
Polyaspartate-derived synthetic antimicrobial polymer enhances the activity of rifampicin against multidrug-resistant Pseudomonas aeruginosa infections
Nalini
Chaudhary
 a,
Bharti
Aggarwal
a,
Varsha
Saini
a,
Prabhu
Srinivas Yavvari
b,
Priyanka
Sharma
c,
Aasheesh
Srivastava
*b and
Avinash
Bajaj
*a
a,
Bharti
Aggarwal
a,
Varsha
Saini
a,
Prabhu
Srinivas Yavvari
b,
Priyanka
Sharma
c,
Aasheesh
Srivastava
*b and
Avinash
Bajaj
*a
aLaboratory of Nanotechnology and Chemical Biology, Regional Centre for Biotechnology, NCR Biotech Science Cluster, 3rd Milestone, Faridabad-Gurgaon Expressway, Faridabad-121001, Haryana, India. E-mail: bajaj@rcb.res.in
bDepartment of Chemistry, Indian Institute of Science Education and Research, Bhopal By-pass Road, Bhauri, Bhopal-462030, India. E-mail: asrivastava@iiserb.ac.in
cDepartment of Microbiology, All India Institute of Medical Sciences, New Delhi-110029, India
First published on 27th June 2022
Abstract
Infections caused by multidrug-resistant Pseudomonas aeruginosa (P. aeruginosa) pose major challenges for treatment due to the acquired, adaptive, and intrinsic resistance developed by the bacteria. Accumulation of mutations, the ability to form biofilms, and the presence of lipopolysaccharides in the outer bacterial membranes are the key mechanisms of drug resistance. Here, we show that a polyaspartate-derived synthetic antimicrobial polymer (SAMP) with a hexyl chain (TAC6) is an effective adjuvant for a hydrophobic antibiotic, rifampicin. Our in vitro studies demonstrated that the combination of TAC6 and rifampicin is effective against clinically isolated multidrug-resistant strains of P. aeruginosa. Membrane permeabilization studies showed that TAC6 allows the permeabilization of bacterial membranes, and the accumulation of rifampicin inside the cells, thereby enhancing its activity. The combination of TAC6 and rifampicin can also degrade the P. aeruginosa biofilms, and therefore can mitigate the adaptive resistance developed by bacteria. We further demonstrated that the combination of TAC6 and rifampicin can clear P. aeruginosa-mediated wound infections effectively. Therefore, our study showed polyaspartate-derived SAMP to be an effective antibiotic adjuvant against P. aeruginosa infections.
Introduction
Infections caused by multidrug-resistant Pseudomonas aeruginosa (P. aeruginosa) pose a serious threat as this pathogen can cause bacteremia, severe pneumonia, urinary tract infections, respiratory tract infections and ocular infections.1,2P. aeruginosa also poses a major threat in skin burns, where it hampers wound healing in patients due to the formation of antibiotic-impenetrable biofilms, thereby leading to chronic infections.3,4 Biofilm-dwelling P. aeruginosa cells tend to have a mutated phenotype with a physiology comparable with that of stationary phase planktonic cells as these cells are deeply embedded under thick biofilms with a lack of nutrients and oxygen.5 The matrix of the biofilms also provides a physiological barrier to support the persister cells in the biofilms.6 Therefore, infections caused by P. aeruginosa can possess acquired resistance due to the accumulation of mutations or adaptive resistance due to biofilm formation, thereby causing multifaceted challenges for antibiotics.7 Therefore, there is a need to develop innovative strategies that can mitigate Pseudomonas aeruginosa mediated multidrug-resistant infections.8Apart from adaptive and acquired resistance, Gram-negative bacteria also exhibit intrinsic antimicrobial resistance due to the presence of an outer cell membrane that is rich in heavily glycosylated lipids known as lipopolysaccharides (LPSs).9,10 LPS molecules are crosslinked by divalent cations (Mg2+) through the establishment of ionic bridges among anionic phosphate groups from lipid A. Therefore, LPSs provide a barrier to the majority of hydrophobic antibiotics, and prevent passive diffusion into the bacterial cell.11,12 As the majority of antibiotics are losing their efficacy over Gram-negative bacteria due to the emergence of resistance, re-sensitisation of Gram-negative bacteria against these antibiotics via targeting bacterial cell membranes provides a riveting alternative.13,14 Antibiotic adjuvants are known to re-sensitize Gram-negative bacteria towards ineffective antibiotics by targeting the efflux pumps, disintegrating bacterial membranes, or enhancing the intracellular accumulation of antibiotics.15,16 Antibiotic adjuvants do not show any antimicrobial activity but can inhibit the intrinsic and adaptive drug-resistance developed by the bacteria and allow the activity of existing antibiotics.17–20
Membrane-permeabilising agents can act as effective antibiotic adjuvants as they can lead to the disruption of bacterial membranes and facilitate the entry of antibiotics into bacterial cells.21,22 Antimicrobial peptides (AMPs) produced by the host organism's innate immunity tend to perform a similar function due to their amphiphilic nature. AMPs execute electrostatic interactions with bacterial membranes, followed by insertion in them, causing membrane disruptions.23 However, the clinical utility of these AMPs as antimicrobial agents is restricted due to their instability and toxicity to mammalian cells.24 Antimicrobial polymers present a suitable alternative to AMPs owing to diversity in their composition and design principles, as they can be derived from a variety of classes including polyamides, polyurethanes, and polycarbonates.25–27 Cationic polymers tend to target the anionic phosphate groups present in lipid A of the outer bacterial membrane, and compete with the divalent cations involved in cross-bridging. However, many polymer scaffolds are non-biodegradable in nature and can cause long-term toxic effects.28–30
Polyaspartic acid is a biodegradable aspartic acid-derived polymer that has been explored for a wide range of biomedical applications including drug and protein delivery, gene therapy, and antimicrobial therapy.31–34 Recently we have shown that polyaspartate-derived synthetic antimicrobial polymers (SAMPs) are effective antimicrobials against mycobacteria species, and can penetrate the mycobacterial membranes to bind with genomic DNA.35 Herein, we tested the interactions of these polyaspartate-derived SAMPs with the bacterial membranes of Pseudomonas aeruginosa, and tested their efficacy as potential antibiotic adjuvants for rifampicin. We show that TAC6 acts as an effective adjuvant to rifampicin to alleviate its intrinsic resistance and establish enhanced bactericidal activity. The combination of TAC6 and rifampicin has also been screened for an auxiliary effect on biofilms and its ability to dissipate wound infections. We demonstrate that the combination of TAC6 and rifampicin is also effective against multidrug-resistant clinical strains of P. aeruginosa.
