
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReactivities of hydrated electrons with organic compounds in aqueous-phase advanced reduction processes†
Rose
Daily
and
Daisuke
Minakata
 *
*
Department of Civil, Environmental, and Geospatial Engineering, Michigan Technological University, 1400 Townsend Drive, Houghton, MI 49931, USA. E-mail: dminakat@mtu.edu; Fax: +1 906 487 2943; Tel: +1 906 487 1830
First published on 28th January 2022
Abstract
Advanced reduction processes (ARPs) that generate reactive electrons in homogeneous solution and heterogeneous electrochemical or catalytic processes are effective in degrading oxidized forms of organic and inorganic contaminants. However, the detailed mechanisms of compounds with multiple functional groups and the effect of those functional groups on the reactivities of these compounds toward electrons have not been elucidated. In this study, we use density functional theory to calculate the aqueous-phase one electron reduction potential  of 251 conventional organic compounds containing a wide variety of functional groups. We investigate three possible elementary reaction mechanisms, namely, the associative, concerted and stepwise cleavage mechanisms, at all possible reactive sites and determine the linear free energy relationships (LFERs) between the experimentally measured rate constants of hydrated electrons (eaq−) and the
of 251 conventional organic compounds containing a wide variety of functional groups. We investigate three possible elementary reaction mechanisms, namely, the associative, concerted and stepwise cleavage mechanisms, at all possible reactive sites and determine the linear free energy relationships (LFERs) between the experimentally measured rate constants of hydrated electrons (eaq−) and the 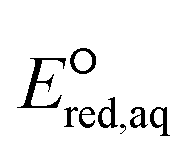 values. In addition, we use the 75 priority per- and polyfluoroalkyl substance (PFAS) subsets from the United States Environmental Protection Agency (U.S. EPA) to calculate the
values. In addition, we use the 75 priority per- and polyfluoroalkyl substance (PFAS) subsets from the United States Environmental Protection Agency (U.S. EPA) to calculate the  values of all possible elementary reactions of each PFAS to determine their dominant reaction mechanisms and reactive sites. LFERs of conventional organic compounds are used to predict the reactivities of eaq− with PFASs, which can be used as a screening tool to evaluate the electron-induced degradability of thousands of PFASs for both homogeneous and heterogeneous reduction processes. Finally, we develop a kinetic model to investigate the impact of an accurate rate constant prediction on the fate of an environmentally relevant organic compound induced by eaq− in a homogeneous aqueous-phase ARP.
values of all possible elementary reactions of each PFAS to determine their dominant reaction mechanisms and reactive sites. LFERs of conventional organic compounds are used to predict the reactivities of eaq− with PFASs, which can be used as a screening tool to evaluate the electron-induced degradability of thousands of PFASs for both homogeneous and heterogeneous reduction processes. Finally, we develop a kinetic model to investigate the impact of an accurate rate constant prediction on the fate of an environmentally relevant organic compound induced by eaq− in a homogeneous aqueous-phase ARP.
Water impactOxidized forms of trace chemical contaminants including per- and poly-fluoroalkyl substances (PFAS) are the group of contaminants of emerging concern. Understanding and predicting the reactivity of solvated electrons enables prediction of the fate of contaminants in the aqueous-phase advanced reduction processes. A computational tool can be used to screen a number of contaminants to prioritize for the reduction processes. |
Introduction
Free radical-based technologies are attractive and promising processes for destroying a wide variety of organic contaminants. Advanced oxidation processes (AOPs) that generate highly reactive oxygenated radical species (e.g., hydroxyl radicals)1,2 and other reactive radicals (e.g., chlorine-,3,4 bromine-5 and nitrogen-derived radicals6,7 and carbonate radicals8) at ambient temperature and atmospheric pressure have been proven to degrade reduced forms of organic contaminants in water at full-scale treatment plants. Advanced reduction processes (ARPs) that generate reactive radicals (e.g., superoxide anion radicals) and electrons in homogeneous solution9,10 and heterogeneous electrochemical11–13 or catalytic14 processes are effective in degrading the oxidized forms of organic and inorganic contaminants. Homogeneous, electrochemical, and a combination of both ARPs have been successfully applied for the degradation of conventional organic contaminants such as alkyl halides and emerging groups of contaminants such as per- and polyfluorinated alkyl substances (PFASs).15While the reactivities of reactive radical species in AOPs have been actively studied and some predictive approaches have been reported in the literature,16 few studies have holistically focused on the reactivities of electrons in aqueous-phase ARPs. The reactivities of aqueous-phase hydrated electrons, eaq−, with a wide variety of individual organic compounds have been experimentally measured, and the second-order rate constants, kexp, have been reported and compiled in the database17 (see Fig. S1 in the ESI† for a box plot of kexp values). However, few studies have developed a predictive tool for the kexp values of eaq− due to a lack of mechanistic understanding of the reactivities with organic compounds.18,19 In general, nucleophilic electrons react at the electron-deficient sites of organic compounds. The three major reaction mechanisms include (1) association with the π bond of a double bond; (2) concerted dissociative cleavage of a carbon halogen (C–X where X = F, Cl, Br or I) bond of haloalkanes or carbon–nitrogen (C–N) bond; and (3) stepwise cleavage of a C–X bond of haloalkanes and haloalkenes, a sulfur–sulfur (S–S) bond or a carbon–sulfur (C–S) bond of sulfides or disulfides.20 Each reaction mechanism depends on the molecular structures and functional groups present in the same molecule. The overall reactivities with eaq− are reduced by electron-donating functional groups and increased by electron-withdrawing functional groups. However, the detailed mechanisms of multiple functional group compounds and the effect of these functional groups on the major reactivities have not been elucidated because of the difficulties in experimental investigations.
The use of quantum mechanics-based methods such as ab initio calculations or density functional theory (DFT) can complement experimental observations of chemical reactivities and provide mechanistic insight into reaction mechanisms. Several DFT-based methods were used to investigate the thermodynamics and kinetics of electron-induced reactions with halogenated compounds such as polychlorinated ethylenes,21,22 polybrominated electrophiles,23 and PFAS.24–26 The dissociation and reductive cleavage of a given molecule were investigated based on the optimized electronic structures, bond dissociation energies and reduction potentials of the corresponding bond. The kexp values represent the overall reactivities, and thus, the elementary reaction mechanisms of the overall reaction cannot be known. Calculating the one-electron reduction potential ( , V) in the aqueous phase of each component in a given molecule can provide quantitative information about all possible reactive sites and help determine the rate-determining reaction mechanism with electrons, which is more advantageous than investigating conventional qualitative molecular descriptors such as lowest unoccupied molecular orbitals.
, V) in the aqueous phase of each component in a given molecule can provide quantitative information about all possible reactive sites and help determine the rate-determining reaction mechanism with electrons, which is more advantageous than investigating conventional qualitative molecular descriptors such as lowest unoccupied molecular orbitals.
In this study, we use DFT to calculate the 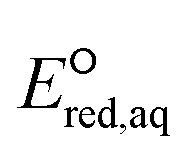 values of conventional organic compounds with a wide variety of functional groups to determine the linear free energy relationships (LFERs) with the experimentally measured rate constants of eaq−. In addition, we use the 75 priority PFAS subset from the U.S. EPA27 and calculate the
values of conventional organic compounds with a wide variety of functional groups to determine the linear free energy relationships (LFERs) with the experimentally measured rate constants of eaq−. In addition, we use the 75 priority PFAS subset from the U.S. EPA27 and calculate the 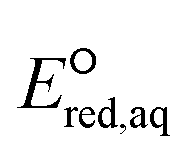 values of all possible elementary reactions of each PFAS to determine its dominant reaction mechanism and reactive sites. The determined LFERs of conventional organic compounds are used to predict the reactivities of eaq− with PFASs, which can be used as a screening tool for thousands of PFASs for electron-induced degradability. Fig. 1 demonstrates the flowchart of methods used in this study from the determination of LFERs for conventional organic compounds to the prediction of kchem values for PFAS. While we demonstrate the prediction of kexp values for eaq− in the homogeneous reduction processes, the reactivities of electrons via direct electron transfer on a heterogeneous-electrode can be extrapolated from the eaq− reactivities and the LFERs are also useful for the heterogeneous processes.
values of all possible elementary reactions of each PFAS to determine its dominant reaction mechanism and reactive sites. The determined LFERs of conventional organic compounds are used to predict the reactivities of eaq− with PFASs, which can be used as a screening tool for thousands of PFASs for electron-induced degradability. Fig. 1 demonstrates the flowchart of methods used in this study from the determination of LFERs for conventional organic compounds to the prediction of kchem values for PFAS. While we demonstrate the prediction of kexp values for eaq− in the homogeneous reduction processes, the reactivities of electrons via direct electron transfer on a heterogeneous-electrode can be extrapolated from the eaq− reactivities and the LFERs are also useful for the heterogeneous processes.
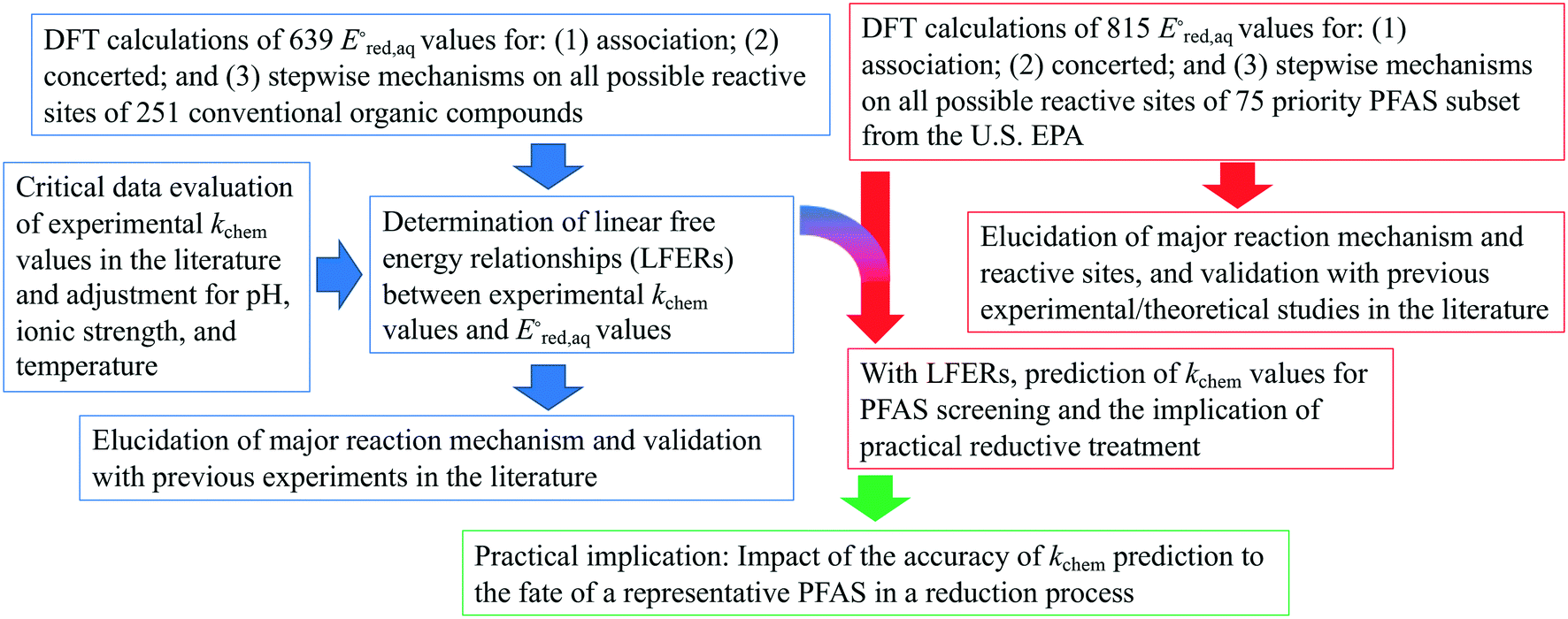 | ||
| Fig. 1 Overall flowcharts of methodologies and logical steps. | ||
Materials and methods
According to the previous experimental studies reported in the literature, three major reaction mechanisms of eaq− include: (1) associative; (2) concerted dissociative; and, (3) stepwise dissociative mechanisms. In the associative mechanism, eaq− reacts with the π bond that can ‘hold’ an extra electron to form an anionic radical species.20 Compounds containing carbonyl functional groups are examples of compounds that undergo associative reactions:RC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O + eaq− → R˙CO− O + eaq− → R˙CO− | (1) |
Both concerted and stepwise mechanisms involve bond cleavage. In the concerted mechanism, single-electron transfer to a parent compound and bond cleavage occur simultaneously, as shown in eqn (2). During the stepwise mechanism, the initial barrierless step of single-electron transfer results in the formation of an intermediate radical anion that has a longer lifetime than the bond vibration time (i.e., 10−13 s).28 The intermediate radical anion then undergoes bond cleavage, as shown in eqn (3). The nonexistence of a radical anion is a sufficient condition for the concerted mechanism to occur, but it is not a necessary condition. Thus, under the concerted mechanism, an intermediate radical anion may have a finite lifetime.29
| RX + eaq− → R˙ + X− | (2) |
| RX + eaq− → [RX]˙− → R˙ + X− | (3) |
S, SS, NO2, CN, CC), strong C–X bonds (e.g., C–F), and/or electron withdrawing groups (e.g., –F, –CN, NO2, –CO) are reduced via the stepwise mechanism.23,30 However, caution should be taken for compounds with strong electron withdrawing groups and halogenated alkenes because a concerted mechanism could possibly occur due to an unstable intermediate radical anion or the requirement of reduced reaction barriers.22,23
To determine which of reduction mechanisms in eqn (1)–(3) is the rate-determining step for a given molecule, we explored the LFER that relates the experimentally measured chemical reaction rate constant, kchem, and the  values relative to the standard hydrogen electrode (SHE) for each mechanism through the relation described by eqn (4). We, then, determined the dominant rate-determining reaction mechanism by investigating the correlation of each LFER. The concept of a LFER may be developed as below. Assuming an elementary reaction proceeds by the same reaction mechanism, the log of the rate constant and the log of the equilibrium constant are linearly related.31 The natural log of the equilibrium constants has a linear relationship with the free energy reaction, ΔGreactaq, which relates to the standard state reduction potential in eqn (5). Combining these two concepts enables the development of the LFER. Upon the calculation of the
values relative to the standard hydrogen electrode (SHE) for each mechanism through the relation described by eqn (4). We, then, determined the dominant rate-determining reaction mechanism by investigating the correlation of each LFER. The concept of a LFER may be developed as below. Assuming an elementary reaction proceeds by the same reaction mechanism, the log of the rate constant and the log of the equilibrium constant are linearly related.31 The natural log of the equilibrium constants has a linear relationship with the free energy reaction, ΔGreactaq, which relates to the standard state reduction potential in eqn (5). Combining these two concepts enables the development of the LFER. Upon the calculation of the 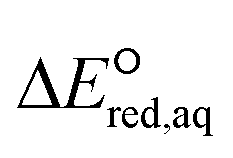 values, all possible eaq− attacking sites for each compound were included, and the largest
values, all possible eaq− attacking sites for each compound were included, and the largest  value (i.e., the smallest free energy of formation) in a given molecule was used for the determination of the LFER. We determined the LFER for each reaction mechanism listed in eqn (1)–(3) to investigate the correlation with kchem values. The procedure to determine the kchem values and the critical evaluation of literature-reported kexp values are provided in Texts S1 in the ESI.†
value (i.e., the smallest free energy of formation) in a given molecule was used for the determination of the LFER. We determined the LFER for each reaction mechanism listed in eqn (1)–(3) to investigate the correlation with kchem values. The procedure to determine the kchem values and the critical evaluation of literature-reported kexp values are provided in Texts S1 in the ESI.†
 | (4) |
 values) and the LFER helps elucidate the dominant reaction mechanism. Determining the aqueous-phase free energies of activation, a parameter that drives the kinetics, for hundreds of reactions involving eaq− by investigating the potential energy surfaces (PES) of reactants and products is not practical for systematic investigation. Thus, we conducted PES scan to determine the reaction mechanism for a few compounds that may undergo more than one reaction mechanism described above.
values) and the LFER helps elucidate the dominant reaction mechanism. Determining the aqueous-phase free energies of activation, a parameter that drives the kinetics, for hundreds of reactions involving eaq− by investigating the potential energy surfaces (PES) of reactants and products is not practical for systematic investigation. Thus, we conducted PES scan to determine the reaction mechanism for a few compounds that may undergo more than one reaction mechanism described above.
For the associative and concerted reaction mechanisms, the 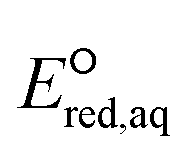 value was determined with eqn (5):
value was determined with eqn (5):
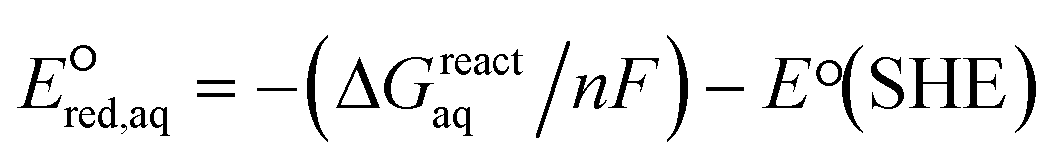 | (5) |
| ΔGreactaq = ΔGreactgas + ΔΔGreactsolv | (6) |
Regarding the stepwise investigation, we calculated  with eqn (7),26,32 which accounts for both the formation of the intermediate radical species and the resulting bond cleavage as an example of an RX bond.26,32
with eqn (7),26,32 which accounts for both the formation of the intermediate radical species and the resulting bond cleavage as an example of an RX bond.26,32
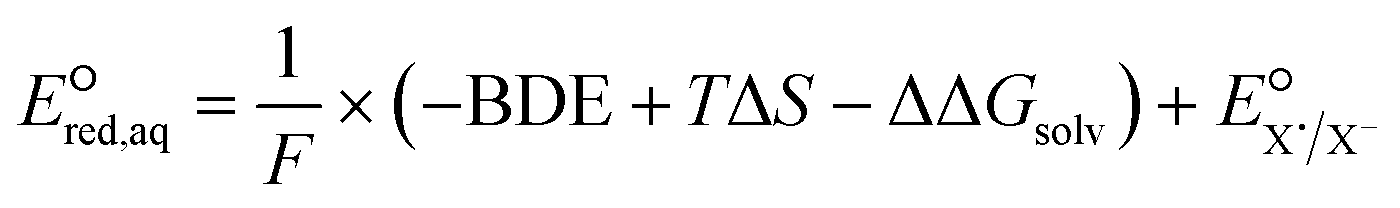 | (7) |
 is the reduction potential of the cleaved aqueous atom.
is the reduction potential of the cleaved aqueous atom.| ΔΔGsolv = ΔGsolv(R˙) + ΔGsolv(X˙) − ΔGsolv(RX) | (8) |
| BDE = −[H(RX) − H(R˙) − H(X˙)] | (9) |
To calculate the 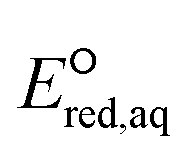 values for the determination of LFERs, single point energy calculations at the M06-2X functional33 and the Aug-cc-pVTZ basis set for all the mechanisms based on the optimized structures determined at M06-2X/cc-pVDZ or Aug-cc-pVTZ, unless detailed method was specified. We used M06-2X/LANL2DZ for compounds that contained iodine because the Dunning's basis set does not cover iodinated compounds. For PFASs, we used M06-2X with a combination of cc-pVDZ or Aug-cc-pVTZ basis set. Table S1 in the ESI† summarizes the method and basis set used for the group of compounds. M06-2X is specifically designed for the accurate treatment of long-distance interaction and/or the stronger electron-acceptor properties of the R˙ fragments resulting from the dissociation of a C–R bond,34 which makes it suitable for this study that investigates nucleophilic reactivity. The M06-2X functional was successfully applied for the reductive dissociation of polybrominated compounds.23 A continuum form of the universal solvation model (SMD)35 was used in the aqueous-phase calculations to account for the impact of an aqueous environment. It is noted that we did not aim to obtain the absolute
values for the determination of LFERs, single point energy calculations at the M06-2X functional33 and the Aug-cc-pVTZ basis set for all the mechanisms based on the optimized structures determined at M06-2X/cc-pVDZ or Aug-cc-pVTZ, unless detailed method was specified. We used M06-2X/LANL2DZ for compounds that contained iodine because the Dunning's basis set does not cover iodinated compounds. For PFASs, we used M06-2X with a combination of cc-pVDZ or Aug-cc-pVTZ basis set. Table S1 in the ESI† summarizes the method and basis set used for the group of compounds. M06-2X is specifically designed for the accurate treatment of long-distance interaction and/or the stronger electron-acceptor properties of the R˙ fragments resulting from the dissociation of a C–R bond,34 which makes it suitable for this study that investigates nucleophilic reactivity. The M06-2X functional was successfully applied for the reductive dissociation of polybrominated compounds.23 A continuum form of the universal solvation model (SMD)35 was used in the aqueous-phase calculations to account for the impact of an aqueous environment. It is noted that we did not aim to obtain the absolute 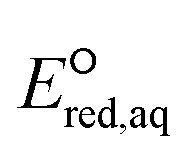 values of each elementary reaction, as they are computationally prohibitive when obtaining highly accurate ΔGreactaq values for a number of compounds. Thus, we used M06-2X to obtain reliable relative
values of each elementary reaction, as they are computationally prohibitive when obtaining highly accurate ΔGreactaq values for a number of compounds. Thus, we used M06-2X to obtain reliable relative  values so that we were able to relatively compare which reactive sites were dominant over other sites under the same reaction mechanism. The dominant reaction mechanism among the three major mechanisms was determined by the LFER with mechanistic insight into the reaction mechanisms, as the direct comparison of the
values so that we were able to relatively compare which reactive sites were dominant over other sites under the same reaction mechanism. The dominant reaction mechanism among the three major mechanisms was determined by the LFER with mechanistic insight into the reaction mechanisms, as the direct comparison of the 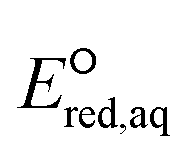 values obtained from eqn (5) and (7) was not possible. The validation of the M06-2X method with various basis sets is provided in Table S2 in the ESI.† All DFT calculations were performed using Gaussian16 (ref. 36) with the Michigan Tech high-performance cluster ‘Superior’ and homemade LINUX workstations.
values obtained from eqn (5) and (7) was not possible. The validation of the M06-2X method with various basis sets is provided in Table S2 in the ESI.† All DFT calculations were performed using Gaussian16 (ref. 36) with the Michigan Tech high-performance cluster ‘Superior’ and homemade LINUX workstations.
Results and discussion
Determination of linear free energy relationships
Inconsistent experimental conditions (e.g., pH, temperature, and ionic strength) were reported to measure kexp values in a number of independent studies reported in the literature. Thus, the critical data evaluation of 268kexp values (Text S1, Fig. S1 and Table S3 in the ESI†) in the literature selected 251kexp values and calculated the chemical reaction rate constants by eliminating the diffusion contribution for the determination of LFERs (Text S1 in the ESI†). This critical data evaluation can potentially eliminate the uncertain kexp values that may indicate significantly larger kexp values that exceed the diffusion rate constant, kD, in eqn (S1) in the ESI.† It should be noted that the diffusion rate constant, kD, value used in eqn (S1)† has the limitations: (1) the Smoluchowski's equation to calculate the kD values does not include either the long range forces between reactants or the diffusive displacement for small molecules; (2) the Smoluchowski's equation assumes the behavior of each reactant like a stationary sink around which a concentration gradient of the other reactant; and (3) the Smoluchowski's equation assumes the continuum structureless treatment of solvent.31 Therefore, the extent of solvation effect may vary depending on the molecules. Thus, the kD values we calculated may not represent the real diffusion rate constants. Nevertheless, the Smoluchowski's treatment has been successfully applied for many radical reactions (e.g., hydroxyl radicals) and predicted the kD in consistent with the experimental values.38 As a consequence, we decided to adapt this approach in our calculations. Fig. 2 displays LFERs between the kchem values and our theoretically calculated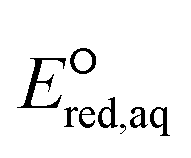 values for 251 organic compounds undergoing three major mechanisms: (a) associative, (b) concerted, and/or (c) stepwise. Table 1 summarizes all the data used to determine the LFERs. Tables S4 and S5 in the ESI† contains all the
values for 251 organic compounds undergoing three major mechanisms: (a) associative, (b) concerted, and/or (c) stepwise. Table 1 summarizes all the data used to determine the LFERs. Tables S4 and S5 in the ESI† contains all the 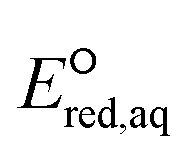 values for all possible reactive sites in a given molecule for the three reaction mechanisms. Regarding the association, we determined the LFER to be ln
values for all possible reactive sites in a given molecule for the three reaction mechanisms. Regarding the association, we determined the LFER to be ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) kchem = 4.43
kchem = 4.43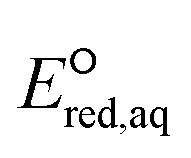 + 31.76 (r2 = 0.72, N = 66, where N is the number of compounds for the development of the LFER) (Fig. 2a). When the carbon of the CO functional group bonds with NH2 or the OR functional group, the mesomeric effect of the –CO–NH2– or –CO–OR– functional group occurs and decreases the double-bond character of the CO functional group, creating new electrophilic centers with lower reactivity.37 While we determined one unified LFER for associative mechanism with both CO of ketones, aldehydes and carboxylate groups (blue dots in Fig. 1) and O of carboxylic acids, alcohols, esters, and amides (red dots in Fig. 1), the functional groups affect the associative mechanism with O in a different way from those with CO functional group (see the next subsection). Compounds 17, 33, 45, and 153, whose kchem values are close to or exceed the diffusion limit (kchem > 2.5 × 1010 M−1 s−1), were not included in either LFER. Compound 39, methyl trifluoroacetate, appear to be slightly off the LFER of the associative mechanism or that of the stepwise mechanism. Our investigation on the PES and spin density distribution supports the associative mechanism (see the detailed discussion in the reaction mechanism section below). Thus, we included this compound in the LFER of the associative mechanism. For the associative mechanism with the CC of alkenes, the LFER was determined to be lnkchem = 7.82
+ 31.76 (r2 = 0.72, N = 66, where N is the number of compounds for the development of the LFER) (Fig. 2a). When the carbon of the CO functional group bonds with NH2 or the OR functional group, the mesomeric effect of the –CO–NH2– or –CO–OR– functional group occurs and decreases the double-bond character of the CO functional group, creating new electrophilic centers with lower reactivity.37 While we determined one unified LFER for associative mechanism with both CO of ketones, aldehydes and carboxylate groups (blue dots in Fig. 1) and O of carboxylic acids, alcohols, esters, and amides (red dots in Fig. 1), the functional groups affect the associative mechanism with O in a different way from those with CO functional group (see the next subsection). Compounds 17, 33, 45, and 153, whose kchem values are close to or exceed the diffusion limit (kchem > 2.5 × 1010 M−1 s−1), were not included in either LFER. Compound 39, methyl trifluoroacetate, appear to be slightly off the LFER of the associative mechanism or that of the stepwise mechanism. Our investigation on the PES and spin density distribution supports the associative mechanism (see the detailed discussion in the reaction mechanism section below). Thus, we included this compound in the LFER of the associative mechanism. For the associative mechanism with the CC of alkenes, the LFER was determined to be lnkchem = 7.82 + 41.25 (r2 = 0.63, N = 13) (Fig. 2b). The reactions of the alkenes with kchem > 5.3 × 109 M−1 s−1 were close to or exceeded the diffusion limit; therefore, the kchem values did not change with an increase in the
+ 41.25 (r2 = 0.63, N = 13) (Fig. 2b). The reactions of the alkenes with kchem > 5.3 × 109 M−1 s−1 were close to or exceeded the diffusion limit; therefore, the kchem values did not change with an increase in the  values. The sample deviation (SD) calculated with eqn (10) was 0.084 for the associative mechanism and 0.13 for the associative mechanism with the CC functional group. The SD values represent the statistical distribution of the experimental kchem values from the predicted values, kpredicted, within the normal distribution.38
values. The sample deviation (SD) calculated with eqn (10) was 0.084 for the associative mechanism and 0.13 for the associative mechanism with the CC functional group. The SD values represent the statistical distribution of the experimental kchem values from the predicted values, kpredicted, within the normal distribution.38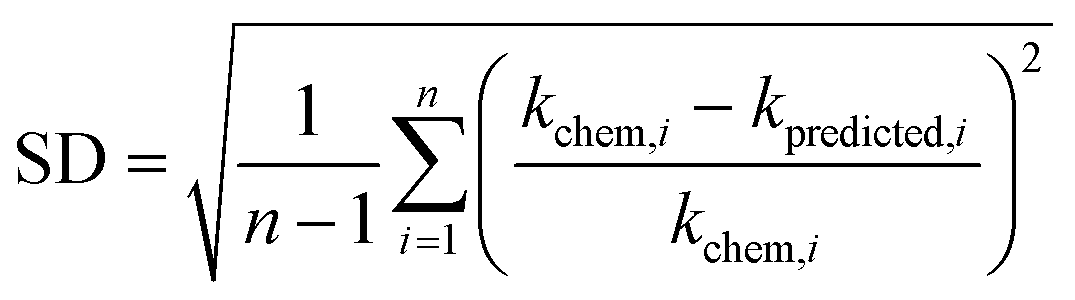 | (10) |
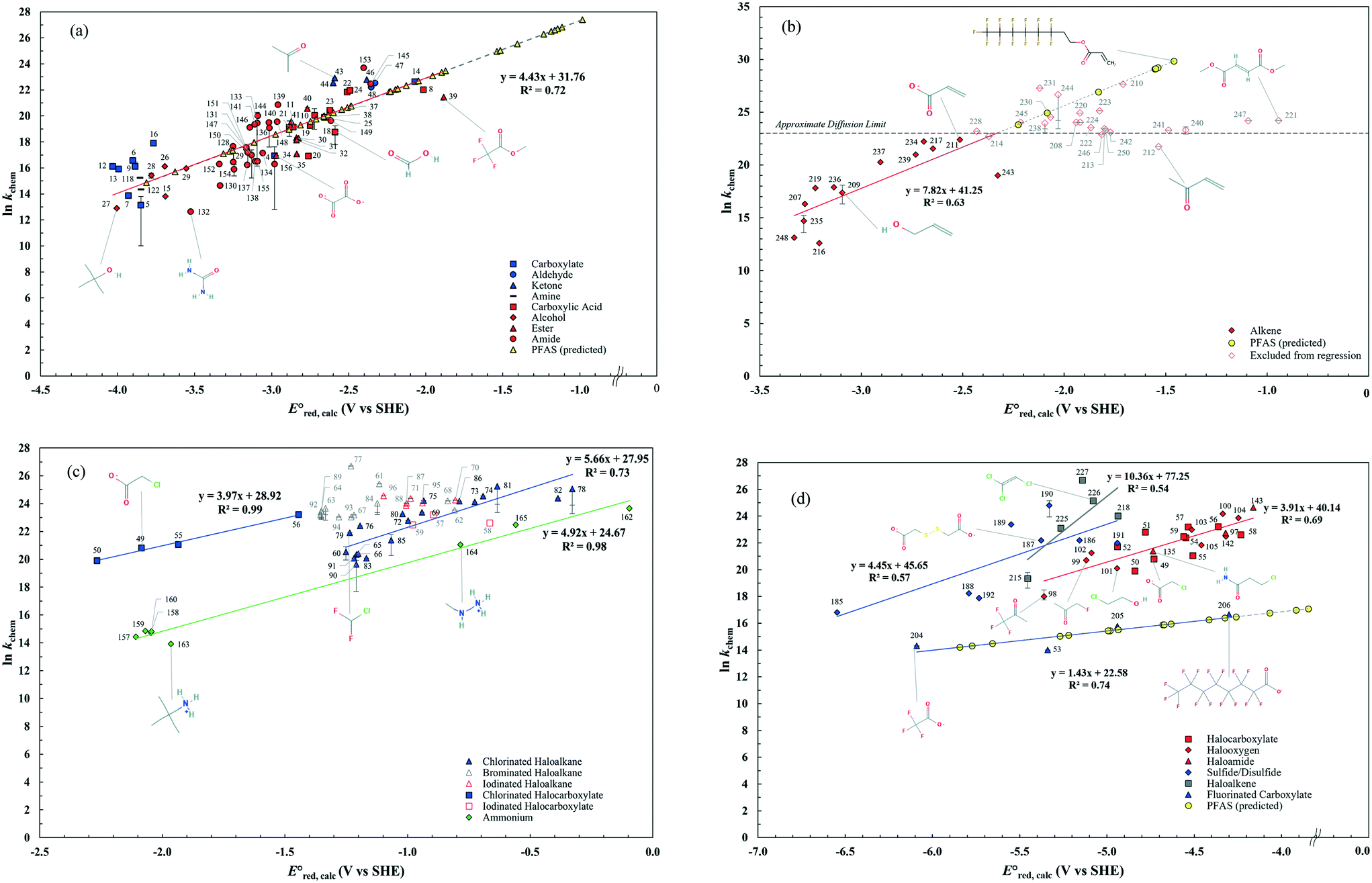 | ||
| Fig. 2 LFERs for the (a) associative mechanism with CO and O, (b) associative mechanism with CC, (c) concerted mechanism, and (d) stepwise mechanism. | ||
 and kchem values of 251 organic compounds used to determine the LFERs. Compounds in regular font are for associative mechanism, those in bold are for concerted mechanism, and those in italic are for stepwise mechanism
and kchem values of 251 organic compounds used to determine the LFERs. Compounds in regular font are for associative mechanism, those in bold are for concerted mechanism, and those in italic are for stepwise mechanism
| Class | No. | Name | Chemical formula |
|
|
k chem (M−1 s−1) | Reference for kexp |
|---|---|---|---|---|---|---|---|
| Alkane | 1 | Methane | CH4 | 25.97 | −5.41 | 1.00 × 107 | 39 |
| 2 | Propane | CH3CH2CH3 | 23.74 | −5.31 | 2.10 × 106 | 40 | |
| 3 | Butane | C4H10 | 22.77 | −5.27 | 2.40 × 106 | 40 | |
| Carboxylate | 4 | Oxalate | −OOCCOO− | −29.94 | −2.98 | 2.28 × 107 | 41–44 |
| 5 | Formate | HCOO− | −9.93 | −3.85 | 5.04 × 105 | 41, 45 | |
| 6 | Succinate | −OOC(CH2)2COO− | −8.72 | −3.90 | 1.59 × 107 | 37, 46 | |
| 7 | Acetate | CH3COO− | −8.07 | −3.93 | 1.05 × 106 | 45, 47 | |
| 8 | Hydrogen oxalate | HOOCCOO− | −52.21 | −2.02 | 3.65 × 109 | 40 | |
| 9 | Malonate | −OOC–CH2–COO− | −9.07 | −3.89 | 1.00 × 107 | 46 | |
| 10 | Malonate(1−) | HOOC–CH2–COO− | −35.93 | −2.72 | 5.06 × 108 | 46, 48 | |
| 11 | Succinate(1−) | HOOC(CH2)2COO− | −17.65 | −3.51 | 2.05 × 108 | 37, 46 | |
| 12 | Lactate | CH3CHOHCOO− | −5.70 | −4.03 | 1.00 × 107 | 39 | |
| 13 | Glycolate | HOCH2COO− | −6.61 | −3.99 | 8.20 × 106 | 49 | |
| 14 | Pyruvate | CH3COCOO− | −50.94 | −2.07 | 6.80 × 109 | 39 | |
| 15 | CID_4134252 | HOCH2(CHOH)4COO− | −13.59 | −3.69 | 1.00 × 106 | 50 | |
| 16 | Malate | −OOCCH2CHOHCOO− | −11.81 | −3.77 | 6.01 × 107 | 51 | |
| Carboxylic acid | 17 | Oxalic acid | HOOCCOOH | −62.94 | −1.55 | 2.50 × 1010 | 52 |
| 18 | Formic acid | HCOOH | −39.00 | −2.59 | 1.41 × 108 | 45 | |
| 19 | Succinic acid | HOOC(CH2)2COOH | −35.30 | −2.75 | 2.30 × 108 | 46, 53 | |
| 20 | Propionic acid | CH3CH2COOH | −35.03 | −2.76 | 2.20 × 107 | 53 | |
| 21 | Acetic acid | CH3COOH | −32.16 | −2.89 | 2.02 × 108 | 45, 54 | |
| 22 | Malonic acid | HOOC–CH2–COOH | −40.83 | −2.51 | 3.03 × 109 | 46, 48, 53 | |
| 23 | Lactic acid | CH3CH(OH)COOH | −38.23 | −2.62 | 7.36 × 108 | 46, 53 | |
| 24 | Malic acid | HOOCCH2CH(OH)COOH | −41.24 | −2.49 | 3.41 × 109 | 55 | |
| 25 | Glycolic acid | HOCH2COOH | −37.42 | −2.66 | 4.38 × 108 | 53 | |
| Alcohol | 26 | Methanediol | CH2(OH)2 | −13.52 | −3.69 | 1.00 × 107 | 45, 56 |
| 27 | tert-Butanol | (CH3)3–C–OH | −6.33 | −4.01 | 4.00 × 105 | 47 | |
| 28 | Butane-1,2,3,4 | HOCH2[CH(OH)]2CH2OH | −11.50 | −3.78 | 5.00 × 106 | 57 | |
| 29 | Mannitol | HOCH2[CH(OH)]4CH2OH | −16.74 | −3.55 | 8.50 × 106 | 57, 58 | |
| Ester | 30 | Methyl acetate | CH3COOCH3 | −33.56 | −2.82 | 8.73 × 107 | 59 |
| 31 | Methyl propionate | C2H5COOCH3 | −33.24 | −2.84 | 9.03 × 107 | 37 | |
| 32 | Ethyl propionate | C2H5COOC2H5 | −33.22 | −2.84 | 7.52 × 107 | 60 | |
| 33 | Dimethyl oxalate | CH3OOCCOOCH3 | −59.03 | −1.72 | 1.04 × 1011 | 48 | |
| 34 | tert-Butyl acetate | (CH3)3CCOOCH3 | −30.20 | −2.97 | 2.30 × 107 | 37 | |
| 35 | 2-Hydroxyethyl acetate | CH3COOCH2CH2OH | −33.27 | −2.84 | 2.60 × 107 | 61 | |
| 36 | Di-tert-butyl peroxide | (CH3)3–COOC(CH3)3 | 44.93 | −6.23 | 1.41 × 108 | 62 | |
| 37 | Methylene glycol monoacetate | HOCH2COOCH3 | −37.37 | −2.66 | 4.90 × 108 | 37 | |
| 38 | Methyl methoxyacetate | CH3OCH2COOCH3 | −38.04 | −2.63 | 4.48 × 108 | 63 | |
| 39 | Methyl trifluoroacetate | CF3COOCH3 | −42.28 | −2.45 | 2.06 × 109 | 37 | |
| 40 | Ethyl glycinate | NH2CH2COOC2H5 | −34.86 | −2.77 | 8.58 × 108 | 64 | |
| 41 | Acetoxymethylamine | H2NCH2COOCH3 | −32.45 | −2.87 | 3.14 × 108 | 37, 65 | |
| Ether | 42 | Diethyl ether | (C2H5)2O | −38.95 | −2.59 | 1.00 × 107 | 20 |
| Ketone | 43 | Acetone | CH3COCH3 | −38.95 | −2.59 | 8.90 × 109 | 66–70 |
| 44 | Methyl ethyl ketone | CH3CH2COCH3 | −38.72 | −2.60 | 6.11 × 109 | 71 | |
| 45 | 2,3-Butanedione | CH3COCOCH3 | −69.05 | −1.29 | 1.67 × 1010 | 48, 72 | |
| 46 | Acetoin | CH3COCH(OH)CH3 | −43.76 | −2.38 | 7.95 × 109 | 72 | |
| Aldehyde | 47 | Acetaldehyde | CH3CHO | −44.97 | −2.33 | 6.11 × 109 | 45, 48 |
| 48 | Propionaldehyde | CH3CH2CHO | −44.42 | −2.35 | 4.43 × 109 | 68, 71 | |
| Halocarboxylate | 49 | Chloroacetate | ClCH 2 COO − | 10.40 | −4.73 | 1.09 × 10 9 | 67, 69, 73–74 |
| 50 | 3-Chloropropanoate | Cl(CH 2 ) 2 COO − | 12.92 | −4.84 | 4.40 × 10 8 | 73 | |
| 51 | Bromoacetate | BrCH 2 COO − | 11.54 | −4.78 | 8.03 × 10 9 | 69 | |
| 52 | 3-Bromopropanoate | Br(CH 2 ) 2 COO − | 15.24 | −4.94 | 2.70 × 10 9 | 69 | |
| 53 | Fluoroacetate | FCH 2 COO − | 66.82 | −7.18 | 1.20 × 10 6 | 69 | |
| 54 | 2-Bromopropanoate | CH 3 CHBrCOO − | 6.18 | −4.55 | 5.30 × 10 9 | 69 | |
| 55 | 2-Chloropropanoate | CH 3 CHClCOO − | 5.26 | −4.51 | 1.40 × 10 9 | 69 | |
| 56 | Trichloroacetate | Cl 3 CCOO − | 1.91 | −4.36 | 1.22 × 10 10 | 69 | |
| 57 | 2-Iodoacetate | ICH 2 COO − | 5.89 | −4.54 | 1.20 × 10 10 | 69 | |
| 58 | 2-Iodopropanoate | CH 3 CHICOO − | −1.08 | −4.23 | 6.60 × 10 9 | 69 | |
| 59 | 3-Iodanylpropanoate | ICH 2 CH 2 COO − | 6.46 | −4.56 | 5.80 × 10 9 | 75 | |
| Haloalkane | 60 | Chloromethane | CH3Cl | −69.84 | −1.25 | 8.33 × 108 | 76–78 |
| 61 | Dibromomethane | CH2Br2 | −73.00 | −1.11 | 1.10 × 1011 | 79 | |
| 62 | Bromoform | CHBr3 | −80.06 | −0.81 | 1.67 × 1010 | 80 | |
| 63 | Bromoethane | CH3CH2Br | −67.93 | −1.33 | 1.89 × 1010 | 80–82 | |
| 64 | Bromopropane | CH3CH2CH2Br | −67.55 | −1.35 | 1.47 × 1010 | 80 , 82 | |
| 65 | Chloropropane | CH3CH2CH2Cl | −70.86 | −1.21 | 6.85 × 108 | 40 , 81, 82 | |
| 66 | Chloroethane | CH3CH2Cl | −71.03 | −1.20 | 7.21 × 108 | 77 | |
| 67 | 1-Bromo-2-chloroethane | CH2ClCH2Br | −70.61 | −1.22 | 1.18 × 1010 | 80 | |
| 68 | Halothane | CF3CHClBr | −79.44 | −0.84 | 3.22 × 1010 | 83 | |
| 69 | 1,1-Dichloroethane | CH3CHCl2 | −77.00 | −0.94 | 1.42 × 1010 | 84 | |
| 70 | Diiodomethane | CH2I2 | −80.13 | −0.81 | 3.40 × 1010 | 79 , 85 | |
| 71 | Iodoethane | CH3CH2I | −75.94 | −0.99 | 3.85 × 1010 | 81 , 82 | |
| 72 | Dichloromethane | CH2Cl2 | −75.69 | −1.00 | 7.95 × 109 | 86 | |
| 73 | Chloroform | CHCl3 | −81.97 | −0.73 | 3.00 × 1010 | 39 | |
| 74 | Trichlorofluoromethane | CCl3F | −82.75 | −0.69 | 4.60 × 1010 | 87 | |
| 75 | Dichlorodifluoromethane | CF2Cl2 | −77.16 | −0.93 | 3.28 × 1010 | 87 | |
| 76 | Chlorotrifluoromethane | CClF3 | −71.19 | −1.19 | 5.36 × 109 | 81 | |
| 77 | Bromotrifluoromethane | CF3Br | −70.32 | −1.23 | 3.93 × 1011 | 81 | |
| 78 | Carbon tetrachloride | CCl4 | −91.15 | −0.33 | 7.61 × 1010 | 68 , 88 | |
| 79 | Chlorodifluoromethane | CHClF2 | −70.22 | −1.24 | 3.29 × 109 | 89 | |
| 80 | 1,1,2-Trichloroethane | ClCH2CHCl2 | −75.17 | −1.02 | 1.27 × 1010 | 84 | |
| 81 | 1,1,1-Trichloroethane | CH3CCl3 | −84.09 | −0.63 | 9.24 × 1010 | 77 , 84 | |
| 82 | Hexachloroethane | CCl3CCl3 | −89.80 | −0.39 | 3.90 × 1010 | 84 | |
| 83 | 2-Chlorobutane | C2H5CH(Cl)CH3 | −71.79 | −1.17 | 5.21 × 108 | 82 | |
| 84 | 1,2-Dibromoethane | BrCH2CH2Br | −72.81 | −1.12 | 2.74 × 1010 | 80 , 84 | |
| 85 | 1,2-Dichloroethane | ClCH2CH2Cl | −74.11 | −1.07 | 1.91 × 109 | 84 , 90 | |
| 86 | 1,1,2-Trichloro-1,2,2-trifluoroethane | ClCF2CCl2F | −80.52 | −0.79 | 3.17 × 1010 | 84 | |
| 87 | 1-Iodopropane | C3H7I | −75.56 | −1.00 | 2.73 × 1010 | 82 | |
| 88 | 1-Iodobutane | CH3(CH2)3I | −75.50 | −1.01 | 2.29 × 1010 | 82 | |
| 89 | 1-Bromobutane | CH3(CH2)3Br | −67.54 | −1.35 | 1.59 × 1010 | 80–82 | |
| 90 | 1-Chlorobutane | CH3(CH2)3Cl | −70.83 | −1.21 | 3.42 × 108 | 40 , 54, 81, 82 | |
| 91 | 1-Chloro-2-methylpropane | (CH3)2CHCH2Cl | −70.62 | −1.22 | 5.21 × 108 | 82 | |
| 92 | 1-Bromopentane | CH3(CH2)4Br | −67.45 | −1.36 | 1.17 × 1010 | 80 | |
| 93 | 2-Bromo-2-methylpropane | (CH3)3CBr | −70.36 | −1.23 | 1.02 × 1010 | 80 | |
| 94 | 2-Bromobutane | CH3CH2CH(Br)CH3 | −69.18 | −1.28 | 1.01 × 1010 | 80 | |
| 95 | Trifluoroiodomethane | CF3I | −77.06 | −0.94 | 2.77 × 1010 | 81 | |
| 96 | Iodomethane | CH3I | −73.39 | −1.10 | 4.64 × 1010 | 81 , 91 | |
| Halooxygen | 97 | Isoflurane | CHF 2 OCHClCF 3 | 0.87 | −4.32 | 5.80 × 10 9 | 84 |
| 98 | 1,1,1-Trifluoroacetone | CF 3 COCH 3 | 24.93 | −5.36 | 6.62 × 10 7 | 37 | |
| 99 | Fluoroacetone | CH 3 COCH 2 F | 19.34 | −5.12 | 9.77 × 10 8 | 37 | |
| 100 | Methoxyflurane | CH 3 OCF 2 CHCl 2 | 1.31 | −4.34 | 3.16 × 10 10 | 84 | |
| 101 | 2-Chloroethanol | ClCH 2 CH 2 OH | 15.25 | −4.94 | 5.34 × 10 8 | 92 | |
| 102 | 2-Bromoethanol | BrCH 2 CH 2 OH | 18.64 | −5.09 | 1.71 × 10 9 | 69 | |
| 103 | Chloroacetic acid | ClCH 2 COOH | 5.40 | −4.51 | 9.60 × 10 9 | 93 | |
| 104 | Chloral hydrate | CCl 3 CH(OH) 2 | −0.79 | −4.25 | 2.31 × 10 10 | 94 | |
| 105 | Enflurane | CHF 2 OCF 2 CHClF | 4.14 | −4.46 | 3.03 × 10 9 | 84 | |
| Cyanide | 106 | Acetonitrile | CH3CN | −14.83 | −3.64 | 3.74 × 107 | 54, 68, 95 |
| 107 | Succinonitrile | NC(CH2)2CN | −21.84 | −3.33 | 1.83 × 109 | 96 | |
| 108 | Trichloroacetonitrile | CCl3CN | −98.67 | 0.00 | 3.20 × 1010 | 84 | |
| 109 | Cyanamide | H2NCN | −21.23 | −3.36 | 1.60 × 109 | 96 | |
| Amine | 110 | Methylamine | CH3NH2 | 19.28 | −5.12 | 9.00 × 105 | 97 |
| 111 | Butylamine | CH3(CH2)3NH2 | 17.07 | −5.02 | 1.10 × 106 | 98 | |
| 112 | Propylamine | CH3CH2CH2NH2 | 19.79 | −5.14 | 1.10 × 106 | 98 | |
| 113 | Ethylamine | CH3CH2NH2 | 20.42 | −5.17 | 1.00 × 106 | 98 | |
| 114 | Isobutylamine | (CH3)2CHCH2NH2 | 18.63 | −5.09 | 1.10 × 107 | 97 | |
| 115 | Isoamylamine | (CH3)2CHCH2CH2NH2 | 20.07 | −5.15 | 1.00 × 106 | 97 | |
| 116 | 1,2-Dimethylhydrazine | CH3NHNHCH3 | 27.98 | −5.49 | 6.10 × 106 | 99 | |
| 117 | Methylhydrazine | CH3NHNH2 | 12.20 | −4.81 | 6.50 × 106 | 99 | |
| 118 | Glycinate | NH2CH2COO− | −9.94 | −3.85 | 1.70 × 106 | 100 | |
| 119 | Ethanolamine | H2NCH2CH2OH | −0.27 | −4.27 | 2.00 × 107 | 101 | |
| 120 | Isopropylamine | (CH3)2CHNH2 | 18.20 | −5.07 | 1.50 × 106 | 97 | |
| 121 | tert-Butylamine | (CH3)3CNH2 | 18.20 | −5.07 | 1.10 × 106 | 97 | |
| 122 | Beta-alaninate | NH2(CH2)2–COO− | −9.80 | −3.85 | 4.20 × 106 | 102 | |
| 123 | N,N-Diethylhydroxylamine | (C2H5)2NOH | −2.51 | −4.17 | 4.81 × 107 | 103 | |
| 124 | N-Methyl-N-tritiohydroxylamine | CH3NHOH | −15.92 | −3.59 | 2.42 × 108 | 65 | |
| 125 | Amylamine | CH3(CH2)4NH2 | 21.11 | −5.20 | 1.00 × 106 | 98 | |
| 126 | Trimethylhydrazine | (CH3)2N–NHCH3 | −16.76 | −3.55 | 1.00 × 108 | 99 | |
| 127 | 1,1-Dimethylhydrazine | (CH3)2NNH2 | 18.28 | −5.07 | 2.40 × 107 | 99 | |
| Amide | 128 | Propionamide | CH3CH2CONH2 | −23.71 | −3.25 | 4.66 × 107 | 100, 104 |
| 129 | N-Ethylacetamide | CH3CONHC2H5 | −23.75 | −3.25 | 1.40 × 107 | 64 | |
| 130 | N-Methylacetamide | CH3CONHCH3 | −21.79 | −3.34 | 2.30 × 106 | 105 | |
| 131 | Acetamide | CH3CONH2 | −25.72 | −3.16 | 3.84 × 107 | 74, 100, 106 | |
| 132 | Urea | H2NCONH2 | −17.40 | −3.53 | 3.10 × 105 | 37, 74 | |
| 133 | Glycinamide | H2NCH2CONH2 | −27.34 | −3.09 | 2.83 × 108 | 65 | |
| 134 | Formamide | HCONH2 | −28.17 | −3.06 | 2.80 × 107 | 73, 100, 106, 107, 108 | |
| 135 | 3-Chloropropionamide | ClCH 2 CH 2 CONH 2 | 10.52 | −4.74 | 1.94 × 10 9 | 104 | |
| 136 | (S)-2-Hydroxypropanamide | CH3CH(OH)CONH2 | −29.16 | −3.02 | 1.91 × 108 | 49 | |
| 137 | Aceturate | CH3CONHCH2COO− | −25.84 | −3.16 | 1.13 × 107 | 65, 109 | |
| 138 | Pivalamide | (CH3)3CCONH2 | −27.03 | −3.11 | 1.50 × 107 | 100 | |
| 139 | Malonamide | H2NCOCH2CONH2 | −30.47 | −2.96 | 1.15 × 109 | 110 | |
| 140 | 2-Hydroxyacetamide | HOCH2CONH2 | −29.10 | −3.02 | 2.93 × 108 | 49 | |
| 141 | Biuret | H2NCONHCONH2 | −26.98 | −3.11 | 2.53 × 108 | 110 | |
| 142 | 2-Chloropropionamide | CH 3 CH(Cl)CONH 2 | 0.91 | −4.32 | 7.58 × 10 9 | 104 | |
| 143 | Iodoacetamide | ICH 2 CONH 2 | −2.75 | −4.16 | 5.00 × 10 10 | 111 | |
| 144 | Hydroxyurea | HONHCONH2 | −27.45 | −3.09 | 4.90 × 108 | 112 | |
| 145 | Oxamate | H2NCOCOO− | −44.35 | −2.36 | 5.70 × 109 | 110 | |
| 146 | Succinamide | H2NCOCH2CH2CONH2 | −26.23 | −3.14 | 2.02 × 108 | 110 | |
| 147 | Asparaginate | H2NCOCH2CH(NH2)COO− | −26.51 | −3.13 | 2.40 × 107 | 113 | |
| 148 | N,N-Dimethylformamide | HCON(CH3)2 | −30.35 | −2.96 | 3.08 × 108 | 100, 107, 108 | |
| 149 | Methyl 2-acetamidoacetate | CH3CONHCH2COOCH3 | −38.38 | −2.62 | 3.34 × 108 | 110 | |
| 150 | 2-Formamidoacetate | HCONHCH2COO− | −25.93 | −3.16 | 2.90 × 107 | 110 | |
| 151 | N-Methylformamide | HCONHCH3 | −25.68 | −3.17 | 4.31 × 107 | 100, 108 | |
| 152 | N-tert-Butylacetamide | CH3CONHC(CH3)3 | −21.69 | −3.34 | 1.20 × 107 | 100 | |
| 153 | Diacetamide | (CH3CO)2NH | −43.29 | −2.40 | 1.98 × 1010 | 110 | |
| 154 | N,N-Diethylacetamide | CH3CON(C2H5)2 | −23.89 | −3.24 | 8.00 × 106 | 100 | |
| 155 | N,N-Dimethylacetamide | CH3CON(CH3)2 | −27.42 | −3.09 | 1.50 × 107 | 100, 105 | |
| 156 | (CH3)3CCON(CH3)2 | −29.95 | −2.98 | 1.20 × 107 | 100 | ||
| Ammonia | 157 | Methyl ammonium hydride | CH3NH3+ | −50.08 | −2.11 | 1.85 × 106 | 97 , 113 |
| 158 | Ethylammonium | C2H5NH3+ | −51.52 | −2.05 | 2.50 × 106 | 98 | |
| 159 | Trideuterio(propyl)azanium | those in bold | −50.99 | −2.07 | 2.80 × 106 | 98 | |
| 160 | Pentylazanium | CH3(CH2)4NH3+ | −51.57 | −2.04 | 2.70 × 106 | 98 | |
| 161 | 2-Methoxy-2-oxoethanaminium | H3COOCCH2NH3+ | −59.49 | −1.70 | 6.80 × 109 | 65 | |
| 162 | Methoxyazanium | CH3ONH3+ | −96.51 | −0.10 | 1.90 × 1010 | 65 | |
| 163 | tert-Butylammonium | (CH3)3CNH3+ | −53.40 | −1.96 | 1.10 × 106 | 97 | |
| 164 | 2-Methylhydrazinium | CH3NHNH3+ | −80.62 | −0.78 | 1.40 × 109 | 99 | |
| 165 | 1,1-Dimethylhydrazinium | (CH3)2NNH3+ | −85.83 | −0.56 | 5.80 × 109 | 99 | |
| 166 | Tetramethylammonium | (CH3)4N+ | −49.22 | −2.15 | 5.60 × 106 | 114 | |
| 167 | Tetraethylammonium | (C2H5)4N+ | −52.94 | −1.98 | 1.20 × 107 | 114 | |
| Hydrogen sulfide | 168 | Cysteaminium | HSCH2CH2NH3+ | −51.15 | −2.06 | 2.25 × 1010 | 115 , 116 |
| 169 | 3-Sulfanylpropylazanium | HS(CH2)3NH3+ | −52.08 | −2.02 | 1.70 × 1010 | 117 | |
| Alkyne | 170 | Acetylene | HC triplet bond CH | −21.82 | −3.33 | 2.00 × 107 | 118 |
| 171 | Propargyl alcohol | HC triplet bond CCH2OH | −24.16 | −3.23 | 2.12 × 108 | 68 | |
| Sulfate | 172 | Ethanesulfonate | C2H5SO3− | 7.65 | −4.61 | 3.50 × 107 | 119 |
| Sulfoxide | 173 | Dibutyl sulphoxide | [CH 3 (CH 2 ) 3 SO(CH 2 ) 3 CH 3 ] | 22.09 | −5.24 | 3.60 × 10 6 | 120 |
| 174 | Di-tert-butyl sulfoxide | [(CH 3 ) 3 C] 2 SO | −63.62 | −1.52 | 1.50 × 10 7 | 120 | |
| 175 | Methyl (methylsulfinyl)methyl sulfide | CH 3 SOCH 2 SCH 3 | 22.05 | −5.24 | 1.31 × 10 8 | 121 | |
| Thiol | 176 | Methanethiol | CH3SH | −47.75 | −2.21 | 1.08 × 1010 | 122 |
| 177 | Thiolactate | CH3(CH)SHCOO− | −58.46 | −1.75 | 2.89 × 109 | 116 | |
| 178 | 2-Mercaptopropionic acid | CH3CH(SH)COOH | −62.50 | −1.57 | 4.08 × 109 | 123 | |
| 179 | Methyl thioglycolate | HSCH2COOCH3 | −56.08 | −1.85 | 1.12 × 1010 | 116 | |
| 180 | Beta-mercaptoethanol | HS(CH2)2OH | −49.88 | −2.12 | 1.73 × 1010 | 115 , 124 | |
| 181 | 2-Methyl-2-propanethiol | (CH3)3CSH | −54.27 | −1.93 | 3.41 × 109 | 122 | |
| 182 | 3-Mercaptopropionic acid | HS(CH2)2COOH | −50.10 | −2.11 | 6.91 × 109 | 123 | |
| 183 | Thioglycolate | HSCH2COO− | −54.30 | −1.93 | 3.03 × 109 | 116 | |
| 184 |
H2NC(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) NH)NHCH2CH2SH NH)NHCH2CH2SH
|
−51.25 | −2.06 | 1.02 × 1011 | 113 | ||
| Sulfide/disulfide | 185 | Dimethylsulfide | CH 3 SCH 3 | 52.27 | −6.55 | 2.00 × 10 7 | 125 |
| 186 | 3,3′-Dithiodipropionate | (SCH 2 CH 2 COO − ) 2 | 20.22 | −5.16 | 4.35 × 10 9 | 126 | |
| 187 | 2,2′-Disulfanediyldiacetate | (SCH 2 COO − ) 2 | 25.32 | −5.38 | 4.30 × 10 9 | 126 | |
| 188 | 2,2′-Sulfanediyldiacetate | S(CH 2 COO − ) 2 | 34.86 | −5.79 | 8.30 × 10 7 | 116 | |
| 189 | N-Acetylcysteamine | CH 3 CONHCH 2 CH 2 SH | 29.28 | −5.55 | 1.43 × 10 10 | 116 | |
| 190 | Cystamine | S 2 (CH 2 CH 2 NH 2 ) 2 | 24.23 | −5.33 | 5.85 × 10 10 | 126 | |
| 191 | L-Cystine anion | S 2 [CH 2 CH(NH 2 )COO − ] 2 | 15.24 | −4.94 | 3.53 × 10 9 | 39 , 115 , 126 , 127 | |
| 192 | 3,3′-Thiodipropanoate | S(CH 2 CH 2 COO − ) 2 | 33.51 | −5.73 | 5.80 × 10 7 | 116 | |
| S− | 193 | 2-Hydroxyethanethiolate | HOCH2CH2S− | −15.20 | −3.62 | 1.80 × 107 | 115 |
| 194 | 2-Lambda1-sulfanylethanamine | H2NCH2CH2S− | −16.84 | −3.55 | 9.55 × 108 | 115 , 116 | |
| 195 | 2-Acetamidoethanethiolate | CH3CONHCH2CH2S− | −16.11 | −3.58 | 1.90 × 109 | 116 | |
| CS | 196 | Carbon disulfide | CS2 | −57.80 | −1.77 | 3.10 × 1010 | 128, 45 |
| 197 | Thiourea | H2NCSNH2 | −18.12 | −3.49 | 3.29 × 109 | 20 | |
| 198 | Thiosemicarbazide | H2NNHCSNH2 | −19.10 | −3.45 | 1.15 × 109 | 129 | |
| 199 | N,N′-Diethylthiourea | CH3CH2NHCSNHCH2CH3 | −19.13 | −3.45 | 5.10 × 108 | 129 | |
| Nitro | 200 | Nitromethane | CH3NO2 | −61.02 | −1.63 | 1.80 × 1011 | 130–131 |
| 201 | 1-Nitropropane | CH3CH2CH2NO2 | −60.85 | −1.64 | 2.70 × 1010 | 132 | |
| 202 | Nitroethane | CH3CH2NO2 | −60.17 | −1.67 | 2.70 × 1010 | 132 | |
| 203 | 2-Methyl-2-nitrosopropane | (CH3)3C(NO) | −63.46 | −1.53 | 8.26 × 109 | 133 | |
| PFAS | 204 | Trifluoroacetate | CF 3 COO − | 76.90 | −7.61 | 1.65 × 10 6 | 69 , 134 |
| 205 | Perfluorobutanoic acid | C 3 F 7 COO − | 57.88 | −6.79 | 7.10 × 10 6 | 134 | |
| 206 | Perfluorooctanoic acid | C 7 F 15 COO − | 42.92 | −6.14 | 1.70 × 10 7 | 134 | |
| Alkene | 207 | Allylamine | H2CCHCH2NH2 |
−23.13 | −3.28 | 1.20 × 107 | 97 |
| 208 | Acrylonitrile | H2CCHCN |
−53.94 | −1.94 | 2.78 × 1010 | 135 | |
| 209 | Allyl alcohol | H2CCHCH2OH |
−27.37 | −3.09 | 3.47 × 107 | 54, 68, 70 | |
| 210 | Acrylic acid | H2CCHCOOH |
−59.27 | −1.71 | 1.03 × 1012 | 136 | |
| 211 | Acrylate | CH2CHCOO− |
−40.74 | −2.51 | 5.30 × 109 | 136 | |
| 212 | Methyl vinyl ketone | H2CCHCOCH3 |
−63.32 | −1.53 | 2.78 × 109 | 137 | |
| 213 | Methyl acrylate | H2CCHCOOCH3 |
−57.17 | −1.80 | 1.52 × 1010 | 138 | |
| 214 | Senecioic acid amide | (CH3)2CCHCONH2 |
−44.00 | −2.37 | 7.23 × 109 | 139 | |
| 215 | Vinyl chloride |
CH
2
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) CHCl CHCl
|
27.10 | −5.45 | 2.53 × 10 8 | 140 | |
| 216 | Ethylene | H2CCH2 |
−24.75 | −3.21 | 3.00 × 105 | 54 | |
| 217 | Ethenesulfonate | CH2CHSO3− |
−37.67 | −2.65 | 2.30 × 109 | 141 | |
| 218 | Tetrachloroethylene |
Cl
2
CCCl
2
|
15.17 | −4.94 | 2.67 × 10 10 | 90 , 140 | |
| 219 | Crotonyl alcohol | CH3CHCHCH2OH |
−24.31 | −3.23 | 5.51 × 107 | 54 | |
| 220 | Crotonic acid | CH3CHCHCOOH |
−54.36 | −1.92 | 6.62 × 1010 | 136 | |
| 221 | Dimethyl fumarate | CH3OOCCHCHCOOCH3 |
−76.95 | −0.94 | 3.30 × 1010 | 110 | |
| 222 | Divinyl sulfone | (H2CCH)2SO2 |
−55.62 | −1.87 | 1.66 × 1010 | 137 | |
| 223 | Methacrylic acid | H2CC(CH3)COOH |
−56.59 | −1.83 | 8.26 × 1010 | 136 | |
| 224 | Methyl methacrylate | H2CC(CH3)COOCH3 |
−54.41 | −1.92 | 2.72 × 1010 | 139 | |
| 225 | trans-1,2-Dichloroethylene |
ClCHCHCl
|
22.70 | −5.26 | 1.08 × 10 10 | 140 | |
| 226 | Trichloroethylene |
ClCHCCl
2
|
18.45 | −5.08 | 8.28 × 10 10 | 140 | |
| 227 | cis-1,2-Dichloroethylene |
H
2
CCCl
2
|
19.86 | −5.14 | 3.86 × 10 11 | 140 | |
| 228 | 1,3-Butadiene | H2CCHCHCH2 |
−42.65 | −2.43 | 1.19 × 1010 | 20 | |
| 229 | Acetaldehyde oxime | CH3CHNOH |
−30.63 | −2.95 | 7.22 × 107 | 37 | |
| 230 | N,N-Dimethylacrylamide | CH2CHCON(CH3)2 |
−51.04 | −2.07 | 4.51 × 1010 | 139 | |
| 231 | Methacrylamide | H2CC(CH3)CONH2 |
−49.80 | −2.12 | 7.10 × 1011 | 139 | |
| 232 | Cyanoguanidine | NCNC(NH2)2 |
−31.89 | −2.90 | 1.96 × 1010 | 142 | |
| 233 | Tetracyanoethylene |
(NC)
2
CC(CN)
2
|
36.90 | −5.88 | 3.74 × 10 10 | 20 | |
| 234 | Methacrylate | CH2C(CH3)COO− |
−36.63 | −2.69 | 4.50 × 109 | 136 | |
| 235 | 3-Buten-1-ol | H2CCHCH2CH2OH |
−22.99 | −3.28 | 2.45 × 106 | 54, 68 | |
| 236 | 3-Buten-2-ol | H2CCHCH(OH)CH3 |
−26.41 | −3.13 | 5.91 × 107 | 54 | |
| 237 | 3-Methylbut-2-enoate | (CH3)2CCHCO2− |
−31.71 | −2.91 | 6.40 × 108 | 143 | |
| 238 | 3,3-Dimethylacrylic acid | (CH3)2CCHCOOH |
−50.40 | −2.09 | 2.53 × 1010 | 136, 143 | |
| 239 | Isocrotonate | CH3CHCHCOO− |
−35.70 | −2.73 | 1.30 × 109 | 136 | |
| 240 | Hydrogen fumarate | HOOCCHCHCOO− |
−66.40 | −1.40 | 1.35 × 1010 | 48, 110 | |
| 241 | Monomethyl fumarate | CH3OOCCHCHCOO− |
−64.43 | −1.49 | 1.30 × 1010 | 110 | |
| 242 | 2-Hydroxyethyl acrylate | CH2CHCOOCH2CH2OH |
−57.85 | −1.77 | 1.08 × 1010 | 144 | |
| 243 | trans-Aconitate(3−) |
−OOCCHC(COO−)CH2COO− |
−45.03 | −2.33 | 1.80 × 108 | 51 | |
| 244 | Acrylamide | H2CCHCONH2 |
−51.89 | −2.03 | 3.81 × 1011 | 45, 106, 107, 139, 145, 146 | |
| 245 | Crotonamide | CH3CHCHCONH2 |
−47.62 | −2.22 | 2.75 × 1010 | 139 | |
| 246 | 4-(Ethylamino)-4-oxobut-2-enoate | C2H5NHCOCHCHCOO− |
−56.87 | −1.81 | 8.50 × 109 | 99 | |
| 247 | cis-Dimethyl fumarate | CH3OOCCHCHCOOCH3 |
−73.51 | −1.09 | 3.20 × 1010 | 110 | |
| 248 | 4-Penten-2-OL | H2CCHCH2CH(OH)CH3 |
−21.90 | −3.33 | 5.00 × 105 | 68 | |
| 249 | Guanidine | H2NC(NH)NH2 |
−4.98 | −4.06 | 2.02 × 108 | 113 | |
| 250 | Ethyl acrylate | H2CCHCOOC2H5 |
−57.33 | −1.79 | 1.34 × 1010 | 138 | |
| 251 | Acetone oxime | (CH3)2CNOH |
−25.74 | −3.16 | 3.29 × 108 | 37, 106 |
For the concerted cleavage of the C–Cl bond of haloalkanes and halocarboxylate, we determined the LFERs to be lnkchem = 5.66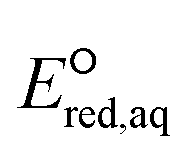 + 27.95 (r2 = 0.73, N = 19) and lnkchem = 3.97
+ 27.95 (r2 = 0.73, N = 19) and lnkchem = 3.97 + 28.92 (r2 = 0.99, N = 4), respectively (Fig. 2c). The SD values were 0.044 and 0.025, respectively. All the kchem values of the haloalkane and halocarboxylate compounds that contain C–Br and C–I bonds were close to or exceeded the diffusion limit; therefore, we did not determine their LFERs. The presence of carboxylate functional groups impacted the
+ 28.92 (r2 = 0.99, N = 4), respectively (Fig. 2c). The SD values were 0.044 and 0.025, respectively. All the kchem values of the haloalkane and halocarboxylate compounds that contain C–Br and C–I bonds were close to or exceeded the diffusion limit; therefore, we did not determine their LFERs. The presence of carboxylate functional groups impacted the 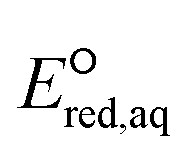 value of the cleavage of the C–Cl bond in the halocarboxylates; thus, a different LFER was determined for the group of chlorinated halocarboxylates. The four chlorinated carboxylates also appeared to adhere to the LFER for stepwise mechanism. According to experimental works, the group of these compounds undergo both concerted and stepwise mechanisms21,22 and thus we keep these compounds in both LFERs. We also determined the LFER for the concerted cleavage of the C–N bond of alkyl ammonium (RNH3+) as lnkchem = 4.92
value of the cleavage of the C–Cl bond in the halocarboxylates; thus, a different LFER was determined for the group of chlorinated halocarboxylates. The four chlorinated carboxylates also appeared to adhere to the LFER for stepwise mechanism. According to experimental works, the group of these compounds undergo both concerted and stepwise mechanisms21,22 and thus we keep these compounds in both LFERs. We also determined the LFER for the concerted cleavage of the C–N bond of alkyl ammonium (RNH3+) as lnkchem = 4.92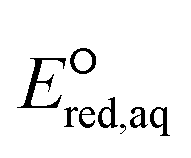 + 24.67 (r2 = 0.98, N = 7) (Fig. 2c) with the exception of tetramethylammonium (no. 166) and tetraethylammonium (no. 167), which contain a different base structure of >N+–C. Additionally, we did not include compounds that contain oxygenated functional groups (no. 161, oxoethanaminium and no. 162, methoxyazanium) because of their dominant associative mechanism. The SD value was 0.051.
+ 24.67 (r2 = 0.98, N = 7) (Fig. 2c) with the exception of tetramethylammonium (no. 166) and tetraethylammonium (no. 167), which contain a different base structure of >N+–C. Additionally, we did not include compounds that contain oxygenated functional groups (no. 161, oxoethanaminium and no. 162, methoxyazanium) because of their dominant associative mechanism. The SD value was 0.051.
For the stepwise mechanism, we determined the LFERs for compounds with (1) haloalkanes that contain CO functional groups (i.e., halocarboxylates, halooxygens and haloamides) or C–F bonds (fluorinated carboxylates), (2) haloalkenes, and (3) sulfides or disulfides for the stepwise mechanism to be the following: (1) lnkchem = 3.96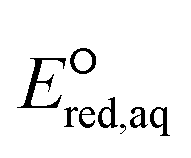 + 40.29 (r2 = 0.69, N = 22) or lnkchem = 1.43
+ 40.29 (r2 = 0.69, N = 22) or lnkchem = 1.43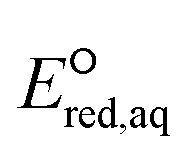 + 22.58 (r2 = 0.74, N = 4), (2) lnkchem = 10.36
+ 22.58 (r2 = 0.74, N = 4), (2) lnkchem = 10.36 + 77.25 (r2 = 0.54, N = 5), and (3) lnkchem = 4.45
+ 77.25 (r2 = 0.54, N = 5), and (3) lnkchem = 4.45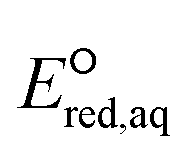 + 45.65 (r2 = 0.57, N = 8) (Fig. 2d). Because of the high strength of a C–F bond, the slope of the LFER for fluorinated carboxylates is significantly smaller than that of other haloalkanes. The SD values were 0.048 for haloalkanes, 0.26, for fluorinated carboxylates, 0.077 for haloalkenes, and 0.089 for disulfides. A detailed mechanistic discussion and prediction of PFAS are given below.
+ 45.65 (r2 = 0.57, N = 8) (Fig. 2d). Because of the high strength of a C–F bond, the slope of the LFER for fluorinated carboxylates is significantly smaller than that of other haloalkanes. The SD values were 0.048 for haloalkanes, 0.26, for fluorinated carboxylates, 0.077 for haloalkenes, and 0.089 for disulfides. A detailed mechanistic discussion and prediction of PFAS are given below.
Impact of functional groups
The functional group(s) in the neighboring position of an eaq− attacking site substantially impact the reactivities with eaq−. In general, electron donating groups such as alkyl and amine functional groups in the neighboring position(s) decrease the nucleophilic reactivity of eaq− by increasing the electron density of the reactive site. For example, the negatively charged oxygen of the COO− functional group acts as an electron donor to the adjacent CO bond due to its lone pair of electrons and hence reduces the reactivity of eaq− in association with CO. In contrast, electron withdrawing functional groups such as ketones and carboxylic acids decrease the electron density of the reactive site and hence increase the reactivity of eaq−. Fig. 3 plots the total sum of the Taft constants,147 σ*, of neighboring functional group(s) against our 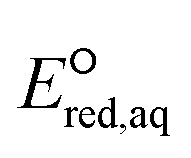 values for all the reaction mechanisms investigated in this study. The Taft constants of functional group(s) located in the neighboring position(s) of an eaq− attacking site are additive.148,149 When the Taft constant of a functional group was not available, we used the value of a structurally similar functional group. Fig. S6 in the ESI† provides all the Taft constant values we used. Overall, we confirm the excellent correlations of all three reaction mechanisms, indicating that our theoretically calculated
values for all the reaction mechanisms investigated in this study. The Taft constants of functional group(s) located in the neighboring position(s) of an eaq− attacking site are additive.148,149 When the Taft constant of a functional group was not available, we used the value of a structurally similar functional group. Fig. S6 in the ESI† provides all the Taft constant values we used. Overall, we confirm the excellent correlations of all three reaction mechanisms, indicating that our theoretically calculated 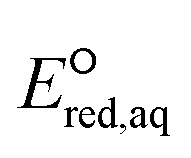 values represent the general electron donating/withdrawing properties of the functional groups of aliphatic compounds. As expected, all correlations exhibit positive slopes, which confirm that larger
values represent the general electron donating/withdrawing properties of the functional groups of aliphatic compounds. As expected, all correlations exhibit positive slopes, which confirm that larger 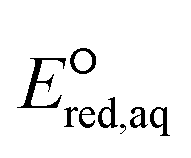 values represent stronger electron-withdrawing functional groups (i.e., larger Taft constants). As shown by the LFERs in Fig. 2, larger
values represent stronger electron-withdrawing functional groups (i.e., larger Taft constants). As shown by the LFERs in Fig. 2, larger  values correlate with larger overall kchem values because of the increase in the nucleophilic reactivities of eaq−. Different correlations with Taft constants developed for the associative mechanism with CO and O confirm the different influence of neighboring functional groups resulting from inductive and mesomeric effects. The smaller slope (1.53) for the associative mechanism with O than that (1.69) for the associative mechanism with CO indicates the inhibition of reactivity resulting from the mesomeric effect. Hart et al.37 examined the negative slope between logkexp and the Taft constants for groups of ketones with a limited number of data (N = 10) and concluded that the slowing effect due to the mesomeric effect was more extensive than expected from inductive electron-donating/withdrawing effects. They also acknowledged that both mesomeric and inductive effects could apply to carboxylic compounds (N = 3), and their data appeared to adhere to both correlations well. Notably, two compounds that we propose for the stepwise cleavage mechanism of the C–F bond of CF3COCH3 (no. 98) and CH3COCH2F (no. 99) (see discussion on the reaction mechanism below) were included in the correlation with the Taft constant for the associative mechanism with CO by Hart. If these compounds and carboxylic compounds were removed from their correlation, a handful of compounds (N = 4) would remain in close proximity, and a negative correlation between the kchem values and the sum of Taft constants would not be observed. Most likely, our extensive analysis of data (N = 66) and the use of
values correlate with larger overall kchem values because of the increase in the nucleophilic reactivities of eaq−. Different correlations with Taft constants developed for the associative mechanism with CO and O confirm the different influence of neighboring functional groups resulting from inductive and mesomeric effects. The smaller slope (1.53) for the associative mechanism with O than that (1.69) for the associative mechanism with CO indicates the inhibition of reactivity resulting from the mesomeric effect. Hart et al.37 examined the negative slope between logkexp and the Taft constants for groups of ketones with a limited number of data (N = 10) and concluded that the slowing effect due to the mesomeric effect was more extensive than expected from inductive electron-donating/withdrawing effects. They also acknowledged that both mesomeric and inductive effects could apply to carboxylic compounds (N = 3), and their data appeared to adhere to both correlations well. Notably, two compounds that we propose for the stepwise cleavage mechanism of the C–F bond of CF3COCH3 (no. 98) and CH3COCH2F (no. 99) (see discussion on the reaction mechanism below) were included in the correlation with the Taft constant for the associative mechanism with CO by Hart. If these compounds and carboxylic compounds were removed from their correlation, a handful of compounds (N = 4) would remain in close proximity, and a negative correlation between the kchem values and the sum of Taft constants would not be observed. Most likely, our extensive analysis of data (N = 66) and the use of 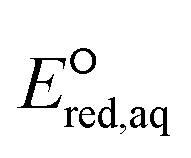 values revealed a better comprehensive picture of the impact of functional groups.
values revealed a better comprehensive picture of the impact of functional groups.
 | ||
Fig. 3 Sum of Taft constants against the theoretically calculated 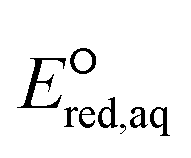 values of each compound. values of each compound. | ||
Neighboring functional groups also affect the concerted cleavage mechanism of each C–Cl, C–NH3+, and S–S bond as well as the stepwise cleavage mechanism of each C–Cl and S–S bond. The slopes of the correlations for C–Cl bond cleavage for both concerted and stepwise mechanisms were found to be significantly steeper than those for C–N and C–S cleavages. The similar slopes of the correlations for C–Cl bond between concerted and stepwise mechanisms indicate that the impact of neighboring functional groups is similar despite of the formation of intermediate species in the stepwise mechanism. While the Taft constants for C–C bond cleavage were substantially different for various functional groups in the neighboring positions, the range of 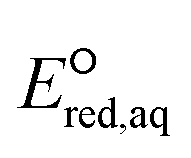 values was within approximately 1 V. These results indicated the higher sensitivity of the
values was within approximately 1 V. These results indicated the higher sensitivity of the 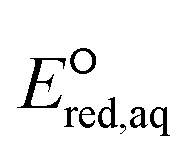 values associated with the properties of the electron-donating/withdrawing functional groups when compared with those examined for C–N and S–S bonds.
values associated with the properties of the electron-donating/withdrawing functional groups when compared with those examined for C–N and S–S bonds.
Reaction mechanisms
O is maintained or the stepwise cleavage of a C–X bond occurs in the group of halooxygens.21,22,152 Our investigation of the PES of methyl trifluoroacetate (no. 39) and the product of radical anion as a function of one of dihedral angles showed the merging point of these PESs at approximately −70 degree (Fig. S2 in the ESI†), indicating the possibility of stepwise dissociative mechanism. However, the spin density distributions of elongated C–F bond structures were located on the acetate carbonyl functional group with an increase in one of the C–F bonds, which confirmed the associative mechanism (Fig. S3 in the ESI†). Our investigation on the lowest unoccupied molecular orbital (LUMO) represents no antibonding orbitals with regard to the C–F bond (Fig. S4 in the ESI†), supporting the initial electron association with the CO functional group.
The mesomeric effect described in the overall results move the reaction center from the CO functional group of carboxylic, ester, and acetamide compounds to the alkoxyl group of O. The partial positive charge generated on OH of carboxylic and alcohol, C–O of ester, C–N of acetamide is the site of eaq− addition in the mesomeric form, which were confirmed by our analysis on the charge distribution (Fig. S5 in the ESI†). Although concerted cleavage of a C–O bond of esters or alcohol could occur, a better correlation of LFER for the association with O than those for the concerted cleavage of a C–O bond is the evidence for the dominant associative mechanism (Fig. S6 in the ESI†).
The kchem values range from 107 to 1012 M−1 s−1 for the group of alkenes and some kexp values are very close to or exceed the diffusion-limited rate. The association of eaq− with one of unsaturated carbons generates a radical anion intermediate described as a 3-electron 2-orbital state of π character22 with carbon atoms that are sp3 hybridized with a dangling lone pair of electrons on one carbon atom and an unpaired radical electron on the other carbon atom. The initial injection of an electron produces a carbon-centered radical that further undergoes the reaction with second eaq− to produce a stable unsaturated carbon after leaving the halogen functional group.
The group of nitro compounds include three kchem values for nitromethane (no. 200), 1-nitropropane (no. 201), and nitroethane (no. 202). The kchem values for all the compounds are greater than 1010 M−1 s−1 exceeding the diffusion-limited. Our 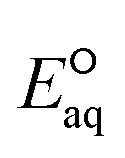 values for the associative mechanism are approximately −1.6 V for all the compounds, whereas those for the concerted cleavage of C–NO2 bond are approximately −2.2 V. This indicates the preference of the associative mechanism with eaq− and is supported by the spin density distribution on the NO2 functional group (Fig. S7 in the ESI†). Due to the few datasets, we were not able to develop the LFER.
values for the associative mechanism are approximately −1.6 V for all the compounds, whereas those for the concerted cleavage of C–NO2 bond are approximately −2.2 V. This indicates the preference of the associative mechanism with eaq− and is supported by the spin density distribution on the NO2 functional group (Fig. S7 in the ESI†). Due to the few datasets, we were not able to develop the LFER.
Haloalkane and halocarboxylate. The concerted dissociative cleavage of a C–X (X = Cl, Br, and I) bond of haloalkanes is supported by the presence of σ* antibonding orbitals with respect to the C–X bond. Injection of an electron into such orbitals is accompanied by the barrierless dissociation of the C–X bond and the reductive cleavage follows the concerted dissociative mechanism.21,22,153 Despite the fact that a carboxylate functional group was present in the given molecular structure of haloalkanes, we observed that the weak C–X (X = Br and I) bond of halocarboxylates underwent concerted reduction due to the inability to hold the eaq−. We investigated the spin density distribution in a given molecule to identify a possible attacking site by eaq−. The spin density distribution on four chlorinated carboxylates indicates the solvated electron was located on the carbon atom in the C–Cl group (Fig. S8 in the ESI†), suggesting that the C–Cl bond cleavage could occur upon the attack by the eaq−. It should be noted that we observed the significant elongation of a C–Cl bond upon the structure optimization for 2-chloropropanoate (3.77 Å of C–Cl) and trichloroacetate (3.58 Å of C–Cl). This bond elongation suggests that the C–Cl bonds may not cleave upon the attack by the solvated electron, and thus the stepwise reduction mechanism may occur. The eaq− was likely held in the σ* antibonding orbital of chlorine, forming an intermediate radical anion species. Because of the uncertainty of the aqueous-phase PES, we were not able to confirm the dominant mechanism on these two chlorinated carboxylates. The detailed investigation on the PES for this group of compounds are underway.
The brominated and iodinated species appeared to undergo concerted reduction. In all radical anion structures, the C–X (X = Br or I) bond was elongated significantly and the eaq− was located on the carbon of the C–X group (Fig. S9 and S10 in the ESI†). Because we did not observe any associative mechanism of eaq− with the CO functional group, the stepwise reduction mechanism seemed to be unlikely. The concerted mechanism is reasonable because the C–Br bond strength is also relatively weak (285 kJ mol−1) compared to C–Cl and C–F, which is consistent with experimental finding.23
The C–N bond in general undergoes concerted dissociative cleavage. We determined two LFERs of ammonium compounds undergoing concerted and associative mechanisms and confirmed the concerted cleavage of a C–N bond of ammonium functional group for the rate determining step (Fig. S11 in the ESI†). The group of cyanide included 3kchem values of acetonitrile (no. 106), succinonitrile (no. 107), and cyanamide (no. 109). While the kchem value of acetonitrile was 107-th order, the other two were 109-th order. Our 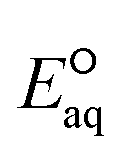 values for the concerted cleavage of C–CN bond for these compounds ranged from −3.33 V to −3.06 V, whereas those for the association were from −3.64 V to −3.33 V. These indicate the preference of concerted cleavage of C–CN bond to form cyanide ion (CN−) and carbon-centered radicals. Our investigation on the spin density of cyanide compounds indicated the high spin density at the cyanide functional group (Fig. S12 in the ESI†). Due to the small number of compounds, we were not able to develop the LFER.
values for the concerted cleavage of C–CN bond for these compounds ranged from −3.33 V to −3.06 V, whereas those for the association were from −3.64 V to −3.33 V. These indicate the preference of concerted cleavage of C–CN bond to form cyanide ion (CN−) and carbon-centered radicals. Our investigation on the spin density of cyanide compounds indicated the high spin density at the cyanide functional group (Fig. S12 in the ESI†). Due to the small number of compounds, we were not able to develop the LFER.
The group of thiol contained 12 compounds containing at least one –SH functional group. As discussed above, the C–S bond is generally the weak point of a molecule because of its bond weakness in comparison to the C–C and C–H bonds. In the thiol compounds, the eaq− likely attacks the C–S group and results in the immediate bond cleavage due to a lack of antibonding σ* orbitals on the –SH functional group to hold the extra electron. Or the eaq− associates with the CO bond and loosens the C–S bond to cleave in the stepwise mechanism. Among all compounds containing thiol functional group, we did not observe any clear LFERs for both mechanisms (Fig. S13 in the ESI†). However, for thiols that do not contain CO functional group, we observed the acceptable LFER for concerted mechanism due to the limited number of data. Therefore, this class of compounds is likely reduced by the concerted mechanism, generating R˙ and HS−.154
Halocarboxylates, halooxygens, chlorinated amides and haloalkenes. We observed the consistent stepwise mechanisms for halocarboxylates (no. 49–59), halooxygens (no. 97–105), and chlorinated amides (no. 135 and no. 142) because of the presence of COO−, OH and CO functional groups, which are consistent with previous experimental observations.20–22 When haloalkanes contain electron-withdrawing and π-acceptor functional groups or other π-fragments, the electrons may initially occupy low-energy π*-orbitals and the reduction of these molecules may result in the transient formation of radical anions.155 For the fluorinated carboxylates (no. 53 and 204–206), we determined the different trend from other halocarboxylates because of the abnormally strong C–F bond. While the optimized structure did not show the elongation of the C–F bond, our spin density observation of fluoroacetate confirms the association of eaq− with the carboxylate functional group (Fig. S14 in the ESI†). In addition, the lowest unoccupied molecular orbital (LUMO) of fluoroacetates confirms antibonding orbitals with regard to the C–F bonds (Fig. S15 in the ESI†). The singly occupied molecular orbitals (SOMO) of the vertically excited radical anions (C–F˙−)* are characterized by essentially the same shapes (Fig. S16 in the ESI†). Geometry optimization of these intermediate radical anion resulted in the significant elongation of one of the C–F bonds and formation of the {C⋯F}˙−. The electronic structure of the radical anion intermediate as a 3-electron 2-orbital state of p character with carbon atoms that are sp3 rather than sp2 hybridized with a dangling lone pair of electrons on one carbon atom and an unpaired radical electron on the other carbon atom.
Electron-withdrawing functional groups adjacent to a CO functional group induce a shortening of the CO bond156 (e.g., 1.30 Å of CF3COCH3 (no. 98) and 1.31 Å of CH3COCH2F (no. 99) compared to 1.32 Å of CH3COCH3 from our optimized structures), which leads to a lower electron density in the π orbitals, resulting in the higher reactivity toward eaq−. However, the kchem values of CF3COCH3 and CH3COCH2F do not appear to follow this trend and show substantially smaller rate constants (i.e., 107–108 M−1 s−1) for associative mechanism. We propose that these two compounds undergo stepwise mechanism where eaq− associates with CO π bond and elongates the C–F bond, followed by the cleavage of the C–F bond. The 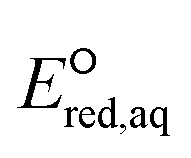 values of these compounds for the stepwise mechanism are −5.36 V for CF3COCH3 and −5.12 V for CH3COCH2F. We confirmed that these
values of these compounds for the stepwise mechanism are −5.36 V for CF3COCH3 and −5.12 V for CH3COCH2F. We confirmed that these 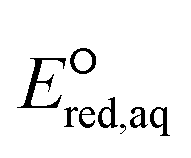 values adhere to the LFER developed for halooxygen/halocarboxylate undergoing stepwise. It should be noted that the aqueous-phase PES of radical anions of these compounds (i.e., intermediate) as a function of dihedral angle has uncertainties in the energy values and we were not able to confirm the stepwise mechanism. Our investigation on the spin density distribution shown in Fig. S17–S20† for both compounds provide the evidence of electron association and elongation of the C–F bond, which support the stepwise mechanism. Furthermore, the LUMO of both compounds (Fig. S21†) was the evidence of stepwise cleavage that holds the electron in one of the C–F bonds. Based on our investigation, we only propose stepwise mechanisms for these two compounds and further study is needed to confirm the reaction mechanism.
values adhere to the LFER developed for halooxygen/halocarboxylate undergoing stepwise. It should be noted that the aqueous-phase PES of radical anions of these compounds (i.e., intermediate) as a function of dihedral angle has uncertainties in the energy values and we were not able to confirm the stepwise mechanism. Our investigation on the spin density distribution shown in Fig. S17–S20† for both compounds provide the evidence of electron association and elongation of the C–F bond, which support the stepwise mechanism. Furthermore, the LUMO of both compounds (Fig. S21†) was the evidence of stepwise cleavage that holds the electron in one of the C–F bonds. Based on our investigation, we only propose stepwise mechanisms for these two compounds and further study is needed to confirm the reaction mechanism.
Sulfides, disulfides, sulfoxide. A total of 8 compounds were investigated for the group of sulfides that contain a –C–S–C– functional group and disulfide that has a –C–S–S–C– functional group. We determined the LFER for the stepwise mechanism that cleave the C–S bond of sulfides and the S–S bond of disulfides. We did not observe any correlation for the concerted mechanism (Fig. S22a in the ESI†). Although some sulfides (no. 186, 187, 191, and 192) contain COO− functional group that implicates the initial association with eaq−, those functional groups are located far from the C–S and S–S sites and do not appear to impact the elongation of those bonds upon the injection of a first electron. Upon the attack by eaq−, the S–S or C–S bond initially elongates which results in a decrease in the energy of the antibonding σ* orbital that localizes over the elongated S–S or C–S bond. This antibonding orbital temporarily holds the eaq− for more than one vibration, creating a three-electron bonded radical anion intermediate structure CSSC˙− or a C-centered radical.157 After the formation of this radical anion, the S–S bond cleaves, resulting in the following products: RS˙ and RS−via a stepwise mechanism.158,159
The sulfoxide class (no. 173–175) has the characteristic of a central SO double bond. The presence of a π-bond which allows for electron localization, accessible antibonding π* orbitals, and two weak C–S bonds suggest that this class of compounds is reduced via the stepwise mechanism. The eaq− likely attacks the sulfur atom, resulting in the elongation of a C–S bond, as was observed in the sulfide and disulfide class. Simultaneously, the π-bond transforms into a σ bond by shifting two electrons to the oxygen atom, creating a negative charge on the oxygen.
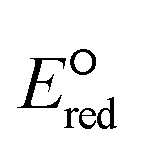 values for the alkane class to be low with the values ranging from −5.27 V to −5.41 V. The kchem values are also significantly small in the range from 106 and 107 M−1 s−1. For these reasons, we do not include any data in the groups of alkanes and amines in the analysis of LFERs.
values for the alkane class to be low with the values ranging from −5.27 V to −5.41 V. The kchem values are also significantly small in the range from 106 and 107 M−1 s−1. For these reasons, we do not include any data in the groups of alkanes and amines in the analysis of LFERs.
Prediction of the reactivities with per- and polyfluoroalkyl substances (PFASs)
We investigate the reactivities of eaq− with the 75 priority PFAS subset from the U.S. EPA. The 75 PFASs were grouped based on the functional groups. The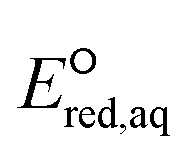 values for (1) the associative mechanism with CO and influenced by
values for (1) the associative mechanism with CO and influenced by 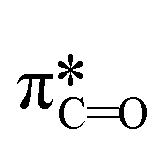 orbitals,160,161 (2) the associative mechanism with O, and (3) the stepwise C–F cleavage mechanism for all possible attacking sites in a given PFAS are summarized in Table 2. Fig. 4 displays the range of
orbitals,160,161 (2) the associative mechanism with O, and (3) the stepwise C–F cleavage mechanism for all possible attacking sites in a given PFAS are summarized in Table 2. Fig. 4 displays the range of 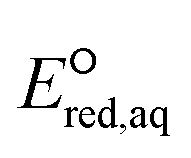 values for the stepwise cleavage of a C–F bond at different positions for the selected groups of PFASs investigated in this study. For this plot, we used M06-2X/cc-pVDZ for both structural optimization and frequency calculations and used the LFER determined from the same method to avoid significant computational time. Notably, we verified that the trend of all energies from representative PFASs was consistent between M06-2X/cc-pVDZ and M06-2X/Aug-cc-pVTZ (Table S6 in the ESI†). The group of polyfluorocarboxylates (PFCAs) has 7 PFASs with
values for the stepwise cleavage of a C–F bond at different positions for the selected groups of PFASs investigated in this study. For this plot, we used M06-2X/cc-pVDZ for both structural optimization and frequency calculations and used the LFER determined from the same method to avoid significant computational time. Notably, we verified that the trend of all energies from representative PFASs was consistent between M06-2X/cc-pVDZ and M06-2X/Aug-cc-pVTZ (Table S6 in the ESI†). The group of polyfluorocarboxylates (PFCAs) has 7 PFASs with 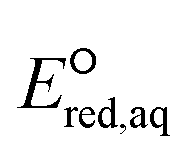 values in the range from −4.1 V to −2.3 V for the associative mechanism with CO and in the range from −7.3 V to −6.0 V for the stepwise C–F cleavage mechanism. From the largest
values in the range from −4.1 V to −2.3 V for the associative mechanism with CO and in the range from −7.3 V to −6.0 V for the stepwise C–F cleavage mechanism. From the largest 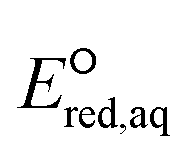 value in each reaction mechanism along with the LFER (lnkchem = 1.82
value in each reaction mechanism along with the LFER (lnkchem = 1.82 + 27.80 in Fig. S23 in the ESI†), the kchem values in neutral solution were predicted to range from 6.9 × 107 M−1 s−1 to 3.8 × 1010 M−1 s−1 for the associative mechanism and 4.5 × 106 M−1 s−1 to 2.1 × 107 M−1 s−1 for the stepwise cleavage mechanism of a C–F bond. The predicted kchem values of perfluorobutanoic acid (3 carbon chains, 6.87 × 107 M−1 s−1), perfluorohexanoic acid (5 carbon chains, 6.66 × 108 M−1 s−1), perfluorooctanoic acid (7 carbon chains, 5.78 × 108 M−1 s−1) and perfluorononanoic acid (8 carbon chains, 7.96 × 108 M−1 s−1) for the associative mechanism with CO were in excellent agreement with the recently reported kexp values of (5.4 ± 1.2) × 108 M−1 s−1 for perfluorobutanoic acid, (5.4 ± 0.1) × 108 M−1 s−1 for perfluorohexanoic acid, (7.1 ± 0.6) × 108 M−1 s−1 for perfluorooctanoic acid, and (6.4 ± 0.4) × 108 M−1 s−1 for perfluorononanoic acid.162 Although this experimental study did not determine the mechanism for those measured rate constants, we believe they measured the rates of the associative mechanism. In contrast, the kexp values for C–F cleavage (106–107 M−1 s−1) were previously reported134 and used for the determination of our LFERs (compound no. 204–206), which confirm the significantly smaller kchem values of the stepwise cleavage mechanism of a C–F bond. The predicted kchem values in three reaction mechanisms for all 75 PFASs are shown in Fig. S24 in the ESI.† The kchem values that exceeded the diffusion limit (3 × 1010 M−1 s−1 and lnk = 24.1) were not included in either figure. The kchem values predicted for the 75 PFASs that undergo the stepwise cleavage mechanism of a C–F bond range from 6.9 × 107 to 3.7 × 108 M−1 s−1. In contrast, 19kchem values range from 4.9 × 107 M−1 s−1 to 3 × 1010 M−1 s−1 for the associative mechanism with CO, 23kchem values range from 1.3 × 106 to 3 × 1010 M−1 s−1 for the associative mechanism with O, and 4kchem values are above the diffusion limit for the CC associative mechanism. We did not determine the LFERs for the functional groups of sulfonic acid (SO3−), phosphinic acid (PO(OH)2), sulfonamide (SO2NH), and sulfonyl (SO2); thus, no rate constant predictions were conducted. The investigation of the
+ 27.80 in Fig. S23 in the ESI†), the kchem values in neutral solution were predicted to range from 6.9 × 107 M−1 s−1 to 3.8 × 1010 M−1 s−1 for the associative mechanism and 4.5 × 106 M−1 s−1 to 2.1 × 107 M−1 s−1 for the stepwise cleavage mechanism of a C–F bond. The predicted kchem values of perfluorobutanoic acid (3 carbon chains, 6.87 × 107 M−1 s−1), perfluorohexanoic acid (5 carbon chains, 6.66 × 108 M−1 s−1), perfluorooctanoic acid (7 carbon chains, 5.78 × 108 M−1 s−1) and perfluorononanoic acid (8 carbon chains, 7.96 × 108 M−1 s−1) for the associative mechanism with CO were in excellent agreement with the recently reported kexp values of (5.4 ± 1.2) × 108 M−1 s−1 for perfluorobutanoic acid, (5.4 ± 0.1) × 108 M−1 s−1 for perfluorohexanoic acid, (7.1 ± 0.6) × 108 M−1 s−1 for perfluorooctanoic acid, and (6.4 ± 0.4) × 108 M−1 s−1 for perfluorononanoic acid.162 Although this experimental study did not determine the mechanism for those measured rate constants, we believe they measured the rates of the associative mechanism. In contrast, the kexp values for C–F cleavage (106–107 M−1 s−1) were previously reported134 and used for the determination of our LFERs (compound no. 204–206), which confirm the significantly smaller kchem values of the stepwise cleavage mechanism of a C–F bond. The predicted kchem values in three reaction mechanisms for all 75 PFASs are shown in Fig. S24 in the ESI.† The kchem values that exceeded the diffusion limit (3 × 1010 M−1 s−1 and lnk = 24.1) were not included in either figure. The kchem values predicted for the 75 PFASs that undergo the stepwise cleavage mechanism of a C–F bond range from 6.9 × 107 to 3.7 × 108 M−1 s−1. In contrast, 19kchem values range from 4.9 × 107 M−1 s−1 to 3 × 1010 M−1 s−1 for the associative mechanism with CO, 23kchem values range from 1.3 × 106 to 3 × 1010 M−1 s−1 for the associative mechanism with O, and 4kchem values are above the diffusion limit for the CC associative mechanism. We did not determine the LFERs for the functional groups of sulfonic acid (SO3−), phosphinic acid (PO(OH)2), sulfonamide (SO2NH), and sulfonyl (SO2); thus, no rate constant predictions were conducted. The investigation of the  values at all possible eaq− attacking sites for all possible eaq− reaction mechanisms highlights the significantly lower reactivity of eaq− for the stepwise cleavage mechanism of a C–F bond and higher reactivity of eaq− with functional groups in a given PFAS structure.
values at all possible eaq− attacking sites for all possible eaq− reaction mechanisms highlights the significantly lower reactivity of eaq− for the stepwise cleavage mechanism of a C–F bond and higher reactivity of eaq− with functional groups in a given PFAS structure.
 values calculated for all possible attacking sites in PFASs
values calculated for all possible attacking sites in PFASs
| Class | Compound name | Attacking site | 2D structure |
|
||
|---|---|---|---|---|---|---|
| Stepwise (C–F cleavage) | Concerted (C–F cleavage) | Association with functional group | ||||
| PFCA | Perfluorobutanoic acid (PFBA) | Alpha |
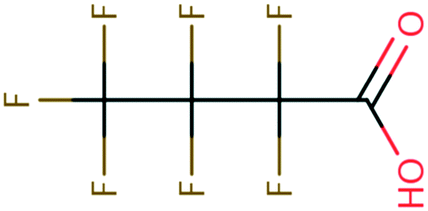
|
−6.86 | −3.45 | −4.10 |
| Beta | −7.13 | −3.69 | ||||
| Terminal | −7.46 | −4.05 | ||||
| Perfluorooctanoic acid (PFOA) | Alpha |

|
−6.14 | −3.48 | −3.50 | |
| Beta | −6.28 | −3.62 | ||||
| Gamma | −6.25 | −3.58 | ||||
| Delta | −6.24 | −3.56 | ||||
| Epsilon | −6.30 | −3.55 | ||||
| Zeta | −6.30 | −3.60 | ||||
| Terminal | −6.66 | −4.01 | ||||
| Perfluorohexanoic acid | Alpha |
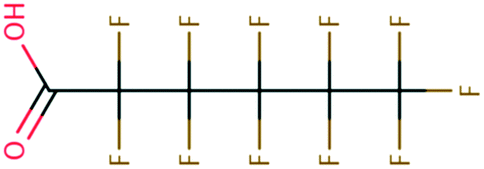
|
−6.49 | −3.49 | −3.46 | |
| Beta | −6.60 | −3.62 | ||||
| Gamma | −6.63 | −3.56 | ||||
| Delta | −6.68 | −3.62 | ||||
| Terminal | −7.02 | −4.02 | ||||
| Perfluorononanoic acid | Alpha |
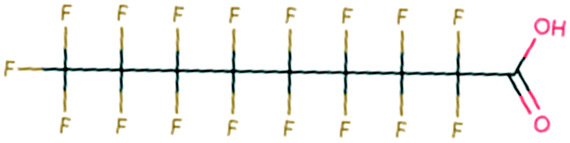
|
−6.01 | −3.53 | −3.41 | |
| Beta | −6.12 | −3.65 | ||||
| Gamma | −6.10 | −3.60 | ||||
| Delta | −6.09 | −3.59 | ||||
| Epsilon | −6.12 | −3.57 | ||||
| Zeta | −6.12 | −3.52 | ||||
| Eta | −6.12 | −3.60 | ||||
| Terminal | −6.55 | −4.08 | ||||
| Ammonium perfluorooctanoate | Alpha |

|
−6.14 | −3.48 | −3.50 | |
| Beta | −6.28 | −3.62 | ||||
| Gamma | −6.25 | −3.58 | ||||
| Delta | −6.24 | −3.56 | ||||
| Epsilon | −6.30 | −3.55 | ||||
| Zeta | −6.30 | −3.60 | ||||
| Terminal | −6.66 | −4.01 | ||||
| Methyl heptafluorobutyrate | Alpha |
|
−6.52 | −3.33 | −2.32 | |
| Beta | −6.89 | −3.65 | ||||
| Terminal | −7.29 | −4.07 | ||||
| Methyl perfluorohexanoate | Alpha |

|
−6.20 | −3.32 | −2.43 | |
| Beta | −6.53 | −3.61 | ||||
| Gamma | −6.48 | −3.54 | ||||
| Delta | −6.51 | −3.57 | ||||
| Terminal | −6.87 | −4.00 | ||||
| PFSA | Perfluorobutanesulfonic acid | Alpha |

|
−6.69 | −3.56 | −2.79 |
| Beta | −6.71 | −3.56 | ||||
| Gamma | −6.60 | −3.48 | ||||
| Terminal | −7.04 | −3.93 | ||||
| Perfluorooctanesulfonic acid | Alpha |

|
−6.08 | −3.64 | −3.31 | |
| Beta | −5.94 | −3.53 | ||||
| Gamma | −5.96 | −3.53 | ||||
| Delta | −5.99 | −3.53 | ||||
| Epsilon | −6.00 | −3.54 | ||||
| Zeta | −6.05 | −3.55 | ||||
| Eta | −6.02 | −3.56 | ||||
| Terminal | −6.58 | −4.40 | ||||
| Potassium perfluorooctanesulfonate | Alpha |
|
−6.08 | −3.64 | −3.31 | |
| Beta | −5.94 | −3.53 | ||||
| Gamma | −5.96 | −3.53 | ||||
| Delta | −5.99 | −3.53 | ||||
| Epsilon | −6.00 | −3.54 | ||||
| Zeta | −6.05 | −3.55 | ||||
| Eta | −6.02 | −3.56 | ||||
| Terminal | −6.58 | −4.40 | ||||
| Potassium perfluorobutanesulfonate | Alpha |
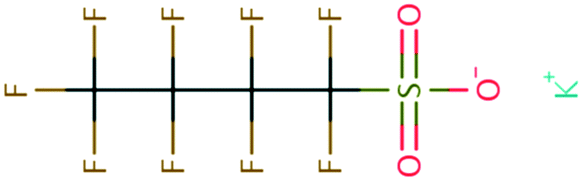
|
−6.69 | −3.56 | −2.79 | |
| Beta | −6.71 | −3.56 | ||||
| Gamma | −6.60 | −3.48 | ||||
| Terminal | −7.04 | −3.93 | ||||
| Potassium perfluorohexanesulfonate | Alpha |

|
−6.45 | −3.65 | −3.43 | |
| Beta | −6.25 | −3.50 | ||||
| Gamma | −6.34 | −3.55 | ||||
| Delta | −6.40 | −3.55 | ||||
| Epsilon | −6.39 | −3.58 | ||||
| Terminal | −6.78 | −4.20 | ||||
| PFPiA & PFPA | Bis(nonafluorobutyl)phosphinic acid | Alpha |
|
−5.66 | −3.11 | −2.81 |
| Beta | −5.77 | −3.28 | ||||
| Gamma | −5.89 | −3.32 | ||||
| Terminal | −6.27 | −3.74 | ||||
| Perfluorohexanephosphonic acid | Alpha |
|
−6.48 | −3.40 | −3.43 | |
| Beta | −6.40 | −3.39 | ||||
| Gamma | −6.54 | −3.45 | ||||
| Delta | −6.47 | −3.37 | ||||
| Epsilon | −6.44 | −3.40 | ||||
| Terminal | −6.88 | −3.86 | ||||
| Perfluorooctanephosphonic acid | Alpha |
|
−6.14 | −3.33 | −3.20 | |
| Beta | −6.12 | −3.35 | ||||
| Gamma | −6.19 | −3.35 | ||||
| Delta | −6.11 | −3.36 | ||||
| Epsilon | −6.27 | −3.40 | ||||
| Zeta | −6.16 | −3.31 | ||||
| Eta | −6.18 | −3.36 | ||||
| Terminal | −6.65 | −3.82 | ||||
| Perfluorodecylphosphonic acid | Alpha |

|
−5.53 | −3.54 | −3.25 | |
| Beta | −5.51 | −3.58 | ||||
| Gamma | −5.49 | −3.50 | ||||
| Delta | −5.53 | −3.51 | ||||
| Epsilon | −5.61 | −3.58 | ||||
| Zeta | −5.49 | −3.51 | ||||
| Eta | −5.50 | −3.48 | ||||
| Theta | −5.44 | −3.45 | ||||
| Iota | −5.52 | −3.55 | ||||
| Terminal | −5.90 | −3.96 | ||||
| P,Pbis(1,1,2,2,3,3,4,4,5,5,6,6,6-tridecafluorohexyl)-phosphinic acid | Alpha |

|
−5.08 | −3.44 | −3.05 | |
| Beta | −5.15 | −3.44 | ||||
| Gamma | −5.22 | −3.56 | ||||
| Delta | −5.23 | −3.54 | ||||
| Epsilon | −5.33 | −3.64 | ||||
| Terminal | −5.65 | −4.01 | ||||
| (Heptadecafluorooctyl)(tridecafluorohexyl)phosphinic acid | Alpha |
|
−4.77 | −3.36 | −3.31 | |
| Beta | −4.95 | −3.54 | ||||
| Gamma | −4.97 | −3.51 | ||||
| Delta | −5.02 | −3.55 | ||||
| Epsilon | −5.03 | −3.57 | ||||
| Zeta | −4.96 | −3.47 | ||||
| Terminal | −5.08 | −3.61 | ||||
| Bis(heptadecafluorooctyl)phosphinic acid | Alpha |
|
−4.44 | −3.46 | −3.39 | |
| Beta | −4.52 | −3.57 | ||||
| Gamma | −4.50 | −3.52 | ||||
| Delta | −4.43 | −3.54 | ||||
| Epsilon | −4.50 | −3.51 | ||||
| Zeta | −4.59 | −3.55 | ||||
| Eta | −4.70 | −3.64 | ||||
| Terminal | −4.98 | −4.02 | ||||
| FTOH | 4:2 fluorotelomer alcohol | Alpha |
|
−6.86 | −3.71 | −4.20 |
| Beta | −6.81 | −3.65 | ||||
| Gamma | −6.80 | −3.64 | ||||
| Terminal | −7.14 | −4.02 | ||||
| 6:2 fluorotelomer alcohol | Alpha |

|
−6.50 | −3.56 | −3.49 | |
| Beta | −6.45 | −3.46 | ||||
| Gamma | −6.40 | −3.38 | ||||
| Delta | −6.32 | −3.37 | ||||
| Epsilon | −6.39 | −3.43 | ||||
| Terminal | −6.78 | −3.87 | ||||
| 4:4 fluorotelomer alcohol | Alpha |
|
−6.60 | −3.68 | −3.93 | |
| Beta | — | — | ||||
| Gamma | −6.54 | −3.59 | ||||
| Terminal | −6.94 | −4.03 | ||||
| 8:2 fluorotelomer alcohol | Alpha |
|
— | — | −3.08 | |
| Beta | −6.07 | −3.32 | ||||
| Gamma | −6.05 | −3.28 | ||||
| Delta | −5.92 | −3.24 | ||||
| Epsilon | −5.92 | −3.24 | ||||
| Zeta | −5.99 | −3.25 | ||||
| Eta | −6.08 | −3.37 | ||||
| Terminal | — | — | ||||
| 11:1 fluorotelomer alcohol | Alpha |
|
−5.70 | −3.20 | −3.02 | |
| Beta | −5.68 | −3.12 | ||||
| Gamma | −5.58 | −3.05 | ||||
| Delta | −5.54 | −3.03 | ||||
| Epsilon | −5.55 | −3.01 | ||||
| Zeta | −5.53 | −3.01 | ||||
| Eta | −5.57 | −3.04 | ||||
| Theta | −5.52 | −3.01 | ||||
| Iota | −5.60 | −3.03 | ||||
| Kappa | −5.61 | −3.09 | ||||
| Terminal | — | — | ||||
| Heptafluorobutanol | Alpha |
|
−7.26 | −3.75 | −3.57 | |
| Beta | −7.24 | −3.70 | ||||
| Terminal | −7.55 | −4.02 | ||||
| 3-(Perfluoropropyl)propanol | Alpha |
|
−7.02 | −3.68 | −2.92 | |
| Beta | −6.97 | −3.63 | ||||
| Terminal | −7.37 | −4.04 | ||||
| 3-(Perfluoro-2-butyl)propane-1,2-diol | Alpha |
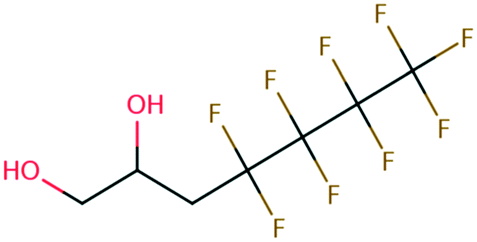
|
−6.64 | −3.69 | −3.93 | |
| Beta | −6.57 | −3.62 | ||||
| Gamma | −6.58 | −3.60 | ||||
| Terminal | −6.98 | −4.04 | ||||
| Fluorotelomer acrylates (FTACs) | 6:2 fluorotelomer acrylate | Alpha |
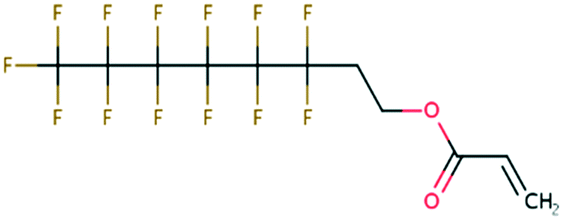
|
−6.15 | −3.55 | −2.08 |
| Beta | −6.13 | −3.47 | ||||
| Gamma | −6.01 | −3.33 | ||||
| Delta | −6.09 | −3.42 | ||||
| Epsilon | −6.13 | −3.51 | ||||
| Terminal | −6.47 | −3.89 | ||||
| 6:2 fluorotelomer methacrylate | Alpha |

|
−6.08 | −3.71 | −1.94 | |
| Beta | −6.03 | −3.54 | ||||
| Gamma | −5.99 | −3.57 | ||||
| Delta | −5.95 | −3.53 | ||||
| Epsilon | −5.97 | −3.57 | ||||
| Terminal | −6.34 | −4.01 | ||||
| Perfluoroalkyl ether carboxylates (PFECAs) | Perfluoro-2-methyl-3-oxahexanoic acid | Alpha |
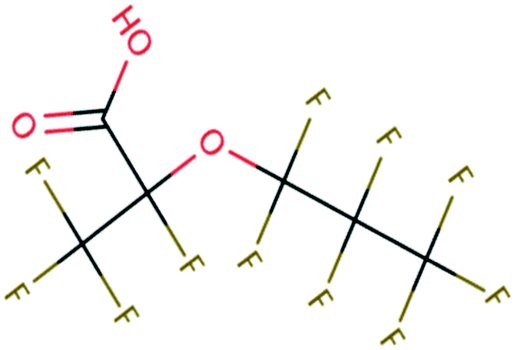
|
−6.22 | −3.42 | −3.57 |
| Beta | −6.88 | −4.16 | ||||
| Gamma | −6.64 | −3.88 | ||||
| Delta | −6.28 | −3.57 | ||||
| Terminal | −6.83 | −4.04 | ||||
| Perfluoro(4-methoxybutanoic) acid | Alpha |
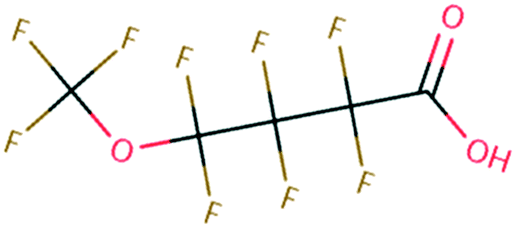
|
−6.46 | −3.36 | −3.08 | |
| Beta | −6.67 | −3.56 | ||||
| Gamma | −6.86 | −3.73 | ||||
| Terminal | −7.24 | −4.17 | ||||
| Perfluoro-3,6-dioxaoctane-1,8-dioic acid | Alpha |

|
−6.87 | −3.71 | −3.43 | |
| Terminal | −7.02 | −3.82 | ||||
| FASA | Perfluorooctanesulfonamide | Alpha |
|
−5.97 | −3.54 | −1.34 |
| Beta | −5.92 | −3.58 | ||||
| Gamma | −5.99 | −3.57 | ||||
| Delta | −6.03 | −3.59 | ||||
| Epsilon | −5.91 | −3.53 | ||||
| Zeta | −6.02 | −3.55 | ||||
| Eta | −6.06 | −3.61 | ||||
| Terminal | −6.41 | −4.02 | ||||
| N-Ethylperfluorooctanesulfonamide | Alpha |
|
−5.82 | −3.57 | −1.33 | |
| Beta | −5.73 | −3.46 | ||||
| Gamma | −5.83 | −3.55 | ||||
| Delta | −5.76 | −3.48 | ||||
| Epsilon | −5.85 | −3.56 | ||||
| Zeta | −5.81 | −3.50 | ||||
| Eta | −5.88 | −3.63 | ||||
| Terminal | −6.24 | −4.03 | ||||
| N-Methylperfluorooctanesulfonamide | Alpha |

|
−5.90 | −3.62 | −1.36 | |
| Beta | −5.80 | −3.50 | ||||
| Gamma | −5.97 | −3.60 | ||||
| Delta | −5.86 | −3.57 | ||||
| Epsilon | −5.95 | −3.59 | ||||
| Zeta | −5.97 | −3.61 | ||||
| Eta | −5.94 | −3.64 | ||||
| Terminal | −6.33 | −4.04 | ||||
| Perfluoroalkane sulfonyl fluorides (PASFs) | Perfluorobutanesulfonyl fluoride | Alpha |
|
−6.61 | −3.50 | −0.46 |
| Beta | −6.58 | −3.51 | ||||
| Gamma | −6.69 | −3.59 | ||||
| Terminal | −7.15 | −4.12 | ||||
| Perfluoroalkane sulfonyl chloride (PASCs) | Perfluoro-1-butanesulfonyl chloride | Alpha |

|
−6.56 | −3.54 | 0.36 |
| Beta | −6.47 | −3.43 | ||||
| Gamma | −6.63 | −3.55 | ||||
| Terminal | −7.10 | −4.12 | ||||
| Perfluoroalkyl acyl fluorides (PAAFs) | Perfluoroglutaryl difluoride | Alpha |

|
−6.33 | −3.13 | −1.70 |
| Beta | −6.83 | −3.58 | ||||
| Fluorotelomer amines | 1H,1H-Perfluoropentylamine | Alpha |

|
−6.96 | −3.73 | −3.95 |
| Beta | −6.85 | −3.60 | ||||
| Gamma | −6.83 | −3.58 | ||||
| Terminal | −7.21 | −4.02 | ||||
| Bis(1H,1H-perfluoropropyl)amine | Alpha |

|
−6.77 | −3.75 | −4.46 | |
| Terminal | −7.10 | −4.12 | ||||
| Perfluoroalkyl amides | Octafluoroadipamide | Alpha |

|
−6.37 | −3.42 | −2.66 |
| Beta | −6.55 | −3.63 | ||||
| Nonafluoropentanamide | Alpha |

|
−6.60 | −3.45 | −2.70 | |
| Beta | −6.77 | −3.58 | ||||
| Gamma | −6.76 | −3.55 | ||||
| Terminal | −7.19 | −4.04 | ||||
| Heptafluorobutyramide | Alpha |
|
−6.59 | −3.18 | −2.78 | |
| Beta | −6.87 | −3.46 | ||||
| Terminal | −7.24 | −3.84 | ||||
| Perfluoroalkyl anhydrides | Pentafluoropropanoic anhydride | Alpha |
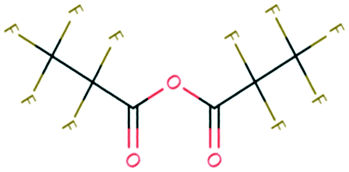
|
−6.26 | −3.20 | −1.35 |
| Terminal | −7.12 | −4.06 | ||||
| Polyfluoroalkyl acyl fluorides | 5H-Octafluoropentanoyl fluoride | Alpha |
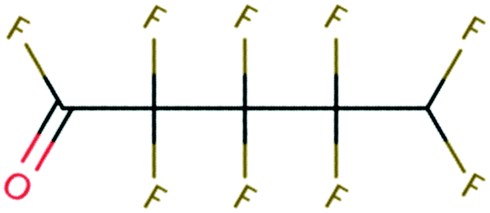
|
−6.35 | −3.17 | −1.74 |
| Beta | −6.80 | −3.59 | ||||
| Gamma | −6.80 | −3.59 | ||||
| Terminal | −6.91 | −3.72 | ||||
| Polyfluoroalkyl aldehydes | 5H-Perfluoropentanal | Alpha |
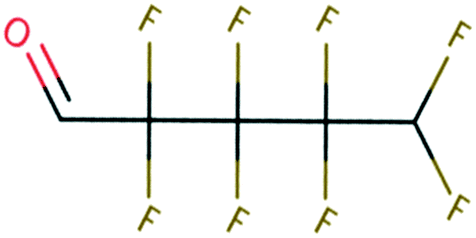
|
−6.34 | −3.13 | −1.74 |
| Beta | −6.81 | −3.56 | ||||
| Gamma | −6.85 | −3.59 | ||||
| Terminal | −6.95 | −3.73 | ||||
| Polyfluoroalkyl ethers | Sevoflurane | Alpha |

|
−7.08 | −3.74 | −2.67 |
| Beta | −7.48 | −4.12 | ||||
| Terminal | −7.50 | −4.14 | ||||
| Flurothyl | Terminal |
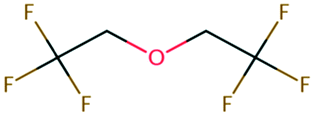
|
−7.66 | −4.14 | −3.07 | |
| Perfluoroisobutyl methyl ether | Alpha |
|
−7.13 | −3.89 | −2.16 | |
| Beta | −6.46 | −3.17 | ||||
| Gamma | −7.27 | −4.04 | ||||
| Terminal | −7.26 | −4.02 | ||||
| Tris(trifluoroethoxy)methane | Terminal |

|
−7.01 | −4.21 | −4.55 | |
| Difluoromethyl 1H,1H-perfluoropropyl | Alpha |

|
−7.32 | −4.07 | −2.11 | |
| Beta | −7.03 | −3.69 | ||||
| Terminal | −7.42 | −4.10 | ||||
| Allyl perfluoroisopropyl ether | Alpha |
|
−6.62 | −3.37 | −1.26 | |
| Beta | −7.30 | −4.07 | ||||
| Terminal | −7.27 | −4.03 | ||||
| Semi-fluorinated alkenes (SFAenes) | 1H,1H,2H-Perfluoro-1-hexene | Alpha |

|
−6.31 | −3.15 | −2.67 |
| Beta | −6.84 | −3.62 | ||||
| Gamma | −6.89 | −3.66 | ||||
| Terminal | −7.22 | −4.03 | ||||
| 6H-Perfluorohex-1-ene | Alpha |
|
−7.20 | −4.15 | −2.28 | |
| Beta | −6.79 | −3.76 | ||||
| Gamma | −6.23 | −3.18 | ||||
| Delta | −6.73 | −3.65 | ||||
| Epsilon | −6.66 | −3.56 | ||||
| Terminal | −6.78 | −3.70 | ||||
| 2-Vinylperfluorobutane | Alpha |

|
−5.85 | −2.66 | −2.27 | |
| Beta | −7.32 | −4.08 | ||||
| Gamma | −6.84 | −3.58 | ||||
| Terminal | −7.25 | −4.02 | ||||
| 1-Propenylperfluoropropane | Alpha |
|
−6.45 | −3.21 | −2.84 | |
| Beta | −6.95 | −3.66 | ||||
| Terminal | −7.30 | −4.05 | ||||
| Perfluoroalkyl alkyl ethers (PFAEs) | Ethyl perfluorobutyl ether | Alpha |

|
−6.97 | −3.88 | −3.90 |
| Beta | −6.75 | −3.64 | ||||
| Gamma | −6.78 | −3.66 | ||||
| Terminal | −7.08 | −4.14 | ||||
| Polyfluorinated alcohols | 1H,1H,5H-Perfluoropentanol | Alpha |

|
−7.01 | −3.71 | −2.18 |
| Beta | −6.89 | −3.57 | ||||
| Gamma | −6.87 | −3.62 | ||||
| Terminal | −6.96 | −3.73 | ||||
| Dodecafluoroheptanol | Alpha |

|
−6.62 | −3.72 | −3.72 | |
| Beta | −6.61 | −3.65 | ||||
| Gamma | −6.44 | −3.54 | ||||
| Delta | −6.58 | −3.59 | ||||
| Epsilon | −6.55 | −3.60 | ||||
| Terminal | −6.58 | −3.71 | ||||
| Hexafluoroamylene glycol | Alpha |

|
−7.05 | −3.66 | −2.73 | |
| Beta | −7.04 | −3.69 | ||||
| 3H-Perfluoro-2,2,4,4-tetrahydroxypentane | Alpha |
|
−6.68 | −3.51 | −2.86 | |
| Terminal | −7.17 | −3.96 | ||||
| 1-Pentafluoroethylethanol | Alpha |

|
−7.21 | −3.72 | −4.02 | |
| Terminal | −7.53 | −4.08 | ||||
| Polyfluoroalkyl carboxylates | 4H-Perfluorobutanoic acid | Alpha |
|
−6.87 | −3.51 | −3.17 |
| Beta | −7.11 | −3.72 | ||||
| Terminal | −7.12 | −3.75 | ||||
| 3,3-Bis(trifluoromethyl)-2-propenoic acid | Alpha |
|
−7.00 | −3.65 | −2.02 | |
| Terminal | −7.03 | −3.67 | ||||
| 3-(Perfluoroisopropyl)-2-propenoic acid | Alpha |
|
−5.88 | −2.58 | −2.62 | |
| Terminal | −7.33 | −4.01 | ||||
| Fluorotelomer carboxylates (FTCAs) | 2H,2H,3H,3H-Perfluorooctanoic acid | Alpha |
|
−6.78 | −3.59 | −3.30 |
| Beta | −6.72 | −3.50 | ||||
| Gamma | −6.63 | −3.39 | ||||
| Delta | −6.65 | −3.43 | ||||
| Terminal | −7.02 | −3.85 | ||||
| Methyl 2H,2H,3H,3H-perfluoroheptanoate | Alpha |
|
−6.59 | −3.71 | −3.25 | |
| Beta | −6.51 | −3.63 | ||||
| Gamma | −6.48 | −3.57 | ||||
| Terminal | −6.88 | −4.03 | ||||
| Fluorotelomer sulfonates (FTSAs) | 4:2 fluorotelomer sulfonic acid | Alpha |
|
−6.70 | −3.77 | −2.94 |
| Beta | −6.58 | −3.62 | ||||
| Gamma | −6.51 | −3.57 | ||||
| Terminal | −6.94 | −4.04 | ||||
| N-Alkyl perfluoroalkane sulfonamidoethanols | N-Ethyl-N-(2-hydroxyethyl)perfluorooctanesulfonamide | Alpha |
|
−5.58 | −3.58 | −1.25 |
| Beta | −5.45 | −3.48 | ||||
| Gamma | −5.60 | −3.56 | ||||
| Delta | −5.57 | −3.56 | ||||
| Epsilon | −5.64 | −3.58 | ||||
| Zeta | −5.68 | −3.58 | ||||
| Eta | −5.62 | −3.62 | ||||
| Terminal | −5.99 | −4.02 | ||||
| Perfluoroalkyl polyether carboxylates (PFPECAs) | Perfluoro-3,6,9-trioxatridecanoic acid | Alpha |
|
−6.13 | −3.48 | −1.51 |
| Beta | −6.28 | −3.57 | ||||
| Gamma | −6.31 | −3.59 | ||||
| Delta | −6.29 | −3.58 | ||||
| Epsilon | −6.36 | −3.61 | ||||
| Zeta | −6.19 | −3.45 | ||||
| Eta | −6.00 | −3.28 | ||||
| Theta | −6.02 | −3.30 | ||||
| Terminal | −6.41 | −3.73 | ||||
| Perfluoro-3,6-dioxaheptanoic acid | Alpha |
|
−6.75 | −3.58 | −3.39 | |
| Beta | −6.93 | −3.72 | ||||
| Gamma | −6.90 | −3.67 | ||||
| Terminal | −7.29 | −4.09 | ||||
| Perfluoroalkyl sulfonamido amines | Perfluorooctanesulfonamido ammonium iodide | Alpha |
|
−5.57 | −3.65 | −1.24 |
| Beta | −5.20 | −3.43 | ||||
| Gamma | −5.27 | −3.45 | ||||
| Delta | −5.23 | −3.41 | ||||
| Epsilon | −5.34 | −3.49 | ||||
| Zeta | −5.43 | −3.51 | ||||
| Eta | −5.51 | −3.62 | ||||
| Terminal | −5.85 | −4.05 | ||||
| Perfluoroalkyl aldehydes (PFALs) | Perfluorobutyraldehyde | Alpha |
|
−6.45 | −3.02 | −1.56 |
| Beta | −7.01 | −3.51 | ||||
| Terminal | −7.43 | −3.96 | ||||
| Perfluoroalkyl amino alcohols | 2-Aminohexafluoropropan-2-ol | Alpha |
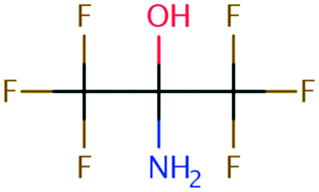
|
−7.60 | −4.12 | −3.63 |
| Terminal | −7.60 | −4.13 | ||||
| Perfluoroalkyl Ketones | Methyl perfluoroethyl ketone | Alpha |

|
−6.73 | −3.25 | −2.05 |
| Terminal | −7.54 | −4.07 | ||||
| Perfluoroalkyl polyether alcohols | 1H,1H,8H,8H-Perfluoro-3,6-dioxaoctane-1,8-diol | Alpha |
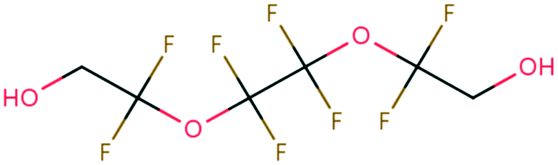
|
−6.92 | −4.00 | −1.77 |
| Terminal | −6.81 | −3.87 | ||||
| Perfluoroalkyl vinyl ethers | Heptafluoropropyl trifluorovinyl ether | Alpha |

|
−7.40 | −3.97 | −3.57 |
| Beta | −7.14 | −3.69 | ||||
| Gamma | −7.16 | −3.62 | ||||
| Delta | −6.83 | −3.36 | ||||
| Terminal | −7.23 | −3.77 | ||||
| Polyfluoroalkane sulfonates | 2,2-Difluoroethyl triflate | Alpha |
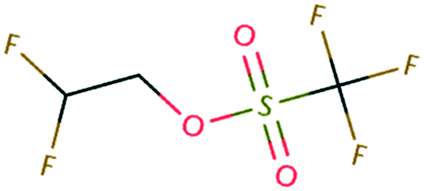
|
−7.28 | −3.95 | −1.11 |
| Terminal | −7.05 | −3.74 | ||||
| Polyfluoroalkyl amides | Perfluoropentanamide | Alpha |
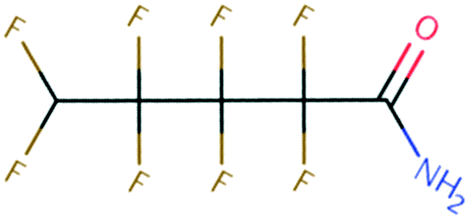
|
−6.63 | −3.46 | −2.78 |
| Beta | −6.77 | −3.60 | ||||
| Gamma | −6.77 | −3.57 | ||||
| Terminal | −6.89 | −3.72 | ||||
| Polyfluoroalkyl Amines | 2-Amino-2H-perfluoropropane | Terminal |

|
−7.68 | −4.16 | −4.24 |
| Polyfluoroalkyl ketones | 3H-Perfluoro-4-hydroxy-3-penten-2-one | Alpha |
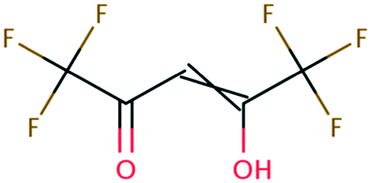
|
−6.88 | −3.56 | −1.20 |
| Terminal | −6.86 | −3.52 | ||||
| Polyfluoroalkyl trifluoromethane-sulfonates | 2-(Trifluoromethoxy)ethyl trifluoromethanesulfonate | Alpha |
|
−7.10 | −3.96 | −0.58 |
| Terminal | −7.34 | −4.09 | ||||
| Semi-fluorinated alkanes (SFAs) | 1,1,1,3,3-Pentafluorobutane | Alpha |

|
−7.48 | −3.79 | −4.94 |
| Terminal | −7.86 | −4.16 | ||||

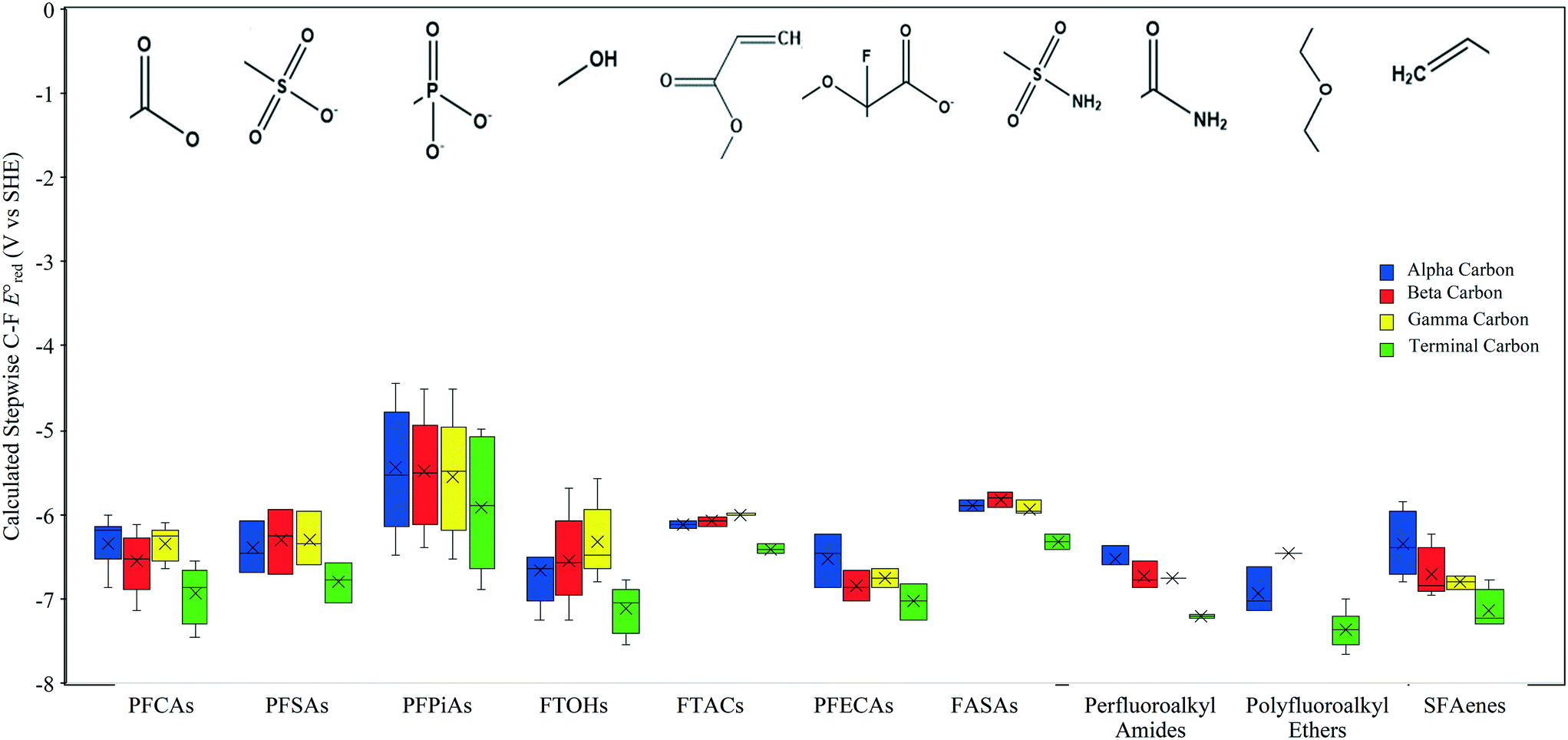 | ||
Fig. 4 Theoretically calculated  values for the cleavage of a C–F bond at different positions of various PFASs. The values for the cleavage of a C–F bond at different positions of various PFASs. The 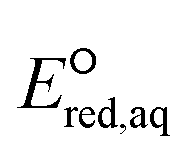 values were calculated based on M06-2X/cc-pVDZ. values were calculated based on M06-2X/cc-pVDZ. | ||
The attachment of an eaq− to the group of PFCAs (N = 8) occurred near the α-carbon, and the resultant intermediate radical dianions were the most stable due to resonance stabilization by the π-system of carboxylate functional groups, which is consistent with previous predictions on PFOA163 and perfluoropentanoic acid (PFPeA).24 Attachment near the α-carbon was also observed for the group of perfluoroalkyl phosphinates (PFPiAs) due to the phosphonate functional group, perfluoroalkyl ether carboxylates (PFECA), perfluoroalkyl amides, polyfluoroalkyl aldehydes and acyl fluorides, and semifluorinated alkenes (SFAenes). In contrast, the group of perfluoroalkane sulfonates (PFSA) (N = 5) showed preferable attachment of an eaq− near the β- or γ-carbon with the largest 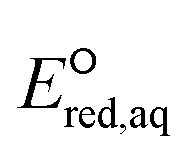 values due to the inability of π-stabilization due to the trigonal geometry of the sulfonate functional group.24 A similar trend was observed for the groups of fluorotelomer alcohols (FTOH) and polyfluorinated alcohols, fluorotelomer acrylates (FTACs), perfluoroalkane sulfonamides (FASAs), perfluoroalkane sulfonyl chlorides (PASCs), and fluorotelomer carboxylates (FTCAs). The investigation of the
values due to the inability of π-stabilization due to the trigonal geometry of the sulfonate functional group.24 A similar trend was observed for the groups of fluorotelomer alcohols (FTOH) and polyfluorinated alcohols, fluorotelomer acrylates (FTACs), perfluoroalkane sulfonamides (FASAs), perfluoroalkane sulfonyl chlorides (PASCs), and fluorotelomer carboxylates (FTCAs). The investigation of the 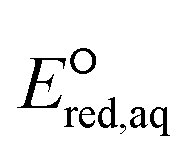 values at all possible eaq− attacking sites for PFASs that contain a wide variety of functional groups highlights significant differences in regard to the most preferable reactive sites of eaq−. While the scope of the current study is on the initial reactivities of eaq− with a wide variety of organic compounds and PFASs, investigating the subsequent degradation pathways is underway. The cleavage of a C–F bond in a PFAS is the major goal for practical PFAS remediation using reductive technologies, and our LFERs and predicted kchem values for the stepwise cleavage mechanism of a C–F bond present significant challenges in cleaving a C–F bond from a kinetics point of view.
values at all possible eaq− attacking sites for PFASs that contain a wide variety of functional groups highlights significant differences in regard to the most preferable reactive sites of eaq−. While the scope of the current study is on the initial reactivities of eaq− with a wide variety of organic compounds and PFASs, investigating the subsequent degradation pathways is underway. The cleavage of a C–F bond in a PFAS is the major goal for practical PFAS remediation using reductive technologies, and our LFERs and predicted kchem values for the stepwise cleavage mechanism of a C–F bond present significant challenges in cleaving a C–F bond from a kinetics point of view.
Impact of the accuracy of rate constant estimation to the fate
To assess the impact of the accuracy of rate constant estimation to the fate of a target organic compound, we develop an unsteady-state kinetic model for the homogeneous aqueous-phase UV/sulfite system to degrade a model compound (e.g., PFAS) with input parameters that are consistent with experimental observations in the literature10 (see the details in Text S4 in the ESI†). It should be noted that the model was used to assess the impact of the initial rate constant prediction accuracy on the time-dependent fate and the predicted results do not necessarily indicate the degradation of a model compound in an environmental matrix. Fig. 5 represents the time-dependent concentration profile of a parent compound that has kchem = 1.0 × 106 M−1 s−1 with eaq−. The profiles were also predicted by changing the kchem values by a difference of a factor of 1.2, 2, and 5. Estimating the rate constant impacted the profile of the parent compound by 33% for kchem values with a factor of 1.2, 77% with a factor of 2, and 100% with a factor of 5.0 at 300 h. While our LFER for the C–F stepwise cleavage mechanism has the ability to predict the kchem values within the difference of a factor of 1.2, this prediction demonstrates the importance of an accurate rate constant of a target compound in estimating the decay of an environmentally relevant contaminant that requires significant amount of time (e.g., PFASs).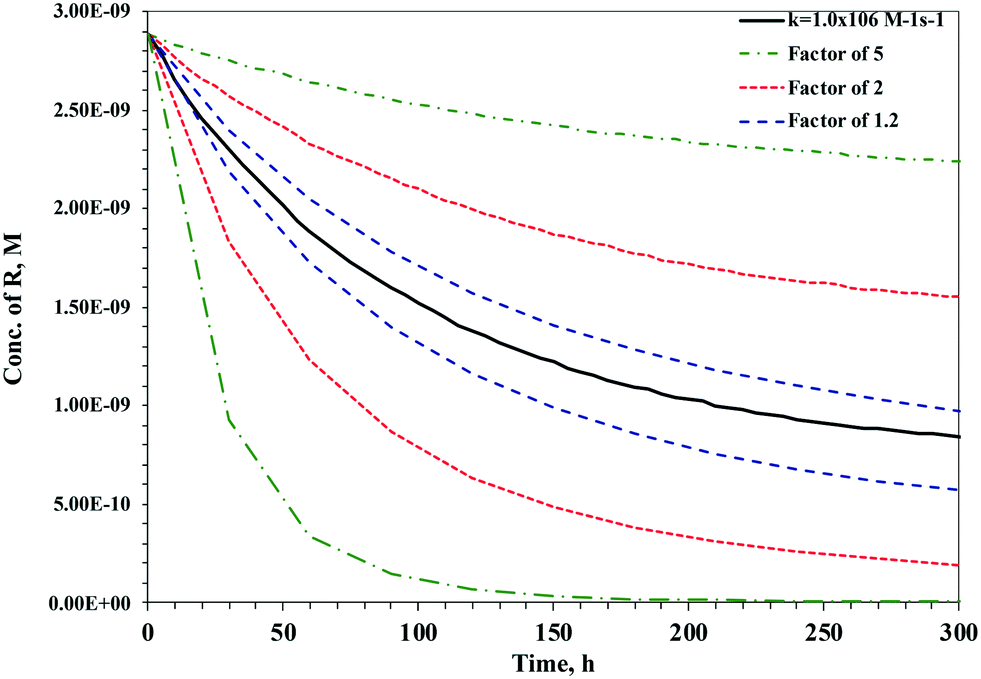 | ||
| Fig. 5 Time-dependent concentration of a target compound with kchem = 1.0 × 106 M−1 s−1 and different predicted values by a difference of a factor of 1.2, 2, and 5 in UV/sulfite process.10 | ||
Conclusion
Reduction of oxidized forms of water contaminants using electrons in the aqueous-phase advanced reduction processes is a novel and attractive approach to destroying the contaminants. The mechanistic insight into the reactivities of solvated electrons with a wide variety of contaminants helps understand and predict the fate of contaminants of emerging concern, and can be extrapolated to the direct electron transfer mechanisms in the heterogeneous electrochemical processes. The determination of linear free energy relationships presented in this study elucidated which reaction mechanisms are the dominant rate-determining step for the reaction of solvated electrons with structurally diverse conventional organic compounds and the 75 priority PFAS subset that contains a wide variety of functional groups. The computational tools determined in this study can be used to predict the reaction rate constants with solvated electrons and screen a number of contaminants and prioritize for the application of advanced reduction processes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Central Chemicals Inc. Any opinions, findings, conclusions, or recommendations expressed in this publication are those of the authors and do not necessarily reflect the view of the supporting organization. R. D. is supported by the NSF Graduate Research Fellowship Program (GRFP). The authors appreciate the support for the use of the Michigan Tech HPC cluster ‘Superior’.References
- W. Glaze, J.-W. Kang and H. Chapin, The chemistry of water treatment processes involving ozone, hydrogen peroxide and ultraviolet radiation, Ozone: Sci. Eng., 1987, 9, 335–352 CrossRef CAS.
- W. Glaze and J.-W. Kang, Advanced oxidation processes. Test of a kinetic model for the oxidation of organic compounds with ozone and hydrogen peroxide in a semibatch reactor, Ind. Eng. Chem. Res., 1989, 28, 1580–1587 CrossRef CAS.
- C. Remucal and D. Manley, Emerging investigators series: the efficacy of chlorine photolysis as an advanced oxidation process for drinking water treatment, Environ. Sci.: Water Res. Technol., 2016, 2, 565–579 RSC.
- D. Minakata, D. Kamath and S. Maetzold, Mechanistic insight into the reactivity of chlorine-derived radicals in the aqueous-phase UV-chlorine advanced oxidation process: Quantum mechanical calculations, Environ. Sci. Technol., 2017, 51, 6918–6926 CrossRef CAS PubMed.
- K. Guo, S. Zheng, X. Zhang, L. Zhao, S. Ji, C. Chen, Z. Wu, D. Wang and J. Fang, Roles of bromine radicals and hydroxyl radicals in the degradation of micropollutants by the UV/bromine process, Environ. Sci. Technol., 2020, 54, 6415–6426 CrossRef CAS PubMed.
- Z. Wu, C. Chen, B.-Z. Zhu, C.-H. Huang, T. An, F. Meng and J. Fang, Reactive nitrogen species are also involved in the transformation of micropollutants by the UV/monochloramine process, Environ. Sci. Technol., 2019, 53, 11142–11152 CrossRef CAS PubMed.
- B. Vellanki, B. Batchelor and A. Abdel-Wahab, Advanced reduction processes: A new class of treatment processes, Environ. Eng. Sci., 2013, 30, 264–271 CrossRef CAS PubMed.
- J. Grebel, J. Pignatello and W. Mitch, Effect of halide ions and carbonates on organic contaminant degradation by hydroxyl radical-based advanced oxidation processes in saline waters, Environ. Sci. Technol., 2010, 44, 6822–6828 CrossRef CAS PubMed.
- X. Li, J. Ma, G. Liu, J. Fang, S. Yue, Y. Guan, L. Chen and X. Liu, Efficient reductive dechlorination of monochloroacetic acid by sulfite/UV process, Environ. Sci. Technol., 2012, 46, 7342–7349 CrossRef CAS PubMed.
- R. Tenorio, J. Liu, X. Xiao, A. Maizel, C. Higgins, C. Schaefer and T. Strathmann, Destruction of per- and polyfluoroalkyl substances (PFASs) in aqueous film-forming foam (AFFF) with UV-sulfite photoreductive treatment, Environ. Sci. Technol., 2020, 54, 6957–6967 CrossRef CAS PubMed.
- E. Mousset and K. Doudrick, A review of electrochemical reduction processes to treat oxidized contaminants in water, Curr. Opin. Electrochem., 2020, 22, 221–227 CrossRef CAS.
- C. Zhang, D. He, J. Ma, W. Tang and T. Waite, Faradic reactions in capacitive deionization (CDI) – problems and possibilities: A review, Water Res., 2018, 128, 314–330 CrossRef CAS PubMed.
- Y. Su, U. Rao, C. Khor, M. Jensen, L. Teesch, B. Wong, D. Cwiertny and D. Jassby, Potential-driven electron transfer lowers the dissociation energy of the C-F bond and facilitates reductive defluorination of perfluooctane sulfonate (PFOS), ACS Appl. Mater. Interfaces, 2019, 11, 33913–33922 CrossRef CAS PubMed.
- Y. Wang and P. Zhang, Photocatalytic decomposition of perfluorooctanoic acid (PFOA) by TiO2 in the presence of oxalic acid, J. Hazard. Mater., 2011, 192, 1869–1875 CrossRef CAS PubMed.
- J. Cui, P. Gao and Y. Deng, Destruction of per- and polyfluoroalkyl substance (PFAS) with advanced reduction processes (ARPs): A critical review, Environ. Sci. Technol., 2020, 54, 3752–3766 CrossRef CAS PubMed.
- P. Tentscher, M. Lee and U. von Gunten, Micropollutant oxidation studied by quantum chemical computations: Methodology and applications to thermodynamics, kinetics, and reaction mechanisms, Acc. Chem. Res., 2019, 52, 605–614 CrossRef CAS PubMed.
- NDRL/NIST Solution Kinetics Database on the Web, NIST Standard Reference Database 40, https://kinetics.nist.gov/solution/, (accessed August 2021) Search PubMed.
- C. Li, S. Zheng, T. Li, J. Chen, J. Zhou, L. Su, Y.-N. Zhang, J. Crittenden, S. Zhu and Y. Zhao, Quantitative structure-activity relationship models for predicting reaction rate constants of organic contaminants with hydrated electrons and their mechanistic pathways, Water Res., 2019, 151, 468–477 CrossRef CAS PubMed.
- S. Zheng, C. Li and G. Wei, QSAR modeling for reaction rate constants of eaq- with diverse organic compounds in water, Environ. Sci.: Water Res. Technol., 2020, 6, 1931–1938 RSC.
- E. Hart, J. Thomas and S. Gordon, A review of the radiation chemistry of single-carbon compounds and some reactions of the hydrated electrons in aqueous solution, Radiat. Res., Suppl., 1964, 4, 75–88 Search PubMed.
- E. Bylaska, M. Dupuis and P. Tratnyek, Ab initio electronic structure study of one-electron reduction of polychlorinated ethylenes, J. Phys. Chem. A, 2005, 109, 5905–5916 CrossRef CAS PubMed.
- E. Bylaska, M. Dupuis and P. Tratneyk, One-electron-transfer reactions of polychlorinated ethylenes: Concerted and stepwise cleavages, J. Phys. Chem. A, 2008, 112, 3712–3721 CrossRef CAS PubMed.
- S. Rosokha, E. Lukacs, J. Ritzert and A. Wasilewki, Mechanism and thermodynamics of reductive cleavage of carbon-halogen bonds in the polybrominated aliphatic electrophiles, J. Phys. Chem. A, 2016, 120, 1706–1715 CrossRef CAS PubMed.
- D. Van Hoomissen and S. Vyas, Early events in the reductive dehalogenation of linear perfluoroalkyl substances, Environ. Sci. Technol. Lett., 2019, 6, 365–371 CrossRef CAS.
- Y. Zhang, A. Moores, J. Liu and S. Ghoshal, New insights into the degradation mechanism of perfluorooctanoic acid by persulfate from density functional theory and experimental data, Environ. Sci. Technol., 2019, 53, 8672–8681 CrossRef CAS PubMed.
- A. Carre-Burritt, D. Van Hoomissen and S. Vyas, Role of pH in the transformation of perfluoroalkyl carboxylic acids by activated persulfate: Implications from the determination of absolute electron-transfer rates and chemical computations, Environ. Sci. Technol., 2021, 55, 8928–8936 CrossRef CAS PubMed.
- G. Patlewicz, A. Richard, A. Williams, C. Grulke, R. Sams, J. Lambert, P. Noyes, M. DeVito, R. Hines, M. Strynar, A. Guiseppi-Elie and R. Thomas, A chemical category-based prioritization approach for selecting 75 per- and polyfluoroalkyl substances (PFAS) for tiered toxicity and toxicokinetic testing, Environ. Health Perspect., 2019, 127, 014501 CrossRef PubMed.
- S. Eldin and W. Jencks, Concerted bimolecular substitution reactions of anilino thioethers, J. Am. Chem. Soc., 1995, 117, 9415–9418 CrossRef CAS.
- C. Andrieux, J.-M. Savéant, A. Tallec, R. Tardivel and C. Tardy, Concerted and stepwise dissociative electron transfers. Oxidability of the leaving group and strength of the breaking illustrated by the electrochemical reduction of a – substituted acetophenones, J. Am. Chem. Soc., 1997, 119, 2420–2429 CrossRef CAS.
- J. Jaworski, M. Cembor and D. Kuck, Solvent effect on reductive bond cleavage of 1-chloro-10-methyltribenzotriquinacene: change from the concerted to the stepwise mechanism, Electrochim. Acta, 2007, 52, 2196–2202 CrossRef CAS.
- P. Brezonik, Chemical Kinetics and Process Dynamics in Aquatic Systems, CRC Press, 1993 Search PubMed.
- A. Isse, C. Lin, M. Coote and A. Gennaro, Estimation of standard reduction potentials of halogen atoms and alkyl halides, J. Phys. Chem. B, 2011, 115, 678–684 CrossRef CAS PubMed.
- Y. Zhao and D. Truhlar, The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: two new functionals and systematic testing of four M06-class functionals and 12 other functionals, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- A. Bauza, I. Alkorta, A. Frontera and J. Elguero, On the reliability of pure and hybrid DFT methods for the evaluation of halogen, chalcogen, and pnicogen bonds involving anionic and neutral electron donors, J. Chem. Theory Comput., 2013, 9, 5201–5210 CrossRef CAS PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- M. J. Frisch, GAUSSIAN 16 (Revision D.01), Gaussian Inc., Wallingford, CT, 2016 Search PubMed.
- E. J. Hart, E. M. Fielden and M. Anbar, Reactions of carbonylic compounds with hydrated electrons, J. Phys. Chem., 1967, 71, 3993–3998 CrossRef CAS.
- D. Minakata, S. P. Mezyk, J. W. Jones, B. R. Daws and J. C. Crittenden, Development of linear free energy relationships for aqueous phase radical-involved chemical reactions, Environ. Sci. Technol., 2014, 48, 13925–13932 CrossRef CAS PubMed.
- E. J. Hart, S. Gordon and J. K. Thomas, The Activation Energy of Hydrated Electron Reactions, J. Phys. Chem., 1964, 68, 1271–1274 CrossRef CAS.
- N. Getoff, Radiation- and photoinduced degradation of pollutants in water. A comparative study, Radiat. Phys. Chem., 1991, 37, 673–680 CrossRef CAS.
- H. A. Schwarz, Reaction of the Hydrated Electron with Water, J. Phys. Chem., 1992, 96, 8937–8941 CrossRef CAS.
- D. R. Prasad, M. Z. Hoffman, Q. G. Mulazzani and M. A. Rodgers, Pulsed-Laser Flash and Continuous Photolysis of Aqueous Solutions of Methyl Viologen, Oxalate, and Their Ion-Pair Complexes, J. Am. Chem. Soc., 1986, 108, 5135–5142 CrossRef CAS.
- Q. G. Mulazzani, M. D'Angelantonio, M. Venturi, M. Z. Hoffman and M. A. J. Rodgers, Interaction of Formate and Oxalate Ions with Radiation-Generated Radicals in Aqueous Solution. Methylviologen as a Mechanistic Probe, J. Phys. Chem., 1986, 90, 5347–5352 CrossRef CAS.
- N. Getoff, S. Schworer, V. M. Markovic, K. Sehested and S. O. Nielsen, Pulse Radiolysis of Oxalic Acid and Oxalates, J. Phys. Chem., 1971, 75, 749–755 CrossRef.
- S. Gordon, E. J. Hart, M. S. Matheson and J. Rabani, Reactions of the Hydrated Electron, Discuss. Faraday Soc., 1963, 36, 193–205 RSC.
- F. A. Peter and P. Neta, The Effect of Ionic Dissociation of Organic Compounds on Their Rate of Reaction with Hydrated Electrons, J. Phys. Chem., 1972, 76, 630–635 CrossRef CAS.
- G. Köhler, S. Solar, N. Getoff, A. R. Holzwarth and K. Schaffner, Relationship between the quantum yields of electron photoejection and fluorescence of aromatic carboxylate anions in aqueous solution, J. Photochem., 1985, 28, 383–391 CrossRef.
- G. Duplatre and C. D. Jonah, Reactions of electrons in high-concentration water solutions – A comparison between pulse-radiolysis and positron annihilation lifetime spectroscopy data, Radiat. Phys. Chem., 1984, 24, 557–565 CrossRef CAS.
- J. A. Bell, E. Grunwald and E. Hayon, Kinetics of Deprotonation of Organic Free Radical in Water. Reaction of HOCHCO2-, HOCHCONH2, and HOCCH3CONH2 with Various Bases, J. Am. Chem. Soc., 1975, 97, 2995–3000 CrossRef CAS.
- G. O. Phillips and N. W. Worthington, Effects of Ionizing Radiations on Glucuronic Acid, Radiat. Res., 1970, 43, 34–44 CrossRef CAS PubMed.
- J. A. D. Stockdale and D. F. Sangster, Relative Reaction Rates of Hydrated Electrons with Krebs Cycle and Other Anions, J. Am. Chem. Soc., 1966, 88, 2907–2910 CrossRef CAS.
- O. Micic and I. Draganic, Some reactions of hydrated electrons in acid medium (pH 0.6-4.0), Int. J. Radiat. Phys. Chem., 1969, 1, 287–295 CrossRef CAS.
- O. I. Micic and V. Markovic, Rates of hydrated electron reactions with undissociated carboxylic acids, Int. J. Radiat. Phys. Chem., 1972, 4, 43–49 CrossRef CAS.
- D. Razem and W. H. Hamill, Electron Scavenging in Ethanol and in Water, J. Phys. Chem., 1977, 81, 1625–1631 CrossRef CAS.
- S. Castillo-Rojas, A. Negron-Mendoza, Z. D. Draganic and I. G. Draganic, The radiolysis of aqueous solutions of malic acid, Radiat. Phys. Chem., 1985, 26, 437–443 CrossRef CAS.
- V. Markovic and K. Sehested, Radiolysis of Aqueous Solutions of Some Simple Compounds Containing Aldehyde Groups: Part I: Formaldehyde, Proceedings of the 3. Tihany Symposium on Radiation Chemistry, 1972, vol. 2, pp. 1243–1253 Search PubMed.
- K. G. Kemsley, J. S. Moore and G. O. Phillips, The effect of molecular size on the rate constants for reaction of hydrated electrons and hydroxyl radicals with carbohydrates, Int. J. Radiat. Biol. Relat. Stud. Phys. Chem. Med., 1979, 36, 429–432 CrossRef CAS PubMed.
- J. V. Davies, W. Griffiths and G. O. Philips, Pulse radiolysis of aqueous carbohydrate solutions, Pulse Radiolysis, 1965, 181–191 Search PubMed.
- M. T. Nenadovic and O. I. Micic, Pulse radiolysis of methyl acetate in aqueous solution, Radiat. Phys. Chem., 1978, 12, 85–89 CrossRef CAS.
- A. Biro and L. Wojnarovits, Pulse radiolysis of ethyl propionate in aqueous solution, J. Radioanal. Nucl. Chem., 1992, 166, 7–14 CrossRef CAS.
- T. Matsushige, G. Koltzenbur and D. Schulte-Frohlinde, Pulse radiolysis of aqueous solutions of acetic acid 2-hydroxylethyl ester. Fast elimination of acetic acid from a primary radical, Ber. Bunsen-Ges., 1975, 79, 657–661 CrossRef CAS.
- W. Bors, D. Tait, C. Michel, M. Saran and M. Erben-Russ, Reactions of Alkoxy Radicals in Aqueous Solution, Isr. J. Chem., 1984, 24, 17–24 CrossRef CAS.
- B. Massaut and B. Tilquin, Reactivity of the "capto-dative" methylmethoxyacetate (MMA) toward radicals, studied by electron pulse radiolysis in neutral aqueous medium, Belgian Chemical Societies, 1988, vol. 97, pp. 1031–1036 Search PubMed.
- R. L. S. Willix and W. M. Garrison, Chemistry of the hydrated electron in oxygen-free solutions of amino acids, peptides, and related compounds, Radiat. Res., 1967, 32, 452–462 CrossRef CAS PubMed.
- M. Simic and E. Hayon, Intermediates produced from the one-electron oxidation and reduction of hydroxylamines. Acid-base properties of the amino, hydroxyamino, and methoxyamino radicals, J. Am. Chem. Soc., 1971, 93, 5982–5986 CrossRef CAS.
- C. C. Lai and G. R. Freeman, Solvent effects on the reactivity of solvated electrons with organic solutes in methanol/water and ethanol/water mixed solvents, J. Phys. Chem., 1990, 94, 302–308 CrossRef CAS.
- G. V. Buxton, C. L. Greenstock, W. P. Helman and A. B. Ross, Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (·OH/·O-) in aqueous solution, J. Phys. Chem. Ref. Data, 1988, 17, 513–886 CrossRef CAS.
- A. M. Afanassiev, K. Okazaki and G. R. Freeman, Effect of Solvation Energy on Electron Reaction Rates in Hydroxylic Solvents, J. Phys. Chem., 1979, 83, 1244–1249 CrossRef CAS.
- M. Anbar and E. J. Hart, The Reactivity of Metal Ions and Some Oxy Anions toward Hydrated Electrons, J. Phys. Chem., 1965, 69, 973–977 CrossRef CAS.
- K. M. Idriss-Ali and G. R. Freeman, Electron behavior in mixed solvents: optical spectra and reactivities in water/alkane diols, Can. J. Chem., 1984, 62, 2217–2222 CrossRef CAS.
- S. P. Mezyk, Rate constant and activation energy determination for reaction of e-(aq) and ·OH with 2-butanone and propanal, Can. J. Chem., 1994, 72, 1116–1119 CrossRef CAS.
- J. Lilie, G. Beck and A. Henglein, Pulsradiolytische Untersuchung des Acetoinradikals und des Diacetylanions in waessriger Loesung, Ber. Bunsen-Ges., 1968, 72, 529–533 CAS.
- M. Anbar and E. J. Hart, The Activation Energy of Hydrated Electron Reactions, J. Phys. Chem., 1967, 71, 3700–3702 CrossRef CAS.
- M. Anbar, Z. B. Alfassi and H. Bregman-Reisler, Hydrated Electron Reactions in View of Their Temperature Dependence, J. Am. Chem. Soc., 1967, 89, 1263–1264 CrossRef CAS.
- J. V. Davies, M. Ebert and M. Quintiliani, Fast intermediate reactions sensitizing alcohol dehydrogenase to radiation, Taylor and Francis, New York NY, 1970 Search PubMed.
- K. H. Schmidt, P. Han and D. M. Bartels, Radiolytic Yields of the Hydrated Electron from Transient Conductivity. Improved Calculation of the Hydrated Electron Diffusion Coefficient and Analysis of Some Diffusion-Limited (e-)aq Reaction Rates, J. Phys. Chem., 1995, 99, 10530–10539 CrossRef CAS.
- N. Getoff, Advancements of Radiation Induced Degradation of Pollutants in Drinking and Waste Water, Appl. Radiat. Isot., 1989, 40, 585–594 CrossRef CAS.
- T. I. Balkas, J. H. Fendler and R. H. Schuler, Radiolysis of aqueous solutions of methyl chloride. The concentration dependence for scavenging electrons within spurs, J. Phys. Chem., 1970, 74, 4497–4505 CrossRef.
- D. Hayes, K. H. Schmidt and D. Meisel, Growth mechanisms of silver halide clusters from the molecule to the colloidal particle, J. Phys. Chem., 1989, 93, 6100–6109 CrossRef CAS.
- M. Lal and H. S. Mahal, Reactions of alkylbromides with free radicals in aqueous solutions, Radiat. Phys. Chem., 1992, 40, 23–26 CrossRef CAS.
- G. Bullock and R. Cooper, Reactions of Aqueous Trifluoromethyl Radicals, Trans. Faraday Soc., 1970, 66, 2055–2064 RSC.
- A. Szutka, J. K. Thomas, S. Gordon and E. J. Hart, Rate Constants of Hydrated Electron Reactions with Some Aromatic Acids, Alkyl Halides, Heterocyclic Compounds, and Werner Complexes, J. Phys. Chem., 1965, 69, 289–292 CrossRef CAS.
- J. Monig, K. Asmus, M. Schaeffer, T. F. Slater and R. L. Willson, Electron transfer reactions of halothane-derived peroxyl free radicals, CF3CHClO2·: Measurement of absolute rate constants by pulse radiolysis, J. Chem. Soc., Perkin Trans. 2, 1983, 1133–1137 RSC.
- M. Lal, C. Schoneich, J. Monig and K. Asmus, Rate constants for the reactions of halogenated organic radicals, Int. J. Radiat. Biol., 1988, 54, 773–785 CrossRef CAS PubMed.
- P. P. Infelta, M. Gratzel and J. K. Thomas, Luminescence Decay of Hydrophobic Molecules Solubilized in Aqueous Micellar Systems. A Kinetic Model, J. Phys. Chem., 1974, 78, 190–195 CrossRef CAS.
- T. I. Balkas, The radiolysis of aqueous solutions of methylene chloride, Int. J. Radiat. Phys. Chem., 1972, 4, 199–208 CrossRef CAS.
- T. I. Balkas, J. H. Fendler and R. H. Schuler, The radiation chemistry of aqueous solutions of CFCl3, CF2Cl2, and CF3Cl, J. Phys. Chem., 1971, 75, 455–466 CrossRef.
- O. I. Micic and B. Cercek, Diffusion-Controlled Reactions in Mixed Solvents, J. Phys. Chem., 1977, 81, 833–837 CrossRef CAS.
- I. M. Salih, T. Soeylemez and T. I. Balkas, Radiolysis of aqueous solutions of difluorochloromethane, Radiat. Res., 1976, 67, 235–243 CrossRef CAS PubMed.
- N. Getoff, Decomposition of biological resistant pollutants in water by irradiation, Radiat. Phys. Chem., 1990, 35, 432–439 CrossRef CAS.
- J. K. Thomas, Pulse radiolysis of aqueous solutions of methyl iodide and methyl bromide. The reactions of iodine atoms and methyl radicals in water, J. Phys. Chem., 1967, 71, 1919–1925 CrossRef CAS.
- J. L. Faria and S. Steenken, Photoionization (.lambda. = 248 or 308 nm) of triphenylmethyl radical in aqueous solution. Formation of triphenylmethyl carbocation, J. Am. Chem. Soc., 1990, 112, 1277–1279 CrossRef CAS.
- E. Hayon and A. O. Allen, Evidence for two kinds of “H atoms” in the radiation chemistry of water, J. Phys. Chem., 1961, 65, 2181–2185 CrossRef CAS.
- T. Eriksen, A. Henglein and K. Stockhausen, Pulse radiolytic oxidation of chloral hydrate in oxygenated and deoxygenated aqueous solutions, J. Chem. Soc., Faraday Trans. 1, 1973, 69, 337–345 RSC.
- N. C. Verma and R. W. Fessenden, Time resolved ESR spectroscopy. IV. Detailed measurement and analysis of the ESR time profile, J. Chem. Phys., 1976, 65, 2139–2155 CrossRef CAS.
- I. Draganic, Z. Draganic, L. Petkovic and A. Nikolic, The Radiation Chemistry of Aqueous Solutions of Simple RCN Compounds, J. Am. Chem. Soc., 1973, 95, 7193–7199 CrossRef CAS.
- N. Getoff and F. Schworer, Pulsradiolytische bestimmung von geschwindigkeitskonstanten der reaktionen einiger amine mit OH und eaq, Int. J. Radiat. Phys. Chem., 1970, 2, 81–89 CrossRef CAS.
- N. Getoff and F. Schworer, Pulse radiolysis of ethyl, n-propyl, n-butyl and n-amyl amine in aqueous solutions, Int. J. Radiat. Phys. Chem., 1973, 5, 101–111 CrossRef CAS.
- E. Hayon and M. Simic, Intermediates Produced from the One-Electron Oxidation of Hydrazine. Evidence for the Formation and Decay of Tetrazane and Triazene, J. Am. Chem. Soc., 1972, 94, 42–47 CrossRef CAS.
- E. Hayon and M. Simic, Free radical intermediates produced in the pulse radiolysis of simple peptides in aqueous solution, Intra-Sci. Chem. Rep., 1971, 5, 357 CAS.
- R. H. Bisby, R. B. Cundall and P. Wardman, A pulse radiolysis study of some free radical reactions with erythrocyte membranes, Biochim. Biophys. Acta, 1975, 389, 137–144 CrossRef CAS.
- P. S. Rao and E. Hayon, Interaction of Hydrated Electrons with the Peptide Linkage, J. Phys. Chem., 1974, 78, 1193–1196 CrossRef CAS.
- B. B. Saunders and R. A. Gorse, Reactions of Diethylhydroxylamine with Radiolytically Produced Radicals in Aqueous Solutions, J. Phys. Chem., 1979, 83, 1696–1701 CrossRef CAS.
- K. W. Chambers, E. Collinson and F. S. Dainton, Addition of e-aq, H· and ·OH to acrylamide in aqueous solution and reactions of the adducts, Trans. Faraday Soc., 1970, 66, 142–162 RSC.
- T. H. Tran-Thi, A. Koulkes-Pujo and J. Sutton, Radiolyse des amides et de leurs solutions aqueuses. Journees d' Etude sur la Chimie des Radiations, 1982, pp. 99–102.
- R. R. H. Farhataziz and E. M. Hansen, Pulse Radiolysis of Liquids at High Pressures. III. Hydrated-Electron Reactions Not Controlled by Diffusion, J. Chem. Phys., 1972, 57, 2959–2963 CrossRef.
- S. K. Kapoor and C. Gopinathan, Studies in mixed solvents: Comparison between solvated electron reactions and quenching of excited states, Int. J. Chem. Kinet., 1995, 27, 535–545 CrossRef CAS.
- N. S. Fel, P. I. Dolin and V. I. Zolotarevskii, Pulsed radiolysis of formamides, High Energy Chem., 1967, 1, 132–138 Search PubMed.
- R. Braams, Rate constants of hydrated electron reactions with peptides and proteins, Radiat. Res., 1967, 31, 8–26 CrossRef CAS PubMed.
- M. Simic and E. Hayon, Interaction of solvated electrons with the amide and imide groups. Acid-base properties of RC(OH)NH2 radicals, J. Phys. Chem., 1973, 77, 996–1001 CrossRef CAS.
- B. B. Singh and A. Kabi, Gamma-ray inactivation of trypsin in solution. Effects of various scavengers, Proc. Natl. Inst. Sci. India, Part B, 1969, 35, 291–297 CAS.
- E. Wold, O. Kaalhus, E. S. Johansen and A. T. Ekse, The electron affinity of some radiotherapeutic agents used in cancer therapy, Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med., 1980, 38, 599–611 CrossRef CAS PubMed.
- R. Braams, Rate Constants of Hydrated Electron Reactions with Amino Acids, Radiat. Res., 1966, 27, 319–329 CrossRef CAS PubMed.
- K. Bobrowski, J. Grodkowski and Z. P. Zagorski, Rate constants of the reactions of tetraalkylammonium cations with eaq- determined by pulse radiolysis method, Radiochem. Radioanal. Lett., 1979, 40, 329–337 CAS.
- S. P. Mezyk, Rate constant determination for the reaction of sulfhydryl species with the hydrated electron in aqueous solution, J. Phys. Chem., 1995, 99, 13970–13975 CrossRef CAS.
- M. Z. Hoffman and E. Hayon, Pulse Radiolysis Study of Sulfhydryl Compounds in Aqueous Solution, J. Phys. Chem., 1973, 77, 990–996 CrossRef CAS.
- S. A. Grachev, E. V. Kropachev, G. I. Litvyakova and S. P. Orlov, Influence of pH on the radiolysis of deaerated aqueous solutions of aminothiols, Russ. Chem. Bull., 1976, 25, 1248–1253 CrossRef.
- R. F. Anderson and D. Schulte-Frohlinde, Reactions induced by hydroxyl radical attack on acetylene in aqueous solution. A pulse radiolysis study, J. Phys. Chem., 1978, 1, 22–26 CrossRef.
- E. A. Balazs, J. V. Davies, G. O. Phillips and D. S. Scheufele, Polyanions and their complexes. Part III. Reactions of heparin, hyaluronic acid, sodium poly(ethylenesulphonate), sodium poly(styrene-sulphonate), and sodium carboxymethylcellulose with hydroxyl radicals and hydrated electrons, J. Chem. Soc. C, 1968, 12, 1420–1423 RSC.
- L. Engman, J. Lind and G. Merenyi, Redox Properties of Diaryl Chalcogenides and Their Oxides, J. Phys. Chem., 1994, 98, 3174–3182 CrossRef CAS.
- T. Sumiyoshi, N. Miura, M. Aikawa and M. Katayama, Pulse Radiolysis Studies on Methyl Methylthiomethyl Sulfoxide in Aqueous Solutions, Bull. Chem. Soc. Jpn., 1982, 55, 2347–2351 CrossRef CAS.
- W. Karmann, A. Granzow, G. Meissner and A. Henglein, Die pulsradiolyse einfacher merkaptane in luft freier wassriger losung, Int. J. Radiat. Phys. Chem., 1969, 1, 395–405 CrossRef CAS.
- T. Tung and R. R. Kuntz, Hydrated electron reactions with thiols in acidic aqueous solutions, Radiat. Res., 1973, 55, 256–264 CrossRef CAS PubMed.
- G. G. Jayson, D. A. Stirling and A. J. Swallow, Pulse- and X-radiolysis of 2-mercaptoethanol in aqueous solution, Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med., 1971, 19, 143–156 CrossRef CAS PubMed.
- G. Meissner, A. Henglein and G. Beck, Pulsradiolytische Untersuchung von Dimethylthioather und Dimethylsulfoxyd in waBriger Losung, Z. Naturforsch., B: Anorg. Chem., Org. Chem., Biochem., Biophys., Biol., 1967, 22, 13–19 CrossRef CAS.
- M. Z. Hoffman and E. Hayon, One-electron reduction of the disulfide linkage in aqueous solution. Formation, protonation, and decay kinetics of the RSSR- radical, J. Am. Chem. Soc., 1972, 94, 7950–7957 CrossRef CAS.
- R. Zhao, J. Lind, G. Merenyi and T. E. Eriksen, Kinetics of one-electron oxidation of thiols and hydrogen abstraction by thiyl radicals from -amino C-H bonds, J. Am. Chem. Soc., 1994, 116, 12010–12015 CrossRef CAS.
- W. Roebke, M. Schoneschofer and A. Henglein, Die y-Radiolyse und Pulsradiolyse des Schwefelkohlenstoffs in wabriger Losung, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1973, 28, 12–22 CrossRef CAS.
- G. R. Dey, D. B. Naik, K. Kishore and P. N. Moorthy, Nature of the transient species formed in the pulse radiolysis of some thiourea derivatives, J. Chem. Soc., Perkin Trans. 2, 1994, 1625–1629 RSC.
- S. C. Wallace and J. K. Thomas, Reactions in Micellar Systems, Radiat. Res., 1973, 54, 49–62 CrossRef CAS PubMed.
- K. Asmus, A. Henglein and G. Beck, Pulsradiolytische Untersuchung der Reaktion des hydratisierten Elektrons mit Nitromethan, Ber. Bunsen-Ges., 1966, 70, 459–466 CAS.
- J. Sutton and T. D. Son, Vitesses de reaction de trois nitroparaffines avec les atomes d'hydrogene et les electrons solvates en milieu aqueux, J. Chim. Phys., 1967, 64, 688–690 CrossRef CAS.
- K. P. Madden and H. Taniguchi, An in Situ Radiolysis Time-Resolved Electron Spin Resonance Study of 2-Methyl-2-nitrosopropane Spin Trapping Kinetics, J. Am. Chem. Soc., 1991, 113, 5541–5547 CrossRef CAS.
- L. Huang, W. Dong and H. Hou, Investigation of the reactivity of hydrated electron toward perfluorinated carboxylates by laser flash photolysis, Chem. Phys. Lett., 2007, 436, 124–128 CrossRef CAS.
- G. V. Buxton, P. G. Ellis and T. F. W. McKillop, Pulse radiolysis study of acrylonitrile in aqueous solution, J. Chem. Soc., Faraday Trans. 1, 1979, 75, 1050–1066 RSC.
- V. Madhavan, N. N. Lichtin and E. Hayon, Protonation Reactions of Electron Adducts of Acrylamide Derivatives. A Pulse Radiolytic-Kinetic Spectrophotometric Study, J. Org. Chem., 1976, 41, 2320–2326 CrossRef CAS.
- M. Kumar, M. J. Rao and P. N. Moorthy, Free-radical species from methyl vinyl ketone in aqueous solution: A pulse radiolysis study, J. Macromol. Sci., Chem., 1990, 27, 299–308 CrossRef.
- P. N. Moorthy, V. Kumar, K. N. Rao and J. Shankar, Rate constants of reactions of photogenerated solvated electrons with monomers, Radiat. Eff., 1971, 10, 129–131 CrossRef CAS.
- V. Madhavan, N. N. Lichtin and E. Hayon, Protonation Reactions of Electron Adducts of Acrylamide Derivatives. A Pulse Radiolytic-Kinetic Spectrophotometric Study, J. Am. Chem. Soc., 1975, 97, 2989–2995 CrossRef CAS.
- R. Koester and K. Asmus, Die Reaktionen chlorierter Aethylene mit hydratisierten Elektronen und OH-Radikalen in waessriger Loesung, Z. Naturforsch., B: Anorg. Chem., Org. Chem., Biochem., Biophys., Biol., 1971, 26b, 1108–1116 CrossRef.
- D. Behar, R. W. Fessenden and J. P. Hornak, Esr and pulse radiolysis investigation of the radiolysis of sodium vinyl sulfonate, Radiat. Phys. Chem., 1982, 20, 267–273 CrossRef CAS.
- Z. D. Draganic, I. G. Draganic and K. Sehested, Radiation Chemistry of Aqueous Solutions of Dicyandiamide, J. Phys. Chem., 1979, 83, 220–224 CrossRef CAS.
- M. Kumar and M. H. Rao, Pulse radiolysis study of initiation, dimerization, and propagation steps of 3,3-dimethylacrylic acid in aqueous medium, J. Macromol. Sci., Chem., 1991, 28, 531–544 Search PubMed.
- A. Safrany and L. Wojnarovits, Radiolysis of hydroxy ethylacrylate in dilute aqueous solutions, Radiat. Phys. Chem., 1993, 41, 531–537 CrossRef CAS.
- B. Cercek, Activation energies for reactions of the hydrated electron, Nature, 1969, 223, 491–492 CrossRef CAS.
- K. W. Chambers, E. Collinson, F. S. Dainton, W. A. Seddon and F. Wilkinson, Pulse radiolysis: Adducts of vinyl compounds and simple free radicals, Trans. Faraday Soc., 1967, 63, 1699–1711 RSC.
- M. Karelson, Molecular Descriptors in QSAR/QSPR, John Wiley & Sons, New York, 2000 Search PubMed.
- J. Shorter, Correlation Analysis of Organic Reactivity with Particular Reference to Multiple Regression, John Wiley & Sons, New York, 1982 Search PubMed.
- O. Exner, Correlation Analysis of Chemical Data, Plenum Press, New York, 1988 Search PubMed.
- S. Gordon, E. J. Hart and J. K. Thomas, The Ultraviolet Spectra of Transients Produced in the Radiolysis of Aqueous Solutions, J. Phys. Chem., 1964, 68(5), 1262–1264 CrossRef CAS.
- S. Gordon, E. J. Hart and J. K. Thomas, The Ultraviolet Spectra of Transients Produced in the Radiolysis of Aqueous Solutions, J. Phys. Chem., 1964, 68, 1262–1264 CrossRef CAS.
- L. Eberson, Problems and Prospects of the Concerted dissociative Electron Transfer Mechanism, Acta Chem. Scand., 1999, 53, 751–764 CrossRef CAS.
- C. Costentin, M. Robert and J.-M. Saveant, Successive removal of chloride ions from organic polychloride pollutants. Mechanisms of reductive electrochemical elimination in aliphatic gem-polychlorides, a,b-polychloroalkenes, and a,b-polychloroalkanes in midly protic medium, J. Am. Chem. Soc., 2003, 125, 10729–10739 CrossRef CAS PubMed.
- M. Z. Hoffman and E. Hayon, Pulse Radiolysis Study of Sulfhydryl Compounds in Aqueous Solution, J. Phys. Chem., 1973, 77(8), 990–996 CrossRef CAS.
- C. P. Andrieux, A. Le Gorande and J. M. Saveant, Electron transfer and bond breaking. Examples of passage from a sequential to a concerted mechanism in the electrochemical reductive cleavage of arylmethyl halides, J. Am. Chem. Soc., 1992, 114, 6892–6904 CrossRef CAS.
- R. N. Jones and C. Sandorfy, Chemical Applications of Spectroscopy, Techniques in Organic Chemistry, Interscience Publishers, New York, N. Y., 1956 Search PubMed.
- C. L. L. Chai, G. A. Heath, P. B. Huleatt and G. A. O'Shea, The first electrochemical study of epidithiopiperazine-2,5-diones, a special class of α,α’-disulfide bridged cyclic dipeptides, J. Chem. Soc., Perkin Trans. 2, 1999, 389–392 RSC.
- S. Antonello, R. Benassi, G. Giovanna, F. Taddei and F. Maran, Theoretical and Electrochemical Analysis of Dissociative Electron Transfers Proceeding through Formation of Loose Radical Anion Species: Reduction of Symmetrical and Unsymmetrical Disulfides, J. Am. Chem. Soc., 2002, 124, 7529–7538 CrossRef CAS PubMed.
- F. Maran, D. D. M. Wayner and M. S. Workentin, Kinetics and mechanism of the dissociative reduction of C-X and X-X bonds (X=O, S), Adv. Phys. Org. Chem., 2001, 36, 85–166 CrossRef CAS.
- S. Wang, Q. Yang, F. Chen, J. Sun, K. Luo, F. Yao, X. Wang, D. Wang, X. Li and G. Zeng, Photocatalytic Degradation of Perfluorooctanoic Acid and Perfluorooctane Sulfonate in Water: A Critical Review, Chem. Eng. J., 2017, 328, 927–942 CrossRef CAS.
- H. Park, C. D. Vecitis, J. Cheng, W. Choi, B. T. Mader and M. R. Hoffmann, Reductive Defluorination of Aqueous Perfluorinated Alkyl Surfactants: Effects of Ionic Headgroup and Chain Length, J. Phys. Chem. A, 2009, 113, 690–696 CrossRef CAS PubMed.
- W. A. Maza, V. M. Breslin, J. C. Owrutsky, B. B. Pate and A. Epshteyn, Nanosecond Transient Absorption of Hydrated Electrons and Reduction of Linear Perfluoroalkyl Acids and Sulfonates, Environ. Sci. Technol. Lett., 2021, 8, 525–530 CrossRef CAS.
- J. Blotevogel, R. J. Giraud and T. Borch, Reductive defluorination of perfluorooctanoic acid by zero-valent iron and zinc: A DFT-based kinetic model, Chem. Eng. J., 2018, 335, 248–254 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional information for 4 text, 7 tables and 24 figures is available in the ESI1 as well as all z-matrix and optimized molecular and radical structures in the ESI2. See DOI: 10.1039/d1ew00897h |
| This journal is © The Royal Society of Chemistry 2022 |