaller size of the hFCD aggregates can be critical to both the lysosomal escape and nucleus entry.
We then performed TEM study to confirm if the fullerene nanocarriers themselves can enter the nucleus. In the TEM images of the cells incubated with FCD (Fig. 5d) and hFCD (Fig. 5e), aggregates of FCD and hFCD (which are completely absent in the TEM of the cell without the nanocarriers, Fig. S17†) were observed dominantly inside the nucleus. Therefore, we conclude that the fullerene nanocarriers did enter the nucleus in our study, at least given long enough time. Meanwhile, a minor number of aggregates were observed in the cell but outside the nucleus. Comparing the results from FCD and hFCD, we found that qualitatively, hFCD has higher ratio of nanocarriers found inside vs. outside the nucleus, which is consistent with our understanding of the better subcellular targeting of hFCD.
Cell viability using the DDSs and doxorubicin loaded DDSs
Viability of HeLa cells upon incubation with the doxorubicin loaded DDSs for 48 and 72 hours were tested to investigate their effect on cell viability (Fig. 6), using DDSs without the drug as controls. There were no reductions in cell viability for all three DDSs up to 72 hours, suggesting none of the DDSs are cytotoxic.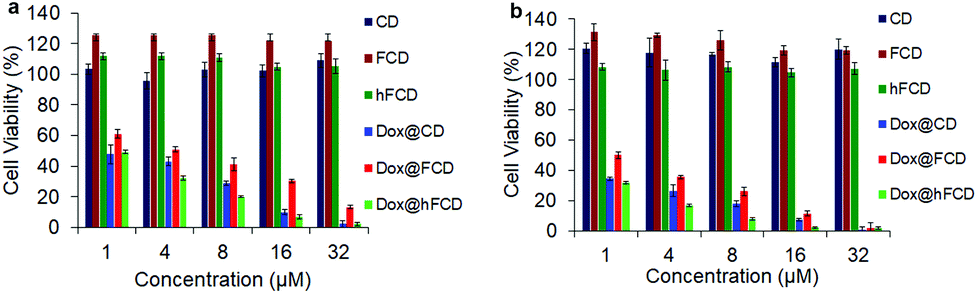 | ||
| Fig. 6 Cell viability of the DDSs and doxorubicin loaded DDSs in HeLa cells after incubation for (a) 48 hours, (b) 72 hours. | ||
The doxorubicin loaded systems, on the other hand, significantly lowered the cell viability, suggesting cells killed by the doxorubicin. All the three systems, Dox@CD, Dox@FCD and Dox@hFCD reduced cell viability to below 5% at 32 μM after incubation for 72 hours. Dox@CD behaves somewhat similar to native doxorubicin drug because CD is not a good DDS except that Dox@CD has a high cellular uptake, which provides a positive control. Dox@FCD takes a longer time and higher concentration to reduce the cell viability compared to the other two systems. This is consistent with the microscopy studies (Fig. 4b) that the doxorubicin signals mainly localize outside the nuclei.
Finally, Dox@hFCD reduced the cell viability with the highest efficacy, even higher than the positive control, consistent with its nuclei targeting capacity. Meanwhile, it continues to have effect on reducing cell viability over the course of 72 hours, allowing for an option to be used in low dose with lasting effect (e.g., 8 μM, 72 hours can reduce the cell viability below 10%). Therefore, hFCD is clearly the most effective as a DDS in delivering the drug specifically to the target with a higher potency when compared to CD and FCD.
