
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChemogenetics of cell surface receptors: beyond genetic and pharmacological approaches
Yuta
Miura
 ,
Akinobu
Senoo
,
Tomohiro
Doura
and
Shigeki
Kiyonaka
*
,
Akinobu
Senoo
,
Tomohiro
Doura
and
Shigeki
Kiyonaka
*
Department of Biomolecular Engineering, Graduate School of Engineering, Nagoya University, Nagoya 464-8603, Japan. E-mail: kiyonaka@chembio.nagoya-u.ac.jp
First published on 27th January 2022
Abstract
Cell surface receptors transmit extracellular information into cells. Spatiotemporal regulation of receptor signaling is crucial for cellular functions, and dysregulation of signaling causes various diseases. Thus, it is highly desired to control receptor functions with high spatial and/or temporal resolution. Conventionally, genetic engineering or chemical ligands have been used to control receptor functions in cells. As the alternative, chemogenetics has been proposed, in which target proteins are genetically engineered to interact with a designed chemical partner with high selectivity. The engineered receptor dissects the function of one receptor member among a highly homologous receptor family in a cell-specific manner. Notably, some chemogenetic strategies have been used to reveal the receptor signaling of target cells in living animals. In this review, we summarize the developing chemogenetic methods of transmembrane receptors for cell-specific regulation of receptor signaling. We also discuss the prospects of chemogenetics for clinical applications.
 Yuta Miura | Yuta Miura received his Bachelor of Engineering from Nagoya University under the guidance of Prof. Shigeki Kiyonaka in 2020. Working in a chemogenetics research group, he spearheaded coordination based chemogenetics efforts on the way to earning his Master of Engineering. |
 Akinobu Senoo | Akinobu Senoo received his PhD degree from University of Tokyo under the guidance of Prof. Kouhei Tsumoto in 2021. He then joined the group of Prof. Shigeki Kiyonaka at Nagoya University as a postdoc researcher. His current research interest is chemogenetic regulation of membrane receptors. |
 Tomohiro Doura | Tomohiro Doura received his PhD from Kyushu University under Prof. Shinsuke Sando in 2012. He then joined the group of Prof. Yasuteru Urano at the University of Tokyo as a postdoc. In 2014-2019, he joined several groups as postdoc and Assistant Professor. In 2019, he joined the group of Prof. Shigeki Kiyonaka as an Assistant Professor in Nagoya University. His current research is focused on the chemogenetic regulation and optical control of receptors for neuroscience and medicine. |
 Shigeki Kiyonaka | Shigeki Kiyonaka obtained his PhD degree from Kyushu University under the guidance of Prof. Seiji Shinkai and Prof. Itaru Hamachi in 2002. He then joined Prof. Yasuo Mori's group as a postdoc and an Assistant Professor in Kyoto University. He was promoted to Associate Professor of Prof. Hamachi's group in 2012 in Kyoto University. In 2020, he started his independent career as full Professor in Nagoya University. His current research interest is chemogenetics regulation of cellular proteins including neurotransmitter receptors. |
1. Introduction
The fate of cells is determined by the integration of extracellular signals in animals. These signals are transmitted from the extracellular space into the intracellular area via various kinds of transmembrane receptors that are mainly categorized into G protein-coupled receptors (GPCRs), receptor tyrosine kinases (RTKs), and ligand-gated ion channels (LGICs). Spatiotemporal regulation of receptor signaling is crucial for cellular functions in the body. Thus, it is highly desired to understand the physiological roles of each receptor not only in cultured cells, but also in living animals.Genetic engineering (i.e., molecular genetics) is widely used to understand the physiological roles of transmembrane receptors. To knockout a target receptor, deletion of the corresponding gene segment or a frameshift mutation has been applied. Alternatively, important amino acid residues in the receptors are mutated to other residues in the gene knock-in strategy. Recent progress in genome-editing techniques, such as CRISPR-Cas9, has facilitated genetic engineering.1 Moreover, cell-specific regulation of target receptors has been achieved using the Cre-loxP system, which is known as the conditional gene regulation approach.2 Although off-target effects can occur in genome editing, most genetic engineering methods allow regulation of a target receptor with high selectivity (Table 1). As a drawback of genetic approaches, it is difficult to control protein functions with high temporal resolution. In most genetic approaches in animals, mutations are introduced in the early embryonic stage. Consequently, the expression of related genes is affected during development to compensate for the loss-of-function of the target receptor.
| Category | Protein selectivity | Cell specificity | Temporal resolution | Non-invasive | Spatial resolution | Natural functiona | Clinical application |
|---|---|---|---|---|---|---|---|
| ++: excellent, +: good or kept, −: poor or lost.a “Natural function” is an index of whether the original receptor function is retained. | |||||||
| Genetic engineering (molecular genetics) | ++ | ++ | − | + | + | − | + or − |
| Optogenetics | ++ | ++ | ++ | − | + | − | + or − |
| Chemical genetics (pharmacology) | + or − | − | + | ++ | − | ++ | ++ |
| Chemogenetics | ++ | ++ | ++ or + | + | + | + or − | + or − |
As a genetic approach to control transmembrane signaling with high temporal resolution, “optogenetics” has been well used recently.3 Optogenetics was first introduced by Deisseroth's group, in which channelrhodopsin, a light-gated cation channel from Chlamydomonas reinhardtti, is expressed in target cells.4 Channelrhodopsin conducts cations upon blue light illumination, which allows depolarization of excitatory cells such as neurons. Instead, halorhodopsin from Natronomonas pharaonis conducts chloride ions into the cytoplasm to hyperpolarize the cells by yellow light.3 Regarding photo control of GPCR signaling, optoXRs have been developed.5 OptoXRs are chimeric GPCRs in which intracellular loops of bovine rhodopsin were replaced with those of Gq-coupled α1a-adrenergic or Gs-coupled β2-adrenergic receptors (β2ARs), which activate G protein signaling after photo-irradiation. Additionally, light-sensitive domains, such as the light-oxygen-voltage-sensing (LOV) domain, would be powerful to control protein function on the transmembrane, which is described in “Section 4.2” in detail. Because of cell-specific expression of photo-responsive proteins using cell type-specific promoters in tissues or animals, optogenetics allows to control cellular signaling with high spatial and temporal resolutions (Table 1), which has led to breakthroughs in neuroscience, cardiology, and cell biology.3 However, considering that cellular signaling was conducted via engineered receptors derived from other species, this technique would be unsuitable to understand the physiological roles of cell surface receptors. Moreover, optical fibers need to be implanted invasively when optogenetics are applied to living animals such as mice and large animals.
Application of chemicals is a potential approach to control receptor functions in a non-invasive fashion. The study of biological systems using small chemicals instead of molecular genetics has been termed “chemical genetics”.6 As is the case with the terminology of genetics, chemical genetics is divided into two approaches: forward and reverse (for details, see comprehensive review articles).7,8 In “reverse chemical genetics”, a small compound that selectively binds to the target gene product is used to regulate receptors endogenously expressed in cells (Table 1). Thus, reverse chemical genetics is synonymous with molecular pharmacology. Although powerful, the selectivity of chemicals to target molecules is insufficient in many cases compared with genetic approaches. Moreover, selective delivery of small chemicals to target cells or tissues in animals is challenging.
To overcome the insufficient selectivity in chemical approaches, the target protein is genetically engineered to interact with a designed chemical partner selectively. The engineered receptor can be used to dissect the function of one member among a highly homologous receptor family, which allows cell-specific regulation by selective expression of the engineered receptor to the target cells. This approach is known as orthogonal chemical genetics, allele-specific chemical genetics, or chemogenetics (Table 1).9–11 We use “chemogenetics” in this review, because this terminology is widely used for expression as a complementary method against optogenetics to manipulate cell surface receptors.12–14 In an early study of chemogenetics, Hwang et al. reported alteration of nucleotide specificity of E. coli elongation factor Tu (EF-Tu), a GTP regulatory protein, by site-directed mutagenesis.15 The EF-Tu mutant has reduced affinity for GTP, and obtained dramatically increased affinity for a designed nucleotide, xanthosine 5′-triphosphate (XTP). In a successful example of chemogenetics to discriminate a target protein among the same protein family, Shokat's group developed a versatile approach for selective regulation of kinases using a bump-and-hole technique.9,16 They mutated a bulky gatekeeper amino acid residue in the ATP-binding pocket with alanine or glycine to form a hole and an appropriately designed bumped inhibitor bound to the mutant selectively through steric complementarity.16 The same concept has been extended to other intracellular proteins such as BET protein,17 histone demethylase,18 and auxin receptor.19 Additionally, chemically induced dimerization,20 ligand-induced destabilization,21 and chemical rescue21 are useful for chemogenetic regulation of intracellular proteins. With regard to transmembrane receptors, various kinds of chemogenetic strategies have been developed for selective regulation of these receptors. Notably, some strategies have been used to reveal receptor signaling in target cells of living animals. In this review, we summarize the development of chemogenetic methods for transmembrane receptors and their application to cell-specific regulation. We also discuss the perspective of chemogenetics for clinical use in the outlook part.
2. Chemogenetic activation of cell surface receptors using designed ligands
Small chemicals that bind to a target receptor are highly desired to control receptor functions in vivo. Mutated receptors activated selectively by artificial ligands are powerful to activate receptors in a cell-specific manner. In particular, receptor mutants that are not activated by intrinsic ligands, but activated by synthetic ligands, are called designer receptors. To obtain designer receptors, bump-and-hole or directed molecular evolution has been successfully applied to GPCRs and LGICs. In this section, we describe the development of designer GPCRs and LGICs.2.1. Designer GPCRs
An agonist binding to a GPCR induces conformational changes of transmembrane regions to activate heterotrimeric G protein complexes that comprise a Gβγ complex and Gα subunit. The Gα subunit is mainly classified into Gαs, Gαi, Gαq, and Gα12 isoforms, each of which has a distinct biological function.22 However, considering that GPCRs have various physiological roles in almost all organ systems,23 selective activation of G protein signaling in a cell-specific manner is required to understand the physiological roles of G protein pathways of cells in tissues or animals. In this context, designer GPCRs, which can activate the target Gα isoform, would be a powerful tool.In a pioneering study to create designer GPCRs by chemogenetics, Strader et al. developed a β2AR mutant, β2AR(D113S).24 β2AR(D113S) is not activated by its endogenous ligand, adrenaline, but is activated by synthetic ligands that are inert at the wildtype (WT) β2AR. However, it was difficult to use β2AR(D113S) in vivo because the affinity of the synthetic agonist was low. Instead, receptors activated solely by synthetic ligands (RASSLs) were developed as the next generation of chemogenetic GPCRs. The first RASSL was Ro1 (RASSL based on opioid receptor, No. 1), which was created by replacing the second extracellular loop (ECL2) of the κ-opioid receptor (KOR) with the ECL2 loop of the δ-opioid receptor.25,26 Unlike β2AR(D113S), Ro1 is activated by the KOR agonist spiradoline at nanomolar concentrations, but not by natural peptide ligands. New RASSLs using other receptors (e.g., β2AR)27 have also been developed (for more details, see a review article for RASSL).28 However, RASSLs have some drawbacks such as the fact that the ligands used for RASSLs also activate endogenous receptors and pathological phenotypes caused by overexpression of RASSLs.29,30
To overcome the above disadvantages, Roth's group developed designer receptors exclusively activated by designer drugs (DREADD) as the second generation of RASSLs.31 They developed M3 DREADD (GqD) by directed molecular evolution of M3 muscarinic acetylcholine receptor using yeast (Saccharomyces cerevisiae), which was genetically modified to promote the growth signaling pathway via activation of exogenously expressed GPCRs (Fig. 1a). GqD is activated by a pharmacologically inert drug-like compound, clozapine-N-oxide (CNO), but not by its endogenous agonist, acetylcholine. Muscarinic M4 DREADD (GiD) was constructed by introduction of corresponding mutations on the basis of amino acid sequence homology (Fig. 1b). GsD32 and G12D33 were created by swapping the intracellular regions of M3 DREADD with that of Gs-coupled β1-adrenergic receptor and G12-coupled GPR132 (or GPR183), respectively (Fig. 1b). In addition to canonical G protein pathways, GPCRs bind to other cytosolic proteins, such as β-arrestin, which elicits G protein-independent signaling through activation of mitogen-activated protein kinase. Nakajima et al. developed a β-arrestin-biased DREADD that was unable to couple to G proteins (Fig. 1b).34 Instead, a G protein-biased DREADD was also constructed by eliminating the β-arrestin-binding affinity of DRAEDD (Fig. 1b).35
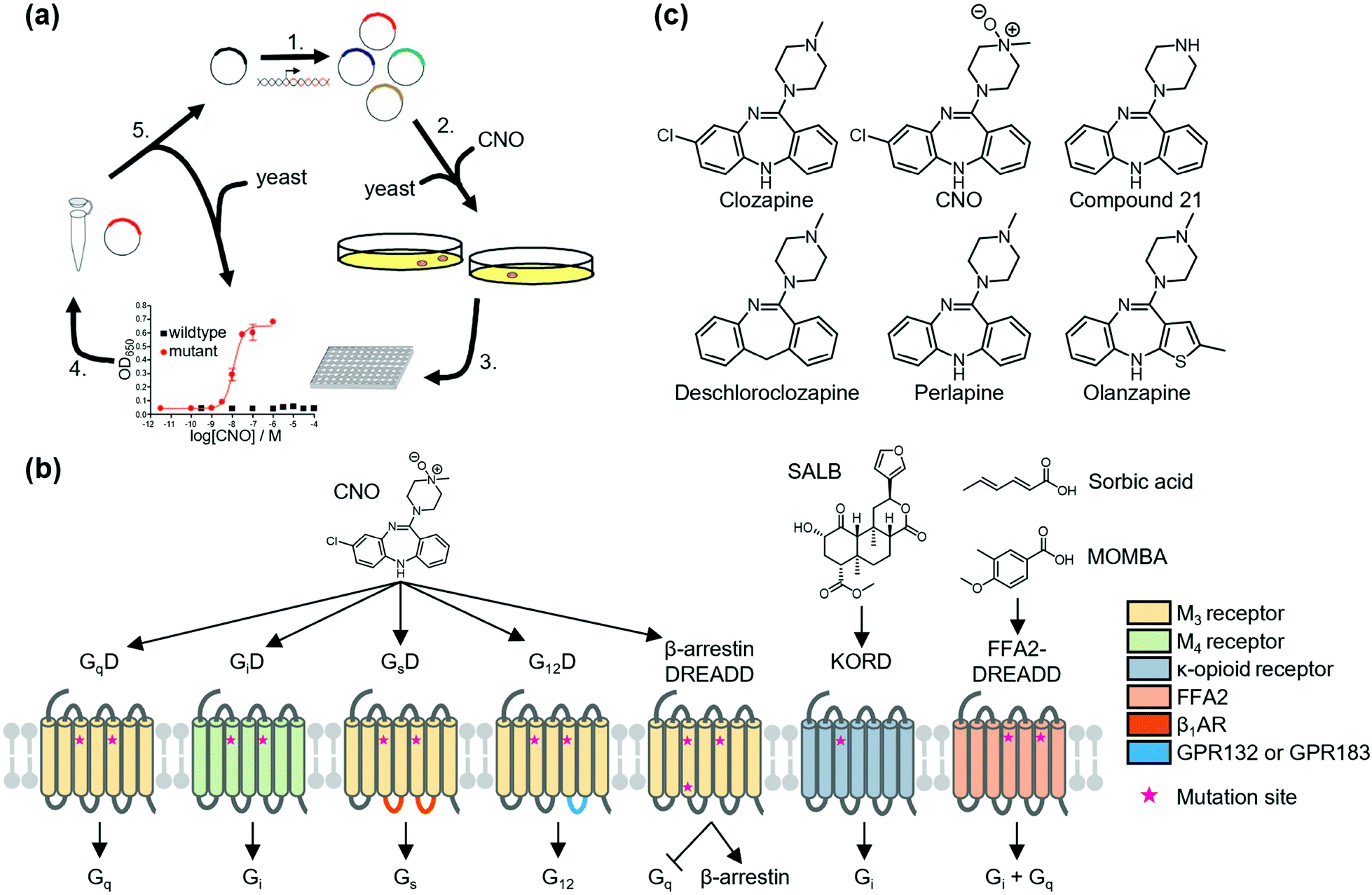 | ||
| Fig. 1 Schematic illustration of DREADDs. (a) Development of DREADD by directed molecular evolution. Reproduced with permission from ref. 31. Copyright 2007 by Proceedings of the National Academy of Sciences. (b) Structure and G protein-coupling properties of DREADDs. All DREADDs are unable to bind to the endogenous agonists, but can be activated by synthetic ligands. (c) Chemical structures of clozapine, CNO, and new muscarinic receptor-based DREADD ligands. | ||
Because of the low cytotoxicity of DREADD proteins and the inert property of the ligand, CNO toward endogenous receptors, the DREADD system has been successfully applied in vivo (for details, see review articles for DREADD).36,37 However, Gomez et al. revealed that CNO is metabolized into clozapine in vivo. They also found that activation of DREADDs was not mediated by CNO but by clozapine, a CNO metabolite in the brain of live animals,38 and clozapine activated not only DREADD proteins but also other GPCRs such as dopamine receptors and serotonin receptors.39 Instead, new DREADD ligands (e.g., compound 21,40 perlapine,40 and deschloroclozapine41) were developed to improve susceptibility to metabolism and decrease potential off-target effects of CNO (Fig. 1c). Notably, Weston et al. found that olanzapine, a Food and Drug Administration (FDA)-approved atypical anti-psychotic, activates GiD at low concentrations (10−9 M order) (Fig. 1c).42 Thus, the use of olanzapine has potential for clinical application of DREADD technology for disease treatment.
New DREADDs activated by chemotypes distinct from CNO or derivatives have been developed for bidirectional or orthogonal chemogenetic control of GPCR signaling. As an orthogonal pair of muscarinic receptor-based DREADDs, Vardy et al. developed KOR-DREADD (KORD) using KOR as a template that is activated solely by salvinorin B (SALB), a drug-like compound (Fig. 1b).43 Hudson et al. also reported FFA2-DREADD using free fatty acid receptor 2 (FFA2) as a template that is exclusively activated by sorbic acid or 4-methoxy-3-methyl-benzoic acid (MOMBA) (Fig. 1b).44,45 Further development of designer ligand and receptor pairs would contribute to clarifying the complex neural activity in living animals.
2.2. Designer LGICs
Chemogenetics has been applied to construct designer LGICs that selectively conduct cations or anions by designer ligands. One of the first chemogenetic LGIC tools was heterologous expression of GABAA-ρ receptors (GABAA-ρRs; also called GABACRs) in hippocampal neurons.46 Because endogenous GABAA-ρRs are expressed in the retina only, a GABAA-ρR selective agonist, cis-4-aminocrotonic acid (CACA), selectively activates transfected GABAA-ρR in hippocampal neurons. However, considering that GABAA-ρR is activated by GABA, its application has limitations. As a next-generation LGIC tool, using a Caenorhabditis elegans glutamate-gated chloride channel (GluCl) mutant and its selective activator ivermectin (IVM), an anthelmintic drug has been developed to control neuronal excitability.47 In the mutant GluCl, the introduction of the mutation in the glutamate-binding site reduces its glutamate response by greater than six-fold. Additionally, an improved version of the GluCl mutant has been reported, in which the original low channel activity and low affinity to IVM have been overcome, and the engineered GluCl is activated by ∼10 nM IVM.48 Another IVM-based chemogenetic tool is based on a human glycine receptor (GlyR) mutant in which two mutations are introduced to eliminate glycine affinity and increase IVM sensitivity.49 The mutant GlyR is activated by 19 nM IVM, which suppresses neurons in vivo and ex vivo. Moreover, Islam et al. converted IVM-sensitive GlyR to a cation channel by introducing mutations in the ion pore domain (IPD) of the channel.50 Although useful, these systems using IVM have some limitations, such as the toxicity and the complex pharmacology of IVM.Magnus et al. created a more sophisticated chemogenetic system on the basis of pharmacologically selective actuator modules (PSAMs) engineered to solely respond to pharmacologically selective effector molecules (PSEMs) (Fig. 2a).51 To design PSAMs, they focused on α7 nicotinic acetylcholine receptor (α7 nAChR) in the Cys-loop LGIC family, because structure–activity relationships of the small chemicals have been well studied. Mutations were introduced into the ligand-binding domain (LBD) for selective binding to designed ligands (PSEM) using the bump-and-hole strategy. Cell-based screening identified three PSAM and PSEM pairs (Fig. 2a). Notably, the LBD can be transplanted onto the transmembrane IPD of other members of Cys-loop LGICs, which allows construction of cation- or anion-conducting designer receptors using the IPD for serotonin receptor 3 or GlyR, respectively. Notably, PSAM and PSEM pairs have been applied to study memory and learning ex vivo and in vivo (for details, see a review article by the authors’ group).52 Recently, Magnus et al. improved this system by mutating three residues of α7 nAChR LBD to develop a novel PSAM termed PSAM4 that is activated by varenicline, an FDA-approved smoking cessation drug.53 Additionally, they developed new designed ligands termed ultrapotent PSEMs (uPSEMs) with subnanomolar affinity for PSAM4. This system has been applied to mice and rhesus monkeys (Fig. 2b).53 These chemogenetic technologies offer opportunities from basic research to therapeutic applications.
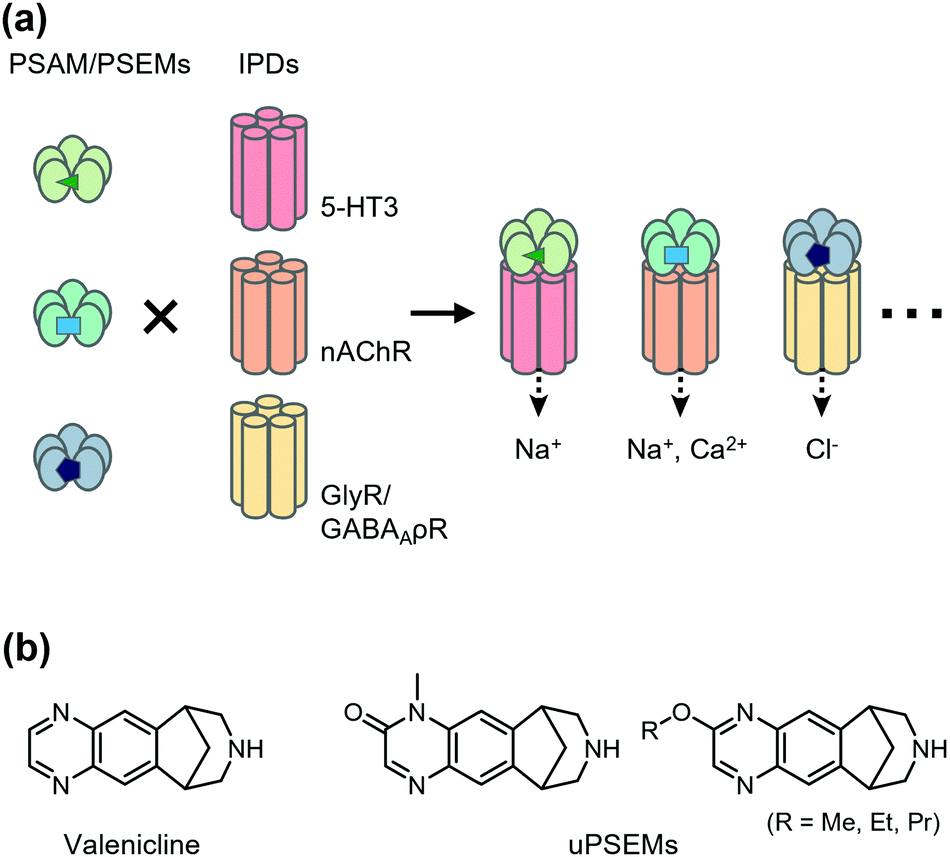 | ||
| Fig. 2 Schematic illustration of PSAM/PSEM system. (a) By combining different IPDs for a given PSAM/PSEM pair, multiple functional outcomes can be achieved, including neuronal activation, regulation of calcium flux, and neuronal silencing. Development of multiple orthogonal PSAM/PSEM pairs has enabled the combinatorial generation of diverse chemogenic ion channel tools. (b) Chemical structures of varenicline and uPSEMs. | ||
3. Chemogenetic regulation of receptor conformation
Ligand-induced receptor activation is accompanied by structural changes of receptor proteins in most cases. Thus, artificial regulation of these structural changes is an approach to control receptor functions. In the 2000s, antibodies were regarded as potential tools to stabilize receptors in active or inactive conformations.54 However, it is still challenging to obtain antibodies that selectively recognize specific structures of a receptor. Instead, chemogenetics using coordination chemistry and genetic engineering has the potential to control the receptor conformation.3.1. Metal coordination to the orthosteric site of receptors
As pioneering research for coordination-based chemogenetics of GPCRs, Elling et al. systematically introduced His mutations into the orthosteric ligand-binding site of tachykinin NK-1 receptor (NK-1R) that belongs to class A GPCRs.55 They designed a double mutant (E193H/Y272H) of NK-1R, in which His residues were introduced at the outer portion of transmembrane (TM) V and VI. The mutant showed high affinity for Zn2+ and the Zn2+ coordination inhibited binding of agonist peptides. Similar approaches have been applied to other class A GPCRs for metal coordination-induced activation. Elling et al. demonstrated artificial activation of β2AR using metal ions.56,57 Based on structural information of the ligand-binding site of β2AR, they introduced mutations of His and Cys as a metal coordination site at TM III and TM VII, and the designed mutant was activated by metal ions (Zn2+ or Cu2+) and metal complexes with aromatic bidentate chelators (Fig. 3). This activation method has been applied to other proteins such as NK-1R58 and melanocortin receptor.59 However, these mutations were introduced at the orthosteric ligand-binding site, which reduced the original affinity for the endogenous agonist, except for a few examples.60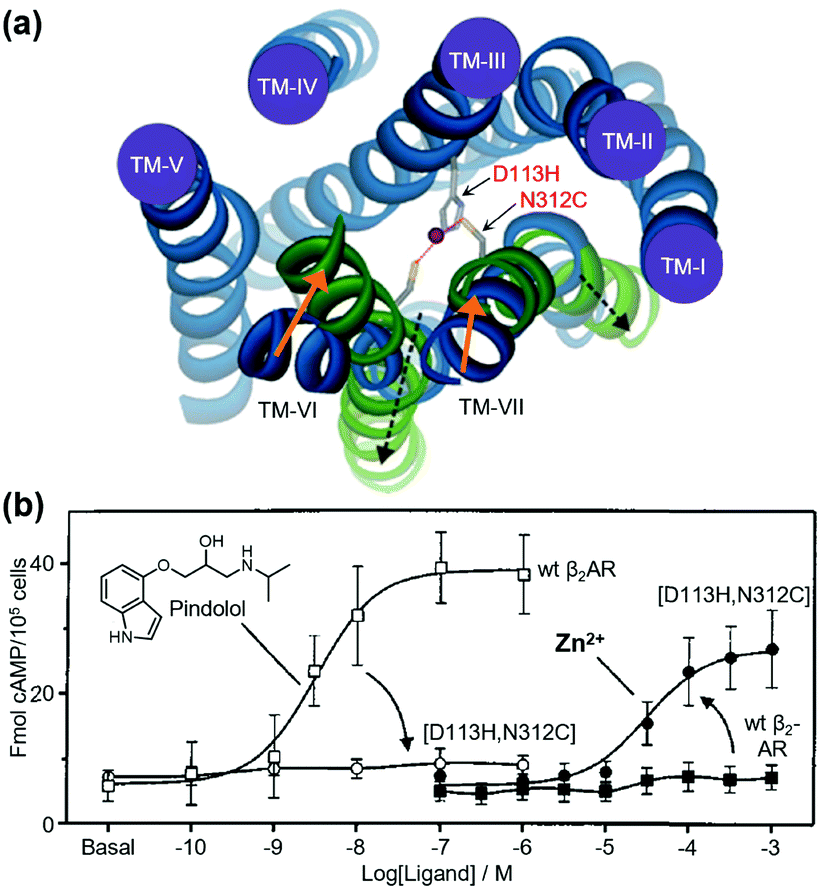 | ||
| Fig. 3 Metal-ion-induced activation of a β2AR mutant. (a) The active form of the receptor is stabilized by the binding of metal ions to the introduced coordination sites (D113H/N312C). (b) β2AR(D113H/N312C) is not activated by normal agonists, but is activated by metal ions. Reproduced with permission from ref. 56 and 57. Copyright 2006 by the American Society for Biochemistry and Molecular Biology and 1999 by Proceedings of the National Academy of Sciences. | ||
3.2. Metal coordination for allosteric regulation of receptors
Glutamate receptors, which include ion channel-type and GPCR-type receptors, have a large LBD in the extracellular region. Glutamate binding to the LBD causes a structural change from an open (inactive) to closed (active) conformation, which activates the receptor (Fig. 4a). Kiyonaka et al. were inspired by the structural changes of glutamate receptors and developed a chemogenetic strategy to control the structural changes by metal coordination, which is termed on-cell coordination chemistry (OcCC) or coordination-based chemogenetics (CBC) (Fig. 4b). In the CBC strategy, His mutations are introduced to both upper and lower lips of the entrance of the glutamate-binding pocket. Metal coordination using Pd(2,2′-bipyridine) [Pd(bpy)] stabilizes the closed (active) conformation (Fig. 4b and c).61,62 In this strategy, the original affinity for glutamate is unaffected because the mutated residues do not join the glutamate binding. They successfully applied this strategy to AMPA-type glutamate receptors (AMPARs) which have essential roles in memory and learning in the brain. In this case, adding Pd(bpy) increased affinity for glutamate, which indicates that the metal coordination acts as a positive allosteric modulator (PAM). It is noteworthy that the function of the receptor can be reversibly controlled by washing out Pd(bpy). They applied this method to chemogenetic activation of AMPARs in live neurons.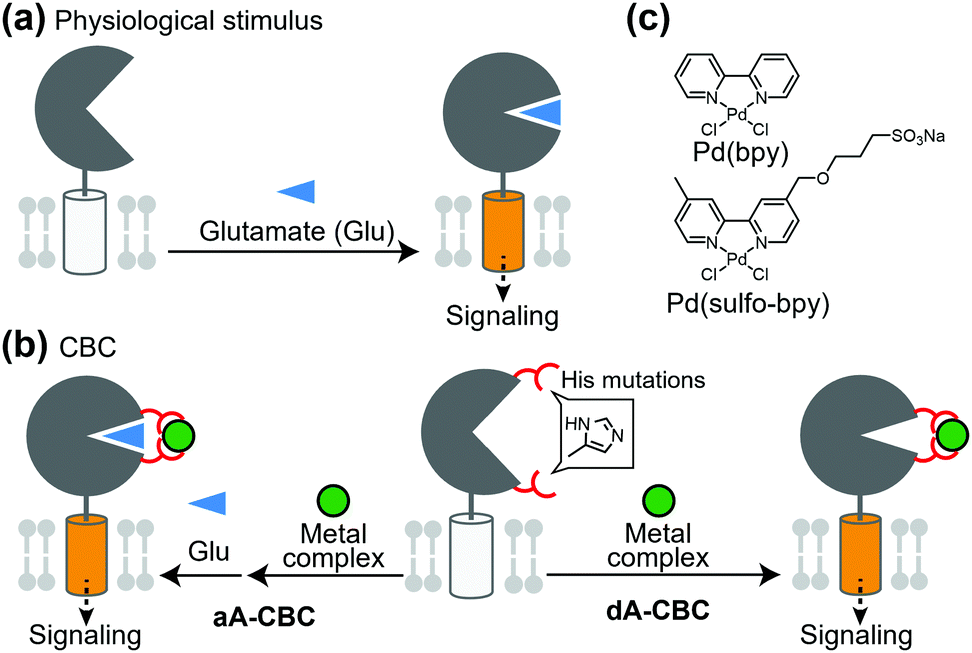 | ||
| Fig. 4 Schematic illustration of coordination-based chemogenetics. (a) Glutamate-binding induces the closing of the LBD for activation of the receptors. (b) Coordination-based chemogenetics (CBC) for glutamate receptors. In aA-CBC (allosteric activation via coordination-based chemogenetics), metal complexes act as PAM. In dA-CBC, metal complexes act as direct activator without affecting sensitivity to glutamate. (c) Chemical structures of Pd(bpy) and its derivative, Pd(sulfo-bpy) showing low cytotoxicity. | ||
PAMs are powerful for treatment of diseases in which the efficacy of endogenous ligands is decreased. However, considering that the concentration of endogenous ligands is not uniform in tissues, the effect of PAMs is highly dependent on the concentration of the ligand in extracellular spaces rather than the receptor concentration. Therefore, direct activators with high subtype selectivity would be useful to understand the physiological roles of receptors. Ojima et al. developed a direct activator to elucidate the mGlu1 function using the CBC concept, which was termed dA-CBC (direct-activation via coordination-based chemogenetics) (Fig. 4b).63 Their screening identified a mGlu1(A59H/N264H) mutant activated by Pd(bpy) without affecting the sensitivity to glutamate. They also developed a derivative of Pd(bpy), Pd(sulfo-bpy), with low cytotoxicity to neurons (Fig. 4c), which allowed application of dA-CBC to neuronal tissues. Notably, chemogenetic activation of mGlu1 induced long-term depression in cerebellar slices prepared from mice with the His mutation in the mGlu1 gene, which revealed the physiological roles of mGlu1 activation in motor learning of mammals. Considering the homologies and similar activation mechanisms of glutamate receptors, the CBC or dA-CBC strategy can be expanded to elucidate the physiological roles of each receptor subtype.
Although useful to control receptor functions, some issues such as cytotoxicity have been indicated for metal complexes. In this context, as described above, Ojima et al. improved cytotoxicity of metal complexes by reducing their cell membrane permeability.63 Thus, chemogenetics using metal coordination has potential to reveal the physiological roles of cell surface receptors.
4. Chemogenetic regulation of protein complexes of receptors
Receptor signaling is strictly regulated by protein complex formation within the transmembrane or in the intracellular side. Thus, regulation of a protein complex is a potential method to control the downstream signaling of receptors. Chemogenetic approaches, such as ligand-induced dimerization and light-induced association, which have been widely accepted to control cytosolic proteins,64,65 can also be applied to receptor proteins such as GPCRs and RTKs.4.1. Ligand-induced dimerization
Protein dimerization using chemical ligands, so-called chemically induced dimerizers (CIDs), is a classical but powerful method of chemogenetics. Theoretically, CID causes homo- or hetero-dimerization of a protein of interest (POI). The first and a landmark study of chemically induced formation of a protein complex was achieved using FK1012, a synthetic dimer of immunosuppressant FK506, by Schreiber's group.66 FK1012 homodimerizes two FKBP (FK506-binding protein) molecules, which is successfully applied to artificially dimerize the ζ domain of T cell antigen receptor to activate downstream signaling. After the success of the first CID, they also succeeded in heterodimerization of a POI using heterodimerizers.67,68 A representative example is rapamycin (Fig. 5a), which binds to both the FKBP and FRB (FKBP12-rapamycin binding) domain of mTOR (mammalian target of rapamycin).69 Rapamycin was successfully used to elucidate the roles of two different TGF-β receptors (Fig. 5b).70 For more details of these and related studies, see a well-organized review article for CID.6,71 Of note, to avoid an interaction of the dimerizer with mTOR and FKBP endogenously expressed in cells, a biologically orthogonal pair of the FRB mutant and a rapamycin derivative,67 known as “rapalog”, have been developed. Regarding orthogonality of the FKBP–rapamycin interaction, rapamycin with a bulky group as a bump was synthesized. This analogue has a low affinity for WT FKBP, but binds to the FKBP(F36V) mutant with a hole in the rapamycin-binding site with high affinity.72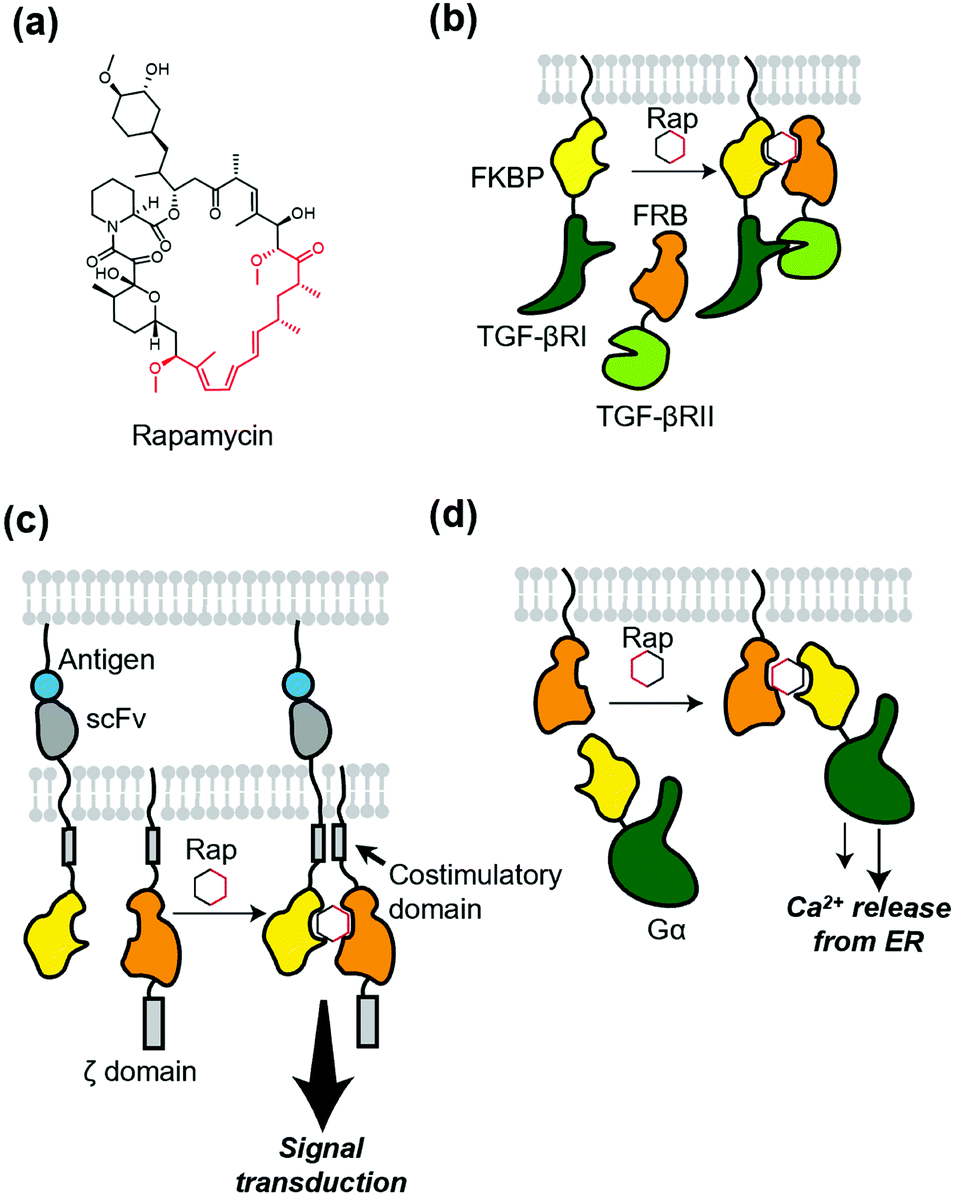 | ||
| Fig. 5 Ligand-induced dimerization and its applications to membrane receptors. (a) Chemical structure of rapamycin (Rap). Black or red region correspond to those interacting with FKBP or FRB, respectively. (b) Schematic illustration of intracellular heterodimerization of TGF-βRI and TGF-βRII by rapamycin. (c) Application of heterodimerization into AND logic gate for the precise control of CAR-T cell. In this system, both antigen-recognition and addition of the dimerizer are required for the signal transduction. (d) Ligand-induced translocation of Gα protein to transmembrane for dissecting its role in the heterotrimeric complex. ER, endoplasmic reticulum. | ||
Because of the versatility of the heterodimerization system that uses rapamycin, many studies have been published. A recent development using the system is precise control of chimeric antigen receptors (CARs). Wu et al. applied the system to split receptor design for CAR-T cells, where the antigen-binding domain (single-chain variable fragment; scFv) and intracellular signaling domain were fused to FKBP and FRB, respectively (Fig. 5c).73 Both antigen binding and the addition of rapalog are required to turn on the signal transduction, which provides the cells with a robust “on switch”. This system is regarded as an AND-logic gate for signal transduction, which can be applied to eliminate inflammatory side effects. In addition, Harris et al. used chemically induced dimerization to quantify persistence in the T-cell signaling network, where the dimerizer was used to initiate physical closeness of T-cells and antigen-presenting cells.74 To dissect G protein-coupled receptor signaling, Putyrski et al. reported FKBP-fused G protein subunits (Fig. 5d).75 Translocation of each subunit of heterotrimeric G proteins to the plasma membrane was triggered by the addition of rapamycin, which was used to evaluate the contribution of each subunit of G proteins to the signaling entities.
Beyond activation of membrane proteins, chemically induced dimerization can regulate various kinds of biological processes such as protein degradation,76 chromatin regulation,77 cell fate regulation,78 design of split protein,79,80 and vesicle contact.81 Furthermore, other protein–ligand pairs for chemically-induced dimerization82,83 and a chemically induced trimerization system84 have been reported. As a dimerization-free system, chemically induced protein localization using self-localizing ligands has been reported, which is termed SLIPT.85,86 These techniques also have the potential to manipulate transmembrane proteins with synthetic ligands.
4.2. Light-induced dimerization and oligomerization
Chemically induced dimerization is useful to control a protein association. Although powerful, methods that use chemical ligands inevitably have issues related to the diffusion of ligands. For reversible regulation, chemical ligands need to be washed out rapidly. Additionally, delivery of ligands is problematic in some cases. In this context, methods that employ light provide reversibility of the protein association by simply turning on or off photo-irradiation. Thus, photo-induced association of a protein complex is a potential strategy to control receptor signaling.Cryptochrome 2 (CRY2) from Arabidopsis thaliana is one of the most commonly used photoactivatable proteins (Fig. 6a). Blue light illumination leads to a conformational change of the PHR domain in CRY2, which results in homo-oligomerization of CRY2 molecules.87 CRY2 also forms a heterodimer with its binding partner CIB induced by photo-illumination (Fig. 6a).87,88 Another well-accepted photoactivatable tool is the photo-responsive domain in LOV domain superfamily proteins. LOV proteins undergo a drastic conformational change upon illumination, and fungal LOV protein VIVID (VVD) is known to form a photo-induced homodimer (Fig. 6a).87 Moreover, Kawano et al. developed a VVD-based heterodimerizer system termed “Magnet”, in which electrostatic interactions are introduced at the interface of VVD dimerization (Fig. 6a).89 “Magnet” is also superior to typical LOV proteins because it has a quick off-rate after turning off the light. As another heterodimerization system utilizing LOV2 protein, Guntas et al. developed an improved light-inducible dimer (iLID), which provides a better dynamic range between the dark and illuminated conditions.90 Regarding the wavelength of light, CRY2 and LOV proteins are mainly regulated by blue light because flavins are used in their light-sensitive domain. Recently, expansion of the wavelength has been studied intensively.65 For example, using the 5′-deoxyadenosylcobalamin (AdoCbl) as a chromophore, the oligomerization state of proteins with cobalamin-binding domains (CBDs) can be regulated by green light. Additionally, some bacterial phytochromes with tetrapyrroles, such as phycocyanobilin (PCB) or biliverdin as chromophores, have properties of red light- or near-infrared light-inducible dimerization. (Fig. 6b).
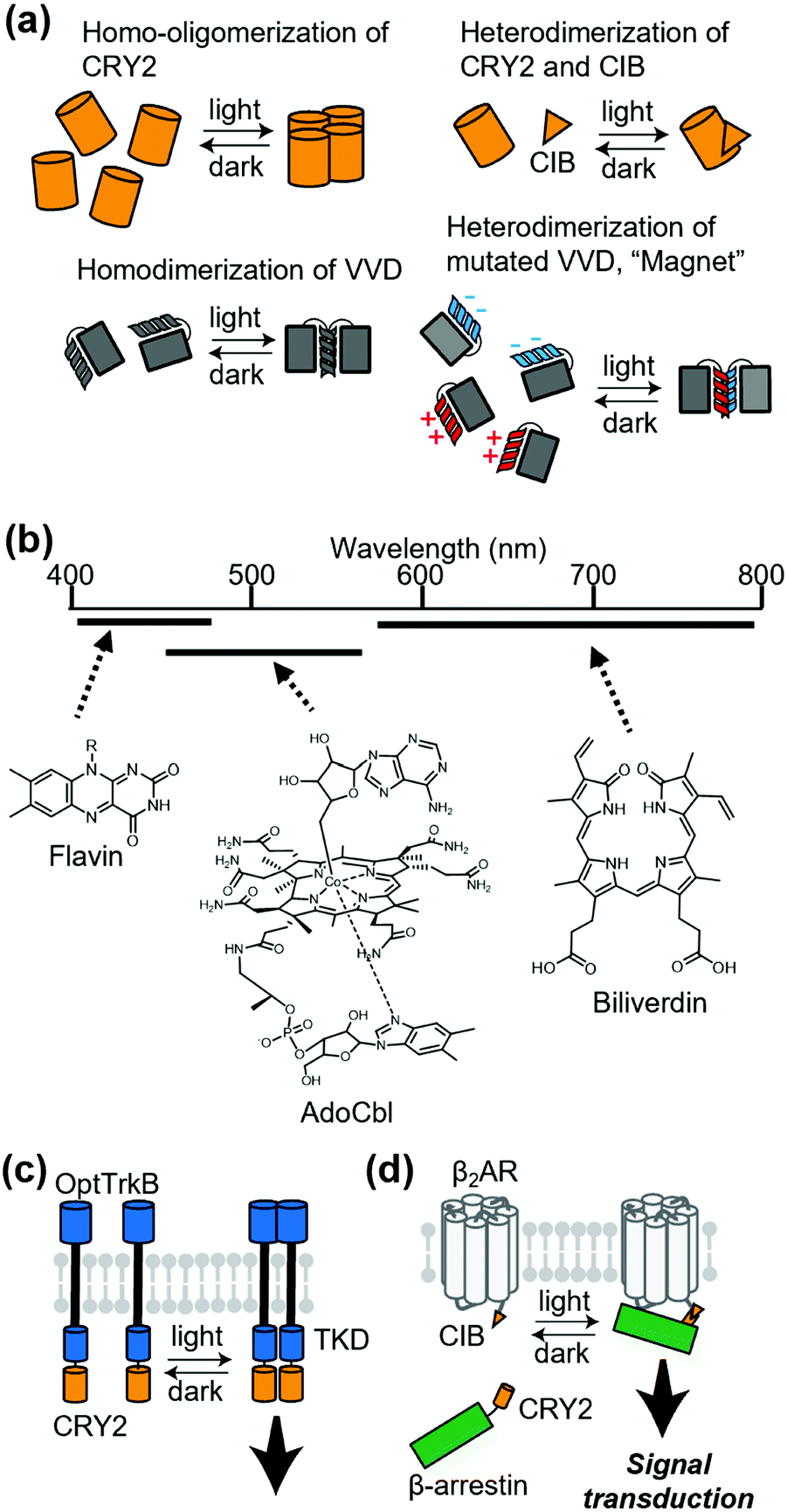 | ||
| Fig. 6 Application of light-induced dimerization or oligomerization to membrane receptors. (a) Representative systems of light-inducible dimerization or oligomerization. (b) Expansion of wavelength available for photoactivate proteins. Three black bars correspond the wavelength covered by each chromophore. (c) Regulation of RTK with light. Intracellular region of RTK is fused with PHR domain of CRY2, which homodimerizes upon photo-irradiation. TKD, Tyrosine kinase domain. (d) Photo-Induced interaction between β2AR and β-arrestin utilizing CRY2-CIB interaction. | ||
The first attempt to apply light-induced homo-interaction to a membrane receptor was a blue-light inducible RTK. Chang et al. fused the PHR domain of CRY2 to the intracellular part of tropomyosin-related kinase (Trk), a kind of RTK, and the fusion protein was termed OptTrkB (Fig. 6c).91 In cultured hippocampal neurons that expressed OptTrkB, repetitive and continuous illumination of blue light induced filopodia and spine formation. In almost the same period of the report of CRY2-fused RTK, LOV-fused RTK was reported by Grusch et al.92 Kainrath et al. developed systems in which green light-inducible inactivation of RTKs is achieved using the CBD of CarH protein, with AdoCbl as a chromophore.93 Additionally, Reichhart et al. and Leopold et al. expanded the wavelength available to red and the near-infrared region to activate RTKs using cyanobacterial phytochrome 1 with PCB and a bacterial phytochrome called DrBphP with biliverdin, respectively.94,95
Light-induced dimerization has been applied to other transmembrane proteins. Takenouchi et al. applied CRY2 fusion to an artificial trigger to induce an interaction between β-arrestin and β2AR.96 They used light-induced heterodimerization between CRY2 and CIB to prepare CRY2-fused β-arrestin and CIB-fused β2AR (Fig. 6d). The system allowed reversible control of the duration of the interaction between β2AR and β-arrestin, which demonstrated that the duration of the interaction is crucial for the decision of the β2AR-trafficking pathway. Sinnen et al. used the same light-inducible CRY2-CIB interaction to manipulate the abundance of AMPARs at the postsynaptic density (PSD).97 They fused CRY2 to a PSD scaffold protein and CIB to GluA1, an AMPAR subunit. Upon blue light treatment, CIB-fused GluA1 was recruited to the PSD site, which increased functional synaptic connections. Beyond dimerization, the oligomerizing property of CRY2 can also be used to regulate various kinds of proteins that include membrane receptors.98–100 For example, Kyung et al. reported the OptoSTIM1 system where photo-induced oligomerization of OptoSTIM1 activates the endogenous Ca2+ release-activated Ca2+ channel in vivo.101
A disadvantage of the slow kinetics of the chemically induced dimerization system can be solved by adding photo-responsive properties to the small chemicals. In this context, photocaged chemical dimerizers102,103 and photoactivatable self-localizing ligands104 have been developed for photo-induced association of POIs. Conversely, photo-induced dissociation of a protein–protein interaction (PPI) has been achieved by Zimmermann et al. through chemically induced dimerization of POIs followed by light-induced dissociation.105 They synthesized a ligand with both a SNAP-tag substrate and Halo-tag substrate linked to a methyl-6-nitroveratryl (MeNV-HaXS). The addition of MeNV-HaXS specifically induces heterodimerization of two POIs and UV irradiation disrupts the PPI in a minute order. Collectively, by selecting or combining some of these tools for protein dimerization, researchers can establish methods tailor-made for the regulation of their target proteins.
5. Chemogenetic regulation of receptors by ligand tethering
Protein-directed ligand tethering approaches enable selective modulation of POIs. Chemogenetic strategies for covalent or non-covalent ligand tethering have been employed to control sensitized receptors or ion channels using photoswitchable ligands.5.1. Covalent tethering of ligands and photoswitching approaches
Ligand-tethered receptors are constructed by tethering reactions between designer ligands and sensitized receptors.106 A POI is sensitized by site-directed introduction of a reactive amino acid such as Cys. Covalent tethering of ligands has been applied to covalent inhibition of a POI. Designer ligands with different spacers between the pore-binding ligand and reactive module have also been used as molecular tapes to estimate the distance between the tethering site and pore-blocking site of a voltage-gated K+ channel.107 Although the covalent tethering of ligand irreversibly inhibits the protein function, covalent tethering of photoswitchable ligands allows its reversible modulation by photo-irradiation.Fine-tuning of the functional switching of receptors has been demonstrated by optimizing the photo-responsive properties of the photoswitchable group of the tethered ligand. Azobenzene is arguably the most acceptable photoswitchable motif and derivatization allows modulation of the photochemical properties in terms of both trans-to-cis and cis-to-trans isomerization (Fig. 7a). For example, although trans-to-cis photoisomerization of azobenzenes is triggered by ultraviolet light (typically 360–380 nm), “push–pull” azobenzenes, which have an electron-donating group and electron-withdrawing group on the 4 and 4′ positions, respectively, are photoisomerized by blue light (typically 445–472 nm) owing to the red shift of the π–π* transition band.108 Additionally, the low energy barrier of thermal isomerization shortens the half-life of the cis form in “push–pull” azobenzenes within a second.108 For in vivo application, electronically asymmetric azobenzenes such as “push–pull” azobenzenes are switched through two-photon (2P) and one-photon stimulation (1P), because of the large 2P absorption cross-section.109,110 Another candidate of visible light-gated azobenzene is the tetra-ortho-substituted azobenzene series. In contrast to “push–pull” azobenzenes, tetra-ortho-substituted azobenzenes can be switched in both trans-to-cis and cis-to-trans directions with visible light because of the bistability of trans and cis isomers.111–113 Additionally, diazocines114,115 and arylazopyrazoles116 are novel photoswitchable groups (Fig. 7b). Diazocine is regarded as a bridged azobenzene whose cis isomer is more stable than the trans isomer in contrast to unbridged azobenzenes. Arylazopyrazoles exhibit high photoswitchability because of well-separated transition bands. These chromophores will expand the repertoire of optical chemogenetic tools.
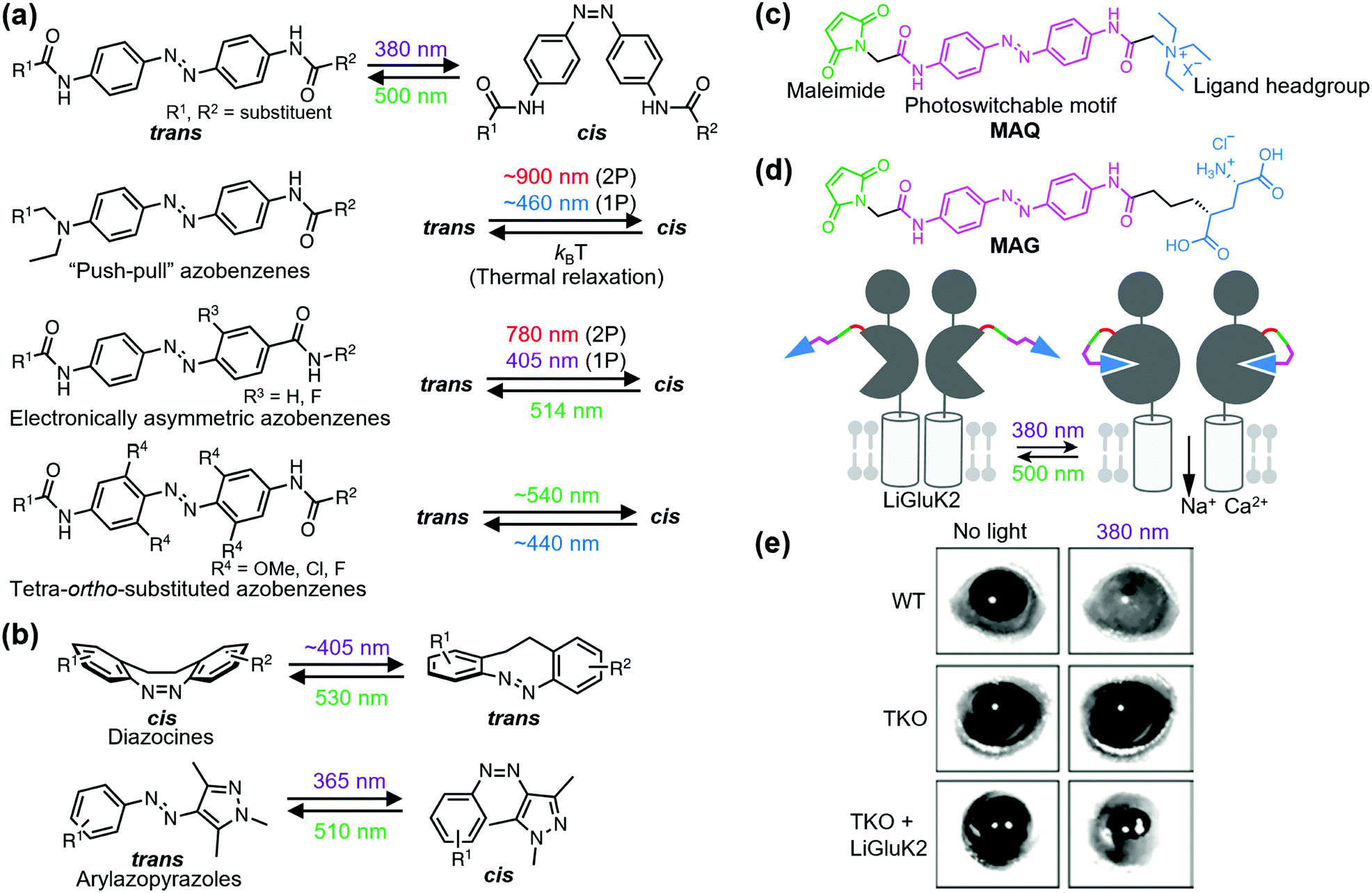 | ||
| Fig. 7 Photoswitching of PTL-tethered receptors. (a) Chemical structures of various azobenzene-based photoswitchable motifs. (b) Chemical structures and the photoisomerization of diazocines and arylazopyrazoles. (c) Chemical structure of a PTL, MAQ. (d) Chemical structure of MAG and schematic illustration of a light-responsive ionotropic glutamate receptor, LiGluK2. (e) Restoration of pupillary reflex in TKO mice lacking phototransduction and pupillary reflexes by expression LiGluK2 (right). Representative infrared images of the pupil area taken in the dark (left column) and under 380 nm light (right column) for WT, TKO, and TKO-LiGluK2 mice are shown. Reproduced with permission from ref. 121. Copyright 2011 by Elsevier, Inc. | ||
5.2. Ligand-tethering near the ligand-binding domain
Optochemogenetic regulation of ligand-tethered receptors was first achieved by Banghart et al. in 2004.117 They demonstrated optical control of the sensitized Shaker K+ channel using maleimide-azobenzene-quaternary ammonium (MAQ) as a photoswitchable tethered ligand (PTL) with three components: a maleimide, photoswitchable motif, and ligand headgroup (Fig. 7c). Sensitized receptors for PTLs are constructed by a single amino acid substitution to Cys near the ligand-binding site. The chemogenetic approach using PTLs has been applied to optical control of various sensitized ligand-gated ion channels, especially ionotropic glutamate receptors, and GPCRs. For details about PTLs, please refer to other excellent reviews.118,119 Here, we present the in vivo and therapeutic applications of PTL-tethered receptors.In the optochemogenetic approach using PTLs, minimal perturbation of sensitized receptors by single point mutations allows optical control of PTL-tethered receptors in vivo. Szobota, et al. generated transgenic zebrafish larvae that expressed sensitized kainate-type glutamate receptor GluK2 in sensory neurons. After tethering the PTL, maleimide-azobenzene-glutamate (MAG), an escape movement in response to touch of the transgenic larvae was controlled optically (Fig. 7d).120 Notably, expression of light-regulated ion channels in surviving retinal neurons can be applied as a treatment strategy of inherited retinal degeneration. Caporale et al. expressed MAG-tethered GluK2 in retinal ganglion cells of the rd1 mouse, a model of retinal degeneration. They demonstrated that the engineered light-responsive GluK2 (LiGluK2) restored a light-avoidance behavior in rd1 mice and the pupillary reflex in the mutant (TKO) mice that lacked phototransduction and pupillary reflexes (Fig. 7e).121 Similarly, in vivo applications of PTL-tethered receptors have been applied to other ligand-gated ion channels such as NMDA receptor GluN2A,122 two-pore domain potassium channel TREK1,123 and GPCRs such as metabotropic glutamate receptor mGlu2.124
The PTL approach has been applied to photo-control of sensitized receptors endogenously expressed in animals. Lin et al. designed and synthesized PTLs with either GABA or its guanidinium analogs as the ligand for optochemogenetic regulation of GABAA receptor.125 They generated knock-in mice with a sensitized point mutant in the GABAA receptor gene and applied a PTL by intracranial infusion. The light-responsive knock-in mice allowed in vivo optical control of the endogenous GABAA receptor function with high spatial, temporal, and biochemical precision.
5.3. Ligand-tethering via self-labeling protein tags
Although site-specific ligand-tethering to engineered Cys is powerful for tethering ligands, this approach has several limitations. Thiol-reactive electrophiles such as maleimide react with native thiol groups, which are abundantly present in cells, and maleimides are susceptible to hydrolysis at physiological pH. To circumvent these limitations, ligand-tethering approaches via self-labeling protein tags, such as SNAP-, CLIP-, and Halo-tags, have been used for ligand-tethering, and protein tags are genetically fused to receptors.126 Additionally, photoswitchable orthogonal remotely tethered ligands (PORTLs), which are composed of ligand headgroups, photoswitchable motifs, flexible linkers, and unique substrates of self-labeling protein tags, are used to covalently label protein-tagged receptors. The PORTL-tethered receptors are optically and reversibly controlled with high spatiotemporal resolution using the photoswitchable motifs. Unlike maleimides, the unique substrates and covalent adducts are inert to water and thiol-containing biomolecules. Because self-labeling protein tags are “single-turnover” enzymes with unique substrate specificities, PORTLs are rapidly labeled on self-labeling protein-tagged receptors with high selectivity.Compared with maleimide-based PTLs, PORTLs are tethered adequately at >100 times lower concentrations because of the reaction kinetics derived from a self-labeling protein tag as a “single-turnover” enzyme and the stability of unique substrates (PTLs: 100–200 μM; PORTLs: ∼1 μM).127 Additionally, the tethering site of PORTLs is separated from the ligand-binding site of the receptors in the range of several nanometers by considering the size of the self-labeling protein tags (SNAP- and CLIP-tags: 19.4 kDa; HaloTag: 33.6 kDa).126 The remote tethering causes a relatively low local concentration of the tethered ligand (<100 μM) around the ligand-binding site.128 The low effective ligand concentration requires high-affinity ligands for the headgroups of PORTLs. Conversely, maleimide-based PTLs are attached next to the ligand-binding site. The proximity leads to relatively high effective molarity of the tethered ligand (>1 mM).128 The high local ligand concentration permits relatively low-affinity ligands for the headgroups of PTLs.
Broichhagen et al. first demonstrated the utility of the covalent-tethering approach using a self-labeling protein tag and PORTLs in 2015.127 They tethered a PORTL, benzylguanine-azobenzene-glutamate with a 12-polyethylene glycol-repeat linker (BGAG12) to SNAP-tagged mGlu2. The PORTL-tagged mGlu2 termed “SNAG-mGlu2” was reversibly regulated by light irradiation (Fig. 8a). Furthermore, substrate specificity of each self-labeling protein tag permits bio-orthogonal tethering of PORTLs to two different kinds of receptors. Levitz et al. demonstrated multiplexed orthogonal optical control of mGlu2 and mGlu7 using orthogonal protein tags (CLIP- and SNAP-tags) and two photoswitchable motifs with different photochemical properties.128 Additionally, Leippe et al. demonstrated that the optochemogenetic approach with PORTLs can be applied to RTKs by swapping the LBD of RTKs with the SNAP-tag fused LBD of mGlu2.129 Indeed, tethering a PORTL, BGAG8, to SNAP-tagged chimeric receptors allowed photo control of tyrosine kinase activities and downstream signaling cascades.
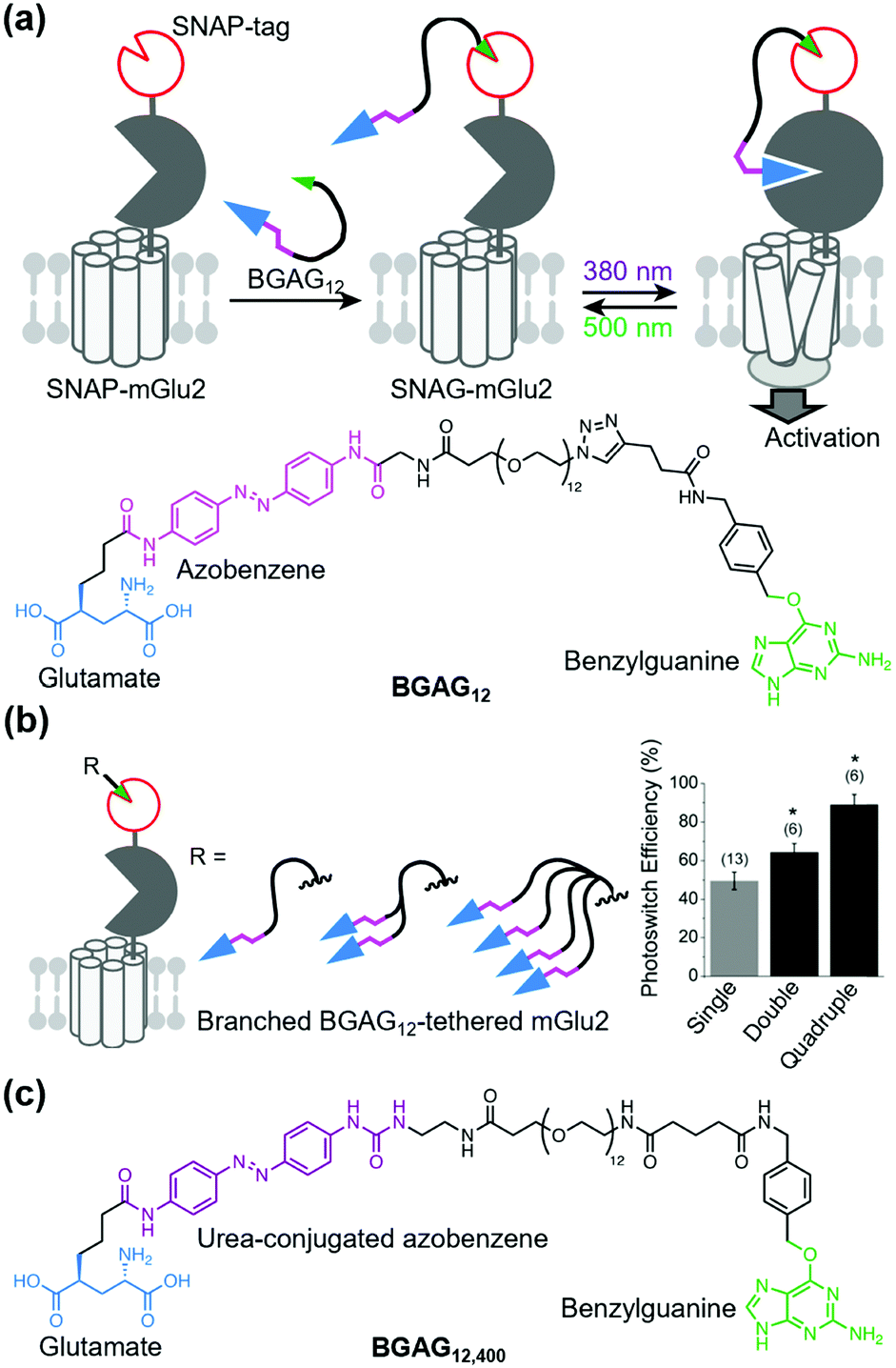 | ||
| Fig. 8 Optical control of a PORTL (BGAG12)-tethered mGlu2 (SNAG-mGlu2). (a) Schematic illustration of PORTL-mediated reversible regulation of SNAG-mGlu2 (top). Chemical structure of the PORTL, BGAG12 (bottom). (b) Schematic illustration of branched BGAG12-tethered mGlu2 (left). Bar plot showing photoswitch efficiencies for different levels of branched BGAG12-tethered mGlu2 (right). Reproduced with permission from ref. 130. Copyright 2020 by Elsevier, Inc. (c) Chemical structure of an optimized PORTL with 4-urea-4′-amide azobenzene, BGAG12,400. | ||
Although useful, an insufficient photoswitching efficiency of SNAG-mGlu2 activity (∼60%) was found. In this context, the efficiency was improved by two strategies: branching the photoswitchable ligand (branched PORTL) and optimizing the photoswitchable motif. Acosta-Ruiz et al. developed branched PORTLs, which permitted optical and reversible control of PORTL-tethered mGlus with high efficiency (typically 80–90%) (Fig. 8b).130 The branched PORTL approach enabled elucidation of the roles of mGlu2 relevant to working memory via in vivo optical control of branched PORTL-tethered mGlu2. As another PORTL, Gutzeit et al. developed BGAG12,400 with 4-urea-4′-amide azobenzene as the fine-tuned photoswitchable motif (Fig. 8c).131 Because of the enhanced molar extinction coefficient of BGAG12,400, the photo-switching efficiency of BGAG12,400-tethered mGlu2 was enhanced to ∼90%, which is comparable to that of branched PORTL-tethered mGlu2.
Biorthogonal tethering of SNAP-tagged receptors with PORTLs has potential therapeutic applications. For treatment of retinitis pigmentosa, Berry et al. expressed SNAP-tagged mGlu2 in vivo via intravitreal injection of an adeno-associated virus (AAV) vector that encoded SNAP-tagged mGlu2 under the control of the neuron-specific hSyn-1 promoter. BGAG12,460 was also administrated by direct injection into eyes to construct SNAG-mGlu2 in vivo.132 Notably, the SNAG-mGlu2 in the retina of retinitis pigmentosa model mice successfully restored light avoidance and patterned vision.
A tethering approach using a self-labeling protein tag also provides chemical switching of receptors of interest. Podewin et al. designed and synthesized a benzylguanine-linked peptide agonist of glucagon-like peptide-1 receptor (GLP-1R) with a cleavable disulfide bridge.133 Covalent tethering of this agonist to SNAP-tagged GLP-1R induced prolonged cAMP generation, cytosolic Ca2+ rises, and internalization of the receptor. These effects were readily attenuated by cleavage of the disulfide bridge using the cell-permeable reducing agent mercaptoethanol. This approach is termed reductively cleavable agonist (RECON), which has been applied to other peptidergic GPCRs for conditional and reversible activation of SNAP-tagged GPCRs.
5.4. Ligand-tethering via non-covalent labeling
In contrast to covalent tethering, non-covalent tethering of ligands provides reversibility at the ligand-tethering step in chemogenetic regulation. Antibodies, nanobodies, and other antibody mimetics are potential tools for non-covalent tethering of ligands. In fact, Farrants et al. established an immunochemical manipulation method of a green fluorescent protein (GFP)-fused GPCR using anti-GFP nanobody–photoswitch conjugates (NPCs).134 The anti-GFP NPCs were synthesized by orthogonal labeling of PORTLs such as BGAG12 to SNAP-tagged anti-GFP nanobodies, which allowed a photoswitch of GFP-fused mGlu2 (Fig. 9a). Considering the high flexibility of nanobodies, this strategy has potential for targeting endogenous receptors.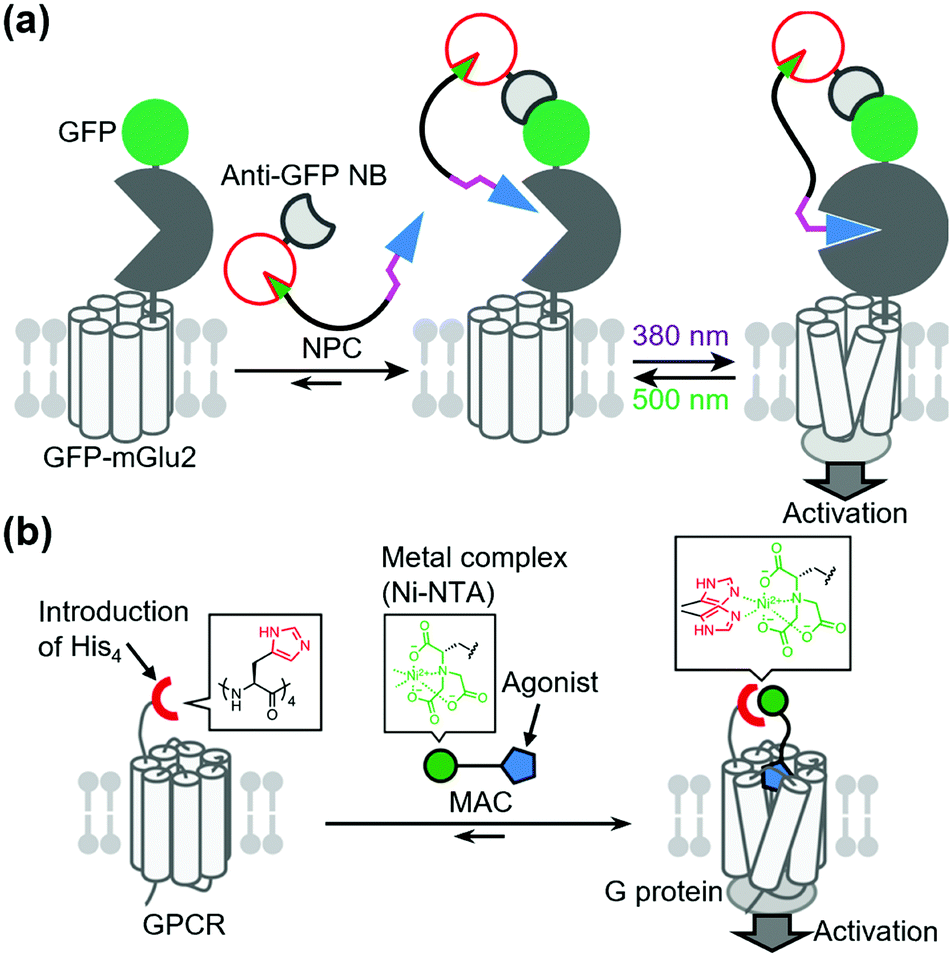 | ||
| Fig. 9 Non-covalent ligand tethering for chemogenetic regulation of receptors of interest. (a) Schematic illustration of non-covalent tethering of photoswitchable ligands using anti-GFP NPCs. NB, nanobody. (b) Schematic illustration of non-covalent agonist tethering using metal coordination for the selective activation of GPCRs of interest. | ||
Another potential method for non-covalent tethering a ligand is introduction of a recognition site into receptors of interest to increase affinity for designer ligands. In this context, Kubota et al. reported a chemogenetic approach for selective activation of class A GPCRs using coordination tethering, in which coordination between a metal complex of the ligand and His-tag of the receptor increases the affinity of the designed ligand for sensitized receptors.135 They demonstrated that His4-tagged β2AR and M1 muscarinic acetylcholine receptor (M1R) were selectively activated by selective ligands termed metal complex–agonist conjugates (MACs) in living cells, thanks to the coordination bond between the His4-tag and a Ni2+–nitrilotriacetic acid (Ni–NTA) complex of MAC (Fig. 9b). In fact, the coordination tethering approach allowed selective activation of His4-tagged M1R over endogenous muscarinic acetylcholine receptors in primary rat cortical astrocytes.
6. Chemogenetic regulation of receptors using genetic code expansion
Site-specific incorporation of unnatural amino acids (UAAs) into POIs using genetic code expansion is a potential approach for functional regulation of proteins.136–139 In this method, engineered tRNA with UAAs is prepared using orthogonal aminoacyl-tRNA synthetase and tRNA pairs. In most cases, the amber stop codon (UAG) is reassigned for site-specific incorporation of the UAA into the target protein (Fig. 10a). As a side effect of genetic code expansion, use of the amber codon produces truncated proteins when UAG is recognized as a stop codon, but not a UAA. Additionally, the amber codon in other endogenous proteins may be misled as a UAA. However, because of the high versatility of genetic code expansion, many successful examples have been reported for functional control of POIs in mammalian cells, primary neurons, and living animals.140,141 In this section, we focus on functional regulation of ion-channels or ion-channel-type receptors using genetic-code expansion as the representative. For details on its application to other cell-surface receptors such as GPCRs and RTKs, see well-organized other review articles.136–139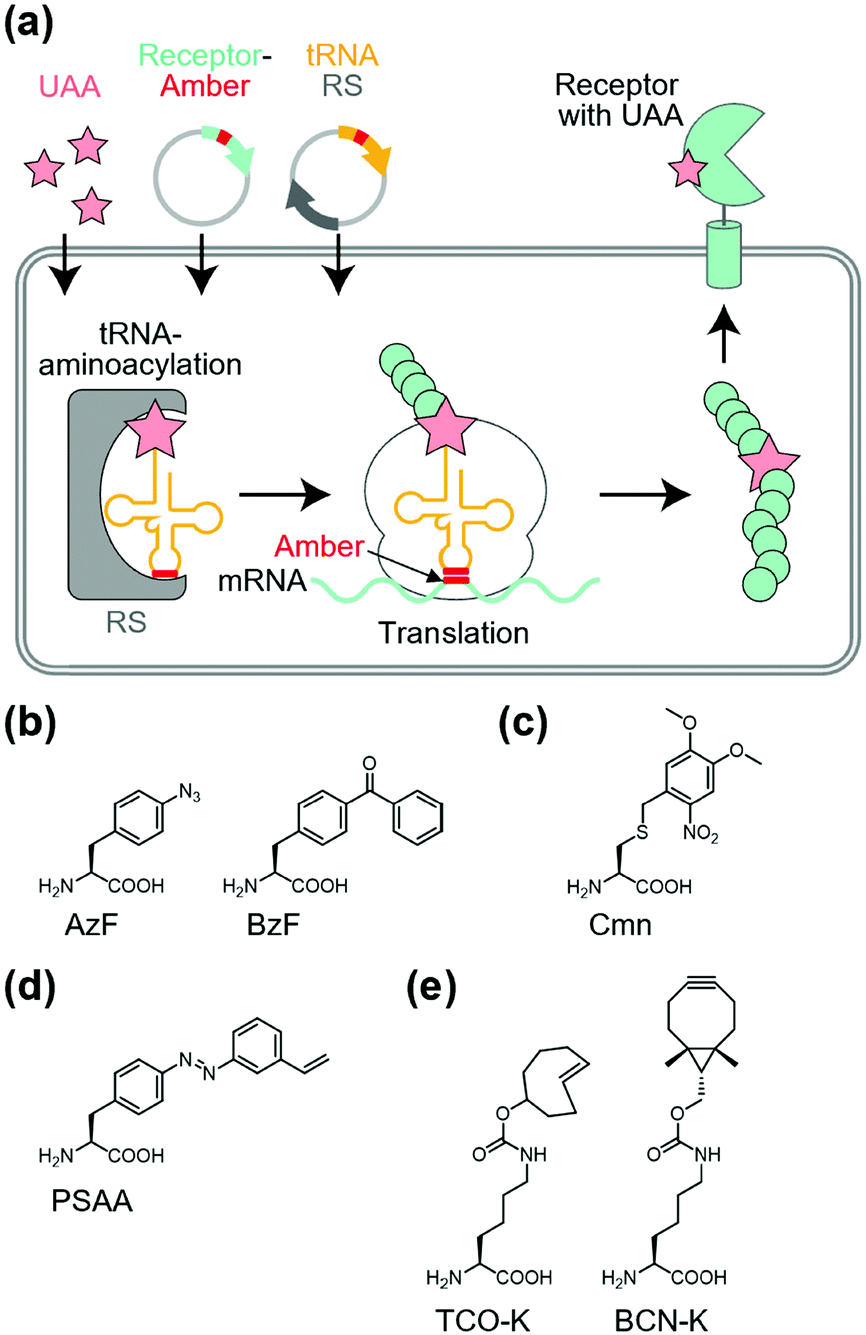 | ||
| Fig. 10 General principle of genetic code expansion. (a) Schematic illustration of the incorporation of UAAs into transmembrane receptors. RS, orthogonal aminoacyl-tRNA synthetase. (b–e) Chemical structures of representative UAA structures for photo-crosslinking (in b), photo-cleavage (in c), photo-switching (in d) and bioorthogonal tethering (in e). | ||
Incorporation of UAAs with photo-reactive, photo-cleavable, or photoswitchable groups at appropriate positions allows functional regulation of receptors by photo-irradiation. Zhu et al. reported incorporation of p-azido phenylalanine (AzF) as a photo-reactive UAA for photo-crosslinking of NMDA-type glutamate receptors (NMDARs) (Fig. 10b).142 The incorporation of AzF in the GluN1 subunit of the NMDAR at the N-terminal domain led to irreversible allosteric inhibition of receptor activity upon UV illumination.142,143 Similarly, AzF or p-benzoly phenylalanine (BzF) have been incorporated into the ligand-binding or transmembrane domain of AMPARs for photo-control of receptor functions.144,145 Additionally, photo-cleavable (i.e., caged) UAAs have been introduced into ion channels and receptors. As a representative example, Kang et al. reported incorporation of a caged UAA (Cmn) into the pore region of inwardly rectifying potassium channel Kir2.1 (Fig. 10c).146 The caged UAA occluded the pore, which rendered the channel to a non-conducting state. After brief UV illumination, the caging group was released, which permitted outward K+ current in cells. Moreover, this approach has been expanded to the embryonic mouse neocortex to express the light-activated K+ channel, which allowed photo-induced suppression of neuronal firing.146 Regarding photo-switching, Klippenstein et al. incorporated a photoswitchable UAA (PSAA) into NMDARs (Fig. 10d).147 Notably, incorporation of the photoswitchable group by genetic code expansion provides site-tolerance, which is in large contrast to the ligand tethering via chemical modification to solvent-accessible regions. In fact, PSAA has been successfully introduced into solvent-inaccessible transmembrane regions as well as solvent-exposed extracellular regions for photo-switching of receptor functions (Fig. 11).147
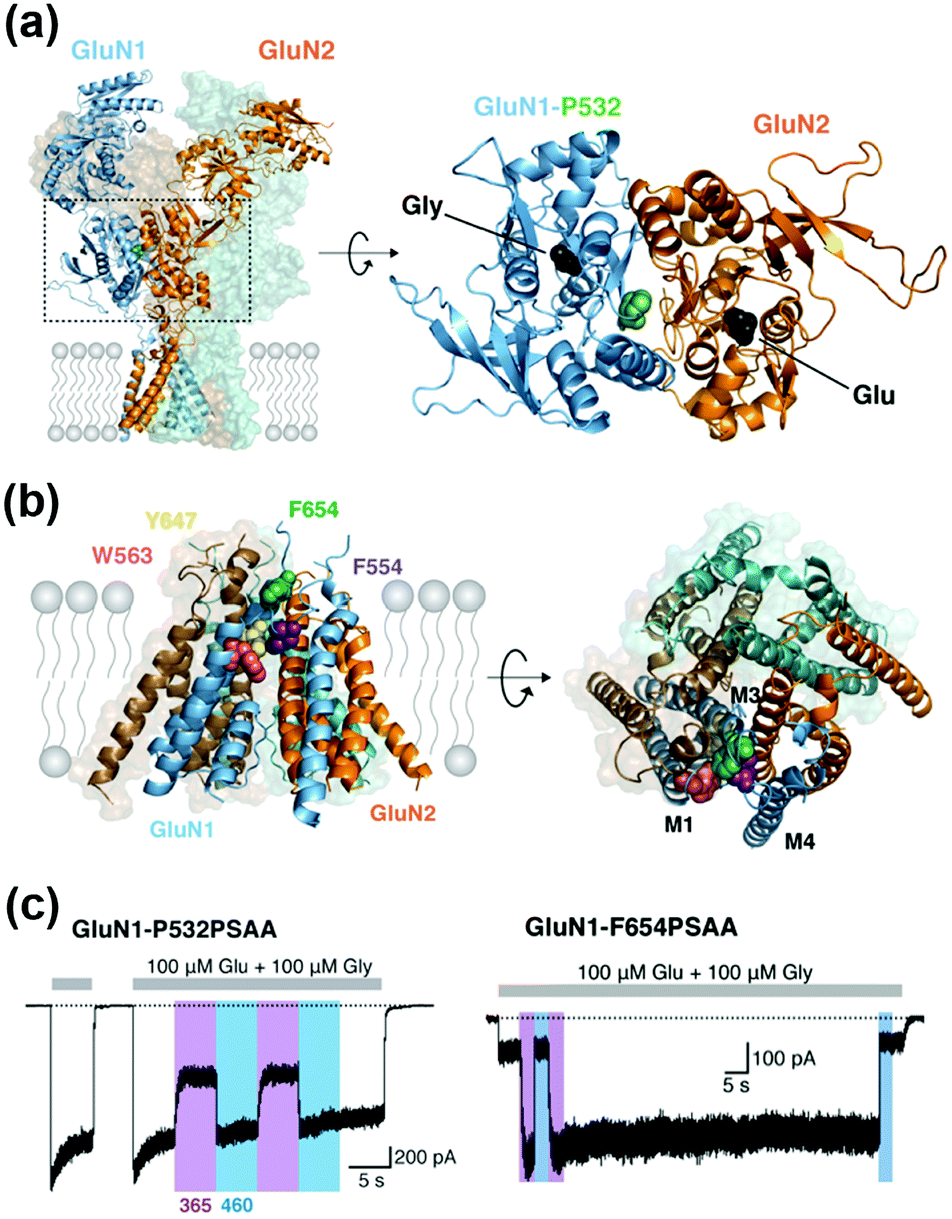 | ||
| Fig. 11 Photoswitching of NMDARs using genetic-code expansion. (a) Structure of NMDARs composed of GluN1 and GluN2 subunits. In the right panel, the top view of the LBD is shown, and mutation site (P523) is highlighted. (b) Structure of the transmembrane domain (TMD) of NMDARs represented by a side (Left) and top view (Right). (c) Representative current traces of NMDARs having an azobenzene-based photo-responsive UAA (PSAA) at P532 in the LBD (in left) or at F654 in the TMD (in right). Reproduced with permission from ref. 147. Copyright 2017 by eLife Sciences Publications, Ltd. | ||
Site-specific incorporation of bio-orthogonal groups by genetic code expansion provides another strategy for tethering chemicals. In this context, many kinds of bio-orthogonal groups, such as azides, alkynes, ketones, alkenes, and tetrazoles, have been successfully incorporated into POIs.139 Considering the high reaction rate and selectivity of inverse electron-demand Diels–Alder reactions between strained alkenes and tetrazines, UAAs with bicyclo[6.1.0]-nonyne (BCN-K) or trans-cyclooxtene (TCO-K) have been used for rapid labeling with tetrazine-conjugated probes (Fig. 10e).148 As successful examples, Neubert et al. demonstrated site-specific tethering fluorophore Cy5 to the GluN1 subunit of the NMDAR, in which TCO-K was incorporated site-specifically to allow super-resolution imaging of NMDARs.149 To the best of our knowledge, functional regulation of receptors has not been demonstrated using bio-orthogonal chemistry. However, considering the successful examples of functional switching of kinase activities in live cells using bio-orthogonal ligand tethering,150,151 this strategy would be applicable to functional switching of transmembrane receptors.
7. Chemogenetic regulation of endogenous receptors
In this section, we introduce chemogenetic methods to target endogenous proteins. Genetic modification is not introduced into the POI itself, which is largely different from the methods described in the other sections. Instead, tagged proteins or enzymes are exogenously expressed in target cells. This feature makes these methods most suitable to investigate the natural function of POIs endogenously expressed in cells.7.1. Membrane tethering of genetically encoded ligands
A classical technique to manipulate transmembrane receptors is the use of membrane-tethered antagonists or agonists. A membrane-tethered antagonist is t-toxin. T-toxin is composed of three components: an antagonist, transmembrane domain or glycosyl-phosphatidyl inositol (GPI) anchor, and linker (Fig. 12a).152 The linker structure needs to be optimized for the antagonist to act on receptors. Thanks to the genetically encodable properties, t-toxin can be expressed in a cell-specific manner. Ibanez-Tallon et al. reported the first study of genetically encoded membrane-tethered toxins,153 where the function of nAChR was suppressed by membrane-tethered α-bungarotoxin, a selective nAChR antagonist using a GPI anchor. In another study, Struzebecher et al. reported tethered μO-conotoxins via a GPI anchor for selective inhibition of voltage-gated sodium channels in a cell-specific manner.154 Moreover, t-toxins have been applied to investigate the function of membrane proteins in vivo.154,155 Although useful to inhibit receptors or ion channels in a cell-specific manner, inhibition is irreversible and persistent after expression of the toxin in this method. To overcome this limitation, Schmidt et al. reported a next-generation membrane-tethered toxin named “lumitoxin” (Fig. 12b).156 Lumitoxin contains membrane-anchored photoreceptor LOV2 connected to a K+-channel-specific peptide toxin (α-dendrotoxin) via a flexible linker. In lumitoxin, unfolding of the N-terminal helix of LOV2 decreases the local concentration of the toxin near the transmembrane. This strategy using LOV2 is theoretically applicable to other receptors and ion channels.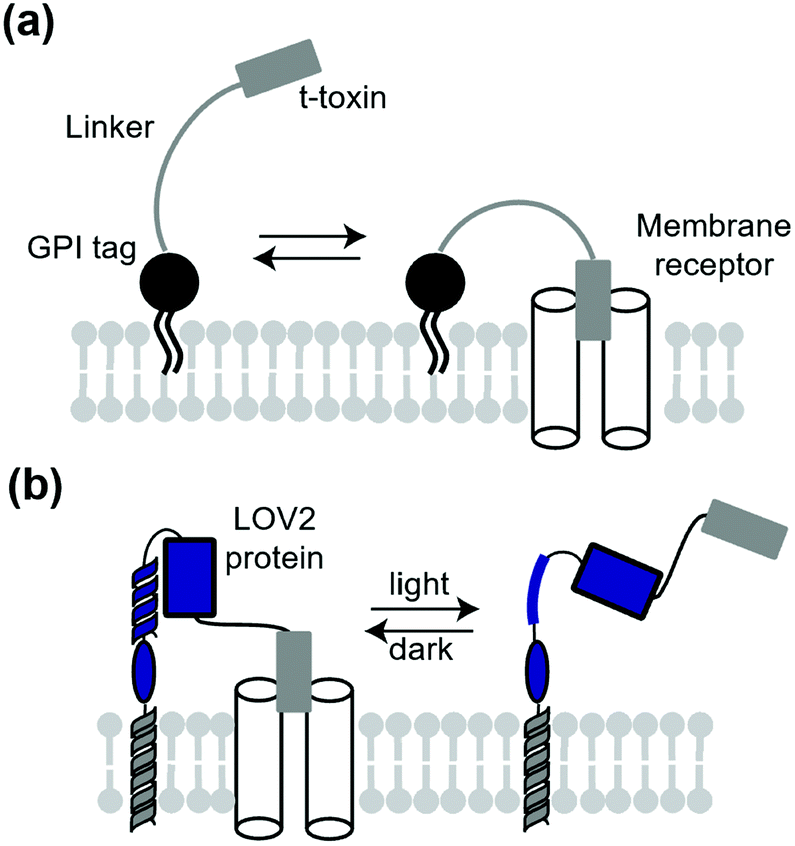 | ||
| Fig. 12 Schematic illustration of t-toxin and lumitoxin. (a) T-Toxin inhibits the target membrane receptor in a cell-specific manner. (b) Lumitoxin having LOV2 domain can reversibly control the target receptor by photo-irradiation. | ||
7.2. Membrane-tethered synthetic ligands
As stated above, a tethered toxin has good compatibility with genetic engineering because the ligand can be transfected in target cells. However, this approach is limited to genetically encodable ligands. To deliver a synthetic small molecule that targets an endogenous protein in a cell-specific manner, Shields et al. developed a method named “DART” (drugs acutely restricted by tethering) (Fig. 13a and b).157 In this method, a membrane-anchored Halo-tag is expressed to capture the synthetic ligand to the defined cell surface. They demonstrated the utility of the DART system using an antagonist of AMPAR conjugated with a Halo-tag ligand via a PEG linker (Fig. 13a and b). By tethering the ligand on the cell surface by the covalent bond to the Halo-tag, the effective molarity was increased, which resulted in a lower IC50 value than that of the diffusible ligand. This approach was successfully applied for functional investigation of endogenous AMPARs in cultured neurons and freely moving mice. The DART system was also applied to the muscarinic acetylcholine receptor. However, a drawback of the DART system is poor reversibility as is the case with t-toxin. To improve this aspect, Donthamsetti et al. developed a system that combines the membrane anchor of a DART with the photoswitchable control of a PORTL (see Section 5-3 for details of PORTL), which is termed “maPORTL” (membrane-anchored photoswitchable orthogonal remotely tethered ligand) (Fig. 13c and d).158 In maPORTL, a membrane-anchored SNAP-tag is used to tether the synthetic ligand to the defined cell surface instead of a Halo-tag in DART. Additionally, photoactivation is tuned by adjusting the length of the PORTL as well as the expression level and geometry of the membrane anchor. Consequently, endogenous mGlu2 was photo-controlled by maPORTL in primary cortical neurons. Notably, they applied the maPORTL system to dopamine D1 receptor (D1R), which allowed photoswitching of endogenous D1R in dorsal striatal medium spiny neurons in vivo.159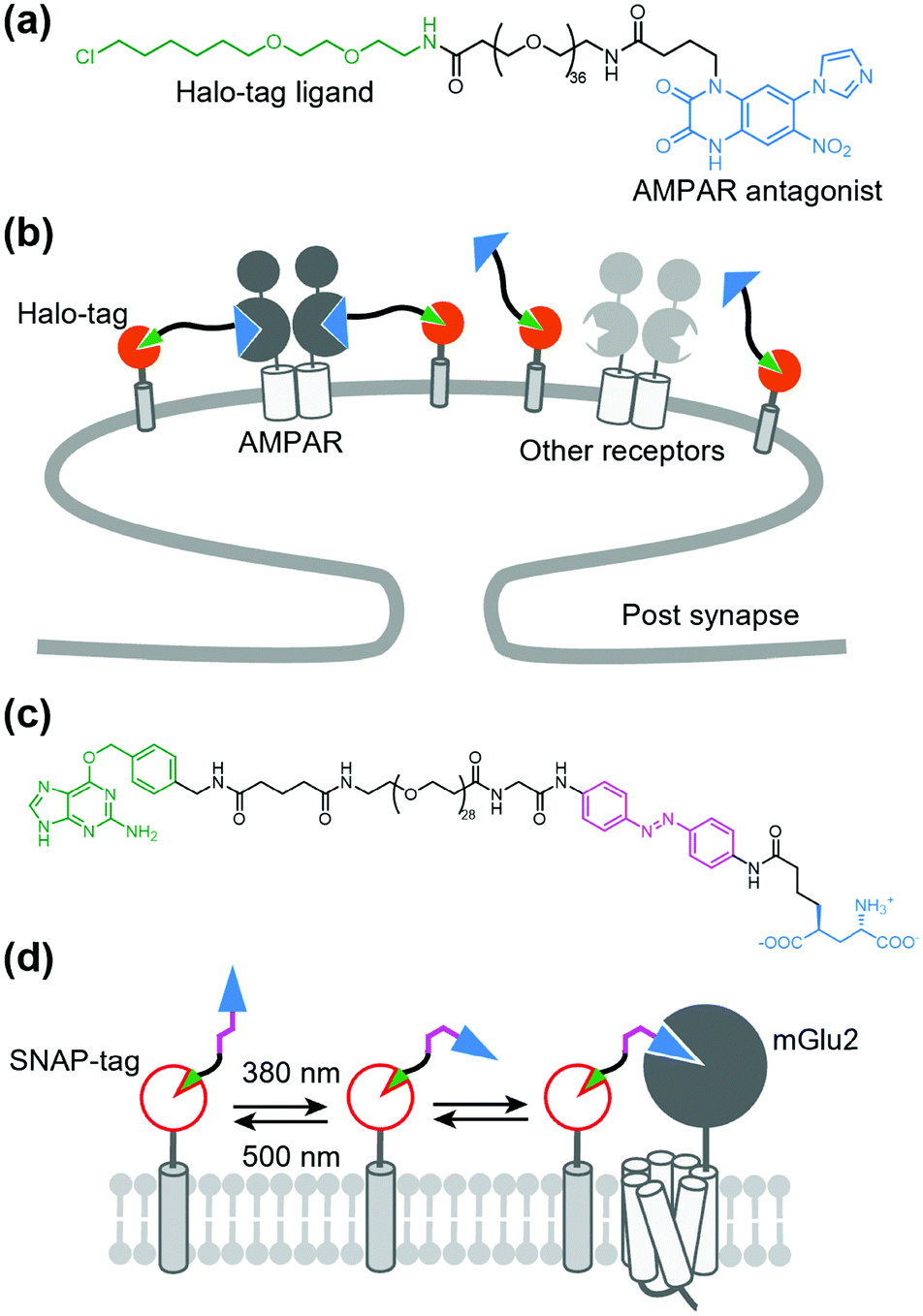 | ||
| Fig. 13 DART and maPORTL system for regulation of endogenous receptors. (a) Chemical structure of a designer ligand for DART system for AMPAR. (b) Schematic illustration of DART system for cell-specific inhibition of endogenous AMPARs. (c) Chemical structure of maPORTL ligand for mGlu2. (d) Schematic illustration of maPORTL system for photoswitching of endogenous mGlu2. | ||
7.3. Prodrug approaches
Cell-specific activation of a prodrug is another method to control endogenous receptors. In this approach, a unique enzyme that converts the inactive prodrug to its active form is expressed in target cells. As a representative study, Yang et al. reported cell-specific inhibition of NMDARs using a unique enzyme, porcine liver esterase (PLE), which selectively hydrolyzes methylcyclopropane carboxylic acid ester (CM) (Fig. 14a).160,161 They synthesized a prodrug, CM-MK801, in which MK801, an antagonist of NMDAR, was masked with CM ester. CM-MK801 was inert to endogenous esterase in neurons, but became active only in neurons that expressed PLE (Fig. 14b). This approach revealed that NMDARs are necessary for cocaine-induced synaptic potentiation in dopamine neurons. In addition to the PLE and CM pair, E. coli nitroreductase and the 2-nitro-N-methylimidazolyl group have been reported as another enzyme and substrate pair,162 which may allow orthogonal regulation of endogenous receptors. However, we should consider that the activated ligand can diffuse and may affect cells other than the target cells, especially in cases where the ligand-binding site is located in the extracellular region.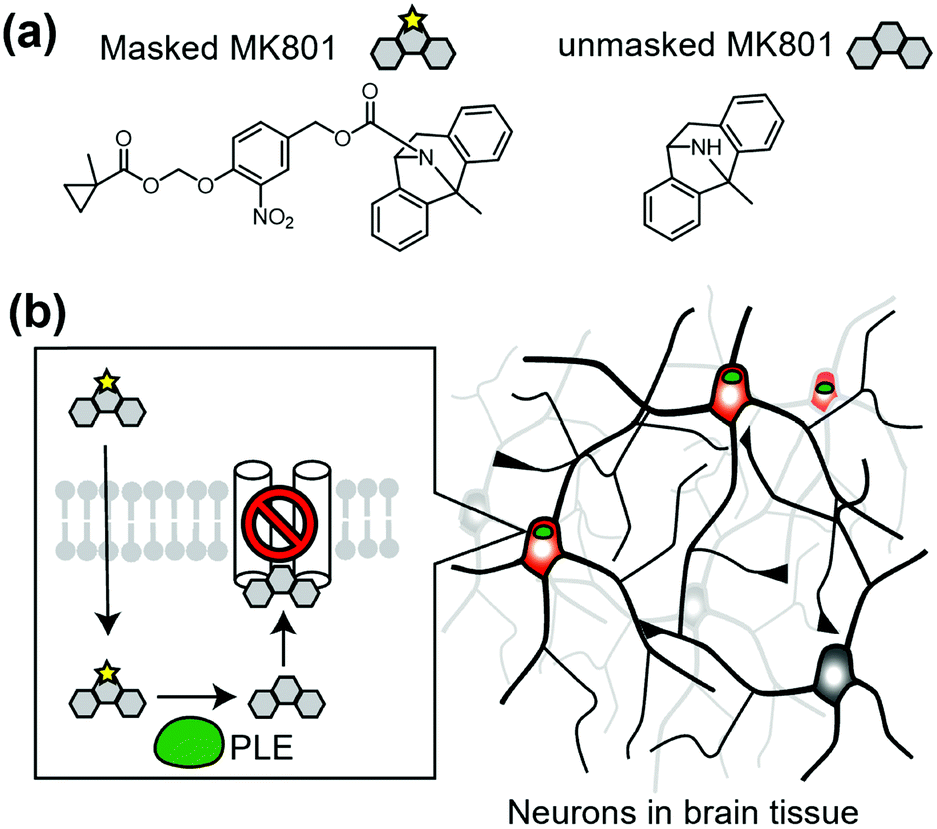 | ||
| Fig. 14 Chemogenetic regulation of endogenous NMDAR using prodrug approach. (a) Chemical structure of a PLE-specific prodrug for NMDAR. (b) Schematic illustration of cell-specific inhibition of NMDAR using prodrug. The prodrug is converted into active inhibitor, MK801 to inhibit NMDAR in a cell-specific manner by PLE. | ||
8. Conclusion and outlook
In this review, we summarized receptor chemogenetics to control receptor functions or downstream signals at the time of adding designer chemicals in a cell-specific manner. We also described functional regulation of receptors with high spatial and temporal resolutions by a combination of chemogenetics and photochromic regulation. If designer chemicals show favorable pharmacokinetics with sufficient selectivity for the designer receptor, chemogenetics can be applied in living animals. In fact, DREADD has been used in the neuroscience field to selectively activate target neurons or glial cells in freely moving animals, which has significantly improved understanding of the relationships between neuronal circuits and the expression of behaviors.163,164 Beyond its application in neuroscience, DREADD has been employed to dissect the role of G protein signaling in vivo to study cardiac functions and metabolic diseases such as type 2 diabetes and obesity.165,166 Although undoubtedly powerful, DREADD and related approaches require ectopic expression of designer receptors. In this context, chemogenetics for endogenous receptors such as the DART technique are useful.157 However, these examples remain limited, especially for in vivo applications. Thus, developing a new methodology for selective modulation of endogenous receptors in target cells in vivo is highly desired as the next direction of chemogenetics.Another direction of chemogenetics is clinical applications. Although chemogenetics have not been applied in human therapeutics or clinical trials, some studies have proceeded in this direction.11,167 A major concern in clinical use is applicability of designer chemicals to human. In this point, as described in Section 2, olanzapine, a clinically approved antipsychotic drug as a DREADD ligand42 and varenicline, an approved anti-smoking drug for PSAM/PSEM, would be potential candidates.53 Another concern is safety issues that originate from the designer receptors. Artificial receptors from other species may induce immune responses. In this context, the use of human-derived receptors with minimal mutations would be better to escape from the immune system. The third concern is delivery of a gene that encodes designer receptors. Considering recent progress in gene therapy studies, clinically acceptable viral vectors such as desirable AAVs would solve this concern.168,169 In summary, although there are obstacles for the clinical use of chemogenetics, some potential solutions have been proposed. Thus, considering the superiority compared with conventional genetic or pharmacological approaches, chemogenetics is an attractive approach for next-generation therapeutics.
Author contributions
All authors contributed to writing and editing the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Mitchell Arico from Edanz (https://jp.edanz.com/ac) for editing a draft of this manuscript. This work was funded by Grants-in-Aid for Scientific Research (KAKENHI) (Grant Number 19H05778, and 20H02877) and JST ERATO (JPMJER1802) to S. K.References
- A. V. Anzalone, L. W. Koblan and D. R. Liu, Nat. Biotechnol., 2020, 38, 824–844 CrossRef CAS PubMed.
- J. J. Gierut, T. E. Jacks and K. M. Haigis, Cold Spring Harb. Protoc., 2014, 2014, 339–349 Search PubMed.
- L. Fenno, O. Yizhar and K. Deisseroth, Annu. Rev. Neurosci., 2011, 34, 389–412 CrossRef CAS PubMed.
- E. S. Boyden, F. Zhang, E. Bamberg, G. Nagel and K. Deisseroth, Nat. Neurosci., 2005, 8, 1263–1268 CrossRef CAS PubMed.
- R. D. Airan, K. R. Thompson, L. E. Fenno, H. Bernstein and K. Deisseroth, Nature, 2009, 458, 1025–1029 CrossRef CAS PubMed.
- S. L. Schreiber, Bioorg. Med. Chem., 1998, 6, 1127–1152 CrossRef CAS.
- D. P. Walsh and Y. T. Chang, Chem. Rev., 2006, 106, 2476–2530 CrossRef CAS.
- D. R. Spring, Chem. Soc. Rev., 2005, 34, 472–482 RSC.
- Z. A. Knight and K. M. Shokat, Cell, 2007, 128, 425–430 CrossRef CAS PubMed.
- K. Islam, ACS Chem. Biol., 2015, 10, 343–363 CrossRef CAS PubMed.
- O. Keifer, K. Kambara, A. Lau, S. Makinson and D. Bertrand, Biochem. Pharmacol., 2020, 175, 113889 CrossRef CAS PubMed.
- J. Bang, H. Y. Kim and H. Lee, Exp. Neurobiol., 2016, 25, 205–221 CrossRef PubMed.
- K. Vlasov, C. J. Van Dort and K. Solt, Methods Enzymol., 2018, 603, 181–196 CAS.
- M. C. Walker and D. M. Kullmann, Neuropharmacology, 2020, 168, 107751 CrossRef CAS PubMed.
- Y. W. Hwang and D. L. Miller, J. Biol. Chem., 1987, 262, 13081–13085 CrossRef CAS.
- A. C. Bishop, J. A. Ubersax, D. T. Petsch, D. P. Matheos, N. S. Gray, J. Blethrow, E. Shimizu, J. Z. Tsien, P. G. Schultz, M. D. Rose, J. L. Wood, D. O. Morgan and K. M. Shokat, Nature, 2000, 407, 395–401 CrossRef CAS PubMed.
- M. G. J. Baud, E. Lin-Shiao, T. Cardote, C. Tallant, A. Pschibul, K. H. Chan, M. Zengerle, J. R. Garcia, T. T. Kwan, F. M. Ferguson and A. Ciulli, Science, 2014, 346, 638–641 CrossRef CAS PubMed.
- M. Breski, D. Dey, S. Obringer, B. Sudhamalla and K. Islam, J. Am. Chem. Soc., 2016, 138, 13505–13508 CrossRef CAS PubMed.
- N. Uchida, K. Takahashi, R. Iwasaki, R. Yamada, M. Yoshimura, T. A. Endo, S. Kimura, H. Zhang, M. Nomoto, Y. Tada, T. Kinoshita, K. Itami, S. Hagihara and K. U. Torii, Nat. Chem. Biol., 2018, 14, 299–305 CrossRef CAS PubMed.
- R. DeRose, T. Miyamoto and T. Inoue, Pflugers Arch., 2013, 465, 409–417 CrossRef CAS PubMed.
- G. M. Burslem and C. M. Crews, Chem. Rev., 2017, 117, 11269–11301 CrossRef CAS PubMed.
- S. R. Neves, P. T. Ram and R. Iyengar, Science, 2002, 296, 1636–1639 CrossRef CAS PubMed.
- H. Kaur, J. Carvalho, M. Looso, P. Singh, R. Chennupati, J. Preussner, S. Gunther, J. Albarran-Juarez, D. Tischner, S. Classen, S. Offermanns and N. Wettschureck, Nat. Commun., 2017, 8, 15700 CrossRef CAS PubMed.
- C. D. Strader, T. Gaffney, E. E. Sugg, M. R. Candelore, R. Keys, A. A. Patchett and R. A. Dixon, J. Biol. Chem., 1991, 266, 5–8 CrossRef CAS.
- P. Coward, H. G. Wada, M. S. Falk, S. D. Chan, F. Meng, H. Akil and B. R. Conklin, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 352–357 CrossRef CAS PubMed.
- C. H. Redfern, P. Coward, M. Y. Degtyarev, E. K. Lee, A. T. Kwa, L. Hennighausen, H. Bujard, G. I. Fishman and B. R. Conklin, Nat. Biotechnol., 1999, 17, 165–169 CrossRef CAS.
- K. M. Small, K. M. Brown, S. L. Forbes and S. B. Liggett, J. Biol. Chem., 2001, 276, 31596–31601 CrossRef CAS PubMed.
- B. R. Conklin, E. C. Hsiao, S. Claeysen, A. Dumuis, S. Srinivasan, J. R. Forsayeth, J. M. Guettier, W. C. Chang, Y. Pei, K. D. McCarthy, R. A. Nissenson, J. Wess, J. Bockaert and B. L. Roth, Nat. Methods, 2008, 5, 673–678 CrossRef CAS PubMed.
- E. J. Sweger, K. B. Casper, K. Scearce-Levie, B. R. Conklin and K. D. McCarthy, J. Neurosci., 2007, 27, 2309–2317 CrossRef CAS PubMed.
- J. Peng, M. Bencsik, A. Louie, W. Lu, S. Millard, P. Nguyen, A. Burghardt, S. Majumdar, T. J. Wronski, B. Halloran, B. R. Conklin and R. A. Nissenson, Endocrinology, 2008, 149, 1329–1337 CrossRef CAS PubMed.
- B. N. Armbruster, X. Li, M. H. Pausch, S. Herlitze and B. L. Roth, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5163–5168 CrossRef PubMed.
- J. M. Guettier, D. Gautam, M. Scarselli, I. Ruiz de Azua, J. H. Li, E. Rosemond, X. Ma, F. J. Gonzalez, B. N. Armbruster, H. Lu, B. L. Roth and J. Wess, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 19197–19202 CrossRef CAS PubMed.
- A. Inoue, F. Raimondi, F. M. N. Kadji, G. Singh, T. Kishi, A. Uwamizu, Y. Ono, Y. Shinjo, S. Ishida, N. Arang, K. Kawakami, J. S. Gutkind, J. Aoki and R. B. Russell, Cell, 2019, 177(1933–1947), e1925 Search PubMed.
- K. Nakajima and J. Wess, Mol. Pharmacol., 2012, 82, 575–582 CrossRef CAS PubMed.
- J. Hu, M. Stern, L. E. Gimenez, L. Wanka, L. Zhu, M. Rossi, J. Meister, A. Inoue, A. G. Beck-Sickinger, V. V. Gurevich and J. Wess, J. Biol. Chem., 2016, 291, 7809–7820 CrossRef CAS PubMed.
- P. D. Whissell, S. Tohyama and L. J. Martin, Front Genet, 2016, 7, 70 Search PubMed.
- J. Meister, L. Wang, S. P. Pydi and J. Wess, J. Neurochem., 2021, 158, 603–620 CrossRef CAS PubMed.
- J. L. Gomez, J. Bonaventura, W. Lesniak, W. B. Mathews, P. Sysa-Shah, L. A. Rodriguez, R. J. Ellis, C. T. Richie, B. K. Harvey, R. F. Dannals, M. G. Pomper, A. Bonci and M. Michaelides, Science, 2017, 357, 503–507 CrossRef CAS PubMed.
- N. Brunello, C. Masotto, L. Steardo, R. Markstein and G. Racagni, Neuropsychopharmacology, 1995, 13, 177–213 CrossRef CAS PubMed.
- X. Chen, H. Choo, X. P. Huang, X. Yang, O. Stone, B. L. Roth and J. Jin, ACS Chem. Neurosci., 2015, 6, 476–484 CrossRef CAS PubMed.
- Y. Nagai, N. Miyakawa, H. Takuwa, Y. Hori, K. Oyama, B. Ji, M. Takahashi, X. P. Huang, S. T. Slocum, J. F. DiBerto, Y. Xiong, T. Urushihata, T. Hirabayashi, A. Fujimoto, K. Mimura, J. G. English, J. Liu, K. I. Inoue, K. Kumata, C. Seki, M. Ono, M. Shimojo, M. R. Zhang, Y. Tomita, J. Nakahara, T. Suhara, M. Takada, M. Higuchi, J. Jin, B. L. Roth and T. Minamimoto, Nat. Neurosci., 2020, 23, 1157–1167 CrossRef CAS PubMed.
- M. Weston, T. Kaserer, A. Wu, A. Mouravlev, J. C. Carpenter, A. Snowball, S. Knauss, M. von Schimmelmann, M. J. During, G. Lignani, S. Schorge, D. Young, D. M. Kullmann and A. Lieb, Sci. Adv., 2019, 5, eaaw1567 CrossRef CAS PubMed.
- E. Vardy, J. E. Robinson, C. Li, R. H. J. Olsen, J. F. DiBerto, P. M. Giguere, F. M. Sassano, X. P. Huang, H. Zhu, D. J. Urban, K. L. White, J. E. Rittiner, N. A. Crowley, K. E. Pleil, C. M. Mazzone, P. D. Mosier, J. Song, T. L. Kash, C. J. Malanga, M. J. Krashes and B. L. Roth, Neuron, 2015, 86, 936–946 CrossRef CAS PubMed.
- B. D. Hudson, E. Christiansen, I. G. Tikhonova, M. Grundmann, E. Kostenis, D. R. Adams, T. Ulven and G. Milligan, FASEB J., 2012, 26, 4951–4965 CrossRef CAS PubMed.
- N. Barki, D. Bolognini, U. Börjesson, L. Jenkins, J. Riddell, D. I. Hughes, T. Ulven, B. D. Hudson, E. R. Ulven, N. Dekker, A. B. Tobin and G. Milligan, bioRxiv, 2020 DOI:10.1101/2020.01.11.902726.
- Q. Cheng, J. C. Kulli and J. Yang, J. Neurosci., 2001, 21, 3419–3428 CrossRef CAS PubMed.
- E. M. Slimko, S. McKinney, D. J. Anderson, N. Davidson and H. A. Lester, J. Neurosci., 2002, 22, 7373–7379 CrossRef CAS PubMed.
- S. J. Frazier, B. N. Cohen and H. A. Lester, J. Biol. Chem., 2013, 288, 21029–21042 CrossRef CAS PubMed.
- T. Lynagh and J. W. Lynch, J. Biol. Chem., 2010, 285, 14890–14897 CrossRef CAS PubMed.
- R. Islam, A. Keramidas, L. Xu, N. Durisic, P. Sah and J. W. Lynch, ACS Chem. Neurosci., 2016, 7, 1647–1657 CrossRef CAS PubMed.
- C. J. Magnus, P. H. Lee, D. Atasoy, H. H. Su, L. L. Looger and S. M. Sternson, Science, 2011, 333, 1292–1296 CrossRef CAS PubMed.
- D. Atasoy and S. M. Sternson, Physiol. Rev., 2018, 98, 391–418 CrossRef CAS PubMed.
- C. J. Magnus, P. H. Lee, J. Bonaventura, R. Zemla, J. L. Gomez, M. H. Ramirez, X. Hu, A. Galvan, J. Basu, M. Michaelides and S. M. Sternson, Science, 2019, 364, eaav5282 CrossRef CAS PubMed.
- C. Blanpain, J. M. Vanderwinden, J. Cihak, V. Wittamer, E. Le Poul, H. Issafras, M. Stangassinger, G. Vassart, S. Marullo, D. Schlndorff, M. Parmentier and M. Mack, Mol. Biol. Cell, 2002, 13, 723–737 CrossRef CAS PubMed.
- C. E. Elling, S. M. Nielsen and T. W. Schwartz, Nature, 1995, 374, 74–77 CrossRef CAS PubMed.
- C. E. Elling, K. Thirstrup, B. Holst and T. W. Schwartz, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 12322–12327 CrossRef CAS PubMed.
- C. E. Elling, T. M. Frimurer, L. O. Gerlach, R. Jorgensen, B. Holst and T. W. Schwartz, J. Biol. Chem., 2006, 281, 17337–17346 CrossRef CAS PubMed.
- B. Holst, C. E. Elling and T. W. Schwartz, Mol. Pharmacol., 2000, 58, 263–270 CrossRef CAS PubMed.
- M. C. Lagerstrom, J. Klovins, R. Fredriksson, D. Fridmanis, T. Haitina, M. K. Ling, M. M. Berglund and H. B. Schioth, J. Biol. Chem., 2003, 278, 51521–51526 CrossRef PubMed.
- J. Mokrosinski, T. M. Frimurer, B. Sivertsen, T. W. Schwartz and B. Holst, J. Biol. Chem., 2012, 287, 33488–33502 CrossRef CAS PubMed.
- S. Kiyonaka, R. Kubota, Y. Michibata, M. Sakakura, H. Takahashi, T. Numata, R. Inoue, M. Yuzaki and I. Hamachi, Nat. Chem., 2016, 8, 958–967 CrossRef CAS PubMed.
- R. Kubota, S. Kiyonaka and I. Hamachi, Methods Enzymol., 2019, 622, 411–430 CAS.
- K. Ojima, W. Kakegawa, M. Ito, Y. Miura, Y. Michibata, R. Kubota, T. Doura, T. Yamasaki, E. Miura, H. Nonaka, S. Mizuno, S. Takahashi, M. Yuzaki, I. Hamachi and S. Kiyonaka, bioRxiv, 2021 DOI:10.1101/2021.10.01.462737.
- B. Z. Stanton, E. J. Chory and G. R. Crabtree, Science, 2018, 359, eaao5902 CrossRef PubMed.
- L. Klewer and Y. W. Wu, Chem. – Eur. J., 2019, 25, 12452–12463 CrossRef CAS PubMed.
- D. M. Spencer, T. J. Wandless, S. L. Schreiber and G. R. Crabtree, Science, 1993, 262, 1019–1024 CrossRef CAS PubMed.
- S. D. Liberles, S. T. Diver, D. J. Austin and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 7825–7830 CrossRef CAS PubMed.
- P. J. Belshaw, S. N. Ho, G. R. Crabtree and S. L. Schreiber, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 4604–4607 CrossRef CAS PubMed.
- J. Choi, J. Chen, S. L. Schreiber and J. Clardy, Science, 1996, 273, 239–242 CrossRef CAS PubMed.
- B. R. Stockwell and S. L. Schreiber, Curr. Biol., 1998, 8, 761–770 CrossRef CAS PubMed.
- A. Fegan, B. White, J. C. Carlson and C. R. Wagner, Chem. Rev., 2010, 110, 3315–3336 CrossRef CAS PubMed.
- T. Clackson, W. Yang, L. W. Rozamus, M. Hatada, J. F. Amara, C. T. Rollins, L. F. Stevenson, S. R. Magari, S. A. Wood, N. L. Courage, X. Lu, F. Cerasoli, Jr., M. Gilman and D. A. Holt, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 10437–10442 CrossRef CAS PubMed.
- C. Y. Wu, K. T. Roybal, E. M. Puchner, J. Onuffer and W. A. Lim, Science, 2015, 350, aab4077 CrossRef PubMed.
- M. J. Harris, M. Fuyal and J. R. James, Mol. Syst. Biol., 2021, 17, e10091 CrossRef CAS PubMed.
- M. Putyrski and C. Schultz, Chem. Biol., 2011, 18, 1126–1133 CrossRef CAS PubMed.
- K. Stankunas, J. H. Bayle, J. E. Gestwicki, Y. M. Lin, T. J. Wandless and G. R. Crabtree, Mol. Cell, 2003, 12, 1615–1624 CrossRef CAS PubMed.
- S. M. G. Braun, J. G. Kirkland, E. J. Chory, D. Husmann, J. P. Calarco and G. R. Crabtree, Nat. Commun., 2017, 8, 560 CrossRef PubMed.
- D. Kashima, M. Kageoka, Y. Kimura, M. Horikawa, M. Miura, M. Nakakido, K. Tsumoto, T. Nagamune and M. Kawahara, ACS Synth. Biol., 2021, 10, 990–999 CrossRef CAS PubMed.
- D. Ma, S. Peng and Z. Xie, Nat. Commun., 2016, 7, 13056 CrossRef CAS PubMed.
- O. Dagliyan, A. Krokhotin, I. Ozkan-Dagliyan, A. Deiters, C. J. Der, K. M. Hahn and N. V. Dokholyan, Nat. Commun., 2018, 9, 4042 CrossRef PubMed.
- A. Y. Karpova, D. G. Tervo, N. W. Gray and K. Svoboda, Neuron, 2005, 48, 727–735 CrossRef CAS PubMed.
- T. Miyamoto, R. DeRose, A. Suarez, T. Ueno, M. Chen, T. P. Sun, M. J. Wolfgang, C. Mukherjee, D. J. Meyers and T. Inoue, Nat. Chem. Biol., 2012, 8, 465–470 CrossRef CAS PubMed.
- S. R. Cutler, P. L. Rodriguez, R. R. Finkelstein and S. R. Abrams, Annu. Rev. Plant Biol., 2010, 61, 651–679 CrossRef CAS PubMed.
- H. D. Wu, M. Kikuchi, O. Dagliyan, A. K. Aragaki, H. Nakamura, N. V. Dokholyan, T. Umehara and T. Inoue, Nat. Methods, 2020, 17, 928–936 CrossRef PubMed.
- M. Ishida, H. Watanabe, K. Takigawa, Y. Kurishita, C. Oki, A. Nakamura, I. Hamachi and S. Tsukiji, J. Am. Chem. Soc., 2013, 135, 12684–12689 CrossRef CAS PubMed.
- A. Nakamura, C. Oki, S. Sawada, T. Yoshii, K. Kuwata, A. K. Rudd, N. K. Devaraj, K. Noma and S. Tsukiji, ACS Chem. Biol., 2020, 15, 837–843 CrossRef CAS PubMed.
- D. Krueger, E. Izquierdo, R. Viswanathan, J. Hartmann, C. Pallares Cartes and S. De Renzis, Development, 2019, 146, dev175067 CrossRef CAS PubMed.
- K. Shao, X. Zhang, X. Li, Y. Hao, X. Huang, M. Ma, M. Zhang, F. Yu, H. Liu and P. Zhang, Nat. Struct. Mol. Biol., 2020, 27, 480–488 CrossRef CAS PubMed.
- F. Kawano, H. Suzuki, A. Furuya and M. Sato, Nat. Commun., 2015, 6, 6256 CrossRef CAS PubMed.
- G. Guntas, R. A. Hallett, S. P. Zimmerman, T. Williams, H. Yumerefendi, J. E. Bear and B. Kuhlman, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 112–117 CrossRef CAS PubMed.
- K. Y. Chang, D. Woo, H. Jung, S. Lee, S. Kim, J. Won, T. Kyung, H. Park, N. Kim, H. W. Yang, J. Y. Park, E. M. Hwang, D. Kim and W. D. Heo, Nat. Commun., 2014, 5, 4057 CrossRef CAS PubMed.
- M. Grusch, K. Schelch, R. Riedler, E. Reichhart, C. Differ, W. Berger, A. Ingles-Prieto and H. Janovjak, EMBO J., 2014, 33, 1713–1726 CrossRef CAS PubMed.
- S. Kainrath, M. Stadler, E. Reichhart, M. Distel and H. Janovjak, Angew. Chem., Int. Ed., 2017, 56, 4608–4611 CrossRef CAS PubMed.
- E. Reichhart, A. Ingles-Prieto, A. M. Tichy, C. McKenzie and H. Janovjak, Angew. Chem., Int. Ed., 2016, 55, 6339–6342 CrossRef CAS PubMed.
- A. V. Leopold, K. G. Chernov, A. A. Shemetov and V. V. Verkhusha, Nat. Commun., 2019, 10, 1129 CrossRef PubMed.
- O. Takenouchi, H. Yoshimura and T. Ozawa, Sci. Rep., 2018, 8, 677 CrossRef PubMed.
- B. L. Sinnen, A. B. Bowen, J. S. Forte, B. G. Hiester, K. C. Crosby, E. S. Gibson, M. L. Dell'Acqua and M. J. Kennedy, Neuron, 2017, 93, 646–660 e645 CrossRef CAS PubMed.
- L. J. Bugaj, D. P. Spelke, C. K. Mesuda, M. Varedi, R. S. Kane and D. V. Schaffer, Nat. Commun., 2015, 6, 6898 CrossRef CAS PubMed.
- M. Endo, M. Hattori, H. Toriyabe, H. Ohno, H. Kamiguchi, Y. Iino and T. Ozawa, Sci. Rep., 2016, 6, 23976 CrossRef CAS PubMed.
- A. Taslimi, J. D. Vrana, D. Chen, S. Borinskaya, B. J. Mayer, M. J. Kennedy and C. L. Tucker, Nat. Commun., 2014, 5, 4925 CrossRef CAS PubMed.
- T. Kyung, S. Lee, J. E. Kim, T. Cho, H. Park, Y. M. Jeong, D. Kim, A. Shin, S. Kim, J. Baek, J. Kim, N. Y. Kim, D. Woo, S. Chae, C. H. Kim, H. S. Shin, Y. M. Han, D. Kim and W. D. Heo, Nat. Biotechnol., 2015, 33, 1092–1096 CrossRef CAS PubMed.
- C. Aonbangkhen, H. Zhang, D. Z. Wu, M. A. Lampson and D. M. Chenoweth, J. Am. Chem. Soc., 2018, 140, 11926–11930 CrossRef CAS PubMed.
- N. Umeda, T. Ueno, C. Pohlmeyer, T. Nagano and T. Inoue, J. Am. Chem. Soc., 2011, 133, 12–14 CrossRef CAS PubMed.
- T. Yoshii, C. Oki, R. Watahiki, A. Nakamura, K. Tahara, K. Kuwata, T. Furuta and S. Tsukiji, ACS Chem. Biol., 2021, 16, 1557–1565 CrossRef CAS PubMed.
- M. Zimmermann, R. Cal, E. Janett, V. Hoffmann, C. G. Bochet, E. Constable, F. Beaufils and M. P. Wymann, Angew. Chem., Int. Ed., 2014, 53, 4717–4720 CrossRef CAS PubMed.
- Y. H. Tsai, T. Doura and S. Kiyonaka, Chem. Soc. Rev., 2021, 50, 7909–7923 RSC.
- R. O. Blaustein, P. A. Cole, C. Williams and C. Miller, Nat. Struct. Biol., 2000, 7, 309–311 CrossRef CAS PubMed.
- M. A. Kienzler, A. Reiner, E. Trautman, S. Yoo, D. Trauner and E. Y. Isacoff, J. Am. Chem. Soc., 2013, 135, 17683–17686 CrossRef CAS PubMed.
- M. Izquierdo-Serra, M. Gascon-Moya, J. J. Hirtz, S. Pittolo, K. E. Poskanzer, E. Ferrer, R. Alibes, F. Busque, R. Yuste, J. Hernando and P. Gorostiza, J. Am. Chem. Soc., 2014, 136, 8693–8701 CrossRef CAS PubMed.
- G. Cabre, A. Garrido-Charles, M. Moreno, M. Bosch, M. Porta-de-la-Riva, M. Krieg, M. Gascon-Moya, N. Camarero, R. Gelabert, J. M. Lluch, F. Busque, J. Hernando, P. Gorostiza and R. Alibes, Nat. Commun., 2019, 10, 907 CrossRef PubMed.
- A. Rullo, A. Reiner, A. Reiter, D. Trauner, E. Y. Isacoff and G. A. Woolley, Chem. Commun., 2014, 50, 14613–14615 RSC.
- S. Samanta, A. A. Beharry, O. Sadovski, T. M. McCormick, A. Babalhavaeji, V. Tropepe and G. A. Woolley, J. Am. Chem. Soc., 2013, 135, 9777–9784 CrossRef CAS PubMed.
- C. Knie, M. Utecht, F. Zhao, H. Kulla, S. Kovalenko, A. M. Brouwer, P. Saalfrank, S. Hecht and D. Bleger, Chem. – Eur. J., 2014, 20, 16492–16501 CrossRef CAS PubMed.
- E. R. Thapaliya, J. Zhao and G. C. R. Ellis-Davies, ACS Chem. Neurosci., 2019, 10, 2481–2488 CrossRef CAS PubMed.
- G. Cabre, A. Garrido-Charles, A. Gonzalez-Lafont, W. Moormann, D. Langbehn, D. Egea, J. M. Lluch, R. Herges, R. Alibes, F. Busque, P. Gorostiza and J. Hernando, Org. Lett., 2019, 21, 3780–3784 CrossRef CAS PubMed.
- N. N. Mafy, K. Matsuo, S. Hiruma, R. Uehara and N. Tamaoki, J. Am. Chem. Soc., 2020, 142, 1763–1767 CrossRef CAS PubMed.
- M. Banghart, K. Borges, E. Isacoff, D. Trauner and R. H. Kramer, Nat. Neurosci., 2004, 7, 1381–1386 CrossRef CAS PubMed.
- K. Hull, J. Morstein and D. Trauner, Chem. Rev., 2018, 118, 10710–10747 CrossRef CAS PubMed.
- W. C. Lin and R. H. Kramer, Bioconjugate Chem., 2018, 29, 861–869 CrossRef CAS PubMed.
- S. Szobota, P. Gorostiza, F. Del Bene, C. Wyart, D. L. Fortin, K. D. Kolstad, O. Tulyathan, M. Volgraf, R. Numano, H. L. Aaron, E. K. Scott, R. H. Kramer, J. Flannery, H. Baier, D. Trauner and E. Y. Isacoff, Neuron, 2007, 54, 535–545 CrossRef CAS PubMed.
- N. Caporale, K. D. Kolstad, T. Lee, I. Tochitsky, D. Dalkara, D. Trauner, R. Kramer, Y. Dan, E. Y. Isacoff and J. G. Flannery, Mol. Ther., 2011, 19, 1212–1219 CrossRef CAS PubMed.
- S. Berlin, S. Szobota, A. Reiner, E. C. Carroll, M. A. Kienzler, A. Guyon, T. Xiao, D. Trauner and E. Y. Isacoff, eLife, 2016, 5, e12040 CrossRef PubMed.
- G. Sandoz, J. Levitz, R. H. Kramer and E. Y. Isacoff, Neuron, 2012, 74, 1005–1014 CrossRef CAS PubMed.
- J. Levitz, C. Pantoja, B. Gaub, H. Janovjak, A. Reiner, A. Hoagland, D. Schoppik, B. Kane, P. Stawski, A. F. Schier, D. Trauner and E. Y. Isacoff, Nat. Neurosci., 2013, 16, 507–516 CrossRef CAS PubMed.
- W. C. Lin, M. C. Tsai, C. M. Davenport, C. M. Smith, J. Veit, N. M. Wilson, H. Adesnik and R. H. Kramer, Neuron, 2015, 88, 879–891 CrossRef CAS PubMed.
- P. Leippe, J. Koehler Leman and D. Trauner, Biochemistry, 2017, 56, 5214–5220 CrossRef CAS PubMed.
- J. Broichhagen, A. Damijonaitis, J. Levitz, K. R. Sokol, P. Leippe, D. Konrad, E. Y. Isacoff and D. Trauner, ACS Cent. Sci., 2015, 1, 383–393 CrossRef CAS PubMed.
- J. Levitz, J. Broichhagen, P. Leippe, D. Konrad, D. Trauner and E. Y. Isacoff, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E3546–E3554 CrossRef CAS PubMed.
- P. Leippe, J. Broichhagen, K. Cailliau, A. Mougel, M. Morel, C. Dissous, D. Trauner and J. Vicogne, Angew. Chem., Int. Ed., 2020, 59, 6720–6723 CrossRef CAS PubMed.
- A. Acosta-Ruiz, V. A. Gutzeit, M. J. Skelly, S. Meadows, J. Lee, P. Parekh, A. G. Orr, C. Liston, K. E. Pleil, J. Broichhagen and J. Levitz, Neuron, 2020, 105, 446–463.e13 CrossRef CAS PubMed.
- V. A. Gutzeit, A. Acosta-Ruiz, H. Munguba, S. Hafner, A. Landra-Willm, B. Mathes, J. Mony, D. Yarotski, K. Borjesson, C. Liston, G. Sandoz, J. Levitz and J. Broichhagen, Cell Chem. Biol., 2021, 28, 1648–1663 CrossRef CAS PubMed.
- M. H. Berry, A. Holt, J. Levitz, J. Broichhagen, B. M. Gaub, M. Visel, C. Stanley, K. Aghi, Y. J. Kim, K. Cao, R. H. Kramer, D. Trauner, J. Flannery and E. Y. Isacoff, Nat. Commun., 2017, 8, 1862 CrossRef PubMed.
- T. Podewin, J. Ast, J. Broichhagen, N. H. F. Fine, D. Nasteska, P. Leippe, M. Gailer, T. Buenaventura, N. Kanda, B. J. Jones, C. M'Kadmi, J. L. Baneres, J. Marie, A. Tomas, D. Trauner, A. Hoffmann-Roder and D. J. Hodson, ACS Cent. Sci., 2018, 4, 166–179 CrossRef CAS PubMed.
- H. Farrants, V. A. Gutzeit, A. Acosta-Ruiz, D. Trauner, K. Johnsson, J. Levitz and J. Broichhagen, ACS Chem. Biol., 2018, 13, 2682–2688 CrossRef CAS PubMed.
- R. Kubota, W. Nomura, T. Iwasaka, K. Ojima, S. Kiyonaka and I. Hamachi, ACS Cent. Sci., 2018, 4, 1211–1221 CrossRef CAS PubMed.
- J. W. Chin, Nature, 2017, 550, 53–60 CrossRef CAS PubMed.
- L. Wang, Acc. Chem. Res., 2017, 50, 2767–2775 CrossRef CAS PubMed.
- D. D. Young and P. G. Schultz, ACS Chem. Biol., 2018, 13, 854–870 CrossRef CAS PubMed.
- A. R. Nodling, L. A. Spear, T. L. Williams, L. Y. P. Luk and Y. H. Tsai, Essays Biochem., 2019, 63, 237–266 CrossRef PubMed.
- W. Wang, J. K. Takimoto, G. V. Louie, T. J. Baiga, J. P. Noel, K. F. Lee, P. A. Slesinger and L. Wang, Nat. Neurosci., 2007, 10, 1063–1072 CrossRef CAS PubMed.
- R. J. Ernst, T. P. Krogager, E. S. Maywood, R. Zanchi, V. Beranek, T. S. Elliott, N. P. Barry, M. H. Hastings and J. W. Chin, Nat. Chem. Biol., 2016, 12, 776–778 CrossRef CAS PubMed.
- S. Zhu, M. Riou, C. A. Yao, S. Carvalho, P. C. Rodriguez, O. Bensaude, P. Paoletti and S. Ye, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6081–6086 CrossRef CAS PubMed.
- M. Tian and S. Ye, Sci. Rep., 2016, 6, 34751 CrossRef CAS PubMed.
- V. Klippenstein, V. Ghisi, M. Wietstruk and A. J. Plested, J. Neurosci., 2014, 34, 980–991 CrossRef CAS PubMed.
- M. H. Poulsen, A. Poshtiban, V. Klippenstein, V. Ghisi and A. J. R. Plested, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 13358–13367 CrossRef CAS PubMed.
- J. Y. Kang, D. Kawaguchi, I. Coin, Z. Xiang, D. D. O'Leary, P. A. Slesinger and L. Wang, Neuron, 2013, 80, 358–370 CrossRef CAS PubMed.
- V. Klippenstein, C. Hoppmann, S. Ye, L. Wang and P. Paoletti, eLife, 2017, 6, e25808 CrossRef PubMed.
- K. Lang, L. Davis, S. Wallace, M. Mahesh, D. J. Cox, M. L. Blackman, J. M. Fox and J. W. Chin, J. Am. Chem. Soc., 2012, 134, 10317–10320 CrossRef CAS PubMed.
- F. Neubert, G. Beliu, U. Terpitz, C. Werner, C. Geis, M. Sauer and S. Doose, Angew. Chem., Int. Ed., 2018, 57, 16364–16369 CrossRef CAS PubMed.
- Y. H. Tsai, S. Essig, J. R. James, K. Lang and J. W. Chin, Nat. Chem., 2015, 7, 554–561 CrossRef CAS PubMed.
- C. W. Morgan, I. L. Dale, A. P. Thomas, J. Hunt and J. W. Chin, J. Am. Chem. Soc., 2021, 143, 4600–4606 CrossRef CAS PubMed.
- I. Ibanez-Tallon and M. N. Nitabach, Curr. Opin. Neurobiol., 2012, 22, 72–78 CrossRef CAS PubMed.
- I. Ibanez-Tallon, H. Wen, J. M. Miwa, J. Xing, A. B. Tekinay, F. Ono, P. Brehm and N. Heintz, Neuron, 2004, 43, 305–311 CrossRef CAS PubMed.
- A. S. Sturzebecher, J. Hu, E. S. Smith, S. Frahm, J. Santos-Torres, B. Kampfrath, S. Auer, G. R. Lewin and I. Ibanez-Tallon, J. Physiol., 2010, 588, 1695–1707 CrossRef PubMed.
- D. J. Christoffel, S. A. Golden, J. J. Walsh, K. G. Guise, M. Heshmati, A. K. Friedman, A. Dey, M. Smith, N. Rebusi, M. Pfau, J. L. Ables, H. Aleyasin, L. A. Khibnik, G. E. Hodes, G. A. Ben-Dor, K. Deisseroth, M. L. Shapiro, R. C. Malenka, I. Ibanez-Tallon, M. H. Han and S. J. Russo, Nat. Neurosci., 2015, 18, 962–964 CrossRef CAS PubMed.
- D. Schmidt, P. W. Tillberg, F. Chen and E. S. Boyden, Nat. Commun., 2014, 5, 3019 CrossRef PubMed.
- B. C. Shields, E. Kahuno, C. Kim, P. F. Apostolides, J. Brown, S. Lindo, B. D. Mensh, J. T. Dudman, L. D. Lavis and M. R. Tadross, Science, 2017, 356, eaaj2161 CrossRef PubMed.
- P. C. Donthamsetti, J. Broichhagen, V. Vyklicky, C. Stanley, Z. Fu, M. Visel, J. L. Levitz, J. A. Javitch, D. Trauner and E. Y. Isacoff, J. Am. Chem. Soc., 2019, 141, 11522–11530 CrossRef CAS PubMed.
- P. Donthamsetti, N. Winter, A. Hoagland, C. Stanley, M. Visel, S. Lammel, D. Trauner and E. Isacoff, Nat. Commun., 2021, 12, 4775 CrossRef CAS PubMed.
- L. Tian, Y. Yang, L. M. Wysocki, A. C. Arnold, A. Hu, B. Ravichandran, S. M. Sternson, L. L. Looger and L. D. Lavis, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4756–4761 CrossRef CAS PubMed.
- Y. Yang, P. Lee and S. M. Sternson, eLife, 2015, 4, e10206 CrossRef PubMed.
- T. D. Gruber, C. Krishnamurthy, J. B. Grimm, M. R. Tadross, L. M. Wysocki, Z. J. Gartner and L. D. Lavis, ACS Chem. Biol., 2018, 13, 2888–2896 CrossRef CAS PubMed.
- B. L. Roth, Neuron, 2016, 89, 683–694 CrossRef CAS PubMed.
- A. Ozawa and H. Arakawa, Behav. Brain Res., 2021, 406, 113234 CrossRef CAS PubMed.
- E. Kaiser, Q. Tian, M. Wagner, M. Barth, W. Xian, L. Schroder, S. Ruppenthal, L. Kaestner, U. Boehm, P. Wartenberg, H. Lu, S. M. McMillin, D. B. J. Bone, J. Wess and P. Lipp, Cardiovasc. Res., 2019, 115, 1052–1066 CrossRef CAS PubMed.
- L. Wang, L. Zhu, J. Meister, D. B. J. Bone, S. P. Pydi, M. Rossi and J. Wess, Annu. Rev. Pharmacol. Toxicol., 2021, 61, 421–440 CrossRef CAS PubMed.
- A. Lieb, M. Weston and D. M. Kullmann, EBioMedicine, 2019, 43, 641–649 CrossRef PubMed.
- D. Wang, P. W. L. Tai and G. Gao, Nat Rev Drug Discovery, 2019, 18, 358–378 CrossRef CAS PubMed.
- C. Li and R. J. Samulski, Nat. Rev. Genet., 2020, 21, 255–272 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |