
Molecular bases for temperature sensitivity in supramolecular assemblies and their applications as thermoresponsive soft materials†
Hongxu
Liu
 a,
Theeraphop
Prachyathipsakul
a,
Thameez M.
Koyasseril-Yehiya
a,
Stephanie P.
Le
a and
S.
Thayumanavan
*abcd
a,
Theeraphop
Prachyathipsakul
a,
Thameez M.
Koyasseril-Yehiya
a,
Stephanie P.
Le
a and
S.
Thayumanavan
*abcd
aDepartment of Chemistry, University of Massachusetts Amherst, Amherst, Massachusetts 01003, USA. E-mail: thai@umass.edu
bDepartment of Biomedical Engineering, University of Massachusetts, Amherst, Massachusetts 01003, USA
cMolecular and Cellular Biology Program, University of Massachusetts, Amherst, Massachusetts 01003, USA
dCentre for Bioactive Delivery, Institute for Applied Life Science, University of Massachusetts, Amherst, Massachusetts 01003, USA
First published on 9th September 2021
Abstract
Thermoresponsive supramolecular assemblies have been extensively explored in diverse formats, from injectable hydrogels to nanoscale carriers, for a variety of applications including drug delivery, tissue engineering and thermo-controlled catalysis. Understanding the molecular bases behind thermal sensitivity of materials is fundamentally important for the rational design of assemblies with optimal combination of properties and predictable tunability for specific applications. In this review, we summarize the recent advances in this area with a specific focus on the parameters and factors that influence thermoresponsive properties of soft materials. We summarize and analyze the effects of structures and architectures of molecules, hydrophilic and lipophilic balance, concentration, components and external additives upon the thermoresponsiveness of the corresponding molecular assemblies.
 Hongxu Liu | Hongxu Liu received his BSc and MSc degrees in Chemistry from Zhengzhou University, China. He then joined the Department of Chemistry at the University of Massachusetts Amherst in 2016 as a PhD student and worked under the guidance of Prof. S. Thayumanavan. He received his PhD degree in Chemistry in 2021. His doctoral dissertation was focused on new chemical tools for the design of stimuli-responsive supramolecular assemblies and functional polymers. |
 Theeraphop Prachyathipsakul | Theeraphop “Theo” Prachyathipsakul graduated with a BA degree in Chemistry from Wesleyan University in 2019. He is currently a chemistry PhD student at University of Massachusetts Amherst under the guidance of Prof. S. Thayumanavan. His research focus is on cancer targeting therapy using antibodies. |
 Thameez M. Koyasseril-Yehiya | Thameez M. Koyasseril-Yehiya is currently a postdoctoral research associate in the Siegrist lab at University of Massachusetts Amherst. He received his BS-MS dual degree in Chemistry from Indian Institute of Science Education and Research (IISER) Pune in 2015. Thameez received his PhD in Chemistry under the guidance of Prof. S. Thayumanavan from the University of Massachusetts Amherst in 2021. His doctoral dissertation was focused on the design and synthesis of stimuli-responsive materials for antibacterial applications. |
 Stephanie P. Le | Stephanie Le received her BSc from the University of California, Irvine with a dual degree in Biological Sciences and Chemistry in 2016. She is currently pursuing her PhD in Chemistry under the supervision of Prof. S. Thayumanavan. Her current research focuses on stimuli responsive materials and catalysis. |
 S. Thayumanavan | S. “Thai” Thayumanavan is a Distinguished Professor of Chemistry and Biomedical Engineering and the Director of the Center for Bioactive Delivery at the University of Massachusetts Amherst. He received his BSc and MSc degrees from the American College in Madurai, India. He received his PhD from the University of Illinois at Urbana-Champaign and did postdoctoral work at Caltech. His research work involves the design and synthesis of stimuli-responsive supramolecular assemblies and materials and utilizing them in a variety of applications, including in the delivery of small molecules and large biologics as targeted therapeutics, self-healing materials, sensing and diagnostics. |
1. Introduction
Stimuli-responsive supramolecular assemblies have been extensively studied in the past few decades due to their potential applications in a variety of areas such as sensing, imaging, diagnosis, drug delivery, catalysis, and tissue engineering.1–4 The controllable responses to specific stimuli provide opportunities to design smart materials to fit practical demands. Among various stimuli, temperature is one of the easiest to manipulate, thus attracting many interests.5–7 For instance, temperature-responsive hydrogels have been designed as injectable drug carriers which can deliver therapeutic drugs in sustainable ways.8 The sol-to-gel transformation ensures injectability as solution at room temperature and durability after gel formation at body temperature (37 °C).9Most of the current temperature-responsive assemblies are designed based on phase transition behaviors either at the lower critical solution temperature (LCST) or upper critical solution temperature (UCST) i.e., cloud point. For assemblies with LCST, the solute molecules are typically well-solubilized under the critical temperature while forming aggregates and undergoing phase separation at higher temperatures.10 At lower temperatures, interactions between the solute molecules and water are favorable due to strong hydrogen bonding, resulting in the hydrated state of the molecules. At elevated temperatures, the increased molecular vibrations weaken hydrogen bonds. Consequently, the polymer–polymer interactions are more favorable, leading to dehydration and aggregation.11,12 In contrast, molecules with UCST are soluble above the critical temperature but form aggregates below this point. These molecules usually have stronger interchain interactions at lower temperatures, preventing molecules from dissolving due to an enthalpic barrier. Elevated temperature enhances the effects of entropy favoring solute–solvent interactions.13 Some of assemblies can exhibit both LCST and UCST depending on the molecular structure, concentration, and external additives.14–17
A variety of molecules have been designed for temperature-responsive assemblies including dendrimers,18 random19 and block20 copolymers, and small molecules such as lipids.21 The temperature-responsiveness can be manipulated by several parameters e.g., molecular structure, functional groups, concentration, and hydrophilic–lipophilic balance (HLB). Previously, thermoresponsive materials have been summarized in some reviews from different perspectives. Most of them focuses on material components9,11,22,23 and applications such as delivery,7,16,24,25 tissue engineering,26,27 and catalysis.28 In this review, we focus on the molecular bases and factors behind the temperature-responsiveness. These molecules are discussed based on different topologies, i.e., linear polymers, polymers with side chains, dendrimers and hyperbranched polymers (Fig. 1). Finally, we will briefly summarize how these systems are applied for designing optimal thermoresponsive materials for different applications.
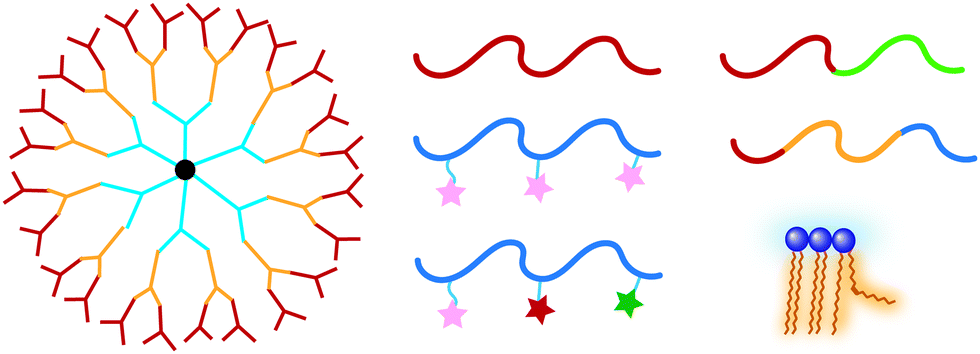 | ||
| Fig. 1 Thermoresponsive molecules with different topology. | ||
2. Temperature responsive linear block copolymers
Macromolecules that undergo physical property changes in response to temperature have gained significant interest in the development of functional thermoresponsive materials and their biomedical applications.8,28–30 Linear block copolymers have been applied as temperature responsive materials for a long time. Most of these polymers contain polyethylene glycol (PEG) as the major temperature responsive moiety. As explained previously, the disruption of hydrogen bonding at higher temperature leads to thermoresponsiveness. For example, surfaces containing a Pluronic block copolymer with PEG fragments were studied at different temperatures. When the temperature increases from 25 to 37 °C, the water contact angle of the polymer changes from ∼35° to 63°, demonstrating an increased hydrophobicity at higher temperatures.31 Similar phenomenon was also reported for other oxyethylene tethered-block copolymers.32 The hydrophilic–hydrophobic transition properties of these linear polymers with temperature have been applied for many thermoresponsive studies such as self-assembly under different temperatures, thermo-triggered morphology transformations, and temperature responsive hydrogels.33,34 In this section, the impact of molecular structures on the thermoresponsiveness of linear block copolymers will be discussed.2.1 Polymer materials and architectures
A variety of hydrophobic moieties have been assembled with PEG in linear polymers as thermoresponsive materials. These hydrophobic segments can be polyethers, polypeptides and polyesters. The common examples include poly(L-alanine) (L-PA),35 polylactic acid (PLA),36 poly(propylene oxide) (PPO),31 polycaprolactone (PCL),37 poly(lactic-co-glycolic acid) (PLGA),38 and poly(ε-caprolactone-co-lactide) PCLA.39 Different structures have distinct hydrophobicity, molecular interactions, and crystalline properties, thus exhibiting unique thermoresponsive properties when co-incorporated with PEG (Fig. 2).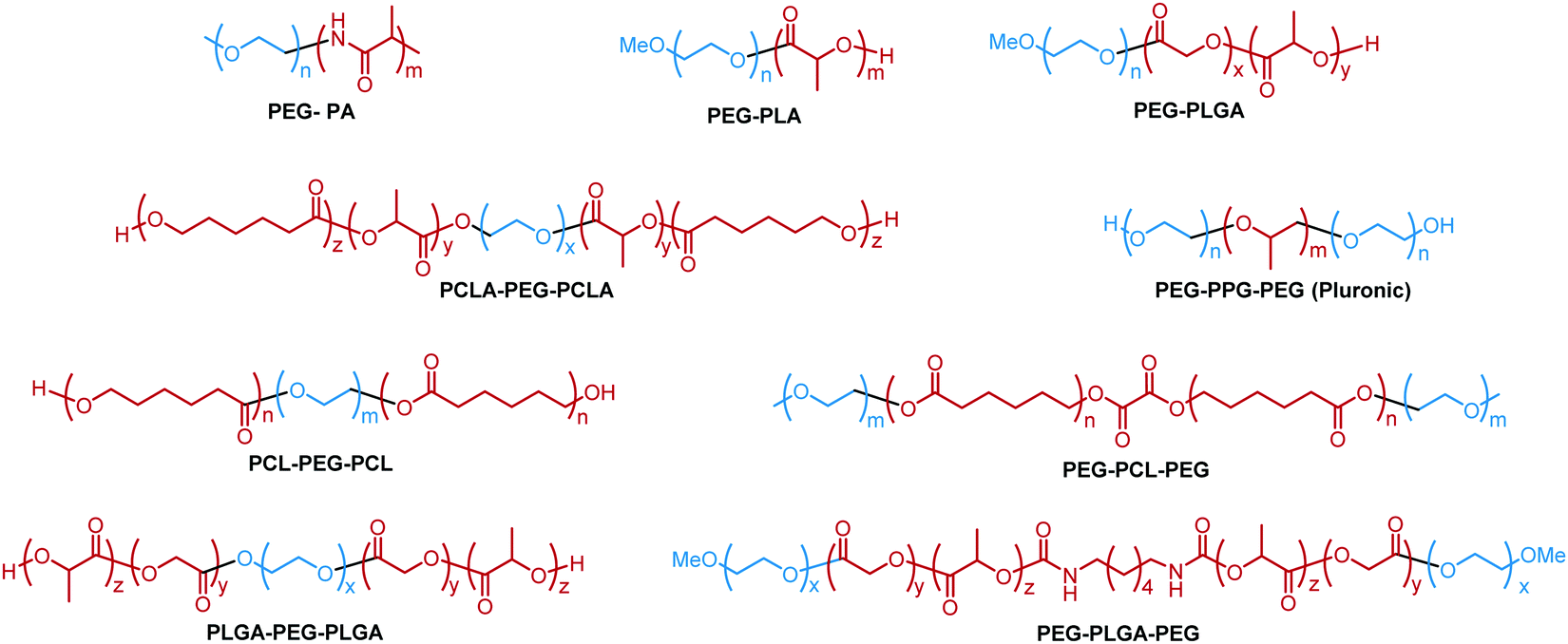 | ||
| Fig. 2 Examples of linear block copolymers with different hydrophobic moieties.31,35–39,45,61 | ||
Linear polymers may have different architectures depending on the position of block polymer segments, such as diblock (A–B), triblock (A–B–A or B–A–B) and even multiblock. The architecture of polymer blocks significantly affects the conformation and assembly of the polymers in solution, leading to different thermoresponsive nature.40–42 For example, aggregation and theromoresponsive properties of the diblock PEG–PLGA and the triblock PLGA–PEG–PLGA copolymers were studied (Fig. 3a–c).38 The two polymers formed micelles in aqueous solution with similar critical aggregation concentration (Fig. 3d). However, the triblock copolymer formed larger assemblies than the diblock at the same polymer concentration (Fig. 3e). Also, the triblock copolymer formed gel at lower concentrations with lower gelation temperature (Tgel) than the diblock copolymer (Fig. 3b). Similarly, thermoresponsive properties of a diblock copolymer PEG–PCL and a triblock copolymer PEG–PCL–PEG were compared. Interestingly, PEG–PCL–PEG had a broader gel window than PEG–PCL, thus enabling gelation at broader concentrations. For example, 25–37 wt% of the polymer PEG–PCL–PEG formed gel at 37 °C, whereas PEG–PCL was always in solution state. This distinct characteristic was applied for the design of reactive oxygen species (ROS) triggered thermogel for drug release.43
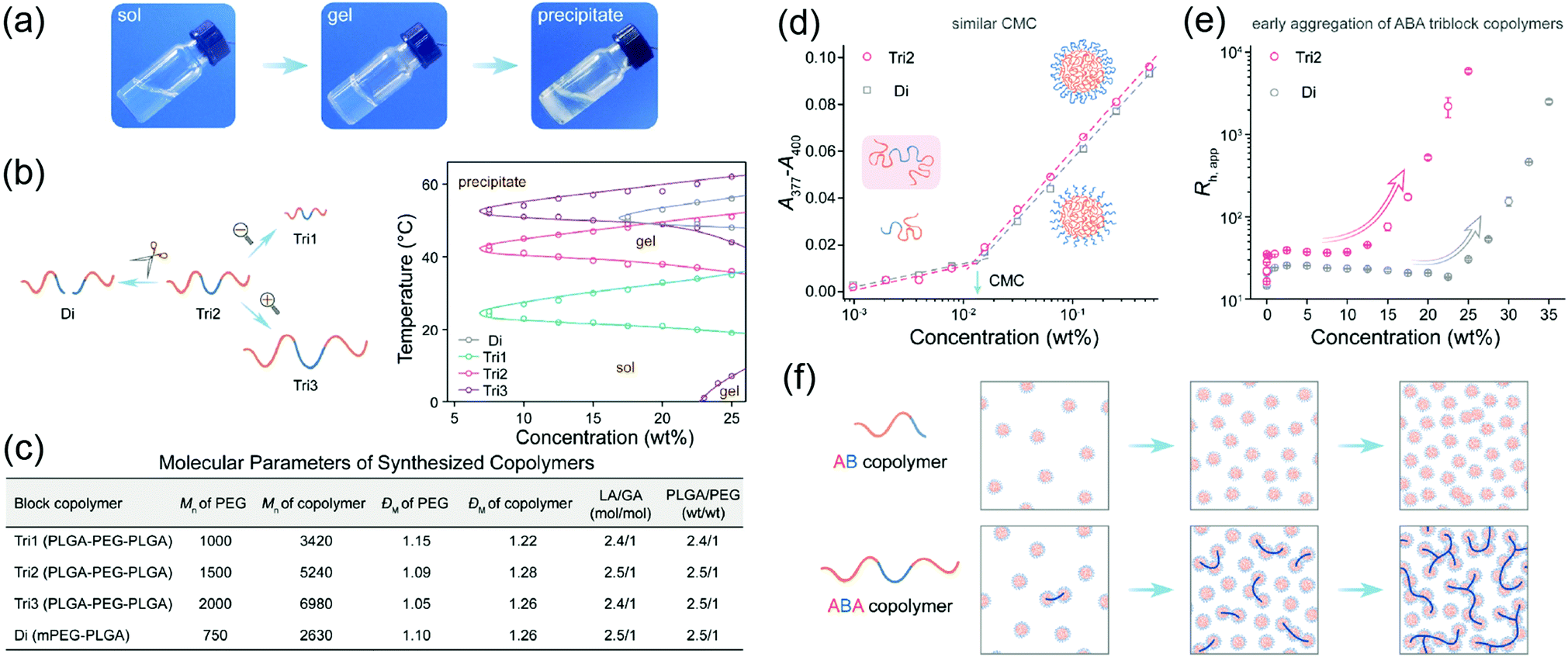 | ||
| Fig. 3 (a) Images of the Tri2 solution (25 wt%) at different temperatures (25, 37, 60 °C). (b) Schematic relationship of block copolymers with different molecular weights and architectures and their state diagrams in aqueous solution. (c) Parameters of the synthesized polymers. (d) CMC of copolymers Di and Tri2 in water at 25 °C. The representative morphologies Di and Tri2 at different concentration regions. (e) Apparent hydrodynamic radii (Rh,app) of Di and Tri2 as a function of concentration measured by 3D DLS at 25 °C. (f) Schematic presentation of the morphology evolutions of AB or ABA copolymer with an increase of concentration in water. Reproduced with permission from ref. 38. Copyright 2019, American Chemical Society. | ||
2.2 Molecular weight and dispersity
Linear polymers with different molecular weight and dispersity exhibit various thermoresponsive properties. For example, when the molecular weight of diblock copolymer PEG–L-PA increased from PEG1000–L-PA795 to PEG2000–L-PA1150, the gelation temperature rose significantly from 7 to 35 °C.35,44 A similar trend was also found in triblock copolymer PLGA–PEG–PLGA.38 As shown in Fig. 3b and c, when the molecular weight of polymer increased from 3420 g mol−1 to 6980 g mol−1 while retaining the lactic acid/glycolic acid (LA/GA) and PLGA/PEG ratio, the gelation temperature (Tgel) of the polymers increases. However, the impact of molecular weight can vary case by case. The Tgel of triblock polymer PCL–PEG–PCL has been reported to decrease significantly when the molecular weight doubled.45 Recently, it was also found that the dispersity of block copolymers is crucial for thermoresponsive properties.36 For example, a discrete structure (oligomer) of poly-L-lactic acid (PLLA) based triblock copolymer PEG–PLLA–PEG had a gelation temperature between 42 to 48 °C, while no gelation was observed for a dispersed polymer with the same components (PDI = 1.2), demonstrating the negative impact of high dispersity on crystallinity, self-assembly and gelation.2.3 Hydrophilic and lipophilic balance
Most commonly, HLB is used to manipulate the temperature responsiveness of linear polymers. This can be realized by either varying the length of hydrophobic and hydrophilic chains or changing chemical moieties. An improper HLB may lead to the loss of thermoresponsiveness, like LCST and gelation temperature. For example, decreasing the hydrophobic PLGA length of a thermogelling polymer PEG750–PLGA from PLGA1870 to PLGA1510 and PLGA980, results in micelle solutions at the experimental temperature and loss of the gelation capability.46 Since HLB can be tuned by both the hydrophobic and hydrophilic segments, PEG length could be another adjustable parameter.38 When the hydrophilic PEG linker in PLGA–PEG–PLGA was too long, no gelation was observed. The impact of HLB is not only limited to thermo-induced gelation but also applied for LCST. Similarly, increase in hydrophobic fragment ratios has been reported to lead to the decrease in the LCST of the polymer PLGA–PEG–PLGA.47 Similar effect was also found for poly(ε-caprolactone-co-lactide)–poly(ethylene glycol)–poly(ε-caprolactone-co-lactide) (PCLA–PEG–PCLA).39,48 It was speculated that PEG linker variation caused the micellar conformation change which can be another reason for the increase in LCST. Similar relationship between LCST and HLB was also found for the PEG-tethered dendritic molecules which will be discussed in the following section.49 Apart from the length of hydrophobic and hydrophilic segments, the manipulation of HLB can also be achieved by varying the structure of the hydrophobic moieties and component ratio. For example, increasing the LA ratio in PLGA resulted in the increase in hydrophobicity of PEG–PLGA–PEG polymer, leading to different thermoresponsive properties.50 This parameter variation can also be applied to tune polymer degradation rate for sustained drug release.51,522.4 Polymer blends
Blending polymers with distinct HLB is a convenient way to tune the thermoresponsive properties of linear polymers (Fig. 4a). To control the gelation temperature of thermogel, PEG1000–PLGA800 was blended with PEG1000–PLGA1600.53 With the increase of PEG1000–PLGA1600 ratio in the blended system, the gelation temperature decreased. Similarly, the gelation window of triblock copolymer PLGA–PEG–PLGA could be broadened by blending two individual polymers,54 which is also dependent on the blend ratio (Fig. 4b).55 In another report, the blending of two PLGA–PEG–PLGA polymers generated thermogel with excellent responsive parameters i.e., gelation temperature (Tgel) < room temperature (Tair) < gel–sol (suspension) temperature (Tsol (suspension)) < body temperature (Tbody). This thermo-triggered gel-to-sol transformation could be applied for rapid release of drugs for photodynamic therapy.56 A thermogel could even be synthesized from blending two copolymers which individually do not have thermogelling behavior (Fig. 4c).55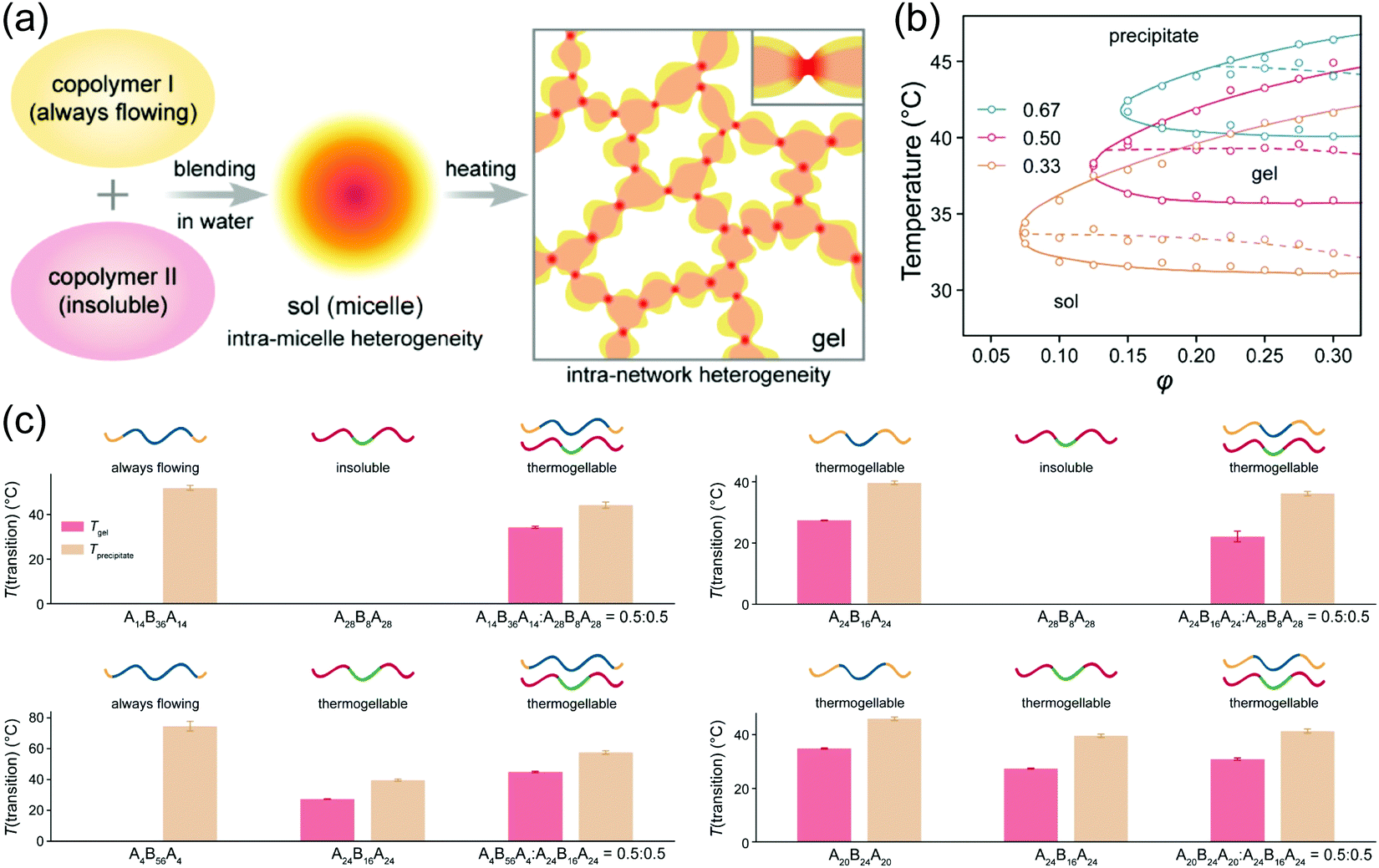 | ||
Fig. 4 (a) Schematic presentation of creating thermogel by blending polymers without thermogelling ability. (b) State diagrams of the blended polymers of PLGA14–PEG36–PLGA14 and PLGA28–PEG8–PLGA28 with various ratios (0.33, 0.50 and 0.67). (c) Thermogelling abilities of blended polymers and their constituent copolymers. For all systems, the blending ratio was 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5, and the total volume fraction φ = 0.25. T(transition) means transition temperature including both Tgel for sol–gel transition and Tprecipitate for sol-precipitate or gel-precipitate transition upon heating. Only Tprecipitate existed for a system with a sol-precipitate transition upon heating, while neither Tgel nor Tprecipitate existed for an insoluble system. Reproduced with permission from ref. 55. Copyright 2020 American Chemical Society. :0.5, and the total volume fraction φ = 0.25. T(transition) means transition temperature including both Tgel for sol–gel transition and Tprecipitate for sol-precipitate or gel-precipitate transition upon heating. Only Tprecipitate existed for a system with a sol-precipitate transition upon heating, while neither Tgel nor Tprecipitate existed for an insoluble system. Reproduced with permission from ref. 55. Copyright 2020 American Chemical Society. | ||
2.5 External additives
Desired thermal parameters can be achieved by manipulating molecular interactions using external additives like metal ions, extra hydrogen bond moieties, and other composites. For example, the gelation temperature of diblock copolymer PEG1000–L-PA795 can be tuned by coordination with Fe3+.57 With the addition of Fe3+, the Tgel of the system significantly decreased from 19 °C to 8 °C. The change was concentration dependent; the higher Fe3+ concentration, the lower Tgel. Similar impact was also discovered in host–guest interactions. As shown in Fig. 5a, addition of α-cyclodextrin (α-CD) carboxylate or phosphate into PEG–L-PA thermogelling led to 3–5 °C decrease in the polymer Tgel.58 Not only components with specific interactions, but also non-specific composites in the system may affect thermoresponsive properties. For example, in a composite of laponites with PEG–PLGA, increase in additive concentration, laponites, resulted in a lower gelation temperature.59 However, there was no clear trend for the effect of random composites to the thermoresponsive behaviors. In another report, it was found that mixing layered double hydroxide (LDH) nanoparticles with triblock PLGA–PEG–PLGA polymer caused the increase of gelation temperature.60 The effect of supramolecular interactions on the thermoresponsive properties of materials has been recently summarized.12 | ||
| Fig. 5 (a) Schematic presentation of supramolecular interaction between α-CD and linear block copolymer PEG-PA and the effect on gelation temperature. Reproduced with permission from ref. 58. Copyright 2020, American Chemical Society. (b) The incorporation of UPy as additional hydrogen bonding moiety and effect on the LCST of triblock copolymer PLGA–PEG–PLGA. Reproduced with permission from ref. 47. Copyright 2020, American Chemical Society. | ||
2.6 Other variations
Even the variation of small moieties of linear block copolymers may have significant impact on their thermoresponsive properties. For instance, the variation of a small linker in PLGA moiety of polymer PEG–PLGA–PEG led to a significant change in their gelation temperature.61 When a flexible hexamethylene was replaced by a rigid para-phenyl linker, the Tgel significantly decreased from 33 °C to 23 °C. The topology of small aromatic linkers also affects the responsive properties. For example, the conjugation of two polymer blocks at the ortho, meta and para positions of a benzene ring, with different substitution angles, led to block copolymers with distinct thermoresponsive properties. In this report, bi(mPEG–PLGA)-o-PC has a lower Tgel than bi(mPEG–PLGA)-m-PC and bi(mPEG–PLGA)-p-PC because of the change in polymer conformation. Similarly, the variation of a small terminal group on block copolymers may result in substantial change of their thermoresponsiveness. Also, the introduction of 2-ureido-4[1H]-pyrimidinone (UPy) to the terminus of PLGA–PEG–PLGA caused a sharp decrease of the polymer LCST (Fig. 5b).47 This is because of the introduction of extra hydrogen bonding moieties (UPy) to the polymer system. Similar phenomenon was also observed for a block copolymer poly(ε-caprolactone-co-p-dioxanone)–poly(ethylene glycol)–poly(ε-caprolactone-co-p-dioxanone), where the introduction of carboxylic acid instead of hydroxyl group led to lower Tgel.62Overall, the thermoresponsive properties of linear block copolymers could be tuned by varying intrinsic molecular bases of polymers, such as different hydrophobic moieties, polymer architectures, molecular weight, polydispersity, and HLB; or by manipulating the molecular interactions and microenvironment using polymer blends or external additives. The variation of these factors could lead to significant changes in the thermoresponsiveness of polymeric materials and even the loss of the responsive properties, e.g. gelation temperature and LCST, leading to the alterations in morphology, rheology, mechanical properties and host–guest properties. Although the specific trend of influences by the factors could be different (e.g. molecular weight) for different polymeric materials, they still provide a direction for tuning the thermoresponse for various application purposes, especially for polymers with the same structural components.
3. Polymers with temperature-sensitive pendant groups
Besides linear block copolymer, polymers with pendant groups are also widely used for thermoresponsive applications. These polymers have advantages from convenient alteration of monomer structures and ratios to optimize the responsive temperature range.11,63–65 The thermoresponsive properties of polymers, i.e. LCST and UCST-type properties, with pendant groups mainly arise from the equilibrium between polymer–polymer and polymer–aqueous solution interactions at different temperatures.11,66,67 The temperature-sensitive range of these polymers can be shifted to match their potential applications by modifying monomer structures, monomer compositions, and degree of polymerization. Additionally, external conditions, such as solvent environment and additives, can be manipulated to realize the optimal HLB. In the following section, different contributions to the alteration of transition temperature will be discussed in detail.3.1 Polymer materials
There are several classes of monomers that are commonly used for synthesizing polymers with thermoresponsive pendants. The structure of these common monomers can be adjusted as a mean to tune HLB and responsive temperature range. Examples of commonly used monomers, yielding thermoresponsive polymer, can be categorized into different groups (Fig. 6). | ||
| Fig. 6 Examples of thermoresponsive polymers with pendant groups. Polymers in the first three rows possessing LCST-type behavior. Polymers in the last row showing UCST-type behavior.11,63,64,88,286 | ||
N-Substituted acrylamide polymers have been studied for their thermoresponsive characteristics since 1967,68 especially poly(N-isopropylacrylamide) (PNIPAAM) known to show a sharp LCST-type transition at 32 °C in aqueous solutions.69 PNIPAAM received a lot of interest for biomedicine applications since its LCST is near body temperature.70–77 The LCST properties of this polymer occur from the hydrophilic amide moiety and the hydrophobic N-alkyl chain, where the hydrophilicity of the polymer dominates at lower temperatures, while higher temperatures lead to favorable hydrophobic interactions.77 Length and shape variations of the hydrophobic N-alkyl chain were studied due to the interest in similar polymers with slightly different thermoresponsive properties, as well as alteration of other physical properties, such as cytotoxicity and limited drug loading capacity for biomedical applications.71,78 For N-substituted acrylamide polymers with similar alkyl chain architecture, longer hydrophobic chains generally lead to lower LCST. Increasing one carbon in the N-alkyl chain of poly(N-ethyl acrylamide) (PNEAM) to poly(N-propyl acrylamide) (PNPAM) led to a drastic drop in the LCST from 70–80 °C to 20–25 °C.
Asides from the N-substituted acrylamide polymers, another class of amide-pendant polymers with LCST properties close to body temperature is poly(N-vinylcaprolactam) (PVCL) with LCST at 35 °C. The main advantages of PVCL over PNIPAAM are lower cytotoxicity and broad LCST transition temperatures, which could be more suitable for some biomedical applications such as solubilizing hydrophobic drugs.78–80
As it was discussed in the previous section, ethylene glycol-based polymers exhibit LCST behaviors at different temperature range depending on the molecular weight and architecture of the polymers. For pendant polymers, ether-pendant, including ones with OEG sidechains, can also cause LCST-type response in polymers regardless of their backbone chemistry.81–83 Similar to the previous examples, the LCST behavior of polymers with OEG sidechain originates from the combination of hydrophobic alkyl backbones and hydrophilic OEG sidechain. The LCST of these polymers are also influenced by OEG length alteration.76,84,85 The general trend suggested that the longer the OEG sidechain, the higher the LCST which can be tuned from around 26 °C of P(EG2MA) to 90 °C of P(EG8MA) as described in a recent review.76 This trend is applicable for other ether-pendant polymers such as the increase in methoxy length in P(EOVE) to P(EOEOVE) leading to ∼20 °C shift in LCST. This observation is analogous to the amide-pendant polymer, in that increase in hydrophilicity resulted in higher LCST.
Poly(2-oxazolines) is classified as synthetic polyamides where the connected N-ethylene served as a backbone of the polymer. The pendant moieties are composed of acyl groups with a choice of alkyl chain or other functional groups.86 This class of polymer can be synthesized via cationic ring-opening polymerization (CROP), resulting in a controllable degree of polymerization with desired end-groups.87 The thermoresponsiveness of poly(2-oxazolines) emerges from the balance between the hydrophilicity of tertiary amide backbone and the hydrophobicity of alkyl side chains. In order to obtain the desired LCST, the length and the hydrophobicity of the pendant group can be adjusted via structural variations of the 2-oxazoline monomers at the 2-position or post-modification of the polymers. For alkyl side chains, it was found that the LCST of the polymer decreased with the increase of chain length.88
A classic example of thermoresponsive zwitterionic polymers is poly(sulfobetaine), a zwitterionic polymer with positively-charged quaternary amine in the middle of the pendants and negatively-charged terminal sulfone moiety. The electrostatic interactions between the opposite charges lead to attraction of side chains.89 Unlike the LCST-analogues, this inter-pendant interactions are more favored at lower temperature. At higher temperatures, the heat causes water molecules to penetrate the interaction networks and disrupt them. Consequently, the soluble forms of the polymer are more favorable.90,91 The UCST comparison between of polysulfopropylbetaine methacrylate (PSPB) and polysulfobutylbetaine methacrylate (PSBB) suggests that increasing the methylene chain length between the ammonium and the sulfonate moieties substantially increased the UCST temperature.64 On the other hand, it has also been shown that lengthening methylene chain length between the backbone and the ammonium groups in poly(sulfobetaines) caused drastic decrease in UCST.92
3.2 Copolymerization of different components
Another approach employed to modulate the temperature-sensitive range of a polymer is co-polymerizing the desired thermoresponsive monomer(s) and other monomers with different hydrophobicity or hydrophilicity. Similar to monomer structural modification approach, incorporating more hydrophobic monomers results in a lower LCST, whereas introducing more hydrophilic monomers mostly gives rise to a higher LCST. For example, randomly mixing a charged 1-(4-vinylbenzyl)-3-methylimidazolium tetrafluoroborate ([VBMI][BF4]) monomer to PNIPAAM caused slightly increased in LCST from 31 °C to 37 °C depending on the ratio between PNIPAAM and P[VBMI][BF4] (Fig. 7a and b). The higher LCST was a result of co-polymerizing a relatively more hydrophilic monomer to PNIPAAM. The LCST-type behavior also vanished, as increase in P[VBMI][BF4] molar fraction resulted in less percent transmittance change.93 Furthermore, in another example, introducing hydrophilic OEGMA300 and hydrophobic butyl methacrylate (BuMA) monomers to thermoresponsive poly(diethylene glycol methacrylate) (PDEGMA) to alter their cloud point temperature. It was shown that the cloud point increased when the content of OEGMA300 increased due to the hydrophilic effect, and the cloud point decreased as the amount of BuMA in the polymer increased due to its hydrophobicity.65 There are several more examples in a recent review that illustrates this concept.94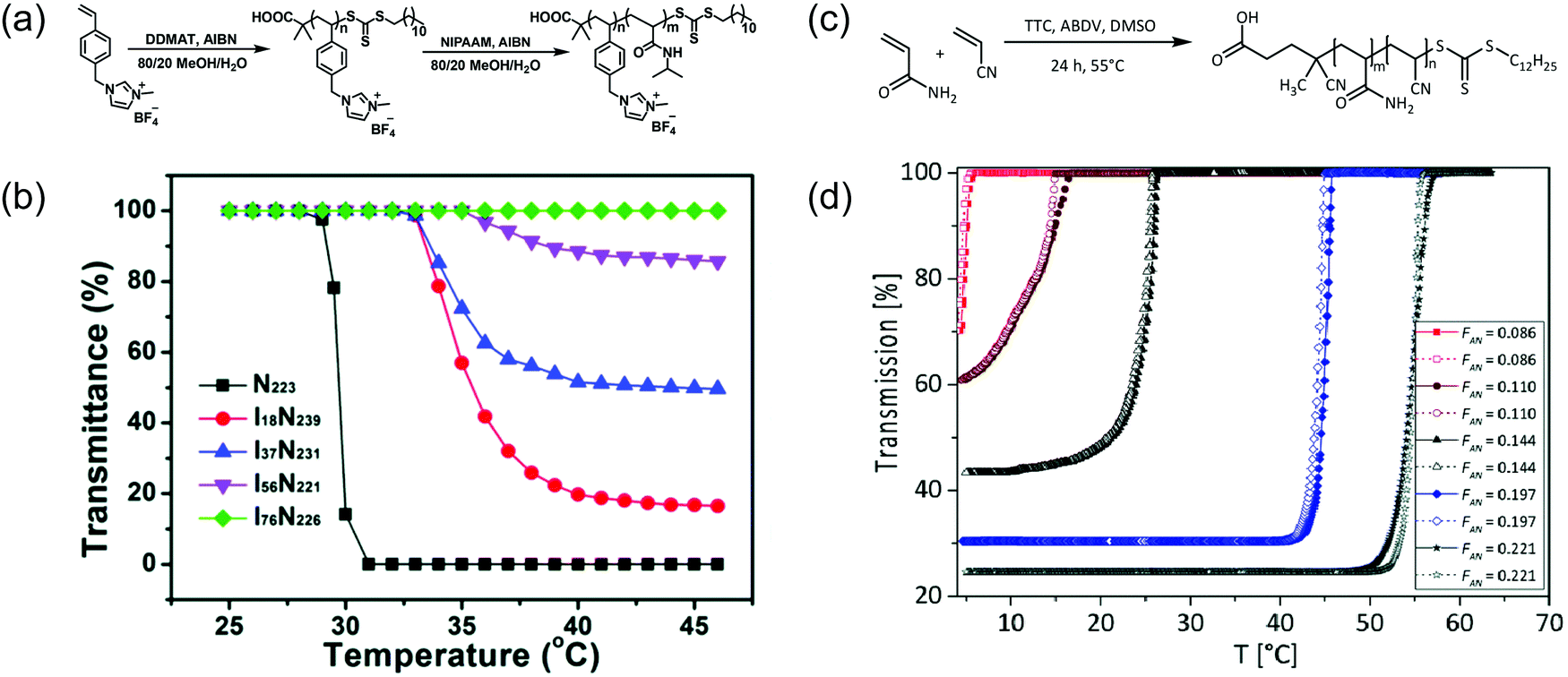 | ||
| Fig. 7 Examples of effects of co-polymerization on thermoresponsive behavior. (a) Synthetic scheme of P(NIPAAM-co-[VBMI][BF4]). (b) Increasing in LCST and diminishing of thermoresponsive behavior illustrated as effects of co-polymerizing NIPAAm with a charged ionic liquid monomers. Reproduced with permission from ref. 93. Copyright 2019, Royal Society of Chemistry. (c) Synthetic scheme of P(AAm-co-AN) (d) elevation of UCST and more pronounced temperature sensitivity shown as impacts of co-polymerizing AAm with AN. Reproduced with permission from ref. 95. Copyright 2017, Royal Society of Chemistry. | ||
The strategy of incorporating a copolymer can also be used for introducing thermoresponsiveness to a water-soluble polymer. One of the most used examples is co-polymerizing acrylonitrile (AN) with the water-soluble acrylamide (AAm), obtaining poly(acrylamide-co-acrylonitrile) (P(AAm-co-AN)) with UCST-type property.89 The UCST behavior of P(AAm-co-AN) was tunable within the range of 5.5 °C to 56.5 °C by altering the AN content in the copolymer (Fig. 7c and d). The obtained trend showed that the increase in AN content corresponded to higher UCST. This observation was a consequence of increasing in hydrophobicity and stronger inter-chain interactions compared to the polymer–solvent interactions.95
3.3 Molecular weight and polymer concentration
The molecular weight of these polymers can also impact the range of responsive temperature, as it also alters the balance between polymer–polymer interactions and polymer–solution interactions. There are several examples that show the influence of molecular weight on the LCST or UCST of thermoresponsive polymers.86,95 Recently, this concept was studied by synthesizing molecular brush support for L-proline catalyst using poly[norbornene-poly(2-methyl-2-oxazoline-b-2-propyl-2-oxazoline)]-graft-poly[norbornene L-proline]. While maintaining the ratio of poly[norbornene-poly(2-methyl-2-oxazoline-b-2-propyl-2-oxazoline)] and poly[norbornene L-proline], the increase in degree of polymerization, ultimately molecular weight, was found to lower the LCST of the polymer. This was attributed to enhance hydrophobic effect of the norbornene backbone.96 On the contrary, it was also demonstrated that increasing molecular weight of poly(N,N′-dimethyl(methacryloylethyl)ammonium propanesulfonate) (PDAMPS) gives rise to higher UCST. This could be explained by considering the increasing inter-chain interactions due to the increasing number of charged pendants. More heat is required to break the polymer–polymer interactions, leading to higher UCST.97Polymer concentration is another important factor in determining the responsive temperature ranges (Fig. 8a). One study conveyed that increase in polymer concentration of poly(ornithine-co-citrulline), regardless of their stereochemistry, showed an elevation in UCST.98 In contrast, another study illustrated that when the concentration of PMEO2MA-b-POEGMA300 rises, LCST decreased.99 As also observed from other studies, increasing in polymer concentration generally promotes polymer–polymer interactions, leading to higher UCST or lower LCST.92,95,100
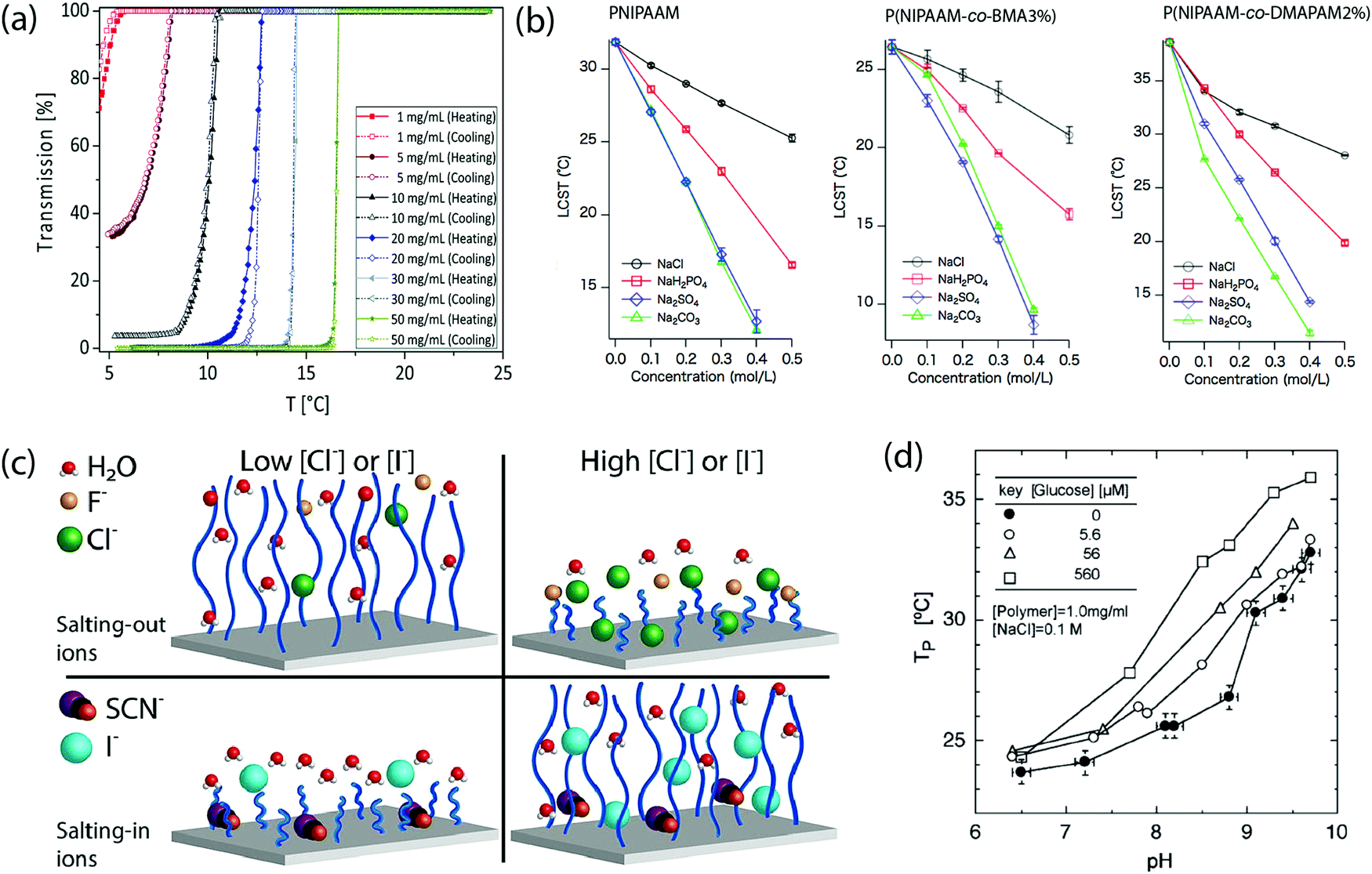 | ||
| Fig. 8 (a) Concentration-dependent alteration of UCST of P(AAm-co-AN). Reproduced with permission from ref. 95. Copyright 2017, Royal Society of Chemistry. (b) Effects of various sodium salts on LCST of PNIPAAM. Different degrees of impact shown when PNIPAAM was co-polymerized with hydrophobic or ionic monomers. Reproduced with permission from ref. 109. Copyright 2015, Elsevier. (c) Schematic representation of ions promoting or worsening the interactions between polymer and water molecules. Reproduced with permission from ref. 111. Copyright 2021, Elsevier. (d) Changes in phase transition temperature of sugar- and temperature-sensitive P(NIPAAM-co-AAPBA) as glucose concentration increases. Reproduced with permission from ref. 113. Copyright 2004, WILEY-VCH. | ||
3.4 Salt concentration
In addition to optimize the intrinsic properties of the polymer (monomer structures, polymer compositions, and degree of polymerization) to obtain the ideal thermoresponsive temperature range, extrinsic conditions of polymer solutions can also be manipulated to achieve the desired responsive range. Several factors can be modified including type and concentration of salt additives, and polymer concentrations. Type of solvents and their mixture can also be altered, but such topics will not be discussed in this review. This provides a convenient approach for modulating transition temperature of polymers without having to re-synthesize the polymers.Hofmeister categorized salt ions based on their impacts on solubility of macromolecules such as proteins and polymers.101–106 The phenomenon in which ions promoting solubility of the macromolecules is known as “salting-in effect”, whereas the exclusion of macromolecules from solvent assisted by ions is called “salting-out effect”.107 Since the thermoresponsiveness of polymers are directly correlated to balance between polymer–polymer and polymer–solvent interactions, ionic additives presented in a solution is critical for determining LCST or UCST. This effect is more pronounced in the case of anions in comparison with cations.66 Polymers with higher ionic content are also reported to be more sensitive to the change of ion concentrations than their non-ionic counterparts (Fig. 8b).89,95,108,109 Increasing the salt concentration in the solution of polysulfobetaines has been shown to result in depression of cloud points, although slight variations are observed based on the monomer structures and the type of salt.92 Furthermore, it was reported the subtle effect of one or multiple salts on thermoresponsiveness of PNIPAAM. Depending on the concentration of hydrated ions and polymer-absorbed ions, the swelling-collapsing state, and ultimately the LCST of PNIPAAM can be altered.110 This observation was due to the presence of anion, leading to water polarization, increasing surface tension around hydrophobic surface, and direct binding of anions to partially negatively-charged of N-amide atom.66 Meanwhile, another study revealed that for OEG-based polymers, the salt can be categorized into “salting-in ions” and “salting-out ions” whose effects were more enhanced as the concentration increased (Fig. 8c).111 Hofmeister effects are not only applicable to soluble polymers, but they also impact LCST of thermoresponsive-polymer-based macromolecules. The work from our group studied Hofmeister effects on nanogels assembled from OEG-based random copolymers. In this case, salting-in ions elevated LCST of the uncrosslinked assemblies and the nanogels as the concentration increased, while salting-out ions had the opposite effects. Beyond the thermoresponsive features, salt types and concentrations also impacted size, encapsulation efficiency, and release kinetic of the assembles and nanogels.112
3.5 Non-ionic additives
Non-ionic additives such as glucose were shown to affect the LCST of thermoresponsive polymers when co-polymerized with a sugar-sensitive moiety, including boronic acid as found in N-acryloyl-3-aminophenylboronic acid (AAPBA). For example, P(NIPAAM-co-AAPBA) showed a rise in cloud point temperature as the sugar content in the solution was enhanced. This response was due to interactions between the boronic acid and the sugar additives, resulting in increasing hydrophilicity of the polymer (Fig. 8d).113 Recently, a mean-field model was proposed to study the effect of multiple sugars on the thermoresponsiveness of PNIPAAM by measuring the degree of swelling at different pH, concentration of sugars, and temperature. Their models suggested that the higher mole fraction of sugars in the solution, the LCST decreased.114 Other non-ionic additives also showed some influence on the LCST of PNIPAAM depending on the concentration and detailed structure of the additives.1153.6 Manipulation of non-covalent interactions
In addition to the intrinsic amphiphilic characteristic of the monomers, thermosensitivity in polymer can be altered by introducing temperature-sensitive non-covalent interactions, such as host–guest interactions, and hydrogen bonds. These interactions can lead to either a shift in HLB or an attraction between polymer chains. As previously discussed, the magnitude of inter-chain interactions, relative to the polymer–solvent interactions, is a crucial factor to determine whether the materials exhibit thermoresponsiveness. For the polymers with intrinsic temperature-sensitive characteristics, instillation of non-covalent interactions can be utilized to tune the range of LCST or UCST.12There are a few demonstrations on the impact of host–guest interactions on the range of LCST or UCST. For example, PNIPAAM system with an adamantane terminal experienced a shift in LCST upon exposing to β-CD-bovine serum albumin (BSA) conjugate. The host–guest interactions between the adamantane terminal and the β-CD generated a shift in HLB by covering up hydrophobic adamantane and introducing hydrophilic BSA to the polymer system. This resulted in a shift of LCST from 29.3 to 30.7 °C.116 Additionally, the host–guest assisting salting-in effects occurred when installing benzo-21-crown-7 (B21C7) to poly(vinyl alcohol-co-vinyl acetate), which did not possess thermoresponsiveness initially. The introduction of B21C7 to the polymer system not only introduced the temperature-sensitive characteristic but also provided the handle for tuning the LCST via the host–guest chemistry of B21C7 and potassium ions. This specific interaction led to a significant salting-in effect (increasing LCST), competing with the typical salting-out effect (decreasing LCST), which could nearly restore the original LCST (27.9 °C compared to 30.2 °C of the salt-free LCST).117
A couple of examples also revealed that by modifying a polymer with moieties that are prone to cause intermolecular non-covalent interactions, such as boronic acid118 and urea,119,120 could be applied for introducing and optimizing the range of thermoresponsiveness. It was illustrated that introduction of a bis-urea terminal group to poly(N,N-dimethylacrylamide) (PDMAc) enabled the temperature-sensitive characteristic to the polymer. A bis-urea free PDMAc was reported to be water-soluble, and the cloud point was not observed below 80 °C. In contrast, the bis-urea modified PDMAcs were shown to exhibit cloud point temperature ranging from 30 to 70 °C depending on the degree of polymerization. It was explained that the bis-urea terminal facilitated the hydrogen bond formation intermolecularly, causing stronger inter-chain interactions and temperature sensitivity.120
Universally, polymers with pendant groups have been studied for thermoresponsive applications. These polymers benefit from modulation of responsive temperature range by simply changing monomer structures, polymer composition, and degree of polymerization. Physical properties of polymer solutions, such as polymer and additive concentrations, are also critical for determining the range of temperature sensitivity. These factors emphasize the importance of HLB, intra-chain polymer interactions, and polymer–solution interactions on thermoresponsiveness. By considering a judicial combination of these factors, polymers with pendant groups can be synthesized with desired temperature-sensitive range, as well as other beneficial features for a plethora of applications.
4. Temperature-responsive dendrimers and hyperbranched polymers
Apart from linear block copolymers and polymers with sidechains, dendrimers have gained attention due to their unique advantages such as monodispersity, stable assembly formation and capability for functionalization at surface, core and the middle region.121,122 In general, thermoresponsive dendrimers are synthesized by (a) directly incorporating PEG and PNIPAAM polymers into the dendritic core or surface;123–126 (b) conjugation of temperature sensitive small molecules such as peptides, oligoethylene glycols (OEG), isobutyl amide onto the dendritic surface;127–131 and (c) building dendrimers with amphiphilic components containing OEGs or β-aminoesters (Fig. 9).18,132–134 | ||
| Fig. 9 Design of different types of thermoresponsive dendrimers (a) conjugation of thermoresponsive moieties (polymer124,128/small molecule139/peptide127) to dendritic surface. Construction of dendrons with amphiphilic components containing thermoresponsive groups (b) biaryl-core facially amphiphilic G2 dendrimer141 and (c) phenylene vinylene core G3 dendrimer.132 | ||
4.1 Thermoresponsive moieties in dendrimers.
Among temperature-responsive dendrimers, PEG and PNIPAAM based systems are extensively studied.29,34,135,136 Recently, PNIPAAM has been incorporated onto the surface of polyamidoamine (PAMAM) dendrimers.137 Water-soluble catalysts were physically encapsulated inside these dendrimers to achieve thermally-controllable catalysis. The authors demonstrated temperature-dependent catalytic activity due to the structural changes in the dendritic host. Similarly, a pH and thermoresponsive polymer, poly(N,N-dimethylaminoethyl methacrylate) (PDMA), was conjugated to the periphery of PAMAM dendrimer.138 This PAMAM-g-PDMA dendrimer exhibited LCST-type property, which was dependent on the graft length of PDMA on PAMAM surface. As the graft length of PDMA increased, the overall hydrophobicity of the dendrimer also increased. This process can prevent PDMA groups from interacting with water, thus causing the decrease in LCST behavior. Since the PDMA moiety is also pH responsive, the LCST of the dendrimer was found to vary with pH. In another study, surface of PAMAM dendrimers were modified with temperature sensitive alkoxy diethylene glycols.127 By controlling the ratios of different alkoxy diethylene glycols in the dendrimer periphery, the LCST behavior of these dendrimers was successfully modulated.Conjugation of thermoresponsive small molecules to the dendritic surfaces has also gained significant interests. In one such effort, an elastin-like oligopeptide (ELP), which has thermoresponsive folding capability was successfully incorporated onto the fourth generation (G4) PAMAM dendritic surface.130 This G4-ELP dendrimer exhibited LCST behavior at the physiological temperature under neutral pH. The LCST was also found to vary with the pH, presumably due to the cooperative interplay between the folding state of peptide and the ionization state of the dendrimer core. In another study, isobutyramide (IBAM) groups known for their thermoresponsive properties were conjugated to each chain of PAMAM dendron-based lipids (Fig. 10a).128 In aqueous solution, these dendrons formed assemblies with IBAM groups exposed on its surface and exhibited LCST around 40 °C. Interestingly, the authors observed temperature-sensitive morphology transformations in G2 and G3 dendron lipids (Fig. 10b). Both dendrons formed vesicular morphologies that destabilized above LCST through a change in hydration of the vesicle surface. The authors speculated that in G2 IBAM dendrons, hydration of dendron moieties led to molecular packing suitable for lamellar phase formation. However, above LCST dehydration of the IBAM groups induced shrinkage of head groups, favoring the truncated cone molecular shape, thereby forming inverted rod-like micelles. In contrast, G3 IBAM dendrons possess larger head groups than that of G2 IBAM. Consequently, after dehydration of head groups in G3 IBAM, dendrons retained cylindrical shapes that formed vesicles. However, dehydration of vesicular surfaces may have increased hydrophobicity of assembly surfaces that aided in aggregation and vesicle fusion.
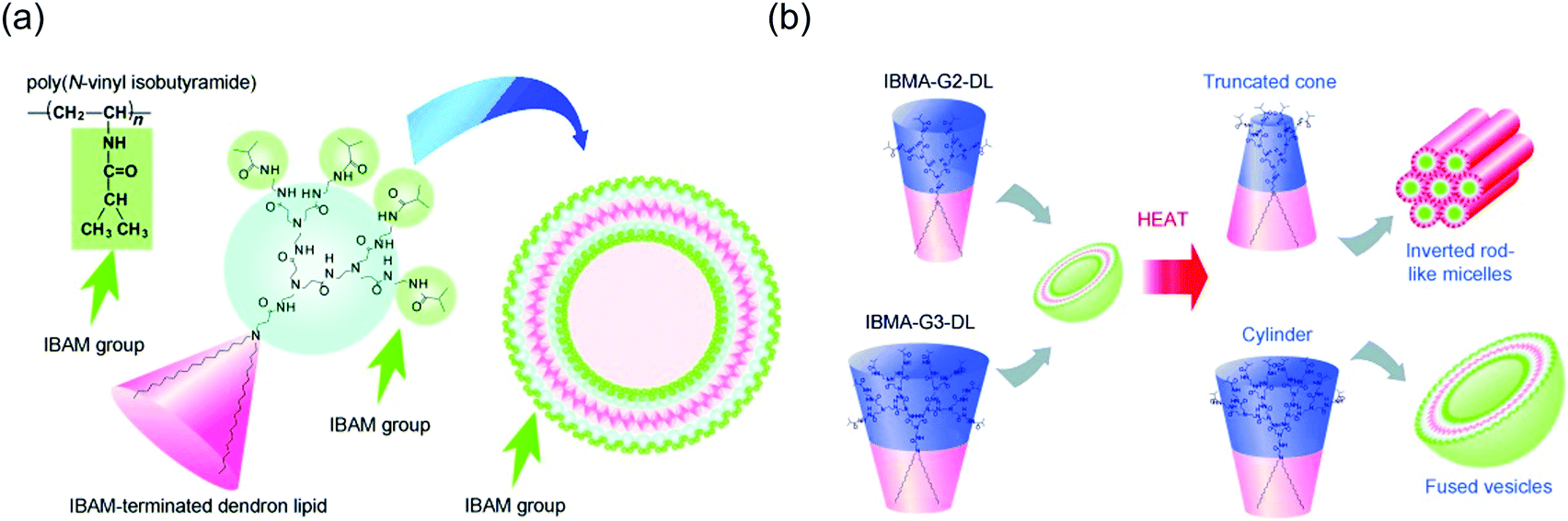 | ||
| Fig. 10 (a) Design of molecular assemblies with temperature-sensitive properties using isobutyramide terminated dendron-based lipids. (b) Mechanism for temperature-responsive structural transition. Reprinted with permission from ref. 128. Copyright 2011, WILEY-VCH. | ||
4.2 Impact of molecular structures on “facially amphiphilic” dendrimers
Our group has developed a new class of dendrimers called “facially amphiphilic” dendrimers by incorporating hydrophilic and hydrophobic functional groups. This modification was performed at either face of the planar building block using a biaryl moiety.139 In aqueous solutions, these dendrimers containing penta(ethylene glycol) units as the hydrophilic moiety and decyl chain as hydrophobic moiety were found to form micelle-like aggregates within nanometer range.140,141 Temperature-sensitivity due to the presence of ethylene glycol units were evaluated in different dendrimer generations.142 A generation-dependent temperature-sensitivity was observed, due to the aggregation size and cooperativity when PEG moieties were tethered together in self-assembled dendrons (Fig. 11a). Additionally, the role of hydrophobic moieties in temperature-responsive behavior were probed in self-assembled dendrons by systematically varying the aromaticity in hydrophobic units, while keeping the hydrophilic component the same.49 Increase in aromaticity could make the assemblies less sensitive to temperature change and even lose the temperature responsiveness. Combined experimental and simulation studies further revealed that the supramolecular structures were less dynamic with the increase in the degrees of aromaticity due to the strong π–π interactions. These findings demonstrated how subtle changes in self-assembled units could have significant impacts on temperature-sensitivity in supramolecular systems.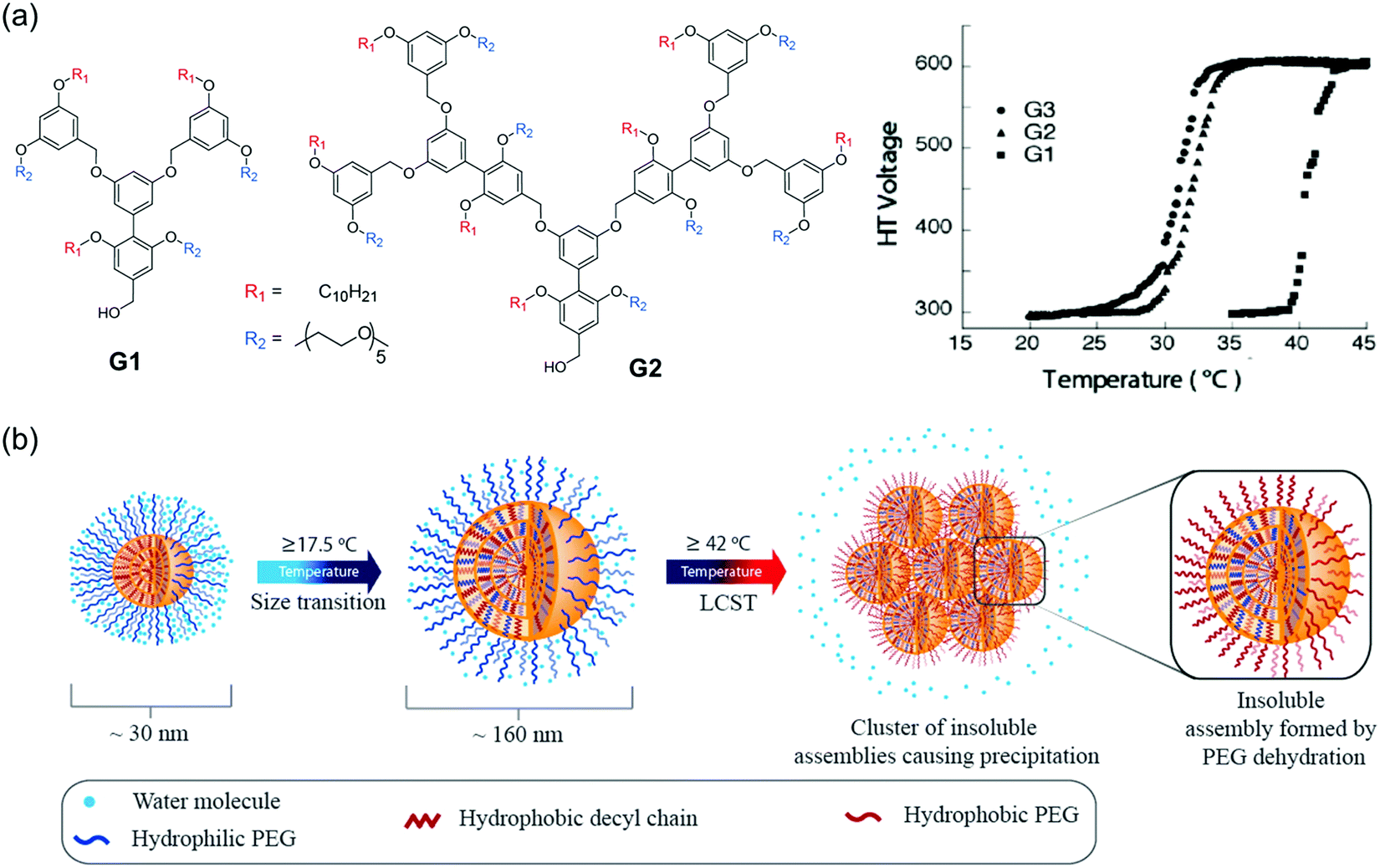 | ||
| Fig. 11 (a) Chemical structure of G1 and G2 dendrons and temperature sensitivity in different generations of facially amphiphilic dendrons. Reproduced with permission from ref. 142. Copyright 2005, American Chemical Society. (b) Schematic representation of the proposed sub-LCST supramolecular transition. Reproduced with permission from ref. 10. Copyright 2013, American Chemical Society. | ||
LCST transitions are based on phase separation in solution i.e., soluble molecules become insoluble in response to an increase in temperature. Interestingly, we have found a temperature-transition well below the LCST (sub-LCST) in facially amphiphilic dendritic assemblies formed by G1 dendrons.10 From dynamic light scattering (DLS), a size transition from ∼160 nm to ∼30 nm was observed at 17.5 °C, which was below the actual LCST (42 °C). Sub-LCST behavior was presumably due to the enhanced hydration of oligoethylene units in the amphiphilic dendrimer at lower temperatures, which makes the dendrons dynamic in the micelle-like aggregates (Fig. 11b). This hypothesis was tested using fluorescence resonance energy transfer technique (FRET) by examining the temperature-dependent guest and host exchange using a pyrene-labeled dendrimer.143 As expected, dendrons in the assembled state rapidly exchanges among each other at lower temperatures whereas, the assemblies were not dynamic at higher temperatures. Interestingly, sub-LCST behavior was found to be unique to G1 dendrons although higher generation dendrimers were structurally similar. We speculated that this could be due to larger energetic penalty for reorganization in the case of G2 and G3 dendron based assemblies containing higher number of amphiphilic units at ambient temperatures.
Interestingly, incorporation of single OEG unit alone or with small molecules do not offer any noticeable thermoresponsive behaviors. However, when OEG units are conjugated to a scaffold that presents these moieties in a multimeric form due to self-assembly, they could exhibit thermal sensitivity. As a result, thermoresponsive oligomers have attracted a great deal of interest.144–147 Our group has designed and synthesized a series of oligomers containing amphiphilic OEG-based side chains (Fig. 12a).148 We found that non-covalent organization of OEG units through aggregation increased the thermoresponsive behavior in oligomers. Additionally, the covalent tethering of amphiphilic units significantly influenced the temperature-sensitivity. With the increase in OEG units, oligomers exhibited increasingly sharp LCST transition, which indicated cooperativity in thermal sensitivity when OEG units are tethered together. In another study, we have investigated the structural requirements for oligomeric amphiphiles to exhibit sub-LCST transition (Fig. 12b).149 Interestingly, the mere presence of OEGs in the oligomer does not guarantee molecules with sub-LCST behavior. However, we found that conformational rigidity in the amphiphilic backbone could impart sub-LCST transition in oligomers. For example, molecule T2 in Fig. 12b, which is conformationally rigid, was found to exhibit sub-LCST transition whereas a relatively flexible molecule T1 does not have. Interestingly, the rigidity can be achieved by intramolecular hydrogen bonding between the amide bonds in the backbone of these oligomers. One example is molecule T3, which can be stabilized using intramolecular hydrogen bonding thereby offering conformational stability whereas amide-methylated molecule T1 lacks such stabilization. Consequently, we found that only molecule T3 exhibited sub-LCST behavior, implying the crucial role of conformational stability for sub-LCST transition. Very recently, we studied the factors controlling the dynamics of such type of thermoresponsive assemblies (Fig. 12c).150 We found both the dehydration of OEGs and thermally promoted molecular motions play roles in assembly dynamics. When temperature increases, the dominant factor transitions from dehydration to thermally-promoted molecular motions. This transition temperature and dynamics dominant factors can be tuned by a single-site mutation with a small hydrophobic group on one of the hydrophilic chains in the oligomer. Apart from these studies, an ethynylhelicene oligomer containing six tri(ethylene)glycol moieties exhibited inverse thermoresponsive behavior.151 This oligomer reversibly changed structure between a double helix and a random coil when subjected to heating and cooling. This indicated that the hydration of tri(ethylene)glycol groups led to the conformation change of triethylamine domains which promoted double helix formation by π–π interactions.
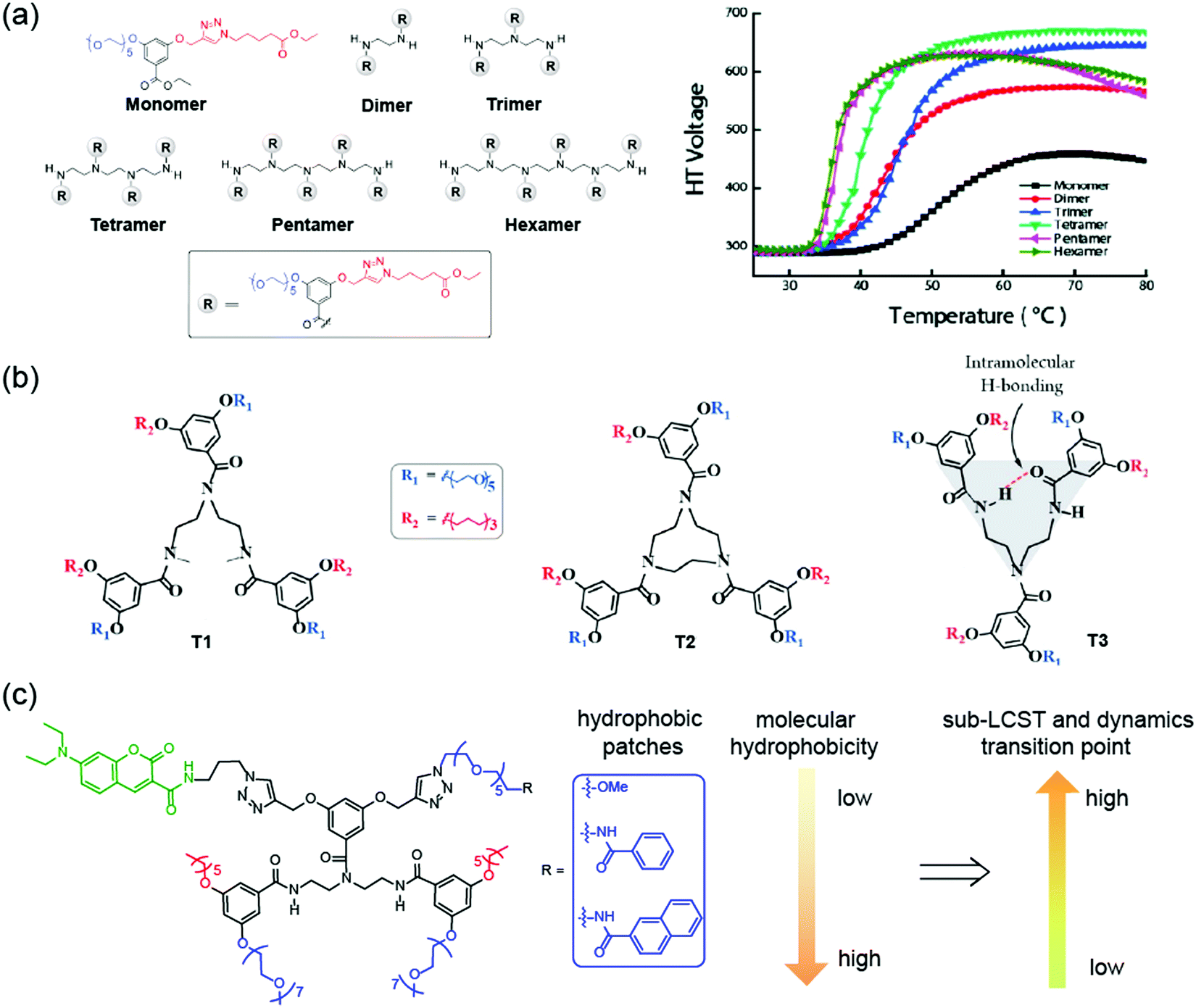 | ||
| Fig. 12 (a) Chemical structures of amphiphilic oligomers containing OEG units and the temperature sensitivity study using turbidity measurement by measuring high-tension voltage response on CD spectrometer. Reproduced with permission from ref. 148. Copyright 2011, American Chemical Society (b) structures of amphiphilic oligomers for probing sub-LCST behavior. Reproduced with permission from ref. 149. Copyright 2015, American Chemical Society. (c) Correlations between amphiphile structure and dynamic transition point. Reproduced with permission from ref. 150. Copyright 2021, Royal Society of Chemistry. | ||
4.3 Modulation of the thermoresponsive properties of hyperbranched dendrimers and dendronized polymers
Macromolecules with confined microenvironments are of significant interest due to their ability to mimic biomacromolecules with well-defined functions and bioactivities.152 Dendritic macromolecules such as hyperbranched dendrimers and dendronized polymers belong to this category since they can create confined microenvironments through cooperative interactions from topological molecules.153–155 Confined microenvironments can also be created by incorporating thermoresponsive features. For example, macromolecules can collapse its hydrophobic parts at temperatures above LCST through enhanced cooperative interactions of the densely crowded dendritic/polymer chains.156–159 Researchers have utilized a combination of thermoresponsive features of OEG and multivalency from the highly branched dendritic architectures to create such confined microenvironments.160–162 For example, OEG-based dendronized polymers have been used to demonstrate sharp thermal transitions with small hysteresis (Fig. 13a and b).161 Additionally, LCSTs can be tuned by changing three key structural parameters in the dendronized polymers: (i) terminal groups, methoxy vs. ethoxy; (ii) length of the OEG chain; and (iii) generation of dendrimer. Although the interior part of the polymer showed minor effects in LCST, the periphery of dendrons wrapped around the dendronized polymer dominated the apparent hydrophobicity for LCST transition. Generally, LCST of thermoresponsive systems are modulated by varying the ratio of hydrophilic and hydrophobic units. Interestingly, it was found that changes in packing arrangements could influence the thermoresponsive behavior in hyperbranched dendrimers.163 In this study, the influence of stereochemical differences on LCST transition was evaluated in hyperbranched polyethyleneimine (HPEI) based system. Hyperbranched polymers with the same chemical composition but different spatial arrangements of functionalities can exhibit remarkable differences in LCST (Fig. 13c). Detailed nuclear magnetic resonance (NMR) studies revealed that differences in distribution of acetamide and isobutyramide groups in the HPEI-based polymers induced different packing arrangements of the same thermoresponsive functional groups. Most of the thermoresponsive systems exhibited a concentration-dependent LCST behavior. Initially, LCST decreased with the increase in polymer or dendrimer concentration, followed by attenuation until saturation.164,165 Interestingly, hyperbranched systems with abnormal thermoresponsive behavior, where LCST increased with rising concentration, have been reported.166 In one such study, hyperbranched polyglycerol (HPG) derivatives were synthesized through esterification reaction between HPG and aliphatic acids with different carbon lengths.167 From turbidity and fluorescence measurements, HPG derivatives with 5–8 aliphatic units exhibited abnormal thermoresponsive behaviors, whereas 2–4 aliphatic units behaved as expected. The existence of relatively strong hydrophobic interactions is crucial for hyperbranched systems to exhibit an abnormal thermoresponsive behavior in water.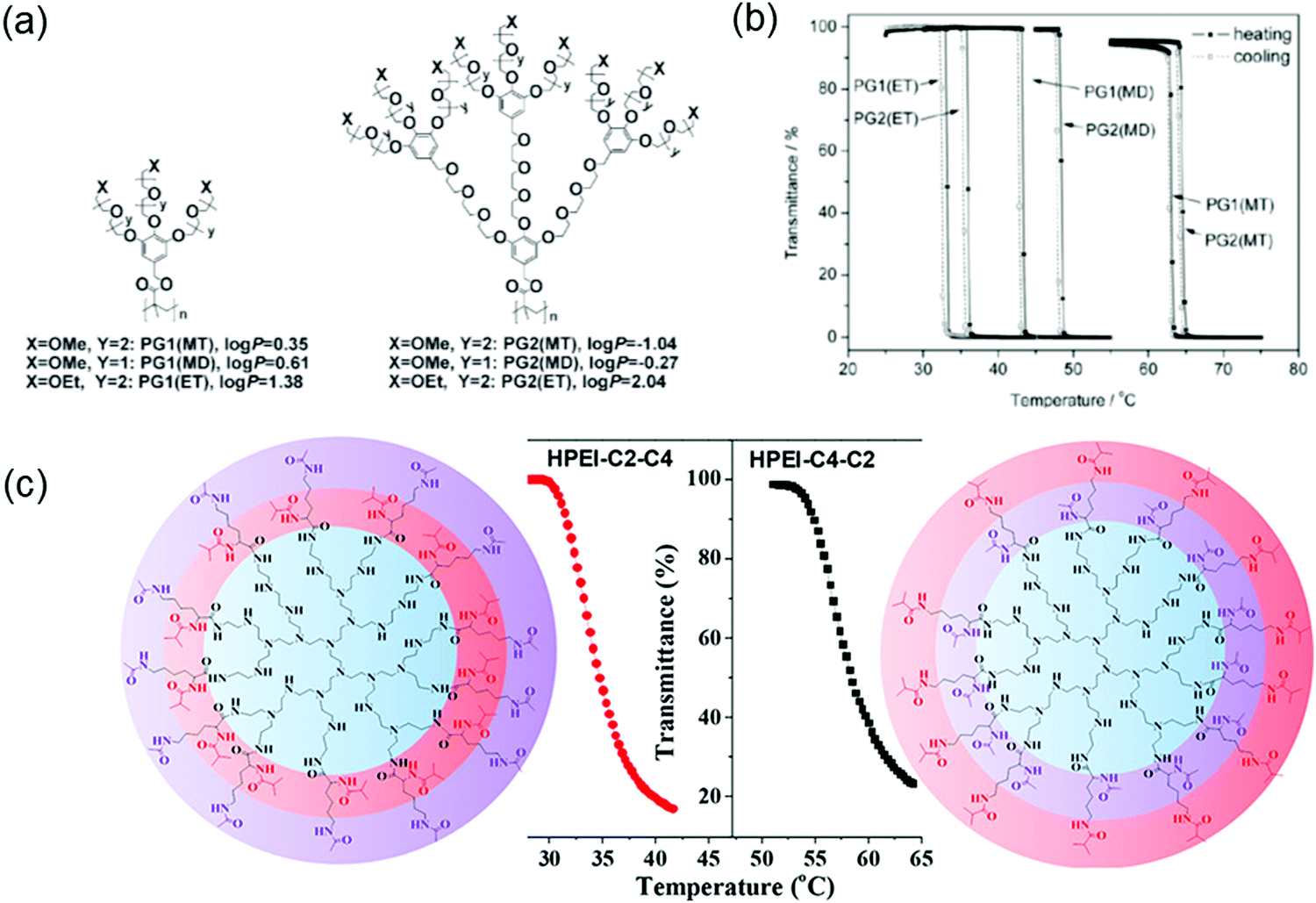 | ||
| Fig. 13 (a) Chemical structures of dendronized polymers and their (b) thermoresponsive behavior. Reproduced with permission from ref. 161. Copyright 2008, Royal Society of Chemistry. (c) Influence of stereochemical differences on the LCST phase transition in hyperbranched polyethyleneimines conjugated with spatially isomerized groups. Reproduced with permission from ref. 163. Copyright 2017, American Chemical Society. | ||
Overall, oligomers or macromolecules with dendritic architectures offer advantages including monodispersity, multivalency and ease in functionalization for the design and synthesis of thermoresponsive materials. Among these materials, the key parameter that determines the thermal sensitivity is HLB. By varying the ratio of hydrophilic and hydrophobic units, the LCST can be rationally tuned. More hydrophilic dendritic molecules show a higher LCST than their hydrophobic counterparts. Apart from the variation in HLB, other factors that affect temperature sensitivity in dendritic macromolecules are dendron generation, aggregation size, length of thermoresponsive units, concentration, rigidity and packing arrangements. Understanding these key factors that modulate the temperature sensitivity behavior will be crucial for the rational design of thermoresponsive dendritic molecules.
5. Other thermoresponsive molecules
Oligo- or polypeptides are a promising class of thermoresponsive polymers that have been used for numerous biomaterials and biomedical applications.168,169 The most studied thermoresponsive polypeptide is elastin-like peptides (ELPs), which is a biomimetic class of protein polymers consisting of five hydrophobic amino acid motifs (Val-Pro-Gly-X-Gly), with X (guest residue) being any amino acid except proline.170–173 Micellar assemblies can be fabricated from either ELP block copolymers or hybrids with other macromolecules. Parameters used to tune the thermoresponsiveness of the assemblies can be categorized into external and internal. External parameters include polymer concentration, salt content, and pH, whereas internal include changing guest residue X, varying the number of repetitive units within ELP, sequence directionality, and conjugation to other molecules.174–180The responsiveness of these assemblies follows an LCST-like phase behavior. Generally, the phase transition temperature decreases when the hydrophobicity of guest residue increases.181,182 Consecutive nonpolar amino acids (NAAs) could suggest thermoresponsiveness to miniaturized elastin-like peptides (MELPs) (∼20 amino acids) through phase transition mechanism mediated by micelle self-assembly. An N-terminal amino acid substitution could also be used for tuning the HLB and thermoresponsiveness.183 Additionally, it was also demonstrated that chemo-selective modifications of methionine thioether as the guest residue in diblock ELP could tune thermal behaviors and lead to transition into micelles depending on the block length.33 Steric effects also played a role if the two groups have similar hydrophobicity. For example, substitution with valine compared to isoleucine lowered transition temperature by almost 20 °C, suggesting that steric effects of linear and branched side chains also impacted transition temperature.184 When short ELPs (4–6 pentads) were conjugated to collagen-like peptides, decrease in transition temperature was found, likely due to local crowding effect which impacted entropic driving forces on the transition.180,185 The LCST of polypeptides can be tuned via external additives depending on peptide concentrations. At low peptide concentrations, divalent cation additives led to lower LCST compared to monovalent ones, but an inverse trend was observed when peptide concentration increased.184 LCST has also been reported to be inversely related to ELP length and concentration due to increasing hydrophobic interactions.175,186 It was recently reported that reversing the sequence direction of poly(VPGVG) resulted in different transition temperatures and hysteresis due to different molecular interactions and aggregate conformations.187 MELPs with four pentad repeats became thermoresponsive with the aid of small hydrophobic compound (9-fluororenylmethoxycarbonyl (Fmoc) group) conjugated at an N-terminus.188 Interestingly, the size of ELP aggregates could be controlled and sustained when conjugated to positively-charged polyelectrolyte, polyethyleneimine (PEI).189 The modification at the side terminal end leads to different conformation during self-assembly process, making it easier to form an inner core of aggregate surrounded by PEI blocks. Additionally, the copolymer solutions displayed lower LCST with higher polymer concentrations and salt content. This core–shell aggregate formed at lower temperatures than uncharged ELPs. Additional information can be found in a recent review about molecular determinants of ELP and ELP-hybrid architecture.190
Apart from ELPs, proline-based peptides are also studied as thermoresponsive materials. Oligo- or polyprolines adopt two helical conformations: compact, right-handed polyproline (PPI) and stretched, left-handed polyproline II (PPII).191 They originally are water-soluble and not responsive to temperature. However, modifications with hydrophobic pendants can introduce thermoresponsive behaviors such that the transition temperature decreases with increasing hydrophobicity of polypeptide.192,193 For instance, hydrophobic units differing in geometry and location on oligo-prolines affected the transition temperature.194 Polyprolines could also be modified with OEG dendrons to elicit thermoresponsive behaviors. Thermally-induced phase transition was found to be dependent on dendron generation and the spatial arrangement along polyproline backbone.195
Comprised of a hydrophilic head group attached to a pair of long hydrophobic fatty acid tail, lipids can spontaneously fold and form bilayers, liposomes, and micelles in water. Unlike the thermosensitive molecules discussed thus far, the temperature-sensitivity of lipids does not follow UCST or LCST mechanism. Rather, the primary factor driving temperature response arises from their reorganization in response to changes in free energy of the system.196 At the phase transition temperature, the orientation of C–C single bonds in hydrocarbon tails changes from trans to gauche state.197 The transition temperature of lipid molecules is determined by differential scanning calorimetry (DSC) where heat absorption is measured when bilayers undergo phase transition from gel to liquid phase.198 Varying the ratio between different lipid molecules can tune the transition temperature of the membrane to have properties such as fluidity, permeability, and curvature.199–201
Most lipid-based formulations incorporate phospholipid 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) due to its appropriate phase transition temperature of 42 °C. Systems containing pure DPPC or pure 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC) (Tm = 55 °C) tend to form liposomes.202,203 Several factors that affect the thermoresponsiveness of lipids include hydrophobic chain length, unsaturation, and incorporating sterols and lysolipids. In theory, phase transition temperature increases by increasing the length of hydrocarbon tails due to stronger intermolecular van der Waals (VDW) interactions, surface area, degrees of freedom, and heat capacity.204 Incorporating unsaturated lipid molecules can weaken VDW interactions between lipid tails and result in the lower transition temperature.205 Double bonds closer to center of alkyl chain cause larger disruptions in packing, compared to those located closer to the head or end of the chain. Additionally, sterols can accumulate in between fatty acid chains, causing decrease in membrane fluidity and lower transition temperature. Lysolipids are a derivative of phospholipid with one of the acyl groups removed by hydrolysis. Due to having a larger hydrophilic head group in relation to hydrocarbon tail, lysolipids tend to form structures with positive curvature and increase permeability of membrane for a rapid cargo release.206–208
The transition temperature of lipids can be modulated by covalent PEGylation and physically mixing with other additives. Previously, pure 1,2-distearoyl-sin-glycero-3-phosphoethanolamine (DSPE) lipid systems was found to form liposomes (Tm = 74 °C), but when PEGylated, as seen in pure 1,2-distearoyl-sin-glycero-3-phosphoethanolamine–PEG(2000) (DSPE–PEG2000) systems, the transition temperature shifted to 12 °C and formed globular micelles and bicelles.209–211 Interestingly, opposite trend was observed in another study where increasing the degree of PEGylation alters fluidity and shape of bilayers in DPPC:DPSE–PEG2000 lipid systems and higher phase temperature.212 This was associated to decrease in overall lateral pressure as fatty acid chains of DPPC and DSPE–PEG2000 became increasingly mismatched. Addition of sterols also led to lower the Tm. In a recent study where low amounts of cholesterol (0–10 mol%) was formulated with DPPC:MSPC:DSPE–PEG2000 liposomes, significant reduction of DOX leakage was observed at 37 °C, while maintaining fast release at Tm of 42 °C.213 The type of encapsulated cargo can also affect the Tm and morphology of lipsomes. For example, the thermosensitivity of lysoplipid-TSLs (LTSLs) was manipulated via DOX crystal modification rather than lipid bilayer compositions.214 For additional information on chemical structures that affect liposomes, the reader can refer to a recent review.215
As discussed, in addition to the previously mentioned linear, block and dendritic polymers, other molecules such as peptides and lipids also display thermoresponsive behaviors. The transition temperatures of ELPs and polyprolines are primarily affected by increasing hydrophobicity. This may include introducing different “X” guest residues, varying the number of repetitive units, changing the sequence directionality, or attaching different hydrophobic pendants. Contrary to polymers and polypeptides, lipids do not follow the standard UCST or LCST mechanism, but respond to changes in the free energy of the system. Alterations to the length of hydrophobic chains and incorporation of different degrees of unsaturation, sterols and lysolipids are the main factors to tune lipid temperature-sensitivity. Considering the parameters discussed thus far, understanding key factors that modulate the temperature sensitivity behavior will be crucial for the development of future thermoresponsive materials for a variety of applications.
6. Applications
In the previous sections, we summarized the recent advances in temperature responsive assemblies based on molecules with different topology and discussed how the molecular bases and different factors can influence thermoresponsiveness. In this section, we focus on the practical utility of thermoresponsive materials in drug delivery, tissue engineering and catalysis, and how the previously mentioned factors were utilized to achieve the optimal properties. As explained before, thermoresponsiveness of molecules originates from molecular level transformations which result in the macroscopic alterations e.g., morphology change, phase separation and rheology alteration of corresponding materials. These macroscopic transformations are taken into various practical uses.6.1 Drug delivery
Therapeutic drug and macromolecule delivery is one of the most extensively investigated areas of thermoresponsive assemblies. Thermoresponsive dendrimers have been widely utilized in drug delivery applications due to their highly tunable LCST and ease in surface functionalization. In one such study, PAMAM dendrimers decorated with alkoxy diethylene glycols with tunable LCST were synthesized.127 By varying the ratios of different alkoxy diethylene glycols on the periphery of dendrimers, the transition temperature could be tuned to body temperature. These dendrimers were found to be noncytotoxic and cellular uptake was enhanced in HeLa cells by increasing their incubation temperature above its LCST. In another study, dendrimers modified with a thermoresponsive collagen model peptide, (Pro–Pro–Gly)5 were synthesized.131 Although these dendrimers did not exhibit phase transition, a thermoresponsive molecular release was observed, which was attributed to the change in the extent of triple helix nature of collagen peptides in the dendrimers. In an effort to utilize dendrimers as drug carriers, thermoresponsive PEG and PNIPAAM units have been grafted onto the surface of PAMAM dendrimers to yield PAMAM-g-PNIPAAM and PAMAM-g-PNIPAAM-co-PEG.125As expected, the unmodified PAMAM dendrimers did not exhibit temperature dependent guest release characteristics whereas, both dendrimers modified using either just PNIPAAM or PNIPAAM co-grafted with PEG units exhibited temperature dependent release profile of indomethacin. More recently, PNIPAAM and phenylboronic acid grafted temperature-responsive polymers were synthesized for the delivery of siRNA (Fig. 14).216 The authors demonstrated that their system could release the loaded siRNA in response to temperature below its LCST. The authors speculated that above LCST, stability of siRNA complexation with polymer increased due to the collapse of PNIPAM moieties, while below LCST, expansion of PNIPAAM groups destabilized the polymer/siRNA complex causing the siRNA release. Additionally, gene silencing efficacy of polymer/siRNA complex was found to significantly increase upon cold treatment after its cellular uptake.
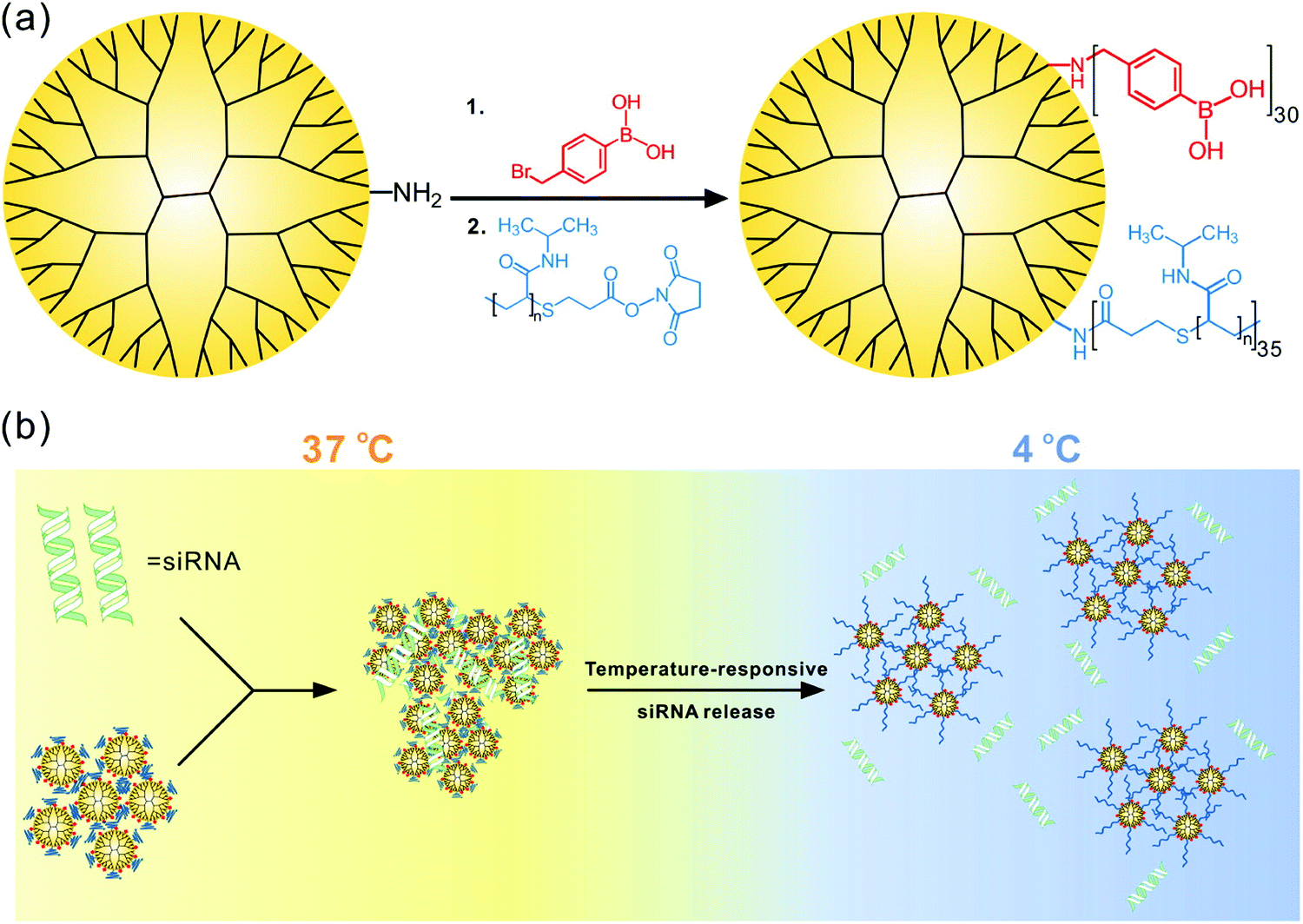 | ||
| Fig. 14 (a) Synthesis of G5 dendrimer containing poly(N-isopropylacrylamide) and phenylboronic acid. (b) Proposed mechanism of temperature-responsive siRNA release. Reproduced with permission from ref. 216. Copyright 2016, American Chemical Society. | ||
Linear block copolymers with different architectures have very distinctive thermoresponsive properties. For example, the difference of gelation temperatures between di- and tri-block copolymers has been utilized to design a ROS-responsive PEG–PCL–PEG which can be cleaved in the middle of the PCL.37 Before ROS-triggered cleavage, the triblock copolymer solution was in a gel state at body temperature and showed relatively slow drug release kinetics. Once it was cleaved by ROS, there was a gel-to-sol transformation because of the generation of a diblock copolymer, resulting in a faster release. The duration of thermogel could also be controlled by the presence of ROS. In another study, two PEG–PLGA polymer fragments were covalently modified to a Pt(IV) prodrug, generating a Pt(IV)-linked triblock thermoresponsive PLGA–PEG–PLGA polymer. This system was then applied to the preparation of micellar drug-loading system. These micelles were used for the encapsulation of another hydrophobic drug, paclitaxel (PTX), for co-delivery of two anticancer drugs. The micellar solution had a sol-to-gel transition at 35 °C, a little lower than body temperature, qualifying it as an injectable delivery system.217 Sun and coworkers dispersed layered double hydroxide (LDH) nanoparticles in a triblock PLGA–PEG–PLGA polymer solution as a drug carrier. The drug-loaded nanoparticle-thermogel system had a sol–gel transition at 38.6 °C, very close to body temperature, and the system exhibited a sustainable release profile compared to the sole nanoparticle for delivery.60 Furthermore, PLGA–PEG–PLGA polymer-based thermoresponsive hydrogel was utilized as a delivery platform for the sustainable release of biomacromolecules (Fig. 15a).218 The authors systematically investigated the impact of LA/GA ratio and concentration of polymers on self-assembly, gel rheology, and guest-release kinetics of hydrogels. According to the results, the polymer with 3:1 LA/GA ratio formed larger micelles (43 nm) than the 94:6 counterpart (24 nm) because of the increased hydrophilicity. This ratio variation also led to the change of rheology of hydrogel and degradation rate of polymer, resulting in distinctive guest-release profile (Fig. 15b and c). As shown in Fig. 15b, 94-6(LA/GA) hydrogel exhibited much lower storage modulus and faster guest release kinetics than 3-1(LA/GA) hydrogel. Besides, the system with higher polymer concentration showed a faster polymer gelation rate, higher storage moduli, and more sustainable release. Interestingly, the addition of excipients, like sodium alginate (ALG) and hyaluronic acid (HA), caused the change of mechanical properties, gelation time, and release rate, thus these factors could be used for fine-tuning the thermoresponsiveness of hydrogels and drug release kinetics.
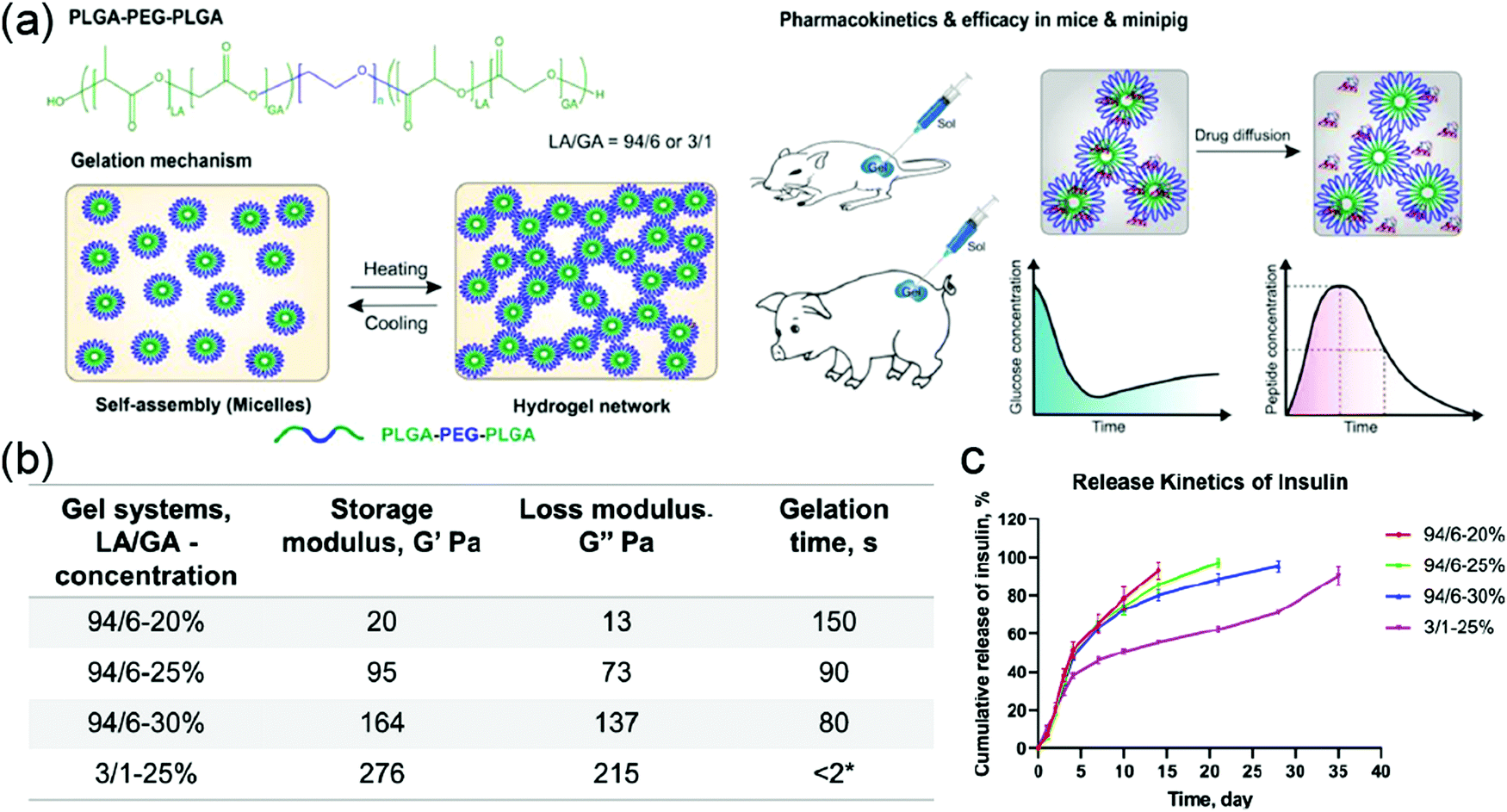 | ||
| Fig. 15 (a) Structure of PLGA–PEG–PLGA polymer and schematic presentation of thermo-induced gelation process and the in vivo test in this work. (b) Summary of the storage and loss moduli, and gelation time for the experimented hydrogel systems at 37 °C. (c) Cumulative release kinetics of insulin from different PLGA–PEG–PLGA thermogels (n = 3). Reproduced with permission from ref. 218 (DOI: 10.1021/acsomega.0c02009). Copyright 2020, American Chemical Society. | ||
Thermoresponsive poly(γ-oligo(ethylene glycol)-ε-caprolactone)-b-poly(γ-benzyloxy-ε-caprolactone) block copolymer has been utilized to prepare micellar nanocarriers for the co-delivery of doxorubicin and quercetin to cancer cells.219 The drug release was based on phase transition of micelles at higher temperatures. The size, LCST, and drug loading capacity of the micelles were tuned by varying the length of OEG moieties. Increasing the OEG length resulted in higher LCST, consistent with many other PEG-attached polymers. Fascinatingly, the loading combination of two different drugs could significantly improve the drug loading capacity, which was due to hydrogen bonding and π–π stacking between the two drugs (Fig. 16). An injectable supramolecular hydrogel from thermoresponsive nanoparticles and α-CD was studied for delivery applications.220 The thermoresponsive properties were highly related to polymer and α-CD concentrations. This thermogel could gradually release 50 nm size nanoparticles in a sustainable way.
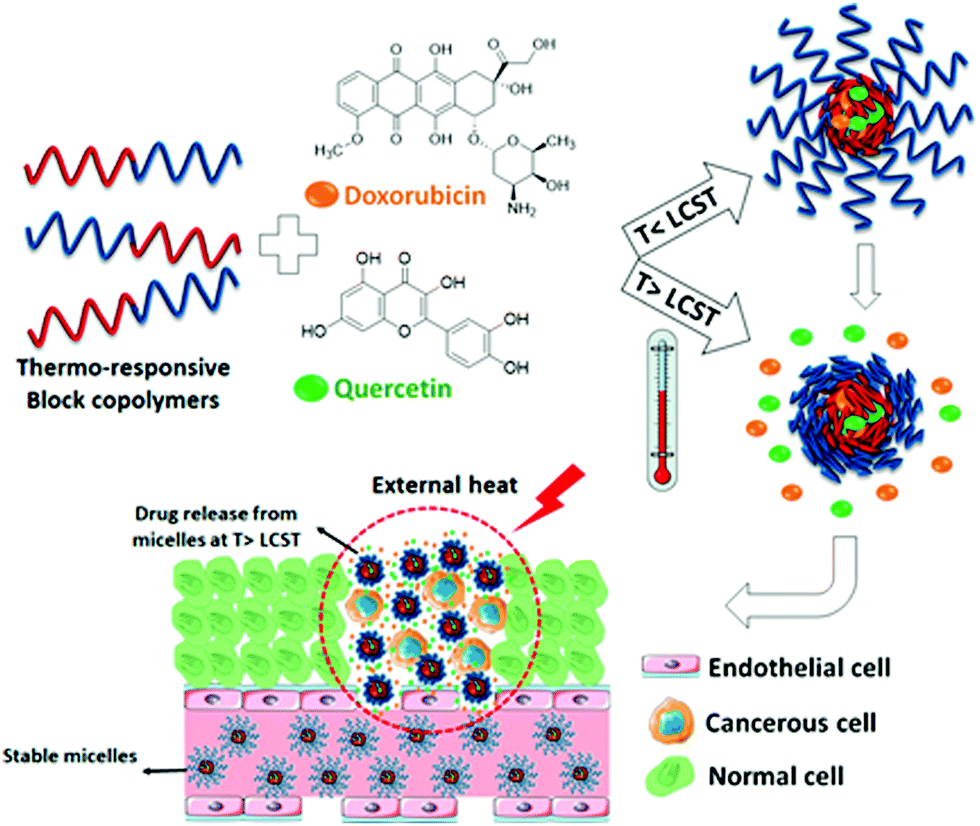 | ||
| Fig. 16 Schematic presentation of the codelivery of anticancer drugs using thermoresponsive micelles. Reproduced with permission from ref. 219. Copyright 2020, American Chemical Society. | ||
The majority of thermoresponsive pendant polymers have been reported within the field of drug delivery. The polymer can be specifically designed to form assemblies with desired morphology. In one study, non-crosslinked PNIPAAM was incorporated as a template for preparation of hollow drug-encapsulated nanoparticles by complexing with a polymer synthesized from NIPAAM, N,N′-bis (acryloyl) cystamine (BAC), sulfated 2-acrylamido-2-methyl-1-propanesulfonic acid (AMPS), and acrylic acid (AAc). The latter polymer was crosslinked to form a core–shell nanoparticle (NP), followed by the temperature depression to release the soluble PNIPAAM core template by using its temperature-sensitive property (Fig. 17a). The NIPAAM units on the hollow shell also assisted with drug encapsulation by introducing the anti-inflammatory peptides when the shell was swollen at lower temperature. The heat was then added to shrink the shell and keep the peptide caged.221 In another study, a block-co-polymer displayed different micelle morphologies and LCST-type behavior depending on the concentration and temperature by simply introducing a galactose-functionalized monomer to a thermoresponsive PDEGMA polymer. The galactose moieties also provided hepatoma-targeting features to the micelle.222 Besides the advantages over morphological control, the LCST of thermoresponsive nanocarriers could be modulated to match desired applications. A PNIPAM-based polymeric NP was applied for targeted delivery of paclitaxel to mitochondria due to the relatively high temperature (∼50 °C) in the organelle. In order to achieve the responsiveness at the temperature of mitochondria, NIPAAM was co-polymerized with poly(ethylene glycol) methyl ether methacrylate with the ratio of 8.5:1, thus increasing the LCST from 32 °C to around 50 °C. The results demonstrated that the thermoresponsive nanocarrier indeed enhanced the ability of mitochondria targeting due to the organelle local heat (Fig. 17b).223,224 Similarly, the ratio between acrylamide and acrylonitrile was optimized in their copolymers to obtain thermoresponsive polymers which functioned at mild hyperthermia conditions for delivering doxorubicin to the tumor site.225 To take advantage of this concept, the tunability in LCST was applied by co-polymerizing NIPAAM with N-methylolacrylamide to obtain polymers with relatively high LCST. These thermoresponsive polymers were then conjugated to hydrophobic cores, which brought down the LCST because of the increased hydrophobicity. With this precise control, they were able to minimize the drug release at normal physiological temperature, while promoting the extrusion of the drug at the tumor site with slightly higher temperatures.226 More examples of thermoresponsive polymeric carriers for delivery applications can be found in previous review articles.74,76–78,227
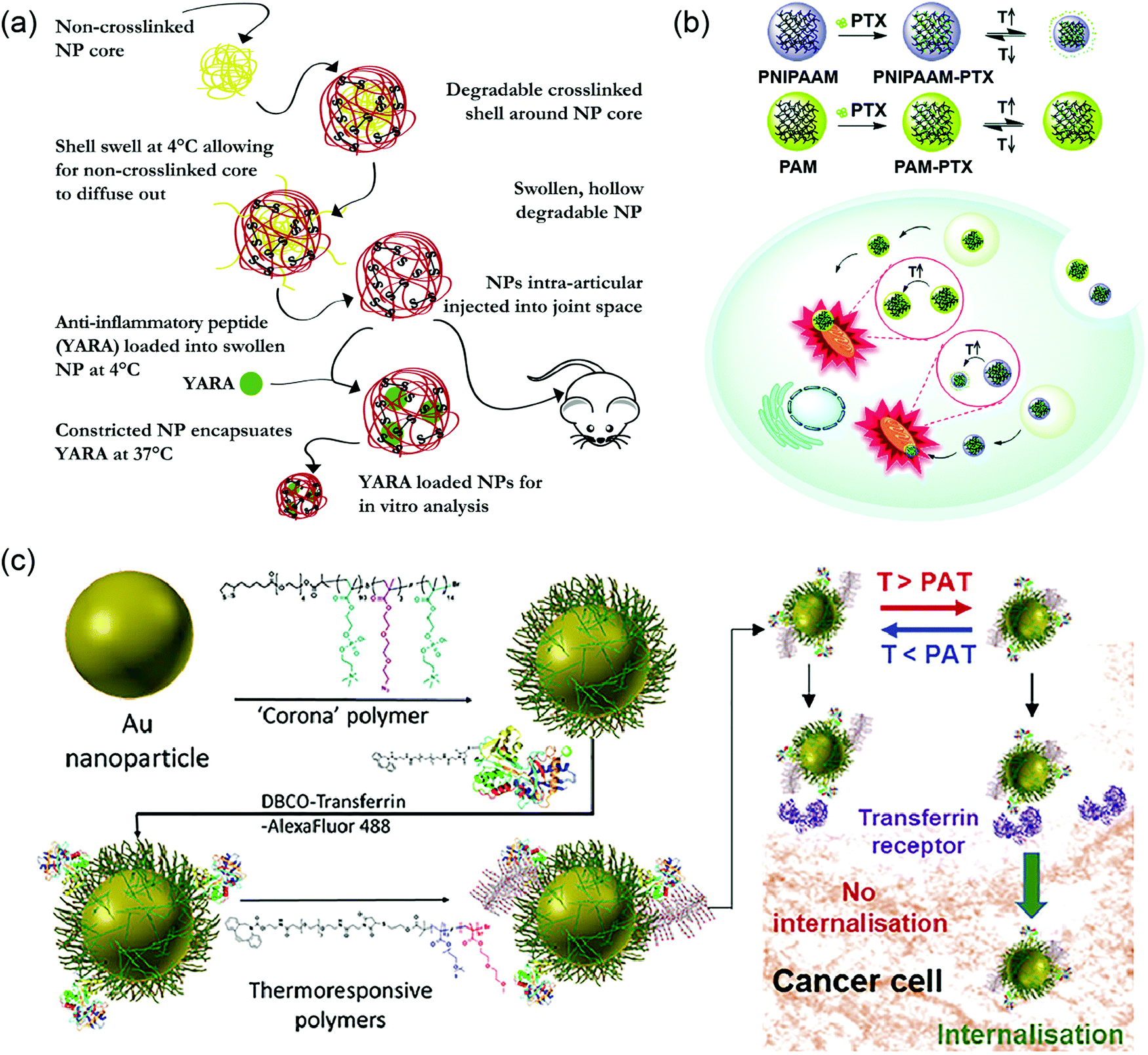 | ||
| Fig. 17 Delivery applications of thermoresponsive nanocarriers (a) PNIPAM core assisting the formation of hollow nanocarriers while PNIPAM co-polymer shell helping with drug encapsulation. Reproduced with permission from ref. 221. Copyright 2020, Elsevier. (b) PNIPAM nanocarriers used for organelle targeting purpose as mitochondria have elevated local temperature. Reproduced with permission from ref. 223. Copyright 2019, Royal Society of Chemistry. (c) Thermoresponsive ethylene glycol-based polymers decorated on AuNPs to control the ligand expose and cellular internalization. Reproduced with permission from ref. 231. Copyright 2018, American Chemical Society. | ||
In addition to directly assembling polymers, thermoresponsive molecules can also be modified onto nano-sized particles to form a core–shell structure. The responsiveness to temperature change can then be used for particle formation and controlled release of drug molecules. In 2018, a thermoresponsive core–shell system was prepared by co-polymerizing OEGMA with MEO2MA on the surface of ZnO quantum dots and applied for imaging and drug delivery applications. The particle platform showed great biocompatibility, yet high toxicity at the temperature above LCST.228 The change in cell viability came from phase transition of the grafted polymers. When the polymers became dehydrated above LCST, DOX encapsulated at the shell of the particles was released more efficiently, leading to cell death. Moreover, PEO–PPO-pendant polyphosphazene could be grafted onto mesoporous silica NPs for pH- and thermoresponsive drug delivery systems.81 Many more thermoresponsive polymer-grafted porous silica nanoparticles has been reviewed.229 Additionally, one study revealed an interesting photothermal system with temperature-controlled release by covalently modifying a thermoresponsive OEGMA and MEO2MA-based copolymer on to the surface of hollow gold nanoparticles (AuNPs).230 Apart from the controlled release applications, thermoresponsive gates can also be applied for controlled cellular uptake by regulating the accessibility of the targeting ligand. One example is decorating AuNPs with transferrin protein ligands and OEG-based thermoresponsive polymers. Upon mild heating, the protein ligands could be revealed to cellular transferrin receptors, inducing internalization of the AuNPs (Fig. 17c).231
ELP sequences are easily modified to include therapeutic peptides, proteins, and small molecule drugs. A library of ELP fusion proteins was constructed to elucidate the impact of molecular weight on pharmacokinetics, biodistribution, and renal localization.232 ELP fusion proteins with higher molecular weight or chain length had lower Tt, consistent with prior observations.175 These results demonstrated that medium molecular weight proteins (37–74 kDa) were found to be the most suited for delivery due to longer plasma half-lives and high total renal accumulation. While a number of light-responsive ELP hydrogels have been developed, it has proved difficult to create stable hydrogels using ELPs with temperature-sensitive characteristics for drug delivery because of their high hydrophobicity and tendency to aggregate heterogeneously.233,234 Recently, genetically engineered temperature-responsive multifunctional protein hydrogels were developed for spatiotemporal control of cellular functions (Fig. 18a).235 ELP (AVGVP)n was attached to both ends of matrix protein and used as crosslinking points. In addition, polyaspartic acid (polyD) and coil-LL peptide were fused to ELP sequence (CUBEs), exhibiting controllable sol–gel transition, superior transparency, tunable mechanical and biofunctional properties, and growth-factor delivering activity. ELP fused with polyD have been shown to form size-controlled nanoparticles due to negative charge repulsion of polyD block and increase in polyD chain length improved hydrogel transparency. These results suggest that the hydrogel formation was promoted by increasing the hydrophobic intermolecular interactions of ELP depending on the concentration. Interestingly, short peptide sequences such as Boc–Phe–Phe–Gly–Gly–OH were reported to also have thermoresponsive behaviors.236,237 When coated onto ZnO@Fe3O4 nanoparticles, peptides acted as nanovalves that “open and close” in response to local heating of the core generated from microwave irradiation. The peptide closes pores by forming β-sheet protofibrils through self-assembly at 37 °C but opens pores at transition temperature of 50 °C via disassembling process. For more details on thermoresponsive peptide-based materials, the reader can refer to recent reviews.238–240
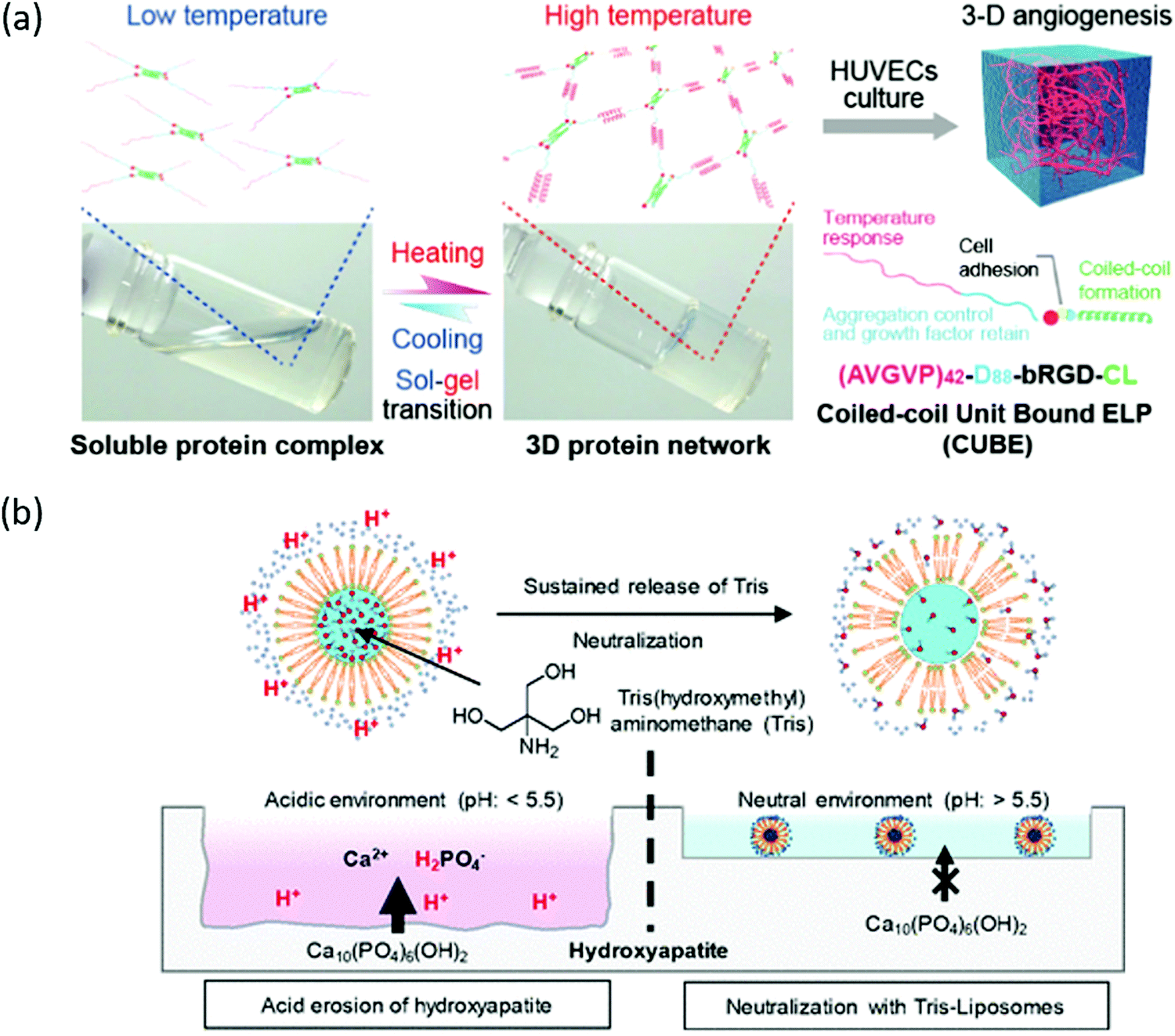 | ||
| Fig. 18 (a) Genetically engineered temperature-responsive multifunctional protein hydrogels for spatiotemporal control of cellular functions. Reproduced with permission from ref. 235. Copyright 2020 American Chemical Society. (b) Thermosensitive DPPC liposomes encapsulating alkalis successfully neutralized environmental acids for up to 3 h, preventing acid erosion of hydroxyapatite matrix for dental oral care. Reproduced with permission from ref. 249. Copyright 2020 American Chemical Society. | ||
Because of their biocompatibility and bioavailability, lipid-based assemblies have mainly been utilized for delivery applications as well. To release cargo within the body, the proper heating temperature must range between 40–42 °C since higher temperatures can result in hemorrhage. Advances in thermoresponsive liposomes are often formulated to respond to mild hyperthermia (43–45 °C) and coupled with radiotherapy and chemotherapy for improved therapeutics. The existence of both solid and liquid lipid domains at the transition temperature leads to leaky regions or increased permeability for drug release.241–246 Recently, thermosensitive liposomal cerasome with specific targeting (c-LIP-WSG) was prepared to reduce side effects and drug leakage, and improve targeting.247 These results indicated that c-LIP-WSG had better stability than most liposomes due to silicon material formed in liposome bilayer, exhibiting excellent structural stability both in storage and in a simulated circulation environment. In vivo data confirmed efficient targeting for SKOV-3 tumor in ovarian carcinoma. Two copolymers, PNIPAAM-b-PLA copolymer (66:34% w/w) and PNIPAAM-b-PLA (50:50% w/w), with the latter being shorter and more hydrophobic, were evaluated on their lyotropic effect on liposomal membrane.248 DSC measurements of chimeric and mixed bilayers and liposomes consisting of DPPC or EPC and PNIPAAM-b-PLA copolymers suggested creation of new functional phase inside membrane which was dependent on both composition and polymer concentration. PNIPAAM-b-PLA (66:34% w/w) had better stability on liposome membrane, while PNIPAAM-b-PLA (50:50% w/w) had no thermoresponsive reduction and lacked transition close to LCST of PNIPAAM. Overall, the length of individual segments of PNIPAAM and PLA and their molecular weights were key factors for insertion and conformation inside membrane that determined final functionality. Although burst release is a single, high-rate release at the target site, this type of release has limited sustained action for drugs. Sustained and slow-releasing drug carriers are less invasive and offer more accessibility as they do not require hyperthermia. Recently, thermosensitive DPPC liposomes encapsulating alkalis successfully neutralized environmental acids for up to 3 hours, preventing acid erosion of hydroxyapatite matrix for dental oral care (Fig. 18b).249 Liposome encapsulating Tris (Tris-Lipo) was prepared in 4.1 M Tris solution, which has significantly higher osmolality than normal human saliva. This large osmolality gap between the inside and the outside of the liposomes caused minor release of cargo below the phase transition temperature of the liposomes (40.3 °C). Tris-Lipo released at 36.5 °C peaked at 2 hours of incubation, while at 25 °C only reached peak release in 3.5 hours, due to the decreased permeability at a lower temperature. This suggested the influence of environment temperature and the difference in pH gradient between the inner and the outer wall of the lipid bilayer membrane. After 3 hours, release was subdued and was not activated again until triggered by a disruption to the osmotic equilibrium (reacidification). Functionalizing Tris-Lipo surfaces with targeting moieties such as tetracycline and alendronate, could have high potential for in vivo as an effective liposomal nanotherapeutic for the prevention of dental cares formation. Thermoresponsive liposomes and their hybrids have been discussed extensively in many reviews;242,250–252 therefore, recent examples listed above were briefly discussed to provide additional insights.
6.2 Tissue engineering
Tissue engineering and 3D cell culture is another hot research area for the applications of thermoresponsive materials, especially thermoresponsive hydrogels, as these are fundamentally important for developing new methods to revert damages from wound and diseases.27 In a recent study, thermoresponsive assemblies were prepared using diblock copolymer poly(glycerol monomethacrylate)–poly(2-hydroxypropylmethacrylate) (PGMA-b-PHPMA) (Fig. 19a). Interestingly, the structure of assembly could be tuned by varying either the length of different blocks or the temperature.253 For example, increasing the fraction of PHPMA led to assembly changes from spheric micelles to worm-like micelles and finally vesicles (Fig. 19b). Reducing the temperature of PGMA54-b-PHPMA140 from 21 to 4 °C could result in a morphology transition from worm-like to spherical micelles (Fig. 19c). In another report, they applied this worm-like thermogel to mimic natural mammalian mucins and used it for 3D cell culture, which could induce stasis in pluripotent stem cells and human embryos (Fig. 19d). Interestingly, cells recovered quickly from the suspended animation state by thermo-induced degelation, demonstrating the superiority of this thermal gel in 3D cell culture and tissue engineering.254 A multiblock polymer synthesized by pyridine-dicarboxylate (PDC) connected poly(ethylene glycol)–poly(propylene glycol)–poly(ethylene glycol) triblock copolymers was applied for tonsil-derived mesenchymal stem cells (TMSCs) culture.255 The gelation temperature of the polymer could be tuned by varying polymer concentration. The mechanical properties of the hydrogel could be modulated by adding different concentrations of Fe3+. A thermoresponsive triblock copolymer (P(NIPAM166-co-nBA9)–PEG–P(NIPAM166-co-nBA9)) was loaded with silver-nanoparticles-decorated reduced graphene oxide nanosheets, Ag@rGO, which had antibacterial activity, and generated a thermoresponsive hydrogel. This hydrogel underwent irreversible sol-to-gel transition at body temperature and was successfully applied for Methicillin-Resistant Staphylococcus aureus (MRSA) infected wound healing.256 | ||
| Fig. 19 (a) The synthesis and structure of PGMA–PHPMA polymer. (b) Digital photographs of three PGMA–PHPMA copolymer dispersions (10 w/w%) at 21 °C, TEM images of diluted polymer solutions and molecular weight of the three polymers. (c) Thermo-induced morphology transformation of PGMA54–PHPMA140 assemblies. Reproduced with permission from ref. 253. Copyright 2012, American Chemical Society. (d) Structure of PGMA55–PHPMA135 polymer for wormlike thermogel and confocal image from 3D cell culture. Reproduced with permission from ref. 254 (DOI: 10.1021/acscentsci.5b00370). Copyright 2016, American Chemical Society. | ||
Thermoresponsive polymers can also be used for selective cell sheets production, which is applicable to many biomedical studies. For instance, PNIPAAM was grafted onto hyperbranched polystyrene or its cationic and anionic derivatives to study the attachment and detachment of mouse 3T3 fibroblast cell sheet.257 For this purpose, the introduction of selectivity into the cell detachment process of cell sheet formation is very important. In a recent study, poly(N,N-dimethylaminopropyl acrylamide)-b-PNIPAAM was grafted from an ATRP-modified glass plate to separate a mesenchymal stem cells from fibroblasts and macrophages, while another report polymerized PDEGMA on the modified gold surface to distinguish stem cells from differentiated cells (Fig. 20a).258,259 These techniques could be used for 3D tissues. The cell sheet used for 3D stacking were obtained from a mold made by UV-cured polyurethane acrylate mixed with glycidyl methacrylate, followed by PNIPAAM grafting (Fig. 20b).260 More tissue engineering applications of thermoresponsive polymers were summarized in a recent review.261
 | ||
| Fig. 20 Tissue engineering applications of PNIPAAM-based polymers grafted surfaces: (a) PNIPAAM-grafted glass plate for cell-selective cell sheet formation. Reprinted with permission from ref. 258. Copyright 2020, WILEY-VCH. (b) PNIPAAM-grafted flexible polymeric molds utilized for 3D cell sheet stacking. Reproduced with permission from ref. 260. Copyright 2020, Elsevier. | ||
6.3 Controlled catalysis
In addition to delivery and tissue engineering applications, thermoresponsive assemblies are also employed for controlled catalysis. Highly branched architecture and multivalent features of dendrimers make them highly attractive for applications in catalysis.262,263 For example, surface of PAMAM dendrimers were modified with thermoresponsive PNIPAAM functional groups and a water-soluble catalyst was physically encapsulated into the interior cavities of these dendrimers.137 In this study, the catalytic activity could be controlled in response to variations in temperature, which was induced by the change in the structure of dendrimers. In another study, a hybrid catalyst based on ruthenium nanoparticles was encapsulated into the networks of poly(propyleneimine) dendrimers crosslinked with PEG diglycidyl ether.264 Additionally, temperature-sensitive catalytic activity of these synthesized hybrid catalysts were tested by using hydrogenation reaction of unsaturated compounds in aqueous conditions. The authors found temperature dependence in catalytic activity, which increased with the temperature before levelling off. More recently, a thermoresponsive nanoreactor from amphiphilic dendrimer-like copolymer has been reported.265 The amphiphilic dendrimer-like copolymer consisted of poly(styrene) in its interior and thermoresponsive PEO as outer segments. Here, the nanoreactor could be regulated using temperature as a stimulus for the activation-and-deactivation of hydrolysis reaction of benzyl chloride (Fig. 21). At temperatures below LCST, nanoreactor was soluble and outer PEO segments were highly hydrated in the reaction media, facilitating the reaction to proceed. However, at temperatures above LCST, due to the dehydration process, PEO segments shrunk thereby causing the aggregation of the unimolecular nanoreactor and sharp decline in the reaction rate. Furthermore, it was found that the activation-and-deactivation process was reversible in nature until the reaction reached the final yield of 99%. | ||
| Fig. 21 (a) Proposed reaction pathway for accelerating reaction by amphiphilic dendrimer-like copolymer in aqueous solution (b) activation/deactivation of nanoreactors based on LCST of the densely grafted peripheral PEO segments. Reproduced with permission from ref. 265. Copyright 2019, American Chemical Society. | ||
It was demonstrated that the introduction of thermoresponsive nanoreactors could introduce the exclusion of water and hydrophobic core formation around the L-proline and L-hydroxyproline. Such confinement led to higher catalysis activity and better enantioselectivity. The nanoreactors were synthesized from 2-oxazoline-based bottlebrush, in which the length of 2-oxazoline could be altered for different thermoresponsiveness and HLB, resulting in tunable catalytic activities (Fig. 22a).96 Furthermore, instillation of the nanoreactors could enable temperature-assisted recyclability to soluble catalysts. Two works published in 2019 illustrated that providing ethylene glycol-based polymers as scaffolds maintained high catalytic activity, in comparison to soluble catalysts, while became recoverable simply by precipitation using additional heat.266,267 Especially, the work with triphenylphosphine pendants showed the tunable thermoresponsiveness by changing ratio between DEGMA and OEGMA300 composition. Likewise, the reusability of enzymatic activities can be installed by covalently grafting polymers with LCST or UCST-type behaviors onto an enzyme. In one study, PNIPAAM was conjugated to B8CYA8 b-glycosidase (Fig. 22b),268 while in another study, P(AAm-co-AN) was attached to Pseudomonas cepacia lipase (PSL) (Fig. 22c).269 In both cases, the enzymes could be easily recovered by heating or cooling the solution, while maintaining decent activities after the recovery.
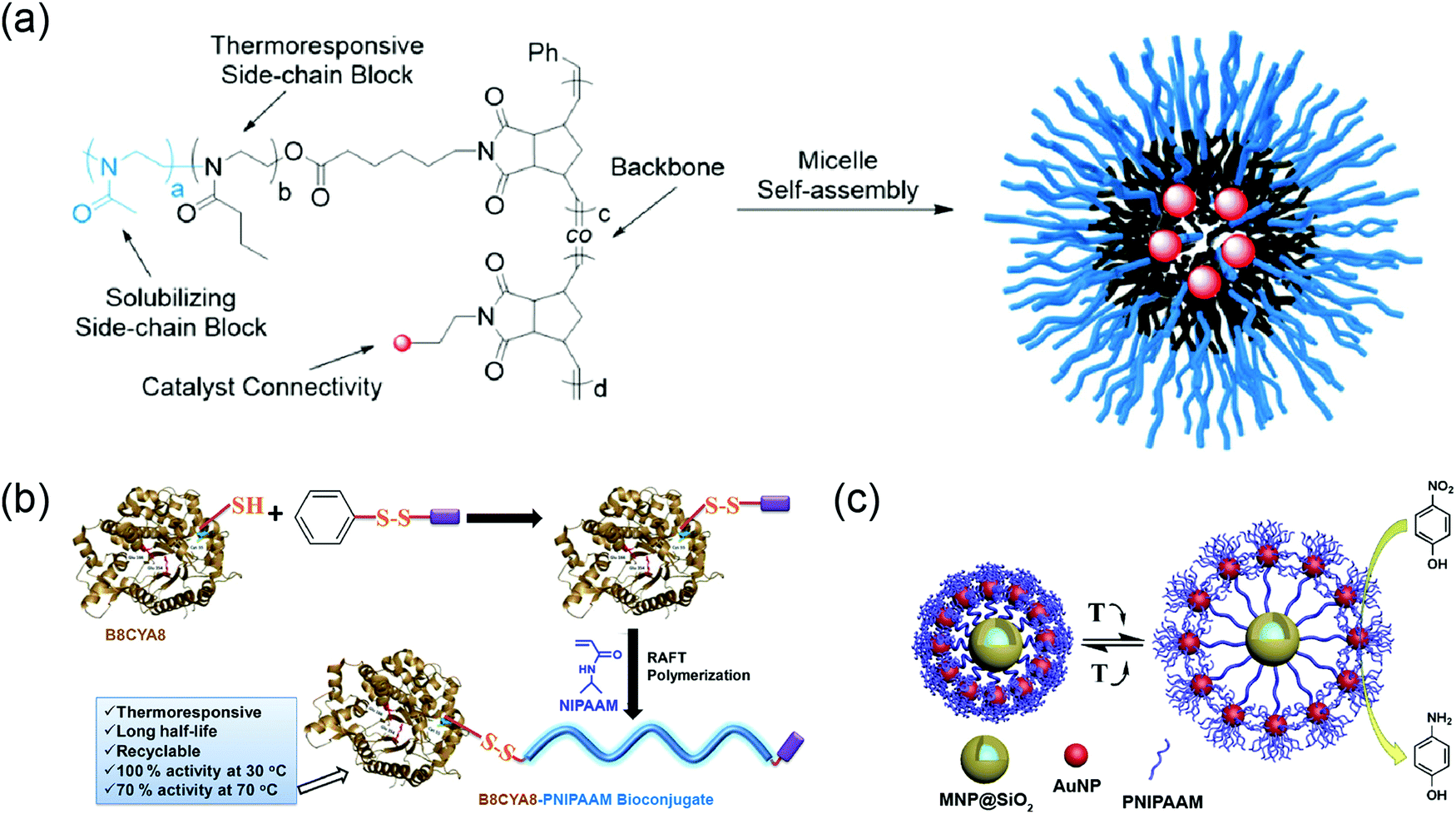 | ||
| Fig. 22 Temperature-controlled catalysis (a) poly(2-oxazoline)-based bottle brush polymers applied for controlled activity and enantioselectivity of L-proline catalyst. Reproduced with permission from ref. 96. Copyright 2021, American Chemical Society. (b) PNIPAAM conjugation to B8CYA8 b-glycosidase for modulable enzymatic activity. Reproduced with permission from ref. 268. Copyright 2018, American Chemical Society. (c) PNIPAAM utilized as linkers and surface decorators for satellite SiO2–AuNPs whose substrate conversion rate was temperature-dependent. Reproduced with permission from ref. 271. Copyright 2019, American Chemical Society. | ||
Thermoresponsive molecules can also be coated onto nanoparticles or proteins as a gate to control their catalytic activities. For example, a study discussed the control of manganese-mediated decomposition of H2O2. This cryptic catalysis system functioned by grafting a temperature-sensitive bottle-brush PEO–PPO–pendant polyphosphazene onto manganese-modified mesoporous silica nanoparticle. The subtle change in temperature could precisely turn on and off the catalysis.81 Another example of gate-controlled catalysis was shown by Guo and coworkers, whose work evolved around attaching thermoresponsive polymers and DNAzymes onto AuNPs. The accessibility of DNAzymes depended on the morphology of the polymers, whether shrinking or expanding.270 Moreover, a core–satellite NP was constructed by conjugating small AuNPs onto a silica nanoparticle core. PNIPAAM was utilized for the conjugation and the shell decoration, introducing temperature-sensitivity to the system, where at elevated temperature, the catalysis activities were reduced.271
7. Conclusions and outlook
In this review, we summarize the recent advances in thermoresponsive assemblies and the molecular basis for the tunability in their temperature sensitivity. Some typical thermoresponsive moieties introduced in this review were PEG, PNIPAAM, polymers with charged moieties, and lipids. Temperature-induced alteration in hydrogen bonds, electrostatic interactions or thermo-induced molecular motions are used as the main driving forces for corresponding thermoresponsiveness. Key parameters used for tuning thermoresponsive behaviors include molecular structure, HLB, molecular weight, concentration, external additives, blended polymer systems, and introduction of small moieties that offers additional molecular interactions.As the major driving forces for thermoresponsiveness, the abrupt changes of hydrogen bonds, electrostatic interactions and molecular conformations are all based on weak interactions that do not disturb the molecular integrity. These weak interactions are customized for different chemical moieties, thus exhibiting different tolerance toward the disturbances from microenvironments due to temperature alteration, which brings the unique thermoresponsiveness for different materials. However, because of these relatively weak interactions, the change of thermoresponsiveness for different materials are usually non-proportional and even inversely correlated to parameters mentioned above. This makes the responsive results less predictable when adjusting parameters for different materials and thus difficult to rationally manipulate material properties. For example, increasing the molecular weight and hydrophobicity of different polymers may lead opposite changes to the thermogelling properties.38,45 In this context, developing thermoresponsive materials relying on different responsive principles is crucial for the design of next generation thermoresponsive supramolecular assemblies with predictable tunability. Many temperature-responsive chemical reactions have been developed in the past few decades.272–274 Thermo-induced chemical bond formation and cleavage can be a robust strategy to tune the molecular integrity, thus robustly altering the HLB and molecular structure in supramolecular assembly.275,276 These changes are based on covalent bond alteration, thus more resistant to microenvironment variations and resulting in more predictable and controllable thermoresponsiveness. We expect more of these covalent bond alteration-based thermoresponsive materials will be designed in the future, enriching the thermoresponsive materials categories, and bringing opportunities for the design of next generation thermoresponsive materials for desired applications.
As summarized before, responsive temperature, mechanical properties, assembly morphology, membrane permeability, and guest release kinetics, can be manipulated by rationally varying the above-mentioned factors. The superior thermoresponsive properties qualify these materials for a wide range of applications. This review discussed the applications of the materials in therapeutic drug delivery, tissue engineering, and thermally controlled catalysis. But the potential utilities are not limited to these three categories. For example, thermoresponsive polymers have also been used for shape memory materials,277–279 temperature and glucose sensors,280,281 smart textiles,282 and thermoresponsive chromatography and electrodes.283–285 We anticipate that thermoresponsive molecules could be applied to more areas as smart materials and contribute a variety of interdisciplinary research.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank U. S. Army Research Office (W911NF-15-1-0568) and National Institutes of Health (GM-136395) for supporting this work.References
- A. K. Das and P. K. Gavel, Soft Matter, 2020, 16, 10065–10095 RSC.
- F. Li, Y. Qin, J. Lee, H. Liao, N. Wang, T. P. Davis, R. Qiao and D. Ling, J. Controlled Release, 2020, 322, 566–592 CrossRef CAS.
- J. Zhuang, M. R. Gordon, J. Ventura, L. Li and S. Thayumanavan, Chem. Soc. Rev., 2013, 42, 7421–7435 RSC.
- Y. Tu, F. Peng, A. Adawy, Y. Men, L. K. E. A. Abdelmohsen and D. A. Wilson, Chem. Rev., 2016, 116, 2023–2078 CrossRef CAS.
- S. Qiao and H. Wang, Nano Res., 2018, 11, 5400–5423 CrossRef CAS.
- C. T. Huynh, M. K. Nguyen and D. S. Lee, Macromolecules, 2011, 44, 6629–6636 CrossRef CAS.
- J. Akimoto, M. Nakayama and T. Okano, J. Controlled Release, 2014, 193, 2–8 CrossRef CAS.
- M. Karimi, P. Sahandi Zangabad, A. Ghasemi, M. Amiri, M. Bahrami, H. Malekzad, H. Ghahramanzadeh Asl, Z. Mahdieh, M. Bozorgomid, A. Ghasemi, M. R. Rahmani Taji Boyuk and M. R. Hamblin, ACS Appl. Mater. Interfaces, 2016, 8, 21107–21133 CrossRef CAS.
- H. J. Moon, D. Y. Ko, M. H. Park, M. K. Joo and B. Jeong, Chem. Soc. Rev., 2012, 41, 4860–4883 RSC.
- J. M. Fuller, K. R. Raghupathi, R. R. Ramireddy, A. V. Subrahmanyam, V. Yesilyurt and S. Thayumanavan, J. Am. Chem. Soc., 2013, 135, 8947–8954 CrossRef CAS.
- D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214–7243 RSC.
- Q. Zhang, S. Dong, M. Zhang and F. Huang, Aggregate, 2021, 2, 35–47 CrossRef.
- L. D. Blackman, P. A. Gunatillake, P. Cass and K. E. S. Locock, Chem. Soc. Rev., 2019, 48, 757–770 RSC.
- A. Das and S. Ghosh, Angew. Chem., Int. Ed., 2014, 53, 1092–1097 CrossRef CAS.
- H. Yoshimitsu, E. Korchagina, A. Kanazawa, S. Kanaoka, F. M. Winnik and S. Aoshima, Polym. Chem., 2016, 7, 2062–2068 RSC.
- M. Naya, K. Kokado and K. Sada, ACS Appl. Polym. Mater., 2020, 2, 4415–4424 CrossRef CAS.
- M. Najafi, M. Habibi, R. Fokkink, W. E. Hennink and T. Vermonden, Soft Matter, 2021, 17, 2132–2141 RSC.
- K. R. Raghupathi, J. Guo, O. Munkhbat, P. Rangadurai and S. Thayumanavan, Acc. Chem. Res., 2014, 47, 2200–2211 CrossRef CAS.
- M. Shibata, T. Terashima and T. Koga, Macromolecules, 2021, 54, 5241–5248 CrossRef CAS.
- S. J. Byard, C. T. O’Brien, M. J. Derry, M. Williams, O. O. Mykhaylyk, A. Blanazs and S. P. Armes, Chem. Sci., 2020, 11, 396–402 RSC.
- K. Sou, L. Y. Chan and C.-L. K. Lee, ACS Sens., 2016, 1, 650–655 CrossRef CAS.
- L. Yu and J. Ding, Chem. Soc. Rev., 2008, 37, 1473–1481 RSC.
- N. Vanparijs, L. Nuhn and B. G. De Geest, Chem. Soc. Rev., 2017, 46, 1193–1239 RSC.
- H. Deng, A. Dong, J. Song and X. Chen, J. Controlled Release, 2019, 297, 60–70 CrossRef CAS.
- E. Haladjova, N. Toncheva-Moncheva, M. D. Apostolova, B. Trzebicka, A. Dworak, P. Petrov, I. Dimitrov, S. Rangelov and C. B. Tsvetanov, Biomacromolecules, 2014, 15, 4377–4395 CrossRef CAS.
- M. Patel, H. J. Lee, S. Park, Y. Kim and B. Jeong, Biomaterials, 2018, 159, 91–107 CrossRef CAS.
- Y. Zhang, J. Yu, K. Ren, J. Zuo, J. Ding and X. Chen, Biomacromolecules, 2019, 20, 1478–1492 CrossRef CAS.
- Z. H. Farooqi, S. R. Khan and R. Begum, Mater. Sci. Technol., 2017, 33, 129–137 CrossRef CAS.
- H. Wang, Q. Huang, H. Chang, J. Xiao and Y. Cheng, Biomater. Sci., 2016, 4, 375–390 RSC.
- Z. Al-Ahmady and K. Kostarelos, Chem. Rev., 2016, 116, 3883–3918 CrossRef CAS.
- D. Lu, H. Wang, T. Li, Y. Li, F. Dou, S. Sun, H. Guo, S. Liao, Z. Yang, Q. Wei and Z. Lei, ACS Appl. Mater. Interfaces, 2017, 9, 16756–16766 CrossRef CAS.
- Y. Seki, A. Kanazawa, S. Kanaoka, T. Fujiwara and S. Aoshima, Macromolecules, 2018, 51, 825–835 CrossRef CAS.
- M. Dai, E. Georgilis, G. Goudounet, B. Garbay, J. Pille, J. C. M. van Hest, X. Schultze, E. Garanger and S. Lecommandoux, Polymers, 2021, 13, 1470 CrossRef CAS.
- D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214–7243 RSC.
- S. J. Kim, M. H. Park, H. J. Moon, J. H. Park, D. Y. Ko and B. Jeong, ACS Appl. Mater. Interfaces, 2014, 6, 17034–17043 CrossRef CAS.
- A. Das, K. Petkau-Milroy, G. Klerks, B. van Genabeek, R. P. M. Lafleur, A. R. A. Palmans and E. W. Meijer, ACS Macro Lett., 2018, 7, 546–550 CrossRef CAS.
- H. J. Lee and B. Jeong, Small, 2020, 16, 1903045 CrossRef CAS.
- S. Cui, L. Yu and J. Ding, Macromolecules, 2019, 52, 3697–3715 CrossRef CAS.
- J. E. Nielsen, K. Zhu, S. A. Sande, L. Kováčik, D. Cmarko, K. D. Knudsen and B. Nyström, J. Phys. Chem. B, 2017, 121, 4885–4899 CrossRef CAS.
- M. A. Ward and T. K. Georgiou, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 2850–2859 CrossRef CAS.
- A. P. Constantinou, N. F. Sam-Soon, D. R. Carroll and T. K. Georgiou, Macromolecules, 2018, 51, 7019–7031 CrossRef CAS.
- A. P. Constantinou, H. Zhao, C. M. McGilvery, A. E. Porter and T. K. Georgiou, Polymers, 2017, 9, 31 CrossRef.
- H. J. Lee and B. Jeong, Small, 2020, 16, 1903045 CrossRef CAS.
- J. H. Hong, H. J. Lee and B. Jeong, ACS Appl. Mater. Interfaces, 2017, 9, 11568–11576 CrossRef CAS.
- N. Y. Steinman, N. Y. Bentolila and A. J. Domb, Polymers, 2020, 12, 2372 CrossRef CAS.
- S. Cui, L. Yu and J. Ding, Macromolecules, 2018, 51, 6405–6420 CrossRef CAS.
- X. Chang, C. Wang, G. Shan, Y. Bao and P. Pan, Langmuir, 2020, 36, 956–965 CrossRef CAS.
- D. A. Prince, I. J. Villamagna, C. C. Hopkins, J. R. de Bruyn and E. R. Gillies, Polym. Int., 2019, 68, 1074–1083 CrossRef CAS.
- O. Munkhbat, M. Garzoni, K. R. Raghupathi, G. M. Pavan and S. Thayumanavan, Langmuir, 2016, 32, 2874–2881 CrossRef CAS.
- M. Qiao, D. Chen, X. Ma and Y. Liu, Int. J. Pharm., 2005, 294, 103–112 CrossRef CAS.
- R. A. Graves, S. Pamujula, R. Moiseyev, T. Freeman, L. A. Bostanian and T. K. Mandal, Int. J. Pharm., 2004, 270, 251–262 CrossRef CAS.
- L. Lipp, D. Sharma, A. Banerjee and J. Singh, ACS Omega, 2019, 4, 1157–1166 CrossRef CAS.
- T. Maeda, M. Kitagawa and A. Hotta, Polym. Degrad. Stab., 2021, 187, 109535 CrossRef CAS.
- S. Cui, Y. Wei, Q. Bian, Y. Zhu, X. Chen, Y. Zhuang, M. Cai, J. Tang, L. Yu and J. Ding, ACS Appl. Mater. Interfaces, 2021, 13, 19778–19792 CrossRef CAS.
- S. Cui, L. Yu and J. Ding, Macromolecules, 2020, 53, 11051–11064 CrossRef CAS.
- D. Cao, X. Chen, F. Cao, W. Guo, J. Tang, C. Cai, S. Cui, X. Yang, L. Yu, Y. Su and J. Ding, Adv. Funct. Mater., 2021, 31, 1–12 Search PubMed.
- M. Patel, H. J. Lee, S. Son, H. Kim, J. Kim and B. Jeong, Biomacromolecules, 2020, 21, 143–151 CrossRef CAS.
- H. Kim, Y. Woo, M. Patel and B. Jeong, Biomacromolecules, 2020, 21, 3176–3185 CrossRef CAS.
- T. Maeda, M. Kitagawa, A. Hotta and S. Koizumi, Polymers, 2019, 11, 250 CrossRef.
- J. Sun, Y. Lei, Z. Dai, X. Liu, T. Huang, J. Wu, Z. P. Xu and X. Sun, ACS Appl. Mater. Interfaces, 2017, 9, 7990–7999 CrossRef CAS.
- J. Luan, S. Cui, J. Wang, W. Shen, L. Yu and J. Ding, Polym. Chem., 2017, 8, 2586–2597 RSC.
- Z.-K. Rao, R. Chen, H.-Y. Zhu, Y. Li, Y. Liu and J.-Y. Hao, Materials, 2018, 11, 338 CrossRef.
- Y. Kotsuchibashi, M. Ebara, T. Aoyagi and R. Narain, Polymers, 2016, 8, 380 CrossRef.
- Y. Zhu, J. M. Noy, A. B. Lowe and P. J. Roth, Polym. Chem., 2015, 6, 5705–5718 RSC.
- A. P. Constantinou, B. Zhan and T. K. Georgiou, Macromolecules, 2021, 54, 1943–1960 CrossRef CAS.
- Y. Zhang, S. Furyk, D. E. Bergbreiter and P. S. Cremer, J. Am. Chem. Soc., 2005, 127, 14505–14510 CrossRef CAS.
- Y. J. Kim and Y. T. Matsunaga, J. Mater. Chem. B, 2017, 5, 4307–4321 RSC.
- J. S. Scarpa, D. D. Mueller and I. M. Klotz, J. Am. Chem. Soc., 1967, 89, 6024–6030 CrossRef CAS.
- A. Halperin, M. Kröger and F. M. Winnik, Angew. Chem., Int. Ed., 2015, 54, 15342–15367 CrossRef CAS.
- S. Lanzalaco and E. Armelin, Gels, 2017, 3, 36 CrossRef.
- X. Xu, Y. Liu, W. Fu, M. Yao, Z. Ding, J. Xuan, D. Li, S. Wang, Y. Xia and M. Cao, Polymers, 2020, 12, 580 CrossRef CAS.
- K. Nagase, M. Yamato, H. Kanazawa and T. Okano, Biomaterials, 2018, 153, 27–48 CrossRef CAS.
- D. W. Dogo, H. Louis, N. I. Iliya, A. U. Ozioma, A. T. Aderemi and B. Stware, J. Med. Chem. Sci., 2019, 2, 162–171 CAS.
- M. Shahriari, V. P. Torchilin, S. M. Taghdisi, K. Abnous, M. Ramezani and M. Alibolandi, Biomater. Sci., 2020, 8, 5787–5803 RSC.
- M. I. Din, R. Khalid, F. Akbar, G. Ahmad, J. Najeeb and Z. U. Nisa Hussain, Soft Mater., 2018, 16, 228–247 CrossRef CAS.
- M. Sponchioni, U. Capasso Palmiero and D. Moscatelli, Mater. Sci. Eng., C, 2019, 102, 589–605 CrossRef CAS.
- A. Bordat, T. Boissenot, J. Nicolas and N. Tsapis, Adv. Drug Delivery Rev., 2019, 138, 167–192 CrossRef CAS.
- V. Kozlovskaya and E. Kharlampieva, ACS Appl. Polym. Mater., 2020, 2, 26–39 CrossRef CAS.
- J. Liu, A. Debuigne, C. Detrembleur and C. Jérôme, Adv. Healthcare Mater., 2014, 3, 1941–1968 CrossRef CAS.
- H. Vihola, A. Laukkanen, L. Valtola, H. Tenhu and J. Hirvonen, Biomaterials, 2005, 26, 3055–3064 CrossRef CAS.
- C. Fiedler, C. Ulbricht, T. Truglas, D. Wielend, M. Bednorz, H. Groiss, O. Brüggemann, I. Teasdale and Y. Salinas, Chem. – Eur. J., 2020, 3262–3267 Search PubMed.
- J. Stanojkovic, J. Oh, A. Khan and M. C. Stuparu, RSC Adv., 2020, 10, 2359–2363 RSC.
- M. Kasprów, J. MacHnik, Ł. Otulakowski, A. Dworak and B. Trzebicka, RSC Adv., 2019, 9, 40966–40974 RSC.
- G. Vancoillie, D. Frank and R. Hoogenboom, Prog. Polym. Sci., 2014, 39, 1074–1095 CrossRef CAS.
- Z. Hu, T. Cai and C. Chi, Soft Matter, 2010, 6, 2115–2123 RSC.
- R. Hoogenboom and H. Schlaad, Polym. Chem., 2017, 8, 24–40 RSC.
- B. Verbraeken, B. D. Monnery, K. Lava and R. Hoogenboom, Eur. Polym. J., 2017, 88, 451–469 CrossRef CAS.
- E. Haladjova, S. Rangelov and C. Tsvetanov, Polymers, 2020, 12, 2609 CrossRef CAS.
- K. K. Bansal, P. K. Upadhyay, G. K. Saraogi, A. Rosling and J. M. Rosenholm, eXPRESS Polym. Lett., 2019, 13, 974–992 CrossRef CAS.
- P. Narang and P. Venkatesu, Adv. Colloid Interface Sci., 2019, 274, 102042 CrossRef CAS.
- P. Banerjee, M. Anas, S. Jana and T. K. Mandal, J. Polym. Res., 2020, 27, 177 CrossRef CAS.
- V. Hildebrand, A. Laschewsky, M. Päch, P. Müller-Buschbaum and C. M. Papadakis, Polym. Chem., 2017, 8, 310–322 RSC.
- G. Luo, Y. Guo, C. Liu, G. Han, X. Ma and W. Zhang, RSC Adv., 2019, 9, 12936–12943 RSC.
- E. M. Frazar, R. A. Shah, T. D. Dziubla and J. Z. Hilt, J. Appl. Polym. Sci., 2020, 137, 1–14 CrossRef.
- A. Asadujjaman, B. Kent and A. Bertin, Soft Matter, 2017, 13, 658–669 RSC.
- M. Kuepfert, E. Ahmed and M. Weck, Macromolecules, 2021, 54, 3845–3853 CrossRef CAS.
- H. Willcock, A. Lu, C. F. Hansell, E. Chapman, I. R. Collins and R. K. O’Reilly, Polym. Chem., 2014, 5, 1023–1030 RSC.
- S. Kuroyanagi, N. Shimada, S. Fujii, T. Furuta, A. Harada, K. Sakurai and A. Maruyama, J. Am. Chem. Soc., 2019, 141, 1261–1268 CrossRef CAS.
- Q. Zhong, C. Chen, L. Mi, J. P. Wang, J. Yang, G. P. Wu, Z. K. Xu, R. Cubitt and P. Müller-Buschbaum, Langmuir, 2020, 36, 742–753 CrossRef CAS.
- M. Glassner, M. Vergaelen and R. Hoogenboom, Polym. Int., 2018, 67, 32–45 CrossRef CAS.
- F. Hofmeister, Arch. für Exp. Pathol. und Pharmakologie, 1888, 24, 247–260 CrossRef.
- R. L. Baldwin, Biophys. J., 1996, 71, 2056–2063 CrossRef CAS.
- Y. Zhang, S. Furyk, L. B. Sagle, Y. Cho, D. E. Bergbreiter and P. S. Cremer, J. Phys. Chem. C, 2007, 111, 8916–8924 CrossRef CAS.
- J. P. Magnusson, A. Khan, G. Pasparakis, A. O. Saeed, W. Wang and C. Alexander, J. Am. Chem. Soc., 2008, 130, 10852–10853 CrossRef CAS.
- S. Z. Moghaddam and E. Thormann, J. Colloid Interface Sci., 2016, 465, 67–75 CrossRef CAS.
- M. Schroffenegger, R. Zirbs, S. Kurzhals and E. Reimhult, Polymers, 2018, 10, 451 CrossRef.
- D. Dupont, D. Depuydt and K. Binnemans, J. Phys. Chem. B, 2015, 119, 6747–6757 CrossRef CAS.
- J. Seuring and S. Agarwal, ACS Macro Lett., 2013, 2, 597–600 CrossRef CAS.
- Y. Hiruta, Y. Nagumo, Y. Suzuki, T. Funatsu, Y. Ishikawa and H. Kanazawa, Colloids Surf., B, 2015, 132, 299–304 CrossRef CAS.
- E. E. Bruce, P. T. Bui, B. A. Rogers, P. S. Cremer and N. F. A. Van Der Vegt, J. Am. Chem. Soc., 2019, 141, 6609–6616 CrossRef CAS.
- H. Robertson, E. C. Johnson, I. J. Gresham, S. W. Prescott, A. Nelson, E. J. Wanless and G. B. Webber, J. Colloid Interface Sci., 2021, 586, 292–304 CrossRef CAS.
- L. Li, J. H. Ryu and S. Thayumanavan, Langmuir, 2013, 29, 50–55 CrossRef CAS.
- K. Shiomori, A. E. Ivanov, I. Y. Galaev, Y. Kawano and B. Mattiasson, Macromol. Chem. Phys., 2004, 205, 27–34 CrossRef CAS.
- A. D. Drozdov and J. Declaville Christiansen, RSC Adv., 2020, 10, 30723–30733 RSC.
- C. Hofmann and M. Schönhoff, Colloid Polym. Sci., 2009, 287, 1369–1376 CrossRef CAS.
- G. Han, J. T. Wang, X. Ji, L. Liu and H. Zhao, Bioconjugate Chem., 2017, 28, 636–641 CrossRef CAS.
- D. Huang, Q. Zhang, Y. Deng, Z. Luo, B. Li, X. Shen, Z. Qi, S. Dong, Y. Ge and W. Chen, Polym. Chem., 2018, 9, 2574–2579 RSC.
- W. M. Wan, F. Cheng and F. Jäkle, Angew. Chem., Int. Ed., 2014, 53, 8934–8938 CrossRef CAS.
- K. Fuchise, R. Kakuchi, S.-T. Lin, R. Sakai, S.-I. Sato, T. Satoh, W.-C. Chen and T. Kakuchi, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6259–6268 CrossRef CAS.
- G. Mellot, J. M. Guigner, J. Jestin, L. Bouteiller, F. Stoffelbach and J. Rieger, J. Colloid Interface Sci., 2021, 581, 874–883 CrossRef CAS.
- C. Park, J. Lee and C. Kim, Chem. Commun., 2011, 47, 12042–12056 RSC.
- D. Astruc, E. Boisselier and C. Ornelas, Chem. Rev., 2010, 110, 1857–1959 CrossRef CAS.
- M. R. Molla, P. Rangadurai, G. M. Pavan and S. Thayumanavan, Nanoscale, 2015, 7, 3817–3837 RSC.
- Q. Zheng and C. Pan, Eur. Polym. J., 2006, 42, 807–814 CrossRef CAS.
- Y. Zhao, X. Fan, D. Liu and Z. Wang, Int. J. Pharm., 2011, 409, 229–236 CrossRef CAS.
- J. H. Kim, E. Lee, J. S. Park, K. Kataoka and W. D. Jang, Chem. Commun., 2012, 48, 3662–3664 RSC.
- X. Li, Y. Haba, K. Ochi, E. Yuba, A. Harada and K. Kono, Bioconjugate Chem., 2013, 24, 282–290 CrossRef CAS.
- K. Kono, E. Murakami, Y. Hiranaka, E. Yuba, C. Kojima, A. Harada and K. Sakurai, Angew. Chem., Int. Ed., 2011, 50, 6332–6336 CrossRef CAS.
- K. Kono, T. Miyoshi, Y. Haba, E. Murakami, C. Kojima and A. Harada, J. Am. Chem. Soc., 2007, 129, 7222–7223 CrossRef CAS.
- T. Koga, M. Iimura and N. Higashi, Macromol. Biosci., 2012, 12, 1043–1047 CrossRef CAS.
- C. Kojima, S. Tsumura, A. Harada and K. Kono, J. Am. Chem. Soc., 2009, 131, 6052–6053 CrossRef CAS.
- Y. Shen, X. Ma, B. Zhang, Z. Zhou, Q. Sun, E. Jin, M. Sui, J. Tang, J. Wang and M. Fan, Chem. – Eur. J., 2011, 17, 5319–5326 CrossRef CAS.
- D. W. Chang and L. Dai, J. Mater. Chem., 2007, 17, 364–371 RSC.
- W. Li, A. Zhang, Y. Chen, K. Feldman, H. Wu and A. D. Schlüter, Chem. Commun., 2008, 5948–5950 RSC.
- S. Ghaeini-Hesaroeiye, H. R. Razmi Bagtash, S. Boddohi, E. Vasheghani-Farahani and E. Jabbari, Gels, 2020, 6, 20 CrossRef CAS.
- K. Kono, Polym. J., 2012, 44, 531–540 CrossRef CAS.
- M. Kimura, M. Kato, T. Muto, K. Hanabusa and H. Shirai, Macromolecules, 2000, 33, 1117–1119 CrossRef CAS.
- H. Hui, F. Xiao-dong and C. Zhong-lin, Polymer, 2005, 46, 9514–9522 CrossRef.
- P. Bharathi, H. Zhao and S. Thayumanavan, Org. Lett., 2001, 3, 1961–1964 CrossRef CAS.
- M. A. Azagarsamy, P. Sokkalingam and S. Thayumanavan, J. Am. Chem. Soc., 2009, 131, 14184–14185 CrossRef CAS.
- T. M. Koyasseril-Yehiya, A. García-Heredia, F. Anson, P. Rangadurai, M. S. Siegrist and S. Thayumanavan, Nanoscale, 2020, 12, 20693–20698 RSC.
- S. V. Aathimanikandan, E. N. Savariar and S. Thayumanavan, J. Am. Chem. Soc., 2005, 127, 14922–14929 CrossRef CAS.
- S. Jiwpanich, J.-H. Ryu, S. Bickerton and S. Thayumanavan, J. Am. Chem. Soc., 2010, 132, 10683–10685 CrossRef CAS.
- J.-H. Lee, Y.-G. Kim, H. S. Cho, J. Kim, S.-C. Kim, M. H. Cho and J. Lee, Biofouling, 2014, 30, 627–637 CrossRef CAS.
- A. Inada, K. Yumiya, K. Kumagai and H. Matsuyama, Colloids Surf., A, 2021, 609, 125659 CrossRef CAS.
- J. Shan, Y. Zhao, N. Granqvist and H. Tenhu, Macromolecules, 2009, 42, 2696–2701 CrossRef CAS.
- Y. G. Takei, T. Aoki, K. Sanui, N. Ogata, T. Okano and Y. Sakurai, Bioconjugate Chem., 1993, 4, 42–46 CrossRef CAS.
- F. Wang, A. Klaikherd and S. Thayumanavan, J. Am. Chem. Soc., 2011, 133, 13496–13503 CrossRef CAS.
- K. R. Raghupathi, U. Sridhar, K. Byrne, K. Raghupathi and S. Thayumanavan, J. Am. Chem. Soc., 2015, 137, 5308–5311 CrossRef CAS.
- H. Liu, C. Lionello, J. Westley, A. Cardellini, U. Huynh, G. Pavan and S. Thayumanavan, Nanoscale, 2021, 13, 11568–11575 RSC.
- N. Saito, H. Kobayashi and M. Yamaguchi, Chem. Sci., 2016, 7, 3574–3580 RSC.
- G. Xu, K. Liu, B. Xu, Y. Yao, W. Li, J. Yan and A. Zhang, Macromol. Rapid Commun., 2020, 41, 2000325 CrossRef CAS.
- W. Li, A. Zhang, K. Feldman, P. Walde and A. D. Schlüter, Macromolecules, 2008, 41, 3659–3667 CrossRef CAS.
- M. Qi, K. Li, Y. Zheng, T. Rasheed and Y. Zhou, Langmuir, 2018, 34, 3058–3067 CrossRef CAS.
- M. L. Ohnsorg, P. C. Prendergast, L. L. Robinson, M. R. Bockman, F. S. Bates and T. M. Reineke, ACS Macro Lett., 2021, 10, 375–381 CrossRef CAS.
- Q. Zhang, C. Weber, U. S. Schubert and R. Hoogenboom, Mater. Horiz., 2017, 4, 109–116 RSC.
- D. Roy, J. N. Cambre and B. S. Sumerlin, Prog. Polym. Sci., 2010, 35, 278–301 CrossRef CAS.
- J. Hu and S. Liu, Macromolecules, 2010, 43, 8315–8330 CrossRef CAS.
- F. D. Jochum and P. Theato, Chem. Soc. Rev., 2013, 42, 7468–7483 RSC.
- W. Li, D. Wu, A. D. Schlüter and A. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 6630–6640 CrossRef CAS.
- W. Li, A. Zhang and A. D. Schlüter, Chem. Commun., 2008, 5523–5525 RSC.
- X. Zhang, W. Li, X. Zhao and A. Zhang, Macromol. Rapid Commun., 2013, 34, 1701–1707 CrossRef CAS.
- B. Wang, T. Xiao, X.-B. Fu, T.-T. Jiang, Y. Chen and Y.-F. Yao, Macromolecules, 2017, 50, 9647–9655 CrossRef CAS.
- S. Sun, H. Wang and P. Wu, Soft Matter, 2013, 9, 2878–2888 RSC.
- M. A. Boerman, H. L. Van der Laan, J. C. M. E. Bender, R. Hoogenboom, J. A. Jansen, S. C. Leeuwenburgh and J. C. M. Van Hest, J. Polym. Sci., Part A: Polym. Chem., 2016, 54, 1573–1582 CrossRef CAS.
- W. Fang, R. Zhang, Y. Yao, H. Liu and Y. Chen, Chin. J. Polym. Sci., 2017, 35, 1035–1042 CrossRef CAS.
- Y. Zhang, R.-C. Wang, H.-J. Liu and Y. Chen, Soft Matter, 2017, 13, 8136–8143 RSC.
- J. E. Gagner, W. Kim and E. L. Chaikof, Acta Biomater., 2014, 10, 1542–1557 CrossRef CAS.
- C. Liu, Q. Zhang, S. Zhu, H. Liu and J. Chen, RSC Adv., 2019, 9, 28299–28311 RSC.
- T. Kowalczyk, K. Hnatuszko-Konka, A. Gerszberg and A. K. Kononowicz, World J. Microbiol. Biotechnol., 2014, 30, 2141–2152 CrossRef CAS.
- A. Ribeiro, F. J. Arias, J. Reguera, M. Alonso and J. C. Rodríguez-Cabello, Biophys. J., 2009, 97, 312–320 CrossRef CAS.
- D. W. Urry, J. Phys. Chem. B, 1997, 101, 11007–11028 CrossRef CAS.
- S. Roberts, M. Dzuricky and A. Chilkoti, FEBS Lett., 2015, 589, 2477–2486 CrossRef CAS.
- D. W. Urry, C. H. Luan, T. M. Parker, D. C. Gowda, K. U. Prasad, M. C. Reid and A. Safavy, J. Am. Chem. Soc., 1991, 113, 4346–4348 CrossRef CAS.
- D. E. Meyer and A. Chilkoti, Biomacromolecules, 2004, 5, 846–851 CrossRef CAS.
- H. Nuhn and H.-A. Klok, Biomacromolecules, 2008, 9, 2755–2763 CrossRef CAS.
- R. L. M. Teeuwen, F. A. de Wolf, H. Zuilhof and J. C. M. van Hest, Soft Matter, 2009, 5, 4305–4310 RSC.
- F. G. Quiroz and A. Chilkoti, Nat. Mater., 2015, 14, 1164–1171 CrossRef CAS.
- Y. Cho, Y. Zhang, T. Christensen, L. B. Sagle, A. Chilkoti and P. S. Cremer, J. Phys. Chem. B, 2008, 112, 13765–13771 CrossRef CAS.
- J. E. Condon, T. B. Martin and A. Jayaraman, Soft Matter, 2017, 13, 2907–2918 RSC.
- D. W. Urry, D. C. Gowda, T. M. Parker, C.-H. Luan, M. C. Reid, C. M. Harris, A. Pattanaik and R. D. Harris, Biopolymers, 1992, 32, 1243–1250 CrossRef CAS.
- D. W. Urry, Prog. Biophys. Mol. Biol., 1992, 57, 23–57 CrossRef CAS.
- W. Jeong, S. Hyun Kwon and Y. Lim, Adv. Funct. Mater., 2018, 28, 1803114 CrossRef.
- F. Aladini, C. Araman and C. F. W. Becker, J. Pept. Sci., 2016, 22, 334–342 CrossRef CAS.
- T. Luo and K. L. Kiick, J. Am. Chem. Soc., 2015, 137, 15362–15365 CrossRef CAS.
- B. Zhao, N. K. Li, Y. G. Yingling and C. K. Hall, Biomacromolecules, 2016, 17, 111–118 CrossRef CAS.
- N. K. Li, S. Roberts, F. G. Quiroz, A. Chilkoti and Y. G. Yingling, Biomacromolecules, 2018, 19, 2496–2505 CrossRef CAS.
- M. Pechar, J. Brus, L. Kostka, Č. Koňák, M. Urbanová and M. Šlouf, Macromol. Biosci., 2007, 7, 56–69 CrossRef CAS.
- J. S. Cobb, A. Engel, M. A. Seale and A. V. Janorkar, Sci. Rep., 2021, 11, 6343 CrossRef CAS.
- S. Saha, S. Banskota, S. Roberts, N. Kirmani and A. Chilkoti, Adv. Ther., 2020, 3, 1900164 CrossRef.
- P. M. Cowan, S. McGavin and A. C. T. North, Nature, 1955, 176, 1062–1064 CrossRef CAS.
- M. Kitamura, S. Kakinoki, Y. Hirano and M. Oka, Polym. Bull., 2005, 54, 303–310 CrossRef CAS.
- M. Kitamura, T. Yamauchi, M. Oka and T. Hayashi, Polym. Bull., 2003, 51, 143–150 CrossRef CAS.
- F. Chen, X. Zhang, W. Li, K. Liu, Y. Guo, J. Yan and A. Zhang, Soft Matter, 2012, 8, 4869–4872 RSC.
- X. Zhang, W. Li, X. Zhao and A. Zhang, Macromol. Rapid Commun., 2013, 34, 1701–1707 CrossRef CAS.
- V. A. Raghunathan and J. Katsaras, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 1996, 54, 4446–4449 CrossRef CAS.
- D. Needham, J.-Y. Park, A. M. Wright and J. Tong, Faraday Discuss., 2013, 161, 515–589 RSC.
- M. H. Chiu and E. J. Prenner, J. Pharm. BioAllied Sci., 2011, 3, 39–59 CrossRef CAS.
- W. Helfrich, Zeitschrift für Naturforsch. C, 1973, 28, 693–703 CrossRef CAS.
- V. A. Raghunathan and J. Katsaras, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 1996, 54, 4446–4449 CrossRef CAS.
- J. Chen, D. Cheng, J. Li, Y. Wang, J. Guo, Z. Chen, B. Cai and T. Yang, Drug Dev. Ind. Pharm., 2013, 39, 197–204 CrossRef CAS PubMed.
- W. Kunz, F. Testard and T. Zemb, Langmuir, 2009, 25, 112–115 CrossRef CAS PubMed.
- C. Arnarez, J. J. Uusitalo, M. F. Masman, H. I. Ingólfsson, D. H. de Jong, M. N. Melo, X. Periole, A. H. de Vries and S. J. Marrink, J. Chem. Theory Comput., 2015, 11, 260–275 CrossRef CAS.
- M. Zein and R. Winter, Phys. Chem. Chem. Phys., 2000, 2, 4545–4551 RSC.
- S. Leekumjorn, H. J. Cho, Y. Wu, N. T. Wright, A. K. Sum and C. Chan, Biochim. Biophys. Acta, Biomembr., 2009, 1788, 1508–1516 CrossRef CAS.
- A. Arouri and O. G. Mouritsen, Prog. Lipid Res., 2013, 52, 130–140 CrossRef CAS.
- J. K. Mills and D. Needham, Biochim. Biophys. Acta, Biomembr., 2005, 1716, 77–96 CrossRef CAS.
- N. Fuller and R. P. Rand, Biophys. J., 2001, 81, 243–254 CrossRef CAS.
- M. C. Sandström, E. Johansson and K. Edwards, Biophys. Chem., 2008, 132, 97–103 CrossRef.
- L. M. Ickenstein, M. C. Sandström, L. D. Mayer and K. Edwards, Biochim. Biophys. Acta, Biomembr., 2006, 1758, 171–180 CrossRef CAS.
- M. Kastantin, B. Ananthanarayanan, P. Karmali, E. Ruoslahti and M. Tirrell, Langmuir, 2009, 25, 7279–7286 CrossRef CAS.
- L. Viitala, S. Pajari, L. Gentile, J. Määttä, M. Gubitosi, J. Deska, M. Sammalkorpi, U. Olsson and L. Murtomäki, Langmuir, 2019, 35, 3999–4010 CrossRef CAS.
- N. Sadeghi, R. Deckers, B. Ozbakir, S. Akthar, R. J. Kok, T. Lammers and G. Storm, Int. J. Pharm., 2018, 548, 778–782 CrossRef CAS.
- A. Ruiz, G. Ma, J. Seitsonen, S. G. T. Pereira, J. Ruokolainen and W. T. Al-Jamal, J. Controlled Release, 2020, 328, 665–678 CrossRef CAS.
- Z. Al-Ahmady and K. Kostarelos, Chem. Rev., 2016, 116, 3883–3918 CrossRef CAS PubMed.
- M. Wang and Y. Cheng, Bioconjugate Chem., 2016, 27, 495–499 CrossRef CAS.
- W. Shen, X. Chen, J. Luan, D. Wang, L. Yu and J. Ding, ACS Appl. Mater. Interfaces, 2017, 9, 40031–40046 CrossRef CAS PubMed.
- K. Dutta, R. Das, J. Ling, R. M. Monibas, E. Carballo-Jane, A. Kekec, D. D. Feng, S. Lin, J. Mu, R. Saklatvala, S. Thayumanavan and Y. Liang, ACS Omega, 2020, 5, 17531–17542 CrossRef CAS PubMed.
- P. Soltantabar, E. L. Calubaquib, E. Mostafavi, M. C. Biewer and M. C. Stefan, Biomacromolecules, 2020, 21, 1427–1436 CrossRef CAS PubMed.
- S. Xu, L. Yin, Y. Xiang, H. Deng, L. Deng, H. Fan, H. Tang, J. Zhang and A. Dong, Macromol. Biosci., 2016, 16, 1188–1199 CrossRef CAS.
- M. Deloney, K. Smart, B. A. Christiansen and A. Panitch, J. Controlled Release, 2020, 323, 47–58 CrossRef CAS PubMed.
- J. Quan, F. W. Shen, H. Cai, Y. N. Zhang and H. Wu, Langmuir, 2018, 34, 10721–10731 CrossRef CAS.
- D. Wang, H. Huang, M. Zhou, H. Lu, J. Chen, Y. T. Chang, J. Gao, Z. Chai and Y. Hu, Chem. Commun., 2019, 55, 4051–4054 RSC.
- L. Ruan, M. Zhou, J. Chen, H. Huang, J. Zhang, H. Sun, Z. Chai and Y. Hu, Chem. Commun., 2019, 55, 14645–14648 RSC.
- A. Bordat, N. Soliman, I. Ben Chraït, K. Manerlax, N. Yagoubi, T. Boissenot, J. Nicolas and N. Tsapis, Eur. J. Pharm. Biopharm., 2019, 142, 281–290 CrossRef CAS PubMed.
- N. Xu, X. Huang, G. Yin, M. Bu, X. Pu, X. Chen, X. Liao and Z. Huang, RSC Adv., 2018, 8, 15604–15612 RSC.
- F. Doberenz, K. Zeng, C. Willems, K. Zhang and T. Groth, J. Mater. Chem. B, 2020, 8, 607–628 RSC.
- E. J. Al Dine, S. Marchal, R. Schneider, B. Hamie, J. Ghanbaja, T. Roques-Carmes, T. Hamieh, J. Toufaily, E. Gaffet and H. Alem, Bioconjugate Chem., 2018, 29, 2248–2256 CrossRef CAS.
- S. A. Jadhav and D. Scalarone, Aust. J. Chem., 2018, 71, 477–481 CrossRef CAS.
- T. Alejo, V. Andreu, G. Mendoza, V. Sebastian and M. Arruebo, J. Colloid Interface Sci., 2018, 523, 234–244 CrossRef CAS PubMed.
- E. J. Sayers, J. P. Magnusson, P. R. Moody, F. Mastrotto, C. Conte, C. Brazzale, P. Borri, P. Caliceti, P. Watson, G. Mantovani, J. Aylott, S. Salmaso, A. T. Jones and C. Alexander, Bioconjugate Chem., 2018, 29, 1030–1046 CrossRef CAS PubMed.
- M. Kuna, F. Mahdi, A. R. Chade and G. L. Bidwell, Sci. Rep., 2018, 8, 7923 CrossRef PubMed.
- D. W. Lim, D. L. Nettles, L. A. Setton and A. Chilkoti, Biomacromolecules, 2007, 8, 1463–1470 CrossRef CAS.
- Y.-N. Zhang, R. K. Avery, Q. Vallmajo-Martin, A. Assmann, A. Vegh, A. Memic, B. D. Olsen, N. Annabi and A. Khademhosseini, Adv. Funct. Mater., 2015, 25, 4814–4826 CrossRef CAS PubMed.
- Y. Mizuguchi, Y. Mashimo, M. Mie and E. Kobatake, Biomacromolecules, 2020, 21, 1126–1135 CrossRef CAS.
- L. Ruan, W. Chen, R. Wang, J. Lu and J. I. Zink, ACS Appl. Mater. Interfaces, 2019, 11, 43835–43842 CrossRef CAS PubMed.
- Z. Shi, C. Yang, R. Li and L. Ruan, J. Mater. Sci., 2020, 55, 6118–6129 CrossRef CAS.
- J. Despanie, J. P. Dhandhukia, S. F. Hamm-Alvarez and J. A. MacKay, J. Controlled Release, 2016, 240, 93–108 CrossRef CAS.
- A. Varanko, S. Saha and A. Chilkoti, Adv. Drug Delivery Rev., 2020, 156, 133–187 CrossRef CAS PubMed.
- E. E. Fletcher, D. Yan, A. A. Kosiba, Y. Zhou and H. Shi, Protein Expr. Purif., 2019, 153, 114–120 CrossRef CAS PubMed.
- P. S. Zangabad, S. Mirkiani, S. Shahsavari, B. Masoudi, M. Masroor, H. Hamed, Z. Jafari, Y. D. Taghipour, H. Hashemi, M. Karimi and M. R. Hamblin, Nanotechnol. Rev., 2018, 7, 95–122 CrossRef CAS PubMed.
- H. Bi, J. Xue, H. Jiang, S. Gao, D. Yang, Y. Fang and K. Shi, Asian J. Pharm. Sci., 2019, 14, 365–379 CrossRef PubMed.
- J. K. Mills and D. Needham, Methods Enzymol., 2004, 387, 82–113 CAS.
- B. Almeida, O. K. Nag, K. E. Rogers and J. B. Delehanty, Molecules, 2020, 25, 5672 CrossRef CAS PubMed.
- E. Mazzotta, L. Tavano and R. Muzzalupo, Pharmaceutics, 2018, 10, 150 CrossRef CAS.
- D. Haemmerich and A. Motamarry, Adv. Cancer Res., 2018, 139, 121–146 CrossRef CAS PubMed.
- S. Li, G. Yin, X. Pu, Z. Huang, X. Liao and X. Chen, Int. J. Pharm., 2019, 570, 118660 CrossRef CAS.
- N. Naziris, A. Skandalis, A. Forys, B. Trzebicka, S. Pispas and C. Demetzos, J. Therm. Anal. Calorim., 2020, 141, 751–766 CrossRef CAS.
- J. R. Chong, D. L. Le, H. Sato and K. Sou, ACS Appl. Mater. Interfaces, 2020, 12, 21463–21469 CrossRef CAS PubMed.
- B. S. Pattni, V. V. Chupin and V. P. Torchilin, Chem. Rev., 2015, 115, 10938–10966 CrossRef CAS PubMed.
- J. Li, X. Wang, T. Zhang, C. Wang, Z. Huang, X. Luo and Y. Deng, Asian J. Pharm. Sci., 2015, 10, 81–98 CrossRef.
- M. Amin, W. Huang, A. L. B. Seynhaeve and T. L. M. Ten Hagen, Pharmaceutics, 2020, 12, 1007 CrossRef CAS.
- A. Blanazs, R. Verber, O. O. Mykhaylyk, A. J. Ryan, J. Z. Heath, C. W. I. Douglas and S. P. Armes, J. Am. Chem. Soc., 2012, 134, 9741–9748 CrossRef CAS.
- I. Canton, N. J. Warren, A. Chahal, K. Amps, A. Wood, R. Weightman, E. Wang, H. Moore and S. P. Armes, ACS Cent. Sci., 2016, 2, 65–74 CrossRef CAS PubMed.
- D. Y. Ko, M. Patel, H. J. Lee and B. Jeong, Adv. Funct. Mater., 2018, 28, 1706286 CrossRef.
- X. Yan, W.-W. Fang, J. Xue, T.-C. Sun, L. Dong, Z. Zha, H. Qian, Y.-H. Song, M. Zhang, X. Gong, Y. Lu and T. He, ACS Nano, 2019, 13, 10074–10084 CrossRef CAS PubMed.
- Y. Sudo, R. Kawai, H. Sakai, R. Kikuchi, Y. Nabae, T. Hayakawa and M. A. Kakimoto, Langmuir, 2018, 34, 653–662 CrossRef CAS.
- K. Nagase, A. Ota, T. Hirotani, S. Yamada, A. M. Akimoto and H. Kanazawa, Macromol. Rapid Commun., 2020, 41, 1–7 CrossRef.
- S. Jiang, M. Müller and H. Schönherr, Angew. Chem., Int. Ed., 2019, 58, 10563–10566 CrossRef CAS.
- N. P. Williams, M. Rhodehamel, C. Yan, A. S. T. Smith, A. Jiao, C. E. Murry, M. Scatena and D. H. Kim, Biomaterials, 2020, 240, 119856 CrossRef CAS PubMed.
- K. J. Hogan and A. G. Mikos, Polymer, 2020, 211, 123063 CrossRef CAS.
- L. J. Twyman, A. S. H. King and I. K. Martin, Chem. Soc. Rev., 2002, 31, 69–82 RSC.
- D. Astruc and F. Chardac, Chem. Rev., 2001, 101, 2991–3024 CrossRef CAS.
- E. Karakhanov, A. Maximov, A. Zolotukhina, Y. Kardasheva and M. Talanova, J. Inorg. Organomet. Polym. Mater., 2016, 26, 1264–1279 CrossRef CAS.
- K. Zheng, J. Ren and J. He, Macromolecules, 2019, 52, 6780–6791 CrossRef CAS.
- T. Chen, Z. Xu, L. Zhou, J. Qiu, M. Wang and J. Wang, Mol. Catal, 2019, 474, 110422 CrossRef CAS.
- T. Chen, S. Zhang, L. Hua, Z. Xu, L. Zhou and J. Wang, Macromol. Res., 2019, 27, 931–937 CrossRef CAS.
- I. Mukherjee, S. K. Sinha, S. Datta and P. De, Biomacromolecules, 2018, 19, 2286–2293 CrossRef CAS PubMed.
- L. L. Lou, H. Qu, W. Yu, B. Wang, L. Ouyang, S. Liu and W. Zhou, ChemCatChem, 2018, 10, 1166–1172 CrossRef CAS.
- F. Li, Q. Gao, M. Yang and W. Guo, Langmuir, 2018, 34, 14932–14939 CrossRef CAS.
- J. Tian, B. Huang and W. Zhang, Langmuir, 2019, 35, 266–275 CrossRef CAS PubMed.
- N. Luisier, K. Schenk and K. Severin, Chem. Commun., 2014, 50, 10233–10236 RSC.
- R. J. Wojtecki, M. A. Meador and S. J. Rowan, Nat. Mater., 2011, 10, 14–27 CrossRef CAS.
- A. W. Jackson and D. A. Fulton, Polym. Chem., 2013, 4, 31–45 RSC.
- Z. Wang, L. Guo, H. Xiao, H. Cong and S. Wang, Mater. Horiz., 2020, 7, 282–288 RSC.
- Y. Amamoto, M. Kikuchi, H. Masunaga, S. Sasaki, H. Otsuka and A. Takahara, Macromolecules, 2010, 43, 1785–1791 CrossRef CAS.
- K. Zhang, X. Feng, C. Ye, M. A. Hempenius and G. J. Vancso, J. Am. Chem. Soc., 2017, 139, 10029–10035 CrossRef CAS PubMed.
- X. Zhang, K. Liu, J. Liu, Y. Ding, W. Li and A. Zhang, Eur. Polym. J., 2020, 141, 110092 CrossRef CAS.
- B. Maiti, A. Abramov, L. Franco, J. Puiggalí, H. Enshaei, C. Alemán and D. D. Díaz, Adv. Funct. Mater., 2020, 30, 1–10 Search PubMed.
- J. He, G. Xiao, X. Chen, Y. Qiao, D. Xu and Z. Lu, RSC Adv., 2019, 9, 23957–23963 RSC.
- L. Sambe, V. R. Delarosa, K. Belal, F. Stoffelbach, J. Lyskawa, F. Delattre, M. Bria, G. Cooke, R. Hoogenboom and P. Woisel, Angew. Chem., Int. Ed., 2014, 53, 5044–5048 CrossRef CAS PubMed.
- Z. S. Huang, J. W. Shiu, T. F. Way and S. P. Rwei, Polymer, 2019, 184, 121917 CrossRef CAS.
- M. Baert, K. Wicht, Z. Hou, R. Szucs, F. Du Prez and F. Lynen, Anal. Chem., 2020, 92, 9815–9822 CrossRef CAS PubMed.
- E. Katz, Electroanalysis, 2016, 28, 1916–1929 CrossRef CAS.
- I. Tan, F. Roohi and M. M. Titirici, Anal. Methods, 2012, 4, 34–43 RSC.
- Z. Wang, J. Guo, X. Liu, J. Sun and W. Gao, J. Controlled Release, 2020, 328, 444–453 CrossRef PubMed.
Footnote |
| † Dedicated to Professor Seth R Marder on the occasion of his 60th birthday. |
| This journal is © The Royal Society of Chemistry 2022 |