Conclusions
We have reported two water-soluble C60-β-cyclodextrin conjugates as fullerene-based DDSs for small molecule drugs. The presence of the C60 cage can facilitate the doxorubicin loading compared to a non-fullerene control. While all DDSs increased doxorubicin uptake in HeLa cells, the hydroxylated hFCD selectively delivered the drug to the cell nucleus, as corroborated by cell uptake, lysosomal staining, TEM and cell viability data. While this work achieves an important drug-delivery task with the intrinsic property of the functionalized fullerene cage itself, the nature of the fullerene cage as a precise organic structural scaffold opens paths towards biological-ligand functionalized molecular nanocarbon (including other fullerenes like C70, metallofullerenes,57 fullertubes58) for new biomedical applications.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Kate Waldie and Enver Izgu for instrumental support, and Dr Rajesh Patel for his help with the TEM. J. Z. thanks the support from the Rutgers Busch Biomedical Grant.Notes and references
- F. Yang, S. S. Teves, C. J. Kemp and S. Henikoff, Biochim. Biophys. Acta, Rev. Cancer, 2014, 1845, 84–89 CrossRef CAS PubMed.
- Y. Pommier, E. Leo, H. Zhang and C. Marchand, Chem. Biol., 2010, 17, 421–433 CrossRef CAS PubMed.
- N. Zhao, M. C. Woodle and A. J. Mixson, J. Nanomed. Nanotechnol., 2018, 9, 100519 Search PubMed.
- S. Senapati, A. K. Mahanta, S. Kumar and P. Maiti, Signal Transduction Targeted Ther., 2018, 3, 7 CrossRef PubMed.
- E. Cukierman and D. R. Khan, Biochem. Pharmacol., 2010, 80, 762–770 CrossRef CAS PubMed.
- D. Rosenblum, N. Joshi, W. Tao, J. M. Karp and D. Peer, Nat. Commun., 2018, 9, 1410 CrossRef PubMed.
- J. Li and D. J. Mooney, Nat. Rev. Mater., 2016, 1, 16071 CrossRef CAS PubMed.
- K. Ulbrich, K. Holá, V. Šubr, A. Bakandritsos, J. Tuček and R. Zbořil, Chem. Rev., 2016, 116, 5338–5431 CrossRef CAS.
- J. Yang, H.-W. An and H. Wang, ACS Appl. Bio Mater., 2021, 4, 24–46 CrossRef CAS.
- M. J. Mitchell, M. M. Billingsley, R. M. Haley, M. E. Wechsler, N. A. Peppas and R. Langer, Nat. Rev. Drug Discovery, 2021, 20, 101–124 CrossRef CAS PubMed.
- Z. Liu, J. T. Robinson, S. M. Tabakman, K. Yang and H. Dai, Mater. Today, 2011, 14, 316–323 CrossRef CAS.
- R. G. Mendes, A. Bachmatiuk, B. Büchner, G. Cuniberti and M. H. Rümmeli, J. Mater. Chem. B, 2013, 1, 401–428 RSC.
- E. Castro, A. H. Garcia, G. Zavala and L. Echegoyen, J. Mater. Chem. B, 2017, 5, 6523–6535 RSC.
- M. Sμnchez-Navarro, A. MuÇoz, B. M. Illescas, J. Rojo and N. Martín, Chem. – Eur. J., 2011, 17, 766–769 CrossRef PubMed.
- P. Compain, C. Decroocq, J. Iehl, M. Holler, D. Hazelard, T. M. Barragán, C. O. Mellet and J.-F. Nierengarten, Angew. Chem., 2010, 122, 5889–5892 CrossRef.
- A. Muñoz, D. Sigwalt, B. M. Illescas, J. Luczkowiak, L. Rodríguez-Pérez, I. Nierengarten, M. Holler, J.-S. Remy, K. Buffet, S. P. Vincent, J. Rojo, R. Delgado, J.-F. Nierengarten and N. Martín, Nat. Chem., 2016, 8, 50–57 CrossRef PubMed.
- J. Ramos-Soriano, J. J. Reina, A. Perez-Sanchez, B. M. Illescas, J. Rojo and N. Martín, Chem. Commun., 2016, 52, 10544–10546 RSC.
- J.-F. Nierengarten, Chem. – Eur. J., 2000, 20, 3667–3670 CrossRef.
- A. Montellano, T. Da Ros, A. Bianco and M. Prato, Nanoscale, 2011, 3, 4035–4041 RSC.
- Y. Hurmach, M. Rudyk, S. Prylutska, V. Hurmach, Y. I. Prylutskyy, U. Ritter, P. Scharff and L. Skivka, Mol. Pharm., 2020, 17, 3622–3632 CrossRef PubMed.
- Y. I. Prylutskyy, M. P. Evstigneev, I. S. Pashkova, D. Wyrzykowski, A. Woziwodzka, G. Gołuński, J. Piosik, V. V. Cherepanov and U. Ritter, Phys. Chem. Chem. Phys., 2014, 16, 23164–23172 RSC.
- J. Wong-Ekkabut, S. Baoukina, W. Triampo, I.-M. Tang, D. P. Tieleman and L. Monticelli, Nat. Nanotechnol., 2008, 3, 363–368 CrossRef CAS.
- S. Zhang, Y. Mu, J. Z. H. Zhang and W. Xu, PLoS One, 2013, 8, e77436 CrossRef CAS.
- M. Bortolus, G. Parisio, A. L. Maniero and A. Ferrarini, Langmuir, 2011, 27, 12560–12568 CrossRef CAS.
- R. Qiao, A. P. Roberts, A. S. Mount, S. J. Klaine and P. C. Ke, Nano Lett., 2007, 7, 614–619 CrossRef CAS.
- Z. Markovic and V. Trajkovic, Biomaterials, 2008, 29, 3561–3573 CrossRef CAS PubMed.
- H. Ma, J. Zhao, H. Meng, D. Hu, Y. Zhou, X. Zhang, C. Wang, J. Li, J. Yuan and Y. Wei, ACS Appl. Mater. Interfaces, 2020, 12, 16104–16113 CrossRef CAS PubMed.
- C. Zhou, M. Zhen, M. Yu, X. Li, T. Yu, J. Liu, W. Jia, S. Liu, L. Li, J. Li, Z. Sun, Z. Zhao, X. Wang, X. Zhang, C. Wang and C. Bai, Sci. Adv., 2020, 6, eabc1586 CrossRef CAS PubMed.
- J. Li, M. Guan, T. Wang, M. Zhen, F. Zhao, C. Shu and C. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 25770–25776 CrossRef CAS PubMed.
- J.-J. Yin, F. Lao, P. P. Fu, W. G. Wamer, Y. Zhao, P. C. Wang, Y. Qiu, B. Sun, G. Xing, J. Dong, X.-J. Liang and C. Chen, Biomaterials, 2009, 30, 611–621 CrossRef CAS.
- T. Li, L. Xiao, J. Yang, M. Ding, Z. Zhou, L. LaConte, L. Jin, H. C. Dorn and X. Li, ACS Appl. Mater. Interfaces, 2017, 9, 17681–17687 CrossRef CAS PubMed.
- Y. Zhou, M. Zhen, M. Guan, T. Yu, L. Ma, W. Li, J. Zheng, C. Shu and C. Wang, Sci. Rep., 2018, 8, 16573 CrossRef PubMed.
- Y. Zhou, J. Li, H. Ma, M. Zhen, J. Guo, L. Wang, L. Jiang, C. Shu and C. Wang, ACS Appl. Mater. Interfaces, 2017, 9, 35539–35547 CrossRef CAS PubMed.
- M. J. Webber and R. Langer, Chem. Soc. Rev., 2017, 46, 6600–6620 RSC.
- G. Yu and X. Chen, Theranostics, 2019, 9, 3041–3074 CrossRef CAS PubMed.
- X. Ma and Y. Zhao, Chem. Rev., 2015, 115, 7794–7839 CrossRef CAS PubMed.
- J. Wankar, N. G. Kotla, S. Gera, S. Rasala, A. Pandit and Y. A. Rochev, Adv. Funct. Mater., 2020, 30, 1909049 CrossRef CAS.
- J. Zhang and P. X. Ma, Adv. Drug Delivery Rev., 2013, 65, 1215–1233 CrossRef CAS PubMed.
- P. Zhou, X. Liang, C. Zhou, J. Qin, C. Hou, Z. Zhu, W. Zhang, S. Wang and D. Zhong, J. Mater. Chem. B, 2019, 7, 5363–5375 RSC.
- S. Filippone, F. Heimann and A. Rassat, Chem. Commun., 2002, 1508–1509 RSC.
- S. Samal and K. E. Geckeler, Chem. Commun., 2000, 1101–1102 RSC.
- X. Zhu, A. Quaranta, R. V. Bensasson, M. Sollogoub and Y. Zhang, Chem. – Eur. J., 2017, 23, 9462–9466 CrossRef CAS PubMed.
- M. Raoof, Y. Mackeyev, M. A. Cheney, L. J. Wilson and S. A. Curley, Biomaterials, 2012, 33, 2952–2960 CrossRef CAS PubMed.
- P. Thirumurugan, D. Matosiuk and K. Jozwiak, Chem. Rev., 2013, 113, 4905–4979 CrossRef CAS PubMed.
- H. Kato, K. Suenaga, M. Mikawa, M. Okumura, N. Miwa, A. Yashiro, H. Fujimura, A. Mizuno, Y. Nishida, K. Kobayashi and H. Shinohara, Chem. Phys. Lett., 2000, 324, 255–259 CrossRef CAS.
- J. Li, A. Takeuchi, M. Ozawa, X. Li, K. Saigo and K. Kitazawa, J. Chem. Soc., Chem. Commun., 1993, 1784–1785 RSC.
- M. H. Lee, J. H. Han, J. H. Lee, N. Park, R. Kumar, C. Kang and J. S. Kim, Angew. Chem., Int. Ed., 2013, 52, 6206–6209 CrossRef CAS PubMed.
- H. Hou, Y. Zhao, C. Li, M. Wang, X. Xu and Y. Jin, Sci. Rep., 2017, 7, 1759 CrossRef PubMed.
- D. M. Guldi, J. Phys. Chem. A, 1997, 101, 3895–3900 CrossRef CAS.
- K. Harano and E. Nakamura, Acc. Chem. Res., 2019, 52, 2090–2100 CrossRef CAS PubMed.
- S. Laus, B. Sitharaman, É. Tóth, R. D. Bolskar, L. Helm, S. Asokan, M. S. Wong, L. J. Wilson and A. E. Merbach, J. Am. Chem. Soc., 2005, 127, 9368–9369 CrossRef CAS PubMed.
- J. Li, M. Zhang, B. Sun, G. Xing, Y. Song, H. Guo, Y. Chang, Y. Ge and Y. Zhao, Carbon, 2012, 50, 460–469 CrossRef CAS.
- J. Tang, R. Zhang, M. Guo, L. Shao, Y. Liu, Y. Zhao, S. Zhang, Y. Wu and C. Chen, Biomaterials, 2018, 167, 205–215 CrossRef CAS PubMed.
- S. Behzadi, V. Serpooshan, W. Tao, M. A. Hamaly, M. Y. Alkawareek, E. C. Dreaden, D. Brown, A. M. Alkilany, O. C. Farokhzad and M. Mahmoudi, Chem. Soc. Rev., 2017, 46, 4218–4244 RSC.
- J. J. Rennick, A. P. R. Johnston and R. G. Parton, Nat. Nanotechnol., 2021, 16, 266–276 CrossRef CAS PubMed.
- C. Perez-Terzic, J. Pyle, M. Jaconi, L. Stehno-Bittel and D. E. Clapham, Science, 1996, 273, 1875–1877 CrossRef CAS PubMed.
- A. A. Popov, S. Yang and L. Dunsch, Chem. Rev., 2013, 113, 5989–6113 CrossRef CAS PubMed.
- R. M. Koenig, H.-R. Tian, T. L. Seeler, K. R. Tepper, H. M. Franklin, Z.-C. Chen, S.-Y. Xie and S. Stevenson, J. Am. Chem. Soc., 2020, 142, 15614–15623 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d2nr00777k |
| This journal is © The Royal Society of Chemistry 2022 |