
Targeted delivery of liver X receptor agonist to inhibit neointimal hyperplasia by differentially regulating cell behaviors†
Jian
Li‡
a,
Fan
Jia‡
 b,
Zhebin
Chen
a,
Jun
Lin
c,
Qingbo
Lv
a,
Yue
Huang
b,
Qiao
Jin
b,
Youxiang
Wang
b,
Guosheng
Fu
*a and
Jian
Ji
*ab
b,
Zhebin
Chen
a,
Jun
Lin
c,
Qingbo
Lv
a,
Yue
Huang
b,
Qiao
Jin
b,
Youxiang
Wang
b,
Guosheng
Fu
*a and
Jian
Ji
*ab
aKey Laboratory of Cardiovascular Intervention and Regenerative Medicine of Zhejiang Province, Department of Cardiology, Sir Run Run Shaw Hospital, Zhejiang University, Hangzhou, 310016, China. E-mail: fugs@zju.edu.cn; jijian@zju.edu.cn
bMOE Key Laboratory of Macromolecule Synthesis and Functionalization of Ministry of Education, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou, 310027, China
cDepartment of Cardiovascular Surgery, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou, 510120, China
First published on 19th August 2022
Abstract
Restenosis induced by neointimal hyperplasia is one of the key reasons limiting the long-term success of cardiovascular interventional therapy. However, it remains a serious challenge to completely overcome restenosis because of the dilemma of simultaneously activating human umbilical vein endothelial cells (HUVECs) and inhibiting human aortic smooth muscle cells (HASMCs). Herein, we developed a targeted nanomedicine encapsulating the liver X receptor (LXR) agonist, T0901317, for differentially regulating the behaviors of HUVECs and HASMCs. The stimulatory effect on HUVEC proliferation/migration and the inhibitory effect on HASMC proliferation/migration were confirmed in vitro, respectively. In the co-culture system, the competitiveness of HUVECs over HASMCs was notably improved after being treated with T0901317-loaded liposomes. Compared to free T0901317 and non-targeted liposomes, the type IV collagen (Col-IV) targeted liposomes could accumulate in the vascular injured area more effectively and inhibit neointimal hyperplasia in a balloon-induced rat carotid artery injury model. Therefore, targeted delivery of LXR agonist might be a very promising therapeutic strategy for anti-restenosis therapy.
1. Introduction
Cardiovascular diseases (CVDs) remain the biggest threat to human life and health worldwide.1 Currently, percutaneous coronary intervention (PCI) is considered the most common and effective strategy for the treatment of acute occlusive vascular diseases.2 However, the long-term therapeutic performance of PCI is limited by various complications, especially restenosis.3,4 To overcome these setbacks, anti-proliferative drugs like paclitaxel and Limus family drugs are introduced to inhibit the overwhelming proliferation and migration of vascular smooth muscle cells (SMCs). This strategy has achieved a great breakthrough in the reduction of restenosis incidence.5–7 However, these drugs also suppress the growth of endothelial cells (ECs), delay the re-endothelialization, and increase the risk of restenosis and late stent thrombosis (LST). As the native defender of blood vessels, the endothelium plays a vital role in maintaining vascular homeostasis. Rapid re-endothelialization is crucial for overall therapeutic performance.8 In some studies, growth factors, such as vascular endothelial growth factors (VEGF), were used to expedite the re-endothelialization process, while the aberrant proliferation of SMCs was also activated.9,10 Therefore, methods that selectively suppress SMC pathologic activity and promote the growth of healthy ECs represent great potential in solving the abovementioned dilemma.11,12Currently, cell-specific delivery of therapeutic molecules is an effective solution for this contradictory situation. Drug nanocarriers are very advantageous in the delivery of chemotherapeutic agents.13–15 Through decorating specific targeting ligands, the drug nanocarriers are endowed with cell-specificity.16–19 For instance, the EC-targeting peptide Arg-Glu-Asp-Val (REDV) has EC specificity, while Val-Ala-Pro-Gly (VAPG) can endow the nanoplatforms with SMC specificity.20,21 However, this strategy requires at least two different nanomedicines for the respective regulation of the behaviors of ECs and SMCs.22 There is an urgent need for a simple, efficient method to inhibit neointimal hyperplasia.
LXR was reported to have a broad role in the regulation of cardiovascular homeostasis.23 For example, LXR can participate in the maintenance of lipid homeostasis and the regulation of pro-inflammatory cytokine secretion, indicating its therapeutic effect on atherosclerosis.24–26 In endothelial progenitor cells, LXR activation can improve these proliferation and migration activities by activating the PI3K/Akt/eNOS pathway and increasing nitric oxide (NO) production.27 However, LXR activation in the SMCs has an inhibitory effect on these proliferation and migration activities by suppressing the G1-S switch in cell cycle progression.28 Considering its various effects on different cells, synthetic LXR agonists, such as T0901317, might be of great promise as an anti-restenosis drug. However, the application of LXR agonists in anti-restenosis has been rarely reported. Besides, in order to fully exert the drug effects, LXR agonists should be specifically delivered to injured vascular sites while being non-specific to cells. Type IV collagen (Col-IV), a main component of the subendothelial basement membrane, is an important target of inflammatory and injured vascular sites. Col-IV targeting peptides, such as KLWVLPKGGGC, were hence developed to effectively bind with Col-IV and subsequently be endocytosed by the nearby ECs and SMCs.19,29
Herein, Col-IV targeting peptide-modified liposomes were proposed to encapsulate the LXR agonist T0901317 to alleviate vascular restenosis after PCI (Fig. 1). The Col-IV targeted liposomes were expected to be selectively accumulated in the vascular injured area via active targeting. T0901317 would improve the proliferation and migration of HUVECs while inhibiting the proliferation and migration of HASMCs. The competitiveness of HUVECs over HASMCs inhibited neointimal hyperplasia in a balloon-induced rat carotid artery injury model. The targeted delivery of the LXR agonist T0901317 provides a new strategy to inhibit neointimal hyperplasia in anti-restenosis therapy.
 | ||
| Fig. 1 Schematic illustration of Col-IV targeted nanomedicine-loaded LXR agonist T0901317 towards the inhibition of neointimal hyperplasia after vascular injury. When the endothelium is injured, the subendothelial basement membrane is exposed. Subsequently, HASMCs are activated and their capability of migration and proliferation is significantly improved, which eventually leads to intimal hyperplasia. Col-IV-targeted liposome-loaded T0901317 can specifically accumulate at the injured sites by binding to the exposed Col-IV and release T0901317 within the lesion. The stimulation of HUVEC proliferation/migration and the inhibition of HASMC proliferation/migration is simultaneously achieved, and intimal hyperplasia is inhibited. | ||
2. Experimental section
2.1 Materials
Lecithin and cholesterol were purchased from A.V.T Co., Ltd. T0901317 and coumarin-6 were purchased from Yuanye Biotech (Shanghai, China). Col-IV targeting peptides (C18-CGGGKPLVWL) and scrambled peptides (C18-CGGGLKVPWKL) were purchased from Yuanpeptide Biotech Co., Ltd (Nanjing, China). The cell cycle assay kit was purchased from Lianke Biotech (Hangzhou, China). The antibody against cyclin D1 was purchased from Cell Signal Technology, Inc. CellTracker Orange and Green dyes were purchased from Thermo Fisher Scientific. The platelet-derived growth factor-BB (PDGF-BB) was purchased from STEMCELL Technologies (Shanghai, China). Water was purified by a Milli-Q Ultrapure Water System (Millipore).2.2 Preparation and characterization of liposome
T0901317-loaded liposomes were prepared by using a filming-ultrasonic dispersion method. Briefly, a mixture of lecithin, cholesterol, T0901317, and C18-CGGGKPLVWLK in a ratio of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:2:3 mg (30 mg in total) was dissolved in 10 ml of dichloromethane (DCM) and was dried into a thin film in a round-bottom flask using a vacuum rotary evaporator. Then, 8 ml of deionized water (DI) was added and the lipid was hydrated and then sonicated for 6 min using an ultrasound probe in an ice bath. Unloaded T0901317 was removed by filtration. Through similar procedures, blank liposomes, T0901317-loaded non-targeting liposomes (T090-nt-lip), T0901317-loaded targeting liposomes (T090-t-lip) and coumarin-6 or Cy5.5-labeled liposomes (Cy5.5-T090-nt-lip and Cy5.5-T090-t-lip) were prepared.
:5:2:3 mg (30 mg in total) was dissolved in 10 ml of dichloromethane (DCM) and was dried into a thin film in a round-bottom flask using a vacuum rotary evaporator. Then, 8 ml of deionized water (DI) was added and the lipid was hydrated and then sonicated for 6 min using an ultrasound probe in an ice bath. Unloaded T0901317 was removed by filtration. Through similar procedures, blank liposomes, T0901317-loaded non-targeting liposomes (T090-nt-lip), T0901317-loaded targeting liposomes (T090-t-lip) and coumarin-6 or Cy5.5-labeled liposomes (Cy5.5-T090-nt-lip and Cy5.5-T090-t-lip) were prepared.
The hydrodynamic size, polydispersity (PDI) and zeta potential of the liposomes were measured by dynamic light scattering (DLS) measurements using a Zetasizer Nano-ZS from Malvern Instruments. The morphology and size of liposomes were characterized by transmission electron microscopy (TEM) using a HT7700 TEM (Hitachi, Japan).
2.3 Measurement of in vitro entrapment efficiency and loading efficiency
To determine the concentration of encapsulated T0901317, the samples were subjected to high-performance liquid chromatography (HPLC). 20 μl of the liposome dispersion was dissolved in 180 μl of methanol. The mobile phase was DI with 0.1% formic acid and the detection wavelength was set at 220 nm. The entrapment efficiency (EE) and loading content (LC) were calculated according to the following equations: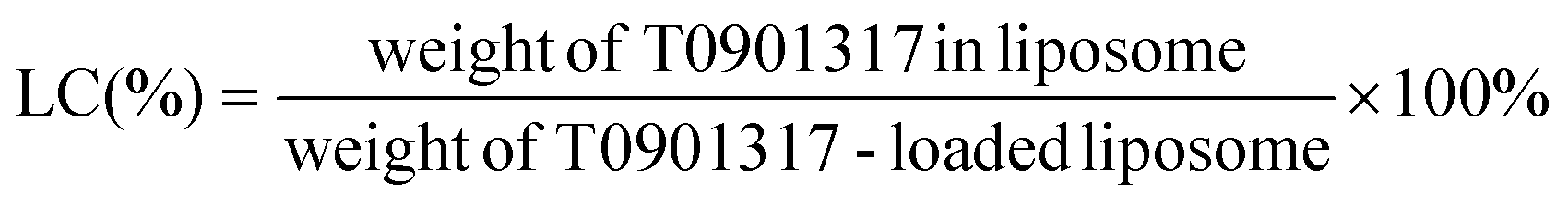

2.4 Cell culture
HUVECs and HASMCs were purchased from Lonza (C2519A, USA) for cell experiments. The cells were cultured in 25 cm2 cell culture flasks at 37 °C under a humidified atmosphere with 5% CO2. Endothelial cell medium (ECM) and smooth muscle cell medium (SMCM) were purchased from ScienCell and specifically used for the culture of HUVECs and HASMCs. HASMCs were treated with PDGF-BB at the final concentration of 20 ng ml−1.2.5 In vitro cellular uptake
HUVECs were seeded in 24-well plates at 1 × 105 cells per well in 500 μl of ECM. After 24 h, the culture medium was replaced with 500 μl of fresh medium containing coumarin-6-labeled liposomes (20 μg ml−1), followed by incubation at 37 °C for 1, 2, and 4 h, respectively. Subsequently, the medium was removed and the cells were washed with PBS 3 times. Fluorescence images were further acquired by using a fluorescence microscope.The quantitative evaluation of internalized liposomes was evaluated by flow cytometry analysis. Briefly, HUVECs were seeded in 24-well plates at 1 × 105 cells per well in 500 μl of ECM. After 24 h, the culture medium was replaced with 500 μl of fresh medium containing coumarin-6-labeled liposomes (20 μg ml−1). After incubating for 1 h, 2 h, and 4 h, the medium was removed, and the cells were washed with PBS 3 times. All cells were trypsinized and 500 μl of cell suspension was measured by flow cytometry (BD FACS-CaliburTM). Cells treated with a blank medium were used as the control. Similarly, HASMC internalization behaviors were observed.
2.6 In vitro cell proliferation assay
HUVECs were seeded in 96-well plates at 5 × 103 cells per well in ECM and incubated at 37 °C. After 24 h of incubation, the medium was replaced with fresh ECM containing 0.5% FBS and treated with PBS, T0901317, T090-nt-lip, T090-t-lip, and blank liposomes at 10 μM of T0901317. After incubation for 24 h, the CCK-8 assay was used to determine the cell viability. Following a similar procedure, the cell viability of HASMCs was determined. In the control group, the cells were treated with SMCM with 0.5% FBS alone, while 20 ng ml−1 PDGF-BB was added in the other groups.2.7 In vitro wound healing assay
HUVECs stained with red live cell dye were seeded in 24-well plates at 1 × 105 cells per well in ECM and incubated at 37 °C. After 24 h of incubation, the cells were scratched and incubated with PBS, T0901317, T090-nt-lip, T090-t-lip, and blank liposome at 10 μM of T0901317 in fresh ECM containing 0.5% FBS. For the evaluation, the cells were photographed using a fluorescence microscope at the indicated time points. HASMCs were stained with the green live cell dye. Similarly, the wound healing assay of HASMCs was carried out.2.8 In vitro transwell assay
The transwell assay of HUVECs was carried out by using a Boyden chamber using the Costar Transwell apparatus with 8.0 μm pore size (Corning, USA).29 Briefly, trypsinized cells were resuspended with ECM containing 0.5% FBS and were seeded in the upper chamber at 5 × 104 cells per well. The lower chamber was filled with 500 μl of ECM containing 10% FBS. PBS, T0901317, T090-nt-lip, T090-t-lip, and blank liposome at 10 μM of T0901317 were added, respectively. After 8 h, all non-migrated cells were gently scratched out from the upper chamber surface using a cotton swab. Thereafter, the migrated cells were fixed in 4% paraformaldehyde, stained with 0.1% crystal violet and quantified by counting the number of stained cells with a microscope.Following a similar procedure, the transwell assay of HASMCs was carried out. The upper chamber medium was replaced with SMCM containing 0.5% FBS. The lower chamber medium was replaced with SMCM containing 0.5% FBS and 20 ng ml−1 PDGF-BB. The cells were treated with SMCM alone in the control group.
2.9 In vitro NO detection
Intracellular NO content was detected using a fluorescence microscope and the Griess kit. Briefly, HUVECs were seeded in 24-well plates at 1 × 105 cells per well in ECM. After 24 h, the cells were incubated with PBS, T090-t-lip, and liposome at 10 μM of T0901317 in fresh ECM containing 0.5% FBS and incubated for another 12 h. BBoxiProbe® O38 solution (a NO indicator, BestBio, BB-47014) was added and the cells were incubated for 30 min. Then the cells were washed 3 times with PBS and photographed by using a fluorescence microscope. For the NO quantitative analysis, the cells were treated with different formulations, harvested and lysed with a lysis buffer to obtain the test liquid. Following the Griess kit protocol (Beyotime, S0024), the intracellular NO content was detected.2.10 In vitro western blot analysis
Western blot analysis was used to evaluate the expression of the relative proteins in HASMCs after different treatments. The cells were seeded in 6-well plates at 2 × 105 cells per well in SMCM. The cells were treated with PBS, T0901317, T090-nt-lip, T090-t-lip, and blank liposome at 10 μM of T0901317 in fresh ECM containing 0.5% FBS and 20 ng ml−1 PDGF-BB, respectively. In the control group, SMCM containing 0.5% FBS alone was added. After 24 h, the cells were lysed and the total protein was quantified using a BCA protein quantification kit. Then, samples containing 60 μg of proteins were separated by 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene fluoride membrane. After blocking nonspecific binding with 5% non-fat milk for 1 h, the membrane was incubated with relevant primary antibodies at 4 °C overnight. After washing 3 times with TBST, the membrane was hybridized at 37 °C for 2 h with horseradish peroxidase-conjugated secondary antibody at a 1:1000 dilution. Finally, the membrane was rinsed with TBST 3 times again and visualized via chemiluminescence using an enhanced chemiluminescence detection kit. Protein expression was normalized to β-actin. The relative densities were quantified using Image J software.
2.11 In vitro cell cycle analysis
HASMCs were seeded in 6-well plates at 2 × 105 cells per well in SMCM. After incubation for 24 h, the medium was replaced by fresh SMCM containing 0.5% FBS and 20 ng ml−1 PDGF-BB. Then, the cells were treated with PBS, T0901317, T090-nt-lip, T090-t-lip, and blank liposome at 10 μM of T0901317, after which the cells were trypsinized and washed with PBS. The cell suspension was acquired by adding 1 ml of staining solutions and 10 μl of permeabilization solution. The cells were assessed by flow cytometry followed by incubation for 30 min at room temperature in the dark. In the control group, the cells were treated with smooth muscle cell medium (SMCM) with 0.5% FBS alone, while 20 ng ml−1 PDGF-BB was added in the other groups.2.12 In vitro HUVECs and HASMCs co-culture
HUVECs and HASMCs pre-stained with red and green live cell dyes, respectively, were trypsinized, mixed together completely in a 1:1 ratio, seeded in 24-well plates at 2 × 104 cells per well in ECM and incubated at 37 °C. After 24 hr of incubation, the medium was replaced with fresh ECM containing 0.5% FBS and 20 ng ml−1 PDGF-BB. PBS, T0901317, T090-nt-lip, T090-t-lip, and blank liposome at 10 μM of T0901317 were added, respectively. For the evaluation, the cells were photographed using a fluorescence microscope at indicated time points. The number of cells was counted by Image J software.
2.13 Animals
All in vivo experiments were performed in accordance with the guidelines for the care and use of laboratory animals and approved by the Animal Care and Use Committee, Zhejiang University. Healthy male Sprague-Dawley rats (400–450 g) were purchased from the animal center of the Zhejiang Academy of Medical Sciences.The injury model in rats was established as described by Clowes et al.30 Twenty male Sprague-Dawley rats were randomly assigned into 5 groups (n = 4), including a sham group (control), a saline-treated group (model), and groups administered with T0901317, T090-nt-lip, or T090-t-lip, respectively. Rat carotid artery balloon injury was induced in all groups except for the sham group. After angioplasty, different formulations were intravenously administered at 7.5 mg kg−1 of T0901317 on days 0, 3, 6, 9, and 12. After 2 weeks, the rats were euthanized. The carotid arteries and major organs were excised for further histological studies.
2.14 In vivo targeting ability and biodistribution
The in vivo targeting ability and biodistribution of liposomes were accessed on the rats with carotid balloon injury. The rats were randomly divided into 2 groups (3 per group). Cy5.5-T090-nt-lip and Cy5.5-T090-t-lip were intravenously injected into the rats. After 8 h, the carotid arteries and major organs were harvested and the ex vivo fluorescence images were taken using an IVIS 50 in vivo imaging system (PerkinElmer, MA).2.15 Histopathological evaluation
For the histopathological evaluation, the injured segment of the left common carotid artery was excised and embedded in paraffin. The cross-sections were prepared from the harvested arteries stained with hematoxylin and eosin (H&E). The images were captured by a microscope.2.16 Cholesterol and triglyceride measurements
Blood samples were centrifuged at 3000g for 10 min, and the supernatant was collected and stored at 4 °C. For the tissue samples, an appropriate amount of liver was weighed and ground in PBS. The mixture was centrifuged at 3000g for 10 min, and the supernatant was collected and stored at 4 °C. Then, the contents of cholesterol and triglyceride were tested by a total cholestenone (TC) content assay kit (PureBio, CH01) and a triglyceride (TG) content assay kit (PureBio, TG01).2.17 Biosafety evaluation in vivo
To evaluate the biosafety of the different formulations in rats, blood samples and major organs were harvested. Blood samples were collected for hematological and biochemical analyses. The major organs were embedded and sliced. The histological sections were stained with H&E.2.18 Statistical analysis
Data were expressed as mean ± SD. The statistical significance was determined using unpaired Student's t-test and one-way ANOVA analysis. A value of p < 0.05 was considered significant.3. Results and discussion
3.1 Preparation and characterization of drug-loaded liposomes
The LXR agonist T0901317-encapsulated liposomes were prepared by a filming-ultrasonic dispersion method.31,32 To achieve selective accumulation of T0901317 in vascular injury sites, a Col IV-targeting peptide-based amphiphilic peptide with a hydrophobic octadecanoic acid molecule tail (C18-CGGGKPLVWL) was synthesized and inserted into the phospholipid bilayer during liposome preparation (Fig. 2A and S1†). A non-targeting C18-CGGGLKVPWKL peptide with a scrambled sequence was applied as the control.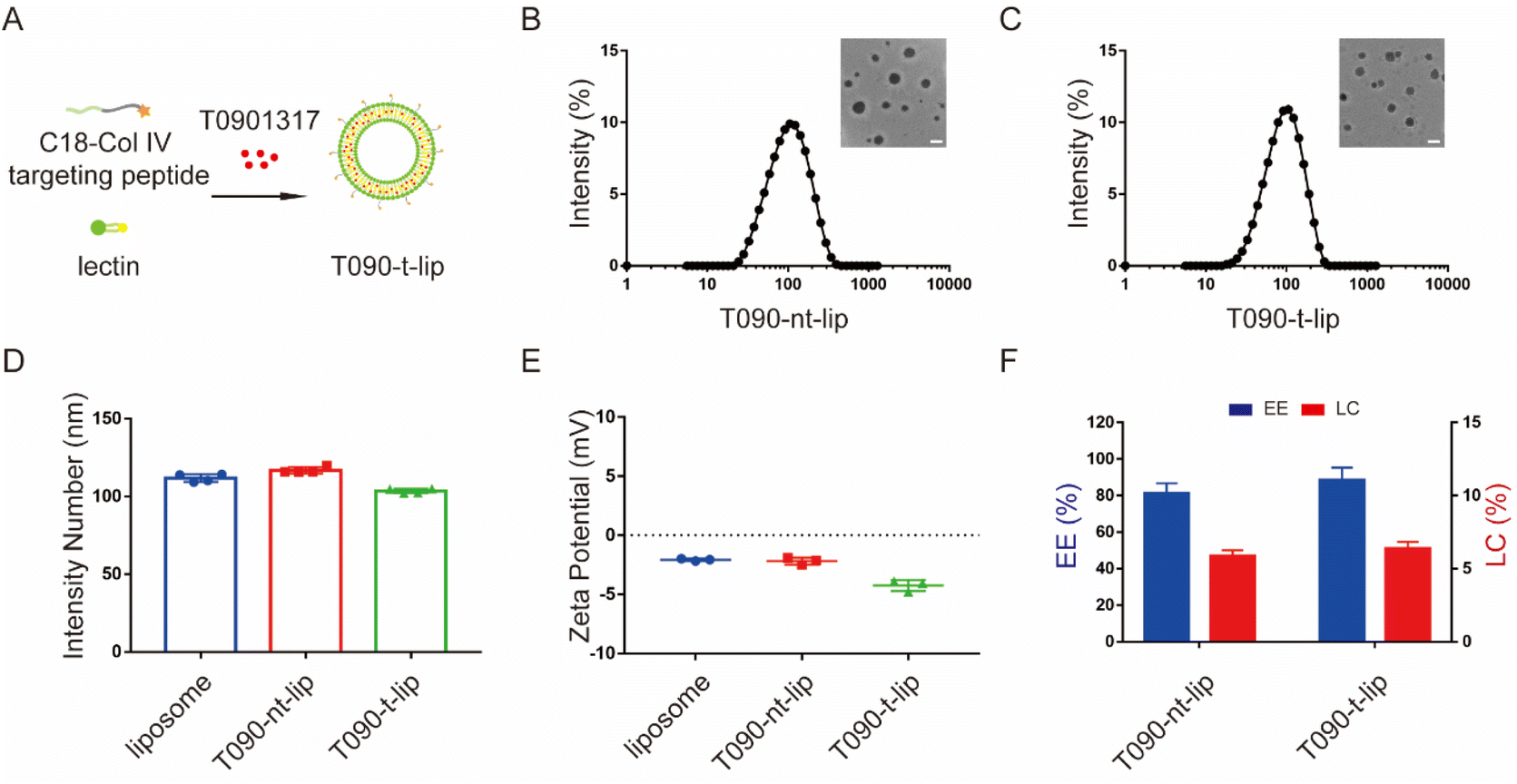 | ||
| Fig. 2 Characterization of different liposomes. (A) Schematic illustration of the Col-IV targeted liposome-loaded T0901317. (B) and (C) The hydrodynamic diameters of T090-nt-lip (B) and T090-t-lip (C); the inset images are TEM results. Scale bars, 200 nm. (D) and (E) The intensity number (D) and zeta potential (E) of different liposomes. (F) The EE and LC values of different liposomes. Data are presented as mean ± SD (n = 3). | ||
The morphology and size of liposomes were evaluated by dynamic light scattering (DLS) and transmission-electron microscopy (TEM) (Fig. 2B–D). All liposomes presented a spherical morphology with hydrodynamic diameters of 111.8 ± 2.5 nm, 116.8 ± 2.0 nm, and 103.6 ± 1.4 nm for blank liposome, T090-nt-lip, and T090-t-lip, respectively. The polydispersity indices (PDI) of T090-nt-lip and T090-t-lip were 0.216 ± 0.009 and 0.194 ± 0.015, respectively, indicating a relatively narrow size distribution of the liposomes. The zeta potential of all liposomes was negative (Fig. 2E), which was typically preferred for intravascular administration to extend the blood circulation time. The LC and EE were measured by HPLC (Fig. 2F). The results indicated that T0901317 was successfully encapsulated within the liposomes with LCs of 5.9 ± 0.4% and 6.4 ± 0.5% for T090-nt-lip and T090-t-lip, respectively. Both liposomes had similar EE (81.1 ± 5.6% for T090-nt-lip and 88.4 ± 6.9% for T090-t-lip). Together, T0901317-loaded liposomes with appropriate size and negative zeta potential were successfully prepared and used for the subsequent bio-experiments.
3.2 In vitro promoting effect of T0901317-loaded liposomes on HUVECs
The cellular uptake of different liposomes was visualized by fluorescence images and quantified by flow cytometry analysis (Fig. 3A, B and S2†), using coumarin-6 loaded liposomes as the model. At the same dose of liposomes, the fluorescence intensity increased over time in all groups. The quantification data by flow cytometry analysis also confirmed that the liposomes can be quickly internalized by HUVECs. Col-IV, as a main component of the subendothelial basement membrane, is a kind of extracellular matrix. The integration of Col IV-targeting peptide in the liposomes was expected to enhance the accumulation of liposomes in vascular injured sites but not the internalization of liposomes by HUVECs. Hence, T090-t-lip and T090-nt-lip exhibited similar cellular internalization abilities without significant difference.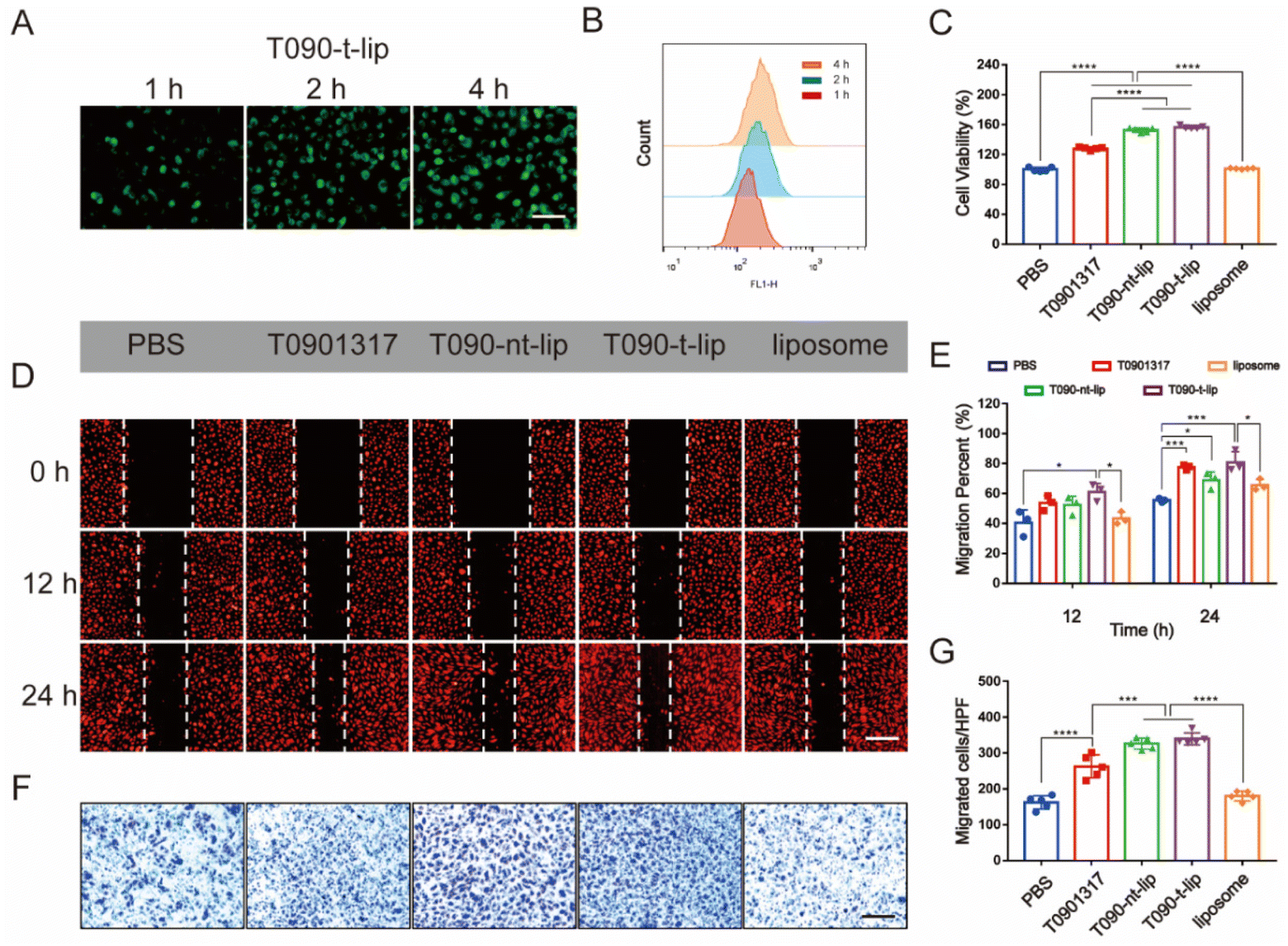 | ||
| Fig. 3 In vitro stimulatory effect on proliferation, migration and NO production of different liposomes in HUVECs. (A) and (B) The cellular uptake of T090-t-lip labeled by coumarin-6 is studied by fluorescence images (A, scale bar, 100 μm) and flow cytometry (B) after different incubation times. (C) Proliferation activity of different treatments in HUVECs by CCK-8 assay. (D)–(G) Migration activity of different treatments in HUVECs by wound healing assay (D and E, scale bars, 25 μm) and transwell assay (F and G, scale bars, 100 μm). HUVECs were incubated by different treatments with T0901317 at the same dose of 10 μM. Data in (E) are presented as mean ± SD (n = 3). Data in (C) and (G) are presented as mean ± SD (n = 5). *p < 0.05, ***p < 0.001, and ****p < 0.0001. | ||
The proliferation of HUVECs after treatment with different liposomes was further evaluated by a CCK-8 assay (Fig. 3C). Blank liposomes did not affect proliferation of HUVECs. Free T0901317 demonstrated moderate promotion of the proliferation of HUVECs. However, the proliferation of HUVECs was much more significantly promoted with treatment of T090-t-lip and T090-nt-lip, which might be attributed to the better uptake of liposomes, thus increasing the intracellular level of drugs. However, as T090-t-lip and T090-nt-lip exhibited similar internalization abilities in HUVECs, T090-t-lip and T090-nt-lip were almost the same in promoting the proliferation of HUVECs.
Migration of HUVECs was vital for the repairing process after vascular injury. Wound healing assay and transwell assay were employed to evaluate the migrating ability of HUVECs (Fig. 3D–G). Free T0901317 or T0901317-loaded liposomes greatly enhanced the migration of HUVECs as indicated by the higher healing percentages in the wound healing assay. In transwell assay, HUVECs treated with T0901317-loaded liposomes had a larger number of traversing cells than that with free T0901317, probably due to the higher internalization ability of the liposomes.
T0901317 was reported to affect the proliferation and migration activity in HUVECs via the NO-related PI3K/Akt/eNOS signaling pathway.27 A fluorescent probe was used to visualize the intracellular level of NO. As shown in Fig. S3A,† the red fluorescence of the NO probe in the T090-t-lip treated group was notably stronger than that of the PBS or liposome-treated groups after 12 h of incubation, which indicated the increased production of NO by T0901317. Furthermore, the intracellular generation of NO was quantitatively evaluated by Griess assay kits (Fig. S3B†), which was consistent with the fluorescence images. The increased production of intracellular NO could promote the proliferation and migration of HUVECs.33–35 These data proved that T0901317-loaded liposomes could be effectively internalized by HUVECs, significantly promoting the proliferation and migration of HUVECs via increasing NO production.
3.3 In vitro inhibiting effect of T0901317-loaded liposomes on HASMCs
Inhibition of abnormal proliferation of vascular SMCs is essential in alleviating restenosis post PCI therapy. The balance of EC and SMC proliferation is vital for vascular injury repair.12,36 Thus, the anti-proliferation and anti-migration ability of different agents against SMCs were further evaluated.Firstly, the internalization of different liposomes by HASMCs was assessed by fluorescence images and flow cytometry analysis using coumarin-6 as a fluorescence tracker (Fig. 4A, B and S4†). At the same dose of liposome, intracellular green fluorescence from coumarin-6 intensified with prolonged incubation time in all the groups. The quantitative data from flow cytometry analysis also confirmed the time-dependent internalization of liposomes by HASMCs. The effective uptake of liposomes was preferred for the subsequential regulation of cell behaviors.
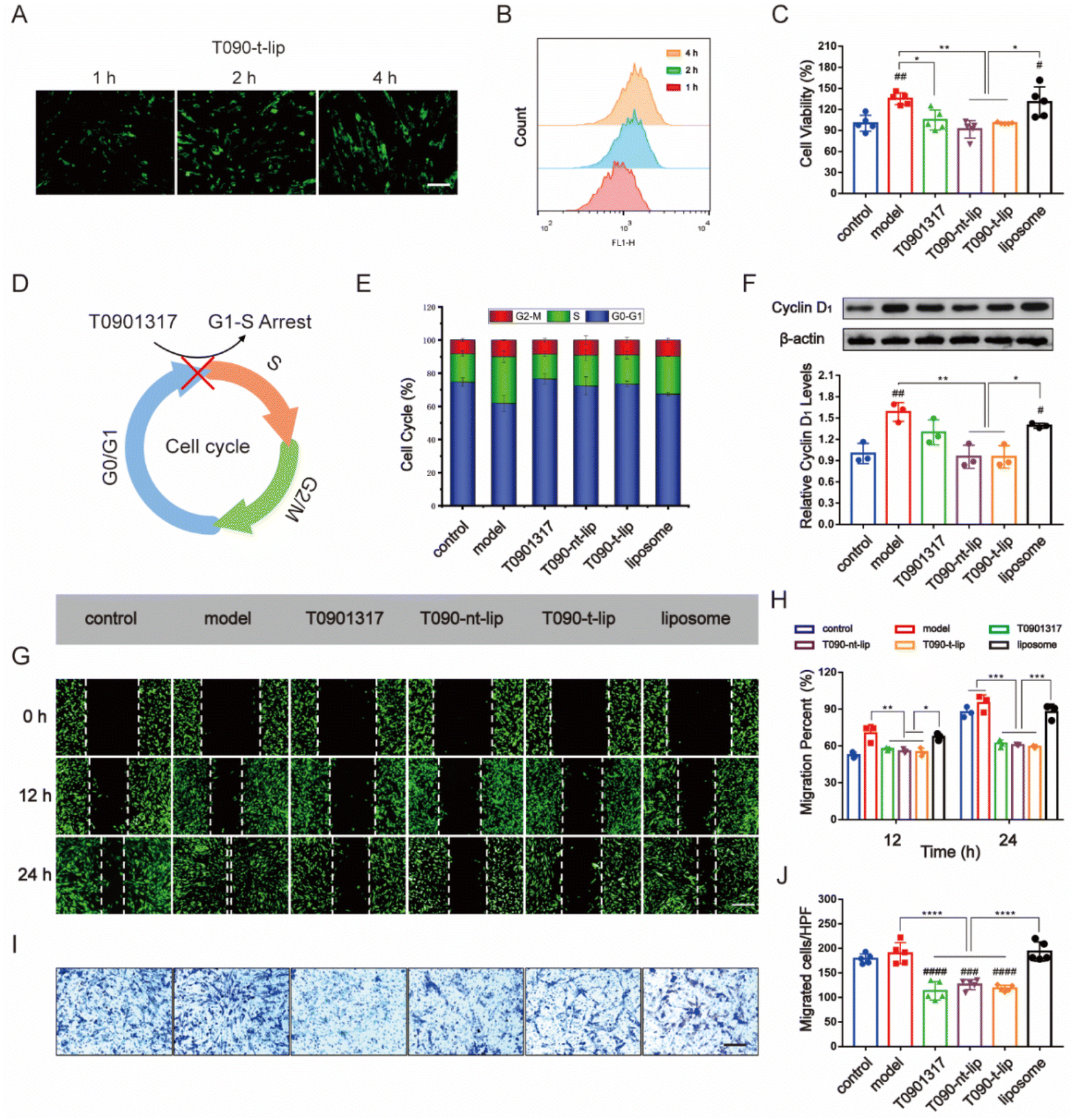 | ||
| Fig. 4 In vitro inhibitory effect on the proliferation/migration and the regulation of the cell cycle of different liposomes in HASMCs. (A) and (B) The cellular uptake of T090-t-lip labeled by coumarin-6 is studied by fluorescence images (A, scale bars, 100 μm) and flow cytometry (B) after different incubation times. (C) Anti-proliferation activity of different treatments in HASMCs by CCK-8 assay. (D) Schematic illustration of the cell cycle progression. The LXR agonist T0901317 can exert the function of G1-S arrest by suppressing the switch of G1 phase to S phase. (E) Quantitative data of the flow cytometric analysis demonstrating the inhibition of G1-S transition in HASMCs. (F) WB band (the upper panel) and quantitative data (the lower panel) showing the levels of cyclin D1 protein in HASMCs. (G)–(J) Anti-migration activity after different treatments in HASMCs by wound healing assay (G and H, scale bars, 25 μm) and transwell assay (I and J, scale bars, 100 μm). Except for the cells in the control, groups were cultured with medium alone, while the other groups were stimulated by 20 ng ml−1 PDGF-BB. HASMCs were incubated by different treatments at the same dose of 10 μM T0901317. Data in (E), (F) and (H) are presented as mean ± SD (n = 3). Data in (C) and (J) are presented as mean ± SD (n = 5). *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. ##p < 0.01 and ####p < 0.0001 vs. control. | ||
The proliferation of HASMCs was evaluated by the CCK-8 assay. HASMCs were pre-treated with PDGF-BB (20 ng ml−1) to promote pathological overgrowth. As expected, PDGF-BB treatment increased the proliferation of HASMCs compared with those incubated with PBS (Fig. 4C). Free T0901317 or T0901317-loaded liposomes inhibited the proliferating effect of PDGF-BB on HASMCs. T090-t-lip and T090-nt-lip were almost the same in inhibiting the proliferation of HASMCs due to the similar internalization abilities of T090-t-lip and T090-nt-lip by HASMCs.
To further verify the mechanism behind such regulating ability, cell cycle progression was determined by flow cytometry analysis. As shown in Fig. 4E and S5,† PDGF-BB pre-treatment reversed the resting effect on HASMCs during serum deprivation, thus promoting more cells entering the S phase than the G0–G1 phase. However, free T0901317 or T0901317-loaded liposomes remarkably restored most of the cells to G1 and inhibited their G1/S transition. Cyclin D1 is a typical cell cycle regulator and is essential for G1/S transition. Western blot (WB) analysis of the expression level of cyclin D1 also indicated an increase in protein level after PDGF-BB pre-treatment and its restoration to normal level after treatment with free T0901317 or drug-loaded liposomes (Fig. 4F). The results suggested that free T0901317 or T0901317 loaded liposomes interfered with the aberrant proliferation of HASMCs via the hindrance of the G1/S transition.
Besides, the over-migration of HASMCs also proved detrimental to vascular repair, as it plays an important role in restenosis. PDGF-BB pre-treatment stimulated the migration of HASMCs. However, free T0901317 and T0901317-loaded liposomes exhibited a great inhibiting effect on the anti-migration of HASMCs as demonstrated by more narrow gaps in the wound healing assay and fewer migrated cells in the transwell assay (Fig. 4G–J). These results can prove that T0901317-loaded liposomes could serve as a potent regulator of HASMC migration.
3.4 In vitro proliferation assay in co-culture of HUVECs and HASMCs
As two major cells in vessels, the interaction between ECs and SMCs is of great significance for the vascular repair process after injury. Thus, co-culture experiments were performed to further evaluate the effect of different treatments on the competitiveness of HUVECs over HASMCs. The cells were pre-treated with PDGF-BB (20 ng ml−1) before the addition of T0901317. HUVECs and HASMCs were distinguishingly pre-stained red and green for visualization by fluorescent dyes, respectively. As shown in the fluorescence images (Fig. 5A), both HUVECs and HASMCs had similar densities and with well adhesion. The cell morphology can intuitively reflect the growth status.37,38 After 24 h of incubation, HUVECs with T0901317 treatment tended to aggregate together with typical pebble and elliptical morphology. HASMCs shrunk significantly after treatment with free T0901317 or T0901317-loaded liposomes, indicating the relatively poor growth status of HASMCs. Therefore, the treatment with T0901317 was more favorable for the growth of HUVECs compared to HASMCs, with the same initial adhesion and state. At the same time, the number of HUVECs and HASMCs was quantitatively counted. The relative number of HUVECs increased by more than 50% after treatment with free T0901317 or T0901317-loaded liposomes (Fig. 5B), which again confirmed that T0901317 could promote the proliferation of HUVECs. Although the relative number of HASMCs was almost unchanged, the spreading area of HASMCs after treatment with free T0901317 or T0901317-loaded liposomes decreased by almost 50% compared to the PBS group (Fig. 5C and D). This might indicate that treatment with T0901317 was disadvantageous for the growth of HASMCs. More importantly, the ratio of cell number between HUVECs and HASMCs in the groups treated with free T0901317 or T0901317-loaded liposomes was significantly higher than that treated with PBS (Fig. 5E). Taking these results into consideration, it can be concluded that HUVECs took the dominant position over HASMCs with T0901317 treatment and the competitiveness of HUVECs over HASMCs was enhanced by the T0901317-loaded liposomes.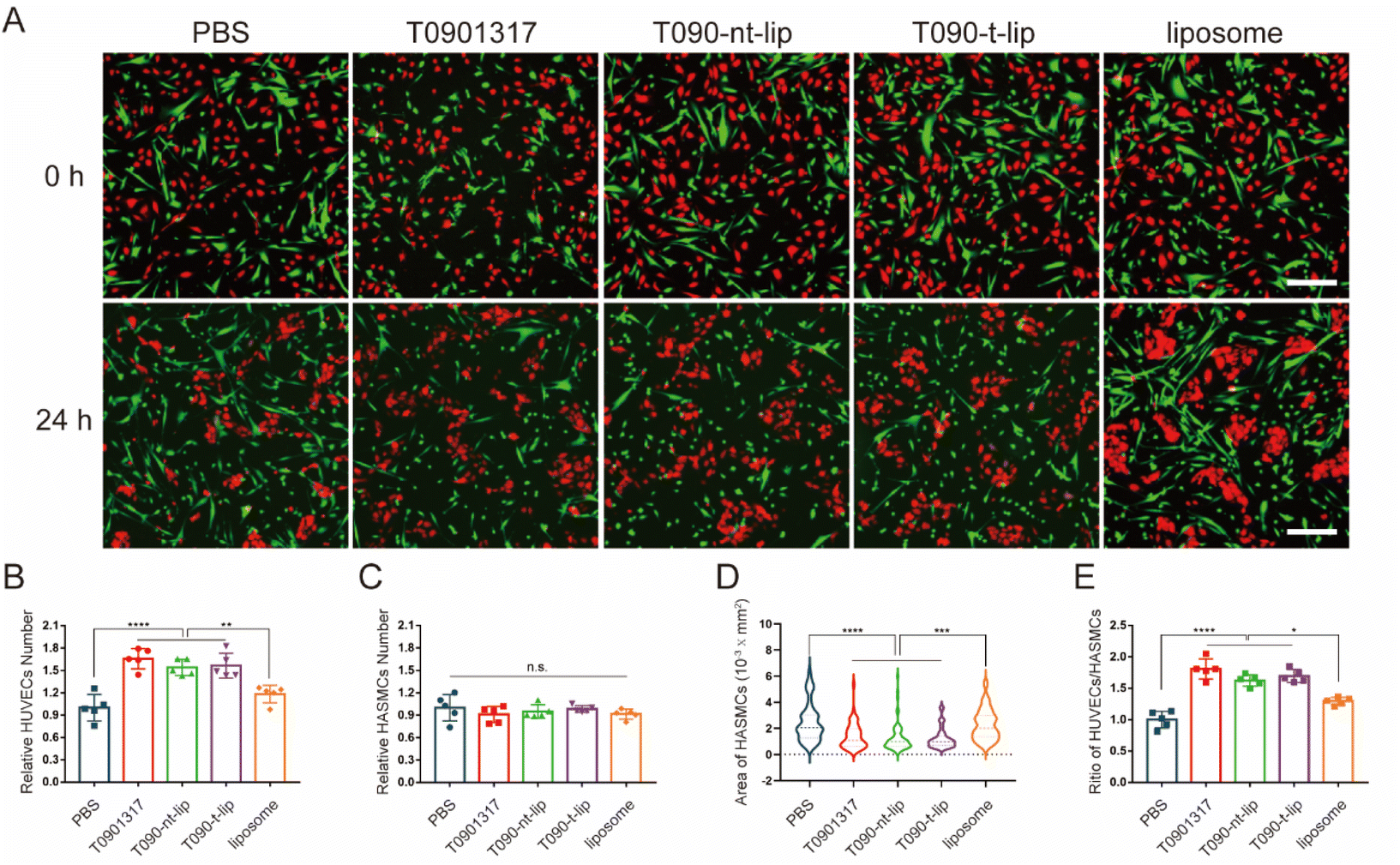 | ||
| Fig. 5 In vitro proliferation assay of HUVECs (red) and HASMCs (green) in the co-culture system. Fluorescence images (A, scale bars, 100 μm) and quantitative results (B)–(E) of relative HUVEC number, relative HASMC number, area of HASMCs and the ratio of HUVECs/HASMCs, respectively. Data in (B), (C) and (E) are presented as mean ± SD (n = 5). A representative 60 cells were selected from each group for statistics (D). *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001. | ||
3.5 In vivo treatment of restenosis by T0901317-loaded liposomes in rats
The in vivo anti-restenosis effect of different treatments was evaluated using a balloon-induced carotid artery injury model in rats. It was reported that the peptide with a sequence of CGGGKPLVWL can selectively bind to Col-IV, which was exposed after vascular injury.19,29 Therefore, the in vivo targeting capability of T090-t-lip was firstly evaluated with Cy5.5-labeled fluorescent liposomes (Fig. 6A and S6†). The Cy5.5-labeled liposomes were intravenously injected immediately after balloon-induced carotid artery injury. After 8 h, the arteries were harvested for ex vivo imaging. The fluorescence intensity of T090-t-lip treated vascular injured sites was much higher than that treated with T090-nt-lip, which indicated that the liposomes decorated with the Col-IV targeting peptide could effectively accumulate at the injured carotid arteries.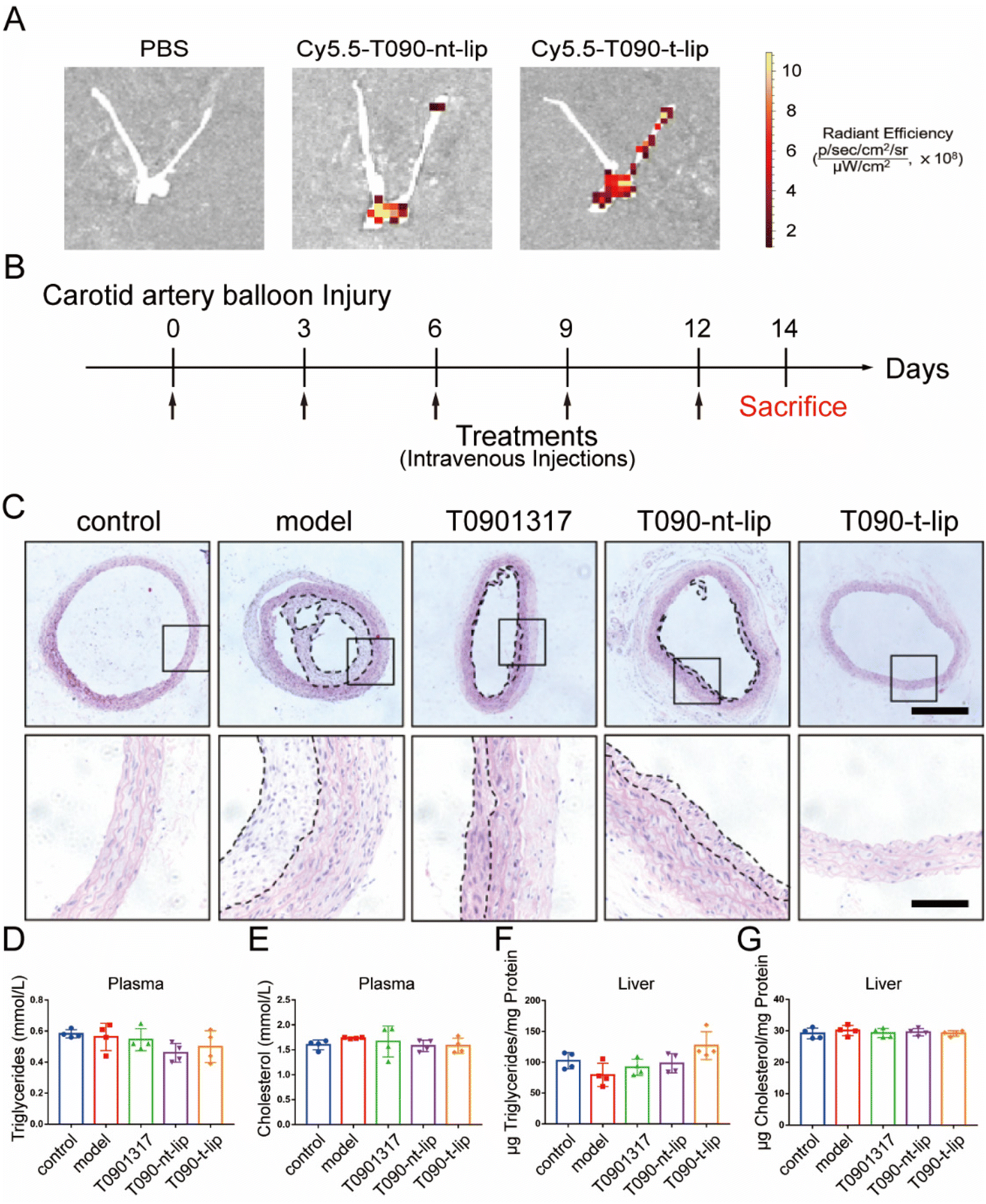 | ||
| Fig. 6 In vivo targeting and anti-restenosis performance of different treatments in rat carotid balloon injury model. Fluorescence images (A) showing the targeting ability of Col-IV liposomes labeled by Cy5.5 to vascular injured sites after intravenous injection. (B) Schematic diagram of the treatment plan. (C) Representative H&E images of carotid arteries after different treatments. Scale bars, upper panels, 4×, 25 μm and lower panels, 10×, 100 μm. Total cholesterol (D) and triglyceride (E) levels were measured in plasma. Total cholesterol (F) and triglyceride (G) levels were measured in liver and normalized to the protein content. Data are presented as mean ± SD (n = 4). | ||
We next tested the anti-restenosis performance of T090-t-lip in vivo. After balloon injury, the rats were injected intravenously with free T0901317, T090-nt-lip, or T090-t-lip (7.5 mg kg−1 T0901317). On day 14, the rats were sacrificed, and the injured carotid arteries were harvested for further analyses. As shown in Fig. 6C, the H&E-staining results demonstrated that neointimal hyperplasia in the model group was more obvious compared to the untreated control group, indicating serious restenosis after injury. Notably, neointimal hyperplasia was greatly inhibited after the rats were treated with free T0901317 or T0901317 loaded liposomes. The T090-t-lip group exhibited the most optimal efficacy against anti-restenosis, as indicated by the thinnest neointimal thickness. These results indicated that Col-IV-targeted liposomes loaded with T0901317 had excellent anti-restenosis capability in rats.
3.6 Biosafety evaluation in vivo
Finally, the biosafety of different treatments was evaluated by hematological analysis. Triglyceride and cholesterol contents in both liver and plasma displayed no significant differences among all groups, which indicated that T0901317-loaded liposomes could exhibit excellent anti-restenosis ability without any adverse effect on serum and liver lipid contents (Fig. 6D–G).39 Due to the targeted design of the liposomes, more drugs could be accumulated at the injured sites. Therefore, compared to the reported literature, a relatively low dose was used in this research (7.5 mg kg−1 once every 3 days for 2 weeks), leading to negligible side effects.40 Besides, the biomarkers of hepatic functions were also unchanged after different treatments, implying a negligible adverse effect on the liver (Fig. S7†). Furthermore, the H&E-stained sections of major organs displayed ignorable injury (Fig. S8†). Consequently, T090-t-lip exhibited excellent biosafety.4. Conclusion
In summary, to overcome the dilemma of simultaneous EC promotion and SMC inhibition, we designed Col-IV targeted liposomes loaded with the LXR agonist T0901317. In vitro experiments demonstrated that T0901317-loaded liposomes can effectively promote the migration and proliferation of HUVECs via promoting NO production. Meanwhile, T0901317-loaded liposomes can suppress the migration and proliferation of restenosis-related HASMCs by arresting cell cycle transition. Such outstanding cell-specific selectivity of T0901317-loaded liposomes improved the competitiveness of HUVECs over HASMCs in the co-culture system. Moreover, T090-t-lip could effectively accumulate at the injured carotid arteries via the Col-IV mediated targeting peptide and display a most excellent anti-restenosis effect in vivo. Besides, the T0901317-loaded liposomes also showed excellent biosafety. Consequently, the Col-IV targeted nanomedicine based on the LXR agonist T0901317 might be a very promising therapeutic strategy for anti-restenosis therapy.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was supported by the National Key Research and Development Program of China (2020YFE0204400), the Natural Key Research and Development Project of Zhejiang Province (2018C03015), Zhejiang Provincial Natural Science Foundation of China (LD22E030002), and the National Natural Science Foundation of China (51933009 and U20A20262).References
- E. J. Benjamin, P. Muntner, A. Alonso, M. S. Bittencourt, C. W. Callaway, A. P. Carson, A. M. Chamberlain, A. R. Chang, S. Cheng, S. R. Das, F. N. Delling, L. Djousse, M. S. V. Elkind, J. F. Ferguson, M. Fornage, L. C. Jordan, S. S. Khan, B. M. Kissela, K. L. Knutson, T. W. Kwan, D. T. Lackland, T. T. Lewis, J. H. Lichtman, C. T. Longenecker, M. S. Loop, P. L. Lutsey, S. S. Martin, K. Matsushita, A. E. Moran, M. E. Mussolino, M. O'Flaherty, A. Pandey, A. M. Perak, W. D. Rosamond, G. A. Roth, U. K. A. Sampson, G. M. Satou, E. B. Schroeder, S. H. Shah, N. L. Spartano, A. Stokes, D. L. Tirschwell, C. W. Tsao, M. P. Turakhia, L. B. VanWagner, J. T. Wilkins, S. S. Wong and S. S. Virani, Circulation, 2019, 139, e56–e528 CrossRef PubMed.
- A. K. Thukkani and S. Kinlay, Circ. Res., 2015, 116, 1599–1613 CrossRef CAS PubMed.
- J. W. Jukema, T. A. Ahmed, J. J. Verschuren and P. H. Quax, Nat. Rev. Cardiol., 2011, 9, 79–90 CrossRef PubMed.
- J. W. Jukema, J. J. Verschuren, T. A. Ahmed and P. H. Quax, Nat. Rev. Cardiol., 2011, 9, 53–62 CrossRef PubMed.
- J. Daemen, P. Wenaweser, K. Tsuchida, L. Abrecht, S. Vaina, C. Morger, N. Kukreja, P. Juni, G. Sianos, G. Hellige, R. T. van Domburg, O. M. Hess, E. Boersma, B. Meier, S. Windecker and P. W. Serruys, Lancet, 2007, 369, 667–678 CrossRef CAS.
- A. K. Hassan, S. C. Bergheanu, T. Stijnen, B. L. van der Hoeven, J. D. Snoep, J. W. Plevier, M. J. Schalij and J. Wouter Jukema, Eur. Heart J., 2010, 31, 1172–1180 CrossRef CAS PubMed.
- T. F. Luscher, J. Steffel, F. R. Eberli, M. Joner, G. Nakazawa, F. C. Tanner and R. Virmani, Circulation, 2007, 115, 1051–1058 CrossRef PubMed.
- S. Godo and H. Shimokawa, Arterioscler., Thromb., Vasc. Biol., 2017, 37, e108–e114 CrossRef CAS PubMed.
- N. Swanson, K. Hogrefe, Q. Javed, N. Malik and A. H. Gershlick, J. Invasive Cardiol., 2003, 15, 688–692 Search PubMed.
- X. Wu, Y. Zhao, C. Tang, T. Yin, R. Du, J. Tian, J. Huang, H. Gregersen and G. Wang, ACS Appl. Mater. Interfaces, 2016, 8, 7578–7589 CrossRef CAS PubMed.
- H. Chang, K. F. Ren, J. L. Wang, H. Zhang, B. L. Wang, S. M. Zheng, Y. Y. Zhou and J. Ji, Biomaterials, 2013, 34, 3345–3354 CrossRef CAS PubMed.
- H. Chang, H. Zhang, M. Hu, X. C. Chen, K. F. Ren, J. L. Wang and J. Ji, Biomater. Sci., 2015, 3, 352–360 RSC.
- Q. Jin, Y. Deng, X. Chen and J. Ji, ACS Nano, 2019, 13, 954–977 CAS.
- J. Ren, X. Shu, Y. Wang, D. Wang, G. P. Wu, X. H. Zhang, Q. Jin, J. Z. Liu, Z. L. Wu, Z. Xu, C. Z. Li and H. Y. Li, Chin. Chem. Lett., 2022, 33, 1650–1658 CrossRef CAS.
- S. T. Wang, Y. Fang, Z. Q. Zhang, Q. Jin and J. Ji, Colloid Interface Sci. Commun., 2021, 40, 100354 CrossRef CAS.
- J. M. Chan, L. Zhang, R. Tong, D. Ghosh, W. Gao, G. Liao, K. P. Yuet, D. Gray, J. W. Rhee, J. Cheng, G. Golomb, P. Libby, R. Langer and O. C. Farokhzad, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2213–2218 CrossRef CAS PubMed.
- Y. Deng, Y. Wang, F. Jia, W. Liu, D. Zhou, Q. Jin and J. Ji, ACS Nano, 2021, 15, 8663–8675 CrossRef CAS PubMed.
- H. Han, Y. Hou, X. Chen, P. Zhang, M. Kang, Q. Jin, J. Ji and M. Gao, J. Am. Chem. Soc., 2020, 142, 4944–4954 CrossRef CAS PubMed.
- N. Kamaly, G. Fredman, J. J. Fojas, M. Subramanian, W. I. Choi, K. Zepeda, C. Vilos, M. Yu, S. Gadde, J. Wu, J. Milton, R. Carvalho Leitao, L. Rosa Fernandes, M. Hasan, H. Gao, V. Nguyen, J. Harris, I. Tabas and O. C. Farokhzad, ACS Nano, 2016, 10, 5280–5292 CrossRef CAS PubMed.
- C. Shi, Q. Li, W. Zhang, Y. Feng and X. Ren, ACS Appl. Mater. Interfaces, 2015, 7, 20389–20399 CrossRef CAS PubMed.
- M. Wen, F. Zhou, C. Cui, Y. Zhao and X. Yuan, Colloids Surf., B, 2019, 182, 110369 CrossRef CAS PubMed.
- X. Wang, B. Gao, X. K. Ren, J. Guo, S. Xia, W. Zhang, C. Yang and Y. Feng, Colloids Surf., B, 2021, 208, 112068 CrossRef CAS PubMed.
- M. V. Cannon, W. H. van Gilst and R. A. de Boer, Basic Res. Cardiol., 2016, 111, 3 CrossRef PubMed.
- N. Benne, R. Martins Cardoso, A. L. Boyle, A. Kros, W. Jiskoot, J. Kuiper, J. Bouwstra, M. Van Eck and B. Slutter, Adv. Healthcare Mater., 2020, 9, 2000043 CrossRef CAS PubMed.
- S. S. Im and T. F. Osborne, Circ. Res., 2011, 108, 996–1001 CrossRef CAS PubMed.
- M. Yu, J. Amengual, A. Menon, N. Kamaly, F. Zhou, X. Xu, P. E. Saw, S. J. Lee, K. Si, C. A. Ortega, W. I. Choi, I. H. Lee, Y. Bdour, J. Shi, M. Mahmoudi, S. Jon, E. A. Fisher and O. C. Farokhzad, Adv. Healthcare Mater., 2017, 6, 1700313 CrossRef PubMed.
- J. Yu, Q. Wang, H. Wang, W. Lu, W. Li, Z. Qin and L. Huang, Vasc. Pharmacol., 2014, 62, 150–161 CrossRef CAS PubMed.
- F. Blaschke, O. Leppanen, Y. Takata, E. Caglayan, J. Liu, M. C. Fishbein, K. Kappert, K. I. Nakayama, A. R. Collins, E. Fleck, W. A. Hsueh, R. E. Law and D. Bruemmer, Circ. Res., 2004, 95, e110–e123 CrossRef CAS PubMed.
- R. Zhang, R. Liu, C. Liu, L. Pan, Y. Qi, J. Cheng, J. Guo, Y. Jia, J. Ding, J. Zhang and H. Hu, Biomaterials, 2020, 230, 119605 CrossRef CAS PubMed.
- A. K. Chan, A. Kalmes, S. Hawkins, G. Daum and A. W. Clowes, J. Vasc. Surg., 2003, 37, 644–649 CrossRef PubMed.
- X. Chen, F. Jia, Y. Li, Y. Deng, Y. Huang, W. Liu, Q. Jin and J. Ji, Biomaterials, 2020, 246, 119999 CrossRef CAS PubMed.
- Y. Li, H. Cong, S. Wang, B. Yu and Y. Shen, Biomater. Sci., 2020, 8, 6442–6468 RSC.
- U. Forstermann and W. C. Sessa, Eur. Heart J., 2012, 33, 829–837 CrossRef PubMed , 837a–837d.
- D. Tousoulis, A. M. Kampoli, C. Tentolouris, N. Papageorgiou and C. Stefanadis, Curr. Vasc. Pharmacol., 2012, 10, 4–18 CrossRef CAS PubMed.
- P. M. Vanhoutte, Y. Zhao, A. Xu and S. W. Leung, Circ. Res., 2016, 119, 375–396 CrossRef CAS PubMed.
- Q. Song, L. Li, K. Xiong, W. Tian, J. Lu, J. Wang, N. Huang, Q. Tu and Z. Yang, Biomater. Sci., 2019, 7, 3741–3750 RSC.
- M. L. Bochaton-Piallat, P. Ropraz, F. Gabbiani and G. Gabbiani, Arterioscler., Thromb., Vasc. Biol., 1996, 16, 815–820 CrossRef CAS PubMed.
- H. K. F. Lau, Cardiovasc. Res., 1999, 43, 1049–1059 CrossRef CAS PubMed.
- T. G. Kirchgessner, P. Sleph, J. Ostrowski, J. Lupisella, C. S. Ryan, X. Liu, G. Fernando, D. Grimm, P. Shipkova, R. Zhang, R. Garcia, J. Zhu, A. He, H. Malone, R. Martin, K. Behnia, Z. Wang, Y. C. Barrett, R. J. Garmise, L. Yuan, J. Zhang, M. D. Gandhi, P. Wastall, T. Li, S. Du, L. Salvador, R. Mohan, G. H. Cantor, E. Kick, J. Lee and R. J. Frost, Cell Metab., 2016, 24, 223–233 CrossRef CAS PubMed.
- C. Ma, K. Feng, X. Yang, Z. Yang, Z. Wang, Y. Shang, G. Fan, L. Liu, S. Yang, X. Li, J. Han, Y. Duan and Y. Chen, Br. J. Pharmacol., 2021, 178, 1620–1638 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2bm01041k |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2022 |