
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Solvatochromism and the effect of solvent on properties in a two-dimensional coordination polymer of cobalt-trimesate†‡
Savannah C.
Zacharias§
*,
Gaëlle
Ramon
and
Susan A.
Bourne
 *
*
Centre for Supramolecular Chemistry Research, Department of Chemistry, University of Cape Town, Rondebosch, Cape Town 7701, South Africa. E-mail: susan.bourne@uct.ac.za
First published on 15th March 2022
Abstract
Stimuli responsive porous materials are being investigated for a plethora of potential applications. These materials have the ability to respond to their physical and chemical environment and thus provide an opportunity to study supramolecular type interactions. We present an investigation into the effects of guest inclusion on framework flexibility in a two-dimensional polymer network, {[Co(btc)(DMF)2]·xDMF}n (1) where btc is benzene-1,3,5-tricarboxylate (trimesate) and DMF N,N-dimethylformamide. Solvents for guest exchange were selected to explore different potential interactions, such as hydrogen bonding (water and alcohols) and π-stacking (benzonitrile) and to cover a wide range of polarities and molecular sizes. Results showed that all solvents used could be exchanged directly for the guest DMF with accompanying visible colour changes. The incorporation of the new guest solvent was confirmed by PXRD and thermal analysis. This process is reversible for most solvents. On removal of the guest DMF, the framework (1-f) is retained, but layers collapse to a less crystalline material. Only water and methanol are capable of penetrating the material to reconstitute the layers. The crystal structure of the guest-exchanged system with benzonitrile (2) showed that the coordination polymer framework is retained, albeit with a change in symmetry and interlayer spacing, making 1 an example of a ‘breathing’ coordination polymer. A survey of related structures in the CSD found that there are four common motifs, which cannot be predicted from either the composition or space group symmetry of the compound.
Introduction
Stimuli responsive materials have attracted attention for use in a wide array of potential applications including catalysis, drug delivery, gas storage, separation and sensing. These “smart” materials include supramolecular gels, metal–organic frameworks (MOFs), covalent organic frameworks (COFs), zeolites and porous coordination networks.1–5 Many of these materials are porous, so they contain voids which are accessible to guest molecules through diffusion. These voids are either intrinsic6,7 or extrinsic8,9 in nature. Systems may have one type exclusively or have a mixture of the two.10,11 These different voids lead to three distinct categories of porosity: conventional porosity, porosity “without pores” and virtual porosity.12The inclusion and exchange of guest molecules may induce changes in the host framework.13–15 In some cases these changes can result in a visible colour change, a phenomenon known as solvatochromism.16 Depending on the flexibility of the host framework, various forms of guest-induced structural flexibility have been observed. These include “gate-opening”17,18 or “breathing” mechanisms.19,20 Dynamic breathing behaviour has been observed in both 3D and 2D metal–organic frameworks (MOFs) and coordination polymers (CPs).21,22
Trimesic acid (benzene-1,3,5-tricarboxylic acid, H3btc) has been widely used in the synthesis of both organic and metal–organic frameworks.23–27 Although the potential for hydrogen bonding through all three carboxylic acid groups might suggest that trimesic acid would form an open pore structure, in most cases it crystallizes as a solvate or co-crystal through incorporation of small molecules. In CPs and MOFs, the coordination of the metal ion typically occurs between carboxylate groups, thus these systems may exhibit similar packing to their purely organic analogues. Here we report the synthesis of a two-dimensional coordination polymer network comprised of cobalt(III) ions and trimesate (btc) linkers that was found to have solvent molecules in cavities between the interdigitated sheets. A survey of related structures in the Cambridge Structural Database was carried out to establish the most common packing motifs observed. We also report the results of guest exchange experiments which were carried out to determine the propensity for this CP to act as a stimuli-responsive material.
Experimental
Materials
Cobalt(III) acetylacetonate (Co(acac)3) and trimesic acid (benzene-1,3,5-tricarboxylic acid, H3btc), each with purity of ca. 98%, were supplied by Sigma-Aldrich. The solvents acetonitrile, benzonitrile, diethyl ether, N,N-dimethylformamide (DMF), ethanol, and methanol were of reagent grade quality and purchased from Sigma-Aldrich. All reagents were used without further purification.Synthesis of {[Co(btc)(DMF)2]·xDMF}n (1)
A solvothermal reaction was carried out using Co(acac)3 (26 mg, 0.073 mmol in 2 mL DMF) and H3btc (22 mg, 0.10 mmol in 3 mL DMF). The solutions were added to an autoclave which was heated in an oven at 105 °C for 20 h followed by a slow cool (10 °C h−1). Compound 1 crystallized as pink plates, which agglomerated into flower shaped crystals (Fig. 1). Crystals of this shape are typically twins,28 and this was indeed the case. | ||
| Fig. 1 Morphology of crystals of 1. The variability in colour under polarised light is an indication of twinning. | ||
Solvent exchange in 1
Freshly prepared crystals of 1 were removed from the mother liquor and placed directly into another solvent. The crystals were left in the solvent for 24 h and then analysed using the various techniques detailed below.To test the reversibility of the solvent exchange, the crystals were placed back into DMF for another 24 h and then analysed.
Thermal analysis
Thermal analysis techniques used were hot stage microscopy (HSM), thermogravimetric analysis (TGA), and differential scanning calorimetry (DSC). All three methods measure a change in the physical properties of the material as a function of temperature.Hot stage microscopy
Hot stage microscopy (HSM) allows for the visualisation of thermal events which can be correlated with the data obtained from TGA and DSC. The sample was heated at a controlled rate (10 °C min−1). A Nikon SMZ-10 stereoscopic microscope, fitted with a Sony Digital Hyper HAD colour video camera, was used to view the samples. Heating took place on a Linkam THMS600 hot stage using a Linkam TP92 temperature control unit. Thermal events were monitored and captured in real-time and viewed using analySIS,29 a Soft Imaging System program. The temperatures at which events occur may differ from those recorded on the TGA and DSC because of the lack of a purge gas and the geometry of the sample holders.Thermogravimetric analysis
Thermogravimetric analysis (TGA) was used to determine the solvent content and stability of the complex. A TA-G500 from TA Instruments was used in conjunction with Universal Analysis 2000 (ref. 30) software. A dry nitrogen flow rate of 50 cm3 min−1 was employed. Typically, 10 mg to 20 mg of material was used with a heating rate of 10 °C min−1.Differential scanning calorimetry
Differential scanning calorimetry (DSC) was used to determine the onset temperature, temperature range, and enthalpy changes for thermal events. Experiments were carried out using either the Q200 or the TA Discovery 25, both TA Instruments. Universal Analysis 2000 (ref. 30) and TRIOS31 were used to analyse the results from the Q200 and TA Discovery 25, respectively. Samples, weighing 1 mg to 2 mg, were analysed in vented Tzero™ aluminium pans at a heating rate of 10 °C min−1. The purge gas was dry nitrogen at flow rates of 60 cm3 min−1 (for the Q200) and 40 cm3 min−1 (for the TA Discovery 25).Powder X-ray diffraction
Powder X-ray diffraction (PXRD) patterns were collected at 298 K on a Bruker D8 Advance diffractometer equipped with a Lynxeye detector using CuKα-radiation (λ = 1.5406 Å). Samples were lightly ground, placed on a zero background sample holder and scanned over a 2θ range of 4° to 40°. A step size of 0.02° to give a total of 1762 steps was used. A current of 40 mA and accelerating voltage of 30 kV were used to generate the X-rays. A receiving slit of 0.6 mm and primary and secondary slits of 2.5 mm were used. Variable temperature powder X-ray diffraction (VT-PXRD) was measured on the same instrument. The X-rays were generated by a voltage of 40 kV and a current of 40 mA. The same step size (0.02°) and total number of steps (1762) as used for PXRD were used.Calculated powder X-ray diffraction patterns were generated using Mercury v3.7 (ref. 32) from single crystal data. The same wavelength (λ = 1.5406 Å) was used in the calculation. These patterns were used to determine phase purity of the bulk material by comparison to the experimental data.
Single crystal X-ray diffraction data collection and structure refinement of {[Co(btc)(DMF)2]·2/3DMF}n (1) and {[Co(btc)(DMF)2]·yPhCN}n (2)
A Bruker KAPPA APEX DUO II diffractometer, Mo radiation (λ = 0.7107 Å), was used to collect the intensity data at 173(2) K. The X-ray diffraction pattern of 1 was determined to have Laue symmetry of 2/m, indicating the monoclinic crystal system. XPREP33 was used to determine the space group and the|E2![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) | value of 0.938 indicated centrosymmetry. Cell-now34 determined that the crystal was a non-merohedral twin with a BASF factor of 0.2. Data scaling and absorption correction were performed using TWINABS,35 and SAINT36 was used for integration. The structure was solved by direct methods in SHELXS and refined by full-matrix least-squares on F2 using SHELXL37 within the XSEED interface.38 Non-hydrogen atoms were located in difference electron density maps and were refined anisotropically while hydrogen atoms were placed in calculated positions and refined using a riding model. The BASF value (which specifies the fractional contribution of the twin components) refined to 0.416 over the course of the structure refinement. We attempted to model the DMF guest molecules, using the TGA results as a guide to site occupancies. While we could identify two sites for guest molecules, the DMFs were disordered and could not be modelled to fully account for the electron density in those regions. Thus the final refinement involved masking the solvent using the SQUEEZE routine in Platon.39 Details of the crystal structure are given in Table 1. Crystals of 2 were obtained after soaking crystals of 1 in benzonitrile for 24 h. A significant loss of crystallinity was observed. 2 crystallizes in the trigonal space group P
| value of 0.938 indicated centrosymmetry. Cell-now34 determined that the crystal was a non-merohedral twin with a BASF factor of 0.2. Data scaling and absorption correction were performed using TWINABS,35 and SAINT36 was used for integration. The structure was solved by direct methods in SHELXS and refined by full-matrix least-squares on F2 using SHELXL37 within the XSEED interface.38 Non-hydrogen atoms were located in difference electron density maps and were refined anisotropically while hydrogen atoms were placed in calculated positions and refined using a riding model. The BASF value (which specifies the fractional contribution of the twin components) refined to 0.416 over the course of the structure refinement. We attempted to model the DMF guest molecules, using the TGA results as a guide to site occupancies. While we could identify two sites for guest molecules, the DMFs were disordered and could not be modelled to fully account for the electron density in those regions. Thus the final refinement involved masking the solvent using the SQUEEZE routine in Platon.39 Details of the crystal structure are given in Table 1. Crystals of 2 were obtained after soaking crystals of 1 in benzonitrile for 24 h. A significant loss of crystallinity was observed. 2 crystallizes in the trigonal space group P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) . Absorption correction was performed using SADABS40 and structure elucidation was carried out as for 1.
. Absorption correction was performed using SADABS40 and structure elucidation was carried out as for 1.
| {[Co(btc)(DMF)2]·xDMF}n (1) | |
|---|---|
| CCDC deposition number | 2133533 |
| Molecular formula | [Co(C9H3O6)(C3H7NO)2]·x(C3H7NO) |
| Molecular mass [g mol−1] | 412.2 |
| Crystal system | Monoclinic |
| Space group | P21/n |
| α [Å] | 16.6748(18) |
| β [Å] | 14.1341(16) |
| γ [Å] | 28.845(3) |
| β [°] | 89.974(2) |
| Volume [Å3] | 6798.3(13) |
| Z, Z′ | 12, 3 |
| Densitycalcd: [g cm−3] | 1.351 |
| μ [MoKα] [mm−1] | 0.804 |
| F (000) | 2864 |
| Temperature [K] | 173(2) |
| Crystal size [mm] | 0.02 × 0.13 × 0.22 |
| Range scanned θ [°] | 1.41 to 26.50 |
| Index range | h: −20 20; k: 0 17; l: 0 36 |
| φ and ω scan angles [°] | 0.5 |
| Dx [mm] | 55 |
| Total number of reflections | 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 520 520 |
| Number of independent reflections | 13520 |
| Number of reflections with I > 2σ(I) | 8945 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0771, wR2 = 0.1941 |
| R indices (all data) | R 1 = 0.1191, wR2 = 0.2162 |
| S | 1.050 |
| Number of parameters | 716 |
| Number of reflections omitted | 0 |
| Parameters a, b in w = 1/[σ2(F02) + (aP)2 + (bP)] | a = 0.0850; b = 22.67 |
| (δ/σ)mean | <0.001 |
| Δρ excursions [e Å3] | 1.316 and −0.715 |
Results and discussion
Crystal structure of {[Co(btc)(DMF)2]·xDMF}n (1)
The pink crystals obtained solvothermally were found to be a 2D coordination polymer comprised of cobalt(III), trimesic acid and N,N-dimethylformamide (DMF). The asymmetric unit of 1 consists of three cobalt(III) metal centres, three fully deprotonated trimesic acid linkers, and six coordinated DMF molecules (ESI‡ Fig. S1). Each Co(III) ion is coordinated to two DMF molecules and three trimesate linkers, two through a η1 mode of binding and the third through a chelating η2 mode, in the equatorial position. Bond lengths and angles for the coordination to the cobalt ions are within the typical range (ESI‡ Table S1).41 The metal centres all display a distorted octahedral geometry with bond lengths and angles within the expected ranges. The structure of 1 is a 2D honeycomb coordination polymer in which each hexagonal pore is framed by three cobalt ions and three trimesate linkers. The coordinated DMF molecules are perpendicular to the planar 2D layers, and result in interdigitated sheets which run parallel to [100] and [001], Fig. 2. The interlayer spacing is 6.8 Å.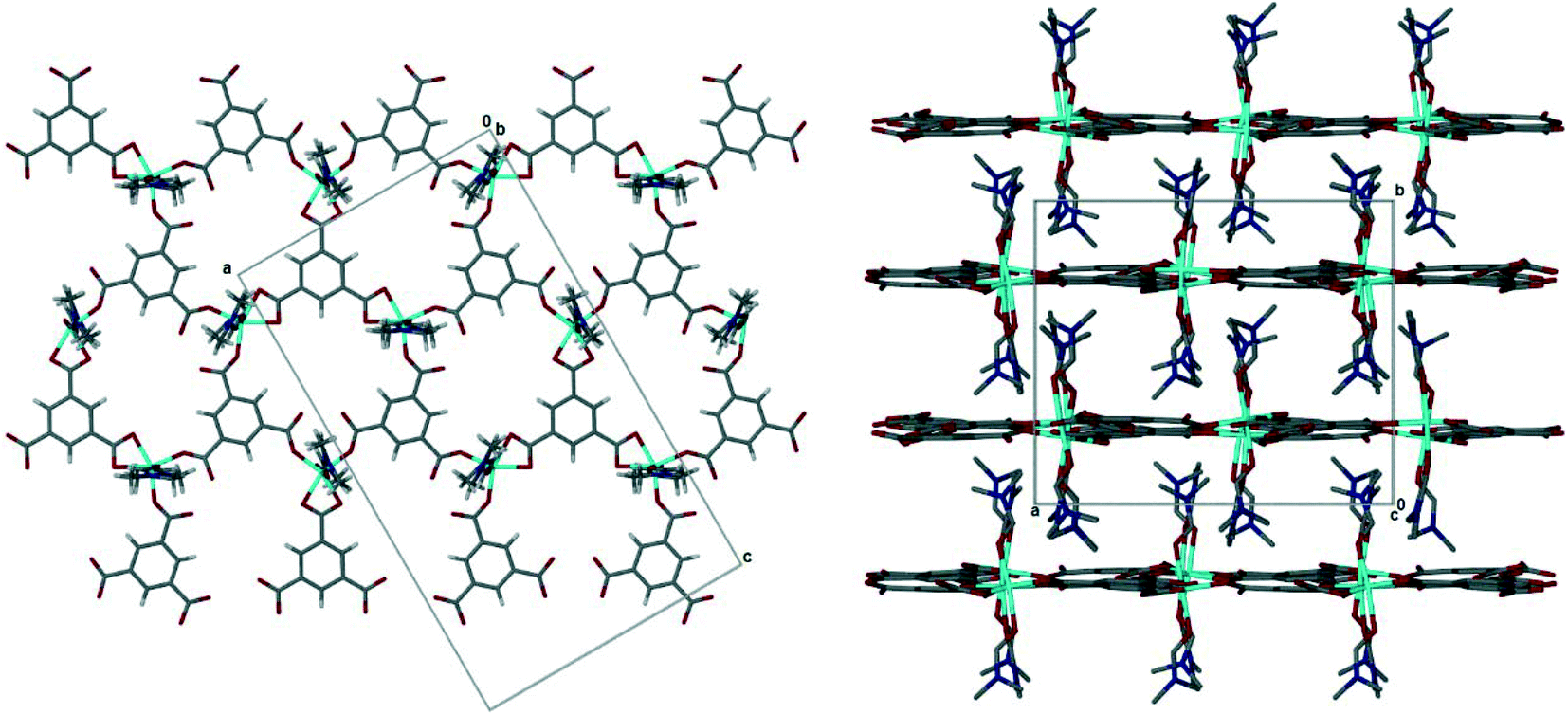 | ||
| Fig. 2 (Left) The 2D honeycomb layer in 1, viewed down [010]. Guest DMF omitted. (Right) Interdigitation of 2D layers in 1, viewed down [001]. Guest DMF and hydrogen atoms omitted for clarity. | ||
Guest DMF molecules are found in the cavities between the layers and the interdigitated coordinated DMFs (ESI‡† Fig. S3), with three sites clearly evident, although the guest molecules are severely disordered. This arrangement results in a large amount of potential void space, should the uncoordinated solvent be removed and the structural identity of [Co(btc)(DMF)2] be retained. Using Mercury v3.7 (ref. 32) potential void space was calculated using a probe radius 1.2 Å with approximate grid spacing of 0.7 Å, this resulted in a contact surface area of 1635 Å3 (24.1% of unit cell volume).
There are fifteen independent hydrogen bonds in 1, all of which are weak C–H⋯O interactions (ESI‡ Table S2). Three distinct types of hydrogen bond can be seen in Fig. 3. The intramolecular hydrogen bonds labelled 1–5 result in 5-membered rings between the coordinated DMF and the oxygen atom from the carboxylate of the trimesic acid ligand within the same layer. 5-Membered rings are favourable as there is little steric strain. One of the uncoordinated DMF solvent molecules is held in place by hydrogen bond 6. It is this supramolecular interaction that may be disrupted to exchange the solvent molecules. The remaining eight hydrogen bonds (labelled 7–15) link the axially coordinated DMF molecules to the carboxylate groups of an adjacent coordination polymer layer.
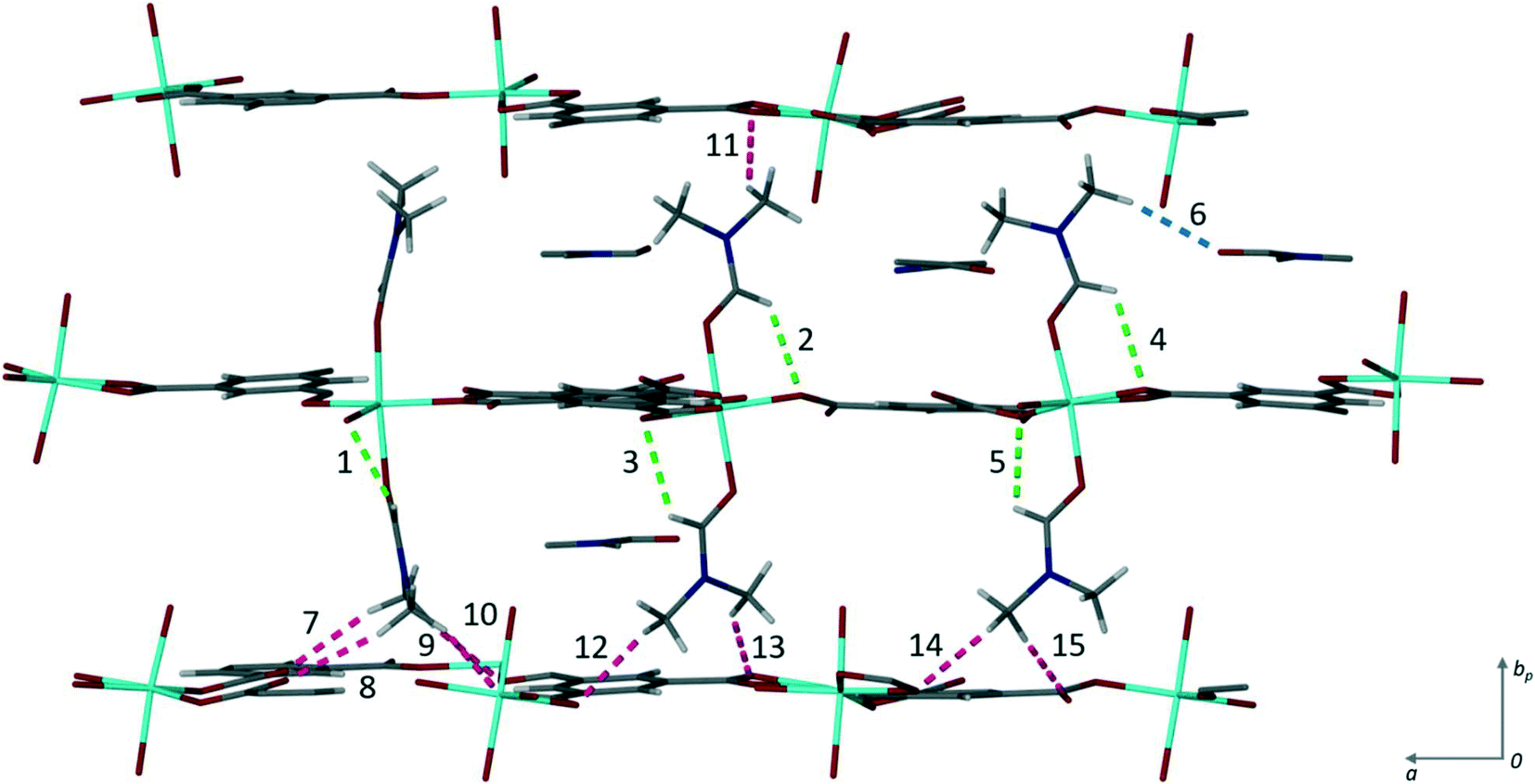 | ||
| Fig. 3 Hydrogen bonding in 1. The three types found within the structure are shown in different colours: blue shows the hydrogen bond (6) between the coordinated DMF and the solvent DMF, green the intramolecular hydrogen bonds (1–5) from the coordinated DMF t to the carboxylate oxygen, and pink the hydrogen bonds (7–15) between the axially bound DMF molecules to the carboxylate oxygen atoms of subsequent layers. The numbers correspond to those given in Table S2.‡ | ||
The agreement between the calculated and experimentally obtained PXRD patterns indicates the homogeneity of the bulk material (ESI‡ Fig. S4), while variable-temperature PXRD (ESI‡ Fig. S5) shows no phase change until the loss of guest DMFs between 75 and 100 °C. At higher temperatures the material loses crystallinity, and this does not change on cooling to 25 °C. This reduction in crystallinity corresponds to the onset of loss of DMF from the material as seen in the thermal analysis. The DMF molecules are instrumental to the retention of the crystalline structure which is not unexpected given that 1 does not have a rigid 3D framework. To estimate the amount of DMF in the channels, we considered the electron count in voids calculated by Platon. The 459 electrons in voids corresponds to 13.5 DMF molecules, or slightly more than 1 DMF per Co-unit. This is slightly higher than the 0.7 guest DMF per Co-unit we obtained from thermogravimetry. The TGA for 1 shows a three-step mass loss of 40.8 ± 0.8% between 88.6 ± 2.7 °C and 248.0 ± 0.6 °C (n = 3) (ESI‡ Fig. S6). This corresponds to the removal of DMF guests in the channels (calc. 10.6%), followed by the loss of the coordinated DMF molecules (calc. 31.7%). A change in colour of the crystals from pink to blue above 200 °C confirms the loss of the coordinated DMFs as the geometry around the Co(III) ions changes from octahedral to tetrahedral (Fig. 4). The small difference in DMF occupancy between the Platon calculations and TGA measurements could be accounted for by the former representing the single crystal selected for analysis, while the latter represents bulk material.
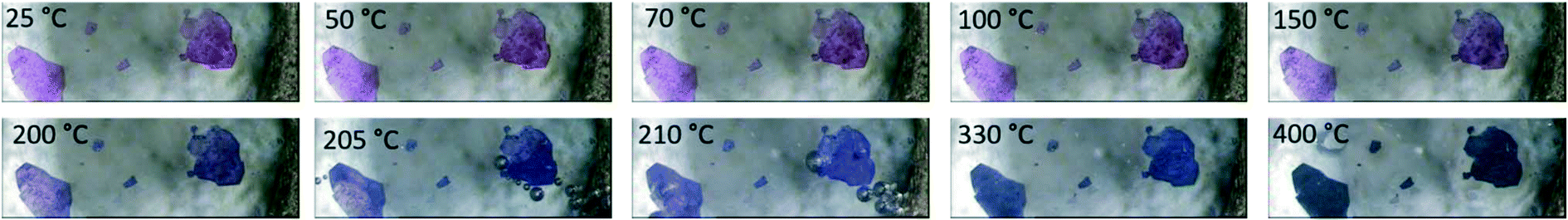 | ||
| Fig. 4 Hot stage images of 1. The crystals darken from pink to purple upon heating. All DMF is removed by 220 °C. | ||
Solvent exchange
To assess the accessibility of the void space, solvent exchange experiments were performed. Crystals of 1 were exposed to other solvents to test whether they would replace the uncoordinated DMF molecules. Solvents used were acetonitrile (MeCN), benzonitrile (PhCN), diethyl ether, ethanol (EtOH), methanol (MeOH), and water (H2O). This range of solvents were selected to cover a variety of potential interactions, such as hydrogen bonding (water and alcohols) and π-stacking (benzonitrile) and for their different polarities ranging from 0.117 for diethyl ether to 1.000 for water (ESI‡ Table S3).42 Co(III) has an affinity for nitrogen donors,43 so solvents with cyano functional groups were included to test if these solvents would replace the coordinated DMF molecules in addition to the guest DMFs.In the first set of experiments, crystals of 1 were soaked directly in solvent. The crystals change from bright pink to differing shades of pink and purple depending on the solvent used (Fig. 5), a phenomenon known as solvatochromism. Water exposure resulted in the lightest shade of pink, and the crystals of 1 turned purple when exposed to ethanol. This colour change may be a result in a change of coordination geometry around the Co(III) centre43 or changes to the channels which alters the electronic spectrum of the compound.
 | ||
| Fig. 5 Crystals of 1 turn different shades of pink and purple after exposure to the solvents indicated in the labels. | ||
TGA was used to estimate the amount of each solvent absorbed by the framework CP (Table 2, and ESI‡ Fig. S8).
| Solvent | Mass loss [%] (observed) | Mass loss [%] (calculated) | Moles solvent per Co-unita |
|---|---|---|---|
| a Calculated from the observed mass loss, assuming only the DMF solvate molecules are replaced. Crystals were surface dried before TGA. | |||
| Acetonitrile (MeCN) | 10.5 ± 0.8 | 11.0 | 1.2 |
| Benzonitrile (PhCN) | 13.7 ± 3.1 | 13.5 | 0.6 |
| Diethyl ether (Ether) | 16.3 ± 1.6 | 14.4 | 1.0 |
| Ethanol (EtOH) | 21.9 ± 3.8 | 21.7 | 2.5 |
| Methanol (MeOH) | 16.9 ± 0.7 | 16.2 | 2.6 |
| Water (H2O) | 38.5 ± 9.9 | 38.5 | 14 |
PXRD traces (ESI‡ Fig. S9) for all sorbed compounds are similar, though the peak at 12° present in 1 is absent in the water, methanol, ethanol and acetonitrile compounds, while the peak at 25° shifts slightly in most compounds. This lends credence to our model for the solvent:CP ratios reported in Table 2, which were calculated on the basis of guest DMFs being replaced, but the framework CP being retained with coordinated DMF in place.
To investigate the reversibility of this solvent exchange, the 1-solvent crystals were soaked again in DMF. TGA (ESI‡ Fig. S10) is not definitive, but PXRD (ESI‡ Fig. S11) indicates that all (except 1-water/MeOH) return to the original phase seen in 1, i.e. the solvents are replaced by DMF in a reversible manner.
In a second set of experiments, we explored the possibility of desolvated powdered 1 to sorb solvents. 1 was dried in a vacuum oven at 85 °C for 24 h to remove the uncoordinated DMF and resulted in a purple powder, 1-f, which retains the 2D CP framework. As noted before, the desolvation of the guest DMF results in a loss of crystallinity. As 1 is not a rigid framework, removal of uncoordinated DMF results in the collapse of the structures and the channels are closed which prevents solvent from entering the structure Powdered 1-f was immersed in the same range of solvents for 24 h at RT. Methanol and water, the smallest of the solvents investigated and those with the highest polarity, were taken up by 1-f, which was confirmed by PXRD (ESI‡ Fig. S31–S33).
Crystal structure of {[Co(btc)(DMF)2]·yPhCN}n (2)
Single crystal X-ray data was collected on a crystal of 1 which had been soaked in PhCN for 24 h as described above. The crystal had high mosaicity and diffracted poorly. As a result the refinement is poor and it is not possible to describe the structure completely.¶ Despite this it is possible to examine the coordination geometry of the Co(II) and to establish that the 2D coordination polymer is retained from 1. The coordinated DMF molecules have not been removed, lending further confidence to the modelling of the solvent ratios sorbed in the solvent exchange experiments. Although there is a change in the space group from monoclinic P21/n to trigonal, P, the coordination polymer network changes only subtly. This is evident in Fig. 6 which shows the 2D hexagonal network and the interdigitation of the coordinated DMFs between layers. The layers in 2 have an interplanar spacing of 7.1 Å, slightly longer than that observed in 1. The intramolecular hydrogen bonding (hydrogen bonds 1–5 in 1) appears to be preserved, but the distances from DMF methyl carbons to trimesate carboxylate oxygens appear to be too long (>3.4 Å) to be likely to form the interlayer hydrogen bonding seen in 1. As the model is incomplete only an approximate estimate of the void space in 2 is possible. However, if one estimates this void space using Mercury (as described for 1 above), voids of ca. 1700 Å3 per unit cell are observed. Unlike the self-contained cavities observed in 1, these appear to form channels running parallel to [100] and [010] (Fig. S12‡) with openings of approximately 4.2 by 3.4 Å. Benzonitrile molecules could fit within these voids.
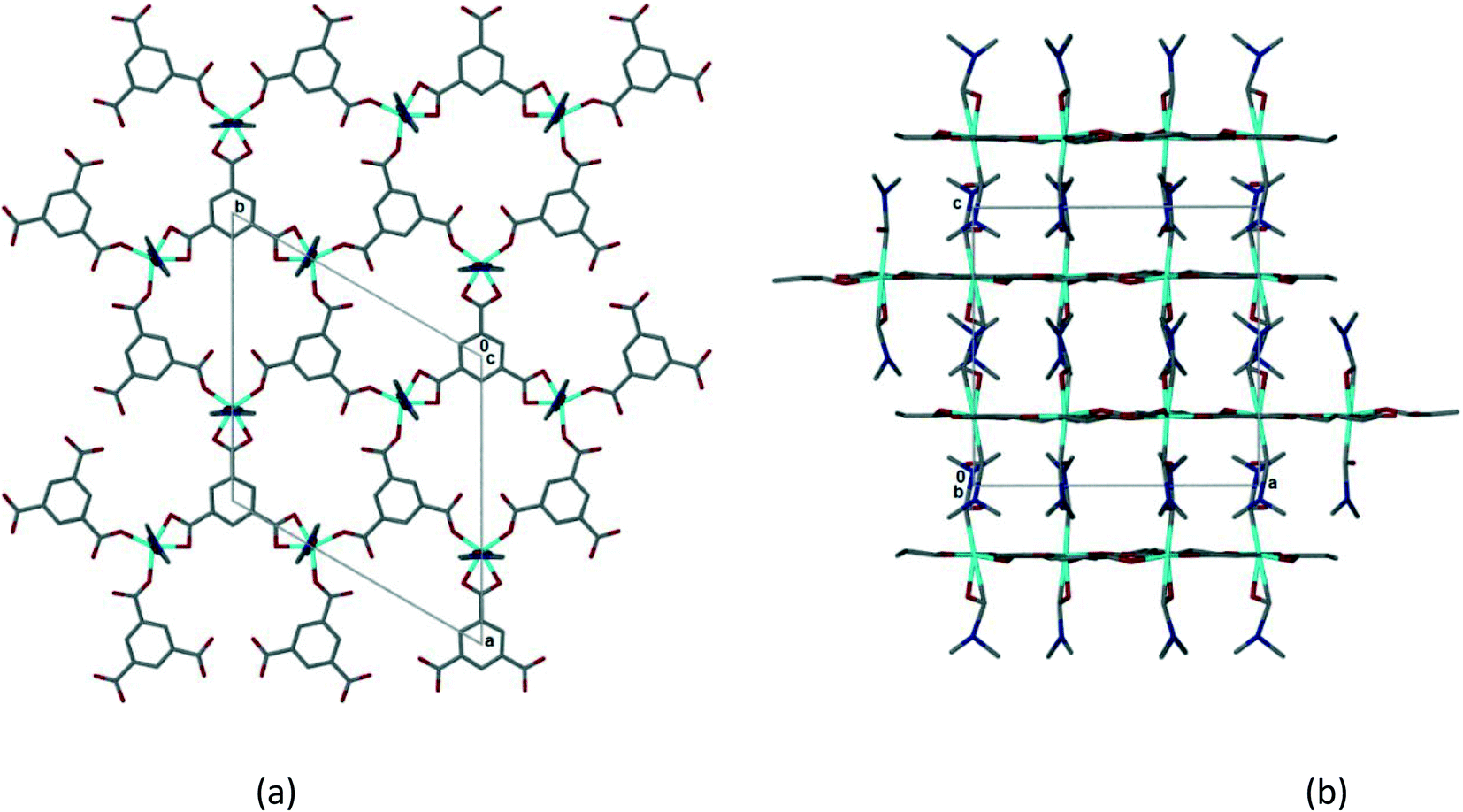 | ||
| Fig. 6 (a) The 2D honeycomb layer in 2, viewed down [001]. (b) Interdigitation of 2D layers in 2, viewed down [010]. | ||
Survey of related trimesic acid crystal structures in the CSD
Trimesic acid (H3btc) has been extensively used in crystal engineering studies.23–27 Ward and Oswald44 described the formation of trimesic acid solvates with ethanol, isopropyl alcohol and dimethylformamide, and reviewed the structures formed by H3btc with alcohols and other solvents. In alcohol structures, H3btc molecules form a R22(8) dimer through hydrogen bonding of the carboxylic acid groups. These dimers then self-assemble through a hydrogen bonding interaction with a free carbonyl of the H3btc and the alcohol solvate, to form an extended tape motif with large rings (R44(32)). They further found a correlation between the length of the alcohol chain and the degree of distortion in the ring framework. On the other hand, the 2:1 H3btc:DMF structure forms a layer where two trimesic acid molecules hydrogen bond to two DMF molecules and an adjacent trimesic acid, to form a 2D tape with no interactions between the layers (Fig. 7).
 | ||
| Fig. 7 (a) H3btc:EtOH solvate which shows typical layer formed in alcohol solvates with [ChemTemp]46); (b) disruption of the layer in the 2:1 H3btc:DMF solvate (XAVPOZ47) (adapted from ref. 44). | ||
Intrigued by the similarity of these layered organic structures with the honeycomb network layer formed in 1 (Fig. 2), we undertook a survey of structures reported in the Cambridge Structural Database (CSD).45 A search of the CSD (v 5.42, September 2021) found 2211 structures which contain trimesic acid (search parameters: 3D coordinates determined, R ≦ 0.1). Of these, there are 174 structures with trimesic acid in its neutral form (H3btc), and a further 461 structures in which the deprotonated trimesic acid (btc) is bound to a transition metal in bidentate coordinating mode, similar to that seen in 1. Eliminating those structures with metal clusters, more than one metallic element or with a second bridging linker left 89 unique structures in this dataset.
Within this dataset we found that there are four common motifs, which are illustrated in Fig. 8(a)–(d). The two most common are (a) the honeycomb network (as seen in 1), and (b) a flat tape which occurs when there are two types of metal coordination environment. Although some structures with the honeycomb motif (a) crystallize in hexagonal space groups, there are other examples which do not. In most cases the interlayer space is interdigitated by other ligands coordinated to the metal (similar to the DMF in 1). Structures with the tape motif in (b) also span a wide range of space groups, with the tapes sometimes interacting with one another through weak interactions such as hydrogen bonding. A small number of structures crystallize with motifs (c) and (d), a discrete 4-sided ring motif or a spiral motif with a 4-sided ring repeating unit. Only one structure in the dataset (MITRUD52) crystallized in a 2D coordination polymer with 4 metals and 4 btc linkers forming a square grid motif (Fig. 8(e)).
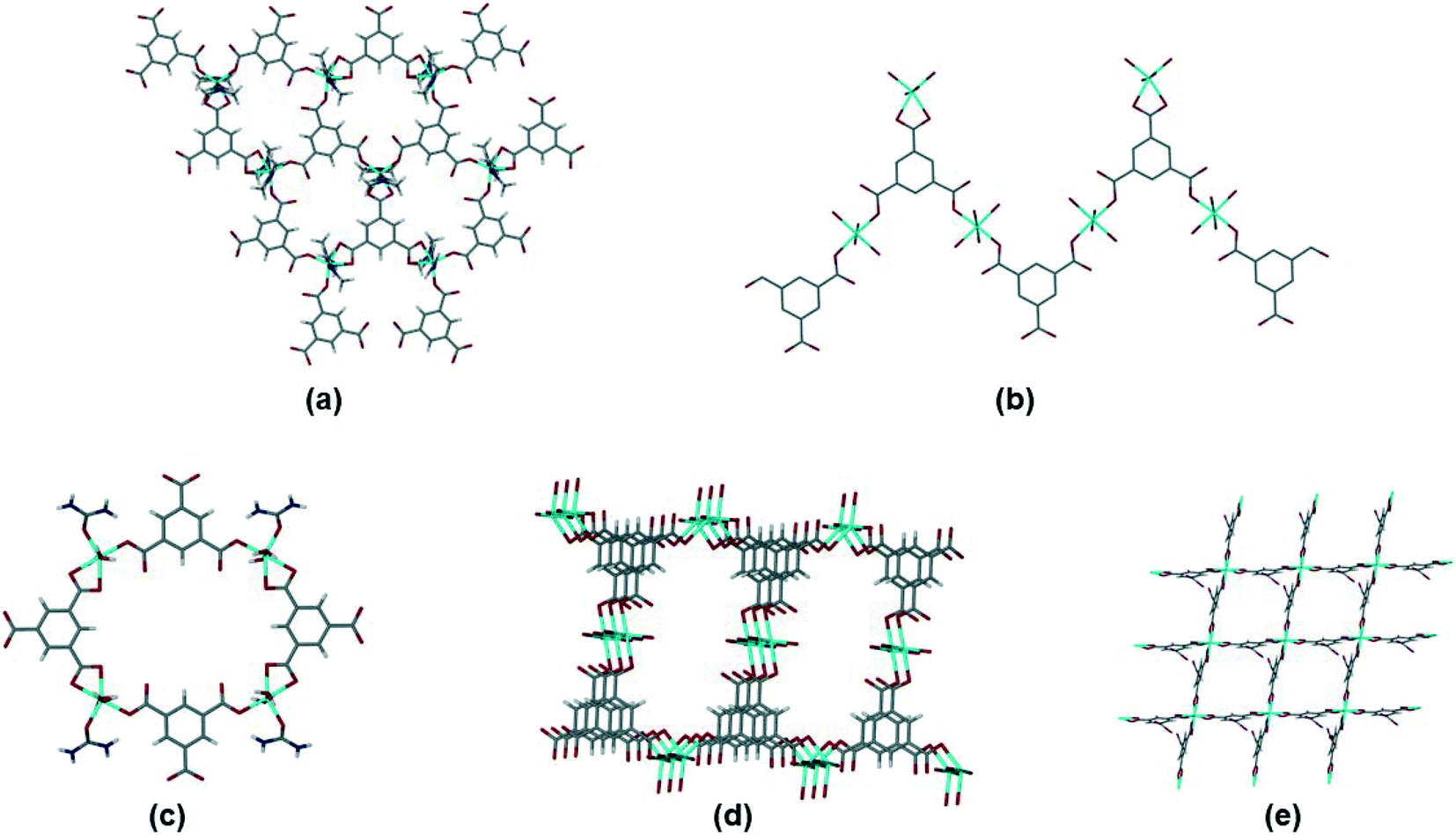 | ||
| Fig. 8 Motifs observed for simple btc coordination polymers. (a) Hexagonal network, illustrated by ADOLUB,48 (b) 2D tape, illustrated by TOPMIU,49 (c) discrete quadrilateral motif, illustrated by OXAWOB,50 (d) quadrilateral spiral network, illustrated by JEQNUP,51 and (e) square-grid network, illustrated by MITRUD.52 | ||
Conclusion
Compound 1, {[Co(btc)(DMF)2]·xDMF}n, is a coordination polymer in which 2D honeycomb layers pack in parallel with interdigitating coordinated DMF moieties. Voids between the DMF moieties allow the CP to act as a host compound, to include small molecule guests. This packing motif has been reported in other coordination complexes with trimesic acid, as confirmed by a survey of the CSD. Interestingly, the composition, unit cells and space groups of these complexes can differ markedly.Heating 1 causes the removal of first the guest DMF and then the coordinated DMF, with a concomitant change in cobalt(III) coordination geometry and crystal colour. On removal of the guest DMF, the structure (1-f) has collapsed layers with a reduction in crystallinity. Of the solvents used, only methanol and water (both the smallest and most polar of the solvents used) were capable of penetrating this structure to reconstitute the layers of the dried material. Intermolecular interactions between the guest solvents and the framework will certainly play an important role in this effect, but it is difficult to specify this role in the absence of fully refined crystal structures. Rather than harsh heating, we found that soaking crystals of 1 in solvents of varying properties allowed exchange of the guests, which could be monitored visually through solvatochromic effects. There is no evidence that competing cyano moieties displaced the coordinated DMF. In fact, crystals of 1 soaked in benzonitrile (2) showed the retention of the framework of 2D honeycomb layers with interdigitated DMF, albeit with a change in crystal symmetry and interlayer spacing.
Compound 1 is an example of a two-dimensional coordination polymer capable of responding to its chemical environment through a breathing-type mechanism. Similar behaviour has been observed in other 2D coordination polymers.21,22 Studies such as this report are useful for designing stimuli-responsive materials, as they give insight into types of potential interactions and how they might disrupt the properties of a system.
Author contributions
Conceptualization S. A. B.; data acquisition and analysis S. C. Z.; assistance with crystallography and analysis S. A. B., G. R.; supervision S. A. B., G. R.; writing – original draft S. C. Z., S. A. B.; all authors assisted with writing, review and editing.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
Funding for this research was provided by: National Research Foundation (grant No. 90495). SCZ thanks the National Research Foundation of South Africa and the University of Cape Town for financial support. Any conclusions expressed in this presentation are those of the authors and the NRF does not accept liability in this regard.References
- S. Panja and D. J. Adams, Chem. Soc. Rev., 2021, 50, 5165 RSC.
- T. Wang, E. Lin, Y.-L. Peng, Y. Chen, P. Cheng and Z. Zhang, Coord. Chem. Rev., 2020, 423, 213485 CrossRef CAS.
- Y. Wang, W. Wang, Z. Zhang and P. Li, Appl. Surf. Sci., 2022, 571, 151355 CrossRef CAS.
- H. Zhang, C. Gu, M.-S. Yao and S. Kitagawa, Adv. Energy Mater., 2021, 2100321 Search PubMed.
- S. Mukherjee and M. J. Zaworotko, Trends Chem., 2020, 2, 506 CrossRef CAS.
- C. M. Kane, A. Banisafar, T. P. Dougherty, L. J. Barbour and K. T. Holman, J. Am. Chem. Soc., 2016, 138, 4377 CrossRef CAS PubMed.
- M. du Plessis, V. I. Nikolayenko and L. J. Barbour, J. Am. Chem. Soc., 2020, 142, 4259 CrossRef PubMed.
- M. Baroncini, S. D'Agostino, G. Bergamini, P. Ceroni, A. Comotti, P. Sozzani, I. Bassanetti, F. Grepioni, T. M. Hernandez, S. Silvi, M. Venturi and A. Credi, Nat. Chem., 2015, 7, 634 CrossRef CAS PubMed.
- J. Lü and R. Cao, Angew. Chem., Int. Ed., 2016, 55, 9474 CrossRef PubMed.
- M. Mastalerz, Chem. – Eur. J., 2012, 18, 10082 CrossRef CAS PubMed.
- Y. Yang, X. He, P. Zhang, Y. H. Andaloussi, H. Zhang, Z. Jiang, Y. Chen, S. Ma, P. Cheng and Z. Zhang, Angew. Chem., 2020, 132, 3707 CrossRef.
- L. J. Barbour, Chem. Commun., 2006, 1163 RSC.
- M. Albrecht, M. Lutz, A. L. Spek and G. van Koten, Nat., 2000, 406, 970 CrossRef CAS PubMed.
- G. Minguez Espallargas, L. Brammer, J. van de Streek, K. Shankland, A. J. Florence and H. Adems, J. Am. Chem. Soc., 2006, 128, 9584 CrossRef CAS PubMed.
- C. A. Ndamyabera, S. C. Zacharias, C. L. Oliver and S. A. Bourne, Chemistry, 2019, 1, 111 CrossRef.
- G. Mehlana and S. A. Bourne, CrystEngComm, 2017, 19, 4238 RSC.
- N. Nijem, H. Wu, P. Canepa, A. Marti, K. J. Balkus, T. Thonhauser, J. Li and Y. J. Chabal, J. Am. Chem. Soc., 2012, 134, 15201 CrossRef CAS PubMed.
- L. Abylgazina, I. Senkovska, S. Ehrling, V. Bon, P. St Petkov, J. D. Evans, S. Krylova, A. Krylov and S. Kaskel, CrystEngComm, 2021, 23, 538 RSC.
- K. Davies, S. A. Bourne and C. L. Oliver, Cryst. Growth Des., 2012, 12, 1999 CrossRef CAS.
- H. Wang, M. Warren, J. Jagiello, S. Jensen, S. K. Ghose, K. Tan, L. Yu, T. J. Emge, T. Thonhauser and J. Li, J. Am. Chem. Soc., 2020, 142, 20088 CrossRef CAS PubMed.
- F.-X. Coudert, C. Mellot-Draznieks, A. H. Fuchs and A. Boutin, J. Am. Chem. Soc., 2009, 131, 3442 CrossRef CAS PubMed.
- G. Mehlana, G. Ramon and S. A. Bourne, CrystEngComm, 2014, 16, 8160 RSC.
- R. E. Melendez, C. V. K. Sharma, M. J. Zaworotko, C. Bauer and R. D. Rogers, Angew. Chem., Int. Ed. Engl., 1996, 35, 2213 CrossRef CAS.
- S. Chatterjee, V. R. Pedireddi, A. Ranganathan and C. N. R. Rao, J. Mol. Struct., 2000, 520, 107 CrossRef CAS.
- N. Sahiner, K. Sel, O. F. Ozturk, S. Demirci and G. Terzi, Appl. Surf. Sci., 2014, 314, 663 CrossRef CAS.
- K. Sel, S. Demirci, E. Meydan, S. Yildiz, O. F. Ozturk, H. Al-Lohedan and N. Sahiner, J. Electron. Mater., 2015, 44, 136 CrossRef CAS.
- S. Konar, P. S. Mukherjee, E. Zangrando, M. G. B. Drew, C. Diaz, J. Ribas and N. R. Chaudhuri, Inorg. Chim. Acta, 2005, 358, 29 CrossRef CAS.
- P. Lof, Minerals of the World, Elsevier Science, Netherlands, 1983 Search PubMed.
- Soft Imaging System GmbH: Digital Solutions for Imaging and Microscopy, Version 3.1 for Windows, Muenster, Germany, 1987–2000 Search PubMed.
- TA Instruments, Universal Analysis 2000 for Windows, Version 47A, TA Instruments-Waters LLC, New Castle, DE, USA, 1998–2009 Search PubMed.
- TA Instruments, TRIOS, TA Instruments – Waters LLC, New Castle, Delaware, 2016 Search PubMed.
- C. F. Macrae, I. J. Bruno, J. A. Chisholm, P. R. Edgington, P. McCabe, E. Pidcock, L. Rodriguez-Monge, R. Taylor, J. van de Streek and P. A. Wood, J. Appl. Crystallogr., 2008, 41, 466 CrossRef CAS.
- XPREP, Data Preparation and Reciprocal Space Exploration, 5.1, Bruker Analytical X-ray Systems, Madison, Wisconsin, USA, USA, 1997 Search PubMed.
- Cell_now, Bruker AXS Inc, Madison, Wisconsin, USA, 2008 Search PubMed.
- G. M. Sheldrick, TWINABS, University of Gottingen, Germany, 2001 Search PubMed.
- SAINT 7.60a, Bruker AXS Inc, Madison, Wisconsin, USA, 2006 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3 Search PubMed.
- L. J. Barbour, J. Appl. Crystallogr., 2020, 53, 1141 CrossRef CAS.
- A. L. Spek, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 9 CrossRef CAS PubMed.
- G. M. Sheldrick, SADABS, Version 2.05, University of Göttingen, Germany, 2007 Search PubMed.
- A. G. Orpen, L. Brammer, F. H. Allen, O. Kennard, D. G. Watson and R. Taylor, J. Chem. Soc., Dalton Trans., 1989, S1–S83 RSC.
- S. Murov, http://murov.info/orgsolvents.htm, Normalised from measurements of solvent shifts of absorption spectra, extracted from C. Reichardt, Solvents and Solvent Effects in Organic Chemistry, Wiley-VCH, 3rd edn, 2003 Search PubMed.
- F. A. Cotton and G. Wilkinson, Advanced Inorganic Chemistry, A Comprehensive Text, Wiley Interscience Publishers, Inc., London, 3rd edn, 1972 Search PubMed.
- M. R. Ward and I. D. H. Oswald, Crystals, 2020, 10, 1098 CrossRef CAS.
- C. R. Groom, I. J. Bruno, M. P. Lightfoot and S. C. Ward, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72, 171 CrossRef CAS PubMed.
- M. R. J. Elsegood and D. M. Collins, CSD Communication, CCDC 2055868, 2021.
- S. Chatterjee, V. R. Pedireddi, A. Ranganathan and C. N. R. Rao, J. Mol. Struct., 2000, 520, 107 CrossRef CAS.
- C. Gao, S. Liu, L. Xie, Y. Ren, J. Cao and C. Sun, CrystEngComm, 2007, 9, 545 RSC.
- O. M. Yaghi, H. Li and T. L. Groy, J. Am. Chem. Soc., 1996, 118, 9096 CrossRef CAS.
- Q.-Q. Xu, B. Liu, L. Xu and H. Jiao, CSD Communication, CCDC 1510315, 2016.
- M. Xue, G.-S. Zhu, L.-F. Wang, Q.-R. Fang, J.-Y. Sun, F.-X. Sun, X.-D. Guo, X.-J. Zhao, G. Tian and S.-L. Qiu, Gaodeng Xuexiao Huaxue Xuebao, 2005, 26, 2211 CAS.
- Q. Fang, G. Zhu, M. Xue, Z. Wang, J. Sun and S. Qiu, Cryst. Growth Des., 2008, 8, 319 CrossRef CAS.
Footnotes |
| † Dedicated to Paul R. Raithby, celebrating a career in inorganic and organometallic chemistry, on the occasion of his 70th birthday. |
| ‡ Electronic supplementary information (ESI) available. CCDC 2133533. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d2ce00039c |
| § Present address: University of Gothenburg, Department of Chemistry and Molecular Biology, Kemivägen 10, SE-412 96, Gothenburg, Sweden. |
| ¶ [Co(C9H3O6)(C3H7NO)2]n, trigonal, P, a = b = 16.623(6), c = 14.071(15) Å, R1 = 0.223. |
| This journal is © The Royal Society of Chemistry 2022 |