
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBi- and trimetallic complexes with macrocyclic xanthene-4,5-diNHC ligands†
Alexander
Kaps
,
Sabine
Foro
and
Herbert
Plenio
 *
*
Organometallic Chemistry, Technical University of Darmstadt, Alarich-Weiss-Str. 12, 64287 Darmstadt, Germany. E-mail: herbert.plenio@tu-darmstadt.de
First published on 20th January 2022
Abstract
Three different types of bimetallic NHC-metal complexes were synthesized, whose NHC units are attached at the 4,5-positions of xanthene. The NHC units are in close proximity and are designed such that each carbene coordinates one ML unit, while the chelation of one metal by two NHC is not possible. Several xanthene-((NHC)ML)2 complexes with ML = RhCl(cod), IrCl(cod), RhCl(CO)2, IrCl(CO)2, AuCl, AgCl, CuCl and Pd(allyl)Cl were synthesized and investigated.
Introduction
The chemistry of N-heterocyclic carbenes has experienced an enormous development in the last decades following the seminal work of Arduengo et al.1 and Herrmann.2 Monographies provide a comprehensive overview of NHC chemistry,3,4 while succinct reviews, such as for example the one by Glorius et al. provide an inspiring, excellent introduction to the field.5 NHC-metal complexes involving transition metals play an essential role in homogeneous catalysis, and are real game changers in ruthenium based olefin metathesis catalysts of the Grubbs type6–8 and in several gold-catalyzed transformations.9 Other highlighted applications of NHC-metal complexes are Pd-and Ni-catalyzed cross coupling reactions10,11 or olefin polymerizations,12 and catalytic reactions involving other M(NHC) complexes with M = Mo and W,13 Fe,14 Pt,15 Co,16 Rh,17 Ir,18–20 coinage metals,21 Zn22 or main group elements.23These applications rely on a correspondingly large variety of different NHC ligands.24–31 Most such ligands contain only a single NHC unit and obviously ligands containing several NHC ligands are less common. Four different motifs involving metal complexes with ligands containing more than a single NHC unit can be distinguished (Scheme 1).
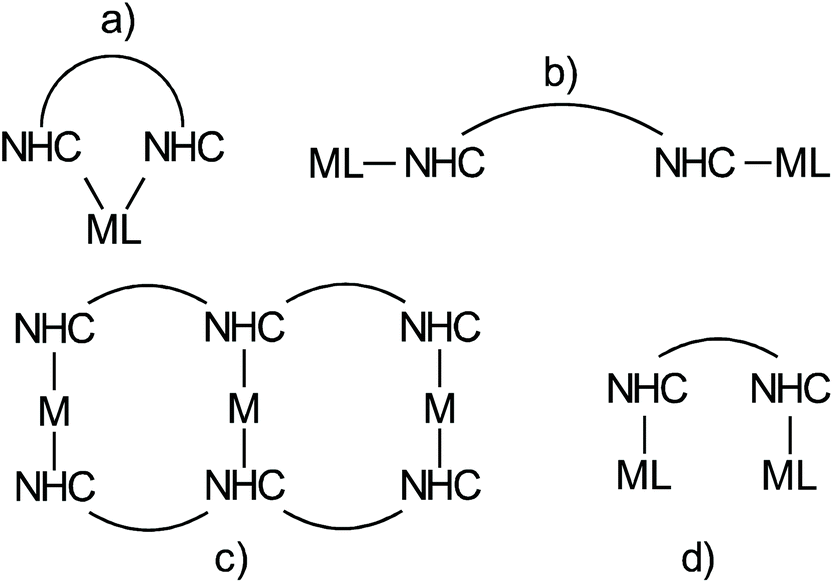 | ||
| Scheme 1 Different structural motifs in metal complexes with bidentate or polydentate NHC ligands. | ||
Structure (a), in which two NHC ligands bind a single metal in a chelating fashion, represents a common theme, which has also been used in numerous catalytic transformations.32 Motif (b) in which two NHC bind different metals individually, which cannot directly interact with each other was established by Bielawski et al.33,34 and used to generate polymeric materials.35 This approach was foremost extended by Peris et al. in numerous Janus type NHC metal complexes.36 Approach (c) leading to complex supramolecular motifs is actively pursued by E. Hahn and Ying-Feng Han37 and the Peris group.38 Typically, diverse arrays of coinage metal complexes of the type M(NHC)2+ are formed in which the coordination sphere of the metal is saturated by the two NHC ligands. The basic concept in motif (d) (Scheme 1) is to synthesize ligands containing two NHC ligands, which are able to bind two metals in close proximity and which in addition also possess ligands other than NHC. The ligands also need to be designed in such a way that the two NHC ligands cannot bind a single metal atom in a chelating fashion like in (a). Accordingly, one metal is bonded to each of the two NHC ligands. Consequently, the two metal centers are in close proximity and are potentially able to interact and to cooperate. To the best of our knowledge such complexes have not been reported before.
Experimental
General experimental
All chemicals were purchased as reagent grade from commercial suppliers and used without further purification, unless otherwise noted. Tetrahydrofuran and toluene were dried with sodium, MeOH/CH3CN/CH2Cl2/DMF and DMSO were dried over CaH2, and distilled under nitrogen or argon. All dried solvents were stored over molecular sieves (4 Å). Cy/EA stands for “cyclohexane/ethyl acetate”. 1H and 13C NMR spectra were recorded with a Bruker AC300 or DRX500 spectrometer. The chemical shifts are given in parts per million (ppm) on the δ scale and are referenced to the residual peak of CDCl3 (δH = 7.26 ppm, δC = 77.16 ppm), DMSO-d6 (δH = 2.50 ppm, δC = 39.52 ppm), CDCl2 (δH = 5.32 ppm, δC = 53.84 ppm) or MeOD (δH = 3.31 ppm, δC = 49.00 ppm). Abbreviations for NMR: s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, quint = quintet, sept = septet, m = multiplet. TLC was performed by using silica 60 F 254 (0.2 mm) on alumina plates. Preparative chromatography was carried out on Merck silica 60 (0.063–0.2 mm). Cyclic voltammograms were recorded in dry CH2Cl2 under nitrogen at ambient temperature. A three-electrode configuration was employed: the working electrode was a Pt disk (diameter 1 mm) sealed in soft glass with a Pt wire as counter electrode, and the pseudo-reference electrode was an Ag wire. Potentials were calibrated internally against the formal potential of octamethylferrocene [E1/2 = −0.01 V (CH2Cl2)]. Bu4NPF6 (0.1 mol L−1) was used as supporting electrolyte.2,3,6,7,9,9-Hexamethylxanthene (2)
2,3,6,7,9,9-Hexamethylxanthene was prepared according to a modified literature procedure.39 A mixture of 3,4-dimethylphenol (210 g, 1.74 mol, 10 eq.), acetone (12.8 mL, 174 mmol, 1.0 eq.) and methanesulfonic acid (1.1 mL, 17.4 mmol, 0.1 eq.) was heated at 110 °C for 5 d and then at 150 °C for 6 h. The product mixture was diluted with 200 mL toluene, washed 3 times with 10% NaOH aq. (300 mL) and water, dried over MgSO4, and concentrated under reduced pressure to give a dark waxy solid. The residue was slurried in boiling methanol and filtered over a fritted funnel. The filtrate was then concentrated under reduced pressure to give an off-white solid. 1H NMR (300 MHz, CDCl3): δ 7.13 (s, 2H), 6.82 (s, 2H), 2.25 (s, 6H), 2.24 (s, 6H), 1.61 (s, 6H). 13C NMR (126 MHz, CDCl3): δ 148.4, 135.8, 130.8, 127.2, 117.2, 33.4, 33.0, 19.5, 19.4. Yield: 32% (14.8 g, 55.6 mmol).4,5-Dibromo-2,3,6,7,9,9-hexamethylxanthene (3)
4,5-Dibromo-2,3,6,7,9,9-hexamethylxanthene was prepared according to a modified literature procedure.40 To a Schlenk flask containing 2,3,6,7,9,9-hexamethylxanthene (13.9 g, 52.2 mmol) and 350 mL DCM was added Br2 (6.15 mL, 120 mmol, 2.3 eq.) dropwise at −78 °C. The reaction mixture was stirred at rt overnight. Na2S2O3 (aq.) was added to quench excess of bromine. The layers were separated and aqueous phase was extracted with DCM. The combined organic phase was washed with water and brine, and was dried over MgSO4. The volatiles were evaporated to afford 4,5-dibromo-2,3,6,7,9,9-hexamethylxanthene as a white solid. 1H NMR (300 MHz, CDCl3): δ 7.10 (s, 2H), 2.40 (s, 6H), 2.33 (s, 6H), 1.59 (s, 6H). Yield: 92% (20.4 g, 48.1 mmol).2,3,6,7,9,9-Hexamethylxanthene-4,5-dicarboxylic acid (4)
2,3,6,7,9,9-Hexamethylxanthene-4,5-dicarboxylic acid was prepared according to the literature procedure for 2,7-di-tert-butyl-9,9-dimethyl-9H-xanthene-4,5-dicarboxylic acid.41 In a three-necked round flask, 4,5-dibromo-2,3,6,7,9,9-hexamethylxanthene (18.7 g, 44.1 mmol) was dissolved in 500 mL dry THF and cooled with an acetone/dry ice bath to −78 °C. A solution of n-butyllithium (1.6 M in n-hexane, 82.7 mL, 132.3 mmol) was slowly added and the mixture stirred for further 30 min at −78 °C. CO2 was produced by evaporating dry ice (excess) in a round flask, bubbled through a drying tube filled with silica desiccant and passed into the solution for approx. 2 h. The reaction was quenched with ice and conc. HCl. The layers were separated and the aqueous phase was extracted with diethyl ether. The organic phases were combined, washed with brine, dried over MgSO4, filtered and the solvent evaporated. The residue was suspended in pentane, stirred and filtered and the solid was washed again with pentane, affording the product as a white solid. 1H NMR (300 MHz, DMSO-d6): δ 12.94 (bs, 2H), 7.35 (s, 2H), 2.23 (s, 6H), 2.12 (s, 6H), 1.56 (s, 6H). 13C NMR (75 MHz, DMSO-d6): δ 168.0, 143.2, 131.2, 127.5, 126.6, 124.4, 33.1, 32.4, 19.2, 15.9. HRMS (ESI): calcd for C21H23O5 (M + H)+ 355.15400. Found 355.15364 (Δ = 0.36 mmu). Yield: 60% (9.4 g, 26.5 mmol).Dibenzyl (2,3,6,7,9,9-hexamethylxanthene-4,5-diyl)dicarbamate (5)
Dibenzyl (2,3,6,7,9,9-hexamethylxanthene-4,5-diyl)dicarbamate was prepared according to a modified literature procedure for dibenzyl (2,7-di-tert-butyl-9,9-dimethyl-9H-xanthene-4,5-diyl)dicarbamate.41 2,3,6,7,9,9-Hexamethylxanthene-4,5-dicarboxylic (6.0 g, 16.9 mmol, 1.0 eq.) was suspended in 120 mL dry toluene under molecular sieve. Then diphenylphosphoryl azide (9.01 mL, 47.4 mmol, 2.8 eq.) was slowly added and the solution stirred for 5 min at rt. Triethylamine (6.61 mL, 47.4 mmol, 2.8 eq.) was added and the mixture was refluxed for 2 h. After this time benzyl alcohol (3.70 mL, 54.2 mmol, 3.2 eq.) was added and refluxed overnight. The solvent was evaporated and the residue purified by column chromatography (Cy/EA 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1→3:1) affording the pure product as a white solid. 1H NMR (300 MHz, CDCl3): δ 7.49–7.25 (m, 10H), 7.09 (s, 2H), 6.52 (s, 2H), 5.15 (s, 4H), 2.27 (s, 6H), 2.16 (s, 6H), 1.57 (s, 6H). 13C NMR (75 MHz, CDCl3): δ 144.0, 136.7, 131.4, 128.6, 128.3, 128.2, 125.0, 67.3, 34.2, 31.8, 20.4, 14.7. Rf = 0.21 (Cy/EA = 10:1). HRMS (ESI): calcd for C35H37N2O5 (M + H)+ 565.26970. Found 565.26979 (Δ = 0.09 mmu). Yield: 62% (3.1 g, 5.49 mmol).
:1→3:1) affording the pure product as a white solid. 1H NMR (300 MHz, CDCl3): δ 7.49–7.25 (m, 10H), 7.09 (s, 2H), 6.52 (s, 2H), 5.15 (s, 4H), 2.27 (s, 6H), 2.16 (s, 6H), 1.57 (s, 6H). 13C NMR (75 MHz, CDCl3): δ 144.0, 136.7, 131.4, 128.6, 128.3, 128.2, 125.0, 67.3, 34.2, 31.8, 20.4, 14.7. Rf = 0.21 (Cy/EA = 10:1). HRMS (ESI): calcd for C35H37N2O5 (M + H)+ 565.26970. Found 565.26979 (Δ = 0.09 mmu). Yield: 62% (3.1 g, 5.49 mmol).
4,5-Diamino-2,3,6,7,9,9-hexamethylxanthene (6)
4,5-Diamino-2,3,6,7,9,9-hexamethylxanthene was prepared according to the literature procedure for 2,7-di-tert-butyl-9,9-dimethyl-9H-xanthene-4,5-diamine.41 A solution of potassium hydroxide (3.43 g, 61.1 mmol, 15.0 eq.) in water (30 mL) was added to a solution of dibenzyl (2,3,6,7,9,9-hexamethylxanthene-4,5-diyl)dicarbamate (2.3 g, 4.07 mmol, 1.0 eq.) in ethanol (80 mL). The mixture was refluxed for 24 h. Ethanol was removed under reduced pressure and the aqueous residue was diluted with water and extracted with diethyl ether. The organic phases were combined, dried with magnesium sulfate, filtered and the solvent evaporated. The residue was purified by column chromatography (Cy/EA 10:1→1:1) affording the pure product as a light brown solid. 1H NMR (300 MHz, CDCl3): δ 6.68 (s, 2H), 3.83 (s, 4H), 2.27 (s, 6H), 2.11 (s, 6H), 1.59 (s, 6H). 13C NMR (75 MHz, CDCl3): δ 136.8, 132.4, 130.6, 127.2, 119.4, 116.5, 33.8, 32.4, 20.5, 13.0. Rf = 0.43 (Cy/EA = 1:1). Yield: 83% (1.0 g, 3.37 mmol).
:1→4:1→1:1). Yield: 16% (0.22 g). (4.7 mmol scale)→no Na2CO3; yield: 40% (0.13 g). (1.2 mmol scale)→with 2 eq. Na2CO3; yield: 36% (0.25 g). (2.4 mmol scale)→with 2 eq. Na2CO3; yield: 43% (0.60 g). (4.7 mmol scale)→with 2 eq. Na2CO3; yield: 38% (1.06 g). (9.4 mmol scale)→with 1 eq. Na2CO3.
4-Amino-2,3,6,7,9,9-hexamethylxanthene (7)
4-Amino-2,3,6,7,9,9-hexamethylxanthene was obtained as side product from the Cu-catalyzed synthesis of 4,5-diamino-2,3,6,7,9,9-hexamethylxanthene. The mixture was purified by column chromatography (Cy/EA 20:1→4:1→1:1) affording the pure product as a light brown solid. 1H NMR (300 MHz, CDCl3): δ 7.15 (s, 1H), 6.89 (s, 1H), 6.66 (s, 1H), 3.90 (s, 2H), 2.27 (s, 3H), 2.26 (s, 3H), 2.25 (s, 3H), 2.11 (s, 3H), 1.61 (s, 6H). 13C NMR (75 MHz, CDCl3): δ 148.3, 136.7, 135.6, 132.5, 130.9, 130.4, 127.4, 127.2, 126.9, 119.2, 117.2, 116.2, 33.6, 32.7, 20.5, 19.5, 19.4, 13.0. Rf = 0.39 (Cy/EA = 10:1). Yield: 40%.
Xanthene diimine (8)
A suspension of 4,5-diamino-2,3,6,7,9,9-hexamethylxanthene (350 mg, 1.18 mmol, 2.0 eq.), glyoxal (40% aq., 135 μL, 1.18 mmol, 2.0 eq.), and 2 mg of p-toluenesulfonic acid monohydrate in n-BuOH (8 mL) was stirred under nitrogen in a Schlenk tube at 50 °C for 24 h. The cooled reaction mixture was filtered and residual solid was washed with cold methanol and pentane to give xanthene diimine 8 as a yellow solid. 1H NMR (500 MHz, CD2Cl2) δ 8.28 (s, 4H), 7.09 (s, 4H), 2.30 (s, 12H), 2.14 (s, 12H), 1.64 (s, 12H). 13C NMR (126 MHz, CD2Cl2) δ 166.7, 138.6, 138.3, 131.8, 128.9, 128.9, 123.9, 34.7, 32.0, 20.2, 14.3. HRMS (ESI): calcd for C42H45N4O2 (M + H)+ 637.35370. Found 637.35397 (Δ = 0.27 mmu). Yield: 85% (0.32 g, 0.50 mmol).General procedure for the synthesis of imidazolium chloride
To a flame dried Schlenk tube containing a solution of the corresponding diimine (1.0 eq.) and paraformaldehyde (1.1–2.4 eq.) in EA was added Me3SiCl (1.5–2.5 eq.). After being stirred at 70 °C overnight, the resulting suspension was cooled to −20 °C, filtered and washed with cold EA, diethyl ether gave the title compound.Xanthene diimidazolium salt (9·2HCl)
According to the general procedure starting materials used were xanthene diimine (8) (150 mg, 0.235 mmol, 1.0 eq.), paraformaldehyde (17 mg, 0.565 mmol, 2.4 eq.), Me3SiCl (74.7 μL, 0.589 mmol, 2.5 eq.) in 2 ml EA. The title compound was obtained as an off-white solid. 1H NMR (300 MHz, DMSO-d6): δ 9.70 (s, 2H), 8.16 (s, 4H), 7.80 (s, 4H), 2.38 (s, 12H), 2.02 (s, 12H), 1.80 (s, 6H), 1.64 (s, 6H). 1H NMR (300 MHz, CDCl3): δ 9.78 (s, 2H), 8.26 (s, 4H), 7.39 (s, 4H), 2.35 (s, 12H), 2.05 (s, 12H), 1.74 (s, 6H), 1.65 (s, 6H). 13C NMR (75 MHz, CDCl3): δ 141.9, 138.8, 133.4, 132.8, 130.5, 128.0, 127.2, 121.3, 33.9, 33.6, 33.0, 20.2, 14.2. HRMS (ESI): calcd for C44H46ClN4O2 [M − Cl]+ 697.33038. Found 697.33064 (Δ = 0.26 mmu). Yield: 85% (146 mg, 0.199 mmol).General procedure for the synthesis of imidazolium tetrafluoroborate
The halide salt (50 mg) was dissolved in acetonitrile, and a water solution with an excess of NaBF4 added. After stirring the solution for 15 min the solvent was removed under reduced pressure. Then water was added and extracted several times with dichloromethane. The organic solution was subsequently collected and dried over MgSO4, filtrated, concentrated under reduced pressure and then diethyl ether was added to precipitate the product as a white crystalline powder.Xanthene diimidazolium salt (9·2HBF4)
According to the general procedure 9·2HCl (50 mg) was used. 1H NMR (500 MHz, CD2Cl2): δ 8.57 (t, J = 1.6 Hz, 2H), 7.75 (d, J = 1.1 Hz, 4H), 7.51 (s, 4H), 2.38 (s, 12H), 2.00 (s, 12H), 1.79 (s, 6H), 1.69 (s, 6H). 13C NMR (126 MHz, CD2Cl2): δ 142.7, 139.0, 133.7, 133.3, 131.1, 128.6, 126.6, 121.6, 34.4, 33.9, 32.8, 20.4, 14.3. Yield: 99% (56 mg, 0.067 mmol).General procedure for the synthesis of (NHC)metal complexes
Method A: A vial was charged with the corresponding NHC salt (1.0 eq.), metal precursor (0.5–1.0 eq.) (M = Ir, Rh, Au) and K2CO3 (3.0 eq.). The resulting mixture was suspended in acetone or acetone/DCM (3:1 v/v) and stirred for 24 h at 50 °C. After this time the solvent was removed in vacuo and dichloromethane was added. The mixture was filtered through a pad of silica. The pad of silica was washed with dichloromethane until filtrate becomes colorless and the solvent was removed in vacuo.
Method B: To a Schlenk tube containing a solution of NHC salt (1.0 eq.) in dry THF was added sodium amylate (2.5 M, 2.0 eq.) at −78 °C. After stirring for 10 min, the cooling bath was removed and the solution stirred for 30 min at rt. After cooling again to −78 °C, [MCl(cod)]2 (1.0 eq.)(M = Rh, Ir) was added and the mixture stirred overnight. The mixture was filtered through a pad of Celite then the Celite was washed with DCM until the filtrate becomes colorless and the solvent was removed in vacuo.
Method C: A vial was charged with the corresponding NHC salt (1.0 eq.) and silver(I) oxide (1.0–1.5 eq.) were dissolved in dry MeOH and stirred at rt in the absence of light for 24 h. After this time metal precursor (1.0–2.0 eq.) (M = Au, Rh) was added, and in the absence of light the mixture was stirred at 50 °C for 2 h, then 12 h at rt. The mixture was filtered through Celite and the solvent was removed under reduced pressure. The residue was then dissolved in a minimum amount of DCM and diethyl ether was added. The precipitate was filtered and dried under reduce pressure.
Method D: A vial was charged with the corresponding NHC salt (1.0 eq.), metal precursor (1.0–2.0 eq.) (M = Ir, Rh), NaOAc (2.0 eq.), K2CO3 (2.0 eq.) and KBr (excess) were dissolved in dry MeCN and stirred at 80 °C for 12 h. After this time the mixture was filtered through Celite and the solvent was removed under reduced pressure.
Method E [MX(CO) 2 (NHC)] complexes (X = Cl, Br) via substitution of cod by CO: The respective [IrX(cod)(NHC)], [RhX(cod)(NHC)] complex was dissolved in CH2Cl2 and cooled to 0 °C. Then CO was bubbled through this solution for 30 min. The solvent was evaporated and the residue washed with pentane to obtain the products as yellow powders. The conversions for all reactions are quantitative, the lower isolated yields result from small losses (several mg) of material during workup.
[(IrCl(cod))
2
(
9
)]. Method B. Starting materials 9·2HCl (100 mg, 0.136 mmol, 1.0 eq.), sodium amylate (2.5 M in toluene, 0.11 mL, 0.273 mmol, 2.0 eq.), [IrCl(cod)]2 (91.5 mg, 0.136 mmol, 1.0 eq.) in 20 mL dry THF. The complex was purified via column chromatography (Cy/EA 8:1, v/v) to obtain the desired complex as yellow solid. (Product partial decomposed upon attempted chromatographic purification.) Single crystals of [(IrCl(cod))2(9)] were obtained from of a MeCN/DCM solution of the Ir-complex at −20 °C. 1H NMR (500 MHz, CDCl3): δ 7.40 (s, 4H), 6.78 (s, 4H), 4.14–4.02 (m, 4H), 3.36–3.20 (m, 4H), 2.37 (s, 12H), 2.24 (s, 12H), 1.95 (s, 6H), 1.56 (s, 6H), 1.53–1.45 (m, 6H), 1.30–1.25 (m, 6H), 1.17–1.08 (m, 4H). 13C NMR (126 MHz, CDCl3): δ 183.8 (CNHC), 146.2, 135.7, 133.0, 129.4, 127.4, 126.2, 124.6, 82.7, 51.9, 34.6, 34.1, 33.7, 29.1, 25.4, 20.5, 17.4. HRMS (ESI): calcd for C44H46N4O2Ir2Cl [M − Cl]+ 1297.42838. Found 1297.42960 (Δ = 1.75 mmu). Yield: 27% (50 mg, 37.3 μmol).
[(IrCl(CO) 2 ) 2 ( 9 )]. Method E. Complex [(IrCl(cod))2(9)] (15.0 mg, 11.2 μmol) was used. 1H NMR (500 MHz, CDCl3): δ 7.38 (s, 4H), 6.90 (s, 4H), 2.35 (s, 12H), 2.10 (s, 12H), 1.90 (s, 6H), 1.46 (s, 6H). 13C NMR (126 MHz, CDCl3): δ 180.0 (CCO), 178.8 (CNHC), 167.2 (CCO), 145.8, 135.0, 132.2, 129.7, 128.8, 127.2, 126.2, 123.9, 34.8, 34.3, 29.9, 28.2, 25.3, 20.5, 16.2. HRMS (ESI): calcd for C48H45Ir2N4O7 [M − 2Cl + OH]+ 1175.25363. Found 1175.25413 (Δ = 1.12 mmu). IR (ν(CO); [cm−1]): 2066/1981. Yield: 99% (14 mg, 11.2 μmol).
[(RhCl(cod)) 2 ( 9 )]. Method B. Starting materials 9·2HCl (30 mg, 40.88 μmol), sodium amylate (2.5 M in toluene, 32.7 μL, 81.77 μmol, 2.0 eq.), [RhCl(cod)]2 (20.2 mg, 40.88 μmol, 1.0 eq.) in 10 mL dry THF. The complex was obtained as crude yellow/brown solid. (Product decomposed upon attempted chromatographic purification.) 1H NMR (300 MHz, CDCl3): δ 7.44 (s, 4H), 6.75 (s, 4H), 4.49 (s, 4H), 3.50 (s, 4H), 2.41 (s, 12H), 2.27 (s, 12H), 1.99 (s, 6H), 1.60 (s, 6H). HRMS (ESI): calcd for C60H68N4O2Rh2Cl [M − Cl]+ 1117.31415. Found 1117.31354 (Δ = 0.61 mmu). Yield: 55% (25 mg, 22.4 μmol, crude product).
[(AuCl) 2 ( 9 )]. Method C. Starting materials 9·2HBF4 (20 mg, 23.9 μmol, 1.0 eq.), silver(I) oxide (6.1 mg, 26.3 μmol, 1.1 eq.), [AuCl(SMe2)] (14.8 mg, 47.8 μmol, 2.0 eq.) 3 mL dry MeOH. The title complex was obtained as a white solid. 1H NMR (500 MHz, CD2Cl2): δ 7.34 (s, 4H), 6.93 (s, 4H), 2.38 (s, 12H), 1.92 (s, 6H), 1.70 (s, 12H), 1.27 (s, 6H). 13C NMR (126 MHz, CD2Cl2): δ 144.6, 133.4, 132.3, 129.6, 128.2, 126.3, 125.2, 125.1, 36.5, 34.8, 24.5, 20.9, 14.2. Au–C could not be observed. HRMS (APCI): calcd for C44H44Au2ClN4O2 [M − Cl]+ 1089.24787 Found 1089.24795 (Δ = 0.08 mmu). Yield: 48% (13 mg 11.6 μmol).
Xanthene diimine (10)
A suspension of 4-amino-2,3,6,7,9,9-hexamethylxanthene (250 mg, 0.89 mmol, 2.0 eq.), glyoxal (40% aq., 51 μL, 0.44 mmol, 1.0 eq.), and 2 mg of p-toluenesulfonic acid monohydrate in EtOH (4 mL) was stirred under nitrogen in a Schlenk tube at 50 °C for 24 h. The cooled reaction mixture was filtered and residual solid was washed with cold methanol and pentane to give xanthene diimine 2 as a yellow solid. 1H NMR (300 MHz, CDCl3): δ 8.53 (s, 2H), 7.15 (s, 2H), 7.07 (s, 2H), 6.85 (s, 2H), 2.33 (s, 6H), 2.25 (s, 12H), 2.21 (s, 6H), 1.64 (s, 12H). 13C NMR (75 MHz, CDCl3): δ 165.9, 148.5, 138.4, 137.8, 135.8, 131.1, 130.9, 128.2, 128.1, 127.3, 126.8, 124.0, 117.5, 34.0, 32.5, 20.2, 19.5, 19.4, 14.4. HRMS (ESI): calcd for C40H45N2O2 (M + H)+ 585.34756. Found 585.34718 (Δ = 0.37 mmu). Yield: 67% (0.35 g, 0.599 mmol).Xanthene imidazolium salt (11·HCl)
Method B. Starting materials: xanthene diimine (10) (160 mg, 0.274 mmol, 1.0 eq.), paraformaldehyde (9 mg, 0.301 mmol, 1.1 eq.), Me3SiCl (52 μL, 0.410 mmol, 1.5 eq.) in 2 mL EA. The title compound was obtained as a white solid. 1H NMR (300 MHz, CDCl3): δ 10.11 (bs, 1H), 7.96 (s, 2H), 7.39 (s, 2H), 7.14 (s, 2H), 6.56 (s, 2H), 2.38 (s, 6H), 2.31 (s, 6H), 2.23 (s, 6H), 2.12 (s, 6H), 1.65 (s, 12H). 13C NMR (75 MHz, CDCl3): δ 146.9, 143.3, 136.3, 132.6, 132.4, 132.2, 130.1, 128.9, 127.3, 126.4, 125.7, 121.3, 117.0, 33.8, 32.8, 20.2, 19.4, 14.9. HRMS (ESI): calcd for C41H45N2O2 [M − Cl]+ 597.34756. Found 597.34806 (Δ = 0.51 mmu). Yield: 63% (110 mg, 0.173 mmol).
[(AuCl)(
11
)]. Method A. Starting materials: 11·HCl (40 mg, 63 μmol), [AuCl(SMe2)] (21 mg, 69 μmol, 1.1 eq.), K2CO3 (34.9 mg, 0.253 mmol, 4.0 eq.) in 2 mL acetone. The complex was purified via column chromatography (Cy/EA 2:1, v/v) to obtain xanthene-gold complex as a white solid. 1H NMR (500 MHz, CDCl3): δ 7.33 (s, 2H), 7.18 (s, 2H), 7.14 (s, 2H), 6.50 (s, 2H), 2.36 (s, 6H), 2.24 (s, 6H), 2.20 (s, 6H), 2.19 (s, 6H), 1.66 (s, 12H). 13C NMR (126 MHz, CDCl3): δ 175.3 (CNHC), 147.6, 144.4, 136.0, 133.1, 131.7, 131.3, 128.7, 128.4, 127.2, 126.8, 125.7, 122.9, 117.0, 33.8, 32.9, 32.7, 20.2, 19.6, 19.4, 14.7. HRMS (ESI): calcd for C43H47AuN3O2 [M − Cl + CH3CN]+ 834.33285. Found 834.33251 (Δ = 0.34 mmu). Yield: 31% (16 mg, 19.3 μmol).
[(IrCl(cod))( 11 )]. Method A. Starting materials: 11·HCl (100 mg, 0.158 mmol, 1.0 eq.), [IrCl(cod)]2 (53.0 mg, 79 μmol, 0.50 eq.), K2CO3 (87.3 mg, 0.632 mmol, 4.0 eq.) in 2 mL acetone. The title complex (syn/anti isomer) was obtained as a yellow solid. 1H NMR (500 MHz, CDCl3): δ 7.33 (s, 2H), 7.18 (s, 2H), 7.06 (s, 2H), 6.70 (s, 2H), 4.04 (d, J = 5.8, 2H), 3.35 (d, J = 5.4, 2H), 2.36 (s, 6H), 2.26 (s, 6H), 2.25 (s, 6H), 2.19 (s, 6H), 1.76 (s, 6H), 1.64 (s, 6H), 1.55–1.47 (m, 2H), 1.35–1.16 (m, 4H), 1.18–0.98 (m, 2H). (Signals shown for main isomer only.) 13C NMR (126 MHz, CDCl3): δ 184.0 (CNHC), 147.6, 144.2, 135.9, 135.5, 131.5, 131.4, 127.6, 127.3, 127.3, 127.0, 123.6, 117.2, 81.9, 52.3, 34.5, 33.7, 33.5, 31.8, 29.2, 20.4, 19.7, 19.5, 16.7. (Signals shown for main isomer only.) HRMS (ESI): calcd for C49H56IrN2O [M − Cl]+ 897.39776. Found 897.39656 (Δ = 0.86 mmu). Yield: 74% (109 mg, 0.12 mmol, mixture of syn/anti isomers).
[(IrCl(CO)
2
)(
11
)]. Method E. Starting materials: [(IrCl(cod))(11)] (26.6 mg, 28.5 μmol). 1H NMR (500 MHz, CDCl3): δ 7.35 (s, 3H), 7.21 (s, 1H), 7.19 (s, 2H), 7.17 (s, 2H), 7.12 (s, 1H), 6.79 (s, 2H), 6.60 (s, 1H), 2.37 (s, 9H), 2.35 (s, 6H), 2.25 (s, 9H), 2.23 (s, 6H), 2.23 (s, 6H), 2.19 (s, 3H), 2.17 (s, 6H), 2.08 (s, 6H), 1.74 (s, 6H), 1.70 (s, 3H), 1.63 (s, 6H), 1.61 (s, 6H) (isomer mixture, assignment unclear). 13C NMR (126 MHz, CDCl3) δ 181.5 (CCO), 181.1 (CCO), 179.3 (CNHC), 168.3 (CCO), 167.7 (CCO), 147.7, 147.6, 144.3, 135.8, 135.7, 134.0, 133.5, 131.5, 131.1, 130.9, 128.8, 128.5, 128.3, 128.1, 127.8, 127.1, 126.9, 126.8, 125.9, 125.8, 124.0, 123.8, 117.8, 117.6, 34.3, 33.8, 33.7, 31.7, 31.7, 29.9, 28.2, 20.3, 19.6, 19.4, 19.4, 19.4, 15.8, 15.4. (Isomer mixture, assignment unclear.) HRMS (ESI): calcd for C43H44IrN2O4 [M − Cl]+ 845.29276. Found 845.29256 (Δ = 0.20 mmu). IR (ν(CO); [cm−1]): 2064/1984. Yield: 99% (25 mg, 28.4 μmol, isomer mixture 70:30), isomers could not be separated.
Xanthene tetraamide (12)
A solution of 4,5-diamino-2,3,6,7,9,9-hexamethylxanthene (0.60 g, 2.0 mmol, 1.0 eq.) and triethylamine (0.56 mL, 4.0 mmol) in 15 mL of DCM was added dropwise to the solution of the acid chloride (0.91 g, 4.0 mmol, 2.0 eq.) in 10 mL of DCM at 0 °C. The resulting mixture was stirred at 0 °C for 1 h then at rt overnight. The reaction was filtered, and the collected solid was washed with diethyl ether to give the desired product. The filtrate was then washed with water several times. The organic phase was dried over MgSO4, and concentrated to give a residue, which was washed with 10% of DCM/diethyl ether. The combined solids were dried under reduced pressure to give xanthene tetraamide as a white solid. 1H NMR (500 MHz, DMSO-d6): δ 10.08 (s, 2H), 9.98 (s, 2H), 7.32 (s, 2H), 6.78 (s, 4H), 2.28 (s, 6H), 2.24 (s, 6H), 2.03 (s, 6H), 1.98 (s, 12H), 1.57 (s, 6H). 13C NMR (126 MHz, DMSO-d6): δ 159.2, 158.2, 143.7, 135.6, 134.8, 132.2, 131.5, 131.1, 128.2, 128.1, 124.8, 122.7, 45.6, 33.8, 31.0, 20.6, 19.9, 18.0, 14.8. HRMS (ESI): calcd for C41H47N4O5 [M + H]+ 675.35410. Found 675.35474 (Δ = 0.64 mmu). Yield: 73% (1.00 g, 1.48 mmol).Xanthene tetramine (13)
Under nitrogen, xanthene tetraamide (12) (0.8 g, 1.19 mmol, 1.0 eq.) was dissolved in BH3·THF (1 M, 15 mL, 15 mmol, 13 eq.) and heated at reflux for 3 d. After cooling to room temp., the solution was poured into a mixture of ice, water, and aq. HCl (1 M). The precipitate was filtered off and dissolved in THF/water (1:2). The solution was basified with sodium hydroxide. The product was extracted with diethyl ether, the combined fractions were washed with brine, dried with MgSO4, and concentrated under reduced pressure. The crude product was then purified by column chromatography (Cy/EA 10:1) to give the diamine as a white solid. 1H NMR (500 MHz, CDCl3): δ 6.87 (s, 2H), 6.75 (s, 4H), 3.79 (s, 4H), 3.22 (t, J = 5.6 Hz, 4H), 3.00 (t, J = 5.6 Hz, 4H), 2.26 (s, 6H), 2.21 (s, 6H), 2.20 (s, 6H), 2.13 (s, 12H), 1.61 (s, 6H). 13C NMR (126 MHz, CDCl3): δ 143.5, 141.3, 135.1, 131.5, 130.2, 129.5, 127.4, 127.3, 120.3, 49.5, 48.9, 34.1, 32.6, 20.7, 20.6, 18.4, 14.7. HRMS (ESI): calcd for C41H55N4O [M + H]+ 619.43704. Found 619.43711 (Δ = 0.07 mmu). Yield: 26% (190 mg, 0.307 mmol).
Imidazolinium chloride
The corresponding diamine (1.0 eq.) and NH4Cl (1.0 eq.) were suspended in HC(OEt)3 and 2 drops of formic acid were added. The reaction mixture was stirred at 110 °C for 12–24 h. After this time the mixture was cooled to rt and poured into water. The aqueous phase was washed with diethyl ether and extracted with DCM The combined organic layers were dried over MgSO4. After drying under reduced pressure, the imidazolinium chloride was obtained as a white solid.Xanthene diimidazolinium salt (14·2HCl)
Starting materials: xanthene tetramine (13) (100 mg, 0.162 mmol, 1.0 eq.), NH4Cl (17.3 mg, 0.323 mmol, 2.0 eq.) in 4 mL triethyl orthoformate. The title compound was obtained as a white solid. 1H NMR (500 MHz, MeOD): δ 7.63 (s, 2H), 7.15 (s, 4H), 5.22–4.88 (m, 4H), 4.79–4.63 (m, 4H), 2.66–2.21 (m, 30H), 1.73 (s, 6H). 13C NMR (126 MHz, MeOD): δ 144.9, 142.3, 136.4, 135.1, 134.9, 131.7, 131.2, 131.2, 129.5, 122.9, 53.1, 52.7, 34.7, 33.8, 21.0, 20.2, 18.2, 15.3. HRMS (ESI): calcd for C43H52ClN4O [M − Cl]+ 675.38242. Found 675.38155 (Δ = 0.86 mmu). Yield: 63% (73 mg, 0.103 mmol).
[(IrCl(cod))
2
(
14
)]. Method A. Starting materials: 14·2HCl (24.6 mg, 34.6 μmol), [IrCl(cod)]2 (23.2 mg, 34.6 μmol), K2CO3 (19.1 mg, 0.138 mmol, 4.0 eq.) in acetone/DCM (3 mL/1 mL) The complex was then purified by column chromatography (Cy/EA 8:1, v/v) to obtain the desired complex as yellow solid. 1H NMR (500 MHz, CD2Cl2): δ 7.40–7.24 (m, 2H), 7.00–6.89 (m, 4H), 5.31–5.12 (m, 1H), 4.43–4.31 (m, 1H), 4.18–3.87 (m, 8H), 3.85–3.55 (m, 2H), 3.25–3.07 (m, 1H), 3.07–2.99 (m, 2H), 2.97–2.88 (m, 1H), 2.55 (s, 3H), 2.45–2.38 (m, 6H), 2.38–2.27 (m, 18H), 2.24 (s, 3), 1.80 (s, 3H), 1.75 (s, 3H), 1.66 (s, 1H), 1.60 (s, 2H), 1.51–1.21 (m, 10H), 1.21–1.13 (m, 1H), 1.08–0.95 (m, 2H). 13C NMR (126 MHz, CD2Cl2): δ 210.0 (CNHC), 209.3 (CNHC), 146.9, 144.7, 139.0, 138.9, 138.8, 138.4, 138.2, 137.6, 136.9, 136.5, 136.4, 135.9, 135.2, 135.1, 134.6, 133.1, 132.3, 131.2, 130.3, 130.2, 130.1, 129.3, 129.2, 129.1, 128.1, 128.0, 127.8, 127.7, 127.5, 127.3, 127.1, 127.0, 86.9, 85.9, 85.5, 85.3, 84.7, 82.9, 80.8, 79.8, 54.8, 53.7, 53.3, 53.1, 52.9, 52.3, 52.2, 51.9, 51.6, 48.3, 36.8, 36.7, 34.6, 34.4, 34.2, 34.1, 34.0, 33.6, 33.2, 32.5, 31.5, 31.4, 31.3, 30.3, 30.2, 29.9, 29.7, 29.2, 29.0, 28.6, 26.7, 26.5, 23.3, 21.3, 20.7, 20.6, 20.5, 20.3, 19.8, 19.1, 18.9, 18.7, 17.7, 17.5, 16.7, 14.5. HRMS (ESI): calcd for C59H74ClIr2N4O [M − Cl]+ 1275.4804. Found 1275.4791 (Δ = 1.3 mmu). Yield: 53% (24 mg, 18.3 μmol).
[(IrCl(CO) 2 ) 2 ( 14 )]. Method E. [(IrCl(cod))2(14)] (10 mg, 10.4 μmol, 1.0 eq.) was used. 1H NMR (300 MHz, CD2Cl2): δ 7.32 (s, 2H), 7.01 (s, 2H), 6.94 (s, 2H), 5.05 (s, 2H), 4.33–4.14 (m, 2H), 4.10–3.91 (m, 4H), 2.44 (s, 6H), 2.34 (s, 12H), 2.31 (s, 12H), 1.65 (s, 6H). 13C NMR (75 MHz, CD2Cl2): δ 203.5 (CNHC), 181.3 (CCO), 169.1 (CCO), 145.6, 139.5, 136.1, 135.5, 134.8, 132.3, 130.2, 130.0, 128.4, 127.9, 126.4, 52.7, 34.0, 33.7, 30.3, 28.6, 21.4, 20.6, 19.5, 18.8, 16.6. HRMS (ESI): calcd for C47H50ClIr2N4O5 [M − Cl]+ 1171.27228. Found 1171.26929 (Δ = 2.15 mmu). IR (ν(CO); [cm−1]): 2070/1982. Yield: 99% (9 mg, 7.5 μmol).
[(AuCl) 2 ( 14 )]. Method A. Starting materials: 14·2HCl (20.5 mg, 28.80 μmol, 1.0 eq.), [AuCl(SMe2)] (17.0 mg, 57.6 μmol, 2.0 eq.), K2CO3 (15.9 mg, 0.115 mmol, 4.0 eq.) in acetone/DCM (3 mL/1 mL). The title complex was obtained as a white solid. Single crystals of [(AuCl)2(14)] were obtained by slow diffusion of diethyl ether into a DCM/MeOH solution of the gold complex at 2–8 °C. 1H NMR (500 MHz, CD2Cl2): δ 7.37 (s, 2H), 7.05 (s, 2H), 6.99 (s, 2H), 4.67–4.57 (m, 2H), 4.27–4.13 (m, 2H), 4.10–3.97 (m, 4H), 2.40 (s, 6H), 2.35 (s, 6H), 2.32 (s, 12H), 2.30 (s, 6H), 1.69 (s, 6H). 13C NMR (126 MHz, CD2Cl2): δ 197.7 (CNHC), 145.0, 140.0, 136.2, 135.3, 135.2, 134.9, 133.3, 130.8, 130.2, 129.1, 128.3, 126.3, 52.2, 51.7, 34.1, 33.9, 21.4, 20.7, 18.9, 18.7, 16.6. HRMS (ESI): calcd for C45H53Au2ClN5O [M − Cl + MeCN]+ 1108.3265. Found 1108.3269 (Δ = 0.6 mmu). Yield: 62% (20 mg, 18.1 μmol).
1,1′-(2,3,6,7,9,9-Hexamethyl-9H-xanthene-4,5-diyl)bis(1H-imidazole) (15)
:1) affording the pure product as a off white solid. 1H NMR (300 MHz, CD2Cl2): δ 7.30 (s, 2H), 7.23 (s, 2H), 7.01 (s, 2H), 6.69 (s, 2H), 2.30 (s, 6H), 1.87 (s, 6H), 1.66 (s, 6H). 13C NMR (75 MHz, CD2Cl2): δ 144.0, 138.0, 134.1, 132.6, 129.6, 128.1, 127.8, 125.0, 120.9, 34.4, 32.9, 20.4, 14.4. HRMS (ESI): calcd for C25H27N4O (M + H)+ 399.21794. Found 399.21831 (Δ = 0.37 mmu). Yield: 46% (432 mg, 1.08 mmol).
:1) to give a off white solid. Yield: 28% (190 mg, 0.477 mmol).
Xanthene diimidazolium salt (16·2HBr)
1,1′-(2,3,6,7,9,9-Hexamethyl-9H-xanthene-4,5-diyl)bis(1H-imidazole) (443 mg, 1.11 mmol, 1.0 eq.) and 1,5-dibromopentane (256 mg, 151.4 μL, 1.11 mmol, 1.0 eq.) were added to 60 mL acetonitrile in a Schlenk flask. The mixture was stirred under reflux for 5 d while a precipitate formed. The mixture was allowed to cool before diethyl ether was added. The precipitate was filtrated, washed with pentane and dried under reduce pressure to obtain the title compound as a white powder. 1H NMR (500 MHz, MeOD): δ 9.31 (s, 2H), 8.03 (d, J = 1.6 Hz, 2H), 7.80 (d, J = 1.6 Hz, 2H), 7.72 (s, 2H), 4.64–4.54 (m, 2H), 4.51–4.43 (m, 2H), 2.44 (s, 6H), 2.24–2.09 (m, 4H), 2.02 (s, 6H), 1.86 (s, 3H), 1.73 (s, 3H), 1.63–1.50 (m, 1H), 0.55–0.42 (m, 1H). 13C NMR (126 MHz, MeOD): δ 143.6, 139.1, 134.9, 134.8, 131.6, 129.5, 126.7, 124.9, 122.6, 50.1, 35.0, 34.5, 32.1, 27.4, 20.7, 19.9, 14.1. HRMS (ESI): calcd for C30H35Br2N4O [M − H]− 625.11825. Found 625.11831 (Δ = 0.21 mmu). Yield: 84% (590 mg, 0.939 mmol).Xanthene diimidazolium salt (16·2HBF4)
According to the general procedure 16·2HBr (50 mg) was used. 1H NMR (500 MHz, CD2Cl2): δ 8.55 (s, 2H), 7.81 (s, 2H), 7.47 (s, 2H), 7.33 (s, 2H), 4.45–4.27 (m, 4H), 2.33 (s, 6H), 2.17–2.01 (m, 4H), 1.83 (s, 6H), 1.75 (s, 3H), 1.68 (s, 3H), 1.56–1.41 (m, 1H), 0.72–0.62 (m, 1H). 13C NMR (126 MHz, CD2Cl2): δ 142.7, 137.6, 134.0, 133.8, 131.1, 128.6, 125.8, 124.2, 121.4, 50.1, 34.2, 32.8, 26.9, 20.3, 20.2, 14.4. Yield: 99% (51 mg, 79.4 μmol).[(AgBr) 2 ( 16 )]. Method C. Starting materials: 16·2HBr (100 mg, 159 μmol, 1.0 eq.) and silver(I) oxide (40.6 mg, 175 μmol, 1.1 eq.) in 3 mL dry MeOH. The title complex was obtained as a white solid. 1H NMR (500 MHz, CD2Cl2): δ 7.69 (s, 2H), 7.44 (s, 2H), 6.90 (s, 2H), 4.17–4.05 (m, 2H), 3.31–3.21 (m, 2H), 2.33 (s, 6H), 1.91 (s, 3H), 1.77 (s, 6H), 1.64–1.56 (m, 2H), 1.55 (s, 3H), 1.35–1.26 (m, 3H), 0.70–0.60 (m, 1H), 0.22–0.12 (m, 1H). 13C NMR (126 MHz, CD2Cl2): δ 181.4 and 181.5 (dd, JAg–C = 195.6 Hz, JAg–C = 195.0 Hz) 143.8, 135.0, 133.3, 128.9, 128.2, 127.1, 123.9, 123.7, 50.3, 37.1, 34.5, 29.5, 27.7, 20.5, 20.4, 15.1. HRMS (APCI): calcd for C30H34Ag2BrN4O [M − Br]+ 759.00124. Found 759.00121 (Δ = 0.04 mmu). Yield: 63% (85 mg, 101 μmol).
[(AuCl) 2 ( 16 )]. Method C. Starting materials: 16·2HBr (40 mg, 63.7 μmol, 1.0 eq.), silver(I) oxide (16.2 mg, 70.0 μmol, 1.1 eq.), [AuCl(SMe2)] (37.5 mg, 127 μmol, 2.0 eq.) in 3 mL dry MeOH. The title complex was obtained as a white solid. 1H NMR (500 MHz, CD2Cl2): δ 7.49 (s, 2H), 7.40 (s, 2H), 6.96 (s, 2H), 3.92–3.84 (m, 2H), 3.47–3.38 (m, 2H), 2.35 (s, 6H), 1.94 (s, 3H), 1.74 (s, 6H), 1.60–1.56 (m, 2H), 1.54 (s, 3H), 1.48–1.39 (m, 2H), 0.80–0.69 (m, 1H), 0.41–0.28 (m, 1H). 13C NMR (126 MHz, CD2Cl2): δ 185.5 (CNHC), 143.8, 135.4, 133.3, 129.1, 128.4, 126.2, 124.3, 122.6, 50.1, 37.3, 34.6, 29.3, 28.4, 21.0, 20.5, 15.1. HRMS (APCI): calcd for C30H34Au2ClN4O [M − Cl]+ 895.17470. Found 895.17477 (Δ = 0.06 mmu). Yield: 46% (27 mg, 29 μmol).
[(CuI) 2 ( 16 )]. [(AgBr)2(16)] (20 mg, 23.8 μmol, 1.0 eq.) and CuI (9.1 mg. 47.5 μmol 2.0 eq.) were dissolved in 3 mL dry DCM and stirred at rt in the absence of light for 24 h. The mixture was filtered through Celite and the solvent was removed under reduced pressure to obtain the desired complex as a white solid. 1H NMR (500 MHz, CD2Cl2): δ 7.49–7.46 (m, 4H), 6.89 (d, J = 1.8 Hz, 2H), 4.00–3.93 (m, 2H), 3.16–3.06 (m, 2H), 2.35 (s, 6H), 1.96 (s, 2H), 1.79 (s, 6H), 1.58 (s, 6H), 1.35–1.27 (m, 2H), 0.57–0.46 (m, 1H), 0.23–0.15 (m, 1H). 13C NMR (126 MHz, CD2Cl2): δ 177.6 (CNHC), 144.1, 135.3, 133.5, 128.7, 128.6, 126.8, 124.1, 122.5, 50.0, 37.2, 34.7, 28.6, 28.3, 20.5, 20.5, 15.2. HRMS (APCI): calcd for C30H34CuN4O2 [M − CuI2 + O]+ 545.19723. Found 545.19762 (Δ = 0.40 mmu). Yield: 35% (7 mg, 8.3 μmol).
[(RhCl(cod)) 2 ( 16 )]. Method C. Starting materials: 16·2HBr (50.0 mg, 79.6 μmol, 1.0 eq.), silver(I) oxide (20.3 mg, 87.5 μmol, 1.1 eq.), [RhCl(cod)]2 (39.2 mg, 79.6 μmol, 1.0 eq.) in 5 mL dry MeOH. The crude product was purified by short column chromatography (pure DCM then switched to pure EA) to obtain the desired complex as yellow solid. Single crystals of [(RhCl(cod))2(16)] were obtained by slow diffusion of diethyl ether into a DCM solution of the Rh-complex at 2–8 °C. 1H NMR (500 MHz, CD2Cl2): δ 7.43 (s, 2H), 6.83 (d, J = 2.0 Hz, 2H), 6.73 (d, J = 2.0 Hz, 2H), 5.25–5.17 (m, 2H), 4.77–4.70 (m, 2H), 4.62–4.54 (m, 2H), 4.03–3.96 (m, 2H), 3.48–3.38 (m, 2H), 2.75–2.66 (m, 2H), 2.50–2.42 (m, 2H), 2.42 (s, 6H), 2.34–2.28 (m, 2H), 2.21 (s, 6H), 2.14–2.06 (m, 2H), 1.89 (s, 3H), 1.87–1.75 (m, 6H), 1.75–1.66 (m, 2H), 1.62–1.55 (m, 2H), 1.49 (s, 3H), 1.38–1.26 (m, 5H), 0.62–0.47 (m, 1H). 13C NMR (126 MHz, CD2Cl2): δ 182.35 (d, J = 51.8 Hz, CNHC), 144.3, 136.3, 132.9, 127.7, 127.5, 127.4, 124.8, 121.3, 97.0, 96.9, 96.0, 95.9, 69.8, 69.7, 67.5, 67.4, 51.0, 35.9, 34.6, 34.0, 31.5, 30.0, 29.1, 29.0, 27.8, 21.3, 20.4, 17.3. HRMS (APCI): calcd for C46H58ClN4ORh2 [M − Cl]+ 923.24012. Found 923.24037 (Δ = 0.26 mmu). Yield: 26% (20 mg, 20.8 μmol).
[(RhBr(cod)) 2 ( 16 )]. Method D. Starting materials: 16·2HBr (30.0 mg, 47.74 μmol, 1.0 eq.), [RhCl(cod)]2 (23.54 mg, 47.7 μmol 1.0 eq.), NaOAc (7.8 mg, 95.5 μmol, 2.0 eq.), K2CO3 (13.2 mg, 95.5 μmol, 2.0 eq.) and KBr (excess) in 6 mL dry MeCN. The crude product was purified by short column chromatography (pure DCM then switched to pure EA) to obtain the desired complex as yellow solid. 1H NMR (500 MHz, CD2Cl2): δ 7.43 (s, 2H), 6.84 (d, J = 1.9 Hz, 2H), 6.74 (d, J = 1.8 Hz, 2H), 5.26–5.16 (m, 2H), 4.86–4.78 (m, 2H), 4.73–4.66 (m, 2H), 4.05–3.96 (m, 2H), 3.56–3.47 (m, 2H), 2.85–2.78 (m, 2H), 2.41 (s, 6H), 2.33–2.27 (m, 2H), 2.22 (s, 6H), 2.11–2.05 (m, 2H), 1.89 (s, 3H), 1.85–1.77 (m, 5H), 1.74–1.68 (m, 4H), 1.61–1.54 (m, 2H), 1.54–1.52 (m, 2H), 1.49 (s, 3H), 1.39–1.29 (m, 4H), 0.59–0.46 (m, 1H). 13C NMR (126 MHz, CD2Cl2): δ 182.3 (d, J = 51.8 Hz, CNHC), 144.6, 136.5, 133.2, 127.8, 127.6, 125.4, 121.2, 96.8, 96.7, 95.8, 95.8, 70.7, 70.6, 68.7, 68.5, 51.8, 36.0, 34.7, 34.3, 31.4, 30.5, 29.2, 28.9, 28.1, 21.5, 20.5, 18.4. HRMS (APCI): calcd for C46H58BrN4ORh2 [M − Br]+ 967.18986. Found 967.18920 (Δ = 0.65 mmu). Yield: 38% (19 mg, 18.1 μmol).
[(IrBr(cod)) 2 ( 16 )]. Method D. Starting materials: 16·2HBr (100 mg, 0.16 mmol, 1.0 eq.), [IrCl(cod)]2 (106.9 mg, 0.16 mmol, 1.0 eq.), NaOAc (26.1 mg, 0.32 mmol, 2.0 eq.), K2CO3 (43.9 mg, 0.32 mmol, 2.0 eq.) and KBr (excess) in 10 mL dry MeCN. The crude product was purified by short column chromatography (pure DCM then switched to pure EA) to obtain the desired complex as yellow/orange solid. Single crystals of [(IrBr(cod))2(16)] were obtained by slow diffusion of diethyl ether into a DCM solution of the Ir-complex at 2–8 °C. 1H NMR (300 MHz, CD2Cl2): δ 7.41 (s, 2H), 6.89 (d, J = 1.9 Hz, 2H), 6.75 (d, J = 1.9 Hz, 2H), 5.09–4.97 (m, 2H), 4.53–4.43 (m, 2H), 4.26–4.14 (m, 2H), 3.95–3.83 (m, 2H), 3.28–3.15 (m, 2H), 2.60–2.47 (m, 2H), 2.35 (s, 6H), 2.29–2.20 (m, 2H), 2.10 (s, 6H), 2.07–2.01 (m, 2H), 1.98–1.92 (m, 2H), 1.88 (s, 3H), 1.83–1.67 (m, 4H), 1.64–1.57 (m, 2H), 1.54–1.51 (m, 2H), 1.48 (s, 3H), 1.37–1.33 (m, 2H), 1.20–1.03 (m, 3H), 1.01–0.89 (m, 2H), 0.68–0.56 (m, 1H). 13C NMR (75 MHz, CD2Cl2): δ 180.3 (CNHC), 144.7, 136.2, 133.1, 127.8, 127.7, 127.4, 125.1, 121.3, 83.3, 81.4, 52.2, 51.3, 35.9, 35.4, 34.3, 32.3, 31.1, 29.0, 28.7, 28.6, 21.7, 20.5, 18.3. HRMS (APCI): calcd for C46H58BrN4OIr2 [M − Br]+ 1147.3047. Found 1147.3019 (Δ = 1.0 mmu). Yield: 44% (86 mg, 70 μmol).
[(RhCl(CO) 2 ) 2 ( 16 )]. Method E. [(RhCl(cod))2(16)] (10 mg, 10.4 μmol, 1.0 eq.) was used. 1H NMR (500 MHz, CD2Cl2): δ 7.42 (s, 2H), 7.00 (d, J = 1.8 Hz, 2H), 6.90 (d, J = 1.8 Hz, 2H), 5.10–5.02 (m, 2H), 4.00–3.93 (m, 2H), 2.43–2.38 (m, 2H), 2.36 (s, 6H), 2.06 (s, 6H), 1.85 (s, 3H), 1.82–1.76 (m, 2H), 1.41 (s, 3H), 1.32–1.27 (m, 1H), 0.51–0.37 (m, 1H). 13C NMR (126 MHz, CD2Cl2): δ 186.4 (d, JRh–C = 54.3 Hz, CCO), 181.9 (d, JRh–C = 74.2 Hz, CNHC), 177.4 (d, JRh–C = 44.8 Hz, CCO), 144.3, 135.6, 133.2, 128.8, 128.6, 126.9, 124.6, 123.8, 50.4, 35.7, 34.4, 29.2, 29.1, 21.0, 20.5, 16.6. HRMS (ESI): calcd for C34H34ClN4O5Rh2 [M − Cl]+ 819.03223. Found 819.03115 (Δ = 1.09 mmu). IR (ν(CO); [cm−1]): 2076/1994. Yield: 90% (8 mg, 9.4 μmol).
[(IrBr(CO) 2 ) 2 ( 16 )]. Method E. [(IrBr(cod))2(16)] (10 mg, 8.2 μmol, 1.0 eq.) was used. 1H NMR (500 MHz, CD2Cl2): δ 7.43 (s, 2H), 7.04 (d, J = 1.9 Hz, 2H), 6.91 (d, J = 1.9 Hz, 2H), 5.11–5.00 (m, 2H), 3.99–3.89 (m, 2H), 2.41–2.35 (m, 2H), 2.34 (s, 6H), 2.00 (s, 6H), 1.86 (s, 4H), 1.81–1.68 (m, 3H), 1.41 (s, 3H), 0.48–0.33 (m, 1H). 13C NMR (126 MHz, CD2Cl2): δ 181.6 (CCO), 176.2 (CNHC), 166.7 (CCO), 144.7, 135.3, 133.2, 129.1, 128.6, 126.6, 124.9, 123.7, 50.9, 35.4, 34.6, 28.8, 28.3, 20.9, 20.5, 17.2. HRMS (APCI): calcd for C34H34BrN4O5Ir2 [M − Br]+ 1043.09656. Found 1043.0936 (Δ = 0.83 mmu). IR (ν(CO); [cm−1]): 2072/1982. Yield: 98% (9 mg, 8.0 μmol).
[(PdCl(allyl)) 2 ( 16 )]. [(AgBr)2(16)] (25 mg, 29.7 μmol, 1.0 eq.) and [PdCl(allyl)]2 (11.5 mg, 31.2 μmol, 1.05 eq.) were dissolved in 6 mL dry DCM and stirred at rt in the absence of light for 24 h. The mixture was filtered through Celite and the solvent was removed under reduced pressure. The crude product was purified by short column chromatography (pure DCM then switched to pure EA) to obtain the desired complex as a pale, yellow solid. 1H NMR (500 MHz, CD2Cl2): 7.34 (s, 2H), 6.98 (s, 2H), 6.87 (s, 2H), 5.15 (s, 1H), 4.96 (s, 2H), 4.60 (s, 1H), 3.90 (s, 4H), 3.13 (s, 1H), 3.08–2.88 (m, 2H), 2.83 (s, 2H), 2.31 (s, 6H), 2.13 (s, 3H), 2.03–1.91 (m, 3H), 1.82 (s, 3H), 1.79–1.58 (m, 4H), 1.52 (s, 3H), 1.20–0.99 (m, 1H), 0.66–0.35 (m, 1H). 13C NMR (126 MHz, CD2Cl2) δ 181.6 (CNHC), 144.1, 143.7, 136.0, 133.0, 128.4, 128.2, 128.0, 127.8, 123.7, 123.4, 123.0, 114.8, 114.4, 111.7, 71.4, 63.2, 49.9, 36.1, 34.1, 30.1, 29.2, 20.9, 20.5, 15.9. HRMS (ESI): calcd for C36H44ClN4OPd [M − Cl]+ 795.12679. Found 795.12789 (Δ = 0.41 mmu). Yield: 52% (13 mg, 15.5 μmol).
[(Au(AuCl) 2 )(Cl)( 16 ) 2 ]. 16·2HBr (20 mg, 31.8 μmol, 1.0 eq.) and silver(I) oxide (11.1 mg, 47.7 μmol, 1.5 eq.) were dissolved in 3 mL dry MeOH and stirred at rt in the absence of light for 24 h. After this time [AuCl(SMe2)] (13.8 mg, 46.7 μmol, 1.5 eq.) was added, the mixture was stirred at 50 °C for 2 h then for 12 h at rt. in the absence of light. The mixture was filtered through Celite and the solvent was removed under reduced pressure. The residue was dissolved in a minimum amount of DCM and diethyl ether was added. The precipitate was filtered and dried under reduce pressure to obtain the title compound as a white solid. Single crystals of [(Au(AuCl)2)(Cl)(16)2] were obtained by slow diffusion of diethyl ether into a DCM solution of the gold complex at 2–8 °C. 1H NMR (500 MHz, CD2Cl2): δ 7.40 (s, 1H), 7.36 (s, 1H), 7.33 (d, J = 1.9 Hz, 1H), 7.27 (d, J = 1.8 Hz, 1H), 6.84 (d, J = 2.0 Hz, 1H), 6.79 (d, J = 1.9 Hz, 1H), 4.63–4.52 (m, 1H), 4.45–4.34 (m, 1H), 4.14–4.07 (m, 1H), 4.07–3.99 (m, 1H), 2.32 (s, 3H), 2.23 (s, 3H), 2.03–1.94 (m, 2H), 1.85 (s, 3H), 1.83–1.77 (m, 1H), 1.73 (s, 3H), 1.64 (s, 3H), 1.43 (s, 3H), 1.37–1.25 (m, 2H), 1.11–0.98 (m, 1H). 13C NMR (126 MHz, CD2Cl2): δ 185.6 (CNHC), 172.8 (CNHC), 143.8, 143.8, 134.8, 134.7, 133.1, 132.3, 129.1, 129.0, 128.2, 128.2, 126.3, 126.0, 123.8, 123.6, 122.5, 122.0, 51.2, 50.6, 34.4, 34.4, 32.1, 28.8, 28.5, 21.4, 20.6, 20.5, 15.1, 14.5. HRMS (APCI): calcd for C30H34Au2ClN4O [M − Cl − C30H34AuClN4O]+ 895.17470. Found 895.17496 (Δ = 0.26 mmu). Yield: 58% (15 mg, 9.2 μmol).
Results and discussion
In order to realize binuclear structures (Scheme 1d), the 4,5-positions at the xanthene scaffold appear to be ideal anchoring points for the N-heterocyclic carbenes. The two 4,5-nitrogen nitrogen atoms belonging to an imidazolylidene or imidazolidinylidene unit are positioned at a distance of almost 500 pm and since the tilt angle between the two heterocyclic rings allows for some variability, the distances of metals bound the carbene carbon can range between approx. 300–700 pm. The coordination of a single metal in a chelating fashion appears to be unfavorable. In order to retain the orthogonality of the planes of the respective NHC units relative to the xanthene plane, additional methyl group next to the 4,5-NHC site at the 3,6-positions of the xanthene are needed. The advantage of the xanthene unit (as opposed to a naphthalene scaffold) is the absence of atoms between the two NHC units, which for naphthalene would host a CH-unit prone to CH-activation reactions. Based on these considerations 3,6,9,9-tetramethyl-9H-xanthene-4,5-diamine appears to be the ideal building block for the construction of the bisNHC ligands. Similar ligands, but without the crucial 3,6-dimethyl units were reported previously by Messerle et al.42 and the Saito group.43Synthesis
The synthesis of 3,6,9,9-tetramethyl-9H-xanthene-4,5-diamine 6 as the key component for macrocyclic NHC ligands is shown in Scheme 2. Hexamethylxanthene was synthesized according to a modified procedure by Caruso and Lee.39 The Takeuchi et al. synthesis of 1,8-diamino-xanthene40 was significantly improved by adding Na2CO3 to the reaction mixture. Nonetheless, a significant amount of monoamine 7 (yield 40%) is still formed. The hazardous azide chemistry and the tedious purification of the diamino-xanthene (chromatographic separation from monoamino-xanthene, which is produced in approx. equimolar amounts) prompted us to develop an alternative approach via carboxylic acid and carbamate and Hofmann rearrangement, since attempts to directly introduce the amino group via Pd- or Cu-catalyzed cross coupling reactions utilizing 1,8-dibromo-xanthene were not successful. In contrast to the azide route, the sequence of reactions to 6via4 and 5 provides exclusively diamine 6.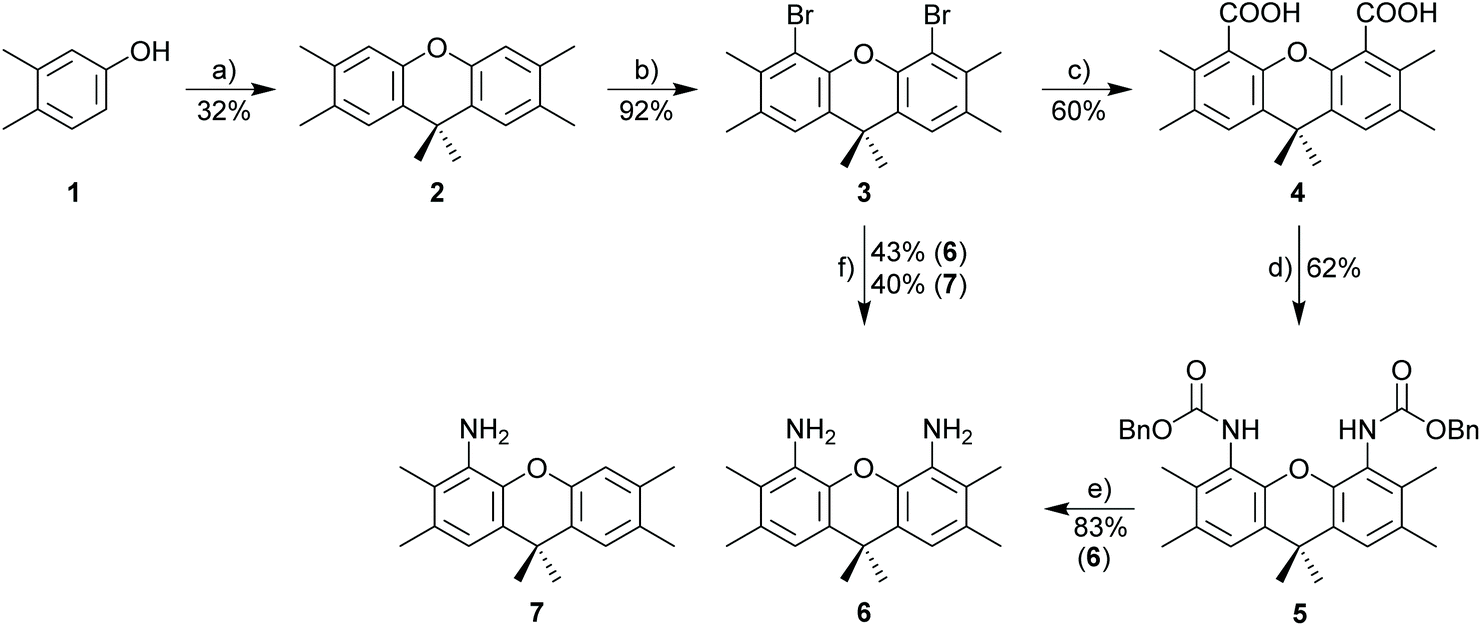 | ||
| Scheme 2 Synthesis of 4,5-diaminoxanthene 6. Reagents and conditions: (a) CH3SO3H, 110 °C for 5 d, then 150 °C for 6 h; (b) Br2, CH2Cl2, −78 °C to rt, 12 h; (c) nBuLi, CO2, THF, −78 °C, 2 h; (d) DPPA, BnOH, NEt3, toluene, 80 °C, 12 h; (e) KOH, EtOH, 90 °C, 24 h; (f) NaN3, CuI, DMEDA, Na2CO3, DMSO, 100 °C, 24 h. | ||
Diamine 6 is converted into diimine 8 according to the general Hintermann procedure44 and then into the respective diimidazolium salt 9·2HCl (Scheme 3)28 in 85% yield (Scheme 3). Initial attempts to synthesize dinuclear NHC metal complexes employing the weak base approach45,46 turned out to be unsuccessful. In the case of the gold complex the transmetalation employing the in situ generated [(AgCl)2(9)] provides a reasonable yield (48%) of [(AuCl)2(9)]. The transfer of the NHC to [MCl(cod)]2 (M = Rh, Ir) using AgNHC, normally is a very reliable method,47 but also fails with the diimidazolium salt 9·HCl. Finally, the complexes [(MCl(cod))2(9)] (M = Rh, Ir) were obtained from the reaction of the free carbene (generated with sodium amylate and diimidazolium salt 9·2HCl) with [(MCl(cod)]2 (M = Rh, Ir). Normally, [MCl(cod)(NHC)] complexes are very stable,29,48–50 however, [RhCl(cod)(9)] turned out to be labile and purification was difficult since the complex suffers from very significant decomposition during chromatographic purification. The closely related iridium complex [(IrCl(cod))2(9)] is slightly more stable and chromatographic purification affords pure complex, albeit in moderate yield since again a significant amount of the complex decomposes during chromatographic purification. The substitution of the cyclooctadiene by CO proceeds cleanly to provide [(IrCl(CO)2)2(9)] in virtually quantitative yield, while the analogous reaction of the Rh-based relative was not successful.
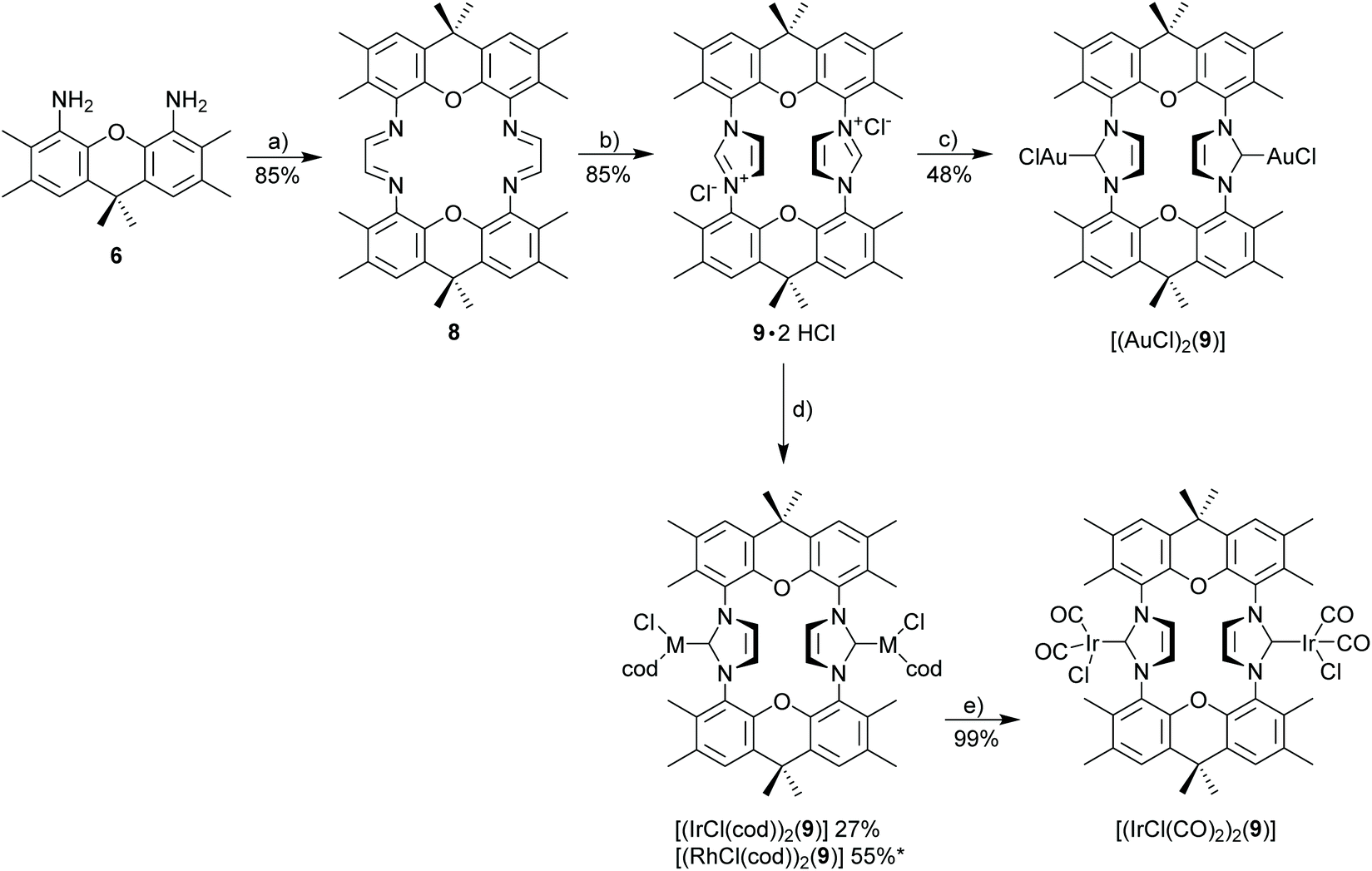 | ||
| Scheme 3 Synthesis of azolium salt 9·2HCl and derived metal complexes. Reagents and conditions: (a) glyoxal, pTsOH, nBuOH, 50 °C; (b) paraformaldehyde, Me3SiCl, EtOAc, 70 °C, 24 h; (c) Ag2O, MeOH, rt, 24 h then [AuCl(SMe2)], 50 °C, 2 h; (d) [MCl(cod)]2 (M = Ir, Rh), sodium amylate, THF, −78 °C; (e) CO, 0 °C, CH2Cl2 30 min; (* yield of crude complex). | ||
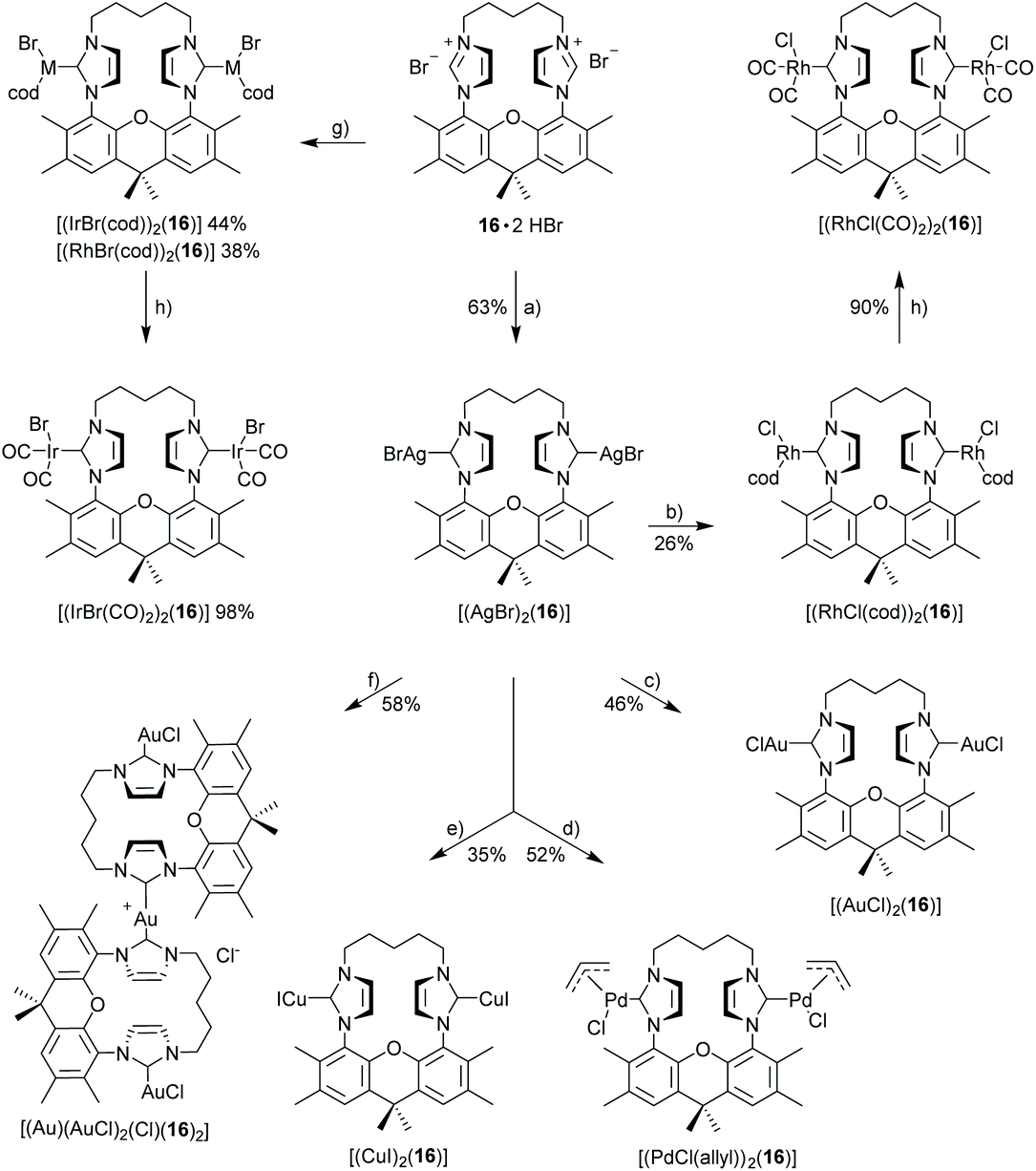
X-Ray crystal structure of [(IrCl(cod))2(9)]51
The bisNHC ligand 9 binds two IrCl(cod) units in a slightly distorted square-planar geometry (Fig. 1). The distance between the two iridium (702.4 pm) is sufficiently large to accommodate the two bulky 1,5-cyclooctadiene ligands between the two metals, while the two smaller chloro ligands are located in the periphery of the molecule. With a view to the structure of [(Au)(AuCl)2(Cl)(16)2] (Fig. 4) the central placement of the two bulky cod-units in the Ir complex and in [(RhCl(cod))2(16)] (Fig. 3) is unexpected (see also discussion of [(Au)(AuCl)2(Cl)(16)2]) but eventually the attractive contribution of dispersion forces between the two cod units are underestimated.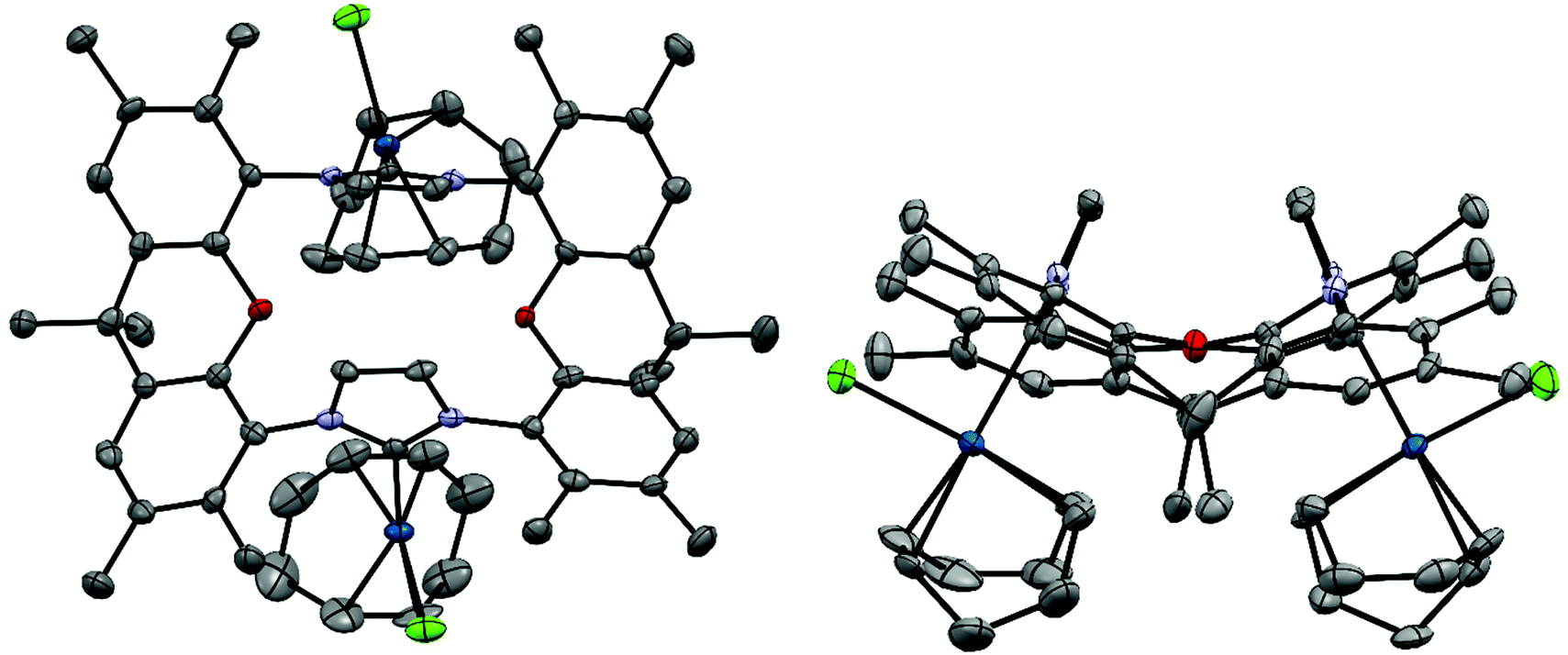 | ||
| Fig. 1 X-ray crystal structure (thermal ellipsoids 80%) of [(IrCl(cod))2(9)] (colour code: Ir blue, Cl green, O red, N blue, H not displayed) plots: perpendicular to xanthene plane and along the oxygen–oxygen axis. Important distances (pm), bond lengths (pm) and angles (°): Ir⋯Ir 702.4, Ir–C(NHC) 205.0(6), 206.1(6), Ir–Cl 238.0(2), 238.1(2), Ir–C(cod) 210.7(6), 214.2(7), 215.0(6), 219.7(7) (av. 214.9), 209.6(6), 211.4(7), 215.7(8), 220.3(8) (av. 214.3), Cl–Ir–C(NHC) 91.8(2), 92.3(2), C(NHC)–Ir–C(cod) 173.6(3), 173.1(3) and 149.9(2), 149.1(2). | ||
The bonds lengths in the coordination sphere of the two distinct iridium atoms are similar to those observed in related complexes.52–55 Typically, the Ir–C(cod) bond lengths of a trans-olefin is 10 pm longer than for the respective cis-olefin due to the trans-effect of the NHC ligand, which is also observed for [(IrCl(cod))2(9)].
The limited stability of metal complexes (Scheme 3) prompted us to attempt the synthesis of the related “open” complexes to better understand the properties of the macrocyclic ligand (Scheme 4). This is convenient since monoamine 7 is formed in significant amounts as a bye-product of diamine 6via the azide route. In order to better understand, why the metal complexes with NHC 9 show untypical behavior, the closely related azolium salt 11·HCl (Scheme 4) was synthesized from amino-xanthene 7. NHC 11 contains only a single NHC ligand and the weak base approach provides good yields (75%) of the respective [IrCl(cod)(11)]. The stability of the complex is comparable to that of the established [(IrCl(cod))(IMes)] complexes.47 Both complexes are stable during chromatographic purification and solutions remain unchanged over several days. Bulky metal complexes with NHC 11 occur as a mixture of syn/anti-isomers, since the bulky ortho-substituents prevent N–C-rotation along which the two isomers (for [IrCl(cod)(11)] ratio 70:30) could interconvert. Temperature-variable NMR-experiments (up to 370 K in toluene-d8) with [IrCl(cod)(11)] did not provide evidence for isomer interconversion.
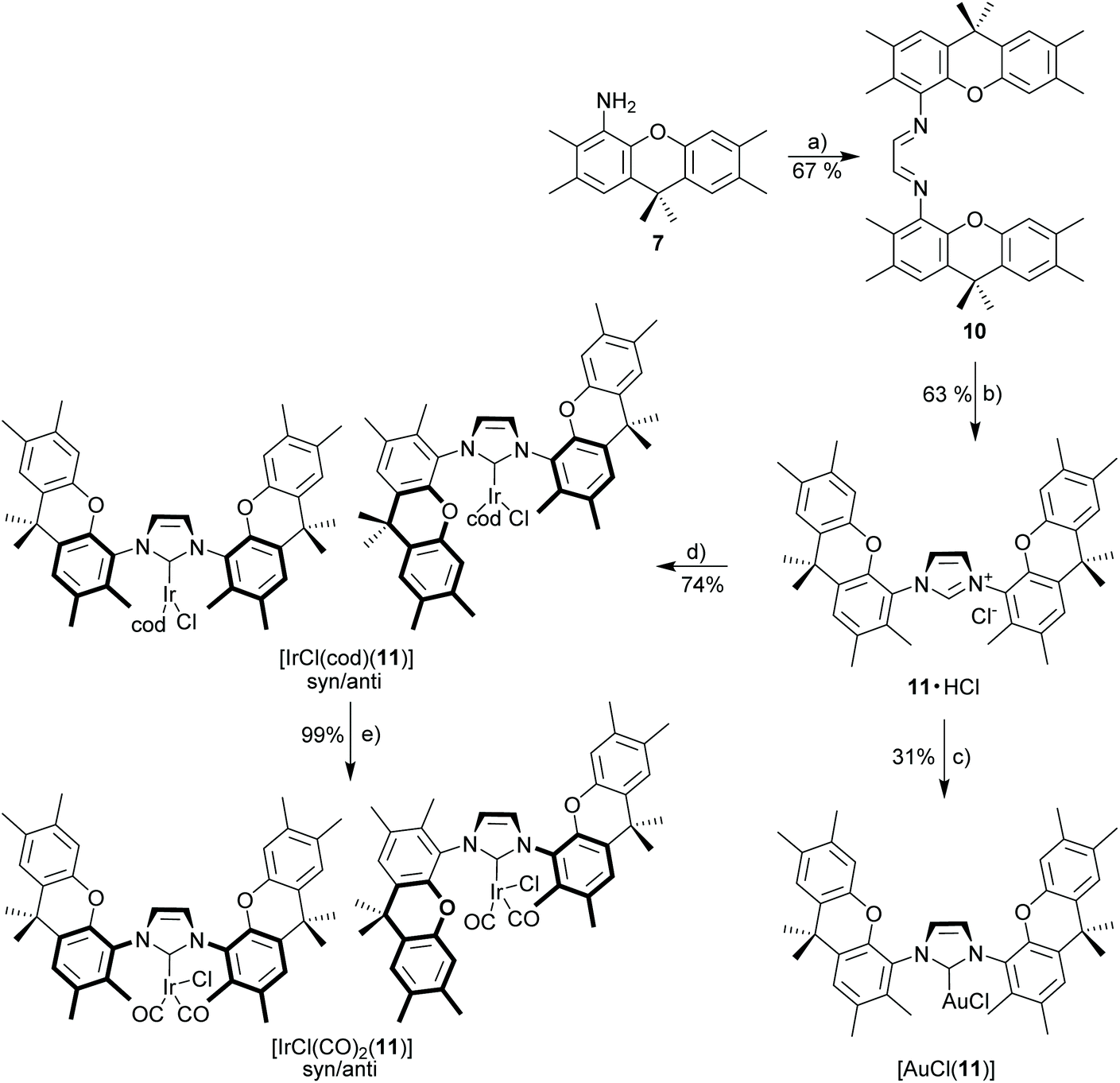 | ||
| Scheme 4 Synthesis of imidazolium salt 11·HCl and derived metal complexes. Reagents and conditions: (a) glyoxal, pTsOH, EtOH, 50 °C; (b) paraformaldehyde, Me3SiCl, EtOAc, 70 °C, 24 h; (c) [AuCl(SMe2)], K2CO3, acetone, 50 °C; (d) [IrCl(cod)]2, K2CO3, acetone, 50 °C; (e) CO, 0 °C, 30 min, CH2Cl2. | ||
In order to better evaluate the properties of binuclear open complexes compared to binuclear, closed (macrocyclic) complexes with NHC 9 (Scheme 3) complexes with the open NHC ligand 14 were synthesized. Azolium salt 14·2HCl containing two NHC units in an open structure was synthesized according to Scheme 5. NHC 14 is similar to NHC 9 concerning the groups directly bonded to the NHC ligands. However, the two NHC units in 8 are part of a macrocyclic structure with an enforced syn-orientation of the two NHC units, while NHC 14 has a flexible structure and the NHC units also have the chance to be in an anti-orientation. NHC metal complexes with NHC 9 were synthesized following established synthetic methodology. Both complexes [(AuCl)2(14)] and [(IrCl(cod))(14)] are stable with properties very similar to those of the monometallic [IrCl(cod)(IMes)] complex. [(IrCl(cod))2(14)] is closely related to complexes reported by Messerle et al.42 The important difference to previously synthesized xanthene-bisNHC complexes are the four ortho-methyl groups, which enforce an approximately orthogonal orientation of the imidazolinium ring relative to the xanthene unit. Based on this, a chelate type complex involving both NHC ligands in 11 and a single metal appears not to be possible, which is intended to enforce the close proximity of two metal centers. In the absence of those methyl groups the xanthene-based NHC ligands were reported to act as tweezer type ligands, with both NHC ligands coordinating the same metal.43,56,57 The derived metal complexes (Scheme 5) are stable and consist of a single isomer.
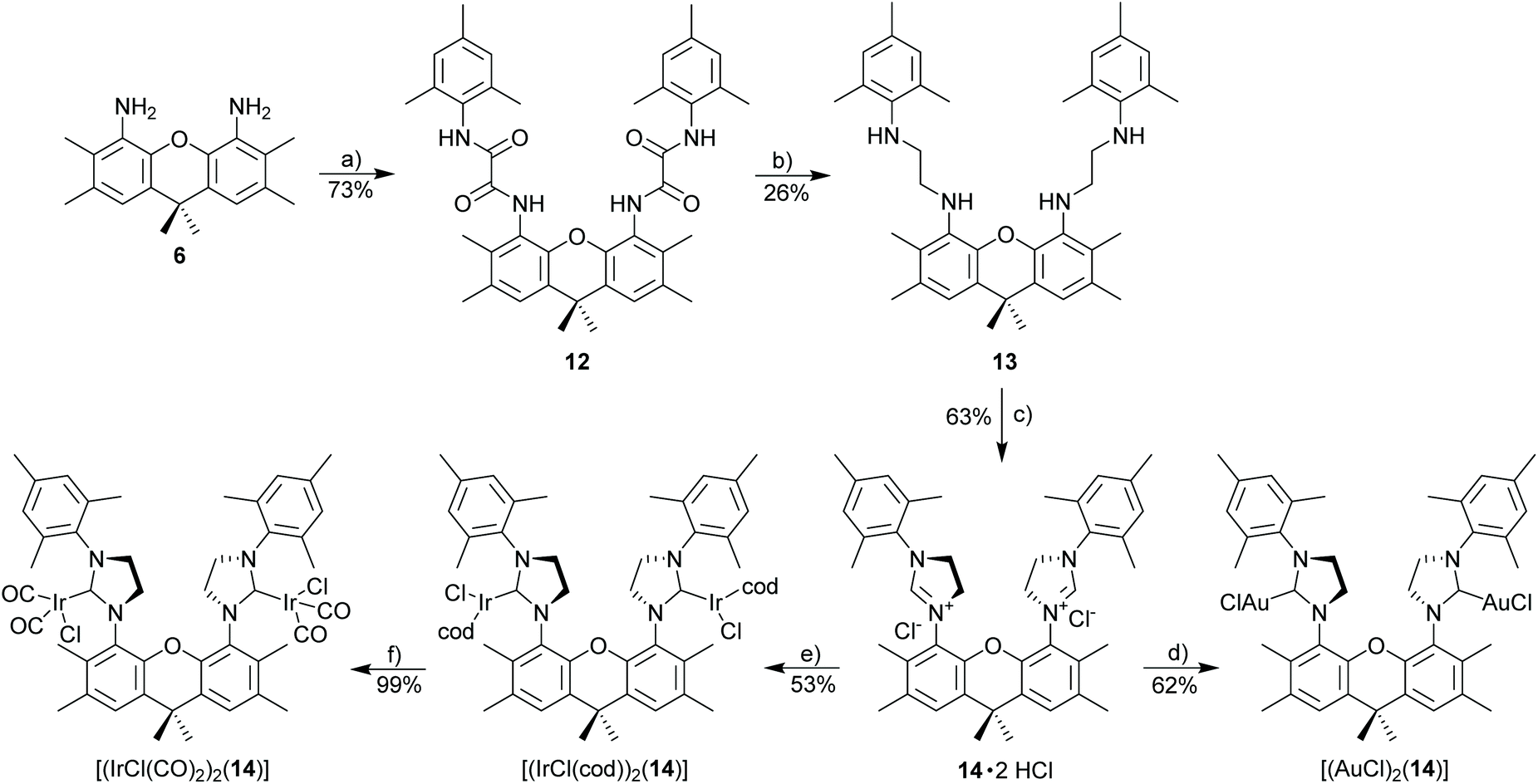 | ||
| Scheme 5 Synthesis of imidazolinium salt 14·2HCl and derived metal complexes. Reagents and conditions: reagents and conditions: (a) MesNHC(O)COCl, NEt3, 0 °C to rt, 1 h; (b) BH3·THF, reflux, 3 d; (c) triethyl orthoformate, NH4Cl, 110 °C, 24 h; (d) [AuCl(SMe2)], K2CO3, acetone/CH2Cl2, 50 °C; (e) [IrCl(cod)]2, K2CO3, acetone/CH2Cl2, 50 °C; (f) CO, 0 °C, 30 min, CH2Cl2. | ||
Due to potentially unfavorable steric interactions of the mesityl groups in the syn-isomer the anti-isomer is expected to be the favored isomer, which is confirmed by the X-ray crystal structure of [(AuCl)2(14)] (Fig. 2).58 The most obvious feature of the crystal structure is the anti-orientation of the two (NHC)AuCl – units resulting in Au⋯Au distance of 904 pm, with gold displaying the typical linear coordination.
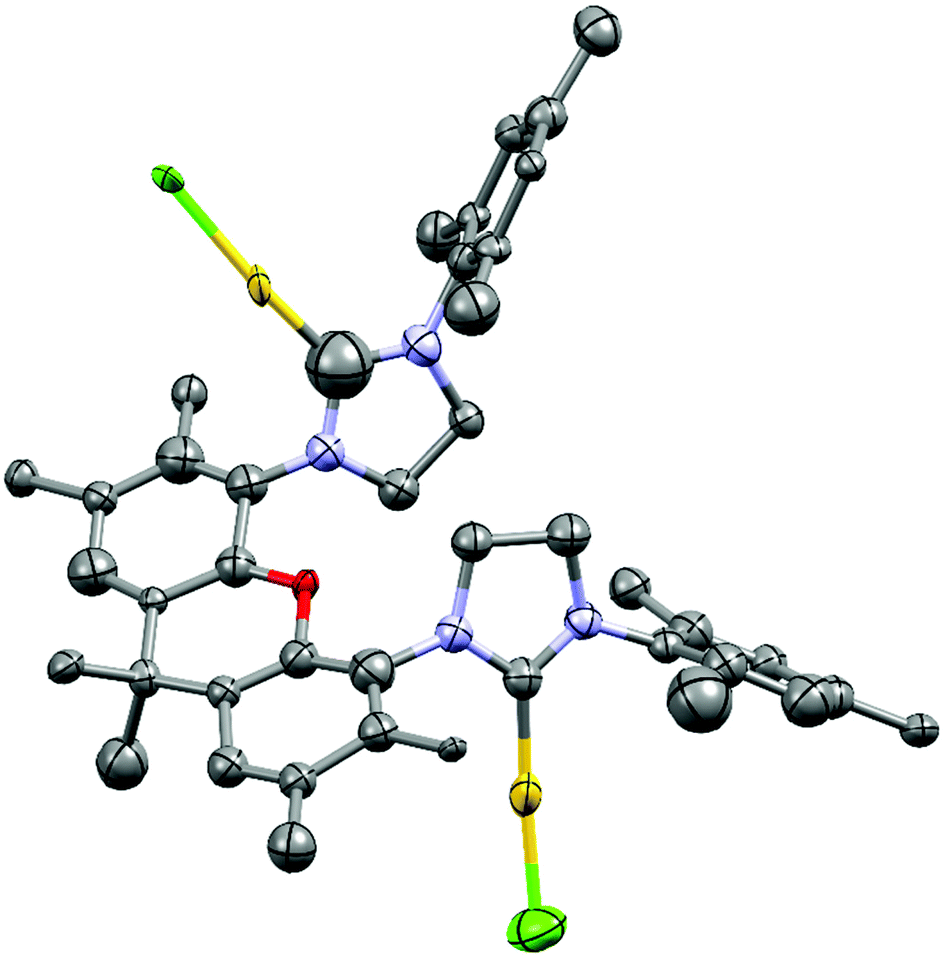 | ||
| Fig. 2 X-ray crystal structure (thermal ellipsoids 80%) of [(AuCl)2(14)] (colour code: Au yellow, Cl green, O red, N blue, H not displayed). Important distances (pm), bond lengths (pm) and angles (°): Au⋯Au 904, Au–C(NHC) 195(2), 196(5), Au–Cl 221(2), 233(3). | ||
So far, our attempts to obtain stable binuclear metal complexes were not entirely satisfactory. Based on this, a different approach was chosen by linking the two imidazoles with an alkyl chain (Scheme 6) to provide macrocyclic diimidazolium salt 16·2HBr. A C5-chain was chosen, since this corresponds to the number of atoms of the xanthene unit linking the two imidazoles and based on this it can be expected to be a strain-free linker.
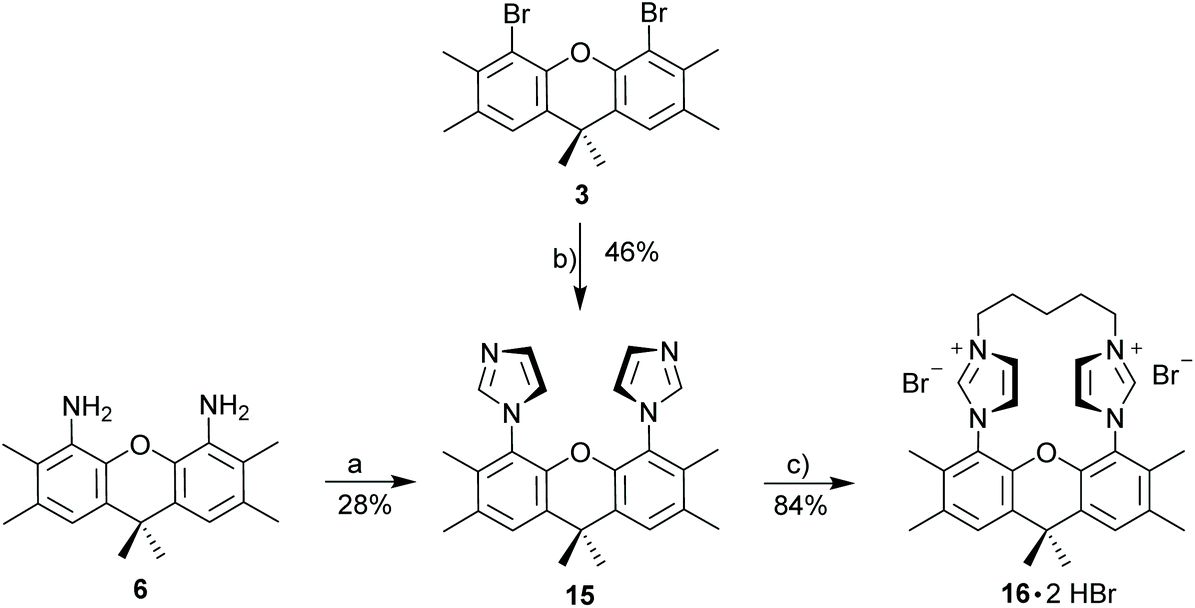 | ||
| Scheme 6 Synthesis of diimidazolium salt 16·2HBr. Reagents and conditions: (a) imidazole, K2CO3, CuI, DMF, 180 °C, 48 h; (b) paraformaldehyde, glyoxal, NH4OAc, AcOH, 80 °C, 12 h; (c) 1,5-dibromopentane, MeCN, reflux; 5 d; (d) MeI, MeCN, 50 °C, 24 h. | ||
The synthesis of 15 is possible starting from 4,5-dibromo-xanthene 3 or from 4,5-diamino-xanthene 6. Both reactions are simple, but since 6 needs to be prepared from 3, the copper-catalyzed reaction is more convenient. However, even though the synthesis of 15 from 3 was successfully carried out more than five times with yields in excess of 40%, it is important to note, that this yield is not always reproducible and in a few instances the reaction fails entirely. We have carefully studied the reaction conditions, but have not been able to identify the factors which sometimes preclude product formation. Utilizing diimidazolium salt 16·2HBr a number of metal complexes with Ag, Au, Cu, Ir, Pd and Rh could be synthesized (Scheme 7). Most complexes were synthesized via [(AgBr)2(16)] – either using the isolated silver complex for the preparation of [(CuI)2(16)] and [(Pd(allyl)Cl)2(16)] or the in situ generated silver complex for the synthesis of [(AuCl)2(16)], [(Au)(AuCl)2(Cl)(16)2] and [(RhCl(cod))2(16)]. However, for the Ir and Rh complexes the direct synthesis using the weak base approach provides better yields than utilizing the silver salt.
 | ||
| Scheme 7 Synthesis of metal complexes with NHC 16. Reagents and conditions: (a) Ag2O, MeOH, rt, 24 h; (b) [RCl(cod)]2, 50 °C, 2 h; (c) [AuCl(SMe2)] (2.0 eq.), 50 °C, 2 h; (d) [PdCl(allyl)]2, DCM, rt, 24 h; (e) CuI, DCM, rt, 24 h; (f) [AuCl(SMe2)] (1.5 eq.), 50 °C, 2 h; (g) [MCl(cod)]2 (M = Ir, Rh), K2CO3, NaOAc, KBr, MeCN, 80 °C, 12 h; (h) CO, 0 °C, CH2Cl2, 30 min. | ||
Depending on the stoichiometric ratio of NHC 16 and the gold precursor [AuCl(SMe2)] different complexes are formed. A 2:1-ratio Au/NHC provides [(AuCl)2(16)] as the only isolable product in 46% yield, while a 3:2-ratio gives [(Au)(AuCl)2(Cl)(16)2]. The latter complex contains two (NHC)AuCl and one (NHC)2Au+ units – both are common motifs in this chemistry, but we are not aware, that both units have been observed in a single complex.
The metal complexes with NHC 16 occur as single isomers – both in the solid state and in solution. The 1H NMR spectra of the respective metal complexes display two different signals for the two bridgehead methyl groups, which is incompatible with the anti-isomer. The syn-orientation is confirmed in the X-ray crystal structures of [(RhCl(cod))2(16)] (Fig. 3) and of [(Au)(AuCl)2(Cl)(16)2] (Fig. 4).51 In the solid state structure of [(RhCl(cod))2(16)] the molecule is bisected by a mirror plane and the two syn-Rh units are symmetry equivalents. The Rh⋯Rh distance of 703.2 pm is very similar to the Ir⋯Ir distance in [(IrCl(cod))2(9)] (702.4 pm).
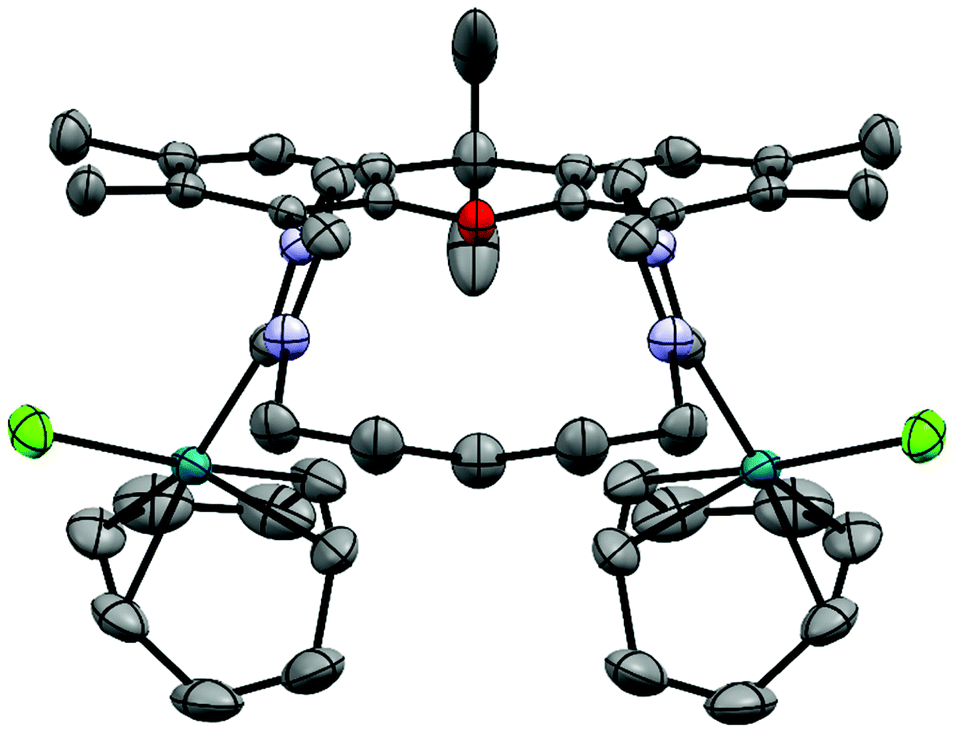 | ||
| Fig. 3 X-ray crystal structure (thermal ellipsoids 80%) of [(RhCl(cod))2(16)] (colour code: Rh turquoise, Cl green, O red, N blue, H not displayed). Important distances (pm), bond lengths (pm) and angles (°): Rh⋯Rh 703.19(5), Rh–Cl 238.4(1), Rh–C(NHC) 203.9(4), Rh–C(cod) 211.7(5), 208.2(5), 218.4(5), 222.4(5); Cl–Rh–C(cod) 168.9(1), 152.2(1), C(NHC)–Rh–C(cod) 169.4(2), 154.7(2). | ||
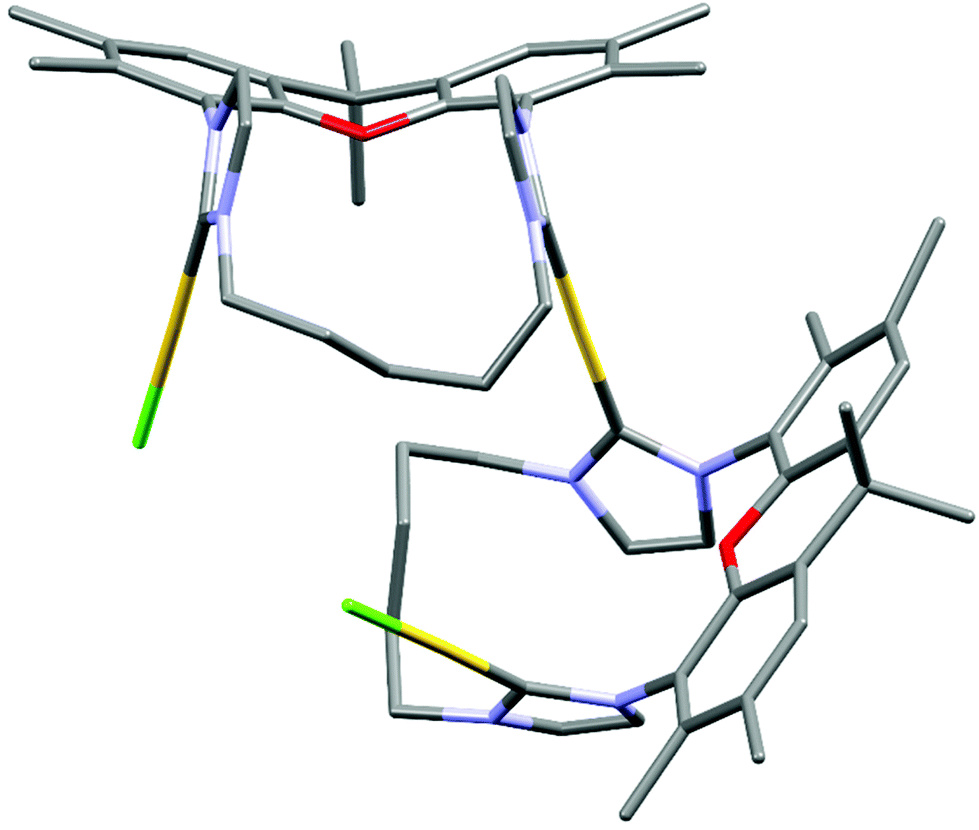 | ||
| Fig. 4 X-ray crystal structure (sticks representation for improved clarity) of [(Au)(AuCl)2(Cl)(16)2] (colour code: Au yellow, Cl green, O red, N blue, H not displayed). Important bond lengths (pm) and angles (°): Au⋯Au 586.2(1), 572.8(1), 660.6(1), Au–C(NHC) 197(2), 198(2), 199(2), Au–Cl 28.4(4), 226.1(5); Cl–Au–C(NHC) 177.2(6), 178.2(6), C(NHC)–Au–C(NHC) 177.3(7). | ||
The crystal structure of [(Au)(AuCl)2(Cl)(16)2] contains two (NHC)AuCl and a single (NHC)2Au+ unit. The chloride counterion is located in a spherical void without any significant contact to the cation. The two peripheral gold atoms are located at distances of 586 pm and 573 pm to the central NHC2Au+-unit, respectively, which is more than 100 pm shorter than the Ir⋯Ir or Rh⋯Rh distances in the other bimetallic complexes [(IrCl(cod))2(9)] and [(RhCl(cod))2(16)]. It seems, that in the absence of steric hindrance, the intermetallic distances tend to be much shorter than in the Ir and the Rh complexes. Again, the question arises, why the cod ligands of the Ir and the Rh complexes are located in the center of the complexes even though the two bulky groups push the metal centers apart. Probably the energy cost of this is relatively small. Alternatively, one could ask, why the two chlorides are not located in the center with the bulkier cod on the periphery. Probably, the square-planar geometry at the d8-metals and the orthogonal orientation of the plane of the imidazolylidene unit relative to the square-planar coordination sphere of Ir and Rh would place the two chlorides too close to each other and lead to even more unfavorable steric repulsion.
The structural flexibility of the xanthene allows a planar xanthene, while the other one displays the butterfly structure tilted at the oxygen and the sp3-CMe2 unit. All gold atoms display an almost linear coordination geometry. The Au–C(NHC) distances in the monoNHC and the bisNHC unit are virtually identical and these and other structural parameters are within expectations for such complexes.59,60
Monocationic complexes
The behavior of the binuclear [(MX(cod))2(NHC)] (X = halide; M = Rh, Ir) in the chromatography purification is unusual, since the observed Rf(CH2Cl2) is approx. 0.1 for such complexes. In contrast, for mononuclear complexes like [MCl(cod)(IMes)] (M = Rh, Ir) chromatographic Rf(CH2Cl2) > 0.9 are normally observed, which is in accord with the covalent, non-polar nature of the complexes. Based on this, the dinuclear complexes may be able to release one chloride forming cationic complexes stabilized by M–X–M (M = Rh, Ir; X = halide) units. This would also lessen steric pressure and allow the demanding cod units to move to the periphery of the complex, previously occupied by the two chloride ions. Such tendency to release anions has also been observed for the related monometallic complexes.61 Based on preliminary DFT calculations the structural flexibility of the complex is sufficient to enable a symmetric structure with a halide bridging the two metal centers. To provide further evidence, a CD2Cl2 solution of complex [(IrCl(cod))2(9)] was treated with one equivalent of AgOTf leading to the precipitation of AgCl and the formation of a cationic complex [(Ir(cod))2Cl(9)](OTf). This complex displays a simple NMR spectrum (Fig. 5), which would be in accord with a symmetric halide bridged complex. It is unlikely, that such complexes are highly dynamic with the structural interconversion being very fast on the NMR time scale. Further evidence for the bridged structure comes from the NMR spectrum, since the 1H NMR resonance of the cod ligands experience significant shifts, which are expected when relocating cod ligands from the interior of the complexes to the periphery. We were not able to obtain the complex in pure state or single crystals, since such complexes tend to decompose upon attempted isolation and purification.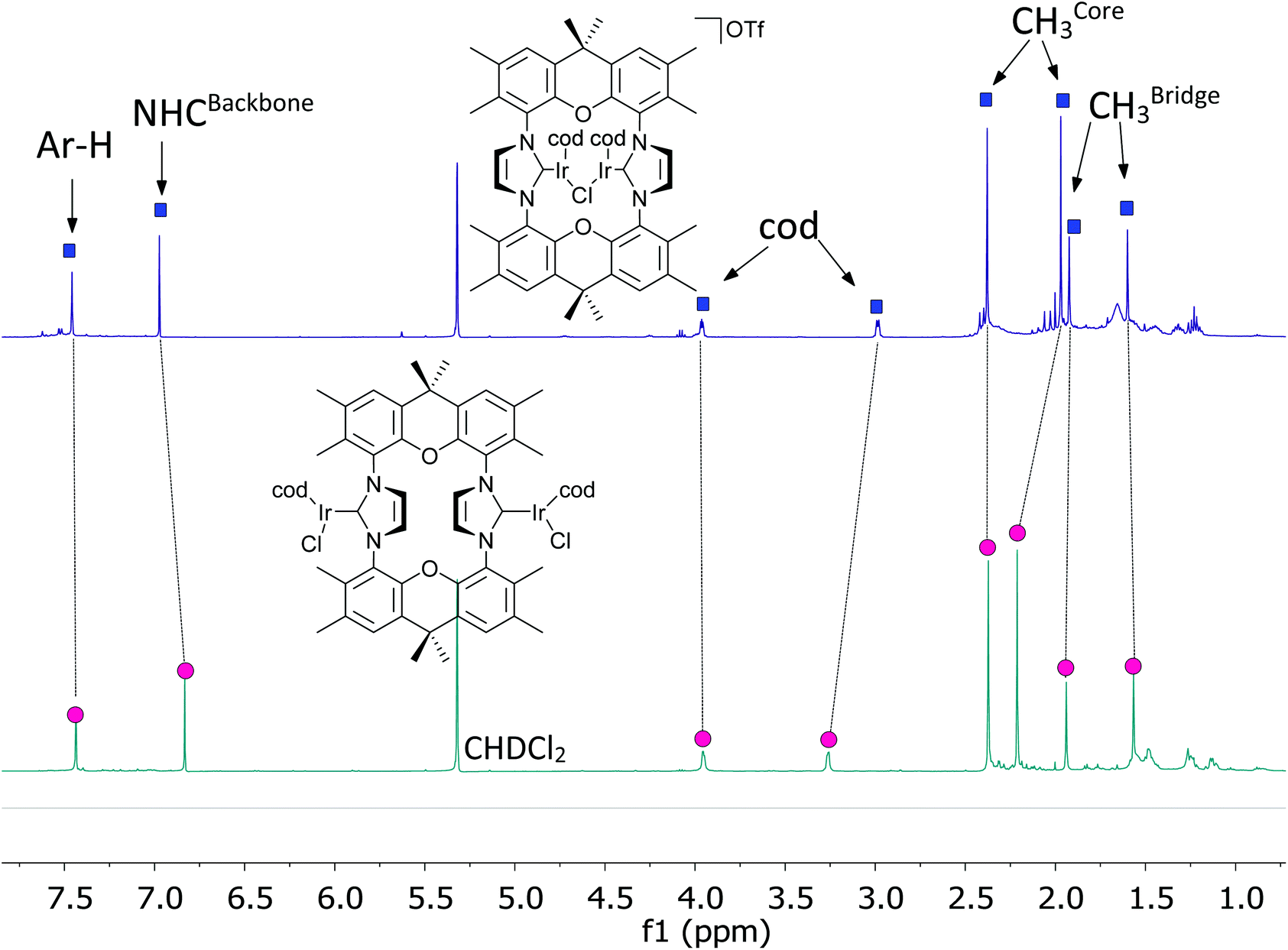 | ||
| Fig. 5 1H spectra of [(IrCl(cod))2(9)] and [(IrCl(cod))2(9)] + AgOTf @24 h in CD2Cl2. | ||
Infrared spectroscopy
The averaged CO-stretch frequencies of the complexes [(MCl(CO)2)2(NHC)] (M = Ir, Rh) provide information on the donor capacity of the respective NHC ligands.47,62 The IR data for the related complexes with the new NHC ligands reported here were determined (Table 1). Despite the constrained geometry of the macrocyclic NHC ligands 9 and 16 the donation of those ligands appears to be nearly the same as that of the reference complexes with IMes ligands. The influence of the anions (chloride, bromide) on the ν(CO) is almost identical. Ligand 14 appears to be slightly less donating than the other NHC ligands studied.| Complex | ν(CO)/cm−1 | ν av(CO)/cm−1 |
|---|---|---|
| [IrCl(CO)2(IMes)] | 2067/1980 | 2023.5 |
| [(IrCl(CO)2)2(9)] | 2066/1981 | 2023.5 |
| [(IrCl(CO)2)(11)] | 2064/1984 | 2024 |
| [(IrCl(CO)2)(14)] | 2070/1982 | 2026 |
| [(IrBr(CO)2)2(16)] | 2072/1982 | 2027 |
| [RhCl(CO)2(IMes)] | 2082/1997 | 2039.5 |
| [(RhCl(CO)2)2(16)] | 2077/2001 | 2039 |
Electrochemistry
The electrochemistry of the mono- and the binuclear complexes were determined to obtain information on the donor properties of the NHC ligands and to learn, whether the metal centers in the binuclear complexes interact with each other. The electrochemistry of the mononuclear metal complexes [(IrCl(cod)(11)] is reversible (E1/2 = 0.689, ΔE = 69 mV), which is typical for such complexes.63 However, the electrochemistry of all binuclear complexes tested [(MCl(CO)2)2(NHC)] (M = Ir, Rh and NHC = 9 or 16) turns out to be irreversible, and do not provide information about potential metal–metal interactions.Conclusions
BisNHC metal complexes containing two distinct (NHC)ML units in close proximity based on a 4,5-substituted xanthene backbone have been synthesized. Four metal complexes were structurally characterized displaying M–M distance as short as 573 pm. Based on the remaining structural flexibility even shorter M–M distance appear to be possible in binuclear complexes. Several of the binuclear metal complexes reported here are significantly less stable than the respective mononuclear complexes, when the coordination sphere contains bulky ligands, like in MX(cod) (M = Ir, Rh) complexes. Binuclear complexes with stability comparable to that of the related mononuclear complexes are those with Cu, Ag and Au metals. Further studies will be directed towards establishing metal–metal cooperativity in catalytic transformations.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the TU Darmstadt.References
- A. J. Arduengo, H. V. R. Dias, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1992, 114, 5530 CrossRef CAS.
- W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS PubMed.
- N-heterocyclic Carbenes, ed. S. P. Nolan, Wiley-VCH, Weinheim, 2014 Search PubMed.
- E. Peris, Chem. Rev., 2018, 118, 9988–10031 CrossRef CAS PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- R. H. Grubbs, Angew. Chem., Int. Ed., 2006, 45, 3760–3765 CrossRef CAS PubMed.
- T. P. Montgomery, T. S. Ahmed and R. H. Grubbs, Angew. Chem., Int. Ed., 2017, 56, 11024–11036 CrossRef CAS PubMed.
- J. Morvan, M. Mauduit, G. Bertrand and R. Jazzar, ACS Catal., 2021, 11, 1714–1748 CrossRef CAS.
- A. S. K. Hashmi, Chem. Rev., 2021, 121, 8309–8310 CrossRef CAS PubMed.
- R. D. J. Froese, C. Lombardi, M. Pompeo, R. P. Rucker and M. G. Organ, Acc. Chem. Res., 2017, 50, 2244–2253 CrossRef CAS PubMed.
- A. J. Nett, S. Cañellas, Y. Higuchi, M. T. Robo, J. M. Kochkodan, M. T. Haynes, J. W. Kampf and J. Montgomery, ACS Catal., 2018, 8, 6606–6611 CrossRef CAS PubMed.
- D.-A. Park, S. Byun, J. Y. Ryu, J. Lee, J. Lee and S. Hong, ACS Catal., 2020, 10, 5443–5453 CrossRef CAS.
- M. R. Buchmeiser, Chem. – Eur. J., 2018, 24, 14295–14301 CrossRef CAS PubMed.
- Q. Liang and D. Song, Chem. Soc. Rev., 2020, 49, 1209–1232 RSC.
- L. M. Martínez-Prieto, L. Rakers, A. M. López-Vinasco, I. Cano, Y. Coppel, K. Philippot, F. Glorius, B. Chaudret and P. W. N. M. van Leeuwen, Chem. – Eur. J., 2017, 23, 12779–12786 CrossRef PubMed.
- Y. Gao, L. Wang and L. Deng, ACS Catal., 2018, 8, 9637–9646 CrossRef CAS.
- J. Lee, H. Hahm, J. Kwak and M. Kim, Adv. Synth. Catal., 2019, 361, 1479–1499 CrossRef CAS.
- G. Sipos and R. Dorta, Coord. Chem. Rev., 2017, 375, 13–68 CrossRef.
- J. Atzrodt, V. Derdau, W. J. Kerr and M. Reid, Angew. Chem., Int. Ed., 2018, 57, 3022–3047 CrossRef CAS PubMed.
- M. Iglesias and L. A. Oro, Chem. Soc. Rev., 2018, 47, 2772–2808 RSC.
- R. Jazzar, M. Soleilhavoup and G. Bertrand, Chem. Rev., 2020, 120, 4141–4168 CrossRef CAS PubMed.
- S. Dagorne, Synthesis, 2018, 50, 3662–3670 CrossRef CAS.
- V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed.
- G. Guisado-Barrios, M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2018, 51, 3236–3244 CrossRef CAS PubMed.
- H. V. Huynh, Chem. Rev., 2018, 118, 9457–9492 CrossRef CAS PubMed.
- A. Gomez-Suarez, D. J. Nelson and S. P. Nolan, Chem. Commun., 2017, 53, 2650–2660 RSC.
- D. Janssen-Muller, C. Schlepphorst and F. Glorius, Chem. Soc. Rev., 2017, 46, 4845–4854 RSC.
- R. Savka, M. Bergmann, Y. Kanai, S. Foro and H. Plenio, Chem. – Eur. J., 2016, 22, 9667–9675 CrossRef CAS PubMed.
- R. Savka, S. Foro and H. Plenio, Dalton Trans., 2016, 45, 11015–11024 RSC.
- S. Roy and H. Plenio, Adv. Synth. Catal., 2010, 352, 1014–1022 CrossRef CAS.
- D. J. Nelson, Eur. J. Inorg. Chem., 2015, 2012–2027 CrossRef CAS.
- M. Poyatos, J. A. Mata and E. Peris, Chem. Rev., 2009, 109, 3677–3707 CrossRef CAS PubMed.
- D. M. Khramov, A. J. Boydston and C. W. Bielawski, Angew. Chem., Int. Ed., 2006, 45, 6186–6189 CrossRef CAS PubMed.
- K. A. Williams and C. W. Bielawski, Chem. Commun., 2010, 46, 5166–5168 RSC.
- A. J. Boydston, K. A. Williams and C. W. Bielawski, J. Am. Chem. Soc., 2005, 127, 12496–12497 CrossRef CAS PubMed.
- M. Poyatos and E. Peris, Dalton Trans., 2021, 50, 12748–12763 RSC.
- Y. Li, Y.-Y. An, J.-Z. Fan, X.-X. Liu, X. Li, F. E. Hahn, Y.-Y. Wang and Y.-F. Han, Angew. Chem., Int. Ed., 2020, 59, 10073–10080 CrossRef CAS PubMed.
- S. Ibáñez, M. Poyatos and E. Peris, Acc. Chem. Res., 2020, 53, 1401–1413 CrossRef PubMed.
- A. J. Caruso and J. L. Lee, J. Org. Chem., 1997, 62, 1058–1063 CrossRef CAS.
- D. Takeuchi, Y. Chiba, S. Takano and K. Osakada, Angew. Chem., Int. Ed., 2013, 52, 12536–12540 CrossRef CAS PubMed.
- F. B. Schwarz, T. Heinrich, J. O. Kaufmann, A. Lippitz, R. Puttreddy, K. Rissanen, W. E. S. Unger and C. A. Schalley, Chem. – Eur. J., 2016, 22, 14383–14389 CrossRef CAS PubMed.
- M. R. D. Gatus, I. Pernik, J. A. Tompsett, S. C. Binding, M. B. Peterson and B. A. Messerle, Dalton Trans., 2019, 48, 4333–4340 RSC.
- S. Saito, T. Kobayashi, T. Makino, H. Yamaguchi, H. Muto, I. Azumaya, K. Katagiri and R. Yamasaki, Tetrahedron, 2012, 68, 8931–8936 CrossRef CAS.
- L. Hintermann, Beilstein J. Org. Chem., 2007, 3, 22–26 Search PubMed.
- E. A. Martynova, N. V. Tzouras, G. Pisanò, C. S. J. Cazin and S. P. Nolan, Chem. Commun., 2021, 57, 3836–3856 RSC.
- R. Savka and H. Plenio, Dalton Trans., 2015, 44, 891–893 RSC.
- S. Leuthäußer, D. Schwarz and H. Plenio, Chem. – Eur. J., 2007, 13, 7195–7203 CrossRef PubMed.
- S. Wolf and H. Plenio, J. Organomet. Chem., 2009, 694, 1487–1492 CrossRef CAS.
- O. Halter, I. Fernández and H. Plenio, Chem. – Eur. J., 2017, 23, 711–719 CrossRef CAS PubMed.
- M. Bergmann, R. Savka, S. Foro and H. Plenio, Eur. J. Inorg. Chem., 2017, 2017, 3779–3786 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- S. Ando, M. Otsuka and T. Ishizuka, Organometallics, 2020, 39, 3839–3848 CrossRef CAS.
- P. J. Rayner, P. Norcott, K. M. Appleby, W. Iali, R. O. John, S. J. Hart, A. C. Whitwood and S. B. Duckett, Nat. Commun., 2018, 9, 4251 CrossRef PubMed.
- H. Valdés, M. Poyatos and E. Peris, Organometallics, 2013, 32, 6445–6451 CrossRef.
- S. Burling, M. F. Mahon, S. P. Reade and M. K. Whittlesey, Organometallics, 2006, 25, 3761–3767 CrossRef CAS.
- T. Makino, R. Yamasaki, I. Azumaya, H. Masu and S. Saito, Organometallics, 2010, 29, 6291–6297 CrossRef CAS.
- T. Makino, H. Masu, K. Katagiri, R. Yamasaki, I. Azumaya and S. Saito, Eur. J. Inorg. Chem., 2008, 4861–4865 CrossRef CAS.
- The plate-like shape of the crystals result in a poor quality of the respective diffraction data, which is the reason why the data were not deposited.
- S. Gaillard, P. Nun, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2010, 29, 5402–5408 CrossRef CAS.
- F. Lazreg, D. B. Cordes, A. M. Z. Slawin and C. S. J. Cazin, Organometallics, 2015, 34, 419–425 CrossRef CAS.
- G. Sipos, P. Gao, D. Foster, B. W. Skelton, A. N. Sobolev and R. Dorta, Organometallics, 2017, 36, 801–817 CrossRef CAS.
- D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC.
- S. Popov and H. Plenio, Eur. J. Inorg. Chem., 2021, 22, 3708–3718 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: NMR spectra (1H and 13C), infrared spectra, hires mass specs, cyclic voltammetry, and X-ray crystal structures. CCDC 212020221202042120205. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1dt03857e |
| This journal is © The Royal Society of Chemistry 2022 |