
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnabling the next steps in cancer immunotherapy: from antibody-based bispecifics to multispecifics, with an evolving role for bioconjugation chemistry
Fabien
Thoreau
* and
Vijay
Chudasama
 *
*
Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: f.thoreau@ucl.ac.uk; v.chudasama@ucl.ac.uk
First published on 22nd October 2021
Abstract
In the past two decades, immunotherapy has established itself as one of the leading strategies for cancer treatment, as illustrated by the exponentially growing number of related clinical trials. This trend was, in part, prompted by the clinical success of both immune checkpoint modulation and immune cell engagement, to restore and/or stimulate the patient's immune system's ability to fight the disease. These strategies were sustained by progress in bispecific antibody production. However, despite the decisive progress made in the treatment of cancer, toxicity and resistance are still observed in some cases. In this review, we initially provide an overview of the monoclonal and bispecific antibodies developed with the objective of restoring immune system functions to treat cancer (cancer immunotherapy), through immune checkpoint modulation, immune cell engagement or a combination of both. Their production, design strategy and impact on the clinical trial landscape are also addressed. In the second part, the concept of multispecific antibody formats, notably MuTICEMs (Multispecific Targeted Immune Cell Engagers & Modulators), as a possible answer to current immunotherapy limitations is investigated. We believe it could be the next step to take for cancer immunotherapy research and expose why bioconjugation chemistry might play a key role in these future developments.
I. Introduction
The general idea of exploiting antibodies as a therapeutic tool is an old concept (19th century).1 However, the incomplete understanding of the immune system, immunogenicity issues and complexity of antibody production held back the development of immunotherapies and the concept was reborn from the ashes only recently.2 The first breakthrough was made with monoclonal antibodies: following the discovery in 1974 – and the related Nobel prizes in 1984 – of the hybridoma technology by C. Milstein and G. J. F. Köhler that allows the production of a large number of monoclonal antibodies, the development of recombinant and fused antibodies was made possible in the mid-1980s.3,4 This progressively led to the production of chimeric, humanized and human monoclonal antibodies, which are less and less immunogenic and more and more efficient. As a consequence, monoclonal antibody FDA approvals and commercialisation took off in the early 2000s, mainly as anticancer treatments.5 The success of monoclonal antibodies is prompted by their binding affinity for tumour antigens that has agonistic, antagonistic or inhibitory effects on the antigens, while their Fc fragment can trigger an Fc-related immune response (antibody-dependent cell-mediated cytotoxicity (ADCC), complement-dependent cytotoxicity (CDC)).6,7 Despite evident beneficial therapeutic outcomes in some cases (e.g. Cetuximab, Bevacizumab, Trastuzumab),8 monoclonal antibodies didn’t improve the therapeutic window enough in other cases, due to their toxicity or lack of efficacy.9 To circumvent these issues, the antibody characteristics were exploited even further.2 One of the strategies adopted was the functionalisation of the antibody with one or several payloads, to generate so-called antibody–drug conjugates (ADCs). ADCs take advantage of the targeting ability of monoclonal antibodies to transport drug effectors such as doxorubicin and MMAE to the tumour site, in order to improve the anti-tumour efficacy. Other payloads such as fluorophores can be used for diagnostics. The ADC field has been reviewed extensively elsewhere.10–13 On the other hand, bispecific antibodies or bispecific constructs were developed and raised high expectations. Indeed, combining two different paratopes on the same full antibody, or combining two different antigen-binding fragments on a construct (through Fab, scFv, or single domain combination), allows the simultaneous targeting of two antigens or two different epitopes of the same antigen, potentially improving affinity, selectivity, and synergistic effects and reducing the risk of antigen loss and on-target toxicity.14,15 Interestingly, the ADC and bispecific concepts can be merged to yield bispecific ADCs. Some examples of this recent but promising approach have already been published and reviewed elsewhere.16A particular class of bispecific antibodies is dedicated to immune response activation (immunotherapy). These antibodies are actually divided into three sub-classes: immune cell redirectors, tumour-targeted immunomodulators, and dual immunomodulators, which can be described as follows. (a) Immune cell redirectors. The idea here is to recruit T cells or Natural Killer cells (NK cells) circulating in the body and redirect them against tumour cells, to trigger tumour destruction through immune activity. To this end, bispecific antibodies/constructs are armed with two different binding sites – one binding site has affinity for a tumour antigen (e.g. CD19, HER2) and the other has affinity for an immune cell antigen (e.g. CD3 for a T cell, forming a Bispecific T cell Engager (BiTE), or CD16 for an NK cell, forming a Bispecific Killer cell Engager (BiKE)). This cytotoxic effector cell redirector strategy represents the majority of bispecifics currently in pre-clinical and clinical trials, with encouraging results as will be discussed later. (b) Tumour-targeted immunomodulators. Their development is driven by the clinical success of inhibitory immune checkpoint inhibitors. Inhibitory immune checkpoints are proteins that are able to down-regulate immune cell activation through different mechanisms of action. For instance, the interaction between the PD-1 receptor on the T cell membrane and its PD-L1 ligand at the surfaces of tumour cells is a down-regulating signal for the T cell. Thus, a bispecific combining a tumour binding site with an antagonistic anti-PD-1 or anti-PD-L1 binding site could locally inhibit the down-regulation of the T cell activation and restore the immune response. Such a bispecific is a tumour-targeted inhibitory immune checkpoint inhibitor. Conversely, stimulatory immune checkpoints such as 4-1BB and OX40, whose stimulation enhances the activation of T cells, can be recruited to the tumour site using a bispecific compound comprising the corresponding ligand (4-1BBL or OX40L) and a tumour associated antigen (TAA). Such a bispecific is a tumour-targeted stimulatory immune checkpoint stimulator. (c) Dual immunomodulators. They are designed to target two immunomodulating targets among the aforementioned inhibitory and stimulatory immune checkpoints. They are thus expected to have a high impact on the immune response stimulation. However, as they are not tumour-targeted, they are more likely to induce associated adverse events such as cytokine release syndrome and are usually used in combination with other therapeutic agents (immunotherapeutic or chemotherapeutic agents).
Some recently published constructs fit in with two of the aforementioned classes: cytotoxic effector cell redirectors and tumour-targeted immunomodulators. Indeed, the TriKE (Trispecific Killer Engager) is a bispecific format engaging CD16 and CD33 to redirect NK cells to myeloid cancer cells,17 with these two binding sites being connected by a modified human IL-15, a cytokine able to induce the maintenance and activation of NK cells (and others). Similarly, Herrmann et al. developed a bifunctional checkpoint inhibitory T cell–engaging (CiTE) antibody, a trispecific construct (αCD3 × αCD33 × αPD-L1) combining a T cell redirection to AML cells with a local PD-L1 blockade ensured by the extracellular domain of PD-1 (PD-1ex) which exhibits a low affinity for PD-L1.18,19 Another concept, named SMITE (simultaneous multiple interaction T cell engaging), consists of a combination of two immune cell engagers (two BiTEs αCD3 × αTAA and αCD28 × αTAA, or two BiTEs αCD3 × αTAA and αCD28 × αPD-L1).20
The field of bispecific immunotherapeutics is wide and promising, and keeps evolving. The arrival of TriKEs, CITEs and SMITEs marks a step ahead, pushing the boundaries of the immunotherapy field, previously limited to dual specificity. It seems this trend can be extended further with a new class of immunotherapeutics: multispecifics. Indeed, given the synergistic effects that immune cell recruitment, immune checkpoint inhibition, immunostimulation and tumour-targeting can have on therapeutic efficacy, it is not surprising that attempts to combine all these aspects in one compound would be the next step to take. For this purpose, multispecific antibodies or constructs have to be designed, comprising 3 or 4 modules including but not limited to a tumour-targeting module, an immune cell engager, an inhibitory immune checkpoint inhibitor, and a stimulatory immune checkpoint stimulator (or immunostimulator). The presence of an additional Fc region could be valuable to keep the related effector function (i.e. ADCC, CDC, longer half-life). Such multispecific antibodies/constructs could co-localise three effectors (immune cell engager, immune checkpoint inhibitor and immunostimulator) in the tumour hotspot thanks to the targeting moiety. We defined this general concept as “MuTICEMs” (Multispecific Targeted Immune Cell Engagers & Modulators). The use of this concept could potentially improve the temporal and localised synergies of immune effectors, as well as reduce immune-related adverse effects such as cytokine release syndrome (CRS) that can arise with non-targeted immunotherapies. Very few examples of compounds falling under the description of MuTICEMs have been reported so far, stemming from the novelty of the concept and the complexity of production of such compounds. After a general overview of the cancer immunotherapy landscape, from monoclonal to bispecific antibodies, this review will focus on the reported compounds that could be categorised as MuTICEMs, and discuss their advantages and limitations as well as the roles that organic chemistry and bioconjugation may play in their production.
II. Cancer immunotherapy definition
According to the Nature definition, “Cancer immunotherapy is a therapy used to treat cancer patients that involves or uses components of the immune system. Some cancer immunotherapies consist of antibodies that bind to, and inhibit the function of, proteins expressed by cancer cells”.21 Cancer immunotherapy can be further divided into two strategies: one uses immune system components such as antibodies to block a cancer cell function, while the other aims at fighting the disease by boosting or reactivating the patient's immune system through immune cell recruitment or immune checkpoint modulation. For the latter, various approaches can be used – vaccines (such as approved Sipuleucel-T®);22,23 monoclonal antibodies, bispecific antibodies, immune checkpoint inhibitors and immunostimulators; adoptive T-cell therapy (ACT) or T cell transfer therapy (T cells are harvested from the patient, cultivated, activated and expanded in vitro, and eventually genetically modified in the case of CAR T cells, before being reinjected into the patient);24,25 oncolytic viruses (viruses preferentially infecting tumour cells, potentially used to directly infect and kill tumour cells, as well as triggering the immune system or being used as vectors).26,27 Cancer immunotherapy, also called immuno-oncology, is experiencing a tremendous development in the clinic and is definitely one of the leading strategies of recent and future years.28 Currently, a large majority of immunotherapies use monoclonal (monospecific) or bispecific antibodies. They will be the focus of the following sections.III. Monoclonal antibodies in immunotherapy
III.1. Tumour-targeted monoclonal antibodies
Whether a monoclonal antibody applied to cancer treatment should be considered as a cancer signal blocker or an immune system activator is not a trivial question as it depends on its mode of action. Indeed, antibodies play major roles in the immune system, as shown by their capacity to bind to a target and trigger Fc-mediated complement dependent cytotoxicity (CDC) and antibody-dependent-cell-cytotoxicity (ADCC). Originally, anti-tumour monoclonal antibodies were essentially selected for their binding affinity for a tumour associated antigen (TAA), in order to block the related signalling pathway and/or induce cell death or tumour growth inhibition.9 We can mention for instance the FDA approved trastuzumab (anti-HER2), rituximab (anti-CD20), and bevacizumab (anti-VEGF) that were selected for their “direct effect”.29–32 Thus, exploiting the antibody's capacity to stimulate the immune system was not the initial purpose. This is reinforced by the fact that most of the antibody–antigen interactions will result in antibody internalisation, potentially reducing the antibody's capacity to induce an immune response, in direct accordance with the internalisation rate. However, it turned out that for such TAA-targeting monoclonal antibodies, not initially used to trigger an immune response, evidence was found that ADCC has an influence on their anti-tumour efficacy.16,17 For instance, removing the Fc fragment from an anti-EGFR antibody conserved the EGFR binding capacity when compared to the original full antibody, but resulted in significantly lower tumour inhibition in vivo.33 It was also demonstrated that ADCC was a key mechanism in the killing activity of rituximab.34 Therefore, it seems that each and every TAA-targeting monoclonal antibody has the potential to activate the immune system, even though its tumour killing activity might be only weakly related to it. This, of course, has to be investigated on a case-by-case basis. Similar conclusions could be drawn for antibody–drug or antibody–protein conjugates, made of monoclonal antibodies conjugated to toxic payloads or another protein respectively. However, depending on the conjugated moiety, direct effects on immune response can be improved. It is notably the expected effect of antibody–cytokine constructs (immunocytokines) where a cytokine like IL-2 is fused to the antibody.35,36III.2. Immune checkpoint inhibitor monoclonal antibodies
A range of mAbs targeting immune checkpoints are specifically purposed to directly regulate, restore or activate the immune system. Inhibitory immune checkpoints down-regulate the immune response (either by a signal inhibiting an activation pathway, or by a signal stimulating a regulatory pathway). On one hand, targeting them with an agonistic antibody can prevent the over-activation of the immune system that can cause serious harmful events such as cytokine storm.37–39 It can notably be used against a wide range of autoimmune diseases that originate from an impaired regulation of the immune system.40,41 On the other hand, targeting inhibitory immune checkpoints with an antagonistic (or blocking) antibody can have positive anti-tumour effects. Indeed, despite the immune system being able to recognize and fight tumour formation, some tumour cells are able to develop an immune resistance by expressing and/or activating the aforementioned immune checkpoints, thus inhibiting the immune response and promoting cancer development. Antibodies blocking this inhibition can restore the anti-tumour immune response. Among the existing immune checkpoints, the most emblematic and first studied are cytotoxic T lymphocyte-associated antigen 4 (CTLA-4, or CD152) and programmed cell death protein 1 (PD-1), both expressed on the surfaces of cytotoxic T cells, as well as the programmed cell death protein 1 ligand (PD-L1), which can be expressed by tumour cells.T cell-activation and related adaptive immune processes can be triggered through the T cell receptor (TCR), a protein complex found on the T cell surface that recognizes antigen fragments presented by the major histocompatibility complex (MHC) found on antigen presenting cells (APCs) (Fig. 1).
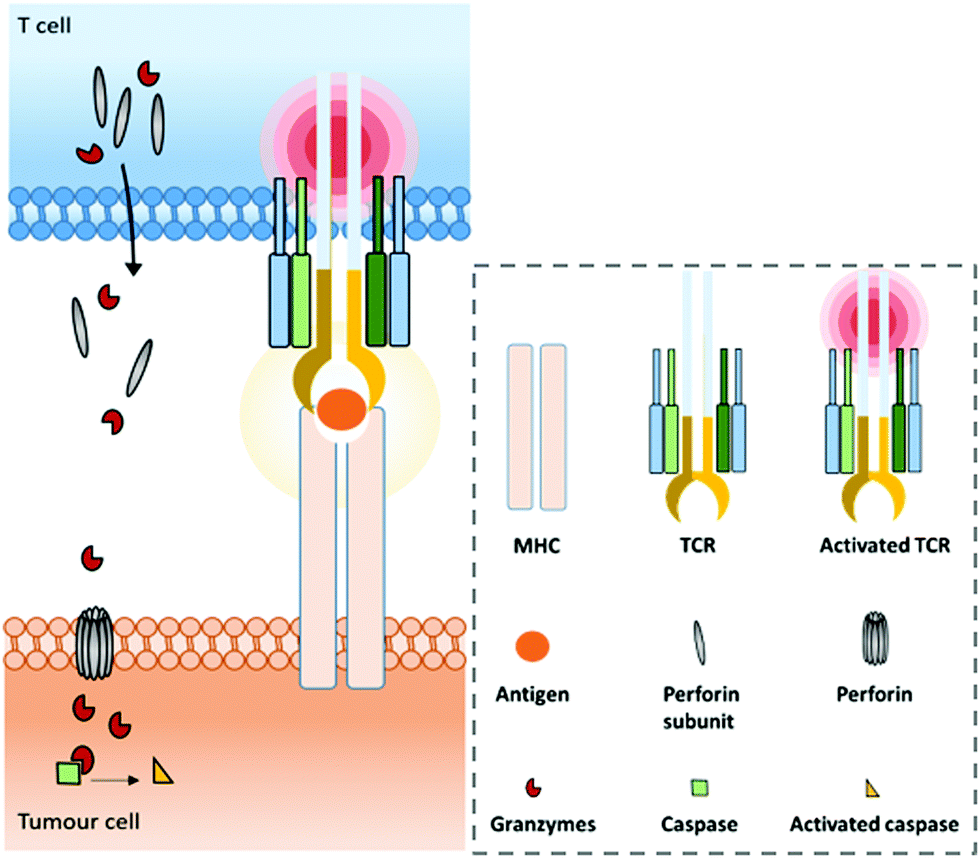 | ||
| Fig. 1 Normal major histocompatibility complex (MHC)-dependent recognition of tumour associated antigens (TAAs) by T cells via their T cell receptor (TCR). In the case of cytotoxic T cells (CD8+), this interaction allows the release of perforin and granzymes to kill the tumour cell. | ||
The CTLA-4 protein acts as an immune checkpoint by competing with CD28, a co-stimulator of the T cell receptor (TCR), as CTLA-4 and CD28 bind the same CD80 (also known as B7.1) and CD86 (also known as B7.2) ligands on the surfaces of APCs. In addition to competing with CD28 activation on T cells, CTLA-4 activation is believed to actively deliver inhibitory signals to T cells.37 The immune checkpoint activity of PD-1 is exerted upon interaction with PD-L1, and inhibits kinases that are involved in T cell activation.37 Thus, employing antagonistic monoclonal antibodies able to bind the CTLA-4 protein, or one of the actors of the PD-1/PD-L1 axis has the potential to inhibit their regulatory effect on T cells and restore the anti-tumour immune response. This approach encountered large success in clinical trials, yielding to the US FDA approval in 2011 of the anti-CTLA-4 monoclonal antibody ipilimumab (Yervoy®) for the first- or second-line treatment of patients with malignant melanoma.42 Some results, however, suggested that the effect of CTLA-4 blockade would be driven by regulatory cells (CTLA-4+ Treg) depletion rather than T cell activation.43 In 2014, the first anti-PD-1 monoclonal antibody, pembrolizumab (Keytruda®), was FDA approved for the treatment of patients with unresectable or metastatic melanoma and disease progression after receiving ipilimumab, and in patients with BRAF V600 mutation melanoma.44 This was the first of a long series. Indeed, from September 2014 to April 2020, the following 6 antibodies targeting the PD-1/PD-L1 axis were FDA approved: anti-PD-1 pembrolizumab (Keytruda®), anti-PD-1 nivolumab, anti-PD-L1 atezolizumab (Tecentriq®), anti-PD-L1 durvalumab, anti-PD-L1 avelumab, and anti-PD-1 cemiplimab, for use in a total of 57 anticancer applications, of which 40 are monotherapies (17 combination therapies).45,46 However, if approved anti-PD-1/PD-L1 antibodies are mainly used in monotherapy, the trend in clinical trials is currently shifting to their use in the context of combination therapies (76% of the active trials in 2019 were testing them as combination regimens in 2019).47 An explanation for this trend might be their lack of cancer-targeting moieties, potentially resulting in on-target, off-cancer activation of T cells and the related toxicity through cytokine release syndrome. The occurrence of a resistance mechanism against anti-PD-L1/PD-1 antibodies is another argument in favour of combination therapies. The FDA approved anti-PD-1/PD-L1 antibodies are applied to a wide range of cancers (14 in total) including melanoma, non-small cell lung carcinoma (NSCLC), renal cell carcinoma, head and neck cancer, small-cell lung carcinoma (SCLC) and classical Hodgkin lymphoma. In the meantime, the EU, Japan and China accorded approvals for the 6 aforementioned as well as 4 additional monoclonal antibodies inhibiting the PD-1/PD-L1 axis: Toripalimab (anti-PD-1), Camrelizumab (anti-PD-1), Sintilimab (Tyvyt®, anti-PD-1), and Tislelizumab (anti-PD-1), for a total of 64 approvals among which 51 were in monotherapy.45
Following these clinical trial successes, many new inhibitory immune checkpoints are now investigated in clinical trials: lymphocyte activation gene-3 (LAG-3),48,49 T cell immunoglobulin and mucin domain containing-3 (TIM-3),50 T cell immunoreceptor with Ig and ITIM domains (TIGIT), V-domain immunoglobulin suppressor of T cell activation (VISTA) (both confirmed inhibitory immune checkpoint activity and possible immune stimulatory roles in some cases have been described),51 B7-H3 (CD276) or BTLA (CD272). However, a majority of the corresponding immune checkpoint inhibitors are evaluated in combination therapies, notably with other checkpoint inhibitors such as ipilimumab, pembrolizumab, and nivolumab for instance. This subject has been recently and thoroughly reviewed elsewhere.52 The search for new immune checkpoints is ongoing and new possibilities such as the tumour glyco-code are being looked at with great promise.53
III.3. Immunostimulator monoclonal antibodies
The other class of immunotherapeutics directly acting on the immune system is stimulatory immune checkpoint stimulators, also shortened as immunostimulators. As opposed to immune checkpoint inhibitors, they are agonists of receptors that stimulate the immune response.54CD28, a TCR co-receptor constitutively expressed on resting lymphocytes, is one of the investigated targets. Indeed, after engagement with its ligand (CD80 or CD86 presented by APCs), CD28 induces signalling cascades that increase proliferation and cytokine secretion, upregulate the expression of anti-apoptotic genes and increase energy metabolism that sustains and supports T cell activation.55 Generally, as a co-stimulatory receptor, ligation of CD28 to its ligand without concomitant engagement of the TCR has no effect on T cells. This characteristic supposedly lowers the risk of a non-specific and uncontrolled immune response, which may induce toxic adverse events. The potential capacity to specifically boost activated T cells makes CD28 a promising immunostimulatory target. Monoclonal antibodies targeting CD28 were thus evaluated. However, the use of superagonist anti-CD28 antibodies (able to activate CD28 without the need for TCR stimulation) in vivo in rodent models resulted in the rapid expansion of TReg cells, which are responsible for regulation of the immune response.56,57 This would have potential application against immune disease but not in the case of an anti-cancer immunotherapy. More importantly and as opposed to rodent experiments, evaluation of a CD28 agonist in humans did induce immune response stimulation, but in a far too elevated manner: a phase I clinical trial of the superagonistic anti-CD28 monoclonal antibody TGN1412 resulted in dramatic clinical toxicity in young healthy volunteers, attributed to cytokine storm, whose first effects were observed only within 90 minutes after receiving the first injection.58 These first results probably mitigated the infatuation with using CD28 agonistic monoclonal antibodies. However, in these cases, agonistic CD28 activation was not TCR dependent. Maybe the discovery of a TCR-dependent agonistic anti-CD28 antibody will focus the immune response to the tumour site and alleviate some of the adverse events, but its efficacy would thus be correlated with the tumour immunogenicity and related TCR activation.
Various members of the TNF receptor (TNFR) family are co-stimulatory receptors and can be targeted with agonists for immunostimulation, such as CD137 (4-1BB), OX40, CD40, GITR and ICOS.54,59–61 Among these, 4-1BB and OX40 are probably the most investigated. 4-1BB is notably expressed on activated T cells (mainly CD8+ and CD4+, but also on TReg cells), cytokine-activated NK cells, and activated DCs.55,62 It has a single characterized ligand, 4-1BBL (also known as CD137L), which is expressed on activated DCs, B cells and macrophages. After binding to its ligand, 4-1BB triggers a co-stimulatory signal that can function independently of CD28 to induce upregulation of the anti-apoptotic genes BCL2, BCL-XL and BFL1, favouring the proliferation and survival of T-cells. Agonistic anti-4-1BB monoclonal antibodies resulting in anti-cancer immunity induction and improvement have been reported in various tumour models and reviewed elsewhere.63,64 However, if promising pre-clinical results strengthened attention to this immunostimulator, the two clinical trials realised so far mitigated this enthusiasm, as urelumab (clinical trial NCT00309023 terminated)62 and utomilumab, two anti-4-1BB monoclonal antibodies, resulted in liver toxicity and weak efficiency respectively.63–66 As for other monoclonal immunotherapies, a complex balance between anti-tumour immunity and auto-immune adverse events probably exists, sustained by an incomplete understanding of the role of 4-1BB.67,68 Noteworthily, Qi et al. demonstrated that the 4-1BB co-stimulation induced by anti-4-1BB monoclonal antibodies is dependent on both their Fab-related affinity for the target and their Fc affinity for FcγRs.69 The 4-1BB/4-1BBL axis is promising but also complex. It is still a hot research topic for immunotherapy but its application is currently being explored more in combination or bispecific therapies rather than monotherapies.63,64,70,71
OX40, another member of the TNFR family, is deeply investigated and holds great promise. The OX40 receptor (CD134) is expressed on CD4+ and CD8+ T cells following activation, but not on resting ones. Its role is thus to function as a late co-stimulatory receptor for CD4+ and CD8+ T lymphocytes. The OX40 expression is also upregulated on Tregs upon activation in humans. Its ligand, OX40L, is not constitutively expressed but is induced on activated APCs such as DCs, B cells and macrophages.55,70 Upon binding to its ligand, OX40 co-stimulates T-cell proliferation and survival. Interestingly, OX40 signalling can prevent Treg-mediated suppression of the anti-tumour immune response. The activation of CD4+ and CD8+ T cells combined with the Treg down-regulation makes OX40 a very interesting target to exploit for immunotherapeutic approaches. So far, autoimmune side effects induced by OX40 stimulation have not been reported. However, and not surprisingly, agonistic anti-OX40 monoclonal antibody administration has very limited impact on the growth of poorly immunogenic tumours, favouring its use in combination immunotherapy.72 This corroborates the fact that despite numerous assets, OX40 is targeted in a small number of agonistic monoclonal monotherapies. According to the National Cancer Institute,73 there are only 4 active clinical trials involving agonistic anti-OX40 antibody monotherapies: one study with BMS-986178 in patients with solid cancers that are advanced or have spread,74 one with ABBV-368 in subjects with locally advanced or metastatic solid tumours,75 one with PF-04518600,76 and one with PF-05082566 in treating patients with relapsed or refractory acute myeloid leukemia.77 However, these 4 aforementioned trials also include the studied antibodies in combination therapies. Other active trials including anti-OX40 antibodies are combination therapies.
Interestingly, Sawada et al. showed that high levels of soluble OX40 (sOX40) in the blood of patients with advanced colorectal cancer were linked to reduced survival times. The hypothesised reason for this observation is that sOX40 could interact with OX40L (and potentially anti-OX40 antibodies), possibly resulting in the reduction of the OX40/OX40L interaction, subsequently reducing T-cell activation and immune response.
Recently another member of the TNFR family, CD40, has aroused high interest for its immunostimulatory capacity through an alternative pathway compared to the aforementioned one. CD40 is notably expressed in dendritic cells (DCs) and its ligand CD40L is expressed by activated CD4+ T cells (and platelets). CD40 has a pivotal role in the adaptive immune response: upon interaction with CD40L presented by activated CD4+ cells, it induces upregulation of MHC molecules, increases the expression of costimulatory molecules such as CD86 (ligand of the CD28 co-receptor on T cells), and induces upregulation of other TNF ligands such as 4-1BBL, GITRL and OX40L. The CD40-induced up-regulation of all these secondary stimulatory molecules in turn enhances antigen presentation and activation of CD8+ T cells.59,78 CD40 activation on DCs has also been shown to increase the levels of cytokines, including IL-12, which are important for CD8+ T cell activation. Based on all these features, CD40 is considered to be a central and initiating factor in the cascade of adaptive immune activation, and its activation via agonistic anti-CD40 monoclonal antibodies appears to be highly valuable. A limitation though is the timing for the DC activation through CD40. Indeed, the role of the dendritic cell subset in fighting or promoting tumour development is variable and complex.79 For instance, plasmacytoid dendritic cells (pDCs) are likely to reduce tumour immunogenicity. Oppositely, conventional dendritic cells (cDCs) are crucial in the anti-tumour response through the recruitment and activation of CD8+ T cells, thanks to the tumour antigen-cross presentation at the DCs’ surfaces. However, CD40-activated DCs have impaired capacity to take up new antigens compared to inactivated DCs, reducing their direct capacity to induce CD8+ T cell activation. Thus, if CD40 activation through anti-CD40 antibodies can boost up the immune response through the second stimulatory cascade described above, it seems that an early administration could be counteractive in a combination context, where tumour antigen-presentation to CD8+ T cells is important. A simultaneous injection or simultaneous presentation of the anti-CD40 and other effectors on the same therapeutic compound could circumvent this limitation. In line with this, administration of agonistic anti-CD40 alone triggered T cell activation and related tumour regression, but only highly immunogenic (strongly antigen expressing) models were responding.59
To the best of our knowledge, 8 agonistic anti-CD40 monoclonal antibodies for cancer treatments were investigated or are being investigated in clinical trials (5 completed and 3 ongoing respectively): ChiLob7/4 (completed); 2141 V-11 (recruiting); HCD122 (completed); CP-870,893 (Secrilumab) (completed); CDX-1140 (recruiting); SEA-CD40 (active, not recruiting); APX005M (completed); and ADC-1013 (JNJ-64457107) (completed).80–88 They are all phase I studies, except for one of the two clinical studies of HCD122 which is a phase I/II study. Some of the latter were also investigated in combination therapy (with Rituximab, Nivolumab notably), while SGN-40 has been evaluated only in combination with other antibodies.89–91 Globally, minimal rates of objective tumour response (ORR) were generated by agonistic anti-CD40 monotherapy clinical trials. What is more, as other immunostimulators, adverse events are observed as soon as a few minutes to hours after infusion (cytokine release syndrome), though as moderate symptoms that can be reversed with care.92 As mentioned before, the optimal use of agonistic anti-CD40 antibodies might be in association with other effectors (in combination or via a multispecific compound). In some tumour models, it has been shown to be synergistic with immune checkpoint inhibitors blocking PD-1/PD-L1 or CTLA-4 (CD40-activated DCs amplify the T cell pool able to respond to immune checkpoint inhibitors, while immune checkpoint inhibitors prevent these CD40+ DC-produced T cells to be down-regulated by immune checkpoints).93,94
Additionally, it has been shown in mice that CD40 activation was responsible for the activation of host macrophages, themselves inducing non-T cell-dependent anti-tumour effects.95,96 In addition, activated macrophages induce fibrosis degradation that is dependent on matrix metalloproteinases (MMPs), resulting in easier access to tumour sites for adjuvant chemotherapy or immunotherapy.97 Hypothetically, a single but multispecific compound encompassing an anti-CD40 fragment could benefit this improved tumour penetration through repeated injections. Altogether, these elements make agonistic anti-CD40 monoclonal antibodies a promising option for immunotherapy, even though their association with other effectors is probably the future direction to take.
III.4. The limitations of monoclonal antibodies
Overall, it is clear that monoclonal immunotherapies have revolutionised the anti-cancer research paradigm of the past two decades, particularly antibodies aiming at blocking inhibitory immune checkpoints or activating stimulatory immune checkpoints. However, while the first preclinical results were really exciting, it seems that the translation to the clinic was only moderately successful. This is likely to be due to: (1) adverse events such as cytokine release syndrome; (2) the lack of efficacy due to low tumour accessibility or low tumour antigen expression; and (3) treatment escape originating in low tumour immunogenicity and immune escape by expressing or up-regulating other immune checkpoints than the one targeted by the therapy. The adverse events are common to almost all immunomodulators (immune checkpoint inhibitors and immune checkpoint stimulators) used as single agents, due to the difficulty in balancing the critical equilibrium between sufficient immune response activation for anti-tumour effects and too strong activation resulting in generalised inflammation. This also highlights the fact that single agent immunomodulators are non-targeted and can activate immune effectors in both tumour and healthy tissues indiscriminately. The expression of the targeted receptors by a subset of cells of the opposite activity compared to the target cells can also result in counter-acting effects, such as activation of both cytotoxic T cells and Tregs.56,57 For some immunomodulatory antibodies themselves, the balance between the Fc affinity for Fc receptors and the paratope affinity for its receptor was also demonstrated to be critical.69To tackle the on-target, off-tumour toxicity, a monoclonal antibody can be converted to a probody format. This strategy confers a conditional activation to the antibody by masking its binding site with a connected substrate that will be released only under specific conditions found in the tumour micro-environment, such as low pH and high protease activity. This concept offering better selectivity and safety has been reviewed elsewhere,98 and proved to be promising since probody formats of anti-CTLA-4 activated by protease cleavage (BMS-986249, CytomX) or reversibly activated by acidic pH (BA3071, BioAtla) have notably been to the clinic.99 However, in addition to systemic toxicity, low expression of targetable receptors and immune escape are crucial bottlenecks to be addressed, ideally in a simultaneous way. For this purpose, it is now evident that combining several effectors is advisable. It can be realised through combination therapy, or by encompassing various effectors in a single compound (e.g. a bispecific). Indeed, the presence of a tumour-targeting moiety could lower the on-target, off-side adverse effects of an immune effector, while combining two targeting moieties could avoid escape due to tumour antigen fading or lack of expression. Combining two immune checkpoint inhibitors (αCTLA-4 and αPD-1, or αCTLA-4 and αPD-L1 for instance) would address some immune escapes, while combining an immune checkpoint inhibitor and an immunostimulator could induce synergistic effects. Additionally, synergistic effects as well as improved tumour infiltration and immunogenicity can be generated via an agonistic anti-CD40 binding module as discussed before. Globally, simultaneously exploiting various effectors could induce synergistic effects and better efficacy while reducing the concentration of the effective dose and lowering toxic effects. Considering the complexity of cancer biology, combining two different effectors might be a valuable prerequisite to reach higher objective response rates (ORRs) and total remission in clinics. In any case, this should ideally be supported by biomarker identification to find the ideal combinations to be evaluated and eventually adapted to patients.
Combination therapies and bispecific antibodies are now developed to simultaneously address multiple effectors (i.e. antibodies or antibody fragments) to various immune checkpoints, or immune cells and/or TAA. Compared to combination therapy, bispecific antibodies present a lower modularity of treatment, but they present the high advantage of being able to direct the immune effector (the displayed checkpoint modulator or the immune cell binder) to the tumour site thanks to a targeting moiety. In the following sections, we will focus on bispecific antibodies or bispecific antibody formats used in cancer immunotherapy and discuss potential future directions.
IV. Bispecific antibodies in immunotherapy
If monospecific antibodies were the hot topic in the beginning of the 21st century, multispecific antibodies (MsAbs), also referred to as polyspecifics (PsAbs), have taken over as the new hot topic. MsAbs are proteins able to simultaneously engage two or more different epitopes. Among multispecifics, the bispecific (BsAb) subset, encompassing two paratopes, is by far the most represented. The BsAb denomination is actually used for either bispecific antibody formats lacking an Fc fragment (BiTEs, DART, F(ab’)2, etc.) or full bispecific antibodies bearing an Fc fragment (IgG-like bispecifics), resulting in bifunctional and trifunctional bispecifics respectively. Noteworthily, depending on the antibody format, a BsAb can be monovalent for both epitopes, bivalent for both, or monovalent for one and bivalent for the other. By containing two different paratopes, bispecifics offer various advantages such as: (1) simultaneously blocking or activating two different pathways in pathogenesis, through TAA targeting (even though this can also be done through combination therapy); (2) redirecting immune cells such as T cells (“BiTEs”) and NK cells (“TriKEs”) to tumour cells and triggering or improving immune-related tumour killing; (3) potentially increasing selectivity and/or avidity by interacting with two different cell surface antigens (or two different epitopes of the same antigen); (4) potentially improving synergistic effects thanks to the improved spatial and temporal colocation of effectors when compared to the combination therapy of monospecific antibodies; (5) reducing costs in terms of development and production when compared to the production of CAR-T cells that share with BiTEs the aim to redirect T cells to the tumour (the subject of CAR-T cells has been widely reviewed elsewhere);100,101 and (6) being used for various other applications such as holding effector proteins together,102 promoting the crossing of biological barriers or fast cell internalisation and subsequent trafficking to lysosomes (one arm acts as a shuttle to allow the other arm to cross into the cell or the biological compartment where its target is located).16,103 The numerous assets of bispecifics are currently making them a leading approach in the field of immunotherapy, as demonstrated by their booming number in clinical trials listed for use in oncology, from 57 BsAbs (for 99 clinical trials) in 2018 to 75 BsAbs (135 active, recruiting, or completed clinical trials) in April 2020.IV.1. BsAb production and formats
Bispecifics have been used to target two different epitopes on the same cell, typically two tumour associated antigens (dual tumour-targeted bispecifics). However, following the clinical success of immunotherapies and in order to overcome their established limitations, it appeared to be of great interest to design bispecifics combining an immune specificity (for an immune cell, or an immunomodulatory checkpoint) with a tumour specificity or a second immune specificity.As a consequence, the research on bispecific production intensified, as reflected by more than a hundred different bispecific formats described so far.104 This variety can be explained by the search for the ideal format combining good production yields and reproducibility, high stability, solubility, optimal serum half-lives, efficient tissue penetration, etc. All these aspects can be summarised as the “developability”. Another contributor to the variability is also the need to adapt the platform to the target by varying the size, the geometry, and the targeting modules of the bispecific. On top of that, the highly competitive atmosphere in the field and the search for intellectual property also contribute to the frenetic development of new formats. The wide landscape of bispecific formats has been thoroughly reviewed by others.15,104,105 Briefly, three methods can be used to produce bispecifics. Originally, and as soon as 1961, BsAb fragments were chemically produced in a mixture after reduction and re-oxidation of two (Fab’)2 species.106 Later, full BsAbs were generated by chemically conjugating two different and isolated monoclonal antibodies. They were also produced by a quadroma cell line – obtained by the fusion of two hybridoma cell lines – able to generate a range of proteins combining the heavy and light chains of the two different antibodies originally produced by the two fused hybridoma cells.107 But the first attempts of chemical conjugation suffered from poor yields and difficulties in purification,108 while the quadroma technology generated many “mismatched species”, randomly combining wrong heavy and light chains (16 different combinations, yielding 10 different molecules), thus generating non-functional proteins that had to be separated from the desired bispecific antibody. The hybridoma method generates “IgG-like bispecifics”, as the constructs present a structure similar to the IgG protein, with two Fabs and one Fc. Later on, genetic engineering enabled the modification of the heavy chains with the “knobs-into-holes” technology (KiH) to favour heterodimerization – mutations in the CH3 domains to replace a small amino acid with a larger one in a heavy chain, creating a “knob”, and to replace a large residue with a smaller one on the other heavy chain, to create a “hole” into which the knob will insert.109 Used in complementarity to the KiH technology, the CrossMAb method, which relies on the crossover of the antibody domains (VH with VL, CH1 with CL, or both) within one of the two Fab-arms of an IgG-like bispecific, allowed satisfactorily tackling the issue of Fab scrambling.110 Recently, Brinkmann et al. described high-throughput technology for BsAb production, the Format Chain Exchange (FORCE). Similar to the knobs-into-holes approach, it uses individual monospecific knob or hole half-antibodies paired with a complementary Fc-dummy chain containing mutations that lead to limited interface repulsions. Upon disulfide reduction, spontaneous chain-exchange is triggered to generate dummy-dummy paired chains and the desired knob-and-hole antibodies, driven by optimal interface complementarity. The BsAb can then be obtained in a one-step purification, and this method offers a wide flexibility in terms of bi- and multi-specific antibody formats.111 Genetic engineering also enabled the production of recombinant proteins where only fragments of antibodies were fused together to generate the so called “antibody formats”. The multitude of existing antibody formats are the result of the fusion of a variety of native or engineered fragments among Fc, Fab and one or more variable fragments, generally linked together by peptide linkers. The more represented of these antibody formats are probably the bispecific F(ab′)2 which is the fusion of two distinct Fab fragments; smaller molecules such as tandem scFv, diabody and DART, which all connect two scFvs in different manners (one scFv is a fusion protein encompassing the variable domains of the heavy and light chains connected together), and even smaller molecules are produced by the fusion of only two single variable domains (tandem dAb).104,112 On the other hand, bigger structures are obtained when antibody fragments are fused to a full antibody, to generate the so called “appended IgG-like” or “modified IgG-like” formats. A direct comparison of all formats’ efficiencies would be valuable but probably an impossible task as every format and every antibody clone would have to be tested and conclusions may vary depending on targeted receptors and their cross-combination in a bispecific context.113
IV.2. BsAb design
In this crowd of bispecific formats, a major distinction that can be made is based on the presence of an Fc fragment (IgG-like and appended IgG-like bispecifics) or lack thereof. Another distinction can be made between active Fc (native or engineered) and engineered inactive Fc fragments. At first glance, the presence of the Fc fragment allows longer serum half-lives due to FcRn-mediated recycling, and can induce Fc related ADCC (antibody-dependent cell-mediated cytoxicity), CDC (complement dependent cytotoxicity) and ADCP (antibody-dependent cellular phagocytosis) effector functions. Conversely, the lack of Fc leads to a smaller size and impaired aforementioned recycling, resulting in higher tissue penetration but shorter half-lives. It also reduces the risk of immunogenicity (undesired and detrimental immune response, which is generally a major cause of adverse events). The choice to include the Fc fragment in the design of the BsAb can be driven by these properties, depending on the final aim. However, the fact is that the underlying layers of complexity are involved. For instance, the presence of an Fc effector function has certain advantages when targeting a solid tumour, as the ADCC and CDC have been shown to be valuable to observe some tumour-killing effects, and was even mandatory for monospecific antibodies rituximab and trastuzumab.114 In the meantime, solid tumours are prone to being poorly infiltrated and adding an Fc fragment could be counter-active regarding the size of the resulting compound and its tumour infiltration. On the other hand, when recruiting immune cells to the tumour site, the presence of the Fc is likely to be detrimental as its effector function could induce the depletion of the immune cells intended to be recruited. The same reflection can be undertaken when targeting immune checkpoints such as PD-1 and 4-1BB, which are present at the surfaces of T cells. Conversely, when the immune checkpoint is present at the surface of the tumour cell, as is the case for PD-L1, then an αPD-L1 × αTAA BsAb would possibly benefit from possessing an Fc fragment to improve the tumour killing. The balance between the positive and negative impacts of the Fc fragment is delicate. Engineering the Fc fragment is a solution to combine assets of both sides – Fc can be inactivated (ineffective) so that only the half-life benefit is conserved, but the immunogenicity due to undesired ADCC effects is avoided. This concept notably drove the generation of DART-Fc, where an engineered Fc fragment was fused to a DART,115 or the development of DuoBody® (GEN3013), an αCD3 × αCD20 BiTE consisting of a full-length bispecific IgG1 immunoglobulin with an effector function-silenced Fc region.116 This approach addresses the inconvenience of using small molecules such as DART and BiTE (∼55 kDa) whose short half-lives impose regular administration to the patient on a daily basis by continuous IV infusion.104,117Furthermore, the complexity arises not only from the presence of but also the affinity of the Fc. Interestingly, it has been shown in several studies that the mode of action of some immune checkpoint inhibitors or immunostimulators might be not only through the blocking of inhibitory signals or the promotion of activation signals on effector T cells, but also (if not mainly) due to the depletion of infiltrating Treg cells,118,119 with the Fc fragment playing a crucial role here. Indeed, Treg cells are T cells that infiltrate the tumour to regulate the immune response. Tregs have been shown to express higher levels of CTLA-4, OX40 or GITR receptors when compared to effector T cells in some tumour cell lines (notably RCC, NSCLC, melanoma).119 Thus, antibodies targeting these receptors are likely to interact with both T cell subsets, but more importantly with Tregs. Bulliard et al. showed that the tumour regression potential of the agonistic anti-OX40 antibody (which presents an Fc) was highly correlated with tumour-infiltrating Treg depletion, itself directly correlated with the expression of activating FcγRs by Treg cells and the capacity of the antibody's Fc to bind these receptors.119 Despite effector T cells (CD4+ FOXP3–) being also depleted, the elevated depletion of tumour infiltrating Treg cells (CD4+ FOXP3+) resulted in a highly improved CD8+/Treg ratio, which is believed to be the main factor for the anti-tumour activity triggered by agonistic anti-OX40 antibodies. Similarly, Arce Vargas et al. evaluated various isotypes of anti-CTLA-4 antibodies, using human IgG variants on mice expressing human FcγRs (hFcγRs), and demonstrated that the anti-tumour activity was directly correlated with Treg depletion with an emphasis on the Fc affinity for FcγRs.118 Antibodies bearing an IgG engineered to present higher binding affinity for activatory FcγRIIIA (CD16a) or to present no binding to hFcγRs, respectively, presented enhanced and poor anti-tumour activities. Their preclinical data strengthen the unifying hypothesis according to which human IgG anti-CTLA-4 mAbs’ anticancer activity originates in promoting a preferential depletion of tumour-infiltrating Treg cells and thus improves the intra-tumoral effector T cell/Treg ratio. However, this Treg depletion is linked to the presence of Tregs themselves, along with innate effector cells expressing FcγRs (such as NK cells, macrophages), which are involved in the ADCC process. As a consequence, this Fc-related anti-tumour activity is likely to be restricted to immunogenic, inflamed and infiltrated tumours. It is the hypothesis developed by Arce Vargas et al. to explain the mitigated clinical results of anti-CTLA-4 monotherapies reported so far, and it was proposed that a prior combination therapy to promote immune infiltration would benefit anti-CTLA-4 therapies.
Qi et al. also demonstrated an influence of the Fc fragment on agonistic anti-4-1BB antibodies, but it was dependent on the Fab affinity for the 4-BB1 receptor. Strongly agonistic antibodies could activate 4-1BB in the absence of FcγRs, while the FcγRs were mandatory for activation by weak agonists. They also demonstrated that strong agonists were linked to liver toxicity. Considering that FcγR interactions could induce detrimental ADCC related depletion of 4-1BB+ T cells, they engineered antibodies to balance the agonistic activity and the strength of FcγR interactions, generating a weakly agonistic humanized 4-1BB mAb-AG with a low activating-to-inhibitory (A/I) FcγR-binding ratio that presented strong anti-tumour activity without liver toxicity.69 This study, along with others,120–122 is another example of the complex impact of the Fc fragment on the anti-tumour activity of agonistic monospecific and bispecific antibodies, and engineering the Fc can potentiate their activity, though requiring deep studies and fine tuning.
Unsurprisingly, the chosen Fab fragments also have a major influence on the function of the bispecific antibody. Several studies have reported that dual targeting alone is not sufficient to enhance target selectivity and that the affinity of individual antigen-binding arms matters. Counter-intuitively, using Fabs with lower affinity for their target in the context of a bispecific construction can be advantageous for better selectivity, efficacy and lower secondary effects. Notably, this observation holds true in cases where the targeted antigens are also expressed by healthy tissues. Indeed, Mazor et al. demonstrated that generating a bispecific antibody with lower affinity Fabs (when compared to the parental monovalent antibody) reduces the binding to cells expressing none or only one of the targeted antigens, but this loss of individual affinity is counter-balanced by the valency of the construct when binding cells expressing both antigens. Thus, the lower affinity of the individual binding arms increases the difference in avidity (overall binding ability of the construct) to the target population over singly expressing populations, improving the selectivity of the BsAb for dually-expressing cells.123,124 This principle has been used for EGFR × C-Met bispecifics, where reduction in the EGFR affinity of the individual αEGFR arm leads to a decrease in toxicity related to basal EGFR expression in the skin.125 The affinity attenuation of individual arms did improve the selectivity of the BsAb for tumour cells that overexpress both antigens and resulted in potent anti-tumour efficacy. In another study, Piccione et al. used a BsAb (αCD20 × αCD46) to induce phagocytosis of B lymphoma cells. Considering the basal expression of CD46 by normal cells (possibly creating an “antigen sink”), they opted for a reduced affinity for CD46, prioritizing the targeting of tumour cells via their CD20 expression. This led to selective binding of the BsAb to tumour cells and effective subsequent phagocytosis.126 Interestingly, Slaga et al. generated a bispecific T cell engager presenting two appended copies of the same low-affinity HER2 binding site. The low affinity of individual binding sites allowed sparing HER2 low-expressing cells, while the avidity generated by the bivalent presentation of the low affinity HER2 binders improved the selectivity for target HER2-overexpressing tumor cells, in accordance with their higher receptor density.127 This bivalent low-affinity binder model could be applied to different targets to improve selectivity for cells with high expression of a particular receptor and reduce on-target off-tumour adverse effects due to the targeting of normal cells with low receptor expression (Fig. 2).
 | ||
| Fig. 2 Summary of some of the main inhibitory and stimulatory immune checkpoints in their expression and interaction patterns. | ||
IV.3. Classification of BsAbs based on their targets
The BsAb landscape is wide as numerous combinations of two targets can be made. However, for the purpose of this review, we tried to classify them to provide a more structured overview. The first distinction can be made between BsAbs that target only cancer cells (by targeting two cancer antigens or two epitopes of the same cancer antigen), or BsAbs targeting a cancer cell and an immune cell. They can be described as “dual tumour-specific BsAbs” and “immunospecific BsAbs”. Secondarily, among immunospecific BsAbs, we can distinguish: (1) those targeting immune cells for their recruitment to the tumour site, called “immune cell engagers”; and (2) those targeting stimulatory immune checkpoints or inhibitory immune checkpoints, described as “immunomodulatory BsAbs”. At the third level, the immune cell engagers group includes not only classical immune cell engagers (αTAA × αCD3 BsAb directly administered) but also BATs (bispecific antibody armed activated T cells) where the immune cell engager is first armed on T cells ex vivo before administering them to the patient. (TCR)-derived immune cell engagers are also a different format as they do not use CD3 to recruit T cells. Concerning immunomodulatory BsAbs, a distinction can be made between BsAbs targeting either one immunomodulatory checkpoint plus a tumour antigen, or two immunomodulatory checkpoints, generating “tumor-targeted immunomodulatory BsAbs” or “dual immunomodulatory bispecifics” respectively. To conclude, all the BsAbs can be used in combination with chemotherapy, monoclonal antibodies or bispecific antibodies. A more detailed description and examples of these different types of BsAbs are provided in Sections IV.3.b to IV.3.d., after a listing of all BsAbs currently investigated in clinical trials for cancer treatment and their classification according to the above mentioned descriptors (Section IV.3.a.). | ||
| Fig. 3 Repartition of the 135 clinical trials (recruiting, active or completed) involving bispecific antibodies referenced on “http://clinicaltrials.gov” for the treatment of cancer, in April 2020. Left dials display dual tumor-specific BsAbs used in monotherapy (top left) and combination therapy (bottom left). Right dials display immunospecific BsAbs (including dual immunomodulators, immune cell engagers, targeted immunomodulators and BATS) used in monotherapy (top dial) and combination therapy (bottom dial). A more detailed discussion of the chosen classification is given in Section IV.3. BATs: bispecific antibody armed activated T cells, ADCs: antibody–drug conjugates. | ||
Among the 20 dual tumour-targeting BsAbs currently in clinical trials (Fig. 3, upper left dial), including 3 bispecific ADCs, 10 are targeting one of the ErbB receptors; EGFR, HER2, or both. It is noteworthy that dual tumour-targeting BsAbs are often employed for pre-targeting strategies for radiotherapy or PET imaging applications. In all 7 corresponding clinical trials listed, the BsAbs contain an anti-CEA binding module and a heptene, being recognized by a subsequently injected compound (generally a peptide) bearing a radioisotope (I,131 Ga,68etc., Fig. 3, upper left dial).
Dual tumour-targeting BsAbs represent a promising application of BsAbs. However, this strategy is not as relevant on its own (i.e. naked BsAbs) as when it is combined with an ADC approach, where the high toxicity of the payload carried by the antibody requires a very high selectivity to reduce potential off-target effects. The current trend is largely in favour of BsAbs targeting at least one immune effector. The immune-specific BsAb subset includes immune cell engagers, tumour-targeted immune checkpoint modulators (inhibitors or stimulators), and dual immunomodulators that are developed in the section below.
Immune cell engager family.
Immune cell engagers. Immune cell engagers are the most common application of BsAb technologies. They are designed to recruit immune cells to the tumour site by combining affinity for an immune cell receptor and a tumour associated antigen (TAA) (Fig. 4a). Recruited immune cells can be NK cells, macrophages or T cells, with the latter being the most exploited. Every bispecific format that recruits T cells to target cells can be defined as a bispecific T cell engager. However, in a global consensus, the “BiTE” acronym (for Bispecific T cell Engager) is more attributed to fusion proteins made of two single chain variable fragments (scFv) connected by a linker, thus lacking the Fc region.
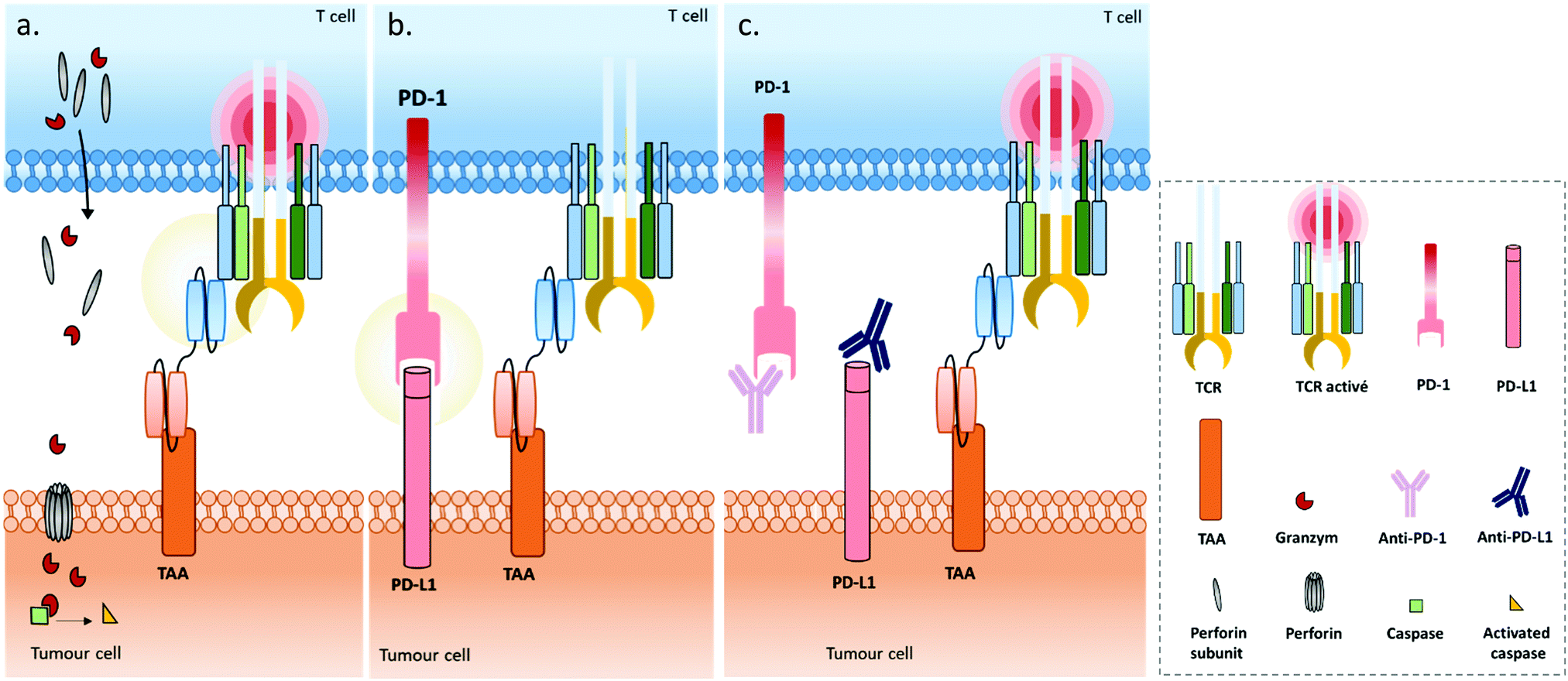 | ||
| Fig. 4 (a) A bispecific T cell engager (BiTE) enables MHC-independent targeting of tumour-associated antigens (TAAs) and T cell activation, by linking the TAA to the CD3ε chain of the TCR complex. The BiTE-induced cytolytic synapse enables perforin and granzyme-mediated destruction of the targeted tumour cell through activation of the proteolytic caspase signalling pathway. (b) Immune escape mechanism involving the PD1/PD-L1 inhibitory immune checkpoint axis during treatment with BiTEs: the tumour cell expresses the programmed cell death ligand 1 (PD-L1) which binds to the programmed cell death protein (PD-1) expressed on activated T cells’ surfaces, resulting in T cell anergy. (c) Combination strategies employing immune checkpoint inhibitors (blocking antibodies) against PD1 and/or PD-L1 proteins in addition to BiTEs, to support their capacity to restore T cell function. | ||
Under physiological conditions, T cell cytotoxic activity is triggered when its T cell receptor (TCR) recognizes a non-self- or neo-antigen loaded onto major histocompatibility class (MHC) molecules on the surface of another cell such as a tumour cell (Fig. 1). However, down-regulation of the MHC class I by cancer cells is a known mechanism of immune escape (among others).131 One of the advantages of T cell engagers is that they are directed against CD3, a T cell co-receptor (and invariant component of the TCR complex) involved in cytotoxic T cell (CD8+) and helper T cell (CD4+) activation independently of the TCR–MHC interaction (Fig. 4a). Thus, they are not affected by escape mechanisms involving down-regulation of antigen expression and presentation, and they can elicit a polyclonal T-cell response.113 Numerous examples of T cell engagers have been reported so far, engaging T cells against tumour cells through affinity for TAAs such as HER2 (mainly for breast cancer),132,133 EGFR (mostly for breast and lung cancer and glioblastomas),134 EpCAM (for colon, pancreas, prostate, and gastric adenocarcinomas),135,136 PSCA (prostate cancer),137 PSMA (prostate cancer),138 CEA (carcinoembryonic antigen positive cancers),139 CD19 (B cell malignancies),140,141 CD20 (B cell malignancies),142 CD33 (acute myeloid leukemia),143 and BCMA (multiple leukemia);144 this list is non-exhaustive. It is noteworthy that Catumaxomab (Revomab®), an αCD3 × αEpCAM BsAb, was the first bispecific antibody approved for a cancer treatment (malignant ascites) in 2009. However, it was voluntarily withdrawn from the US market in 2013 and in 2017 from the EU market for commercial reasons. Blinatumomab (Blincyto™), an αCD3 × αCD19 BsAb, is the only BsAb on the market approved for cancer treatment. It was approved by the FDA in 2014 and by the AMM in 2015 for patients with Philadelphia chromosome-negative (Ph−) relapsed or refractory B-cell precursor acute lymphoblastic leukemia.
Immune engagers also include NK cell and macrophage engagers. Indeed, some examples of BsAbs targeting the high affinity Fc receptor (FcγRI) – also called CD64 – overexpressed by macrophages, monocytes and neutrophils have been reported. The tumour targeting is ensured through affinity for EGFR,145 HER2,146 CD19 (4G7xH22)147 or CD33.148 However, the only positive results published were from the early 2000s, and the only clinical trial currently referring to the use of an αCD64 × αCD19 was terminated.149 This possibly indicates that global and/or long-term inefficacy or toxicity may have impaired the further development of this strategy. Conversely, some BsAbs engaging NK cells are still under evaluation in the clinic (NCT04101331, NCT03192202, NCT04259450) and several studies have reported that they are a good alternative to T cell engagers. NK cell recruitment by BsAbs is generally realised through affinity for CD16a (FcγRIIIa, the A isoform of a low-affinity receptor for the IgG Fc domain, also expressed on macrophages), which has been combined with paratopes with affinity for various TAAs such as CD30,150,151 BCMA, and EGFR.152
Use of immune cell engagers represents a leading strategy in BsAb research, and more generally for cancer immunotherapy, as evidenced by their high proportion of the current BsAbs in clinical trials.153–155 This trend was catalysed by, first, the convincing clinical results and then the approval of Catumaxomab (which was withdrawn since then) and Blinatumomab. Immune cell engagers were primarily evaluated for hematologic cancer treatments but are now almost equally studied in the context of solid tumours. However, despite being considered as a breakthrough in the treatment of some cancers, immune cell engager immunotherapies can suffer from toxicity and efficacy drawbacks. Indeed, T cell activation can occur beyond the tumour site and induce a systemic inflammation response, with high levels of cytokine expression. Known as cytokine release syndrome (CRS), this event can have heavy and even fatal adverse effects on patients. Agents treating CRS (corticosteroids) are administered to patients to manage these issues. Besides, the treatment efficacy can be reduced through different escape mechanisms, mainly: (1) up-regulation of checkpoint inhibitors such as PD-1 and PD-L1, or down-regulation/blocking of co-stimulatory molecules in response to a CD3 targeted treatment, resulting in impaired T cell function (Fig. 4b); (2) antigen escape; through the selection pressure of the treatment, cells expressing the targeted TAA are killed but cells that don’t express the TAA are not, and thus these can keep proliferating; and/or (3) the suppression of the immune response through activation of regulatory T cell expression.155–157 To circumvent the escape mechanisms, addition of checkpoint inhibitors, immunostimulators, or activating cytokines through combined therapy (e.g. co-infusion of anti-PD-1 or anti-PD-L1, see Fig. 4c) or by including them in the same compound (multispecific constructs such as TriKE) is possible, as well as combining several TAAs (to limit antigen escape) or recruiting different types of immune cells (NK cells, macrophages). Compounds reducing the Treg influence are also an interesting option. Such combination therapies including BsAbs are discussed later in this section, while multispecific antibodies (MsAbs) are discussed in the next section.
BATs (bispecific antibody armed activated T cells). BATs, for “Bispecific antibody Armed activated T cells”, are a field of application of BsAbs that is attracting growing interest. Instead of administering the BsAb directly to the patient, the BsAb exhibiting affinity for CD3 and a TAA is incubated with activated T cells ex vivo. Through αCD3/CD3 interaction, the T cell is “armed” with the BsAb, thus conferring a tumour affinity to the resulting BAT. The BAT is then administered to the patient. Various benefits are attributed to this strategy: (1) T cells are already activated when they arrive to the tumour site, thus affording a quicker and more efficient response; (2) T cells (from the patient or healthy donor) are activated and selectively directed to tumour cells; (3) T cells are cultured ex vivo and multiplied before being armed, thus offering an increased pool of available T cells after administration, particularly for patients with low T cell levels; (4) the total amount of BsAb required is lower when used with armed activated T cells (up to 200 times lower) when compared with the BsAb used as a single agent, potentially reducing the adverse events commonly seen with direct BsAb administration; and (5) a better pharmacokinetic profile can be obtained as clearance is reduced when the BsAb is attached to an effector cell and it can also benefit from the natural T cell capacity to extravasate and travel between endothelial barriers to more easily reach the tumour.158–160 Currently, 17 clinical trials are referenced that use BATs (clinicaltrials.gov), among which 13 are using BATs as single agents (4 are used in combination) and 15 of them are evaluated for solid tumour treatment, probably illustrating the better efficacy and tumour penetration of BATs when compared to classical BsAbs (Fig. 3, top and bottom right dials). Still, similarly to BsAbs, the secondary effects of BATs can include CRS and neurotoxicity. In addition, isolating and handling T cells is not trivial and represents a constraint inherent to this approach. Not being the focus of this review, BATs can, however, be considered as BsAbs used in immunotherapy. Nevertheless, we have listed them apart from the other “classical” immunospecific BsAbs that are directly administered to the patient.
(TCR)-derived immune cell engagers. The ImmTAC, a (TCR)-derived bispecific construct, is a bit different from classical BsAbs. ImmTAC molecules are soluble TCRs stabilised using a disulphide bond and fused to an anti-CD3 scFv. Thanks to its TCR portion, the ImmTAC can recognise peptides derived from intracellular tumour targets that are presented by the MHC (or HLA), where a classical antibody binding site does not interact with the MHC but only recognises extracellular antigens on the cell surface. Engineered to present a high antigen-affinity TCR, the ImmTAC thus gives access to the targeting of a broader range of tumour cells, including “cold” cells presenting low immunogenicity. However, their construction is challenging as soluble TCRs are unstable when not embedded in a membrane and tend to aggregate in solution.161
Dual immunomodulatory bispecifics. The main pitfall of immunomodulatory antibodies, either immune checkpoint inhibitors or immunostimulators, is that they are not directed toward tumour cells and can induce a systemic immune response resulting in adverse events. Initially, they were administered in combination with tumour-targeted treatments such as monospecific antibodies and immune-cell engagers. For instance, PD-1/PD-L1 axis inhibition was shown to improve anti-tumour efficacy by reversing related immune resistance when used in combination with various types of T cell engagers, including αHER2 × αCD3,162 αCEA × αCD3,163 αCD33 × αCD3,164 αTrop-2 × αCD3 and αCEACAM5 × αCD3 BsAbs.165 Numerous other examples of immunomodulatory antibodies used in combination therapy, not only with BsAbs, have been reported and reviewed, notably by Patel et al.166 Two immunomodulators can also be combined into the same BsAb, generating a dual immunomodulatory BsAb that has the potential to: (1) avoid immune escape by blocking two different inhibitory immune checkpoints (such dual immune checkpoint inhibitory BsAbs include αPD-1 × αCTLA-4, αPD-1 × αTIM-3, and αPD-1 × αLAG3 bispecifics167–169); (2) induce a strong stimulation and expansion of T cells when combining two immunostimulators (e.g. dual agonistic BsAb αCD137 × αOX40);170 and (3) induce a synergistic effect when blocking an inhibitory immune checkpoint and activating a stimulatory immune checkpoint – this type of BsAb is sometimes defined as an agonist redirected checkpoint (αPD-1 × αOX40, αCTLA-4 × αOX40).171,172 This strategy can enhance T cell expansion, helping in converting “cold” tumours into “hot” ones. It is noteworthy that advantages mentioned in points (1) to (3) might also be attributed to the combination therapy of the corresponding individual mAbs and generate similar results. However, only BsAbs are able to (4) convert an inhibitory signal into a stimulating one at the tumour cell surface (αPD-L1 × αOX40).173 In addition, for any of the cited dual immunomodulatory BsAbs, choosing PD-L1 – which is expressed on tumour cells – as one of the two targeted immune checkpoints can potentially improve the selectivity of the BsAb for the tumour site when compared to a monospecific antibody targeting an immune checkpoint expressed on immune cells.
Similar to monoclonal immunomodulatory antibodies, dual immunomodulatory BsAbs can suffer from their lack of tumour targeting that induces toxicity. 14 clinical trials evaluating dual immunomodulatory BsAbs as a monotherapy are referenced (http://clinicaltrials.gov, accessed April 2020). This number represents more than 10% of all the clinical trials concerning BsAbs (14/135 studies), and 21% of all studies evaluating immunospecific BsAbs in monotherapy (14/66 studies in total, with studies concerning BATs not being included). Interestingly, all studies concern the treatment of solid tumours (Fig. 3, upper right dial).
Tumour-targeted immunomodulatory BsAbs. A way to exploit the immune boosting capacity while alleviating the secondary effects of immunomodulators is to include them in a BsAb targeted to the tumour site. For instance, in a HER2+ TUBO mouse tumour model (moderately resistant to anti-HER2 monotherapy), an αPD-L1 immunomodulator was included in an αHER2 × αPD-L1 BsAb (with a mouse IgG2a Fc backbone) that reduced tumour growth and increased tumour rejection when compared to the monovalent anti-PD-L1 and monovalent anti-HER2 monotherapies or their combination in vivo. Interestingly, in vitro results were less encouraging as the BsAb could bind to HER2 and PD-L1 and block the PD-1/PD-L1 axis but without affecting tumour cell proliferation. As expected, this enhanced anti-tumour effect of the αHER2 × αPD-L1 BsAb was dependent on the presence of CD8+ T lymphocytes and IFN-γ (IFN-γ notably regulates expression of PD-1).174 PRS-343, an αHER2 × α4-1BB BsAb, resulted in tumour growth inhibition and a dose-dependent increase of tumour-infiltrating lymphocytes in a HER2+ SKOV3 tumour model (engrafted with human peripheral blood mononuclear cells (PBMCs) in a humanized mouse model). The authors found that combining the targeting of the T cell co-stimulatory receptor 4-1BB with the HER2 tumour antigen enabled a more localized activation of the immune system, resulting in higher efficacy and reduced toxicity when compared to a monospecific approach.175 In another approach, a BsAb combines affinity for PD-1 and for RANKL, which is a member of the tumour necrosis factor receptor family, expressed by not only some tumour cells but also some immune cells. The corresponding Fab sequences were fused onto an IgG1 backbone and the αRANKL/αPD-1 BsAb monotherapy resulted in anti-tumour activity in αPD-1 resistant tumours and showed equivalent or superior anti-tumour response to the combination of the parent αRANKL and αPD-1 monospecific antibodies, depending on the tumour model. Similarly to other PD-1 targeting BsAbs, the anti-tumour activity is dependent not only on CD8+ T cells but also on host PD-1 and IFN-γ expression.176
BsAb combination therapy also includes the use of two different BsAbs (Fig. 5). This is probably the most extensive use of BsAbs reported so far. Claus et al. recently reported two combinations of two BsAbs, employing an αFAP × 4-1BBL plus an αCEA × αCD3 in one case, or an αCD19 × 4-1BBL plus an αCD20 × αCD3 in another (FAP is fibroblast activation protein, expressed on the tumour stroma).71 Previously, the targeting of the co-stimulatory receptor 4-1BB with a monospecific mAb did not advance to phase 3 in the clinical trial as its efficacy relied on Fcγ receptor-mediated hyper-clustering, which also induced hepatotoxicity. Thus, the authors developed two bispecific proteins: (1) having a mutated Fc that avoids their clustering through FcγR interaction and (2) either targeting CD19 (expressed on normal and malignant B cells) and 4-1BB, or targeting the FAP and 4-1BB. Thus, the T cell co-stimulatory activity of the 4-1BBL × αFAP or 4-1BBL × αCD19 bispecifics was strictly dependent on FAP or CD19-mediated clustering. The 4-1BBL × αFAP was administered in combination with a CEA-targeted T cell engager (αCEA × αCD3) in a gastric solid tumour in vivo model. Synergistic action was expected, as the αCEA × αCD3 BsAb was shown to induce 4-1BB up-regulation in CD8+ T cells. The treatment resulted in tumour growth inhibition, where parent monotherapies or control combination (using untargeted αDP47 × 4-1BBL protein) did not. Similarly, the 4-1BBL × αCD19 bispecific was administered in combination with a B cell-targeting αCD20 × αCD3 T cell engager, in an aggressive human lymphoma in vivo model. The treatment resulted in complete tumour eradication correlated with T cell accumulation in the tumour that was not obtained with the parent monotherapies. A similar strategy was adopted as soon as 2010 by Liu et al., where a combination of a 4-1BBL × αCD20 fusion protein and an αCD3 × αCD20 diabody resulted in more potent inhibition of human B lymphoma xenografts in SCID (severe combined immunodeficient) mice when compared to parental monotherapy, suggesting that the local delivery of 4-1BBL could effectively potentiate the anti-tumour activity of the T cell engager.185
 | ||
| Fig. 5 (a) Some strategies combine a BiTE (bispecific T cell engager) and a tumour targeted immunomodulator. In this example, both BsAbs target the same TAA, and an immunostimulator is used to sustain and support the T cell function through activation of the stimulatory immune checkpoint CD28 (TCR co-receptor). (b) The same combination strategy as 5(a) can be used but with two BsAbs targeting two different TAAs. (c) The SMITE (simultaneous multiple interaction T cell engaging) concept is another example of a combination strategy. It relies on the use of a BiTE and a tumour-targeted immunomodulator that targets a different TAA than the BiTE as well as the PD-L1 immune checkpoint to block its inhibitory activity. | ||
Earlier, Willems et al. described the combination of hPLAP × αCD28- and hPLAP × αCD3-targeting proteins. However, the generated constructs could be classed as trispecific molecules as they were, respectively, containing a peptide TAG P and an scFv specific to the P-peptide. The design of the generated Cri-BsAb (cross-interacting bispecific antibody) was made to induce a covalent interaction between the two proteins at the T cell and the tumour cell surfaces, in order to generate a more stable complex on the tumour cell as well as force the close localisation of CD3 and CD28 receptors on T cells, ensuring their correct activation.186 The authors proposed a sequential administration of the co-stimulating Cri-BsAb (αhPLAP × αCD28 × TAG P) followed by a low dose of activating Cri-BsAb (αhPLAP × αCD3 × αP) once the non-tumour-bound co-stimulating Cri-BsAb is sufficiently cleared. This is consistent with their results, showing that low concentrations of activating Cri-BsAb were needed to trigger T cell activation if a minimal amount of co-stimulating Cri-BsAb was bound to the T cell. However, despite the interesting strategy, only in vitro results were reported, and in vivo evaluation would be a necessary next step to further assay the safety and efficacy of this technique.
Recently, Correnti et al. also reported the use of a pair of BsAbs, and named this strategy SMITE (simultaneous multiple interaction T cell engaging).20 Actually, five different BiTE combinations were evaluated: in the first strategy, the two BiTEs were targeting the same ROR1 or the same CD19 tumour antigen, and either CD3 or CD28 (Fig. 5a), in order to engage and co-activate T cells (αROR1 × αCD3 plus αROR1 × αCD28, or αCD19 × αCD3 plus αCD19 × αCD28). In both cases, the CD28 BiTEs were ineffective alone but augmented the cytotoxic effects of CD3 BiTEs in a dose-dependent manner. Interestingly, the T cell activation was cancer cell binding-dependent when using the combination of two ROR1-targeting BiTEs since T cell activation by the αCD3 × αROR1 BiTE was dependent on ROR1 display on cancer cells, and the αCD28 × αROR1 BiTE on its own could not activate T cells and required preliminary T cell activation to exert co-activation. Conversely, a monoclonal αCD3 antibody (clone: OKT3) activated T cells equally in ROR− and ROR1+ tumour cells. The second SMITE strategy was reliant on a pair of BiTEs targeting different antigens on tumour cells (Fig. 5b), by combining Blinatumomab (αCD3 × αCD19) with an αCD28 × αROR1 BiTE, or an αCD3 × αROR1 BiTE with an αCD28 × αCD19 BiTE. In both cases, the combination of BiTEs resulted in a significant enhancement of cancer cell killing when compared to CD3 BiTE monotherapy, illustrating the possibility of targeting cancers expressing two independent antigens, possibly giving access to a more selective and less toxic treatment. In line with these results, the final SMITE strategy was evaluated, using the inhibitory immune checkpoint PD-L1 as a tumour antigen (Fig. 5c). Indeed, being expressed on tumour cells’ surfaces, PD-L1 can be considered as a tumour antigen. In some particular cases, PD-L1 was shown to be increased following blinatomumab treatment in some refractory cases. Targeting PD-L1 with an αCD28 × αPD-L1 BiTE allows both tumour targeting and the conversion of an inhibitory signal into a stimulatory one. Additionally, the potential toxicity of the αCD28 × αPD-L1 BiTE related to a wide expression of PD-L1, including in healthy tissues, is lowered due to the fact that CD28 activation is dependent on previous CD3 activation. This makes it well suited for combination therapy. Similarly to other SMITEs, the BiTE combinations (αCD28 × αPD-L1) and (αCD3 × αCD19) resulted in T cell co-activation strictly dependent on the expression of PD-L1 antigen by the tumour cell and the engagement of CD3 through the CD19/CD3 interaction. Interestingly, this SMITE not only reversed the PD-L1-mediated resistance in cells expressing PD-L1, but also increased cytotoxic effects when compared to parental cells treated with the (αCD19 × αCD3) BiTE antibody alone, probably resulting from the effective conversion of an inhibitory signal into a stimulatory one due to the (αCD28 × αPD-L1) BiTE. These results confirm the potential of a dual BsAb combination for cancer immunotherapy (Fig. 3, bottom left and right dials). However, as with the aforementioned publications, in vivo results are still necessary to fully validate this promising approach.
V. Multispecific Targeted Immune Cell Engagers & Modulators (MuTICEMs)
V.1. BsAb limitations – toward Multispecific Targeted Immune Cell Engagers & Modulators (MuTICEMs)?
Despite their undeniable positive impact on cancer treatment, BsAb antibodies and fragments can suffer limitations, as toxic adverse events and resistance mechanisms are observed in some cases.39,69,157 Notably, Kato et al. reported that PD-1/PD-L1 inhibitor resistance is linked to overexpression of multiple checkpoints, with VISTA, TIM-3 and macrophage associated marker CD68 being particularly involved, thus supporting the idea that targeting multiple pathways might be a valuable strategy.187 Of course, the BsAbs field is still making progress and all options may have not been explored yet; however, other approaches are still worth investigating. Notably, personalized therapies to answer distinct and complex immune profiles among patients would probably be the ultimate goal, but this requires important technical and financial investments.Decreasing toxicity through better selectivity, circumventing resistance mechanisms with immune checkpoint blockade, and further boosting the anti-tumour efficacy with immunostimulators require an approach with multi-effectors. Accumulating various effectors on a construct and/or at the tumour site is a way to improve treatment, and combination therapies tend to fulfil this requirement. However, when combined with a BsAb, immunomodulatory antibodies retain their potential toxicity as they are still not targeted to a tumour-specific antigen. Dual BsAb combinations (such as SMITEs) answer the need for targeting each effector to tumour cells when both BsAbs contain a tumour–antigen binding motif. However, if the same antigen is targeted by the two BsAbs, then: (1) counter-productive competition between the two BsAbs for binding to the target antigen can occur; (2) the effector/targeting module ratio is 1/1; and (3) the simultaneity of the BsAb actions can be hard to control. When combined BsAbs target different tumour-specific antigens, the competition issue is avoided, but the effector/targeting module ratio is still 1/1 and the spatio-temporal colocation remains a potential issue. Thus, combining multiple effectors (more than two) on the same construct really seems to be a valuable strategy to be evaluated and the next step to take in immunotherapeutic antibody development. Indeed, combining a tumour-targeting module (αTAA), an immune cell engaging module (e.g. αCD3) and one or two checkpoint immunomodulators (e.g. OX40L and αPD-L1) would allow a higher effector/targeting module ratio (2/1, 3/1, etc.) and a spatio-temporal co-localisation of effectors and potentially exert a strong synergic effect. For ease of use, we labelled this concept as MuTICEMs (Multispecific Targeted Immune Cell Engagers & Modulators). Adding two tumour targeting modules and including an Fc region are possible options. As discussed earlier, the PD-L1 receptor on tumour cells may also be considered as a secondary tumour antigen if combined with another TAA that promotes tumour selectivity. Clearly, combining multiple effectors on the same construct represents a higher level of complexity for production, and offers less adaptability of administration when compared to combination therapy (where one effector administration can be stopped while continuing the other one, different dosages, etc.), and including for instance affinity for both αCD3 and a co-stimulator like OX40L or CD28 might be considered risky toward on-target, off-tumour T cell activation and related adverse events. That is why in a MuTICEM approach, the effectors’ affinity for their targets would have to be very carefully designed and evaluated, with a leading affinity for the TAA probably being the best option to get lower toxicity, and the use of two lower-affinity TAA-targeting effectors being recommended when possible. The production, evaluation, safety and efficiency of MuTICEM immunotherapies raise complex challenges and questions. But the potential efficacy, synergy, resistance-overcoming, safety and broader application they could offer deserve to be investigated. In this section, we will discuss recent MuTICEM formats that have been developed, including tri- and tetra-specific antibody formats. We will also discuss the role that organic/bioconjugation chemistries could play in this potential breakthrough shift in the immunotherapy landscape, notably fast and adaptable production of MuTICEM formats for preliminary studies.
V.2. MuTICEMs – trifunctional formats
Only a few examples of trifunctional MuTICEMs have been reported so far. One of them is a checkpoint inhibitory T cell engaging (CiTE) antibody format.18,188,189 In order to circumvent the PD-1/PD-L1-induced adaptive immune resistance of the CD33-targeting BiTE, the authors developed a trifunctional antibody format (αCD33 × αCD3 × PD-1ex) by fusing a high-affinity CD33 scFv to a CD3 scFv and the extracellular domain of PD-1 (PD-1ex). The purpose was to combine T cell redirection to CD33 on AML cells with a locally restricted immune checkpoint blockade (Fig. 6a). Interestingly, PD-1ex shows low-affinity for PD-L1, thus affording the αCD33/CD33 affinity to drive the global selectivity of the CiTE and reducing on-target, off-side toxicity. Especially, the hypothesis was made by authors that the PD-1ex domain is not sufficient to directly target PD-L1-expressing cells and consequently does not block PD-1/PD-L1 interactions unspecifically. Nevertheless, following CiTE interaction with CD33, the proximity and the avidity effect would allow the PD-1ex/PD-L1 interaction. This αCD33-mediated and AML tumour cell-restricted immune checkpoint blockade would in turn potentiate local T cell activation and tumour killing while avoiding broadly distributed immune-related adverse events. For in vitro evaluation, control compounds were generated, including a single-chain triplebody (sctb) in which the PD-1ex module was replaced by an αPD-L1 scFv (αCD33 × αCD3 × αPD-L1, high affinity control); a BiTE-like compound (αCD33 × αCD3); an (αPD-1ex × αCD3) BsAb; and an (αPD-L1 × αCD3) BsAb. Compounds were incubated with HD (Healthy Donor) T cells and MOLM-13 or MOLM-13:PD-L1 cells expressing high levels of CD33, or HD T cells and OCI-AML3 or OCI-AML3:PD-L1 cells which express low CD33 levels.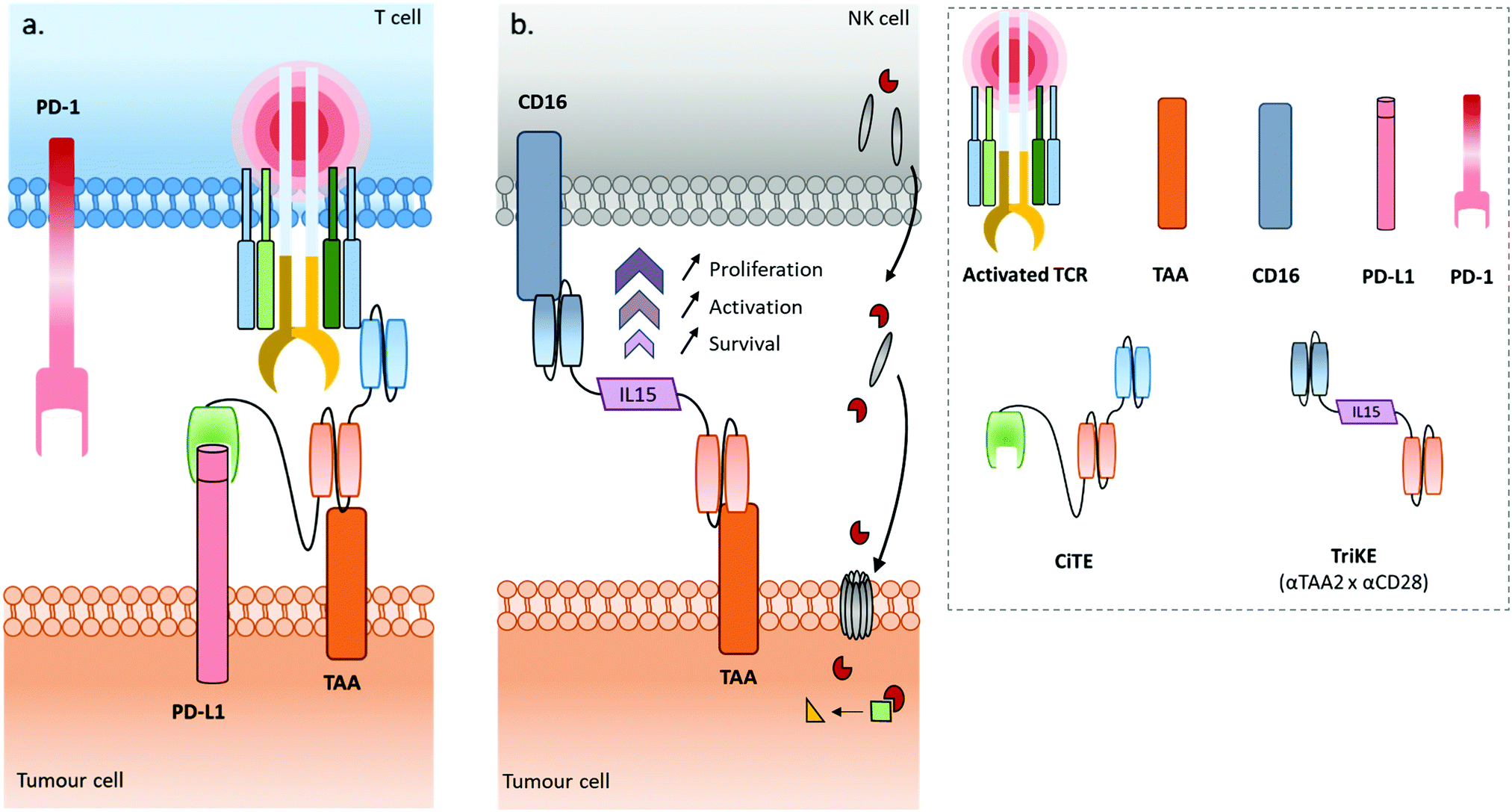 | ||
| Fig. 6 (a) The checkpoint-inhibitory T cell engager (CiTE), a bispecific antibody construct fused to the extracellular domain of PD-1 (PD-1ex), results in a trispecific compound that simultaneously targets a TAA, CD3 and PD-L1 to circumvent inhibitory immune checkpoint anergy that can be encountered when using BiTEs (αTAA × αCD3). (b) A TriKE (Trispecific Killer cell Engager) is a different trispecific construct that recruits NK cells to kill tumour cells through the targeting of CD16 and TAA. These two binding motifs are connected by an IL15 protein, which induces proliferation, activation and survival of the NK cells. | ||
On each of these four cell lines and in the presence of non-stimulated HD T cells, at a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 effector to target cell ratio (E:T), the dose-dependent lysis of targeted cells obtained with the CiTE was similar to that with the sctb. Yet, for both compounds on both high and low CD33-expressing cell lines, the cell expression of PD-1 was correlated with a lower EC50 (effective concentration for 50% of cell lysis). Thus, the inhibitory PD-1/PD-L1 axis was not only countered, but its blockade also improved the cytotoxicity, demonstrating a possible synergy resulting from the PD-1ex/PD-L1 or αPD-L1/PD-L1 involvement on the CiTE and sctb respectively. Surprisingly, the EC50s were lower on both the PD-1+/CD33low and PD-1−/CD33low cell lines when compared to the PD-1+/CD33high and PD-1−/CD33high cell lines respectively. The BiTE (αCD33 × αCD3) cytoxicity was dependent on CD33 expression, but independent of PD-L1 expression (while PD1/PD-L1 adaptive immune resistance could have been expected). Conversely, the CiTE and the sctb cytotoxicities were dependent on both CD33 and PD-L1 expressions. Interestingly, on MOLM-13 (CD33high), at 50 pM and an E:T ratio of 2:1, the BiTE was almost two times more cytotoxic than the CiTE and sctb, but the latter were more cytotoxic when PD-L1 was expressed (MOLM-13:PD-L1 cell line); no rationale was proposed by the authors for this result. A possible explanation might be that in the absence of PD-L1 expression by the CD33high cells, the presence of the fused PD-1ex protein on the original BiTE core (to form a CiTE) could have steric effects that reduce the interaction of the CiTE construct with the CD33 receptors when compared to the original BiTE, resulting in a counteractive effect of the PD-1ex on the therapeutic efficacy in this case. For CD33low cell lines, the BiTE had weak cytotoxic independently of PD-L1 expression, while both the CiTE and sctb were inefficient in the absence of PD-L1 expression but demonstrated marked cytotoxicity when PD-L1 was expressed. This means that despite the low affinity for PD-L1, the CiTE can potently target PD-L1 expressing cells. However, the fact that the (PD-1ex × αCD3) BsAb showed no toxicity on the same CD33low/PD-L1+ cell line confirms that PD-1ex alone is not strong enough to induce cytotoxicity and that CD33 expression, even at a low level, is mandatory. This ensures selectivity for cells expressing both receptors and restricts the PD-1 blockade to a local, CD33-targeted activity. Taken together, these results indicate that a BiTE might continue to be the best option to kill cancer cells with high CD33 and no PD-L1 expressions, but as soon as PD-L1 is expressed, the CiTE and the sctb seem to be better choices to kill cells with high or low CD33 expression. In vitro, the affinity for PD-L1 had an influence on bispecific formats, with the (αPD-L1 × αCD3) BsAb exhibiting a significantly lower EC50 than the (αPD-1ex × αCD3) BsAb when PD-L1 was expressed, independent of CD33 expression. On the other hand, the PD-L1 affinity had barely any influence on CiTE (low PD-L1 affinity) and sctb (high PD-L1 affinity) cytotoxicity – the EC50s are similar from one compound to the other, and from one cell line to the other when PD-L1 is expressed. This tends to indicate that, if the scbt's and CiTE's affinity for PD-L1 is mandatory to target CD33low/PD-L1+, the cytotoxicity of these compounds is probably mainly governed by the affinity for CD33 rather than the affinity for PD-L1, on both CD33high/PD-L1+ and CD33low/PD-L1+. These in vitro results question the interest in using PD-1ex rather than αPD-L1. However, the in vivo experiments (on NSG mice engrafted with MOLM-13:PD-L1 cells receiving in vitro activated human HD T cells before the treatment) confirmed the authors’ choice as the CiTE induced leukemia-eradication without on-target off-leukemia events, while the sctb resulted in body weight loss when compared to the other groups, possibly resulting from a sctb-mediated T cell redirection to PD-1+ cells and related on-target off-leukemia events. Opting for a lower affinity PD-1ex seems to be the rational choice for better safety results. All in all, the preclinical results of Herrmann et al. validate the use of a CiTE, a trispecific MuTICEM. Preclinical and clinical confirmation is needed, but while a BiTE might be a better option to treat non-resistant CD33+ AML cancers, it seems that the use of a trifunctional trispecific construct with a low-affinity PD-L1 binding motif (PD-1ex) does induce local restricted checkpoint inhibition, improved T cell activation and efficient treatment of CD33high and CD33low AML cancer cells expressing PD-L1, with reduced systemic toxicity.
:1 effector to target cell ratio (E:T), the dose-dependent lysis of targeted cells obtained with the CiTE was similar to that with the sctb. Yet, for both compounds on both high and low CD33-expressing cell lines, the cell expression of PD-1 was correlated with a lower EC50 (effective concentration for 50% of cell lysis). Thus, the inhibitory PD-1/PD-L1 axis was not only countered, but its blockade also improved the cytotoxicity, demonstrating a possible synergy resulting from the PD-1ex/PD-L1 or αPD-L1/PD-L1 involvement on the CiTE and sctb respectively. Surprisingly, the EC50s were lower on both the PD-1+/CD33low and PD-1−/CD33low cell lines when compared to the PD-1+/CD33high and PD-1−/CD33high cell lines respectively. The BiTE (αCD33 × αCD3) cytoxicity was dependent on CD33 expression, but independent of PD-L1 expression (while PD1/PD-L1 adaptive immune resistance could have been expected). Conversely, the CiTE and the sctb cytotoxicities were dependent on both CD33 and PD-L1 expressions. Interestingly, on MOLM-13 (CD33high), at 50 pM and an E:T ratio of 2:1, the BiTE was almost two times more cytotoxic than the CiTE and sctb, but the latter were more cytotoxic when PD-L1 was expressed (MOLM-13:PD-L1 cell line); no rationale was proposed by the authors for this result. A possible explanation might be that in the absence of PD-L1 expression by the CD33high cells, the presence of the fused PD-1ex protein on the original BiTE core (to form a CiTE) could have steric effects that reduce the interaction of the CiTE construct with the CD33 receptors when compared to the original BiTE, resulting in a counteractive effect of the PD-1ex on the therapeutic efficacy in this case. For CD33low cell lines, the BiTE had weak cytotoxic independently of PD-L1 expression, while both the CiTE and sctb were inefficient in the absence of PD-L1 expression but demonstrated marked cytotoxicity when PD-L1 was expressed. This means that despite the low affinity for PD-L1, the CiTE can potently target PD-L1 expressing cells. However, the fact that the (PD-1ex × αCD3) BsAb showed no toxicity on the same CD33low/PD-L1+ cell line confirms that PD-1ex alone is not strong enough to induce cytotoxicity and that CD33 expression, even at a low level, is mandatory. This ensures selectivity for cells expressing both receptors and restricts the PD-1 blockade to a local, CD33-targeted activity. Taken together, these results indicate that a BiTE might continue to be the best option to kill cancer cells with high CD33 and no PD-L1 expressions, but as soon as PD-L1 is expressed, the CiTE and the sctb seem to be better choices to kill cells with high or low CD33 expression. In vitro, the affinity for PD-L1 had an influence on bispecific formats, with the (αPD-L1 × αCD3) BsAb exhibiting a significantly lower EC50 than the (αPD-1ex × αCD3) BsAb when PD-L1 was expressed, independent of CD33 expression. On the other hand, the PD-L1 affinity had barely any influence on CiTE (low PD-L1 affinity) and sctb (high PD-L1 affinity) cytotoxicity – the EC50s are similar from one compound to the other, and from one cell line to the other when PD-L1 is expressed. This tends to indicate that, if the scbt's and CiTE's affinity for PD-L1 is mandatory to target CD33low/PD-L1+, the cytotoxicity of these compounds is probably mainly governed by the affinity for CD33 rather than the affinity for PD-L1, on both CD33high/PD-L1+ and CD33low/PD-L1+. These in vitro results question the interest in using PD-1ex rather than αPD-L1. However, the in vivo experiments (on NSG mice engrafted with MOLM-13:PD-L1 cells receiving in vitro activated human HD T cells before the treatment) confirmed the authors’ choice as the CiTE induced leukemia-eradication without on-target off-leukemia events, while the sctb resulted in body weight loss when compared to the other groups, possibly resulting from a sctb-mediated T cell redirection to PD-1+ cells and related on-target off-leukemia events. Opting for a lower affinity PD-1ex seems to be the rational choice for better safety results. All in all, the preclinical results of Herrmann et al. validate the use of a CiTE, a trispecific MuTICEM. Preclinical and clinical confirmation is needed, but while a BiTE might be a better option to treat non-resistant CD33+ AML cancers, it seems that the use of a trifunctional trispecific construct with a low-affinity PD-L1 binding motif (PD-1ex) does induce local restricted checkpoint inhibition, improved T cell activation and efficient treatment of CD33high and CD33low AML cancer cells expressing PD-L1, with reduced systemic toxicity.
Miller et al. described another type of trifunctional MuTICEM, devoted to the recruitment and activation of NK cells to CD33+ tumour cells.17,190,191 For this purpose, they modified a previous (αCD33 × αCD16) BiTE they reported by introducing an IL-15 cytokine serving as a linker to connect the anti-CD16 and anti-CD33 scFvs (Fig. 6b). They named this concept TriKE (for trispecific killer cell engager). (In the context of this review, we would describe it as a trifunctional bispecific construct, as only two binding antibody fragments are used. However, the authors might have considered IL15 as a binding motif for the IL15 receptor and thus considered the TriKe as a trispecific construct. In any case, it falls under our description of MuTICEMs.) IL15 was chosen for its capacity to induce the development, proliferation, activation and survival of NK cells. Thus, the resulting TriKE is able to recruit NK cells to CD33-expressing tumour cells and locally improve and sustain NK cell activity, resulting in higher ADCC-related anti-tumour activity and lower toxicity. In vitro, the TriKE (αCD33 × IL15 × αCD16) demonstrated a better ability than the BiTE (αCD33 × αCD16) to kill CD33+ HL-60 cells in the presence of healthy donor PBMCs, while a single anti-CD16 or anti-CD33 scFv did not increase HL-60 killing when compared to untreated PBMC control. Nevertheless, co-injection of the BiTE and IL15 yielded similar results to the TriKE. Importantly, the TriKE (αCD33 × IL15 × αCD16) was ineffective in killing CD33− HT-29 cells, demonstrating a specific CD33-mediated NK cell-induced killing of CD33+ HL-60 cells. Interestingly, the modularity of the strategy was demonstrated by replacing the αCD33 paratope with an αEpCAM paratope, generating an (αEpCAM × IL15 × αCD16) TriKE that induced potent killing of CD33−EpCAM+ HT-29 cells. The TriKE (αCD33 × IL15 × αCD16) was also shown to significantly increase IFNγ and TNFα expression when compared to the BiKE (αCD33 × αCD16). The higher levels of pro-inflammatory cytokines and chemokines by NK cells certainly seemed to contribute to the better anti-tumour activity. The in vivo evaluation on a murine xenograft model incorporating human NK cells and CD33+ tumour cells (HL-60) confirmed the in vitro results – intraperitoneal administration of 20 μg of TriKE or BiTE for 10 days resulted in similar control tumour burden, significantly better than that in the untreated group on day 14, while only the TriKE resulted in a significantly reduced tumour burden on day 21. Noteworthily, the level of circulating human NK cells in peripheral blood was 350-fold higher for mice treated with the TriKE when compared to those treated with the BiKE, strongly supporting that the IL15 linker within the TriKE delivers strong proliferation, survival and activation of the NK cells and is deeply implied in improved anti-tumour activity. A limitation of the strategy could be the ambiguous role of IL15, which was notably shown to take part in the development of leukemias and solid tumours, inhibit apoptosis of tumour cells, or promote their proliferation in some cases.17 However, the fact that IL15 is embodied in a trifunctional bispecific construct and targeted to the CD33+ cells potentially restricts its action to the tumour site, reducing off-site toxicity, as evidenced by the in vivo experiments for which no body weight loss was observed in mice treated with the TriKE. Taken together, these results support the MuTICEM strategy to exert local action for better efficacy and reduced toxicity. Interestingly, this TriKE strategy was recently applied in a preclinical study to chronic lymphocytic leukemia (CLL) by replacing the CD33 targeting with a CD19 targeting module (αCD19 × IL15 × αCD16), resulting in reversion of the inflammatory dysfunction in CLL and induction of NK cell-mediated CLL killing.192 This further illustrates the adaptability of the TriKE concept to treat various cancer types. A patent related to the generation and comparison of a CD16/IL-15/CD33 TriKE and a CD16/IL-15/CLEC12A TriKE for therapy of AML was recently filed (WO2020081841).193
Other NK cell engagers (NKCEs) consisting of trifunctional bispecific (αCD16 × αNKp46 × αTAA) antibody formats, with the anti-CD16 (anti-FcγRIII) actually being an Fc fragment (we didn’t consider Fc fragments as specific binders for the purpose of this review), were recently reported by Vivier et al.194 However, this bispecific antibody format is rather “outside of the box” and could be considered in a way as a trispecific as the Fc fragment was introduced together with an agonistic αNKp46 to target NK cells in particular and trigger their full activation and related ADCC. At least, this prompted us to classify it among the MuTICEMs for the purpose of this review, as NKp46 was described as an immune checkpoint and because specific recruitment and stimulation of NK cells to tumour cells is pursued here.195 Several bifunctional antibody formats harbouring a silent Fc fragment (no ADCC activity) were initially generated by authors, first by varying the agonistic NKp46-targeting epitopes for optimisation, then by varying TAA (CD19 or CD20) and binding motif formats (scFv, Fab or cross-mAb) for in vitro evaluation (NKCEs incubated with Daudi cells in the presence of NK cells as effectors). All the generated silent-Fc NKCEs were functional for NK-cell activation and presented strong anti-tumour activity, with the better cytotoxicity obtained for the candidate showing normal affinity for FcRn (other candidates had lower affinity for FcRn). In vivo, an (αNKp46 × αCD20) generated an NK-cell-dependent control of tumour progression on a human Raji B-cell lymphoma model, accompanied by significantly improved NK cell infiltration and/or proliferation in the tumour bed. Two trifunctional antibody formats were then generated, targeting CD19 or CD20 (against Daudi human B cell lymphoma cells) or EGFR (against A549 human lung carcinoma cells), and able to co-engage NKp46 and CD16 at the surfaces of NK cells, through either a wild-type Fc portion or an optimized CD16/Fc receptor binding (with the S239D and I332E mutations). An in vitro comparison of Fc-silent (bifunctional), Fc-competent (trifunctional) and Fc-optimized (trifunctional) NKCEs revealed that, independently from the targeted TAA, co-engagement of NKp46 and CD16 increased both the potency of tumour cell lysis and NK cell activation, when compared to the Fc-silent NKCE. Better cytotoxicity and NK cell activation were obtained with the Fc-optimized CD16 binding. Importantly, the co-engagement of NKp46 and CD16 with trifunctional NKCEs was more potent than a mixture of the corresponding bispecific reagents separately activating NKp46 and CD16, illustrating the importance of a simultaneous interaction with the two receptors and validating the construct design. Additionally, the cytotoxicity was TAA-dependent, and neither fratricidal NK-cell killing (NK-versus-NK toxicity) nor improved cytokine release were observed for the CD20-targeted NKCE in the presence of PBMCs, as opposed to obinutuzumab (Fc-engineered anti-CD20 mAb used in clinical practice). These results were confirmed in vivo where the trifunctional NKCEs (Fc-wild and Fc-optimized) significantly reduced tumour progression (but did not induce full eradication 30 days post-injection) when compared to the Fc-silent bifunctional NKCE and obinutuzumab on a solid tumour model (s.c. injection of Raji B lymphoma cells), and rescued all mice in an invasive tumour model (i.v. injection of Raji tumour cells) when obinutuzumab resulted in only 60% rescue at the same dose (50 mg of antibody per kilogram body weight). The improved in vivo tumour progression control of the trifunctional NKCEs thus validates both the targeting and activation of NK cells through simultaneous binding to NKp46 and CD16 receptors and the use of a trifunctional construct to engage and restrict NK cell activation to the tumour site. Notably, the fact that NKCEs’ affinity for NKp46 is 70 to 100 times greater than that of regular Fc for CD16 probably induces valuable selectivity for NKp46+ CD16+ NK cells toward NKp46− CD16+ myeloid cells, while still being able to activate NKp46+ CD16low NK tumour-infiltrating lymphocytes from tumour patients. Overall, the developed NKCEs represent an additional successful example of MuTICEMs. Clinical results are now awaited to confirm their potential as efficient and safer anti-tumour agents.
To the best of our knowledge, only a few other trispecific MuTICEMs have been described, all in patents and without related scientific publications, illustrating both the novelty of the strategy, and the competition for intellectual property in the field. Notably, patent WO2017081101 (“Trispecific Molecule Combining Specific Tumour Targeting and Local Immune Checkpoint Inhibition”, 2017) claims the design and functional characterisation of a TriKE (αCD16 × αCD47 × αCD33), engaging NK cells (via CD16) to CD33+ acute myeloid leukemia cells and simultaneously blocking CD47 (via the N-terminal Ig domain of SIRP-α), a “don’t eat me signal to macrophages” acting as an immune checkpoint.196 The MuTICEM is thus expected to generate local immune checkpoint inhibition and NK cell-related anti-tumour activity. Patent WO2018120843 (“Trifunctional Molecule and Application Thereof”, 2018) refers to the development and use thereof of a trispecific antibody format (recombinant protein peptide chain) made of three domains that may simultaneously target CD19, CD3 and a T cell co-stimulating receptor such as CD28, CTLA-4, and TIGIT. A trispecific antibody format (αCEA × αCD3 × agonistic αCD28) is mentioned as well.197 The clear goal here is to generate a T cell engager promoting local T cell activation, for improved T cell-induced anti-tumour activity and reduced systemic toxicity. Other trispecific MuTICEMs were claimed in patent WO2019166650 (“Trispecific antigen binding protein”, 2019). The aim here was to combine CD3 binding, tumour antigen binding and immune checkpoint pathway blocking, such as PD-1/PD-L1 blocking. Notably, in one example, in order to prevent the immune system from attacking cells indiscriminately, the blocking of the PD-1/PD-L1 axis was realised through low-affinity binding to PD-L1, to afford a rapid dissociation from PD-L1 surface proteins. However, the avidity and higher affinity generated by a simultaneous binding to both the TAA and PD-L1 can result in better selectivity for both protein-expressing tumour cells.198 Various trispecific formats were claimed, with the targeted immune checkpoint being selected from among CD40, CD47, CD80, CD86, GAL9, PD-L1 and PD-L2; the targeted tumour cell surface protein being selected from among BCMA, CD19, CD20, CD33, CD123, CEA, LMP1, LMP2, PSMA, FAP and HER2; and with the third binding domain targeting CD3, TCRαβ, CD16, NKG2D, CD89, CD64 and CD32a for the recruitment of immune cells.
Some other examples of trispecific antibody formats have also been reported: those simultaneously targeting three immune checkpoints, such as the orthogonal Fab-based (αPD-1 × α4-1BB × αCTLA-4) trispecific construct,199 simultaneously targeting three different antigens,200 and simultaneously targeting two tumour antigens and NK cells through a CD16-specific scFv.201 However, they do not combine tumour targeting, immune cell engagement and checkpoint immunomodulation, excluding them from the MuTICEM ensemble we defined and consequently from the focus of this review.
V.3. MuTICEMs – tetrafunctional formats
Very few tetrafunctional MuTICEM antibody formats have been reported so far. Following their work on TriKEs cited above, Miller et al. reported the production and in vitro evaluation of a TetraKE, a tetrafunctional trispecific construct containing an anti-EpCAM scFv (for carcinoma recognition), an anti-CD133 scFv (for cancer stem cell recognition), an anti-CD16 scFv for NK cell engagement and an IL-15 protein, responsible for NK cell survival and proliferation and used as a linker between anti-EpCAM and anti-CD16 entities.202 The purpose of such an antibody format is to simultaneously target two different TAAs to improve selectivity for tumours expressing both receptors. Moreover, CD133 is expressed by cancer stem cells (CSCs) which have been reported to induce chemo- and radiotherapy resistance and have tumour initiating and self-renewal abilities. As such, CSCs play a critical role in the development and/or relapse of cancers, and their destruction is of great importance for tumour eradication. The simultaneous targeting of the two tumour receptors allows a selective recruitment of NK cells to the tumour site in order to induce the NK cell-dependent killing of tumour cells through ADCC. Additionally, the presence of IL-15 on the TetraKE, and thus on the immune synapse, allows NK cell expansion and in turn was hypothesised to sustain the anti-tumour ADCC effect. The concept was evaluated and validated in vitro on various cell lines. Notably, binding competition assays on HT-29 (EpCAM+, CD133−) and Caco-2 (EpCAM+, CD133+) cell lines using EpCAM scFv or CD133 scFv or their combination confirmed the specific binding of an (αCD16 × IL-15 × αEpCAM × αCD133) TetraKE, validating its dual targeting ability. On purified NK cells, the TetraKE induced a significant cell culture expansion, to a greater degree than IL-15 alone, whereas anti-CD16, anti-EpCAM, anti-CD133 scFvs nor a (αEpCAM × αCD16) BiKE did. This demonstrated the ability of the TetraKE to efficiently induce NK cell proliferation, which was accompanied by a significant improvement in the survival of NK cells when compared to the αEpCAM × αCD16 BiKE. The TetraKE ability to induce the NK cell killing of cancer cells was evaluated at increasing effector/target (E:T) ratios on Caco-2 (CD133+, EpCAM+) and HT-29 (CD133−, EpCAM+) with NK cells freshly isolated from two donors. Convincingly, in the presence of either of the donors’ NK cells, TetraKE and BiKE (αEpCAM × αCD16), resulted in significantly superior killing on both cancer cell types when compared to other controls (no treatment, IL-15 alone, αCD133 × αCD16 BiKE, and anti-CD16, anti-EpCAM, anti-CD133 scFvs), with the TetraKE having an activity equivalent to or higher than the BiKE (αEpCAM × αCD16). Overall, these results prove the need for both the binding to one or both of the TAA and the CD16 to generate an immune synapse and the related ADCC tumour killing. However, a few interesting questions remain. (1) On the Caco-2 cell line (EpCAM+, CD133+), the TetraKE demonstrated higher killing than the BiKE (αEpCAM × αCD16) in the presence of NK cells from Patient 1 (cytolytic activity of ∼59% and ∼43% respectively) but equivalent killing in the presence of NK cells from Patient 2 (cytolytic activity of ∼39% and ∼38% respectively). This result indicates that the additional CD33 targeting and/or activity of IL-15 of the TetraKE may not always have a direct impact on tumor cell killing when compared to the αEpCAM × αCD16 BiKE. (2) On the HT-29 cell line (EpCAM+, CD133−), the TetraKE resulted in higher killing than the αEpCAM × αCD16 BiKE in the presence of NK cells from either Patient 1 (cytolytic activity of ∼58% and ∼52% respectively) or Patient 2 (cytolytic activity of ∼63% and ∼54% respectively). Since CD33 receptors are not expressed by HT-29 cells, the increased killing related to the TetraKE could be solely attributed to the presence (and activity) of IL-15 in the construct. Thus, the results on both cell lines can question the real impact of the CD33 binder: the improved TetraKE's activity on Caco-2 cells with NK cells from Patient 1 could not be due to the presence of CD33 binder and only be due to the presence of IL-15, as it presumably is on the HT-29 cell line. As a consequence, we can anticipate that evaluating a TriKE (αCD16 × IL-15 × αEpCAM) on both Caco-2 and HT-29 cell lines would have been of valuable interest to more accurately distinguish the influence of the CD33 binder and the IL-15, as well as confirming that the presence of both CD33 and IL15 in the construct confers an added value; we also appreciate that there are also many more complicating factors at play. Still, it has to be noted that an αCD133 × αCD16 BiKE was evaluated and resulted in effective cytolytic killing of Caco-2 cells in the presence of NK cells of both Patient 1 and Patient 2 (∼17% and 20% respectively), though to a lesser extent than the αEpCAM × αCD16 BiKE and the TetraKE, proving that CD133 is an effective target. Surprisingly, despite expressing both EpCAM and CD133, the cytolytic activity of the TetraKE was not superior on the Caco-2 cell line in comparison to the HT-29 cell line for both patients (the TetraKE induced a cytolytic NK activity of ∼59% in “Caco2 Patient 1”, ∼39% in “Caco2 Patient 2”, ∼58% in “HT59 Patient 1” and ∼63% in “HT59 Patient 2”). No explanation is provided for this observation, but a hypothesis could be that the total number of EpCAM receptors expressed by HT-29 cells is higher than the total of EpCAM and CD133 receptors expressed by Caco-2 cells. This is only speculation however and would have to be investigated for solid conclusions to be made. IFN-γ production and NK cell degranulation (related to surface expression of CD107a) were also evaluated. The TetraKE demonstrated a dose-dependent and specific degranulation capacity as well as a higher INF-γ production than in controls, in several cell lines. Concerning cytokine production (GM-CSF, IL-6, IL-8, TNFα), an important parameter to evaluate potential systemic toxicity, the TetraKE demonstrated similar results to the BiTE, notably a significant augmentation of GM-CSF and TNFα when compared to no treatment. This might be a pitfall when translating to in vivo as some BiTEs, despite their proven efficacy, have been limited in dose administration due to their toxicity.
In summary, these results indicate that the TetraKE (αCD16 × IL-15 × αEpCAM × αCD133) takes advantage of the dual targeting to induce specific ADCC in vitro, providing results equivalent to or better than related BiTEs (αCD16 × αEpCAM and αCD133 × αCD16). The presence of IL-15 results in specific NK cell proliferation, activation and survival, thus potentiating the ADCC anti-tumour effect. Combining the four effectors in the same compound allows their simultaneous action and restricts the IL-15 activity to the tumour site, potentially reducing toxicity. In addition, targeting CSCs is an additional advantage, allowing attack on the cells originating from and replenishing the tumour. However, a direct comparison between a TriKE (αCD16 × IL-15 × αEpCAM) and the TetraKE (αCD16 × IL-15 × αEpCAM × αCD133) would be interesting to confirm whether the targeting of two tumour receptors instead of one can improve the killing of cells that express both receptors. In vivo evaluation of this promising compound is necessary, notably to ensure that the moderately improved cytokine expression observed in vitro does not result in toxicity in vivo. Nonetheless, encouraging results are obtained with the TetraKE that addresses the needs of current BsAb therapies, and this illustrates that a strategic shift toward the use of MuTICEMs is a valuable option for future developments in immunotherapy research.
In addition to the above, a recent patent application (WO2019191120, 2019) describes Guidance and Navigation Control (GNC) proteins that typically fall under the description of a MuTICEM.203 Indeed, in several claimed embodiments, tetraspecific proteins comprise a binding domain for a T cell activating receptor, a binding domain for a tumour associated antigen, an antagonistic binding domain for an inhibitory immune checkpoint receptor, and an agonistic binding domain for a T cell co-stimulating receptor. Thus, the described proteins have the potential to recruit T cells to the cancer site and potentiate their activity by blocking inhibitory pathways and by activating stimulatory pathways. Some embodiments include the possibility to contain an Fc fragment to connect the different binding modules. Patents have to cover a maximum of applications; thus several GNCs were claimed for application in a broad range of cancer treatment, and with a variety of possible formats (1) including bi- to octo-specific GNCs; (2) recruiting immune cells among T cells, NK cells, macrophages and dendritic cells; and (3) possibly targeting a wide variety of TAAs (ROR1, CD19, EGFRγIII, BCMA, CD20, CD33, CD123, CD22, CD30, CEA, HER2, EGFR, LMP1, LMP2A, Mesothelin, PSMA, EpCAM, glypican-3, gpA33, GD2, TROP2, etc.), inhibitory immune checkpoints (PD-L1, PD-1, TIGIT, TIM-3, LAG-3, CTLA4, BTLA, VISTA, PDL2, CD160, LOX-1, siglec-15, CD47, etc.) and stimulatory immune checkpoints (4-1BB, CD28, OX40, GITR, CD40L, ICOS, Light, CD27, CD30, etc.). However, only tetraspecific GNCs were exemplified, all of them encompassing an anti-PD-L1 as the immune checkpoint inhibitor, an anti-CD3 as a T cell activator, an anti-4-1BB as the co-stimulator and either an anti-ROR1, anti-CD19 or anti-EGFRγIII binding motif as the tumour-targeting module. Globally, in vitro tests of exemplified GNCs demonstrated re-directed T cell toxicity (RTCC), specific T cell proliferation and TNFα expression. Interestingly, the proximity of the binding sites with each other was shown to have an impact on the granzyme B production by PBMCs in response to treatment with EGFRγIII targeting tetra-specific GNC antibodies. For instance, the IC50 of granzyme production for an (α4-1BB × αPD-L1 – Fc – αTAA × αCD3) GNC was 61 pM, while it was only 0.006 pM for an (αPD-L1 × αCD3 – Fc – α4-1BB × αTAA) GNC. This result indicates that not only the presence of multiple effectors but also their spatial organisation inside a MuTICEM can influence its activity, being an additional parameter to evaluate. Here again, in vivo evaluation is needed to make further insightful conclusions.
V.4. MuTICEMs – whether or not to include an Fc fragment in the design
The rules previously outlined for the construction of bi and tri-functional bispecific antibodies (see Section IV.2. BsAb design) are likely to be different for multispecific antibodies. For instance, for a tetrafunctional tetraspecific antibody combining a tumour-targeting motif, an immunostimulator, an immune checkpoint inhibitor and engaging T cells (e.g. αTAA × αOX40 × αPD-1 × αCD3), adding an Fc fragment could have beneficial impact through ADCC tumour killing (via recruitment of immune cells to TAA expressing cells) and Treg depletion (through Fc interaction with OX40-expressing Tregs) but possible negative impact through effector T cell depletion (via ADCC killing of CD3+/PD-1− and CD3+/PD-1+ expressing T cells). In addition, considering T-cell redirection is already pretty effective, it is not certain Fc-mediated killing would provide a large increase in cytotoxicity. On these particularly complex cases, experimentations are needed, especially as it is such a technically complex field.V.5. MuTICEMs – a potential application supported by organic and bioconjugation chemistry
To the best of our knowledge, no other tri- and tetrafunctional MuTICEMs have been reported so far, probably illustrating the novelty and the complexity of their production. However, the aforementioned publications and patents constitute a proof of concept for the feasibility of such antibody formats. Pre-clinical and clinical in vivo evaluations will have to be completed in order to confirm if better and sustained anti-tumour efficacy, lower toxicity and a broader spectrum of cancer treatment can be achieved with MuTICEMs. Theoretically, combining more synergistic effectors holds the potential for better treatment. The preliminary results of the publications cited above confirm this trend. However, it is important that the resulting strong activation of targeted immune cells is restricted to the tumour site to avoid systemic inflammation, which is already a pitfall of some current immunotherapies.The other bottleneck inherent to the combination of multiple effectors in a single antibody format is their complex, time-consuming and cost-effective production, as they are mainly produced through DNA and protein engineering (this was the case for all the MuTICEM compounds cited above). These issues are strengthened by the low modularity of such production techniques – for each new antibody format, the whole production design has to be modified. Considering that not only the presence but also the spatial distribution of effectors can have an influence on the anti-tumour activity; possibilities are numerous, and the production time and costs can grow depending on the proportion of possible formats that are to be tested. So far, progress in the immunotherapy landscape has been generally supported by technological or scientific improvements toward the ease of production of therapeutic antibodies, as well as reduction of the time and financial costs required for it. While the difficulties in the production of MuTICEMs will probably be, and might already be, an obstacle to their development, organic/bioconjugation chemistry might partially alleviate these obstacles, by allowing the production of MuTICEMs directly from native (or facilely engineered from native) proteins. Indeed, recent progress in the field including fast metal-free click chemistry reactions, site-selective protein functionalisation, and improved purification methods allows fast, controlled and easy-to-adapt production of modified proteins.204–209 Starting from native proteins, the most recent procedures to chemically generate a multispecific antibody format rely on digesting parent antibodies and isolate the binding motifs of interest (Fab fragments and eventually Fc fragments) to subsequently assemble them directly with each other or on a chemical platform by using fast, orthogonal and chemo- and/or site-selective ligation reactions. Smaller units such as scFvs and small binding sites can also be produced or purchased and chemically modified in order to include them in a multispecific antibody format. But it is also possible to chemically attach two native full antibodies to each other.210 Developed from seminal studies by Sharpless and Bertozzi on click chemistry and copper-free click chemistry, respectively,211,212 popular click reactions for protein assembly include strain-promoted azide–alkyne cycloaddition (SPAAC),213,214 strain-promoted alkyne–nitrone cycloaddition (SPANC),215 and inverse-electron-demand Diels–Alder (iEDDA) reactions involving partners such as trans-cyclooctyne/tetrazine, strained alkyne/tetrazine, and strained alkyne/fluorosydnone partners.216–218 However, native proteins generally have to be modified to introduce click chemistry functionalities. This can be done through protein engineering that allows the site-selective introduction of unnatural amino acids in the protein sequence, providing bio-orthogonal handles such as azides, alkynes and tetrazines to the non-native generated protein.219–221 It is also possible to directly modify native proteins following a strategy sometimes described as “plug-and-play”, where site-selective reactions are used to introduce click chemistry handles on proteins and allow their subsequent chemo-selective assembly.222,223 An ideal site-selective modification presents high efficiency, repeatability, and selectivity for a small number of protein sites (amino-acids or a group of amino-acids), in order to ensure very fine control of the protein modification.224,225 The field of site-selective modification of native proteins, especially antibodies, has been strongly investigated in recent years and includes a wide range of strategies such as endogenous amino acid-selective modification,226 kinetically and template-directed lysine modification,227 enzymatic and chemo-enzymatic modification of either antibody glycans228 or antibody amino-acid residues (with enzymes such as sortase A and transglutaminase, which allow site-selectively connecting peptide-containing substrates on the antibody, or the formylglycine generating enzyme which introduces an aldehyde group exploited in subsequent coupling reactions),229 multi-component reaction modification,230 and nucleotide binding site (NBS) modification.231 However, cysteine alkylation, Michael-addition, and cysteine rebridging constitute the more investigated and exploited approaches.12,232–237 A majority of the site-selective protein modifications and plug-and-play modifications were described for antibody–drug conjugate production.232,238–241 However, similar strategies can be used for protein–protein conjugation to generate multispecific antibody formats, even though overcoming steric hindrance is an additional and important issue to be taken into consideration. For instance, a linker with two terminal “next generation maleimide” (NGM) moieties was used to create an Fab-ScFv bispecific, relying on sequential rebridging of the Fab and scFv motifs, with a 52% yield even though homodimerisation is a drawback of the method.242 The Kauffman group described a plug-and-play approach combining NGMs and click chemistry, consisting of Fab reduction and rebridging with an NGM linker containing either an azide or a dibenzylcyclooctyne (DBCO), to generate the corresponding azide-bearing Fab and DBCO-bearing Fab that were connected through SPAAC to yield a BsAb.223 The same strategy was used to functionalise and connect two full IgG2 units, yielding a full length IgG2-IgG2 BsAb.243 The Chudasama and Baker groups recently reported a dually functionalised bispecific antibody format generated from two Fab fragments isolated from native antibodies and modified with either a bicyclo[6.1.0]nonyne (BCN) or a tetrazine functionality for their subsequent conjugation through the inverse-electron-demand Diels–Alder reaction.222 The click handles were introduced via reduction and rebridging of the disulfide bonds of each Fab with a functionalised dibromopyridazinedione. Subsequent modification with up to two different fluorophore payloads was possible through CuAAC when additional alkyne handles were introduced as well, paving the way for modification with proteins (Fab, scFv, etc.) to yield tri- or tetra-specific antibody formats. The dibromo-pyridazinedione used in this approach can be considered as a chemical platform because this compound can contain two different click handles, thus allowing the connection of three different proteins/payloads through the combination of one disulfide rebridging and two orthogonal click reactions. Such multifunctional chemical platforms displaying several orthogonal click handles might be a key element in generating multispecific antibody formats in a selective, controlled and versatile manner. Some tri- and tetrafunctional linkers, benzene derivatives, and cyclic peptides have been reported and proven to be modular chemical platforms that can be orthogonally functionalised with ligands, payloads and/or proteins.244–247 Further developments introducing handles for faster click and more site-selective reactions would optimize this type of structure for use in multispecific antibody format production, including MuTICEMs.
Considering the selectivity, speed, purity, modularity and reaction scale that bioconjugation and bioorganic chemistries offer, they could be used for fast and small-scale production of a wide variety of tri-, tetra-, or penta-specific antibody formats, quickly available for in vitro and pre-clinical in vivo evaluations (Fig. 7). The quicker results could enable faster feed-back and subsequent tuning of produced antibody formats to improve their activity, powering the wheel of a virtuous circle. Bioconjugation and bioorganic chemistry could thus be used for efficient screening of the best effector-combinations for MuTICEM formats, as has been proposed for BsAbs,248 and be complementary to engineering methods, the use of which would be limited to high-scale production for clinical trials once effector screening has been successful. However, since the format matters, a chemical construct may not have the same effect as an engineered construct even if they both contain the same species. Thus, even if high-scale production is currently limited to engineering methods, one could imagine that developing large-scale chemical production of multispecific constructs, possibly exploiting automated flow reactors and SEC columns, could be a valuable strategy. Moreover, bioconjugation permits the facile functionalisation of proteins with drugs or imaging agents. Thus, MuTICEM–drug conjugates can also be envisioned to improve treatment, and MuTICEM–contrast agent conjugates can be used to study pharmacokinetics/pharmacodynamics profiles and/or treatment response in a theranostic approach.
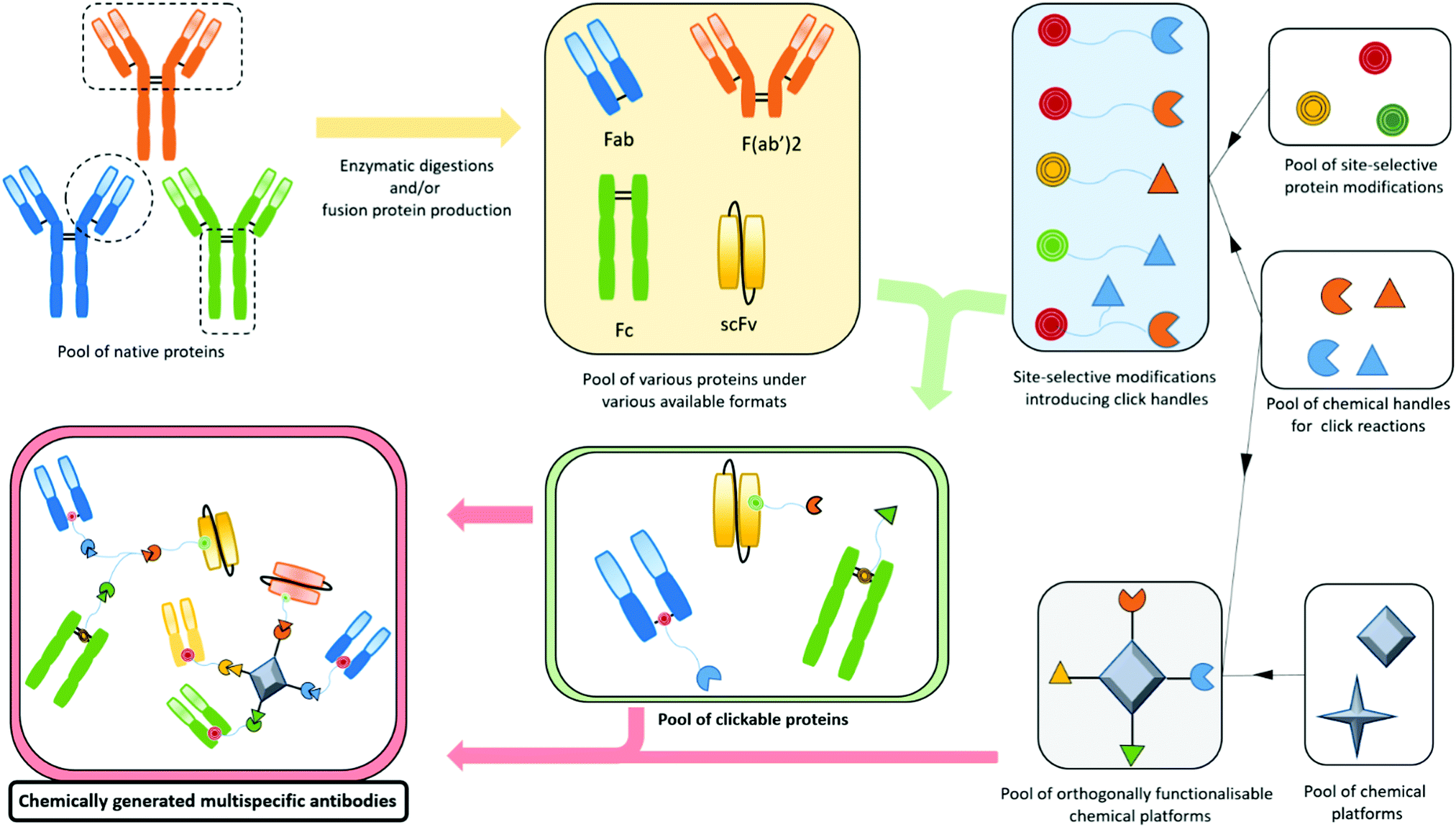 | ||
| Fig. 7 Scheme of processes allowing production of multispecific antibody formats from native proteins by employing site-selective modifications, click chemistry and bioconjugation. Protein engineering can be used, but is not necessary in any step, making the chemically generated multispecific antibody formats a fast, flexible and economic alternative to protein-engineered multispecific formats. | ||
VI. Conclusion
Since the beginning of the 2000s, the development of immunotherapy for cancer treatment has been a booming field of research. This was notably due to the success of immune checkpoint blockade strategies in clinical trials. However, while monoclonal antibodies generated satisfactory results, some resistance and toxicity were observed and oriented the research to the development of bispecific antibodies that enable the dual binding of two targets among tumour receptor, immune checkpoint, and effector cells such as T cells and NK cells. The recruitment of immune cells turned out to be a further breakthrough in the immunotherapy landscape as it generated very positive results and notably led to the commercialisation of Blinatumomab (BlincytoTM) for treatment of leukemia. Prompted by these two decades of progress, current research in immunotherapy mainly focuses on the development of new immune cell engager BsAbs, cancer-targeted BsAbs addressing new immune checkpoints, BsAbs targeting two immune checkpoints, and combinations thereof. While some encouraging results have been obtained, toxicity and resistance are still observed in some cases, probably partly originating from the fact that all the effector activities (cancer cell targeting, immune cell engagement and immune checkpoint modulation) are not tumour site-restricted and simultaneous. In this review, after an overview of the immunotherapy landscape, we focused on the idea that including more than two effectors in a single antibody format to generate a MuTICEM would enable scientists to direct and restrict multiple immune checkpoint-modulations and immune cell engagement to the tumour site, possibly resulting in lower toxicity and better efficacy, notably due to simultaneous and synergistic effects. Clearly, such multispecific antibody formats are more complex structures that would require more effort to develop. However, some examples of MuTICEMs have already been published, proving the feasibility of this approach. These initial results are encouraging but more research on the topic and the assaying of more examples are needed. As they combine all the current leading strategies in cancer immunotherapy, MuTICEM compounds are (in our opinion) likely to be the next step to take in the field of cancer immunotherapy research. Their production through bio-engineering might not be the optimal strategy for development, especially because a lot of constructs would have to be generated and evaluated to define the compounds with the best effector combination, which would depend on the particular cancer's characteristics. However, we believe that organic/bioconjugation chemistry could have a key role to play in this new field. Indeed, recent developments in site-selective protein modification, fast biocompatible click reactions, versatile functionalizable chemical platforms and protein purification could help in producing multispecific antibody formats on a low to medium scale with modularity, speed and cost-effectiveness. We believe that the easier access to a wide variety of effector combinations in multispecific antibody formats would speed up their in vitro and in vivo screening. This in turn would facilitate the transfer to the clinical stage where bio-engineering might still be the method of choice to produce the selected multispecific antibody formats on a high-scale, unless more efforts are put into the development of their high-scale chemical production.Author contributions
The review was written, and the literature analysed, through contributions by both authors. Both authors gave approval to the final version of the manuscript. F. T. and V. C. contributed equally.Conflicts of interest
There are no conflicts to declare but we do highlight that VC is a Director of ThioLogics.Acknowledgements
This project received funding from the European Union's Horizon 2020 research and innovation programme under grant agreement no. 838703. The authors would also like to thank Peter Szijj for useful discussions.Notes and references
- B. S. Graham and D. M. Ambrosino, Curr. Opin. HIV AIDS, 2015, 10, 129–134 CrossRef CAS PubMed.
- P. J. Carter and G. A. Lazar, Nat. Rev. Drug Discovery, 2018, 17, 197–223 CrossRef CAS PubMed.
- G. Köhler and C. Milstein, Nature, 1975, 256, 495–497 CrossRef PubMed.
- P. Chames, M. Van Regenmortel, E. Weiss and D. Baty, Br. J. Pharmacol., 2009, 157, 220–233 CrossRef CAS PubMed.
- P. A. Scolnik, MAbs, 2009, 1, 179–184 CrossRef PubMed.
- A. Saxena and D. Wu, Front. Immunol., 2016, 7, 580 Search PubMed.
- H. Liu, A. Saxena, S. S. Sidhu and D. Wu, Front. Immunol., 2017, 8, 38 Search PubMed.
- J. M. Reichert, C. J. Rosensweig, L. B. Faden and M. C. Dewitz, Nat. Biotechnol., 2005, 23, 1073–1078 CrossRef CAS PubMed.
- E. Cruz and V. Kayser, Biologics, 2019, 13, 33–51 CAS.
- K. J. O. Jr and S. A. Hurvitz, Antibody-Drug Conjugates: Fundamentals, Drug Development, and Clinical Outcomes to Target Cancer, John Wiley & Sons, 2016 Search PubMed.
- P. Khongorzul, C. J. Ling, F. U. Khan, A. U. Ihsan and J. Zhang, Mol. Cancer Res., 2020, 18, 3–19 CrossRef CAS PubMed.
- V. Chudasama, A. Maruani and S. Caddick, Nat. Chem., 2016, 8, 114–119 CrossRef CAS PubMed.
- H. Yao, F. Jiang, A. Lu and G. Zhang, Int. J. Mol. Sci., 2016, 17, 194 CrossRef PubMed.
- R. Stewart, S. A. Hammond, M. Oberst and R. W. Wilkinson, J. ImmunoTher. Cancer, 2014, 2, 29 CrossRef.
- R. E. Kontermann and U. Brinkmann, Drug Discovery Today, 2015, 20, 838–847 CrossRef CAS PubMed.
- A. Maruani, Drug Discovery Today, 2018, 30, 55–61 CrossRef PubMed.
- D. A. Vallera, M. Felices, R. McElmurry, V. McCullar, X. Zhou, J. U. Schmohl, B. Zhang, A. J. Lenvik, A. Panoskaltsis-Mortari, M. R. Verneris, J. Tolar, S. Cooley, D. J. Weisdorf, B. R. Blazar and J. S. Miller, Clin. Cancer Res., 2016, 22, 3440–3450 CrossRef CAS PubMed.
- M. Herrmann, C. Krupka, K. Deiser, B. Lindl, R. Mocikat, K. H. Metzeler, K. Spiekermann, S. Kobold, N. Fenn, K.-P. Hopfner and M. Subklewe, Blood, 2018, 132, 4069 CrossRef.
- M. Herrmann, C. Krupka, K. Deiser, B. Brauchle, A. Marcinek, A. Ogrinc Wagner, F. Rataj, R. Mocikat, K. H. Metzeler, K. Spiekermann, S. Kobold, N. C. Fenn, K.-P. Hopfner and M. Subklewe, Blood, 2018, 132, 2484–2494 CrossRef CAS PubMed.
- C. E. Correnti, G. S. Laszlo, W. J. de van der Schueren, C. D. Godwin, A. Bandaranayake, M. A. Busch, C. J. Gudgeon, O. M. Bates, J. M. Olson, C. Mehlin and R. B. Walter, Leukemia, 2018, 32, 1239–1243 CrossRef CAS PubMed.
- Cancer immunotherapy – Latest research and news|Nature, https://www.nature.com/subjects/cancer-immunotherapy, (accessed March 25, 2021).
- E. Anassi and U. A. Ndefo, Pharm. Ther., 2011, 36, 197–202 Search PubMed.
- R. E. Hollingsworth and K. Jansen, npj Vaccines, 2019, 4, 1–10 CrossRef PubMed.
- S. Srivastava and S. R. Riddell, Trends Immunol., 2015, 36, 494–502 CrossRef CAS PubMed.
- A. N. Miliotou and L. C. Papadopoulou, Curr. Pharm. Biotechnol., 2018, 19, 5–18 Search PubMed.
- Oncolytic Virus Therapy: Using Tumor-Targeting Viruses to Treat Cancer – National Cancer Institute, https://www.cancer.gov/news-events/cancer-currents-blog/2018/oncolytic-viruses-to-treat-cancer (accessed April 29, 2020).
- P. Kleinpeter, L. Fend, C. Thioudellet, M. Geist, N. Sfrontato, V. Koerper, C. Fahrner, D. Schmitt, M. Gantzer, C. Remy-Ziller, R. Brandely, D. Villeval, K. Rittner, N. Silvestre, P. Erbs, L. Zitvogel, E. Quéméneur, X. Préville and J.-B. Marchand, Oncoimmunology, 2016, 5(10), e1220467 CrossRef PubMed.
- J. X. Yu, V. M. Hubbard-Lucey and J. Tang, Nat. Rev. Drug Discovery, 2019, 18, 899–900 CrossRef PubMed.
- T. Cerny, B. Borisch, M. Introna, P. Johnson and A. L. Rose, Anti-Cancer Drugs, 2002, 13, S3 CrossRef CAS PubMed.
- T. Vu and F. X. Claret, Front. Oncol., 2012, 2, 62 Search PubMed.
- S. Trivedi, H.-B. Jie and R. L. Ferris, Semin. Oncol., 2014, 41, 678–684 CrossRef CAS PubMed.
- X. Geng, X. Kong, H. Hu, J. Chen, F. Yang, H. Liang, X. Chen and Y. Hu, Hum. Vaccin. Immunother., 2015, 11, 2769–2776 CrossRef PubMed.
- Z. Fan, H. Masui, I. Altas and J. Mendelsohn, Cancer Res., 1993, 53, 4322–4328 CAS.
- M. J. Glennie, R. R. French, M. S. Cragg and R. P. Taylor, Mol. Immunol., 2007, 44, 3823–3837 CrossRef CAS PubMed.
- K. L. Gutbrodt, G. Casi and D. Neri, Mol. Cancer Ther., 2014, 13, 1772–1776 CrossRef CAS PubMed.
- D. Neri, Cancer Immunol. Res., 2019, 7, 348–354 CrossRef CAS PubMed.
- D. M. Pardoll, Nat. Rev. Cancer, 2012, 12, 252–264 CrossRef CAS PubMed.
- P. Darvin, S. M. Toor, V. S. Nair and E. Elkord, Exp. Mol. Med., 2018, 50, 1–11 CrossRef PubMed.
- J. A. Marin-Acevedo, B. Dholaria, A. E. Soyano, K. L. Knutson, S. Chumsri and Y. Lou, J. Hematol. Oncol., 2018, 11, 39 CrossRef PubMed.
- C. Paluch, A. M. Santos, C. Anzilotti, R. J. Cornall and S. J. Davis, Front. Immunol., 2018, 9, 2306 CrossRef PubMed.
- M. Yasunaga, Semin. Cancer Biol., 2020, 64, 1–12 CrossRef CAS PubMed.
- F. Cameron, G. Whiteside and C. Perry, Drugs, 2011, 71, 1093–1104 CrossRef PubMed.
- X. Du, F. Tang, M. Liu, J. Su, Y. Zhang, W. Wu, M. Devenport, C. A. Lazarski, P. Zhang, X. Wang, P. Ye, C. Wang, E. Hwang, T. Zhu, T. Xu, P. Zheng and Y. Liu, Cell Res., 2018, 28, 416–432 CrossRef CAS PubMed.
- L. A. Raedler, Am. Health Drug Benefits, 2015, 8, 96–100 Search PubMed.
- FDA Approval Timeline of Active Immunotherapies, https://www.cancerresearch.org/scientists/immuno-oncology-landscape/fda-approval-timeline-of-active-immunotherapies (accessed April 30, 2020).
- K. M. Hargadon, C. E. Johnson and C. J. Williams, Int. Immunopharmacol., 2018, 62, 29–39 CrossRef CAS PubMed.
- J. X. Yu, J. P. Hodge, C. Oliva, S. T. Neftelinov, V. M. Hubbard-Lucey and J. Tang, Nat. Rev. Drug Discovery, 2019, 19, 163–164 Search PubMed.
- S. Ghosh, G. Sharma, J. Travers, S. Kumar, J. Choi, H. T. Jun, M. Kehry, S. Ramaswamy and D. Jenkins, Mol. Cancer Ther., 2019, 18, 632–641 CrossRef CAS PubMed.
- E. Ruffo, R. Wu, T. C. Bruno, C. J. Workman and D. A. A. Vignali, Semin. Immunol., 2019, 42, 101305 CrossRef CAS PubMed.
- A. Friedlaender, A. Addeo and G. Banna, ESMO Open, 2019, 4, e000497 CrossRef PubMed.
- X. Huang, X. Zhang, E. Li, G. Zhang, X. Wang, T. Tang, X. Bai and T. Liang, J. Hematol. Oncol., 2020, 13, 83 CrossRef PubMed.
- S. Qin, L. Xu, M. Yi, S. Yu, K. Wu and S. Luo, Mol. Cancer, 2019, 18, 155 CrossRef PubMed.
- E. RodrÍguez, S. T. T. Schetters and Y. van Kooyk, Nat. Rev. Immunol., 2018, 18, 204–211 CrossRef PubMed.
- M. F. Sanmamed, F. Pastor, A. Rodriguez, J. L. Perez-Gracia, M. E. Rodriguez-Ruiz, M. Jure-Kunkel and I. Melero, Semin. Oncol., 2015, 42, 640–655 CrossRef CAS PubMed.
- I. Melero, S. Hervas-Stubbs, M. Glennie, D. M. Pardoll and L. Chen, Nat. Rev. Cancer, 2007, 7, 95–106 CrossRef CAS PubMed.
- X. He, R. L. Smeets, E. van Rijssen, A. M. H. Boots, I. Joosten and H. J. P. M. Koenen, Sci. Rep., 2017, 7, 1–11 CrossRef PubMed.
- C.-H. Lin and T. Hünig, Eur. J. Immunol., 2003, 33, 626–638 CrossRef CAS PubMed.
- G. Suntharalingam, M. R. Perry, S. Ward, S. J. Brett, A. Castello-Cortes, M. D. Brunner and N. Panoskaltsis, N. Engl. J. Med., 2006, 355, 1018–1028 CrossRef CAS PubMed.
- R. H. Vonderheide, Annu. Rev. Med., 2020, 71, 47–58 CrossRef CAS PubMed.
- M. Croft, Nat. Rev. Immunol., 2009, 9, 271–285 CrossRef CAS PubMed.
- T. H. Watts, Annu. Rev. Immunol., 2005, 23, 23–68 CrossRef CAS PubMed.
- D. S. Vinay and B. S. Kwon, BMB Rep., 2014, 47, 122–129 CrossRef PubMed.
- D.-T. Chu, N. D. Bac, K.-H. Nguyen, N. L. B. Tien, V. V. Thanh, V. T. Nga, V. T. N. Ngoc, D. T. Anh Dao, L. N. Hoan, N. P. Hung, N. T. Trung Thu, V.-H. Pham, L. N. Vu, T. A. V. Pham and D. B. Thimiri Govinda Raj, Int. J. Mol. Sci., 2019, 20, 1822 CrossRef CAS PubMed.
- C. Chester, M. F. Sanmamed, J. Wang and I. Melero, Blood, 2018, 131, 49–57 CrossRef CAS PubMed.
- N. H. Segal, T. F. Logan, F. S. Hodi, D. McDermott, I. Melero, O. Hamid, H. Schmidt, C. Robert, V. Chiarion-Sileni, P. A. Ascierto, M. Maio, W. J. Urba, T. C. Gangadhar, S. Suryawanshi, J. Neely, M. Jure-Kunkel, S. Krishnan, H. Kohrt, M. Sznol and R. Levy, Clin. Cancer Res., 2017, 23, 1929–1936 CrossRef CAS PubMed.
- N. H. Segal, A. R. He, T. Doi, R. Levy, S. Bhatia, M. J. Pishvaian, R. Cesari, Y. Chen, C. B. Davis, B. Huang, A. D. Thall and A. K. Gopal, Clin. Cancer Res., 2018, 24, 1816–1823 CrossRef CAS PubMed.
- C. Wang, G. H. Y. Lin, A. J. McPherson and T. H. Watts, Immunol. Rev., 2009, 229, 192–215 CrossRef CAS PubMed.
- M. Compte, S. L. Harwood, I. G. Muñoz, R. Navarro, M. Zonca, G. Perez-Chacon, A. Erce-Llamazares, N. Merino, A. Tapia-Galisteo, A. M. Cuesta, K. Mikkelsen, E. Caleiras, N. Nuñez-Prado, M. A. Aznar, S. Lykkemark, J. Martínez-Torrecuadrada, I. Melero, F. J. Blanco, J. B. de la Serna, J. M. Zapata, L. Sanz and L. Alvarez-Vallina, Nat. Commun., 2018, 9, 1–13 CrossRef CAS PubMed.
- X. Qi, F. Li, Y. Wu, C. Cheng, P. Han, J. Wang and X. Yang, Nat. Commun., 2019, 10, 1–11 CrossRef CAS PubMed.
- X. Han and M. D. Vesely, Int. Rev. Cell. Mol. Biol., 2019, 342, 1–25 CAS.
- C. Claus, C. Ferrara, W. Xu, J. Sam, S. Lang, F. Uhlenbrock, R. Albrecht, S. Herter, R. Schlenker, T. Hüsser, S. Diggelmann, J. Challier, E. Mössner, R. J. Hosse, T. Hofer, P. Brünker, C. Joseph, J. Benz, P. Ringler, H. Stahlberg, M. Lauer, M. Perro, S. Chen, C. Küttel, P. L. B. Mohan, V. Nicolini, M. C. Birk, A. Ongaro, C. Prince, R. Gianotti, G. Dugan, C. T. Whitlow, K. K. S. Sai, D. L. Caudell, A. G. Burgos-Rodriguez, J. M. Cline, M. Hettich, M. Ceppi, A. M. Giusti, F. Crameri, W. Driessen, P. N. Morcos, A. Freimoser-Grundschober, V. Levitsky, M. Amann, S. Grau-Richards, T. von Hirschheydt, S. Tournaviti, M. Mølhøj, T. Fauti, V. Heinzelmann-Schwarz, V. Teichgräber, S. Colombetti, M. Bacac, A. Zippelius, C. Klein and P. Umaña, Sci. Transl. Med., 2019, 11, eaav5989 CrossRef CAS PubMed.
- J. Kjærgaard, J. Tanaka, J. A. Kim, K. Rothchild, A. Weinberg and S. Shu, Cancer Res., 2000, 60, 5514–5521 Search PubMed.
- Intervention Dynamic Trial Listing Page, https://www.cancer.gov/about-cancer/treatment/clinical-trials/intervention/anti-ox40-monoclonal-antibody (accessed May 1, 2020).
- An Investigational Immuno-therapy Study of Experimental Medication BMS-986178 by Itself or in Combination With Nivolumab and/or Ipilimumab in Patients With Solid Cancers That Are Advanced or Have Spread – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02737475 (accessed May 1, 2020).
- A Study of the Safety, Tolerability and Pharmacokinetics of ABBV-368 as a Single Agent and Combination in Subjects With Locally Advanced or Metastatic Solid Tumors – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03071757 (accessed May 1, 2020).
- Study of OX40 Agonist PF-04518600 Alone and in Combination with 4-1BB Agonist PF-05082566 – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02315066 (accessed May 1, 2020).
- OX40, Venetoclax, Avelumab, Glasdegib, Gemtuzumab Ozogamicin, and Azacitidine in Treating Patients With Relapsed or Refractory Acute Myeloid Leukemia – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03390296 (accessed May 1, 2020).
- M. Piechutta and A. S. Berghoff, ESMO Open, 2019, 4, e000510 CrossRef PubMed.
- B. Wylie, C. Macri, J. D. Mintern and J. Waithman, Cancers, 2019, 11(4), 521 CrossRef CAS PubMed.
- A Phase I Study of the Chimeric Anti-CD40 Monoclonal Antibody ChiLob 7/4 to Treat Advanced Malignancies Refractory to Conventional Anti-cancer Treatment – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT01561911 (accessed May 5, 2020).
- A Study Investigating the Safety and Tolerability of an Immune Treatment in Cancer Patients With Lesions to the Skin – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT04059588 (accessed May 5, 2020).
- Safety, Efficacy, Dose-finding Study of a Monoclonal Antibody in Patients With Multiple Myeloma – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT00231166 (accessed May 5, 2020).
- Dose-finding Trial of HCD122 in Patients With Chronic Lymphocytic Leukemia (CLL) That is Relapsed or Non-responsive to Prior Fludaribine Therapy – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT00108108?term=CD40+AND+Anti-CD40+Monoclonal+Antibody&draw=3&rank=9 (accessed May 5, 2020).
- Phase 1, Open-Label, Dose-Escalation Study of CP-870,893 in Patients With Solid Tumors – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02225002 (accessed May 5, 2020).
- A Study of CDX-1140 as Monotherapy or in Combination in Patients With Advanced Malignancies – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03329950 (accessed May 5, 2020).
- Safety Study of SEA-CD40 in Cancer Patients – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02376699 (accessed May 5, 2020).
- Phase I Study of APX005M in Pediatric CNS Tumors – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03389802 (accessed May 5, 2020).
- ADC-1013 First-in-Human Study – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT02379741 (accessed May 5, 2020).
- A Randomized Phase IIb Placebo-Controlled Study of R-ICE Chemotherapy With and Without SGN-40 for Patients With DLBCL – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT00529503 (accessed May 5, 2020).
- A Study of SGN-40 in Combination With Rituximab in Patients With CD20-Positive, Follicular and Marginal Zone B-Cell Non-Hodgkin's Lymphoma – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT00556699 (accessed May 5, 2020).
- A Study of the Safety and Pharmacology of SGN-40 Administered in Combination With Bortezomib in Patients With Relapsed or Refractory Multiple Myeloma – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT00664898 (accessed May 5, 2020).
- R. H. Vonderheide, K. T. Flaherty, M. Khalil, M. S. Stumacher, D. L. Bajor, N. A. Hutnick, P. Sullivan, J. J. Mahany, M. Gallagher, A. Kramer, S. J. Green, P. J. O’Dwyer, K. L. Running, R. D. Huhn and S. J. Antonia, J. Clin. Oncol., 2007, 25, 876–883 CrossRef CAS PubMed.
- A. Zippelius, J. Schreiner, P. Herzig and P. Muller, Cancer Immunol. Res., 2015, 3, 236–244 CrossRef CAS PubMed.
- S. F. Ngiow, A. Young, S. J. Blake, G. R. Hill, H. Yagita, M. W. L. Teng, A. J. Korman and M. J. Smyth, Cancer Res., 2016, 76, 6266–6277 CrossRef CAS PubMed.
- I. N. Buhtoiarov, H. Lum, G. Berke, D. M. Paulnock, P. M. Sondel and A. L. Rakhmilevich, J. Immunol., 2005, 174, 6013–6022 CrossRef CAS PubMed.
- A. L. Rakhmilevich, I. N. Buhtoiarov, M. Malkovsky and P. M. Sondel, Cancer Immunol. Immunother., 2008, 57, 1151–1160 CrossRef PubMed.
- K. B. Long, W. L. Gladney, G. M. Tooker, K. Graham, J. A. Fraietta and G. L. Beatty, Cancer Discovery, 2016, 6, 400–413 CrossRef CAS PubMed.
- K. A. Autio, V. Boni, R. W. Humphrey and A. Naing, Clin. Cancer Res., 2020, 26, 984–989 CrossRef CAS PubMed.
- M. Gutierrez, G. V. Long, C. F. Friedman, D. A. Richards, B. Corr, B. R. Bastos, M. I. Uemura, P. R. Conkling, V. Moreno, W. J. Edenfield, C. R. Becerra, S. A. Piha-Paul, A. C. Peterson, M. Brown, L. P. James, M. Zheng, J. Jiang, G. Kollia, A. Swijter and P. A. Ascierto, J. Clin. Oncol., 2020, 38, 3058 Search PubMed.
- S. Rafiq, C. S. Hackett and R. J. Brentjens, Nat. Rev. Clin. Oncol., 2020, 17, 147–167 CrossRef PubMed.
- S. Depil, P. Duchateau, S. A. Grupp, G. Mufti and L. Poirot, Nat. Rev. Drug Discovery, 2020, 19, 185–199 CrossRef CAS PubMed.
- A. Reyes, C. Révil, M. Niggli, S. Chebon, S. Schlagmüller, J.-P. Flacke, M. Zortel, I. Paz-Priel, E. Asikanius, R. Hampton, A. Mahajan, E. Schmidt and S. C. Edwards, Curr. Med. Res. Opin., 2019, 35, 2079–2087 CrossRef CAS PubMed.
- Y. J. Yu, J. K. Atwal, Y. Zhang, R. K. Tong, K. R. Wildsmith, C. Tan, N. Bien-Ly, M. Hersom, J. A. Maloney, W. J. Meilandt, D. Bumbaca, K. Gadkar, K. Hoyte, W. Luk, Y. Lu, J. A. Ernst, K. Scearce-Levie, J. A. Couch, M. S. Dennis and R. J. Watts, Sci. Transl. Med., 2014, 6, 154 Search PubMed.
- U. Brinkmann and R. E. Kontermann, mAbs, 2017, 9, 182–212 CrossRef CAS PubMed.
- C. Spiess, Q. Zhai and P. J. Carter, Mol. Immunol., 2015, 67, 95–106 CrossRef CAS PubMed.
- C. Spiess, Q. Zhai and P. J. Carter, Mol. Immunol., 2015, 67, 95–106 CrossRef CAS PubMed.
- C. Milstein and A. C. Cuello, Nature, 1983, 305, 537–540 CrossRef CAS PubMed.
- N. Fischer and O. Léger, Pathobiology, 2007, 74, 3–14 CrossRef CAS PubMed.
- J. B. B. Ridgway, L. G. Presta and P. Carter, Protein Eng., Des. Sel., 1996, 9, 617–621 CrossRef CAS PubMed.
- C. Klein, W. Schaefer and J. T. Regula, MAbs, 2016, 8, 1010–1020 CrossRef CAS PubMed.
- S. Dengl, K. Mayer, F. Bormann, H. Duerr, E. Hoffmann, B. Nussbaum, M. Tischler, M. Wagner, A. Kuglstatter, L. Leibrock, C. Buldun, G. Georges and U. Brinkmann, Nat. Commun., 2020, 11, 1–11 Search PubMed.
- A. Bates and C. A. Power, Antibodies, 2019, 8, 28 CrossRef CAS PubMed.
- D. Ellerman, Methods, 2019, 154, 102–117 CrossRef CAS PubMed.
- C. Kellner, A. Otte, E. Cappuzzello, K. Klausz and M. Peipp, Transfus. Med. Hemother., 2017, 44, 327–336 CrossRef PubMed.
- A. R. Root, W. Cao, B. Li, P. LaPan, C. Meade, J. Sanford, M. Jin, C. O’Sullivan, E. Cummins, M. Lambert, A. D. Sheehan, W. Ma, S. Gatto, K. Kerns, K. Lam, A. M. D’Antona, L. Zhu, W. A. Brady, S. Benard, A. King, T. He, L. Racie, M. Arai, D. Barrett, W. Stochaj, E. R. LaVallie, J. R. Apgar, K. Svenson, L. Mosyak, Y. Yang, G. R. Chichili, L. Liu, H. Li, S. Burke, S. Johnson, R. Alderson, W. J. J. Finlay, L. Lin, S. Olland, W. Somers, E. Bonvini, H.-P. Gerber, C. May, P. A. Moore, L. Tchistiakova and L. Bloom, Antibodies, 2016, 5, 6 CrossRef PubMed.
- P. J. Engelberts, I. H. Hiemstra, B. de Jong, D. H. Schuurhuis, J. Meesters, I. Beltran Hernandez, S. C. Oostindie, J. Neijssen, E. N. van den Brink, G. J. Horbach, S. Verploegen, A. F. Labrijn, T. Salcedo, J. Schuurman, P. W. H. I. Parren and E. C. W. Breij, EBioMedicine, 2020, 52, 102625 CrossRef PubMed.
- C. Caux, Bull. Cancer, 2020, 107, 6–9 CrossRef PubMed.
- F. Arce Vargas, A. J. S. Furness, K. Litchfield, K. Joshi, R. Rosenthal, E. Ghorani, I. Solomon, M. H. Lesko, N. Ruef, C. Roddie, J. Y. Henry, L. Spain, A. Ben Aissa, A. Georgiou, Y. N. S. Wong, M. Smith, D. Strauss, A. Hayes, D. Nicol, T. O’Brien, L. Mårtensson, A. Ljungars, I. Teige, B. Frendéus, M. Pule, T. Marafioti, M. Gore, J. Larkin, S. Turajlic, C. Swanton, K. S. Peggs and S. A. Quezada, Cancer Cell, 2018, 33, 649–663 CrossRef CAS PubMed.
- Y. Bulliard, R. Jolicoeur, J. Zhang, G. Dranoff, N. S. Wilson and J. L. Brogdon, Immunol. Cell Biol., 2014, 92, 475–480 CrossRef CAS PubMed.
- F. Li and J. V. Ravetch, Science, 2011, 333, 1030–1034 CrossRef CAS PubMed.
- Final Report Summary – IACT (Immunostimulatory Agonist Antibodies for Cancer Therapy) | Report Summary | IACT | FP7 | CORDIS | European Commission, https://cordis.europa.eu/project/id/602262/reporting (accessed May 18, 2020).
- F. Li and J. V. Ravetch, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 19501–19506 CrossRef CAS PubMed.
- Y. Mazor, A. Hansen, C. Yang, P. S. Chowdhury, J. Wang, G. Stephens, H. Wu and W. F. Dall’Acqua, MAbs, 2015, 7, 461–469 CrossRef CAS PubMed.
- Y. Mazor, K. F. Sachsenmeier, C. Yang, A. Hansen, J. Filderman, K. Mulgrew, H. Wu and W. F. Dall’Acqua, Sci. Rep., 2017, 7, 40098 CrossRef CAS PubMed.
- C. Sellmann, A. Doerner, C. Knuehl, N. Rasche, V. Sood, S. Krah, L. Rhiel, A. Messemer, J. Wesolowski, M. Schuette, S. Becker, L. Toleikis, H. Kolmar and B. Hock, J. Biol. Chem., 2016, 291, 25106–25119 CrossRef CAS PubMed.
- E. C. Piccione, S. Juarez, J. Liu, S. Tseng, C. E. Ryan, C. Narayanan, L. Wang, K. Weiskopf and R. Majeti, MAbs, 2015, 7, 946–956 CrossRef CAS PubMed.
- D. Slaga, D. Ellerman, T. N. Lombana, R. Vij, J. Li, M. Hristopoulos, R. Clark, J. Johnston, A. Shelton, E. Mai, K. Gadkar, A. A. Lo, J. T. Koerber, K. Totpal, R. Prell, G. Lee, C. Spiess and T. T. Junttila, Sci. Transl. Med., 2018, 10, eaat5775 CrossRef PubMed.
- E. R. Ahn and C. L. Vogel, Breast Cancer Res. Treat., 2012, 131, 371–383 CrossRef PubMed.
- B. E. C. G. de Goeij, T. Vink, H. ten Napel, E. C. W. Breij, D. Satijn, R. Wubbolts, D. Miao and P. W. H. I. Parren, Mol. Cancer Ther., 2016, 15, 2688–2697 CrossRef CAS PubMed.
- J. Andreev, N. Thambi, A. E. P. Bay, F. Delfino, J. Martin, M. P. Kelly, J. R. Kirshner, A. Rafique, A. Kunz and T. Nittoli, et al. , Mol. Cancer Ther., 2017, 16, 681–693 CrossRef CAS PubMed.
- E. Reeves and E. James, Immunology, 2017, 150, 16–24 CrossRef CAS PubMed.
- S. Yu, J. Zhang, Y. Yan, X. Yao, L. Fang, H. Xiong, Y. Liu, Q. Chu, P. Zhou and K. Wu, J. Exp. Clin. Cancer Res., 2019, 38, 355 CrossRef PubMed.
- J. Liu, X. Wu, L. Lin, H. Pan, Y. Wang, Y. Li, Y. Zhao and Z. Wang, Mol. Ther. – Oncolytics, 2019, 14, 66–73 CrossRef CAS PubMed.
- M. Sebastian, P. Kiewe, W. Schuette, D. Brust, C. Peschel, F. Schneller, K.-H. Rühle, G. Nilius, R. Ewert, S. Lodziewski, B. Passlick, W. Sienel, R. Wiewrodt, M. Jäger, H. Lindhofer, H. Friccius-Quecke and A. Schmittel, J. Immunother, 2009, 32, 195–202 CrossRef CAS PubMed.
- S. Bellone, J. Black, D. P. English, C. L. Schwab, S. Lopez, E. Cocco, E. Bonazzoli, F. Predolini, F. Ferrari, E. Ratner, D.-A. Silasi, M. Azodi, P. E. Schwartz and A. D. Santin, Am. J. Obstet. Gynecol., 2016, 214, 99 CrossRef PubMed.
- P. T. H. Went, A. Lugli, S. Meier, M. Bundi, M. Mirlacher, G. Sauter and S. Dirnhofer, Hum. Pathol., 2004, 35, 122–128 CrossRef CAS PubMed.
- A. Feldmann, C. Arndt, K. Töpfer, S. Stamova, F. Krone, M. Cartellieri, S. Koristka, I. Michalk, D. Lindemann, M. Schmitz, A. Temme, M. Bornhäuser, G. Ehninger and M. Bachmann, J. Immunol., 2012, 189, 3249–3259 CrossRef CAS PubMed.
- P. Bühler, E. Molnar, E. P. Dopfer, P. Wolf, D. Gierschner, U. Wetterauer, W. W. A. Schamel and U. Elsässer-Beile, J. Immunother., 2009, 32, 565–573 CrossRef PubMed.
- K. Mølgaard, M. Compte, N. Nuñez-Prado, S. L. Harwood, L. Sanz and L. Alvarez-Vallina, Gene Ther., 2017, 24, 208–214 CrossRef PubMed.
- E. Y. Jen, Q. Xu, A. Schetter, D. Przepiorka, Y. L. Shen, D. Roscoe, R. Sridhara, A. Deisseroth, R. Philip, A. T. Farrell and R. Pazdur, Clin. Cancer Res., 2019, 25, 473–477 CrossRef CAS PubMed.
- L. Liu, C.-Y. K. Lam, V. Long, L. Widjaja, Y. Yang, H. Li, L. Jin, S. Burke, S. Gorlatov, J. Brown, R. Alderson, M. D. Lewis, J. L. Nordstrom, S. Koenig, P. A. Moore, S. Johnson and E. Bonvini, Clin. Cancer Res., 2017, 23, 1506–1518 CrossRef CAS PubMed.
- F. R. Schuster, M. Stanglmaier, W. Woessmann, B. Winkler, M. Siepermann, R. Meisel, P. G. Schlegel, J. Hess, H. Lindhofer, A. Borkhardt and R. Buhmann, Br. J. Haematol., 2015, 169, 90–102 CrossRef CAS PubMed.
- U. Reusch, K. H. Harrington, C. J. Gudgeon, I. Fucek, K. Ellwanger, M. Weichel, S. H. J. Knackmuss, E. A. Zhukovsky, J. A. Fox, L. A. Kunkel, J. Guenot and R. B. Walter, Clin. Cancer Res., 2016, 22, 5829–5838 CrossRef CAS PubMed.
- S. Hipp, Y.-T. Tai, D. Blanset, P. Deegen, J. Wahl, O. Thomas, B. Rattel, P. J. Adam, K. C. Anderson and M. Friedrich, Leukemia, 2017, 31, 1743–1751 CrossRef CAS PubMed.
- M. G. Fury, A. Lipton, K. M. Smith, C. B. Winston and D. G. Pfister, Cancer Immunol. Immunother., 2008, 57, 155–163 CrossRef CAS PubMed.
- N. D. James, P. J. Atherton, J. Jones, A. J. Howie, S. Tchekmedyian and R. T. Curnow, Br. J. Cancer, 2001, 85, 152–156 CrossRef CAS PubMed.
- J. Honeychurch, A. L. Tutt, T. Valerius, I. A. F. M. Heijnen, J. G. J. Van de Winkel and M. J. Glennie, Blood, 2000, 96, 3544–3552 CrossRef CAS PubMed.
- L. Balaian and E. D. Ball, Leuk. Res., 2001, 25, 1115–1125 CrossRef CAS PubMed.
- Antibody Therapy in Treating Patients With Refractory or Relapsed Non-Hodgkin's Lymphoma or Chronic Lymphocytic Leukemia – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT00014560 (accessed May 27, 2020).
- S. Wingert, U. Reusch, A. Beez, J. Pahl, A. Cerwenka, J. Koch and M. Treder, Blood, 2018, 132, 1111 CrossRef.
- A. Rothe, S. Sasse, M. S. Topp, D. A. Eichenauer, H. Hummel, K. S. Reiners, M. Dietlein, G. Kuhnert, J. Kessler, C. Buerkle, M. Ravic, S. Knackmuss, J.-P. Marschner, E. Pogge von Strandmann, P. Borchmann and A. Engert, Blood, 2015, 125, 4024–4031 CrossRef CAS PubMed.
- R. Asano, M. Nakayama, H. Kawaguchi, T. Kubota, T. Nakanishi, M. Umetsu, H. Hayashi, Y. Katayose, M. Unno, T. Kudo and I. Kumagai, FEBS J., 2012, 279, 223–233 CrossRef CAS PubMed.
- Z. Shah, M. N. Malik, S. S. Batool, S. Kotapati, A. Akhtar, O. ur Rehman, M. Ghani, M. Sadiq, A. Akbar, A. Ashraf, A. A. A. Farooqui, M. A. Yousaf, M. E. Ahmad, H. M. Fazeel and F. Anwer, Blood, 2019, 134, 5567 CrossRef.
- E. Wolf, R. Hofmeister, P. Kufer, B. Schlereth and P. A. Baeuerle, Drug Discovery Today, 2005, 10, 1237–1244 CrossRef CAS PubMed.
- J. Duell, P. E. Lammers, I. Djuretic, A. G. Chunyk, S. Alekar, I. Jacobs and S. Gill, Clin. Pharmacol. Ther., 2019, 106, 781–791 CrossRef PubMed.
- F. V. Suurs, M. N. Lub-de Hooge, E. G. E. de Vries and D. J. A. de Groot, Pharmacol. Ther., 2019, 201, 103–119 CrossRef CAS PubMed.
- M.-E. Goebeler and R. C. Bargou, Nat. Rev. Clin. Oncol., 2020, 17, 418–434 CrossRef PubMed.
- D. Bhutani and L. G. Lum, Curr. Opin. Hematol., 2015, 22, 476–483 CrossRef CAS PubMed.
- J. Ma, J. Ge, X. Xue, W. Xiu, P. Ma, X. Sun and M. Zhang, Oncol. Rep., 2018, 39, 1245–1252 CAS.
- M. K. Gleason, M. R. Verneris, D. A. Todhunter, B. Zhang, V. McCullar, S. X. Zhou, A. Panoskaltsis-Mortari, L. M. Weiner, D. A. Vallera and J. S. Miller, Mol. Cancer Ther., 2012, 11, 2674–2684 CrossRef CAS PubMed.
- K. L. Lowe, D. Cole, R. Kenefeck, I. OKelly, M. Lepore and B. K. Jakobsen, Cancer Treat. Rev., 2019, 77, 35–43 CrossRef CAS PubMed.
- T. T. Junttila, J. Li, J. Johnston, M. Hristopoulos, R. Clark, D. Ellerman, B.-E. Wang, Y. Li, M. Mathieu, G. Li, J. Young, E. Luis, G. L. Phillips, E. Stefanich, C. Spiess, A. Polson, B. Irving, J. M. Scheer, M. R. Junttila, M. S. Dennis, R. Kelley, K. Totpal and A. Ebens, Cancer Res., 2014, 74, 5561–5571 CrossRef CAS PubMed.
- T. Osada, S. P. Patel, S. A. Hammond, K. Osada, M. A. Morse and H. K. Lyerly, Cancer Immunol. Immunother., 2015, 64, 677–688 CrossRef CAS PubMed.
- C. Krupka, P. Kufer, R. Kischel, G. Zugmaier, F. S. Lichtenegger, T. Köhnke, B. Vick, I. Jeremias, K. H. Metzeler, T. Altmann, S. Schneider, M. Fiegl, K. Spiekermann, P. A. Bauerle, W. Hiddemann, G. Riethmüller and M. Subklewe, Leukemia, 2016, 30, 484–491 CrossRef CAS PubMed.
- C.-H. Chang, Y. Wang, R. Li, D. L. Rossi, D. Liu, E. A. Rossi, T. M. Cardillo and D. M. Goldenberg, Cancer Res., 2017, 77, 5384–5394 CrossRef CAS PubMed.
- S. A. Patel and A. J. Minn, Immunity, 2018, 48, 417–433 CrossRef CAS PubMed.
- S. J. Dovedi, Y. Mazor, M. Elder, S. Hasani, B. Wang, S. Mosely, D. Jones, A. Hansen, C. Yang, Y. Wu, I. Achour, N. Durham, G. Browne, T. Murray, J. Hair, M. Morrow, G. Rainey, M. J. Kunkel, J. Gooya, D. Freeman, R. Herbst and R. Wilkinson, Cancer Res., 2018, 78, 2776 Search PubMed.
- H.-L. Roche, An Open Label, Multicenter, Dose Escalation and Expansion, Phase 1 Study to Evaluate Safety, Pharmacokinetics, and Preliminary Anti-Tumor Activity of RO7121661, a PD-1/TIM-3 Bispecific Antibody, in Patients With Advanced and/or Metastatic Solid Tumors – clinicaltrials.gov, https://clinicaltrials.gov/ct2/show/NCT03708328 (accessed May 2020).
- M. Wierz, S. Pierson, L. Guyonnet, E. Viry, A. Lequeux, A. Oudin, S. P. Niclou, M. Ollert, G. Berchem, B. Janji, C. Guérin, J. Paggetti and E. Moussay, Blood, 2018, 131, 1617–1621 CrossRef CAS PubMed.
- M. Gaspar, J. Pravin, L. Rodrigues, S. Uhlenbroich, K. L. Everett, F. Wollerton, M. Morrow, M. Tuna and N. Brewis, Cancer Immunol. Res., 2020, 8, 781–793 CrossRef CAS PubMed.
- G. Fromm, S. de Silva, K. Johannes, A. Patel, J. C. Hornblower and T. H. Schreiber, J. Immunother. Cancer, 2018, 6, 149 CrossRef PubMed.
- A. M. Kvarnhammar, N. Veitonmäki, K. Hägerbrand, A. Dahlman, K. E. Smith, S. Fritzell, L. von Schantz, M. Thagesson, D. Werchau, K. Smedenfors, M. Johansson, A. Rosén, I. Åberg, M. Winnerstam, E. Nyblom, K. Barchan, C. Furebring, P. Norlén and P. Ellmark, J. Immunother. Cancer, 2019, 7, 103 CrossRef PubMed.
- M. D. Oberst, S. Kasturirangan, C. Sachs, C. Auge, J. Moynihan, R. Rothstein, J. Hair, F. Neal, S. Mamidi, S. Hanabuchi, A. Watkins, Y. Zheng, K. Rosenthal, D. J. Freeman and S. Hammond, Cancer Res., 2018, 78, 2775 CrossRef PubMed.
- D. Mittal, D. Vijayan, J. Neijssen, J. Kreijtz, M. M. J. M. Habraken, H. Van Eenennaam, A. Van Elsas and M. J. Smyth, OncoImmunology, 2019, 8, e1648171 CrossRef PubMed.
- M. J. Hinner, R. S. B. Aiba, T. J. Jaquin, S. Berger, M. C. Dürr, C. Schlosser, A. Allersdorfer, A. Wiedenmann, G. Matschiner, J. Schüler, U. Moebius, C. Rothe, L. Matis and S. A. Olwill, Clin. Cancer Res., 2019, 25, 5878–5889 CrossRef CAS PubMed.
- W. C. Dougall, A. Roman Aguilera and M. J. Smyth, Clin. Transl. Immunol, 2019, 8, e1081 Search PubMed.
- S. Kobold, S. Pantelyushin, F. Rataj and J. vom Berg, Front. Oncol., 2018, 8, 285 CrossRef PubMed.
- Regeneron Pharmaceuticals. A Phase 1 Study to Assess Safety and Tolerability of REGN1979, an Anti-CD20 x Anti-CD3 Bispecific Monoclonal Antibody, and REGN2810, an Anti-Programmed Death-1 Monoclonal Antibody, in Patients With B-Cell Malignancies – clinicaltrials.gov, https://clinicaltrials.gov/ct2/show/NCT02651662 (accessed June 1, 2020).
- National Cancer Institute (NCI). A Phase 1 Study of Blinatumomab in Combination With Checkpoint Inhibitor(s) of PD-1 (Nivolumab) or Both PD-1 (Nivolumab) and CTLA-4 (Ipilimumab) in Patients With Poor-Risk, Relapsed or Refractory CD19+ Precursor B-Lymphoblastic Leukemia -clinicaltrials.gov, https://clinicaltrials.gov/ct2/show/NCT02879695 (accessed June 1, 2020).
- Pembrolizumab and Blinatumomab in Treating Participants With Recurrent or Refractory Acute Lymphoblastic Leukemia – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03512405 (accessed June 1, 2020).
- Open Label Study Investigating the Safety and Efficacy of Blinatumomab in Combination With Pembrolizumab (KEYNOTE-348) – Full Text View – ClinicalTrials.gov, https://clinicaltrials.gov/ct2/show/NCT03340766 (accessed June 1, 2020).
- J. Feucht, S. Kayser, D. Gorodezki, M. Hamieh, M. Döring, F. Blaeschke, P. Schlegel, H. Bösmüller, L. Quintanilla-Fend, M. Ebinger, P. Lang, R. Handgretinger and T. Feuchtinger, Oncotarget, 2016, 7, 76902–76919 CrossRef PubMed.
- G. S. Laszlo, C. J. Gudgeon, K. H. Harrington and R. B. Walter, Blood Cancer J, 2015, 5, e340 CrossRef CAS PubMed.
- E. Poon, M. Gaspar, L. N. Rodrigues, J. Pravin, A. S. Lind, J. Winnewisser, A. Koers, E. McConnell, M. Tuna, M. Morrow and N. Brewis, Presented in part at the Proceedings of the American Association for Cancer Research Annual Meeting 2019, Atlanta, Mar 29–Apr 3, 2019.
- R. Liu, W. Jiang, M. Yang, H. Guo, Y. Zhang, J. Wang, H. Zhu, R. Shi, D. Fan, C. Yang, Z. Zhu, Y. Xie and D. Xiong, J. Immunother., 2010, 33, 500–509 CrossRef CAS PubMed.
- A. Willems, S. Schoonooghe, D. Eeckhout, G. D. Jaeger, J. Grooten and N. Mertens, Cancer Immunol. Immunother., 2005, 54, 1059–1071 CrossRef CAS PubMed.
- S. Kato, R. Okamura, Y. Kumaki, S. Ikeda, M. Nikanjam, R. Eskander, A. Goodman, S. Lee, S. T. Glenn, D. Dressman, A. Papanicolau-Sengos, F. L. Lenzo, C. Morrison and R. Kurzrock, OncoImmunology, 2020, 9, 1708065 CrossRef PubMed.
- M. Herrmann, C. Krupka, K. Deiser, B. Brauchle, A. Marcinek, A. O. Wagner, F. Rataj, R. Mocikat, K. H. Metzeler, K. Spiekermann, S. Kobold, N. C. Fenn, K.-P. Hopfner and M. Subklewe, Blood, 2018, 132, 2484–2494 CrossRef CAS PubMed.
- M. P. Rettig and J. F. DiPersio, Blood, 2018, 132, 2425–2427 CrossRef CAS PubMed.
- H. Don Yun, M. Felices, D. A. Vallera, P. Hinderlie, S. Cooley, M. Arock, J. Gotlib, C. Ustun and J. S. Miller, Blood Adv., 2018, 2, 1580–1584 CrossRef PubMed.
- S. S. Tay, H. Carol and M. Biro, Hum. Vaccines Immunother., 2016, 12, 2790–2796 CrossRef PubMed.
- M. Felices, B. Kodal, P. Hinderlie, M. F. Kaminski, S. Cooley, D. J. Weisdorf, D. A. Vallera, J. S. Miller and V. Bachanova, Blood Adv., 2019, 3, 897–907 CrossRef CAS PubMed.
- J. Miller, M. Felices, D. Vallera and T. Lenvik, NK Engager Molecules and Methods of Use Thereof, WO Pat., Patent Application, WO2020081841 (A1), 2020 Search PubMed.
- L. Gauthier, A. Morel, N. Anceriz, B. Rossi, A. Blanchard-Alvarez, G. Grondin, S. Trichard, C. Cesari, M. Sapet, F. Bosco, H. Rispaud-Blanc, F. Guillot, S. Cornen, A. Roussel, B. Amigues, G. Habif, F. Caraguel, S. Arrufat, R. Remark, F. Romagné, Y. Morel, E. Narni-Mancinelli and E. Vivier, Cell, 2019, 177, 1701–1713 CrossRef CAS PubMed.
- P. M. Lanuza, C. Pesini, M. A. Arias, C. Calvo, A. Ramirez-Labrada and J. Pardo, Front. Immunol., 2020, 10, 3010 CrossRef PubMed.
- K.-P. Hopfner, M. Subklewe, N. Moritz and N. Fenn, Trispecific Molecule Combining Specific Tumor Targeting and Local Immune Checkpoint Inhibition, EP3165536 (A1), 2017 Search PubMed.
- S. Chen, H. Zhu and Y. Liao, Trifunctional Molecule and Application Thereof, WO Pat., Patent Application, WO2018120843 (A1), 2018 Search PubMed.
- L. Borras, D. Escher, C. V. V. Leisner, F. Scheifele, T. Schleier and P. R. Richle, Trispecific Antigen Binding Proteins, WO Pat., Patent Application, WO2019166650 (A1), 2019 Search PubMed.
- X. Wu, R. Yuan, M. Bacica and S. J. Demarest, Protein Eng., Des. Sel., 2018, 31, 249–256 CrossRef CAS PubMed.
- N. Dimasi, R. Fleming, C. Hay, R. Woods, L. Xu, H. Wu and C. Gao, Mol. Pharmaceutics, 2015, 12, 3490–3501 CrossRef CAS PubMed.
- M. Kügler, C. Stein, C. Kellner, K. Mentz, D. Saul, M. Schwenkert, I. Schubert, H. Singer, F. Oduncu, B. Stockmeyer, A. Mackensen and G. H. Fey, Br. J. Haematol., 2010, 150, 574–586 CrossRef PubMed.
- J. U. Schmohl, M. Felices, D. Todhunter, E. Taras, J. S. Miller and D. A. Vallera, Oncotarget, 2016, 7, 73830–73844 CrossRef PubMed.
- Y. Zhu, O. Olsen, D. Xia, D. Jellyman, K. Bykova, A.-M. Rousseau, B. Brady, B. Renshaw, B. Kovacevich, Y. Liang, C. Wang, Z. Gao and H. Huang, Guidance and Navigation Control Proteins and Method of Making and Using Thereof, WO Pat., Patent Application, WO2019191120, 2019 Search PubMed.
- C. S. McKay and M. G. Finn, Chem. Biol., 2014, 21, 1075–1101 CrossRef CAS PubMed.
- N. K. Devaraj, ACS Cent. Sci., 2018, 4, 952–959 CrossRef CAS PubMed.
- S. J. Walsh, J. D. Bargh, F. M. Dannheim, A. R. Hanby, H. Seki, A. J. Counsell, X. Ou, E. Fowler, N. Ashman, Y. Takada, A. Isidro-Llobet, J. S. Parker, J. S. Carroll and D. R. Spring, Chem. Soc. Rev., 2021, 50, 1305–1353 RSC.
- E. A. Hoyt, P. M. S. D. Cal, B. L. Oliveira and G. J. L. Bernardes, Nat. Rev. Chem., 2019, 3, 147–171 CrossRef CAS.
- M. Baalmann, L. Neises, S. Bitsch, H. Schneider, L. Deweid, P. Werther, N. Ilkenhans, M. Wolfring, M. J. Ziegler, J. Wilhelm, H. Kolmar and R. Wombacher, Angew. Chem., Int. Ed., 2020, 59, 12885–12893 CrossRef CAS PubMed.
- P. Szijj and V. Chudasama, Nat. Rev. Chem., 2021, 5, 78–92 CrossRef CAS.
- M. Sen, D. M. Wankowski, N. K. Garlie, R. E. Siebenlist, D. Van Epps, A. V. LeFever and L. G. Lum, J. Hematother. Stem Cell Res., 2001, 10, 247–260 CrossRef CAS PubMed.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- J. Dommerholt, F. P. J. T. Rutjes and F. L. van Delft, Top. Curr. Chem., 2016, 374, 16 CrossRef PubMed.
- J. Dommerholt, O. van Rooijen, A. Borrmann, C. F. Guerra, F. M. Bickelhaupt and F. L. van Delft, Nat. Commun., 2014, 5, 5378 CrossRef CAS PubMed.
- X. Ning, R. P. Temming, J. Dommerholt, J. Guo, D. B. Ania, M. F. Debets, M. A. Wolfert, G.-J. Boons and F. L. van Delft, Angew. Chem., Int. Ed., 2010, 49, 3065 CrossRef CAS PubMed.
- A.-C. Knall and C. Slugovc, Chem. Soc. Rev., 2013, 42, 5131–5142 RSC.
- B. L. Oliveira, Z. Guo and G. J. L. Bernardes, Chem. Soc. Rev., 2017, 46, 4895–4950 RSC.
- H. Liu, D. Audisio, L. Plougastel, E. Decuypere, D.-A. Buisson, O. Koniev, S. Kolodych, A. Wagner, M. Elhabiri, A. Krzyczmonik, S. Forsback, O. Solin, V. Gouverneur and F. Taran, Angew. Chem., Int. Ed., 2016, 55, 12073–12077 CrossRef CAS PubMed.
- A. R. Nödling, L. A. Spear, T. L. Williams, L. Y. P. Luk and Y.-H. Tsai, Essays Biochem., 2019, 63, 237–266 CrossRef PubMed.
- J. Y. Axup, K. M. Bajjuri, M. Ritland, B. M. Hutchins, C. H. Kim, S. A. Kazane, R. Halder, J. S. Forsyth, A. F. Santidrian, K. Stafin, Y. Lu, H. Tran, A. J. Seller, S. L. Biroc, A. Szydlik, J. K. Pinkstaff, F. Tian, S. C. Sinha, B. Felding-Habermann, V. V. Smider and P. G. Schultz, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 16101–16106 CrossRef CAS PubMed.
- C. H. Kim, J. Y. Axup and P. G. Schultz, Curr. Opin. Chem. Biol., 2013, 17, 412–419 CrossRef CAS PubMed.
- A. Maruani, P. A. Szijj, C. Bahou, J. C. F. Nogueira, S. Caddick, J. R. Baker and V. Chudasama, Bioconjugate Chem., 2020, 31, 520–529 CrossRef CAS PubMed.
- J. T. Patterson, J. Isaacson, L. Kerwin, G. Atassi, R. Duggal, D. Bresson, T. Zhu, H. Zhou, Y. Fu and G. F. Kaufmann, Bioorg. Med. Chem. Lett., 2017, 27, 5490–5495 CrossRef CAS PubMed.
- A. Sadiki, S. R. Vaidya, M. Abdollahi, G. Bhardwaj, M. E. Dolan, H. Turna, V. Arora, A. Sanjeev, T. D. Robinson, A. Koid, A. Amin and Z. S. Zhou, Antibody Ther., 2020, 3, 271–284 CrossRef CAS PubMed.
- K. Yamada and Y. Ito, ChemBioChem, 2019, 20, 2729–2737 CrossRef CAS PubMed.
- O. Koniev and A. Wagner, Chem. Soc. Rev., 2015, 44, 5495–5551 RSC.
- G. Chaubet, F. Thoreau and A. Wagner, Drug Discovery Today Technol., 2018, 30, 21–26 CrossRef PubMed.
- P. Dennler, E. Fischer and R. Schibli, Antibodies, 2015, 4, 197–224 CrossRef.
- G. Falck and K. M. Müller, Antibodies, 2018, 7, 4 CrossRef PubMed.
- C. Sornay, S. Hessmann, S. Erb, I. Dovgan, A. Ehkirch, T. Botzanowski, S. Cianférani, A. Wagner and G. Chaubet, Chem. – Eur. J., 2020, 26, 13797–13805 CrossRef CAS PubMed.
- D. Lac, C. Feng, G. Bhardwaj, H. Le, J. Tran, L. Xing, G. Fung, R. Liu, H. Cheng and K. S. Lam, Bioconjugate Chem., 2016, 27, 159–169 CrossRef CAS PubMed.
- A. J. Counsell, S. J. Walsh, N. S. Robertson, H. F. Sore and D. R. Spring, Org. Biomol. Chem., 2020, 18, 4739–4743 RSC.
- B. Bernardim, P. M. S. D. Cal, M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, I. S. Albuquerque, E. Perkins, F. Corzana, A. C. B. Burtoloso, G. Jiménez-Osés and G. J. L. Bernardes, Nat. Commun., 2016, 7, 13128 CrossRef CAS PubMed.
- N. Forte, I. Benni, K. Karu, V. Chudasama and J. R. Baker, Chem. Sci., 2019, 10, 10919–10924 RSC.
- M. Kasper, M. Glanz, A. Stengl, M. Penkert, S. Klenk, T. Sauer, D. Schumacher, J. Helma, E. Krause, M. C. Cardoso, H. Leonhardt and C. P. R. Hackenberger, Angew. Chem., 2019, 131, 11751–11756 CrossRef.
- F. F. Schumacher, J. P. M. Nunes, A. Maruani, V. Chudasama, M. E. B. Smith, K. A. Chester, J. R. Baker and S. Caddick, Org. Biomol. Chem., 2014, 12, 7261–7269 RSC.
- E. Robinson, J. P. M. Nunes, V. Vassileva, A. Maruani, J. C. F. Nogueira, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, RSC Adv., 2017, 7, 9073–9077 RSC.
- I. Dovgan, S. Ursuegui, S. Erb, C. Michel, S. Kolodych, S. Cianférani and A. Wagner, Bioconjugate Chem., 2017, 28, 1452–1457 CrossRef CAS PubMed.
- L. Xu, M. Raabe, M. M. Zegota, J. C. F. Nogueira, V. Chudasama, S. L. Kuan and T. Weil, Org. Biomol. Chem., 2020, 18, 1140–1147 RSC.
- S. J. Walsh, S. Omarjee, W. R. J. D. Galloway, T. T.-L. Kwan, H. F. Sore, J. S. Parker, M. Hyvönen, J. S. Carroll and D. R. Spring, Chem. Sci., 2019, 10, 694–700 RSC.
- A. Maruani, M. E. B. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 6645 CrossRef CAS PubMed.
- E. A. Hull, M. Livanos, E. Miranda, M. E. B. Smith, K. A. Chester and J. R. Baker, Bioconjugate Chem., 2014, 25, 1395–1401 CrossRef CAS PubMed.
- J. T. Patterson, E. Gros, H. Zhou, G. Atassi, L. Kerwin, L. Carmody, T. Zhu, B. Jones, Y. Fu and G. F. Kaufmann, Bioorg. Med. Chem. Lett., 2017, 27, 3647–3652 CrossRef CAS PubMed.
- G. Viault, S. Dautrey, N. Maindron, J. Hardouin, P.-Y. Renard and A. Romieu, Org. Biomol. Chem., 2013, 11, 2693–2705 RSC.
- M. Forner, S. Defaus and D. Andreu, J. Org. Chem., 2020, 85, 1626–1634 CrossRef CAS PubMed.
- F. Thoreau, L. Vanwonterghem, M. Henry, J.-L. Coll and D. Boturyn, Org. Biomol. Chem., 2018, 16, 4101–4107 RSC.
- B. Thomas, M. Fiore, G. C. Daskhan, N. Spinelli and O. Renaudet, Chem. Commun., 2015, 51, 5436–5439 RSC.
- S. Patke, J. Li, P. Wang, D. Slaga, J. Johnston, S. Bhakta, S. Panowski, L. L. Sun, T. Junttila, J. M. Scheer and D. A. Ellerman, mAbs, 2017, 9, 430–437 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |