
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOrganocatalyst based cross-catalytic system†
Marieke J.
Veenstra
 and
Syuzanna R.
Harutyunyan
*
and
Syuzanna R.
Harutyunyan
*
Stratingh Institute for Chemistry, University of Groningen, Nijenborgh 4, Groningen, 9747 AG, The Netherlands. E-mail: s.harutyunyan@rug.nl
First published on 18th November 2022
Abstract
We present our design of a cross-catalytic system based on organocatalysis. The system features two organic reactions, namely a deprotection reaction of Fmoc protected proline and a Mannich reaction between acetone and dihydroisoquinoline. The products of these two reactions, proline and a tetrahydroisoquinoline, respectively, are capable of reciprocal reaction rate enhancement. Detailed kinetic studies of the system and seeding experiments support the cross-catalytic relationship in the reaction network.
Complex chemical reaction networks with multiple interconnections are at the heart of biological systems. Such networks give rise to the vast majority of biological functions and allow biological systems to adapt and respond rapidly to the environment. With the goal of better understanding and mimicking biological systems, as well as building synthetic machinery, there has been significant effort in exploiting synthetic chemistry to study chemical reaction networks and to build complex systems with emergent properties.1 Molecular self-replication is one such property that can emerge from complex reaction networks and has attracted a lot of interest.2 Since autocatalytic and cross-catalytic processes are often invoked in rationalising molecular self-replication, significant effort has also been spent in designing non-enzymatic analogues of such reactions.3–5
The first reported examples of non-enzymatic auto-catalysis were based on oligonucleotides,6 RNA7 and peptides.8 More recently, dynamic covalent libraries in combination with supramolecular chemistry have emerged as efficient tools for constructing replicating system with peptides.9 Small molecule-based auto-catalysis was also described.10–13
Apart from autocatalytic reactions, cross-catalytic systems have been described as well, for example using nucleotide14 and peptide based chemistries.15 Furthermore, self-replicating macrocycles have been shown to be capable of parasitic behaviour.16 Cross-catalytic reaction networks involving small organic molecules have also been realised using synthetic molecules, namely cross-catalytic amide formation17 and cycloaddition reactions.18
The reported examples of cross-catalytic systems commonly make use of reactive sites and template effects to drive catalytic processes, similar to the majority of examples describing auto-catalytic behaviour.3 Template effects and rate accelerations are achieved thanks to the presence, both in the reactants and the resulting products, of recognition sites, which thus are critical elements in such systems. The necessity of such complementary recognition sites, as well as orthogonal reactive centers, results in synthetically complex systems that are challenging and difficult to design and synthesise. In contrast, simple cross-catalytic systems that rely on direct electronic activation of the reaction partners and make use of two orthogonal reactions are scarce.19
Here, we describe our design of a small-molecule, non-templating, cross-catalytic system where reciprocal catalytic rate accelerations are achieved through two different mechanistic pathways: enamine and base catalysis.
In order to design a non-templating cross-catalytic system, two orthogonal reactions are required that form products capable of inducing a rate acceleration for each other's formation. Once one of the two products (i.e. catalysts) is formed, it will increase the rate of formation of the second product, which in turn increases the rate of the first reaction.
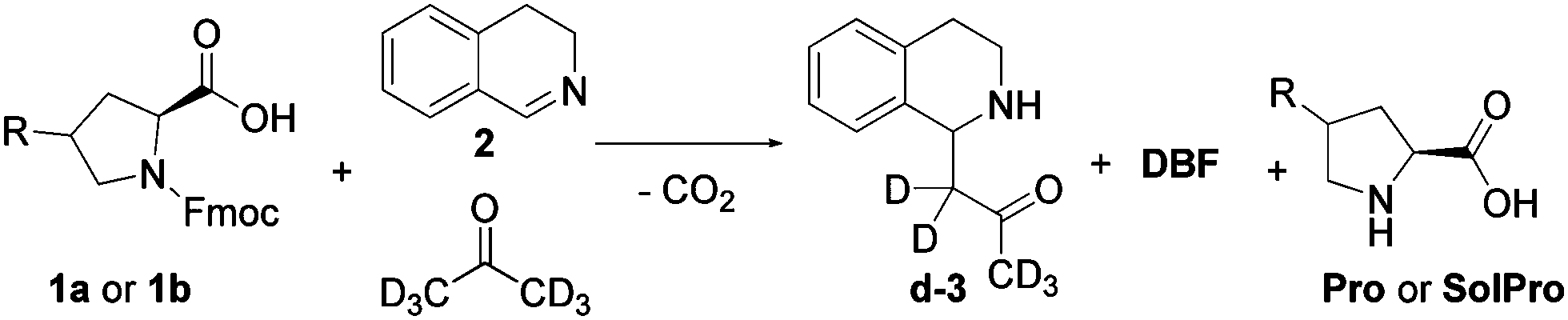
To build the cross-catalytic system (Scheme 1a) we selected as a first reaction the base catalysed deprotection reaction of Fmoc protected amino acids, in this case of proline (Pro).20 We selected Pro because it is known to be an excellent catalyst for various types of transformations, such as aldol reactions, Michael additions and, most importantly, for Mannich type reactions via the enamine mechanism.21,22
 | ||
| Scheme 1 (a) Design of the cross-catalytic system used in this study; (b) soluble analogue of proline SolPro and its Fmoc-protected analogue 1b; (c) the Mannich reaction: addition of acetone-d6 to imine 2 catalysed by Pro. | ||
Especially the latter reaction is attractive for designing a cross catalytic system, because the Mannich reaction, in addition to being catalysed by Pro, results in a secondary amine product, the basicity of which can be tuned. Because the first reaction, Fmoc deprotection generating Pro, is known to be catalysed by amine bases23 we anticipated that the Mannich product might be able to catalyse the first reaction (Scheme 1a, top). For the Pro catalysed reaction, we chose the addition of acetone-d6 to 3,4-dihydroisoquinoline (2) to generate the Mannich product d-3 (Scheme 1a, bottom). Deuterated acetone was used as the solvent in order to directly follow the reaction by 1H-NMR. For the cross-catalytic network to work, a trigger that starts the reaction is needed. We hypothesised that substrate 2 of the Mannich reaction can serve as this trigger as it is basic as well, but less so than Mannich product d-3. The difference in basicity is expected to translate in a much lower deprotection rate enabled by 2, allowing deprotection induced by d-3 to start dominating as its concentration increases over the course of the reaction. Thus, the deprotection rate was anticipated to be very slow at first, when mostly 2 is present, and gradually increase due to the formation of the more basic d-3.
Since the solubility of Pro in organic solvents, in this case acetone, is limited and might result in non-homogeneous reaction mixtures that could influence our reaction network, we also studied the cross-catalytic network using a more soluble analogue of proline. For this purpose, trans-hydroxy proline was modified by diisopropyloctylsiloxy (DIPOS) to generate SolPro and the corresponding Fmoc protected derivative 1b (Scheme 1b).
Before investigating the cross-catalytic system, we analysed the individual reactions. First, the Mannich reaction (Scheme 1c) between 2 and acetone-d6 was performed to see if the reaction takes place without a catalyst, as a spontaneous reaction would defeat the purpose of our cross catalytic system. However, no product formation was observed after 48 hours of stirring at room temperature. On the other hand, in the presence of 10 mol% of Pro (not all the catalyst was solubilised) or SolPro the Mannich product d-3 was formed, with full substrate consumption in 1–2 days depending on the catalyst. The isolated yields of the Mannich product were always around 60%, most likely due to side product formation or retro-Mannich. Next, we studied the reaction kinetics for both catalysts, Pro and SolPro, using 500 MHz 1H-NMR spectroscopy. In the kinetic experiments the reaction partners ([2] = 133 mM) were allowed to react in acetone-d6 at 25 °C and the reaction progress was continuously monitored in situ every 5 or 10 minutes over a period of 12 hours. When 10 mol% of Pro was used, the reaction proceeded slowly, resulting in only 72% conversion of imine 2 after 12 hours and only 22% NMR yield of product d-3. Using 10 mol% of the more soluble SolPro, a higher reaction rate was observed with 95% conversion after only 4 hours. However, the NMR yield of product d-3 was still only 29% due to side reactions caused by the decomposition of the Mannich product d-3 in time. In order to compare the catalytic performance of Pro and SolPro more accurately, the solubility of Pro was determined and found to be 2.3 mM (±0.1). With this value in hand, we carried out the reactions with Pro and SolPro at the concentration range in which Pro is soluble (2.3 mM, 2 mol%) and observed similar catalytic performance in terms of the rate of conversion of substrate 2, although the rate of formation of product d-3 was slightly higher with Pro (Fig. 1a and b).
 | ||
| Fig. 1 Kinetic experiments examining individual reactions involved in cross-catalytic system at 25 °C in acetone-d6 as the solvent. (a) and (b) Mannich reaction between acetone-d6 and 2 (133 mM) catalysed by 2 mol% of Pro (red circles) and 2 mol% of SolPro (blue squares); (c) the deprotection of 1a (red circles) and 1b (blue squares) (133 mM) with 1 equiv. of d-3.24 | ||
Subsequently, we examined the individual deprotection reactions of the Fmoc protected proline-type catalysts ([1a–b] = 133 mM) with 1 equivalent of the different bases. For this deprotection reaction, only the formation of dibenzofulvene (DBF) can be followed by 1H-NMR (singlet at 6.25 ppm) as all characteristic peaks of 1a in 1H-NMR overlap with the signals corresponding to the protons of DBF and Pro. As the deprotection with imine 2 in acetone-d6 would be equivalent to the cross-catalytic network itself, it was studied in DMSO-d6 at 25 °C. Both the imine 2 (1 equiv.) and Pro/SolPro (1 equiv.) were capable of deprotecting 1a and 1b in this solvent, although very slowly with approximately 2% conversion after 12 hours. When 1 equiv. of the Mannich product d-3 was used, the deprotection was faster, affording a conversion of approximately 10% after 2 hours, followed by a rate decrease. We believe that the rate decreases because the Mannich product d-3, which serves as the base catalyst, is unstable and known to undergo a retro-Mannich reaction in DMSO-d6 under these conditions (see ESI†). Changing the solvent to acetone-d6 resulted in slower formation of DBF compared to the reaction in DMSO-d6, but the rate was observed to increase over time (Fig. 1c). We observed an induction period for the deprotection of 1a and 1b in the presence of d-3 (Fig. 1c), which could indicate that the process is autocatalytic. To verify this, we added the reaction product (Pro) from the start of the reaction as this should result in a rate increase and a shortening of the lag period in case of autocatalysis. For this experiment the system with SolPro was used. Deprotection of 1b by SolPro in acetone-d6 was not observed, as no DBF was formed even after 12 hours. Having excluded the possibility of autocatalysis, we hypothesised that a pre-equilibrium might induce the lag period. As 1a and 1b both have a free carboxylic acid, an acid-base equilibrium should be established when the base (d-3) is added (Scheme 2a). The acid-base reaction will take place at the start of the reaction and once the equilibrium is established a small amount of d-3 which has not been protonated will start the deprotection. As Pro can exist as a zwitterion, the acid–base equilibrium will be shifted as soon as it is produced, resulting in increased availability of the free base (d-3). To test this hypothesis, different Fmoc protected amino acids were deprotected with 1 equivalent of d-3 (Scheme 2b). Amino acids and amino acid derivatives like glycine and proline-tetrazole are expected to show similar behaviour as Pro, whereas a disappearance of the lag period is expected for the methyl esters of these species.
 | ||
| Scheme 2 (a) Hypothesised acid-base equilibrium. (b) Deprotection of various Fmoc-protected amino acids in the presence of equimolar amounts of d-3. Conversion to DBF after 12 hours is indicated. | ||
The deprotection of Fmoc-glycine (1c) and 1a does indeed show similar behaviour, but the deprotection of Fmoc-Proline tetrazole (1d) catalyst was slightly faster. The latter can be attributed the tetrazole derivative being less acidic than proline itself. When the methylated analogue of 1c (1c′) was used, the deprotection did not show a lag period and full conversion to DBF was observed after 12 hours.
For the methylated product 1a′ a lag period of 1 hour was observed with full conversion to DBF after 10 hours. As the synthesis of acid free methyl L-prolinate proved difficult, we cannot ascertain whether the lag period observed is due to traces of acid in 1a′. The lag period in the deprotection of 1a′ with d-3 was increased to 4 hours when traces of sulphuric acid were added, showing the importance of acidic protons on the deprotection rate and the duration of the lag period and further supporting the hypothesis of the involvement of an acid-base equilibrium.
Having studied the individual reactions, the deprotection and Mannich reaction, we shifted our focus to the study of the cross-catalytic system constructed from these reactions (Scheme 3). First, 1a and 2 were allowed to react in acetone-d6 at 25 °C in equimolar amounts. The reaction progress was checked continuously by in situ monitoring every 5 or 10 minutes by 500 MHz 1H-NMR spectroscopy over a period of 12 hours, with the reaction concentrations determined relative to the internal standard, HMDSO. After this time solids were observed in the NMR-tube. Filtration of the sample and analysis of the solid confirmed that this was Pro, which, as mentioned before, is poorly soluble in acetone. Due to the ongoing deprotection the concentration of Pro continuously increases, causing precipitation when the solubility threshold is reached.
 | ||
| Scheme 3 Reaction conditions for the cross-catalytic reaction used in this study. 1a or 1b (133 mM) is reacted with 2 (133 mM) in acetone-d6 at 25 °C for 12 hours. | ||
The conversion of 2 in the cross-catalytic reaction with 1a was found to show a small lag period of approximately 30 minutes before the reaction rate starts to increase (Fig. 2a). After approximately 6 hours the conversion of 2 is full, which is faster than expected based on the reaction rate of the individual Mannich reaction. In the control experiments, we found that the reaction rate of the Mannich reaction was significantly increased when 1 equivalent of AcOH was added. The 1 equivalent of acidic protons present in the cross-catalytic reaction (from 1a) can therefore explain this faster reaction rate. As the peak of d-3 shifts over time and increasingly overlaps with a peak of a side product, the formation of the Mannich product could be followed only for the first 4 hours. We observed a lag period in the formation of d-3 (Fig. 2b), followed by rate enhancement. Furthermore, the NMR yield of Mannich product d-3 decreases after longer reaction times, potentially due to decomposition or further reactions. The formation of DBF was found to be slow and only reached 12% NMR yield after 12 hours (Fig. 2c).
 | ||
| Fig. 2 Kinetic experiments examining the cross catalytic reactions of 1a and 1b (133 mM) with 2 (133 mM) and the corresponding seeding reactions in acetone-d6 at 25 °C. Concentrations over time were determined by 1H-NMR. | ||
Next, we examined the cross-catalytic reaction between 1b and 2 (Fig. 2c and d). Here the lag period was found to be shorter compared to the lag period with 1a. In addition, the reaction reached higher reaction rates. Similar kinetic behaviour was observed for the formation of Mannich product d-3. Deprotection of 1b under these reaction conditions is slightly faster than deprotection of 1a under the same conditions. As the catalytic performance of Pro and SolPro was shown to be similar and the deprotection rates are not significantly different for 1a and 1b (vide supra), the faster reaction observed in the cross-catalytic deviates from the characteristics of the individual reactions. As 1a was always used from a commercial source while 1b was synthesised in-house, we decided to synthesise 1a as well and repeat the experiment.
The kinetics of the reaction between self made 1a and 2 were followed for 12 hours (Fig. S7, ESI†) and a significant increase in rate was observed, comparable to the reaction rate in the reaction with 1b. These results suggest the presence of traces of Pro and SolPro in 1a and 1b respectively, which was subsequently confirmed by ninhydrin stain.
In order to corroborate cross-catalytic behaviour, seeding experiments with both proposed catalysts were performed in the cross-catalytic systems. When 10 mol% of Pro was added at the start of the cross-catalytic reaction with 1a (not all the catalyst is solubilised), we were pleased to see that the lag period in both the conversion of 2 and the production of d-3 was shortened. Although no significant effect on the formation of DBF is observed under these conditions (Fig. 2a–c, orange circles), a small decrease in rate could be noted. When 20 mol% of d-3 (proline free) was added at the start of the reaction with 1a, the lag periods for the conversion of 2 and the formation of d-3 shortened (Fig. 2a–c, green circles) further confirming the occurrence of cross-catalysis. A small increase in the rate of formation of DBF was also observed.
The same seeding experiments were performed for the reaction of 1b with 2 (Fig. 2d–f) and increased Mannich reaction rates were observed for the SolPro seed (purple squares). The formation of DBF was clearly slower due to the introduction of more acidic protons at the start of the reaction. No significant change in rate was observed upon seeding with 20 mol% of d-3. The effect of the addition of 20 mol% of d-3 is negligible since the rate of this process is already higher without any additional seeding. Nevertheless, when 100 mol% of d-3 was added, a clear increase in rate was observed, albeit with the formation of several side products.
To conclude, we have shown the first cross-catalytic system based on direct activation of the reaction partners using mechanistic principles of organocatalysis. We thank Gravity program 024.001.035 for funding.
Conflicts of interest
There are no conflicts to declare.Notes and references
- (a) R. F. Ludlow and S. Otto, Chem. Soc. Rev., 2008, 37, 101 RSC; (b) G. Ashkenasy, T. M. Hermans, S. Otto and A. F. Taylor, Chem. Soc. Rev., 2017, 46, 2543 RSC.
- (a) A. Vidonne and D. Philp, Eur. J. Org. Chem., 2009, 593 CrossRef CAS; (b) E. Mattia and S. Otto, Nat. Nanotechnol., 2015, 10, 111 CrossRef CAS PubMed.
- A. J. Bisette and S. P. Fletcher, Angew. Chem., Int. Ed., 2013, 52, 12800 CrossRef PubMed.
- A. I. Hanopolskyi, V. A. Smaliak, A. I. Novichkov and S. N. Semenov, ChemSystemsChem, 2021, 3, e2000026 CrossRef CAS.
- D. G. Blackmond, Angew. Chem., Int. Ed., 2009, 48, 386 CrossRef CAS PubMed.
- (a) G. von Kiedrowski, Angew. Chem., Int. Ed. Engl., 1986, 25, 932 CrossRef; (b) W. D. Zielinski and L. E. Orgel, Nature, 1987, 327, 346 CrossRef CAS PubMed.
- N. Paul and G. F. Joyce, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12733 CrossRef CAS PubMed.
- D. H. Lee, J. R. Granja, J. A. Martinez, K. Severing and M. R. Ghadri, Nature, 1996, 382, 525 CrossRef CAS PubMed.
- (a) J. M. A. Carnall, C. A. Waudby, A. M. Belenguer, M. C. A. Stuart, J. J. P. Peyralans and S. Otto, Science, 2010, 327, 1502 CrossRef CAS PubMed; (b) B. Rubinov, N. Wagner, M. Matmor, O. Regev, N. Ashkenasy and G. Ashkenasy, ACS Nano, 2012, 6, 7893 CrossRef CAS PubMed.
- (a) T. Tjivikua, P. Ballester and J. Rebek Jr., J. Am. Chem. Soc., 1990, 112, 1249 CrossRef CAS; (b) V. Rotello, J. Hong and J. Rebek Jr., J. Am. Chem. Soc., 1991, 113, 9422 CrossRef CAS.
- B. Wang and I. O. Sutherland, Chem. Commun., 1997, 1495 RSC.
- Selected examples: (a) V. C. Allen, D. Philp and N. Spencer, Org. Lett., 2001, 3, 777 CrossRef CAS PubMed; (b) J. M. Quayle, A. M. Z. Slawin and D. Philp, Tetrahedron Lett., 2002, 43, 7229 CrossRef CAS; (c) E. Kassianidis and D. Philp, Angew. Chem., Int. Ed., 2006, 45, 6344 CrossRef CAS PubMed; (d) T. Kosikova, N. I. Hassan, D. B. Cordes, A. M. Z. Slawin and D. Philp, J. Am. Chem. Soc., 2015, 137, 16074 CrossRef CAS PubMed; (e) Y. Feng and D. Philp, J. Am. Chem. Soc., 2021, 143, 17029 CrossRef CAS PubMed.
- (a) K. Soai, S. Niwa and H. Hori, J. Chem. Soc. Chem. Commun., 1990, 14, 982–983 RSC; (b) K. Soai, T. Shibata, H. Morioka and K. Choji, Nature, 1995, 378, 767 CrossRef CAS; (c) K. Soai, I. Sato, T. Shibata, S. Komiqy, M. Hayashi, Y. Matsueda, H. Imamura, T. Hayase, H. Morioka, H. Tabira, J. Yamamoto and Y. Kowata, Tetrahedron: Asymmetry, 2003, 14, 185 CrossRef CAS.
- (a) D. Sievers and G. von Kiedrowski, Chem. – Eur. J., 1998, 4, 629 CrossRef CAS; (b) D. Kim and G. F. Joyce, Chem. Biol., 2004, 11, 1505 CrossRef CAS PubMed.
- (a) S. Yao, I. Ghosh, R. Zutshi and J. Chmielewski, Nature, 1998, 396, 447 CrossRef CAS PubMed; (b) K. Severin, D. H. Lee, J. A. Martinez, M. Vieth and M. R. Ghadiri, Angew. Chem., Int. Ed., 1998, 37, 126 CrossRef CAS.
- (a) M. Altay, Y. Altay and S. Otto, Angew. Chem., Int. Ed., 2018, 57, 10564 CrossRef CAS PubMed; (b) Y. Altay, M. Altay and S. Otto, Chem. – Eur. J., 2018, 24, 11911 CrossRef CAS PubMed.
- (a) R. J. Pieters, I. Huc and J. Rebek Jr., Angew. Chem., Int. Ed. Engl., 1994, 33, 1579 CrossRef; (b) R. J. Pieters, I. Huc and J. Rebek Jr., Tetrahedron, 1995, 51, 485 CrossRef CAS.
- Selected examples: (a) K. Eleftherios and D. Philp, Chem. Commun., 2006, 4072 Search PubMed; (b) C. C. Robertson, H. W. Mackenzie, T. Kosikova and D. Philp, J. Am. Chem. Soc., 2018, 140, 6832 CrossRef CAS PubMed; (c) J. Huck, T. Kosikova and D. Philp, J. Am. Chem. Soc., 2019, 141, 13905 CrossRef CAS PubMed.
- P. Hui, M. Branca, B. Limoges and F. Mavré, Chem. Commun., 2021, 57, 11374 RSC.
- M. Bodanszky and A. Bodanszky, The Practice of Peptide Synthesis, Springer-Verlag, Berlin, 1994 Search PubMed.
- B. List, Chem. Commun., 2006, 819 RSC.
- B. List and K. Maruoka, Asymmetric Organocatalysis 1: Lewis Base and Acid Catalysts, Georg Thieme, Stuttgart, 2012 Search PubMed.
- P. G. M. Wuts and T. W. Greene, Green's Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., 2007 Search PubMed.
- The data points in Fig. 1c in the first 30 minutes of the experiment are spuriously high and not representative of the actual DBF concentration; this is caused by a broad peak close to the singlet of DBF at 6.25 ppm which is partly integrated together with this singlet.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2cc05610k |
| This journal is © The Royal Society of Chemistry 2022 |