Results and discussion
Polyaspartate-derived synthetic antimicrobial polymers (SAMPs) are effective permeabilizers
We used five polyaspartate-derived SAMPs (TAC1, TAC2, TAC4, TAC6, and TAC8) having tetra-alkyl ammonium head groups tethered to the carboxylic acid units through amide bonds. These SAMPs possess varied alkyl chain substitutions like methyl (TAC1), ethyl (TAC2), butyl (TAC4), hexyl (TAC6), and octyl (TAC8) (Fig. 1A). We first determined the minimum inhibitory concentration (MIC99) of the SAMPs against the wild type strain of P. aeruginosa and five multidrug-resistant clinical strains of P. aeruginosa. The antimicrobial activities of SAMPs revealed that none of them is an effective antimicrobial against these strains until a concentration of 64–128 mg mL−1 is reached (Fig. 1B). To decipher the antibiotic adjuvant ability of these SAMPs against the intrinsic resistance of P. aeruginosa, we tested the outer membrane permeabilization abilities of these SAMPs against P. aeruginosa using an N-phenyl naphthalene (NPN) based fluorescence assay. Quantification of NPN fluorescence on the incubation of NPN-stained P. aeruginosa cells with SAMPs (8 μg mL−1) showed a sharp increase in fluorescence in the case of TAC6 and TAC8, whereas there was no significant change in NPN fluorescence on treatment with the other SAMPs (Fig. 1C). These results clearly showed that TAC6 and TAC8 can permeabilize the outer bacterial membranes, and can act as effective antibiotic adjuvants.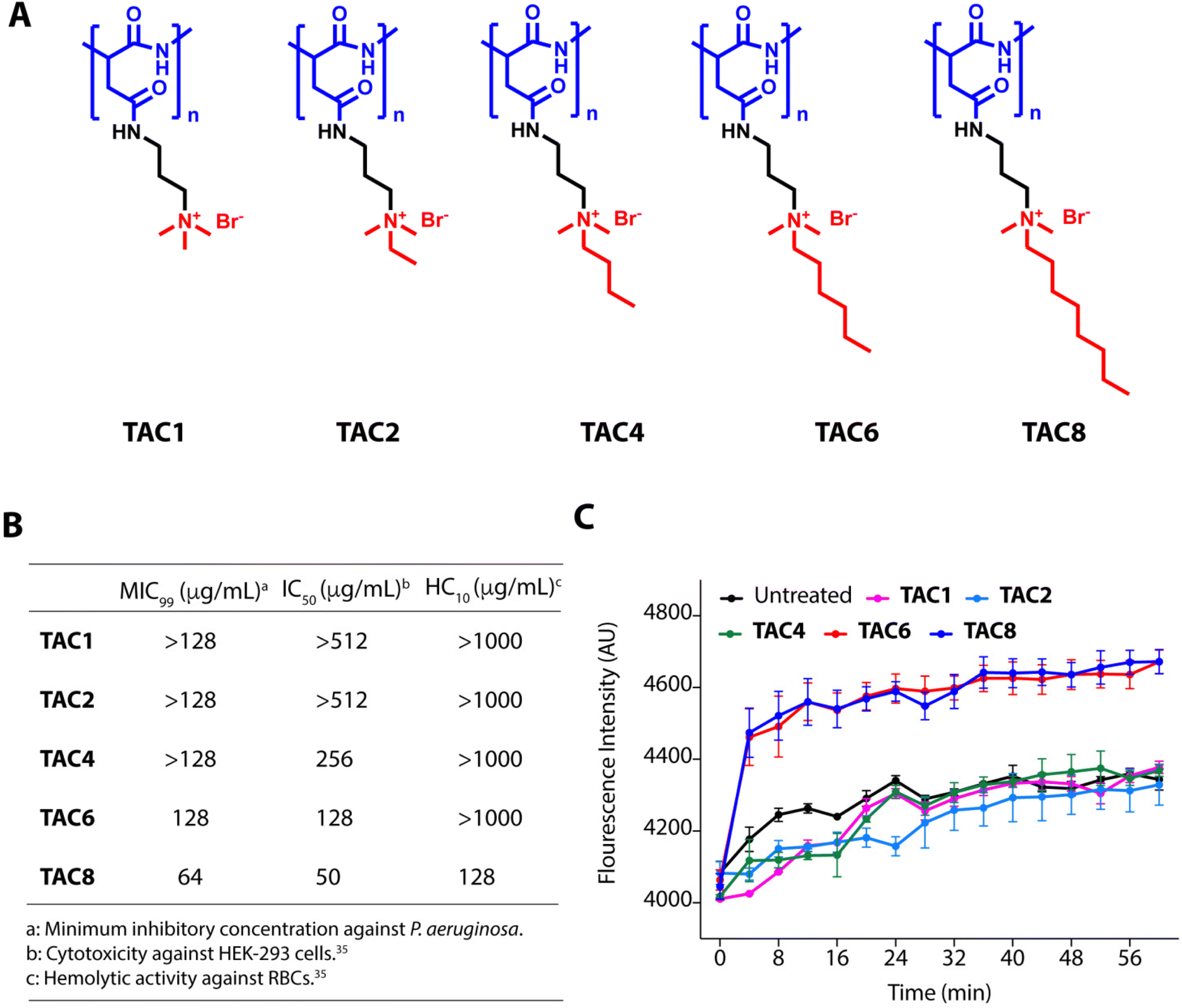 | ||
| Fig. 1 (A) Molecular structures of polyaspartic acid-derived SAMPs with varied hydrophobicity. (B) Minimum inhibitory concentrations (MIC99), cytotoxicity, and hemolytic activity of polyaspartic acid-derived SAMPs against P. aeruginosa, HEK-293 cells, and human RBCs respectively. (C) Change in NPN fluorescence on incubation of NPN-stained P. aeruginosa cells with polyaspartic acid-derived SAMPs. Data in (B) are the means of three independent replicates, and data in (C) are presented as the means ± SDs of three independent replicates. | ||
Rifampicin is a hydrophobic antibiotic that prevents RNA synthesis by the inhibition of DNA-dependent RNA polymerase.36 Rifampicin is widely used for the treatment of Mycobacterium infections in different combination therapeutic regimens owing to its highly hepatotoxic nature.37 Rifampicin is an effective antibiotic against Gram-positive cocci as well but is less effective against Gram-negative bacteria.38 Therefore, there is a need to develop strategies to enhance the antimicrobial activity of rifampicin against Gram-negative bacteria, especially P. aeruginosa. The hemolytic activities showed that TAC8 is highly hemolytic with an HC10 of 128 μg mL−1, whereas the other polymers are non-hemolytic even at 1000 μg mL−1 (Fig. 1B).35 Similarly, the cytotoxicity results against HEK-293 cells witnessed that the TAC8 polymer is highly toxic (Fig. 1B).35 Therefore, we pursued further studies with TAC6 to test its antibiotic adjuvant potential.
TAC6 acts as a potential adjuvant for rifampicin
To test the efficacy of TAC6 as an antibiotic adjuvant, we performed a chequerboard assay, and screened combinations of different concentrations of TAC6 and rifampicin for their synergistic antimicrobial activity against P. aeruginosa (MTCC1688). The synergy was assessed using the fractional inhibitory concentration (FIC) index where a FIC index value <0.5 indicates synergy. We observed that TAC6 and rifampicin alone exhibited MIC99 values of 128 and 8 μg mL−1 respectively against P. aeruginosa (Table 1). The screening of different combinations of TAC6 and rifampicin showed that a combination of TAC6 (8 μg mL−1) and rifampicin (1 μg mL−1) was sufficient to clear the microbial culture (Fig. 2A). To elucidate the synergy of TAC6 and rifampicin across different multidrug-resistant strains, we screened the combination of TAC6 and rifampicin against five different multidrug-resistant clinical strains of P. aeruginosa. We observed that TAC6 (8 μg mL−1) was able to enhance the antimicrobial activity of rifampicin against other multidrug-resistant clinical strains as well (Fig. 2B–F). The synergy curves show a decrease in the MIC99 of rifampicin on increasing the concentration of TAC6 against all P. aeruginosa strains (Fig. 3). We observed that 8 μg mL−1 of TAC6 was sufficient to decrease the MIC99 of rifampicin from 8 μg mL−1 to 1–2 μg mL−1 (Fig. 3). A comparison of the antimicrobial activity of the combination of TAC6 and rifampicin against different strains witnessed that TAC6 is an effective adjuvant against five of the six strains with FIC values of <0.5, and showed an additive effect in only one P. aeruginosa (6/264) strain (Table 1). We also observed 4–8 fold increase in the potentiation of rifampicin on using it in combination with TAC6 (Table 1). Therefore these results clearly demonstrated that TAC6 is a potential antibiotic adjuvant that enhances the activity of rifampicin against multidrug-resistant clinical P. aeruginosa strains.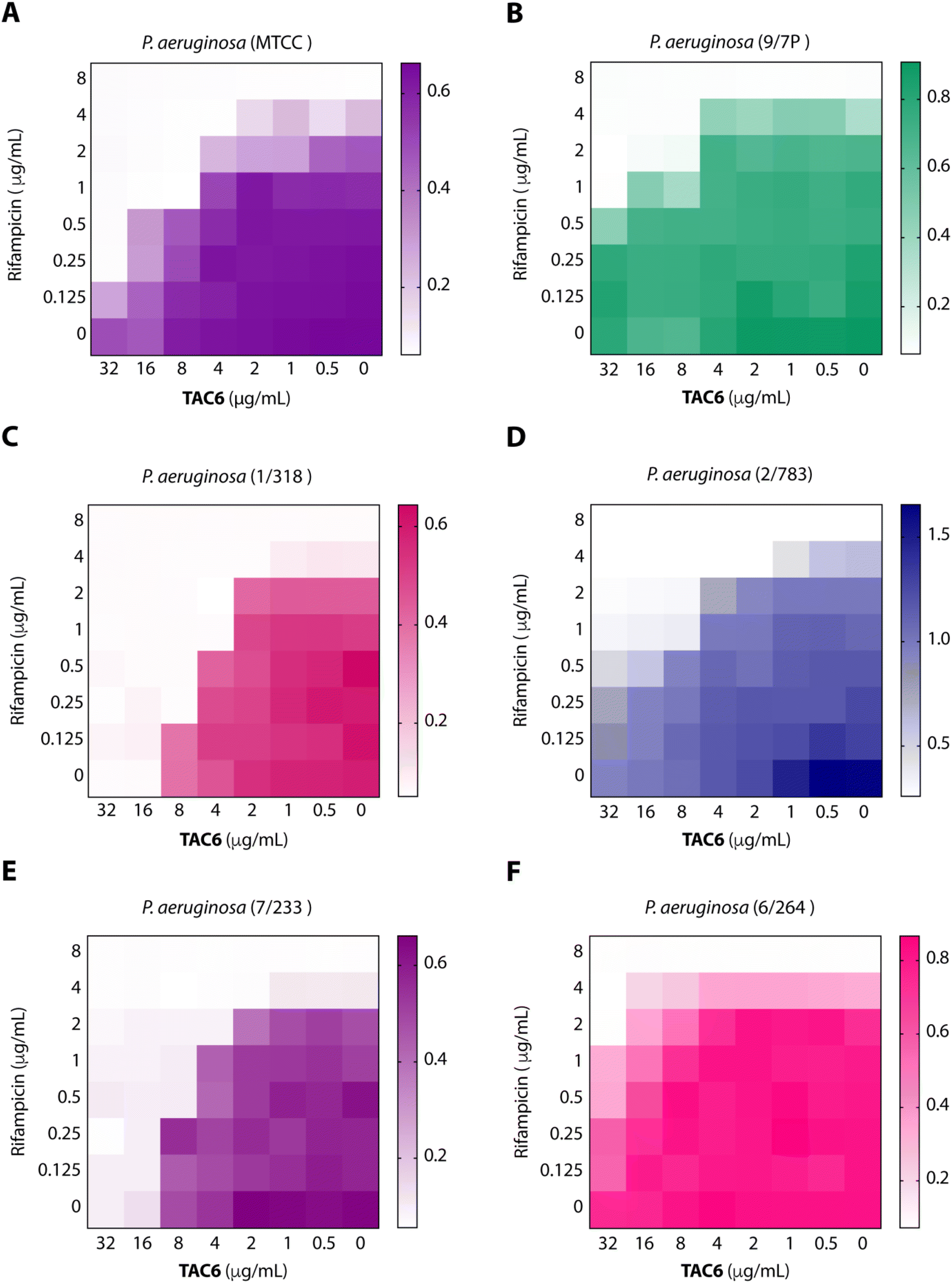 | ||
| Fig. 2 Heat maps (A–F) showing the effect of TAC6 on antimicrobial activity of rifampicin against different P. aeruginosa strains. | ||
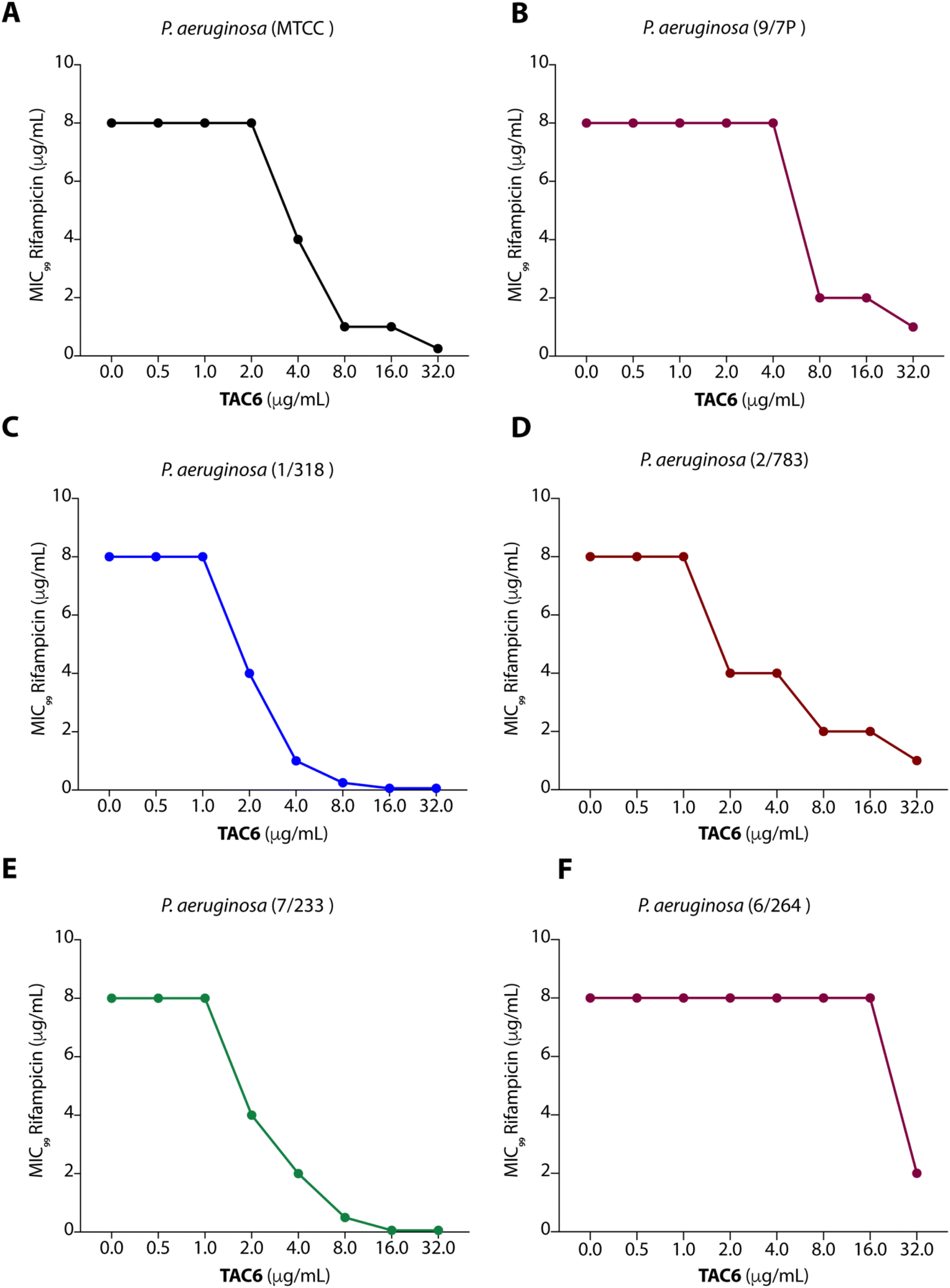 | ||
| Fig. 3 Synergy curves (A–F) showing the effect of TAC6 on the antimicrobial activity of rifampicin against different P. aeruginosa strains. Data are presented as the means of three independent replicates. | ||
| MIC99 (μg mL−1) | FIC index | Potentiation | ||||
|---|---|---|---|---|---|---|
| When used alone | When used in combination | |||||
| TAC6 | RIF | TAC6 | RIF | |||
| P. aeruginosa | 128 | 8 | 8 | 1 | 0.1875 | 8 |
| P. aeruginosa (9/7P) | 128 | 8 | 8 | 2 | 0.3125 | 4 |
| P. aeruginosa (1/318) | 128 | 8 | 4 | 1 | 0.15625 | 8 |
| P. aeruginosa (2/783) | 128 | 8 | 4 | 1 | 0.1875 | 8 |
| P. aeruginosa (7/233) | 128 | 8 | 4 | 2 | 0.28125 | 4 |
| P. aeruginosa (6/264) | 128 | 8 | 32 | 2 | 0.5 | 4 |
TAC6 permeabilizes the bacterial membrane for rifampicin
To evaluate the efficacy of a combination of TAC6 and rifampicin against bacterial growth, we performed time-dependent growth and killing assays. The growth kinetics of untreated bacterial cells evaluated by absorbance at 600 nm for 12 h showed a typical sigmoidal curve, and treatment with a combination of TAC6 (8 μg mL−1) and rifampicin (1 μg mL−1) witnessed no bacterial growth (Fig. 4A). In contrast, treatment with only TAC6 (8 μg mL−1) or only rifampicin (1 μg mL−1) did not show any significant decrease in bacterial growth (Fig. 4A). These findings were further substantiated by a 24 h time–kill assay, where bacterial cell death upon different treatments was evaluated by colony forming units at each time point. The results signified a complete abolition of the bacterial population within 8 h of treatment with a combination of TAC6 (8 μg mL−1) and rifampicin (1 μg mL−1) (Fig. 4B). Next, we used dye-based assays to clarify the hypothesis of outer membrane disruptions as a possible route for the potentiation of rifampicin activity. An NPN dye assay was performed for the scrutiny of membrane permeabilization upon treatment with a combination of TAC6 (8 μg mL−1) and rifampicin (1 μg mL−1). Significant increases in NPN fluorescence upon treatment with the combination therapy confirmed the successful permeabilization facilitating the entry of rifampicin into bacterial cells, thereby leading to bactericidal action (Fig. 4C).39 The permeabilization of the inner membrane was investigated using a membrane potential-sensitive dye, DiSC2(5) (3,3-diethylthiacarbocyanine iodide), that accumulates within the inner membrane and gets quenched.40 Treatment of TAC6 alone or in combination with rifampicin caused a sharp increase in fluorescence intensity (Fig. 4D). We also demonstrated the effect of combination therapy on membrane integrity using the propidium iodide (PI) uptake assay, and observed 40% more PI-positive cells using a combination of TAC6 (8 μg mL−1) and rifampicin (1 μg mL−1) compared with treatment with TAC6 alone (Fig. 4E). We validated the bactericidal action using a combination of TAC6 (8 μg mL−1) and rifampicin (1 μg mL−1) by quantifying the reactive oxidative species (ROS) using DCFH-DA (2′,7′-dichlorofluorescin diacetate). The ROS generated after cellular damage oxidise DCFH-DA to the fluorescent 2′,7′-dichlorofluorescein (DCF).41 We observed a ∼2-fold increase in fluorescence intensity upon treatment with a combination of TAC6 and RIF, confirming the bactericidal effect (Fig. 4F). Therefore, these results clearly demonstrate the membrane-permeabilizing ability of TAC6 that enhances the uptake of rifampicin by bacteria, causing bactericidal action.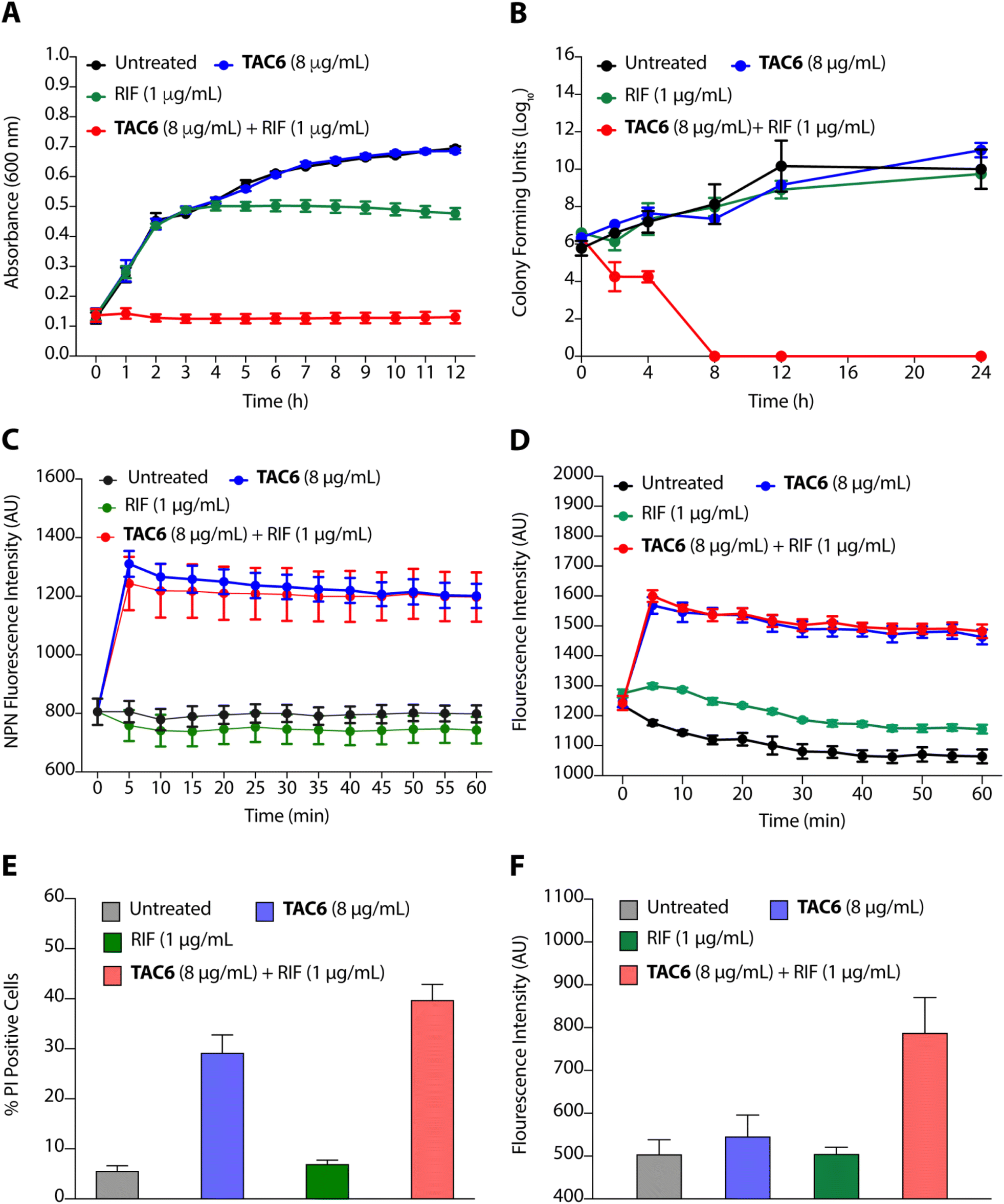 | ||
| Fig. 4 (A) Growth kinetic assay of P. aeruginosa in the presence of TAC6, rifampicin, and a combination of TAC6 and rifampicin. (B) Time–kill kinetic assay upon treatment of P. aeruginosa with TAC6, rifampicin, and a combination of TAC6 and rifampicin. (C and D) Change in NPN (C) and DiSC2(5) (D) fluorescence upon treatment of NPN- and DiSC2(5)-stained P. aeruginosa with TAC6, rifampicin, and a combination of TAC6 and rifampicin. (E) Percentage of PI-positive P. aeruginosa cells upon treatment of P. aeruginosa with TAC6, rifampicin, a and combination of TAC6 and rifampicin. RIF in the figure represents rifampicin. (F) Quantification of DCF fluorescence indicating the ROS levels upon treatment of P. aeruginosa with TAC6, rifampicin, and a combination of TAC6 and rifampicin. Data in above figures are presented as means ± SDs of three independent replicates. | ||
Lipopolysaccharides of outer bacterial membranes are a potential target for TAC6
The outer leaflet of the outer membrane of Gram-negative bacteria consists of a lipopolysaccharide layer (LPSs) comprising lipid A with cross-linkages of divalent cations like Mg2+ and Ca2+.42 As LPSs are the main cause of intrinsic resistance developed by Gram-negative bacteria, we deciphered the effect of LPSs (50 μg mL−1) on the antimicrobial activity of a combination of TAC6 and rifampicin against P. aeruginosa. We observed that the incubation of LPSs inhibits the synergistic antimicrobial effect of a combination of TAC6 and rifampicin as bacteria need 16 μg mL−1 (an 8-fold increase) of rifampicin in combination with 2 μg mL−1 of TAC6 in the presence of LPSs (Fig. 5A). Quantification of PI-positive cells treated with a combination of TAC6 and rifampicin further witnessed a >8-fold decrease in the percentage of PI-positive cells in the presence of LPS (Fig. 5B). Therefore, these results suggest that LPSs interact with TAC6 and inhibit the membrane permeabilization.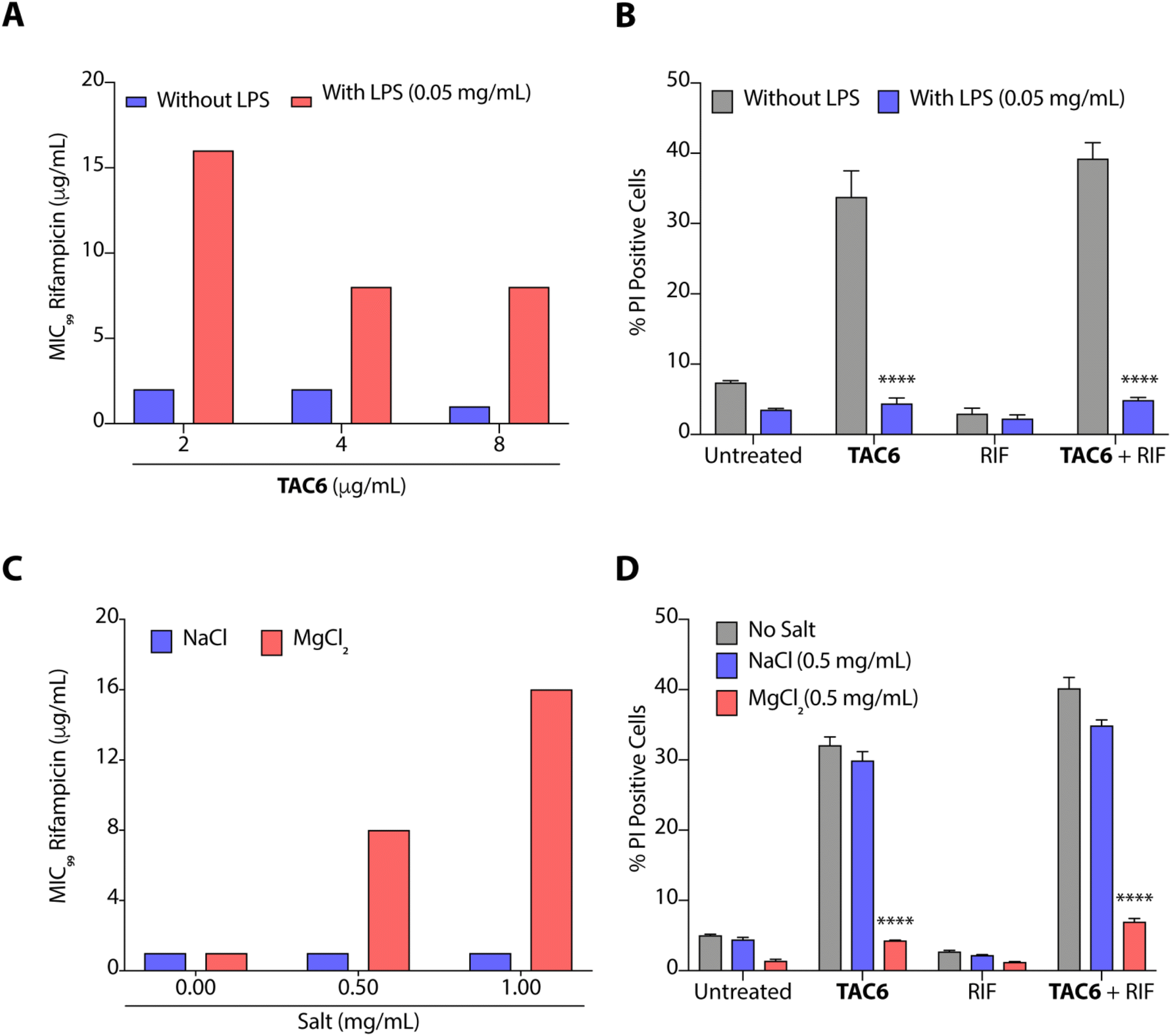 | ||
| Fig. 5 (A) Change in MIC99 of rifampicin in the absence and presence of LPSs upon using it in combination with TAC6 against P. aeruginosa. (B) Change in % PI-positive P. aeruginosa cells upon treatment with TAC6 (8 μg mL−1), rifampicin (1 μg mL−1) or a combination of TAC6 (8 μg mL−1) and rifampicin (1 μg mL−1) in the absence and presence of LPSs. (C) Change in the MIC99 of rifampicin in the absence and presence of Na+ and Mg2+ ions upon using it in combination with TAC6 against P. aeruginosa. (D) Change in % PI-positive P. aeruginosa cells upon treatment with TAC6 (8 μg mL−1), rifampicin (1 μg mL−1) or a combination of TAC6 (8 μg mL−1) and rifampicin (1 μg mL−1) in the absence and presence of Na+ and Mg2+ ions. Data in (A) and (C) are the means of three independent replicates, and data in (B) and (D) are presented as the means ± SD of three independent replicates. | ||
We also tested for the effect of monovalent (Na+) and divalent (Mg2+) cations on the synergistic activity of TAC6 and rifampicin against P. aeruginosa. Monovalent (Na+) cations induced no significant change in the activity of a combination of TAC6 and rifampicin, whereas divalent (Mg2+) cations inhibited the synergy as we observed an 8-fold increase in the MIC99 of rifampicin in the presence of divalent (Mg2+) cations (Fig. 5C). The PI uptake assay further demonstrated that the presence of divalent (Mg2+) cations inhibits the TAC6-mediated uptake of PI by P. aeruginosa cells (Fig. 5D). Therefore, these results clearly confirm that TAC6 interacts with the LPSs of Gram-negative bacteria, displaces the Mg2+ ions, and permeabilizes the bacterial membranes. This TAC6-mediated permeabilization of bacteria allows the influx of rifampicin, and is responsible for the synergistic therapeutic effect of the combination of TAC6 and rifampicin.
The combination of TAC6 and rifampicin can clear bacterial biofilms
Bacterial biofilms are assemblies of bacterial cells within a polysaccharide matrix, and pose a serious challenge for treatment due to the decreased penetration of antibiotics into the matrix.43 A significant effect of biofilm formation is experienced in clinical settings where medical devices and other surfaces provide a great opportunity for biofilm formation, presenting a cause of nosocomial infections.43 Therefore, we quantified the biofilm eradication on using the combination of TAC6 with rifampicin. We assessed the time-dependent eradication of P. aeruginosa biofilms upon treatment of biofilms with only TAC6, only rifampicin, and using a combination of TAC6 and rifampicin. The quantification of colony forming units at various time points post treatment witnessed a time-dependent decrease (Fig. 6A). We observed a ∼3-log reduction after 8 and 12 h of treatment with a combination of TAC6 and rifampicin as compared with untreated biofilms (Fig. 6A). The quantification of CFUs after 12 h of treatment showed ∼1-log decrease on treatment with TAC6 or rifampicin alone as compared with untreated biofilms, and no effect as compared with the initial biofilm load (Fig. 6A). We observed that a combination of TAC6 and rifampicin caused a >4-log decrease in CFUs as compared with untreated biofilms after 12 h of treatment (Fig. 6A and B).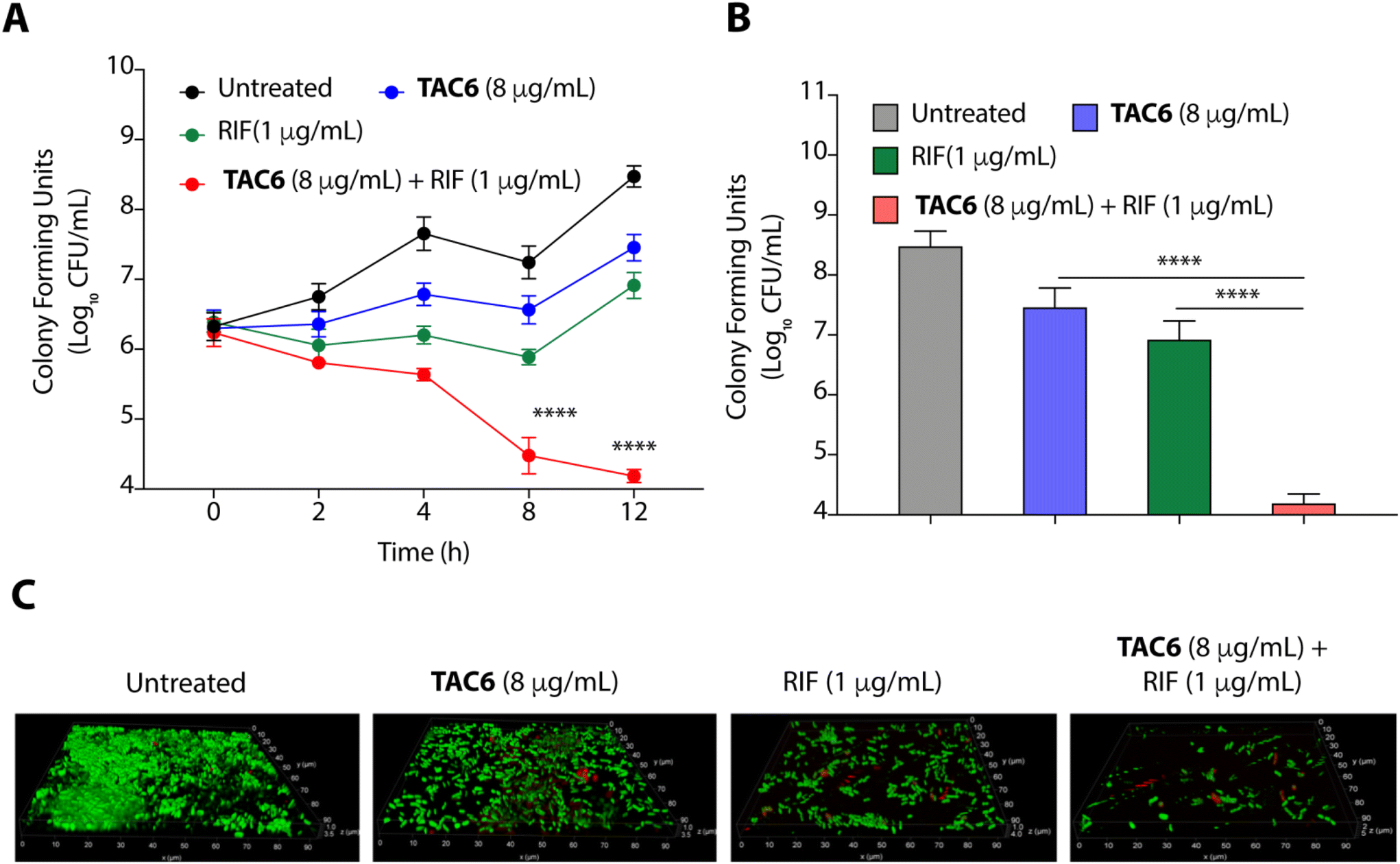 | ||
| Fig. 6 (A) Time-dependent change in CFUs of preformed P. aeruginosa biofilms on treatment with TAC6, rifampicin, and a combination of TAC6 and rifampicin witnesses the synergistic bactericidal effect on biofilms. (B) Change in CFUs after 12 h of treatment of P. aeruginosa biofilms with TAC6, rifampicin, and a combination of TAC6 and rifampicin confirms the anti-biofilm effect of combination therapy. (C) Fluorescence micrographs of SYTO9/PI-stained untreated and treated P. aeruginosa biofilms demonstrate the bactericidal effect. Data in (A) and (B) are presented as means ± SD of three independent replicates. | ||
The biofilm degradation properties of combination of TAC6 and rifampicin were further validated by staining of the untreated and treated P. aeruginosa biofilms upon SYTO9/PI staining.44 Untreated biofilms showed a thick mass of biofilms having live SYTO9-stained cells (Fig. 6C). Fluorescence micrographs of treated biofilms witnessed that a combination of TAC6 and rifampicin can degrade the pre-formed P. aeruginosa biofilms successfully (Fig. 6C). Therefore, these results demonstrate that the combination therapy of TAC6 and rifampicin is an effective strategy for clearing biofilms.
The combination of TAC6 and rifampicin can clear wound infections
P. aeruginosa is one of the key pathogens responsible for wound infections along with S. aureus.45,46 Therefore, we tested the effect of the combination of TAC6 and rifampicin in a wound infection model using P. aeruginosa in BALB/c mice. Circular skin wounds on BALB/c male mice were infected with P. aeruginosa for 24 h, and infected mice were randomized into four groups. Among the different groups, group 1 mice were left untreated, and a second group of mice were treated with TAC6 (10 mg kg−1) three times a day for three days. The mice in the third group were treated with rifampicin (5 mg kg−1) for three days, and group 4 mice were treated with a combination of TAC6 (10 mg kg−1) and rifampicin (5 mg kg−1) for three days (Fig. 7A). On the fourth day, the wound sites were homogenised with a post-surgical incision and scrutinised for colony forming units by plating on LB agar plates. Quantification of CFUs showed a >2-log fold reduction on treatment with the combination of TAC6 and rifampicin as compared with the control group and rifampicin treatment (Fig. 7B). Of note, the combination treatment of rifampicin (5 mg kg−1) and TAC6 (10 mg kg−1) showed potent synergistic activity, while rifampicin alone, which is able to clear the microbes at 8 μg mL−1 (1× MIC) in an in vitro setting, fails in animal studies. This could be due to the tolerance of P. aeruginosa to rifampicin at this concentration. Moreover, TAC6 (10 mg kg−1) shows minor reductions in infection, suggesting that TAC6 is sensitizing the bacterial cells. These results clearly validate the efficacy of the combination treatment against P. aeruginosa in wound infections.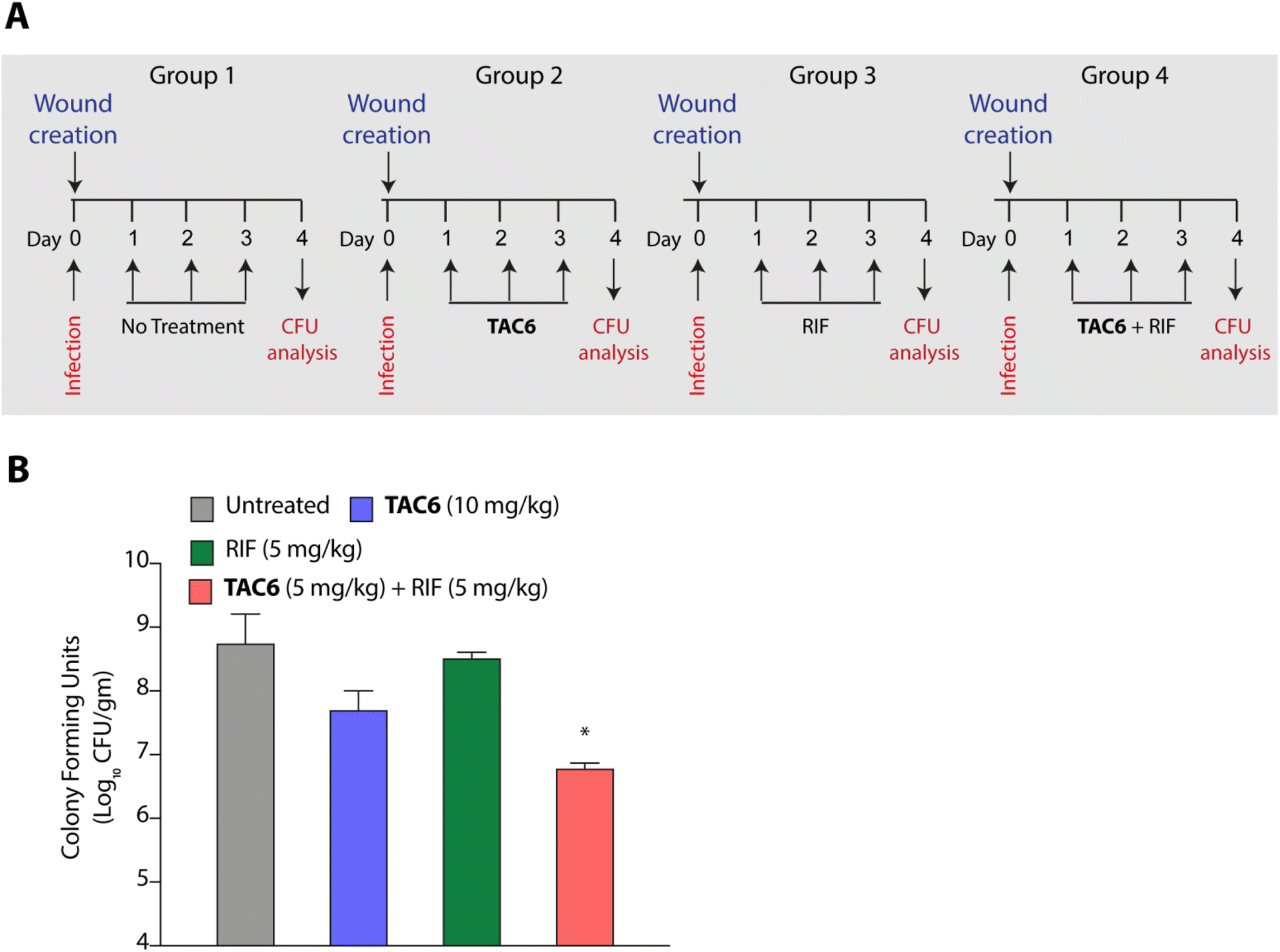 | ||
| Fig. 7 (A) Schematic showing the experimental plan to test the efficacy of a combination of TAC6 and rifampicin against P. aeruginosa wound infections. (B) Quantification of bacterial burden on different treatments. Data in (B) are presented as means ± SD (n = 4 animals per group). | ||
Conclusions
Rifampicin hampers the DNA-dependent RNA polymerase activity of most bacterial genera including mycobacteria and Gram-positive bacteria. The efficacy of rifampicin is restricted against Gram-negative bacteria because of the impermeable LPS outer membrane. To overcome this coldness of rifampicin against Gram-negative bacteria and sensitize these bacterial cells towards rifampicin, various studies have used adjuvant molecules. In one study, the authors screened a library of short linear antibacterial peptides (SLAPs) in combination with different antibiotics like rifampicin, tetracycline, vancomycin, cefepime, and ofloxacin using a chequerboard assay, and disclosed that MDR E. coli B2, which has 25 antibiotic-resistant genes, was effectively killed by a synergistic combination of SLAP-S25 and antibiotics at sub-MIC.21 Our study also showed that rifampicin with TAC6 molecule efficiently inhibits multidrug-resistant P. aeruginosa cells. The membrane disrupting effects of the adjuvant TAC6 are not limited to the outer membrane but TAC6 also depolarizes the inner bacterial membrane without causing any cell death. An anti-protozoal drug, pentamidine, also perturbs the Gram-negative outer membrane through its interactions with LPSs, and allows the entry of hydrophobic drugs like rifampicin and colistin.22 As addition of exogenous LPSs and Mg2+ ions abolishes the TAC6-dependent potentiation of rifampicin, these results suggest that the TAC6-like pentamidine targets LPSs and competes with Mg2+ ions to disrupt the tight joint between the two side-by-side lipid chains of LPSs, and increases the permeability of outer membrane.47 In another study, a lysine-based small molecule has been shown to cause a synergistic antimicrobial effect with rifampicin and tetracycline in mitigating Gram-negative bacterial cells and fully grown biofilms, and to remain active under in vivo conditions.13 Similarly, our results confirmed that a combination of TAC6 and rifampicin can cause the degradation of fully grown biofilms and clear wound infections. Therefore, this study reveals the antibiotic adjuvant effect of polyaspartate-derived polymers that can enhance the activity of rifampicin against multidrug-resistant Gram-negative P. aeruginosa bacteria (Fig. 8), and will help in the design of future effective antibiotic adjuvants.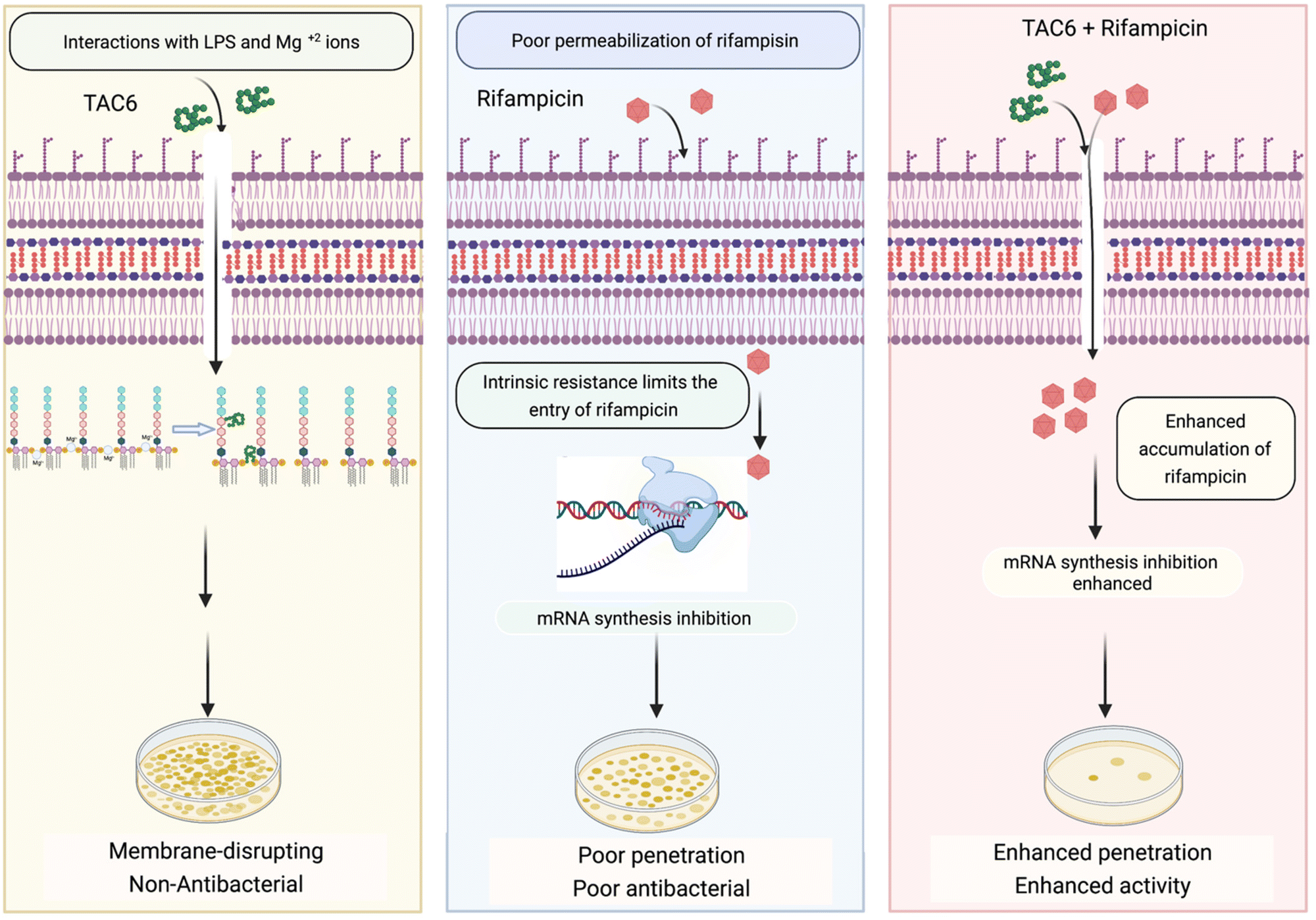 | ||
| Fig. 8 Schematic showing the ability of TAC6 to enhance the therapeutic efficacy of rifampicin and enhance its permeation through bacterial membranes. | ||
Experimental section
Materials
The tryptone soya broth (TSB) (Cat # M011), LB broth (Cat # M1245), rifampicin (Cat # CMS1889) and bacteriological agar (Cat # GRM026) used for culturing different strains were purchased from HiMedia, India. Ciprofloxacin (Cat # 17850), propidium iodide (Cat # P1470), lipopolysaccharides from Escherichia coli O111:B4 (Cat # 1_4130), 3,3-diethylthiadicarbocyanine iodide (Cat # 17375), magnesium chloride (Cat # M8266), sodium chloride (Cat # 106404), and 2′,7′-dichlorofluorescein diacetate (Cat # 35845) were procured from Sigma-Aldrich, USA. The LIVE/DEAD® BacLightTM bacterial viability and counting kit was from Invitrogen, USA, and Pseudomonas aeruginosa (MTCC1688) were purchased from the MTCC (Chandigarh, India).To check the effect of LPSs on synergy, the TAC6 solution was incubated with LPSs (50 μg mL−1) for 30 min before dilution in a 96-well plate along the abscissa. Similarly, to check the effect of monovalent and divalent cations, bacterial suspension cells were incubated with 0.5 mg mL−1 of salts before dilution along the ordinate.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 2 min. The supernatant was transferred to fresh tubes, and treated with 10 μM of DCFH-DA (dichlorofluorescin diacetate) for 1 h at 37 °C. ROS production was analyzed by measuring the fluorescence with an excitation/emission wavelength of 485 nm/535 nm using a Spectramax M5 multimode microplate reader (Molecular Devices, Sunnyvale, CA, USA).
:1 mixture of SYTO-9 and PI from the LIVE/DEAD Baclight bacterial viability and counting kit. The biofilm images were then obtained under a confocal microscope, Leica TCS SP8, Germany.
000 rpm for 2 min. The supernatant was transferred to fresh tubes, and treated with 10 μM of DCFH-DA (dichlorofluorescin diacetate) for 1 h at 37 °C. ROS production was analyzed by measuring the fluorescence with an excitation/emission wavelength of 485 nm/535 nm using a Spectramax M5 multimode microplate reader (Molecular Devices, Sunnyvale, CA, USA).
:1 mixture of SYTO-9 and PI from the LIVE/DEAD Baclight bacterial viability and counting kit. The biofilm images were then obtained under a confocal microscope, Leica TCS SP8, Germany.
Ethical statement
Animal experimental protocols were reviewed and approved by the Institutional Animal Ethics Committee of the Regional Centre for Biotechnology (RCB/IAEC/2019/054). The experiments were carried out as per the guidelines issued by the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Govt of India.Author contributions
NC and BA performed all in vitro and animal experiments. VS supervised the bacterial experiments. PSY and AS provided the compounds. AB conceived the idea and supervised the project.Conflicts of interest
Authors declare no competing financial interest.Acknowledgements
We thank RCB for intramural funding, and the Department of Biotechnology (DBT) Govt of India for supporting the work in AB's laboratory. The students thank the funding agencies CSIR, UGC, and RCB for the research fellowship. The animal work in the small animal facility of the Regional Centre for Biotechnology is supported by BT/PR5480/INF/22/158/2012 (DBT). We acknowledge the support of the DBT e-Library Consortium (DeLCON) for providing access to e-resources. Fig. 8 and the TOC figure were prepared using BioRender.com.Notes and references
- J. P. Horcajada, M. Montero, A. Oliver, L. Sorlí, S. Luque, S. Gómez-Zorrilla, N. Benito and S. Grau, Epidemiology and treatment of multidrug-resistant and extensively drug-resistant Pseudomonas aeruginosa infections, Clin. Microbiol. Rev., 2019, 32, 31–19 CrossRef PubMed.
- S. L. Gellatly and R. E. Hancock, Pseudomonas aeruginosa: new insights into pathogenesis and host defenses, Pathog. Dis., 2013, 67, 59–173 Search PubMed.
- R. Serra, R. Grande, L. Butrico, A. Rossi, U. F. Settimio, B. Caroleo, B. Amato, L. Gallelli and S. de Franciscis, Chronic wound infections: The role of Pseudomonas aeruginosa, and Staphylococcus aureus, Expert Rev. Anti-Infect. Ther., 2015, 13, 605–613 CrossRef CAS PubMed.
- K. H. Turner, J. Everett, U. Trivedi, K. P. Rumbaugh and M. Whiteley, Requirements for Pseudomonas aeruginosa acute burn and chronic surgical wound infection, PLoS Genet., 2014, 10, e1004518 CrossRef PubMed.
- N. M. Maurice, B. Bedi and R. T. Sadikot, Pseudomonas aeruginosa biofilms: Host response and clinical implications in lung infections, Am. J. Respir. Cell Mol. Biol., 2018, 58, 428–439 CrossRef CAS PubMed.
- P. K. Taylor, A. T. Yeung and R. E. Hancock, Antibiotic resistance in Pseudomonas aeruginosa biofilms: Towards the development of novel anti-biofilm therapies, J. Biotechnol., 2014, 191, 121–130 CrossRef CAS PubMed.
- S. Wagner, R. Sommer, S. Hinsberger, C. Lu, R. W. Hartmann, M. Empting and A. Titz, Novel strategies for the treatment of Pseudomonas aeruginosa infections, J. Med. Chem., 2016, 59, 5929–5969 CrossRef CAS PubMed.
- M. Bassetti, A. Vena, A. Croxatto, E. Righi and B. Guery, How to manage Pseudomonas aeruginosa infections, Drugs Context., 2018, 7, 212527 Search PubMed.
- Z. Breijyeh, B. Jubeh and R. Karaman, Resistance of Gram-negative bacteria to current antibacterial agents and approaches to resolve it, Molecules, 2020, 25, 1340 CrossRef CAS PubMed.
- H. Nikaido, Molecular basis of bacterial outer membrane permeability revisited, Microbiol. Mol. Biol. Rev., 2003, 67, 593–656 CrossRef CAS PubMed.
- B. W. Simpson and M. S. Trent, Pushing the envelope: LPS modifications and their consequences, Nat. Rev. Microbiol., 2019, 17, 403–416 CrossRef CAS PubMed.
- B. D. Needham and M. S. Trent, Fortifying the barrier: The impact of lipid A remodelling on bacterial pathogenesis, Nat. Rev. Microbiol., 2013, 11, 467–481 CrossRef CAS PubMed.
- M. M. Konai and J. Haldar, Lysine-based small molecule sensitizes rifampicin and tetracycline against multidrug-resistant Acinetobacter baumannii and Pseudomonas aeruginosa, ACS Infect. Dis., 2020, 6, 91–99 CrossRef CAS PubMed.
- V. B. Hubble, K. R. Bartholomew, A. W. Weig, S. M. Brackett, S. L. Barlock, A. E. Mattingly, A. M. Nemeth, R. J. Melander and C. Melander, Augmenting the activity of macrolide adjuvants against Acinetobacter baumannii, ACS Med. Chem. Lett., 2020, 11, 1723–1731 CrossRef CAS PubMed.
- M. Tyers and G. D. Wright, Drug combinations: A strategy to extend the life of antibiotics in the 21st century, Nat. Rev. Microbiol., 2019, 17, 141–155 CrossRef CAS PubMed.
- R. J. Worthington and C. Melander, Combination approaches to combat multidrug-resistant bacteria, Trends Biotechnol., 2013, 31, 177–184 CrossRef CAS PubMed.
- E. E. Gill, O. L. Franco and R. E. Hancock, Antibiotic adjuvants: diverse strategies for controlling drug–resistant pathogens, Chem. Biol. Drug Des., 2015, 85, 56–78 CrossRef CAS PubMed.
- H. Douafer, V. Andrieu, O. Phanstiel IV and J. M. Brunel, Antibiotic adjuvants: Make antibiotics great again, J. Med. Chem., 2019, 62, 8665–8681 CrossRef CAS PubMed.
- G. D. Wright, Antibiotic adjuvants: Rescuing antibiotics from resistance, Trends Microbiol., 2016, 24, 862–871 CrossRef CAS PubMed.
- Y. Liu, R. Li, X. Xiao and Z. Wang, Antibiotic adjuvants: An alternative approach to overcome multi-drug resistant Gram-negative bacteria, Crit. Rev. Microbiol., 2019, 45, 301–314 CrossRef CAS PubMed.
- M. Song, Y. Liu, X. Huang, S. Ding, Y. Wang, J. Shen and K. Zhu, A broad-spectrum antibiotic adjuvant reverses multidrug-resistant Gram-negative pathogens, Nat. Microbiol., 2020, 5, 1040–1050 CrossRef CAS PubMed.
- J. M. Stokes, C. R. MacNair, B. Ilyas, S. French, J. P. Côté, C. Bouwman, M. A. Farha, A. O. Sieron, C. Whitfield, B. K. Coombes and E. D. Brown, Pentamidine sensitizes Gram-negative pathogens to antibiotics and overcomes acquired colistin resistance, Nat. Microbiol., 2017, 2, 17028 CrossRef PubMed.
- R. E. Hancock and G. Diamond, The role of cationic antimicrobial peptides in innate host defences, Trends Microbiol., 2000, 8, 402–410 CrossRef CAS PubMed.
- K. A. Brogden, Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria?, Nat. Rev. Microbiol., 2005, 3, 238–250 CrossRef CAS PubMed.
- F. Nederberg, Y. Zhang, J. P. Tan, K. Xu, H. Wang, C. Yang, S. Gao, X. D. Guo, K. Fukushima, L. Li and J. L. Hedrick, Biodegradable nanostructures with selective lysis of microbial membranes, Nat. Chem., 2011, 3, 409–414 CrossRef CAS PubMed.
- C. Tantisuwanno, F. Dang, K. Bender, J. D. Spencer, M. E. Jennings, H. A. Barton and A. Joy, Synergism between rifampicin and cationic polyurethanes overcomes intrinsic resistance of Escherichia coli, Biomacromolecules, 2021, 22, 2910–2920 CrossRef CAS PubMed.
- Z. Si, H. W. Lim, M. Y. Tay, Y. Du, L. Ruan, H. Qiu, R. Zamudio-Vazquez, S. Reghu, Y. Chen, W. S. Tiong and K. Marimuthu, A glycosylated cationic block poly (β-peptide) reverses intrinsic antibiotic resistance in all ESKAPE Gram-negative bacteria, Angew. Chem., Int. Ed., 2020, 59, 6819–6826 CrossRef CAS PubMed.
- S. Barman, S. Mukherjee, S. Ghosh and J. Haldar, Amino-acid-conjugated polymer–rifampicin combination: Effective at tackling drug-resistant Gram-negative clinical isolates, ACS Appl. Bio Mater., 2019, 2, 5404–5414 CrossRef CAS PubMed.
- L. Zhai, Z. Zhang, Y. Zhao and Y. Tang, Efficient antibacterial performance and effect of structure on property based on cationic conjugated polymers, Macromolecules, 2018, 51, 7239–7247 CrossRef CAS.
- S. Zhao, W. Huang, C. Wang, Y. Wang, Y. Zhang, Z. Ye, J. Zhang, L. Deng and A. Dong, Screening and matching amphiphilic cationic polymers for efficient antibiosis, Biomacromolecules, 2020, 21, 5269–5281 CrossRef CAS PubMed.
- H. Adelnia, H. D. Tran, P. J. Little, I. Blakey and H. T. Ta, Poly (aspartic acid) in biomedical applications: From polymerization, modification, properties, degradation, and biocompatibility to applications, ACS Biomater. Sci. Eng., 2021, 7, 2083–2105 CrossRef CAS PubMed.
- P. S. Yavvari, S. Gupta, D. Arora, V. K. Nandicoori, A. Srivastava and A. Bajaj, Clathrin-independent killing of intracellular mycobacteria and biofilm disruptions using synthetic antimicrobial polymers, Biomacromolecules, 2017, 18, 2024–2033 CrossRef CAS PubMed.
- B. D. Quan, M. Wojtas and E. D. Sone, Polyaminoacids in biomimetic collagen mineralization: Roles of isomerization and disorder in polyaspartic and polyglutamic acids, Biomacromolecules, 2021, 22, 2996–3004 CrossRef CAS PubMed.
- S. Zakharchenko, E. Sperling and L. Ionov, Fully biodegradable self-rolled polymer tubes: A candidate for tissue engineering scaffolds, Biomacromolecules, 2011, 12, 2211–2215 CrossRef CAS PubMed.
- P. S. Yavvari, A. K. Awasthi, A. Sharma, A. Bajaj and A. Srivastava, Emerging biomedical applications of polyaspartic acid-derived biodegradable polyelectrolytes and polyelectrolyte complexes, J. Mater. Chem. B, 2019, 7, 2102–2122 RSC.
- K. Richardson, O. T. Bennion, S. Tan, A. N. Hoang, M. Cokol and B. B. Aldridge, Temporal and intrinsic factors of rifampicin tolerance in mycobacteria, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8302–8307 CrossRef CAS PubMed.
- M. Toosky and B. Javid, Novel diagnostics and therapeutics for drug-resistant tuberculosis, Br. Med. Bull., 2014, 110, 129–140 CrossRef PubMed.
- G. L. Mandell, The antimicrobial activity of rifampin: emphasis on the relation to phagocytes, Rev. Infect. Dis., 1983, 5, S463–S467 CrossRef CAS PubMed.
- I. M. Helander and T. Mattila-Sandholm, Fluorometric assessment of Gram-negative bacterial permeabilization, J. Appl. Microbiol., 2000, 88, 213–219 CrossRef CAS PubMed.
- J. D. Te Winkel, D. A. Gray, K. H. Seistrup, L. W. Hamoen and H. Strahl, Analysis of antimicrobial-triggered membrane depolarization using voltage sensitive dyes, Front. Cell Dev. Biol., 2016, 29 Search PubMed.
- E. Eruslanov and S. Kusmartsev, Identification of ROS using oxidized DCFDA and flow-cytometry, in Advanced protocols in oxidative stress II, 2010, vol. 594, pp. 57–72 Search PubMed.
- L. A. Clifton, M. W. Skoda, A. P. Le Brun, F. Ciesielski, I. Kuzmenko, S. A. Holt and J. H. Lakey, Effect of divalent cation removal on the structure of Gram-negative bacterial outer membrane models, Langmuir, 2015, 31, 404–412 CrossRef CAS PubMed.
- D. Sharma, L. Misba and A. U. Khan, Antibiotics versus biofilm: an emerging battleground in microbial communities, Antimicrob. Resist. Infect. Control, 2019, 8, 1–10 CrossRef PubMed.
- S. Gupta, J. Thakur, S. Pal, R. Gupta, D. Mishra, S. Kumar, K. Yadav, A. Saini, P. S. Yavvari, M. Vedantham and A. Singh, Cholic acid–peptide conjugates as potent antimicrobials against interkingdom polymicrobial biofilms, Antimicrob. Agents Chemother., 2019, 63, e00520–e00519 CrossRef CAS PubMed.
- B. A. Lipsky and C. Hoey, Topical antimicrobial therapy for treating chronic wounds, Clin. Infect. Dis., 2009, 49, 1541–1549 CrossRef PubMed.
- R. S. Howell-Jones, M. J. Wilson, K. E. Hill, A. J. Howard, P. E. Price and D. W. Thomas, A review of the microbiology, antibiotic usage and resistance in chronic skin wounds, J. Antimicrob. Chemother., 2005, 55, 143–149 CrossRef CAS PubMed.
- R. J. Worthington, C. A. Bunders, C. S. Reed and C. Melander, Small molecular suppression of carbapenem resistance in NDM-1 producing Klebsiella pneumoniae, ACS Med. Chem. Lett., 2012, 3, 357–361 CrossRef CAS PubMed.
- J. H. Jorgensen, J. F. Hindler, L. B. Reller and M. P. Weinstein, New consensus guidelines from the Clinical and Laboratory Standards Institute for antimicrobial susceptibility testing of infrequently isolated or fastidious bacteria, Clin. Infect. Dis., 2007, 44, 280–286 CrossRef PubMed.
- G. Orhan, A. Bayram, Y. Zer and I. Balci, Synergy tests by E test and checkerboard methods of antimicrobial combinations against Brucella melitensis, J. Clin. Microbiol., 2005, 43, 140–143 CrossRef CAS PubMed.
- K. Yadav, S. Kumar, D. Mishra, M. Asad, M. Mitra, P. S. Yavvari, S. Gupta, M. Vedantham, P. Ranga, V. Komalla, S. Pal, P. Sharma, A. Kapil, A. Singh, N. Singh, A. Srivastava, L. Thukral and A. Bajaj, Deciphering the role of intramolecular networking in cholic acid–peptide conjugates (CAPS) at lipopolysaccharide surface in combating Gram-negative bacterial infections, J. Med. Chem., 2019, 62, 1875–1886 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |