
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Pre-clinical non-viral vectors exploited for in vivo CRISPR/Cas9 gene editing: an overview
Nadia
Rouatbi
,
Tasneem
McGlynn
and
Khuloud T.
Al-Jamal
 *
*
Institute of Pharmaceutical Science, Faculty of Life Sciences & Medicine, King's College London, Franklin-Wilkins Building, 150 Stamford Street, London SE1 9NH, UK. E-mail: khuloud.al-jamal@kcl.ac.uk
First published on 23rd May 2022
Abstract
Clustered regulatory interspaced short palindromic repeats or CRISPR/Cas9 has emerged as a potent and versatile tool for efficient genome editing. This technology has been exploited for several applications including disease modelling, cell therapy, diagnosis, and treatment of many diseases including cancer. The in vivo application of CRISPR/Cas9 is hindered by poor stability, pharmacokinetic profile, and the limited ability of the CRISPR payloads to cross biological barriers. Although viral vectors have been implemented as delivery tools for efficient in vivo gene editing, their application is associated with high immunogenicity and toxicity, limiting their clinical translation. Hence, there is a need to explore new delivery methods that can guarantee safe and efficient delivery of the CRISPR/Cas9 components to target cells. In this review, we first provide a brief history and principles of nuclease-mediated gene editing, we then focus on the different CRISPR/Cas9 formats outlining their potentials and limitations. Finally, we discuss the alternative non-viral delivery strategies currently adopted for in vivo CRISPR/Cas9 gene editing.
1. Introduction
In the past decades, great breakthroughs have been made in the development of molecular therapies to treat genetic disorders, neuropathies, cancer, and viral infections. These therapeutic approaches utilise molecular probes (i.e., small interfering RNAs (siRNAs), antisense oligonucleotides (ASOs), and messenger RNAs (mRNAs)) acting at the genetic level to delete, replace, or alter the expression of specific genes, with the final aim of offering a more efficient and durable treatment.1 With the US Food and Drug Administration (FDA) approval of Mipomersen sodium (Kynamro™) (2013),2 a second-generation ASO based therapy for the treatment of Transthyretin amyloidosis (ATTR), substantial efforts have been made towards the designing of vectors that can improve the bioavailability of these molecular probes and promote their clinical translation. This, in turn, stimulated the development of different genetic medicines that are currently being used in the clinic3,4 including the most recently approved vaccines5–8 against COVID-19.Recently, considerable attention has shifted towards a new generation of molecular probes characterized by site-specific nucleases capable of performing precise gene editing. Among these machineries, the first to be discovered were Zinc Finger Nucleases (ZFNs)9 followed by Transcription Activator-Like Effector Nucleases (TALEN),10 and more recently the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) and its CRISPR associated Cas protein (CRISPR/Cas) (Fig. 1).11
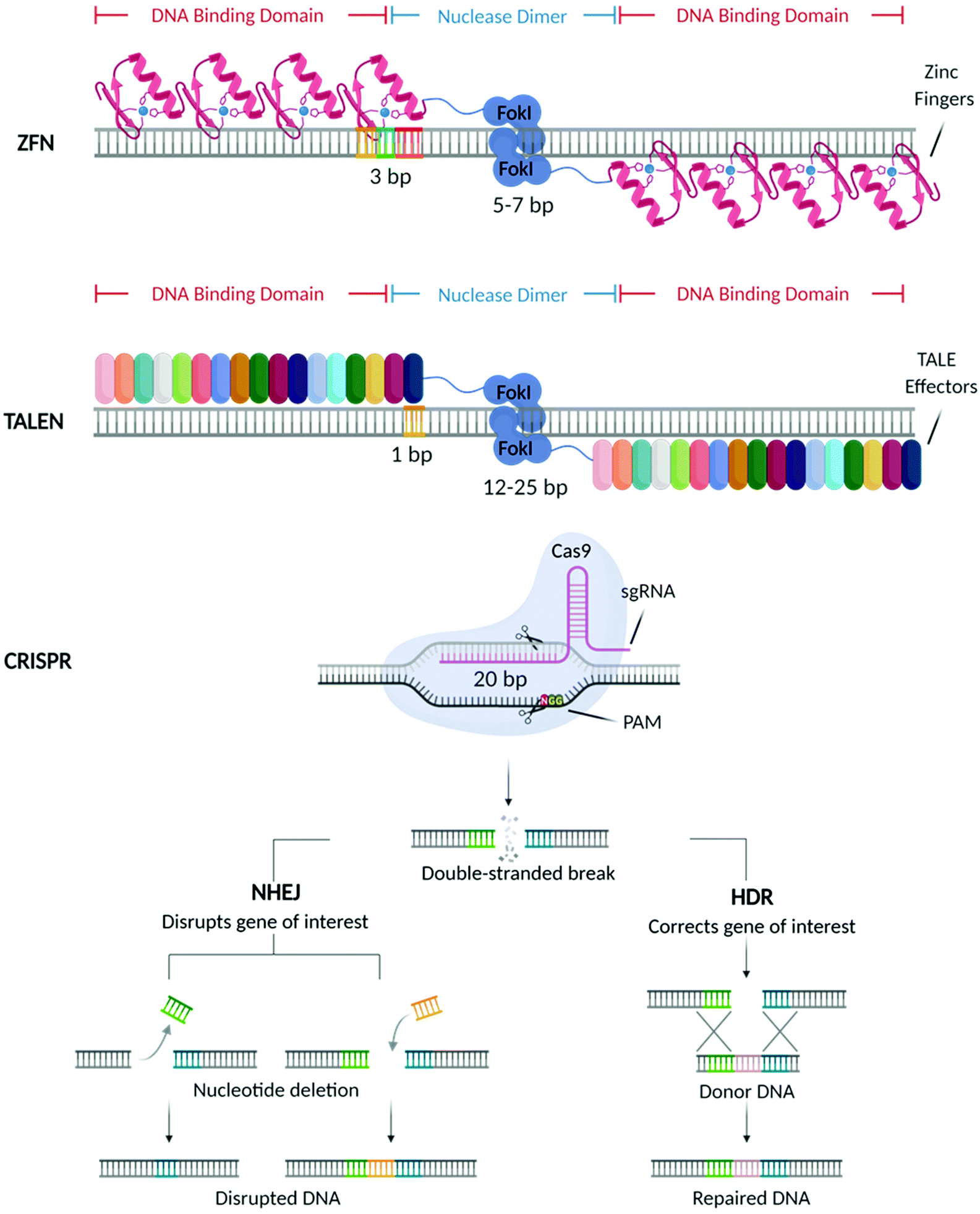 | ||
| Fig. 1 Schematic diagram of nuclease-driven gene editing technologies. Zinc Finger Nucleases (ZFN) were the first nuclease-driven gene editing technology to be discovered, followed by Transcription Activator-Like Effector Nucleases (TALEN), and most recently by Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR). The three types of nucleases can induce double-strand breaks that are repaired by either one of two DNA repair processes: non-homologus end joining (NHEJ) or homology-directed repair (HDR). The first, NHEJ, is an error prone mechanism that causes insertions and deletions (indels) which results in target gene disruption. HDR, is a more accurate process, by which, in the presence of a donor DNA, precise DNA modifications can be introduced to repair a certain gene. Scheme adapted from Karim Shalaby and Esmée Dragt by BioRender.com (2021). Retrieved from https://app.biorender.com/biorender-templates. | ||
As shown in Fig. 1, mechanistically, these nucleases create double-strand breaks (DSBs) in the locus of interest, which are subsequently repaired by host cellular machinery via either non-homologous end-joining (NHEJ) or homology-directed repair (HDR),12 resulting in the deletion, correction and modification of the target gene.
From an early stage, CRISPR/Cas has quickly gained momentum as a versatile tool for relatively easy and fast gene editing in a multitude of cell types,12,13 and organisms.14,15 Its application has been exploited for different purposes, including the generation of genetically engineered mouse models,16 identifying genes underlying drug resistance17 and providing a better understanding of cancer biology.18 Furthermore, there has been great enthusiasm in developing the CRISPR/Cas toolbox for use in clinical medicine, as a potential screening and therapeutic tool. While its application in vitro is relatively easy, its use in humans comes with multiple hurdles, which may include potential off-target effects, instability in the bloodstream and immunogenicity.19 To mitigate these limitations, many different strategies have been employed. One such method is to bypass these obstacles entirely and perform ex vivo gene editing, whereby autologous or allogenic cells are edited in vitro before being infused into patients.20 The safety and feasibility of this method is evinced by the explosive increase in the number of clinical trials that utilise this technique for the treatment of different types of malignancies, sickle cells disease and β-thalassemia, which have been reviewed recently.20–23 In cases where ex vivo gene editing is problematic, CRISPR/Cas may be delivered in vivo via viral and non-viral vectors. While viral vectors have been demonstrated to be highly efficient at in vivo gene editing in multiple disease models, such as Duchenne muscular dystrophy (DMD),24 cardiovascular diseases25 and various malignancies,26 their application in humans raises concerns associated with safety and immunogenicity. For this reason, much effort has been invested in designing non-viral carriers for in vivo gene editing.
Besides being stable, biocompatible, and efficient, non-viral vectors should guarantee the following: (1) physiochemical stability of the cargos to ensure protection from enzymatic degradation in the extracellular, cellular, and intracellular compartments, (2) facilitate cellular internalization and endosomal escape of the cargos, (3) limit off-target effects, and (4) reduce immunogenic side effects.
These features could be achieved only by the incorporation of chemical modifications of the different CRISPR/Cas cargoes as well as by exploiting several types of biomaterials to formulate efficient and safe delivery systems.
Among non-viral vectors, the most commonly utilised are based on lipid, polymeric, organic or hybrid nanoparticles, as well as cell-penetrating peptides. These vectors have proven to efficiently and safely deliver CRISPR/Cas components in vivo to different target genes in multiple tissues such as eyes,27 adipocytes,28 muscles,29 brain,30 liver,31 as well as superficial32 and internal33 malignancies. Herein, we provide an overview of the CRISPR/Cas9 technology, followed by an outline of different physical and viral methods used for CRISPR/Cas9 delivery. We will then, describe the latest advances in the implementation of non-viral approaches for in vivo gene editing while discussing the landscape and challenges for clinical translation.
2. CRISPR
CRISPR was first observed in Escherichia coli in 1987 by Nakata et al.,34 and subsequently identified in an increasing number of prokaryotic organisms including bacteria and archaea.35 It was not until 2007 that its function in bacteria as an adaptive immune system was elucidated36 and it was further adapted as a gene editing tool.The bacterial genomic construct of CRISPR arrays features two key elements, the first of which is the CRISPR cassette. Cassettes consist of conserved, repeating sequences of DNA, typically 21 to 48 base pairs in length with unique non-repeating DNA sequences known as spacers interspersed between them.35 Spacers of individual cassettes are similar in length, often between 26–72 base pairs. They are derived from DNA of previous infective agents such as bacteriophages, invasive plasmids and other mobile genetic elements.37 Through the incorporation of pathogenic-DNA into their genome, bacteria and archaea have evolved a mechanism of heritable acquired immunity, which aids in recognition and defence against future pathogens. The second key element of CRISPR arrays are CRISPR-associated (Cas) genes, which encode numerous, highly diverse Cas proteins.37,38
Mechanistically, CRISPR immunity utilises a self-non-self-discrimination principle37 which can be broadly divided into three phases: (1) immunization, (2) generation of CRISPR RNA (crRNA), and (3) interference.39 In phase 1, foreign nucleic acids from invading pathogens are selected, processed and integrated into the CRISPR array as spacer sequences.40 This provides a ‘memory’ of prior infections that allows the host to defend against future infections by the same pathogen. In phase 2, CRISPR arrays are transcribed to generate precursor crRNA (pre-crRNA). Pre-crRNAs are further processed via endonucleolytic cleavage into mature crRNAs, short sequences of approximately 20 nucleotides that are complementary to the foreign DNA (or RNA).37,40,41 The 5′ end of the crRNAs encodes the spacer sequence, whilst the 3′ end encodes part of the CRISPR-array repeat sequence. When re-infection occurs (phase 3), crRNAs are loaded onto effector modules, structures that consist of either a single Cas protein or a complex of Cas proteins. Guided by crRNAs, the effector modules cleave complementary sequences (proto-spacers) in the pathogenic genome. Invading DNA is then identified via recognition of protospacer adjacent motifs (PAMs). These are 2–6 base pair nucleotides sequences that are typically found 3–4 base pairs downstream of protospacer sequences in the pathogenic genome.42
Two classes of CRISPR systems have been established (1 and 2), with class 1 systems divided into types I, III and IV, and class 2 systems divided into types II, V and VI.38 The different CRISPR systems are classified based upon the structural composition of their effector modules, which are responsible for the processing and transcription of CRISPR arrays, as well as interrogation and cleavage of foreign DNA or RNA.37,43 In class 1, effector modules contain multiple Cas proteins, whereas class 2 systems employ a singular Cas protein.43 The type II CRISPR system (CRISPR/Ca9) is the simplest and most commonly used for genome editing, as it requires the fewest components to induce DSBs.44
In CRISPR/Ca9, a second RNA sequence, trans-activating CRISPR RNA (tracrRNA), a non-coding RNA sequence, is transcribed from a genomic locus upstream of the CRISPR locus. In type II systems, this is essential for the maturation of pre-crRNA, as well as for binding of crRNA with Cas9 protein.44 tracrRNA forms a complex with pre-crRNA by base-pairing with the repeat sequence present in the pre-crRNA. This results in a double-stranded RNA, which is subsequently cleaved by RNase II enzyme. The end result is a functional single guide RNA (sgRNA) complex containing a single spacer sequence.45 As shown in Fig. 1, the sgRNA guides the Cas9 protein to the target site, where the Cas9 nuclease upon recognition of the PAM sequence (5′-NGG-3′) downstream of the target site, induces DNA cleavage. Unlike its predecessors, ZFNs and TALENs, target recognition in CRISPR/Ca9 is not directed by protein DNA-binding domains, but by the 20-nucleotide sgRNA sequence complementary to the target DNA.46 This offers many advantages over ZFNs and TALENs. Namely, by eliminating the need to engineer individual protein domains for each DNA target, CRISPR/Ca9 has better scalability for large-scale genomic manipulation.47 Though CRISPR/Cas9 holds great promise in performing simple and precise gene editing, its safe and efficient translation for in vivo applications is still limited, due to multiple factors that will be outlined later in this review (Table 1).
| Plasmid | mRNA | RNP | |
|---|---|---|---|
| Cas9 expression rapidity | Low | Moderate | High |
| Editing efficiency | Moderate/high | Moderate | Moderate/high |
| Stability | High | Moderate | Moderate |
| Persistence in cells | High | Moderate | Low |
| Immunogenicity | High | Moderate | Moderate |
| Cytotoxicity | Moderate | Low | Low |
| Insertional mutagenesis | High | — | — |
| Production speed | High | Moderate | Low |
| Cost | Low | Moderate | High |
3. CRISPR/Cas9 gene editing formats
As shown in Fig. 2, there are 3 common formats into which CRISPR/Cas9 technology can be incorporated: plasmid DNA (pDNA), messenger RNA (mRNA), and ribonucleoprotein (RNP) complexes. The choice of which CRISPR editing format to use relies on several factors, including but not limited to transfection efficiency, the required speed/duration of expression, and safety.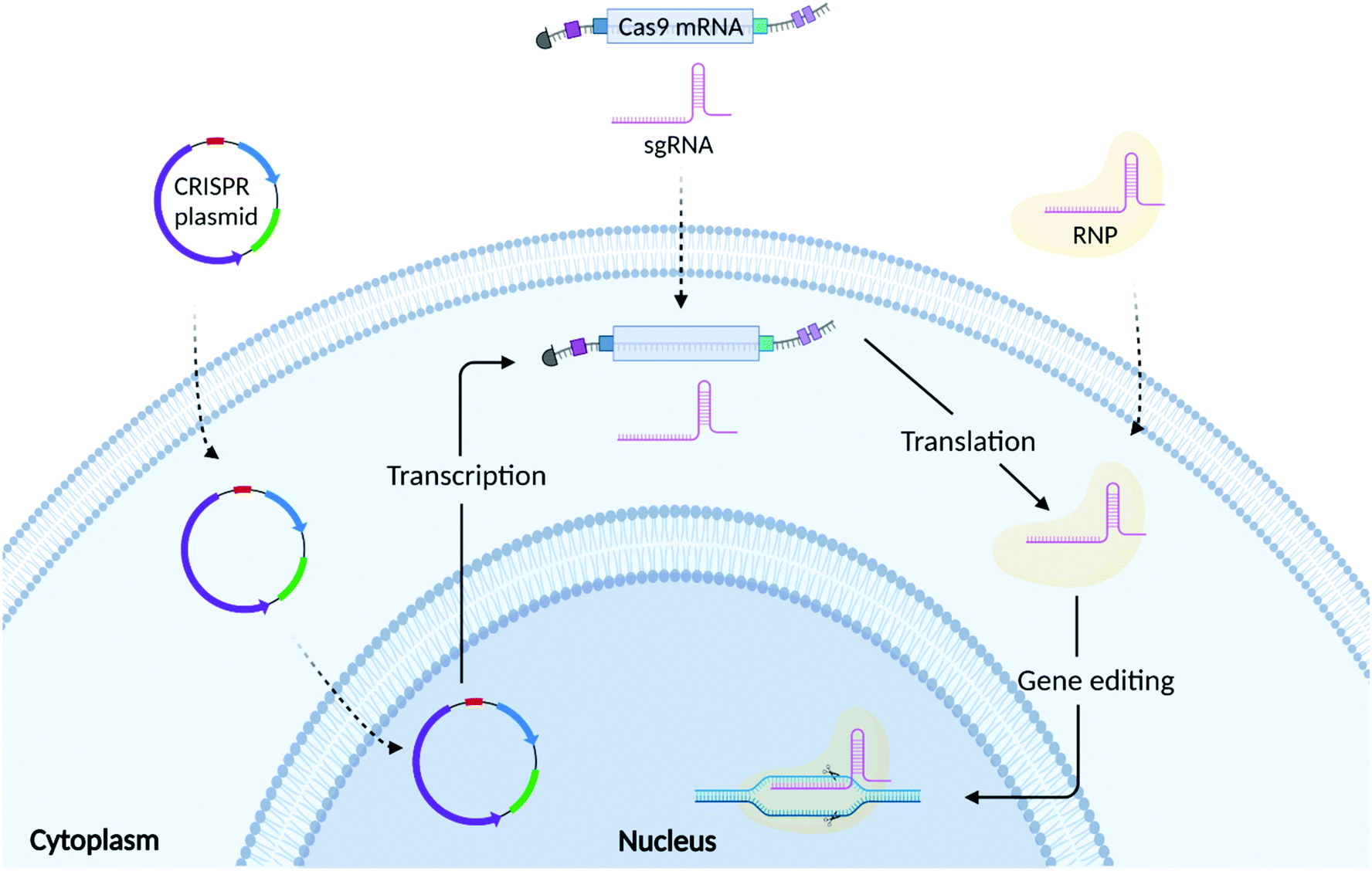 | ||
| Fig. 2 Schematic representation of CRISPR/Cas9 delivery to the cell using three different formats: plasmid DNA, RNA (Cas9 mRNA/sgRNA) and ribonucleic protein (RNP) complexes. Once the plasmid enters the cells it is translocated from the cytoplasm to the nucleus, where transcription into Cas9 mRNA and sgRNA happens. Then, once into the cytoplasm, the Cas9 mRNA is translated into Cas9 protein that complexes with the sgRNA forming the RNP. Afterwards, the RNP are internalized into the nucleus to target a specific DNA sequence and induce gene insertions or deletions. Created with BioRender.com. | ||
3.1 Plasmid mediated CRISPR/Cas9 gene editing
Both Cas9 protein and sgRNA can be encoded into single or multiple plasmids.32 As shown in Fig. 2, plasmid DNA requires nuclear translocation and transcription to Cas9 mRNA (mCas9) prior to Cas9 protein translation.41 Plasmids are easy to design and do not require high manufacturing costs making them an appealing approach for gene editing. Compared to mRNA and RNP, plasmids exhibit greater stability, offering a prolonged expression of the CRISPR components.48 This, however, could increase the likelihood of off-target effects and immunogenic reactions.49 Plasmid DNA is also prone to random integration into host-cell DNA (insertional mutagenesis), which can lead to unintended mutations of genes and subsequent phenotypic changes.50 Plasmid DNA is physically larger than both the mRNA and RNP formats, which presents an additional challenge in the formulation of delivery vectors that can effectively encapsulate them.513.2 RNA mediated CRISPR/Cas9 gene editing
RNA based transfection methods utilise a strategy whereby mRNA encoding for Cas9 protein is delivered alongside a sgRNA, avoiding the need for the transcription step, but still requiring translation of the Cas9 mRNA into its functional protein. Compared to DNA, mRNA turnover in the cell is faster, limiting the persistence of the protein, thus reducing potential off-target effects. However, mCas9RNA can be subject to enzymatic degradation resulting in poor stability. For this reason, Cas9 mRNA is often synthesized incorporating an Anti-Reverse Cap Analog (ARCA) at the 5′ end and a poly(A) tail at the 3′ end.52 These modifications can ensure more efficient mRNA translation as well as increasing mRNA stability in the cytosol.53 Another stumbling block, is represented by the recognition of the mCas9 by Toll-like receptors (TLRs), which can activate a cascade of events triggering moderate to severe immune responses.54 Uridine depletion and incorporation of pseudo bases during Cas9 mRNA synthesis have been used to further increase mRNA stability as well as reduce immunogenicity.52,54 Genome editing research mainly employs a chemically synthesized sgRNA containing a fusion loop between the tracrRNA and crRNA.55 Further chemical modifications have also been implemented for sgRNA synthesis with the final aim of increasing their stability, RNP complexation ability, shelf-life, thermostability and reducing their immunogenicity.56 Although these chemical modifications can increase the stability of both Cas9 mRNA and sgRNA, it is of the utmost importance that the RNA is formulated with a delivery system that can both protect it from enzymatic degradation as well as guarantee its efficient cellular uptake and endosomal escape.57,583.3 RNP mediated CRISPR/Cas9 gene editing
RNP mediated gene editing involves the delivery of Cas9 protein and sgRNA to the cell. Compared to pDNA and mRNA, delivery of RNP bypasses both transcription and translation and only requires the formation of the RNP complex and its subsequent translocation to the nucleus.42 As the RNP is delivered directly to the cell, it guarantees a high gene editing efficiency while limiting the probability of potential off-target effects given their shorter half-life.12,59 However, Cas9 protein is susceptible to enzymatic degradation once injected into the bloodstream. Furthermore, its large size limits trafficking through the cell membrane, requiring the need for formulation with a complex delivery vector.60 The delivery system should also help to mask the presence of the Cas9 protein, which can elicit immune responses. Several independent studies have reported the presence of antibodies against Cas9 protein in both humans and mice.61,62 In depth in vitro and in vivo studies showed that pre-exposure to Cas9 was responsible for the enrichment of different lineage of Cas9 reactive T cells.63–65 In immunized mice, it was observed that CRISPR/Cas9 gene edited liver tissues elicited a strong CD8+ T cell response, which induced a rapid clearance of gene edited hepatocytes, without compromising the regeneration of the liver through proliferation of unedited cells.62 Additionally, whilst Cas9 protein can be easily obtained from several suppliers, the protein purification process is costly and laborious.4. The need for a non-viral delivery vector
In vivo delivery of all three of these CRISPR/Ca9 formats presents both general and specific problems requiring delivery vectors that can guarantee stability and specificity for target cells. Whilst many CRISPR/Cas9 delivery strategies have been developed,66 they are typically physical interventions51 (e.g., electroporation,67 hydrodynamic injection,17 microinjection) or virally vectored delivery68 (e.g., adeno-associated virus (AAVs),69 lentivirus (LVs)70). Although there has been some success with these delivery methods, there are still questions to address concerning their safety and clinical applicability. Physical approaches often rely on techniques that are not translatable for in vivo use. For example, the microinjection technique involves microscopic-guided injection of the gene editing cargo into the target cells and is technically difficult to perform, often resulting in mechanical damage.71 Electroporation is another example of a physical delivery approach, in which an electrical field is applied to temporarily induce pores in the membrane of target cells, facilitating the permeability of the gene editing cargos into the cytoplasm.34 The need for large voltages to achieve the desired levels of gene transfer can induce significant cell apoptosis, limiting its application both in vitro and in vivo. Hydrodynamic injection via the tail vein has been used successfully to induce in vivo gene editing of hepatocytes in animal models,72,73 but is limited to preclinical murine models.74 In contrast, saphenous vein injection was used for the delivery of both siRNA and pDNA to muscle cells.75 This process has been successful in gene transfer in both small animals and primates,76,77 however, the need for a surgical procedure represents a limiting factor for safe and easy clinical translation.Viral vectors have been extensively used to deliver CRISPR/Cas9 constructs resulting in efficient in vitro and in vivo gene editing.78 Despite this, their potential clinical translation is hampered by several concerns. LVs are single-stranded RNA viruses capable of efficiently infecting both dividing and non-dividing cells. However, LVs can integrate their genome into host cells, potentially resulting in persistent expression of CRISPR/Cas9 components, the generation of unwanted off-target effect, insertional mutagenesis and persistent immune reactions.78 AAVs are single-stranded DNA vectors, with a reduced packaging capacity compared to LVs (4.7 kb vs. 8 kb), this often necessitates the need to use multiple vectors, one for the Cas9 protein and one for the sgRNA. AAVs do not induce persistent genomic integration, but instead deliver the genetic material in an episomal form, which reduces unwanted off-target effects and potential immunogenic responses.79,80 While AAVs have been extensively utilised, there is evidence to suggest that their application for in vivo gene editing might still be limited due to the presence of multifunctional immunological responses against CRISPR-AAVs.61,62 Mice receiving CRISPR-AAVs not only showed humoral and cell-mediated immunity against Cas9, but also anti AAV capsid-specific antibodies could be detected.61 Epitope mapping further confirmed the presence of B cell epitopes in the three different isoforms of viral proteins VP1/2/3, with higher prevalence of VP3 which is expressed on the surface of the capsids. These findings are in line with other research studies investigating the immune response to AAVs in humans and which have reported the manifestation of both cell-mediated and humoral immunity against different AAVs.81,82 Combined, these in vivo immunogenicity findings have raised concerns about the feasibility of the clinical translation of AAV mediated gene editing.
By exploiting different biomaterials, non-viral vectors offer a powerful and safer alternative in delivering genetic payloads, drugs, and proteins. While the in vivo gene editing efficiency of non-viral approaches is arguably lower than viral vectors, there is an increased interest in their implementation due to their perceived benefits and safety profile. Successful non-viral vectors should, minimally, sequester the nucleic acids from enzymatic degradation and mask them from recognition by TLR receptors on immune cells,83 increase their bioavailability in specific organs, promote cellular internalization and aid in endosomal escape. In addition, the use of nanocarriers may also have many unique benefits over viral vectors. For example, the nanocarrier may be designed to carry therapeutic cargos of different physico-chemical characteristics, ranging from small drugs to large biologicals such as CRISPR plasmids and RNP. This affords the opportunity to simultaneously deliver multiple therapeutic modalities which may synergise with the therapeutic outcome of gene-editing.
5. Non-viral vectors for in vivo gene editing
An advanced literature search was carried out on PubMed for publications published prior to September 2021, the following search terms were utilised: “CRISPR”, “CRISPR/Cas9”, “CRISPR-Cas9”, “RNP”, “plasmid”, “RNA”, “Cas9 mRNA”, “sgRNA”, “in vivo gene editing”, “in vivo delivery”, “non-viral delivery”, “non-viral vector”, “nanoparticles”, “nanocomplex”. Articles that did not include in vivo investigation or included non-clinically translatable routes of administrations (i.e., hydrodynamic injection) were excluded. The studies extracted explored the delivery of CRISPR/Cas9 as a potential therapeutic approach when used as a single agent or in combination with other modalities in animal disease models. As can be seen in Fig. 3A, cancer was the most investigated disease (47.5%), followed by diabetes (15%) and hypercholesteremia (7.5%). In these studies, the most common platform for CRISPR/Cas9 delivery was plasmid, followed by RNP and RNA. As shown in Fig. 3B and C, different routes of administration and delivery systems were employed for each CRISPR/Cas9 format. In the sections below, a critical overview of the delivery systems utilised for in vivo gene editing will be provided. For each study, the key information, including the nature of the nanoparticles, target genes, animal model and findings are summarized in Tables 2–4.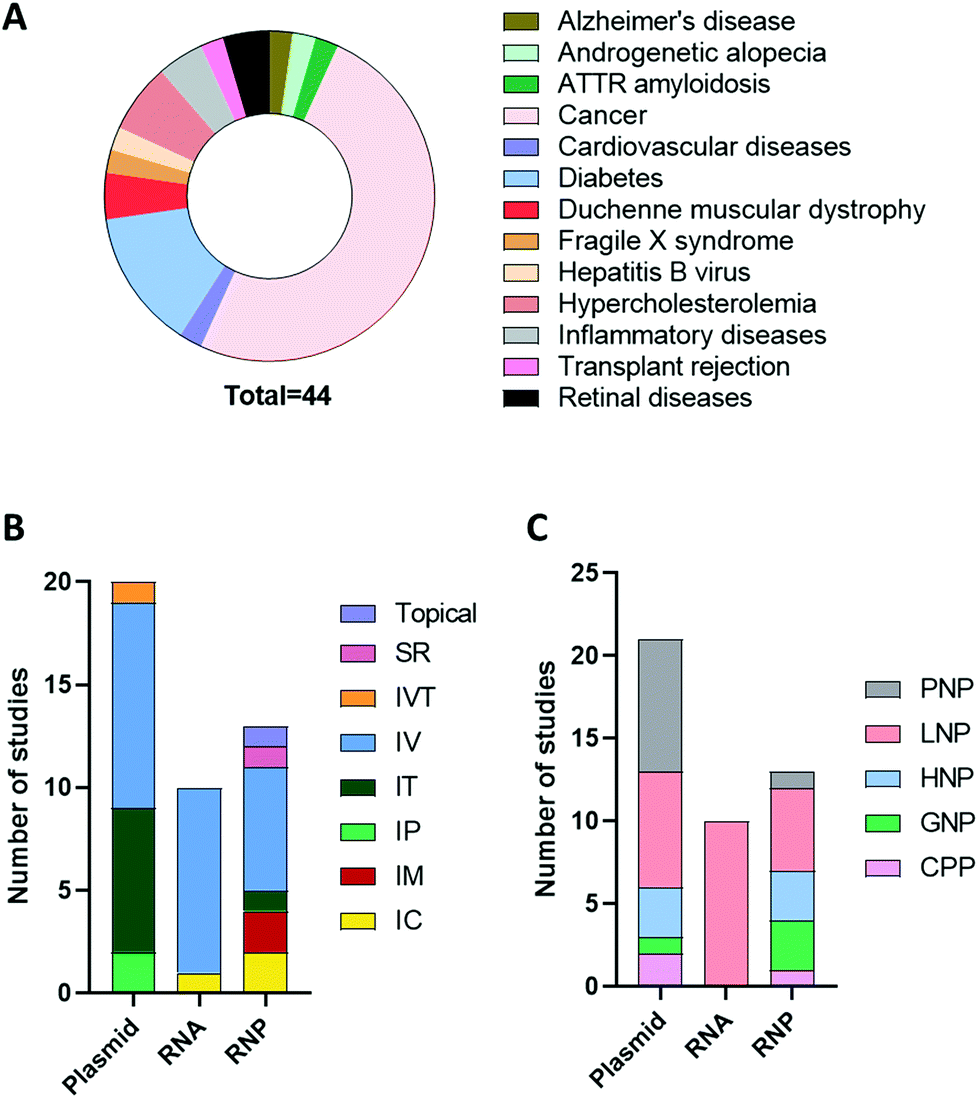 | ||
| Fig. 3 Overview of the studies exploring in vivo non-viral delivery of CRISPR/Cas9. Representative pie chart of the diseases for which CRISPR/Cas9 in vivo non-viral gene editing have been investigated (A). Representative bar charts of the number of studies delivering CRISPR/Cas9 in the form of plasmid, RNA or RNP and the corresponding route of administration and vectors utilized (B and C). Abbreviations: SR = subretinal, IVT = intravitreal, IV = intravenous, IT = intratumoural, IP = intraperitoneal, IM = intramuscular, IC = intracranial, PNP = polymeric nanoparticles (NP), LNP = lipid NP, HNP = hybrid NP, GNP = gold NP, CPP = cell penetrating peptide. | ||
| Disease model | Target | Vector | Mouse model | Dose | Admin. route | Findings | Ref. |
|---|---|---|---|---|---|---|---|
| ALT = alanine aminotransferase, aPBAE NP = 1-(3-aminopropyl)-4-methylpiperazine (AMP)-modified poly-beta-amino ester (PBAE) nanoparticle, AST = aspartate transaminase, ATS-9R oligoplex = adipose tissue targeting sequence (ATS) peptide conjugated to D-form of 9-mer arginine (9R), BCR-ABL = fusion gene of breakpoint cluster region protein (BCR) and tyrosine-protein kinase ABL1 (ABL1) genes, CCL2 = chemokine (C–C motif) ligand 2, CD80/86/40 = cluster of differentiation 80/86/40, CDC6 = cell division control protein 6 homolog, Cdk5 = cyclin dependent kinase 5, CHO-PGEA = cholesterol (CHO)-terminated ethanolamine-aminated poly(glycidyl methacrylate), CTGF = connective tissue growth factor, CXCL1 = chemokine (C–X–C motif) ligand 1, Fabp4 = fatty acid-binding protein 4, Fbn1 = fibrillin-1, GFP = green fluorescent protein, H&E = haematoxylin and eosin histological stains, HIF1A = hypoxia-inducible factor 1-alpha, HP16E7 = human papilloma virus type 16 E7 oncogene, HPPCs PBAE = poly(amide-amine)-poly(β-amino ester) hyperbranched copolymer, HPV16 = high-risk human papillomavirus subtype 16, IHC = immunohistochemical analysis, IL-1β = interleukin 1-beta, IL-6 = interleukin 6, IP = intraperitoneal, IT = intratumoural, IV = intravenous, IVT = intravitreal, KO = knock out, LBP = lactose-derived branched cationic biopolymer, Lcn2 = lipocalin 2, MMP-2 = matrix metalloproteinase 2, MMP-9 = matrix metallopeptidase 9, mPEG-CDM = methoxy poly(ethylene glycol) (mPEG) carboxydimethyl-maleic anhydride (CDM) nanoparticle, MTH1 = MutT homolog 1, NE = neutrophil elastase, Ntn1 = netrin-1, CLAN = cationic-lipid assisted PEG-b-PLGA nanoparticle, PBS = phosphate buffered saline, PD-L1 = programmed death ligand 1, PPCNP = PEG-PEI-Cholesterol nanoparticles, P-HNP = PEGylated helical polypeptide nanoparticle, Plk1 = polo-like kinase 1, PLNP = polyethylene-glycol-phospholipid modified cationic lipid nanoparticle, PTX = paclitaxel, R8-dGR-Lip = R8-dGR-peptide modified cationic liposome, RRPHC = artificial virus composed of fluorinated polymer (PF33)/CRISPR/Cas9 core coated with RGD-R8-PEG-HA polymer, RS1 = retinoschisin 1, SMNP = supramolecular nanoparticle, TAT-GN = HIV-1-transactivator of transcription peptide (TAT peptide) modified cationic gold nanoparticle, TG = triglycerides, tLNG = noncationic deformable tumour-targeted nanolipogel system, TNF-α = tumour necrosis factor alpha, VEGF = vascular endothelial growth factor. | |||||||
| Osteosarcoma | VEGFA | PPCNP | BALB/c nude | 0.75 mg kg−1 of plasmid | IV | - Significantly higher indel formation in osteosarcoma tissues and lung metastasis compared to liver tissues was confirmed. | 84 |
| - IHC showed reduced expression of VEGFA and other relevant markers for proliferation, metastasis and invasion. | |||||||
| - Successful gene editing of VEGFA resulted in significant reduction of the tumour size as well as reduction in the number of lung metastasis. | |||||||
| Ovarian cancer | MTH1 | RRPHC | BALB/c nude | 10 injections of 5 μg of plasmid once every 3 days for 30 days | IP | - IHC analysis showed significant reduction in MTH1 expression following CRISPR treatment. | 85 |
| - Successful gene editing resulted in significant reduction of proliferative marker, Ki-67, and significantly inhibited tumour growth. | |||||||
| - Safety of the complex was confirmed by the low (0.4%) mutation frequencies in potential off-target sites. | |||||||
| Melanoma | Plk1 | TAT-GN | BALB/c nude | 10 μg pCas9 + 10 μg psgPlk1 once every other day for 15 days | IT | - Successful gene editing was confirmed with T7EI assay resulting in significant reduction of Plk1 protein expression as detected by western blot (WB). | 86 |
| - Plk1 knockdown resulted in significant suppression of tumour growth compared to control groups, without inducing any relevant toxicity. | |||||||
| Melanoma | Plk1 | PLNP | BALB/c | 10 μg plasmid once daily for 15 days | IT | - Successful PLK1 knockdown was confirmed by WB. | 87 |
| - Treatment with PLNP-CRISPR significantly inhibited tumour growth by 67% of the PBS group and 50% the PLNP-siRNA group. | |||||||
| Type 2 diabetes | Ntn1 | CLAN | C57BL/6 | 1 or 2 mg kg−1 pCas9/sgNtn1 every other day for 14 days | IV | - Incorporation of the CD68 promoter ensured specific macrophages/monocytes targeting, as confirmed by the Cas9 expression detected with WB. | 88 |
| - Ntn1 gene editing was confirmed by the surveyor assay, with indels frequency of 8.8% and 18.1% for the 1 mg kg−1 and 2 mg kg−1 dose respectively and subsequent reduction of the Ntn1 protein expression as detected by WB. | |||||||
| - CLAN treatment with 2 mg kg−1 dose produced glucose-response profile similar to glyburide positive control, further improving insulin sensitivity. | |||||||
| Cervical cancer | Plk1 | P-HNP | BALB/c nude | 20 μg pCas9 + 20 μg psgPlk1 on alternate days for 10 days | IT | - Treatment with P-HNP-pCas9-psgPlk1 significantly inhibited tumour growth by 71%, which improved the overall survival rate by 60%. | 89 |
| - Deep sequencing confirmed 35% Plk1 gene editing around the target site, which resulted in 67% reduction in the Plk1 protein expression as detected by WB. | |||||||
| Type 2 diabetes | NE | CLAN | C57BL/6 | 2 mg kg−1 pCas9/sgNE once every other day for 14 days | IV | - High uptake of CLAN by Kupffer cells and neutrophils of liver (83.9% and 94.6% respectively). | 90 |
| - T7EI assay on neutrophils sorted from eWAT and the liver revealed indel frequencies of 19.5% and 27.3% respectively. Minimal (<1%) indel frequency detected in potential off-target sites. | |||||||
| - WB analysis confirmed significant reduction in NE protein expression in both eWAT and liver. | |||||||
| - CLAN treatment improved glucose tolerance and enhanced insulin sensitivity with efficiency comparable to that of positive controls metformin and Sivelestat. | |||||||
| - CLAN treatment limited neutrophils infiltration and reduced the concentration of pro-inflammatory factors (TNF-α, CCL2, CXCL1, IL-1β) in the serum. | |||||||
| Melanoma | Plk1 | Lipid-coated TAT-GN | BALB/c nude | 10 μg pCas9-sgPlk1 every other day for 20 days [±Irradiation] | IT | - Enhanced gene KO mediated by the lipid-coated TAT-GN was confirmed in vitro using WB. | 32 |
| - Treatment with lipid-coated TAT-GN + irradiation was significantly more efficient in reducing tumour growth than non-coated TAT-GN + irradiation. | |||||||
| Leukaemia | BCR-ABL | CLAN | NOD/SCID | 1.6 mg kg−1 CLAN (pCas9/gBCR-ABL) once every other day for 14 days | IV | - Indel frequency at target locus was 19.3% and 16.1% in tumour cells sorted from blood and bone marrow respectively of CLAN treated animals. | 91 |
| - CLAN treatment ensured significant reduction of the BCR-ABL protein expression in sorted tumour cells. | |||||||
| - CLAN treatment significantly increased the survival rate compared to the control groups. Further therapeutic improvement was confirmed by the reduction in white blood cell levels as well as reduced splenomegaly. | |||||||
| Pancreatic cancer | HIF1A | R8-dGR-Lip | BALB/c nude | Antitumour effect: 10 μg of pCas9 + 10 μg of psgHIF1A 3 times a week for 3 weeks | IV | - R8-dGR conjugated LNP significantly inhibited tumour growth more efficiently than naked LNP. | 92 |
| Antimetastatic effect: 10 μg of pCas9 + 10 μg of psgHIF1A and 2.5 mg kg−1 PTX every 3 days for 5 times | - Efficient in vivo gene editing was confirmed by HIF1A mRNA analysis and protein analysis via WB. | ||||||
| - The best survival rate was observed in the R8-dGR conjugated LNP group. Codelivery of PTX further increased survival time and showed enhanced antiproliferative effect indicated by increased apoptosis and necrosis of tumour cells. | |||||||
| - Reduction of metastasis was confirmed by IHC analysis of the MMP-9 expression in liver and lungs tissues. | |||||||
| Type 2 diabetes | Fabp4 | ATS-9R oligoplexes | C57BL/6J | 0.35 mg kg−1 of pCas9-sgFabp4 twice a week for 6 weeks | IP | - Fabp4 mRNA levels in mature adipocytes was significantly downregulated following treatment. | 28 |
| - Blood glucose levels reduced with repeated treatment. | |||||||
| - Mean body weights in treatment group decreased by 20%. | |||||||
| - Serum levels of free fatty acid, TG, ALT, and AST reduced by up to ∼50%. IHC further confirmed reduced liver steatosis of treated animals. | |||||||
| Breast cancer | Lcn2 | tNLG | Nude | 1 mg kg−1 pCas9-sgLcn2 once a week for 4 weeks | IV | - Significant reduction of tumour growth was observed in tNLG-sgLcn2 treated group. | 93 |
| - qRT-PCR confirmed that treatment ensured ∼81% Lcn2 gene editing. IHC analysis further confirmed significant reduction in Lcn2 and Ki67 protein expression in tumour tissues. | |||||||
| Hepatocellular carcinoma | Survivin | LBP | BALB/c nude | 20 μg pCas9-sgSurvivin every 2 days for 35 days [±Sorafenib] | IV | - In vivo gene editing efficiency of LBP treated animals (26.4%) was confirmed by Sanger sequencing. This resulted in significant reduction of Survivin protein expression as confirmed by IHC. | 94 |
| - Concomitant in vivo gene editing and chemotherapy significantly induced reduction in tumour growth and the number of tumour nodules. | |||||||
| Type 1 diabetes | CD80 | CLAN | NOD/LtJ | 2 mg kg−1 pCas9 + sgCD80 (or sgCD56, sgCD40) + 0.25 mg kg−1 autoimmune diabetes-relevant peptide twice a week for 5 weeks | IV | - Successful gene editing of the three target loci of DCs in the lymph nodes, blood and spleen was confirmed via T7EI assay, with indel frequency between 3.5 and 10%. Antibody staining showed significant reduction in the expression of CD80, CD56 and CD40. | 95 |
| CD86 | - Diabetes development was significantly inhibited in the group receiving concomitant CLAN and peptide treatment. In this group histopathology analysis revealed decreased infiltration of pro-inflammatory cells and reduced insulitis in the pancreas. | ||||||
| CD40 | |||||||
| X-linked juvenile retinoschisis | RS1 | SMNP | BALB/c | Single-injection of 50 ng pCas9-sgRNA + 50 ng donor RS1-GFP plasmid. | IVT | - Optical coherence tomography and fundus camera confirmed RS1-GFP gene integration at day 18 post IVT. | 27 |
| - PCR amplification and Sanger sequencing further confirmed the integration of the RS1 gene in the excised treated eyes. | |||||||
| - No histological abnormalities in the retinas were observed following gene knock-in. | |||||||
| Colon carcinoma | Cdk5 | mPEG-CDM | BALB/c | 20 μg pCas9-sgCdk5 ± 4 mg kg−1 PTX once every 3 days for 12 days. | IV | - WB analysis confirmed successful Cdk5 KO, which resulted in the reduction of PDL-1 protein expression as hypothesized by the authors. | 96 |
| - Concomitant Cdk5 KO and PTX treatment significantly reduced tumour growth compared to single gene editing or PTX treatment only (85.8% vs. 69.3% vs. 51.7% respectively). This success was also reflected in the survival study (36 vs. 30 vs. 26 days). | |||||||
| Melanoma | Cdk5 | mPEG-CDM | C57BL/6 | 20 μg pCas9-sgCdk5 ± 4 mg kg−1 PTX once every 3 days for 12 days. | IV | - As for the melanoma model dual gene editing and chemotherapy outperformed single treatments in inhibiting the tumour growth (88% vs. 58% vs. 72.8% respectively). | 96 |
| Melanoma | Cdk5 | aPBAE NP | C57BL/6 | 5 μg pCas9-sgCdk5 once every 3 days for 12 days. | IT | - mRNA analysis and WB confirmed successful Cdk5 gene KO, which resulted in downregulation of the PD-L1 expression. | 97 |
| - In vivo gene KO resulted in significant suppression of the tumour growth compared to the other control groups. This was also reflected in extending the overall survival from 26 days for negative control group (PBS), to 32 days for anti-PD-L1 treatment to 40 days for the aPBAE NP. | |||||||
| Triple negative breast cancer | Cdk5 | aPBAE NP | BALB/c | 5 μg pCas9-sgCdk5 once every 3 days for 12 days. | IT | - Similar to the effects observed with the melanoma model, the aPBAE NP outperformed anti-PD-L1 treatment in reducing tumour growth progression. | 97 |
| - KOaPBAE NP group showed superior inhibition of lung metastasis. Fewer tumour nodules were observed in the lungs of the treatment group (5 ± 3 nodules) compared to anti-PD-L1 antibody (25 ± 5 nodules) and PBS control (36 ± 4 nodules) controls. | |||||||
| Breast cancer | CDC6 | PAR–Lipo | Nude | 1.25 mg kg−1 pSpCas9-sgCDC6 | IV | - Animals injected with PAR–Lipo exhibited greater tumour reduction compared to Lipo2000 positive control. | 98 |
| - PAR-Lipo also induced stronger downregulation of cancer cell proliferation and metastasis as determined by Ki67 staining assay. | |||||||
| - CDC6 downregulation was confirmed with WB in MCF-7 tumour tissues. | |||||||
| Cervical cancer | HPV16E7 | HPPCs PBAE | Nude | 10 μg of pCas9/4 doses every 3 days. | IT | - Treatment of animals with PBAE alone or in the presence of HPPCs revealed significant reduction in tumour growth compared to saline control. | 99 |
| - IHC showed significant reduction in HPV16E7 expression following gene KO compared to saline group. | |||||||
| Disease model | Target | Vector | Mouse model | Dose | Admin. route | Findings | Ref. |
|---|---|---|---|---|---|---|---|
| ALT = alanine aminotransferase, AST = aspartate transaminase, ATTR = transthyretin amyloidosis, BAMEA-O16B = irreducible lipid nanoparticle with integrated disulphide bonds, CD40 = cluster of differentiation protein 40, CLAN = cationic-lipid assisted PEG-b-PLGA nanoparticle, H&E = haematoxylin and eosin histological stains, HBV = hepatitis B virus, ICAM-2 = intracellular adhesion molecule 2, IL-1β = interleukin 1-beta, IT = intratumoural, IV = intravenous, KO = knockout, LNP = lipid nanoparticle, MCP-1 = monocyte chemoattractant protein 1, NLRP3 = NLR family pyrin domain containing 3, Pcsk9 = proprotein convertase subtilisin kexin type 9, Plk1 = polo-like kinase 1, SORT-LNP = selective organ targeting lipid nanoparticle, TNF-α = tumour necrosis factor alpha, TT3-LLN = N1,N3,N5-tris(2-aminoethyl) benzene-1,3,5-tricarboxamide derived lipid-like nanoparticle, Ttr = transthyretin. | |||||||
| Hepatitis B virus | HBV DNA | TT3-LLN | C57BL/6 | 0.56 mg kg−1 Cas9 mRNA+, 0.25 mg kg−1 sgRNA 6 hours post Cas9 mRNA injection | IV | - HBV viral loads were significantly reduced (analysis carried on liver and serum HBV antigens as well as RNA and DNA levels). | 100 |
| - DNA sequencing confirmed indel formation at the sgRNA target site. | |||||||
| Hypercholesterolemia | Pcsk9 | LNP | C57BL/6 | 1.2 mg kg−1 Cas9 mRNA + 0.5 mg kg−1 sgPcsk9 | IV | - In vivo gene editing was confirmed by T7EI assay of liver tissues and further screening of Pcsk9 protein in the serum which was undetectable 5 days post-injection. | 101 |
| - Efficient Pcsk9 gene editing resulted in 30–45% reduction of total cholesterol. | |||||||
| Inflammatory diseases | ICAM-2 | LNP (7C2, 7C3) | C57BL/6J | 2 mg kg−1 total RNA on day 0 and 2 (3/1 mCas9/sgICAM-2 w/w) | IV | - 7C3 LNP outperformed 7C2 in inducing ICAM-2 gene editing of endothelial splenic cells. | 102 |
| Type 2 diabetes | NLRP3 | CLAN | C57BL/6 | 0.5, 2 and 2 mg kg−1 Cas9 mRNA/sgNRLP3 | IV | - In vivo gene editing of peritoneal macrophages was shown to be dose and time-dependent with the highest indel frequency (∼47%) obtained at 24 h. This was also reflected in the proteomics data showing ∼57% reduction in NLRP3 protein expression compared to PBS group. | 103 |
| - Treatment of T2D animals with CLAN, induced an improved glucose tolerance and insulin sensitivity comparable to positive control group treated with glyburide. | |||||||
| Cardiovascular disease | Pcsk9 | BAMEA-O16B | C57BL/6 | 0.6 mg kg−1 Cas9 mRNA + 0.8 mg kg−1 sgRNA | IV | - BAMEA-O16B treatment resulted in efficient Pcsk9 gene KO as demonstrated by 80% reduction in Pcsk9 serum levels (ELISA). | 104 |
| - H&E staining revealed no obvious hepatocellular injury after treatment. | |||||||
| Transplant rejection | CD40 | CLAN | BALB/c (graft donor) | 3 mg kg−1 Cas9 mRNA/sgCD40 once every other day, 3 times prior to transplant and 3 times post-transplant | IV | - Efficient CD40 KO in dendritic cells confirmed with T7EI and WB. | 105 |
| C57BL/6 (graft recipient) | - CD40 disruption significantly inhibited T cell activation resulting in a lower graft rejection. This effect was more efficient than Rapamycin positive control. | ||||||
| Glioma tumour | Plk1 | LNP | C57BL/6JOlaHsd | 0.05 mg kg−1 total RNA (mCas9/sgRNA 3/1 w/w) | IC | - NGS confirmed efficient gene editing (∼68%) of Plk1 locus in tumour cells. | 33 |
| - Plk1 KO, significantly reduced tumour growth and increased the average survival time from 32.5 days to >48 days; with 30% treated mice surviving for 60 days. | |||||||
| Ovarian cancer | Plk1 | Anti-EGFR coated LNP | 0.75 mg kg−1 total RNA (mCas9/sgRNA 3/1 w/w), 2 injections 7 days apart. | IP | - NGS analysis confirmed 82% of editing in the PLK1 locus. | 33 | |
| - Tumour growth was significantly inhibited, and the overall survival was increased by ∼80%. | |||||||
| Familial hypercholesterolaemia | Pcsk9 | SORT-LNP | C57BL/6J | 2.5 mg kg−1 total RNA (mCas9/sgPcsk9 4/1 w/w), 3 doses every other day. | IV | - High indel formation at Pcsk9 locus in liver tissues. | 31 |
| - KO of Pcsk9 gene induced ∼100% reduction of Pcsk9 protein in the liver (WB) and serum (ELISA). | |||||||
| ATTR amyloidosis | Ttr | LNP (INT01) | CD-1 | 0.3, 1 and 3 mg kg−1 total RNA (mCas9/sgTtr) | IV | - Liver editing levels were dose dependent. More than 97% reduction of serum Ttr and ∼70% editing in the liver was observed at the highest dose. | 106 |
| - Editing rates and serum reduction of Ttr remained stable for at least 12 months. | |||||||
| Disease model | Target | Vector | Mouse model | Dose | Admin. route | Findings | Ref. |
|---|---|---|---|---|---|---|---|
| ArgNP = Arginine-functionalized gold nanoparticles, Aβ = amyloid beta, Bace1 = beta-secretase 1, CTX = cardiotoxin, DLNP = modified dendrimer lipid nanoparticles, DMD = dystrophin, DPP-4 = dipeptidyl peptidase 4, EGFP = Enhanced Green Fluorescent Protein, GLP-1 = glucagon-like peptide 1, GNP = gold nanoparticle, IC = intracranial, ICP-MS = inductively coupled plasma mass spectrometry, IHC = immunohistochemical analysis, IM = intramuscular injection, IT = intratumoural injection, IV = intravenous, LHNP = liposome-templated hydrogel nanoparticles, mGluR5 = metabotropic glutamate receptor 5, MR = molar ratio, NC = Self assembled nanoclews, NL = lecithin-based liposomal nanocarrier particle, Nrf2 = nuclear factor erythroid 2-related factor 2, NTA-NP = NTA-disulfanediyldipropionate-poly(ethyleneglycol)-b-poly(caprolactone) nanoparticle, Pcsk9 = proprotein convertase subtilisin kexin type 9, PD-L1 = programmed death ligand 1, Plk1 = Polo-like kinase 1, PTEN = phosphatase and tensin homolog, R7L10-NC = R7L10-peptide-based nanocomplex, SMOF NP = PH-responsivesilica–metal–organic framework hybrid nanoparticle, SR = subretinal injection, SRD5A2 = steroid 5 alpha-reductase 2, Tregs = regulatory T cells, US-activated MB-NL = ultrasound-activated microbubble nanoliposome, VEGF = vascular endothelial growth factor, VLN = virus-like nanoparticle. | |||||||
| Fragile X syndrome | mGluR5 | CRISPR-Gold | Fmr1 | 50 pmol Cas9 and 50 pmol sg mGluR5 | IC | - TIDE analysis identified 14.6% indel formation at the target site, and no significant off-target effects were detected. | 30 |
| - mGluR5 mRNA and protein levels were reduced by ∼50%. | |||||||
| - Reduced hyper locomotor activities were observed in CRISPR-Gold treated animals. | |||||||
| Glioma | Plk1 | LHNP | Athymic nude | 1 mg LHNP (containing Cas9 protein/sgPlk1) 3 times a week for 3 weeks | IV | - WB analysis showed that Plk1 expression was significantly inhibited in both subcutaneous (∼30%) and intracranial (∼60%) U87 model. | 107 |
| - LHNP treatment significantly reduced the tumour volume of subcutaneous tumours. | |||||||
| - LHNP treatment significantly prolonged the overall animal survival compared to animals treated with PBS (40 vs. 29 days). | |||||||
| Macrophage-related diseases | PTEN | ArgNP | BALB/c | 0.9 nmol Cas9E20 + 0.9 nmol sgPTEN for 4 consecutive days | IV | - ICP-MS analysis confirmed higher uptake of ArgNP by macrophages rather than B and T cells (liver and spleen). This was also reflected by T7EI assay showing higher PTEN indel formation in the macrophages (8% vs. 4% for B/T cells). | 108 |
| Alzheimer's disease | Bace1 | R7L10-NC | 5XFAD | 10 μL of nanocomplex containing 14.3 μM of RNP, and R7L10 peptide (1/3 sgRNA/R7L10 w/w) | IC | - T7EI assay showed effective gene editing, which was confirmed by WB, showing 70% reduction in Bace1 expression in CA3 hippocampal region. | 109 |
| - IHC revealed a significant reduction of Aβ plaque accumulation and decreased Aβ42 secretion. | |||||||
| - Improved spatial working memory. | |||||||
| Type 2 diabetes | DPP-4 | NL | db/db | 1.6 μgCas9 + 0.55 μg sgDPP-4 once daily for 28 days | IV | - NL treatment resulted in reduced levels of DPP-4 mRNA and protein levels in the liver. | 110 |
| - GLP-1 serum concentration of NL treated animals (21%) increased to levels comparable to that of positive control Sitagliptin (24%). As a consequence, glucose tolerance increased, and insulin resistance was reduced. | |||||||
| Androgenetic alopecia | SRD5A2 | US-activated MB-NL | C57BL/6 | 1 ml of 5% glucose solution containing 0.75 μM Cas9 and 1.25 μM sgRNA daily for 7 weeks [±US] | Topical | - US-activated MB-NL treated animals showed up to 90% hair regeneration. | 111 |
| - US activated treatment induced efficient gene editing (71.6%), which resulted in significant reduction of SRD5A2 mRNA and protein levels. | |||||||
| - As a result of SRD5A2 suppression VEGF expression increased 2-fold in US treated skin (ELISA). | |||||||
| Nasopharyngeal carcinoma | Nrf2 | NTA-NP | nu/nu | 1.5 mg kg−1 Cas9/sgRNA every 2 days for 9 days. [±1 mg kg−1 Ce6, ±NIR] | IV | - The inclusion of targeting peptide and NIR ensured a more efficient knockout of the Nrf2 gene (31.2% vs. 15.4%, T7EI), which resulted in reduced protein expression (IFC, WB). | 112 |
| - NIR mediated in vivo gene editing with concomitant Ce6 reduced tumour growth by 87.7% compared to NTA/NIR/Ce6 treatment alone (41.8%) which resulted in significant extension of the overall animal survival. | |||||||
| Duchenne muscular dystrophy | DMD | DLNP | ΔEx44 DMD | 1 mg kg−1 sgDMD, once weekly for 3 weeks. (Cas9/sgRNA MR 1/3) | IM | - Efficient gene editing (5.7%) was confirmed by T7EI assay in the treatment group. | 113 |
| - Treatment with DLNP restored dystrophin protein expression as confirmed by IHC and WB (+4.2%). | |||||||
| Hypercholesterolemia | Pcsk9 | DLNP | C57BL/6 | 2.5 mg kg−1 sgPcsk9, once weekly for 3 weeks. (Cas9/sgRNA MR 1/3) | IV | - Pcsk9 protein levels were reduced in both serum and liver tissue. | 113 |
| Melanoma | PD-L1 | VLN | C57BL/6 | 10 μg Cas9 every 3 days for 19 days. (Cas9/sgRNA MR 1/1) [±0.55 mg kg−1 Axitinib] | IV | - WB revealed efficient (45.1% PD-L1 knockout) in significant tumour growth suppression and increase of the survival time. | 114 |
| - The tumour growth inhibition and increase in survival was further improved with combinatorial Axitinib. This combinatorial treatment ensured significant increase in CD8+cells infiltration and reduced Tregs population. | |||||||
| Duchenne muscular dystrophy | DMD | GNP | C57BL/10ScSn-Dmdmdx/J | 120 μg Cas9 + 30 μg sgDMD + 6.75 pmol of donor DNA. [±CTX] | IM | - HDR frequency in animals treated with GNP was significantly higher than naked RNP + donor DNA treated group (5.4% vs. 0.3%). | 29 |
| - Tissue immunofluorescence showed relevant dystrophin levels comparable to wild-type animals. | |||||||
| - GNP treatment improved animals’ strength and agility even without CTX. | |||||||
| Osteosarcoma | EGFP | NC | Nude | 40 μg of Cas9 (Cas9/sgRNA MR 1/1) | IT | - Immunofluorescence staining of U20S.EGFP subcutaneous tumours showed reduction of ∼25% of EGFP expression at 10 days post intratumoural injection of NC compared to untreated group or group treated with negative sgRNA. | 115 |
| Retinal diseases | Td - Tomato | SMOF NP | Ai14 mice | 4 μg of RNP (Cas9/sgRNA MR 1/1) | SR | - Confocal laser scanning microscopy images of the retinas excised 13–14 days post gene editing showed strong tdTomato signal confirming successful targeting of the Ai14 stop cassette. | 116 |
5.1 Lipid-based nanoparticles
In the past decades, lipid nanoparticles (LPNs) have been used to deliver a plethora of cargos, including small molecules, drugs117 and nucleic acids.118 Recently, their application for CRISPR research has shown great progress in enhancing the stability of the CRISPR machinery as well as improving its bioavailability in vivo. Lipid nanoparticles are typically formulated through rapid mixing of a water phase containing the negatively charged nucleic acids and a positively charged organic phase made of different lipids: a sterol (e.g. cholesterol), a helper lipid (e.g. 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPE)), a cationic lipid (i.e., 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP)) and a polyethylene glycol (PEG)-lipid (e.g. α-(3′-{[1,2-di(myristyloxy)propanoxy]carbonylamino}propyl)-ω-methoxy, polyoxymethylene (PEG2000-C-DMG)). Cationic lipids electrostatically complex with the anionic nucleic acids to form the nanoparticles and guarantee high encapsulation efficiency of the cargoes.58 Cholesterol and helper lipids confer structural stability to the lipid bilayer and enhance transfection efficacy via membrane fusion and endosomal escape.119 PEGylated lipids are arrayed on the outer layer to reduce LNPs aggregation and opsonization in the bloodstream.120A number of studies have employed LNPs for the delivery of CRISPR plasmids to disrupt different genes (Nf1, CD80, CD86, CD40, Plk1, BCR-ABL, DNMT1) and have significantly enhanced the therapeutic outcomes in animal models for type 1 and 2 diabetes (T1D, T2D),88,90,95 melanoma,87 chronic leukaemia91 and ovarian cancer.121 While promising, plasmid delivery, like viral vectors, still carries a risk of genomic integration, as well as immunogenicity, this has shifted the attention towards developing RNA-based gene editing (Cas9 mRNA/sgRNA) approaches.
Cationic lipid assisted nanoparticles (CLAN) encapsulating mCas9/sgRNA were utilised to knockout NLRP3 in macrophages and alleviate LPS-induced septic shock, monosodium urate crystal-induced peritonitis and high-fat diet-induced T2D.103 In another study, the same group used CLAN to deliver mCas9/sgCD40 to knockout CD40 in dendritic cells (DCs) to limit allogenic immune response caused by antigen presentation to T cells responsible for transplants rejection.105 Others have employed the use of different types of LNPs for the treatment of hypercholesterolemia,31,101 transthyretin amyloidosis,106 glioblastoma and ovarian cancer.33 As reported in Tables 2 and 3, CLANs have been used in multiple studies for the in vivo delivery of CRISPR plasmids and RNAs. CLANs were originally designed for siRNA delivery122 and later adapted for the delivery of CRISPR plasmids88,95 and RNAs, they are fabricated by a double emulsion method and comprise an inner core of cationic lipid N,N-bis(2-hydroxyethyl)-N-methyl-N-(2-cholesteryloxycarbonyl aminoethyl) ammonium bromide (BHEM-Chol), and an external layer of polyethylene glycol-block-poly(D,L-lactic-co-glycolic acid) (PEG-b-PLGA). Both surface charge and PEGylation can have effects on the uptake of the LNPs by specific subsets of cells. For example, Xu, C. et al. in their screenings found that a higher surface charge and moderate PEG density could ensure a better cellular internalization by macrophages.103
A significant step forward in the formulation of LNPs was the substitution of permanently cationic lipids with ionizable lipids. Ionizable lipids can electrostatically bind nucleic acids at low pH, ensuring high encapsulation efficiency and stability of the complex. At physiological pH, the ionizable lipid confers a near neutral charge on the LNP, which reduces interactions with serum proteins in the bloodstream. Within the acidic lumen of the endosome, the lipid head becomes charged interacting with the negatively charged membrane, while the non-polar tail facilitates fusion and endosomal escape, releasing the nucleic acid content to the cytosol.123 Progress in lipid design and LNPs formulation led to the FDA approval of ONPATTRO™ in 2018, a siRNA-based drug, formulated by the incorporation of an ionizable lipid 4-(dimethylamino)-butanoic acid, (10Z,13Z)-1-(9Z,12Z)-9,12-octadecadien-1-yl-10,13-nonadecadien-1-yl ester (DLin-MC3-DMA).124
Many groups have designed and incorporated different ionizable lipids into their formulations to further improve transfection efficiency, reduce toxicity and increase the bioavailability of the encapsulated cargos.58 For example, to allay concerns associated with the bioaccumulation of lipids following systemic delivery of LNPs, Intellia Therapeutics developed a branched ionizable lipid (LP01), that due to the inclusion of liable ester linkages, is rapidly cleared from the liver when compared to LNPs formulated with DLin-MC3-DMA (6 h vs. 24 h).106 Rosenblum et al. formulated LNPs with a novel ionizable lipid (L8) which, unlike DLin-MC3-DMA, possesses an ethanolamine linker group between the linoleic fatty acid chain and the ionizable amine head group in place of the ester linker moiety.33 In their experiments, it was shown that although mCas9/sgRNA LNPs formulated with L8 or DLin-MC3-DMA shared similar physicochemical properties and encapsulation efficiency, L8-LNPs were significantly superior at reducing green fluorescent protein (GFP) expression in vitro. Using confocal microscopy, Ramishetti et al. in their previous studies designed different ionizable lipids for the delivery of siRNA and demonstrated that LNPs formulated with L8 were better internalized by cells than other lipids.125 It could be speculated that this improved internalization capacity is responsible for the improved gene editing efficiency. Many groups synthesised libraries of lipids with the aim of enhancing the properties of ionizable lipids to deliver nucleic acids in vivo. For example, the group of Anderson screened a range of ionisable lipids for the delivery of CRISPR RNAs to knockout proprotein convertase subtilisin/kexin type 9 (Pcsk9), resulting in the development of branched lipopeptide 3,6-bis(4-(bis(2-hydroxydodecyl)amino)butyl)piperazine-2,5-dione (cKK-E12).101,126 In more recent studies they showed that LNPs formulated with ckk-E12 enhanced serum stability and albumin corona formation which promoted LNP uptake by hepatocytes.101,127
In the majority of the studies identified in Tables 2 and 3, intravenous (IV) injection was utilised as the route of administration. Following IV injection, it is reported that LNPs are mainly internalised by hepatic tissues.128 Cheng and co-workers reported a Selective Organ Targeting (SORT) strategy where nanoparticles are engineered to target specific organs, by adjusting the percentage of a 5th lipid added into the traditional four component mix.31 Cheng et al. demonstrated that by increasing the percentage of 1,2-dioleoyl-3-trimethylammonium-propane (DOTAP), a permanently cationic lipid, from 20% DOTAP to 50%, SORT LNP could be redirected from the liver to the lungs. With a similar logic they incorporated into the formulation 10 to 30% of 1,2-dioleoyl-sn-glycero-3-phosphate (18PA), a permanently negative lipid, and observed that the delivery of luciferase mRNA (mLuc) was selectively confined to the spleen. Then they studied the effect of incorporating and additional ionizable lipid such 1,2-dioleoyl-3-dimethylammonium propane (DODAP). Interestingly in this case they did not observe a change in the biodistribution profile, but instead showed that addition of 20% DODAP resulted in 10-fold higher liver delivery as compared to SORT LNP with lower or higher % of DODAP. They used this model to deliver Cas9 mRNA/sgRNA against Cre gene after intravenous administration in Cg-Gt(ROSA)26Sortm9(CAGtdTomato)Hze/J mice. Optical imaging of ex vivo organs ten days post injection confirmed tdTomato expression, as a result of successful Cre recombination, in both liver and lungs consistent with their hypothesis. As a therapeutic proof of concept, they utilized 20% DODAP SORT LNP to target Pcsk9. Following three IV injections, they confirmed efficient gene editing and achieved 100% reduction in Pcsk9 serum levels.
Many studies attempted to perform gene editing in for cancer therapy. Rosenblum et al. have developed anti-EGFR-antibody functionalised LNPs, following peritoneal administration (IP), LNPs were demonstrated to accumulate in OV8 ovarian cancer.33 The same group utilised direct intracranial administration to perform gene editing of E00 GBM tumours and reduce the off-target effects.33 To enhance the tumour accumulation of the CRISPR/Cas9 plasmid and reduce off target effects, Li et al. formulated cationic liposomes conjugated to R8-dGR (Cys-RRRRRRRRdGR), a cell penetrating peptide displaying a dual affinity to integrin αvβ3 and neuropilin-1 (NRP-1) receptors, both highly expressed on pancreatic cancer cells.92 Liposomes consisting of cholesterol, DOTAP and DSPE-PEG200− R8-dGR were formulated by film hydration method. The chemo drug paclitaxel (PTX) and CRISPR plasmid targeting hypoxia-inducible factor-1α (HIF-1α) were encapsulated into the liposomes using the post-insertion method. In vivo biodistribution studies confirmed higher accumulation of the R8-dGR-liposomes into BxPC-3 xenografts compared to the non-functionalized ones. These results were also reflected in the therapy studies where R8-dGR liposomes outperformed untargeted liposomes in inhibiting the tumour growth. Moreover, it was encouraging to see that applying gene editing and chemotherapy modalities outperformed single therapeutic approaches in suppressing metastasis and prolonging animal survival.
While in the studies described above different groups have used either a passive or active targeted LNP administered parentally, Ryu et al. have developed a unique multicompartment ultrasound-guided lipid-based vector for topical use in the treatment of alopecia.111 First RNP targeting steroid type II 5-alpha-reductase (SRD5A2) were complexed and encapsulated into nanoliposomes (NL) following lyophilization, rehydration and then extrusion. Next, lipidic microbubbles (MB) were generated by Sulphur hexafluoride (SF6) gas bubbling. NL were then conjugated onto the MB following pyridyl disulfide reaction chemistry. The microbubbles conjugated nanoliposome (MB-NL) system, when applied with concomitant ultrasound, showed promising results in enhancing the formation of the hair follicle and hair growth compared to the control groups (Table 4).
5.2 Polymer-based nanoparticles
Polymeric nanoparticles (PNPs) hold the promise of being a potential alternative for safer non-viral delivery of genetic cargos. PNPs are commonly synthesized using cationic polymers that can electrostatically bind the negatively charged phosphate moieties of the DNA/RNA backbone. Once complexed, PNPs can ensure that the nucleic acids are protected from nucleases found in the body, reduce immunogenicity and improve cellular uptake of the genetic cargos.129 Common polymers used in the synthesis of polymer-based nanoparticles include poly(lactic-co-glycolic acid) (PLGA), polyethyleneimine (PEI), poly-beta-amino ester (PBAE), poly L-lysine (PLL), chitosan and poly(amidoamine) (PAMAM).130Zhang et al. formulated polymeric nanoparticles using cholesterol (CHO)-terminated ethanolamine–aminated poly (glycidyl methacrylate) (CHO-PGEA) rich in hydroxyl groups to deliver a plasmid cassette encoding Cas9 and sgRNA against fibrillin-1 (Fbn1), which mutations are responsible for Marfan syndrome carrying several complications including aortic aneurysms.131 CHO-PGEA was selected based on previous findings suggesting that the presence of hydroxyl groups in both lipid-132 and polymer-133 based transfection reagents improved their safety, biocompatibility and transfection efficiency. Du et al. showed how the formulated nanoparticles significantly reduced the protein adsorption and weakened the haemolytic properties as compared to polyethyleneimine (PEI)-CRISPR conjugates. Because accumulation of nanoparticles is difficult to achieve in the aorta, Du et al. treated the animals daily for 7 days with a pressor dose of Angiotensin II (Ang II) prior to the administration of the nanoparticles. They hypothesized that this process would increase vascular wall pressure, enhancing the vascular permeability, thus facilitating nanoparticles accumulation in the aorta. This was confirmed by ex vivo optical images showing that, following intravenous administration, Sulfo-Cyanine5 (Cy5) labelled CHO-PGEA NPs accumulated in the aorta with a higher significance in animals pre-treated with Ang II. In further studies, the authors confirmed successful gene editing of Fbn1, with no signs of toxicity.
Qi et al. formulated lactose-derived branched cationic biopolymers (LBP) delivering plasmid encoding for Cas9 and sgRNA against Survivin to treat orthotopic hepatocellular carcinoma (HCC).94 The authors hypothesised that lactose is likely to be a highly efficient carrier for HCC therapy, due to the great affinity between asialoglycoprotein receptor (ASGPr), expressed prevalently on parenchymal liver cells and hepatic cancer cells and the galactose residues present in the lactose. First, lactose was reacted with cystamine to produce amine lactose (L-NH2). Then, an open-ring reaction between L-NH2 and triglycidyl iso-cyanurate was performed to obtain LBP. The highly cationic LBP were then electrostatically complexed with the LBP. In the formulation process, cysteamine was used to introduce disulphide linkages that can be easily degraded in the reductive intra-cellular space. In fact, in the cytoplasm and nucleus, the high concentrations of redox molecules, such glutathione, facilitate the cleavage of the disulphide bonds and the release of the nucleic acids.134 To confirm their hypothesis on LBP affinity to liver cells via galactose and ASGPr interaction, the authors intravenously administered LBP or negative control PEI labelled with Cyanine7-N-hydroxysuccinimide ester (Cy7-NHS). Optical imaging of liver tissues four hours post injection showed significantly higher Cy7 signal in the animals injected with LBP as compared to those injected with PEI. Therapy studies revealed that animals receiving LBP had reduced tumour growth compared to untreated controls. Efficacy of Survivin gene editing was further enhanced by co-administration of sorafenib, a multi-kinase inhibitor clinically used for the treatment of HCC.
In another study, Tu et al. utilised PEG-coated PEI-PLGA PNPs for the co-encapsulation of CRISPR/Cas9-Cdk5 plasmid and paclitaxel to trigger immunogenic cell death in tumour models for melanoma and colon carcinoma.96 Given the hydrophobicity of PXT, the drug was firstly encapsulated into PEI-PLGA. Then, the CRISPR/Cas9-Cdk5 plasmid was incubated with PXT-PEI-PLGA nanoparticles (NPPXT) to facilitate electrostatic condensation of the nucleic acids onto the positively charged nanoparticles. The PXT-CRISPR nanoparticles (NPPXT-Cdk5), were then coated with PEG conjugated to maleic acid amide linker (PEG-CDM) to protect the plasmid from enzymatic degradation and reduce opsonization and phagocytosis of the PNPs. Once the PEG-CDM coated NPPXT-Cdk5, reach the acidic tumour microenvironment the CDM linker is cleaved, and the cationic NPPXT-Cdk5 are taken up by cancer cells through endocytosis. IV injection of these nanoparticles caused efficient Cdk5 gene KO, which resulted in the reduction of the programmed death-ligand 1 (PD-L1). Immunohistochemical (IHC) analysis of tumour tissues identified the presence of different damage-associated molecular patterns (DAMPs) in the groups treated with paclitaxel, confirming PTX triggered immunogenic cell death. Animals receiving combination treatment showed significantly reduced tumour growth as compared to untreated controls and animals treated with either gene editing or chemotherapy. Cdk5 targeting using PNPs was also adopted by Deng et al. for the treatment of melanoma.97 To address toxicity concerns caused by the polymer PEI, Deng et al. utilized the biodegradable cationic polymer poly(β-amino esters) (PBAEs). Intravenous injection of these PNPs resulted in successful gene editing leading to significantly reduced tumour growth compared to anti-PD-L1 antibody treatment controls in both B16F10 and 4T1 tumour models.
Conjugation of tumour-specific aptamers to NPs is often utilized to increase tumour accumulation of the NPs with the final aim of improving treatment efficacy and concomitant reduction of overall toxicity. Liang et al. designed PEG-PEI-Cholesterol (PPC) NPs encapsulating CRISPR plasmid targeting VEGFA and conjugated with an osteosarcoma cell-specific aptamer.84 These NPs showed an improved tumour-specific biodistribution compared to their non-functionalized counterpart. The delivery system could induce significant VEGFA KO, which resulted in reductions in tumour growth and reduced lung metastasis.
To encapsulate pCas9/sgRNA and donor Retinoschisin 1 (RS1)-GFP-plasmid, Chou et al. developed supramolecular nanoparticles (SMNP) generated through the self-complexation of three different polymers, namely β-cyclodextrin (CD)-grafted branched polyethyleneimine (CD-PEI), adamantane (Ad)-grafted poly-amidoamine dendrimer (Ad-PAMAM), and Ad-grafted poly(ethylene glycol) (Ad-PEG), as an application for X-linked juvenile retinoschisis treatment.27 Following a high-throughput approach based on two microfluidic systems135 the authors screened different formulations in vitro in B16 cells and identified the best formulation which was then intravitreally injected into the mice eyes. SMNP treatment resulted in successful RS1/GFP expression at 18 post injection as detected by optical coherence tomography, Sanger sequencing and IHC analysis, with no evidence of histological abnormalities.
5.3 Cell-penetrating peptides
Cell-penetrating peptides (CPPs) are short lengths of amino acids (typically 5–30 amino acid residues in length) that can be cationic, amphipathic, or non-polar. They can enhance transport of proteins, drugs, nucleic acids and imaging agents directly across the cell membrane or following clatherin-mediated endocytosis.54 CPPs have been safely used in vivo in several pre-clinical and clinical research studies for imaging and therapeutic purposes.136Wang et al. formulated PEGylated helical polypeptide nanoparticles (P-HNPs) which consisted of poly(γ-4-((2-(piperidin-1-yl)ethyl)amino-methyl)benzyl-L-glutamate) (PPABLG) surrounded by PEG-PEIencapsulating Plk1 targeting CRISPR plasmid.89 PPABLG was chosen based on previous findings by the Cheng group,137 who proposed a core of α-helical polypeptide with side-chains featuring a cationic terminus to facilitate effective gene transfer. Due to the non-natural amino acid sequence in the PPABLG backbone and the long hydrophobic side chains, P-HPNs featured improved stability against nucleases. Following three intratumoural injections of P-HNPs, successful in vivo PLK1 gene knockout was confirmed by Western Blot. Reduction in Plk1 expression significantly inhibited HeLa xenograft growth and improved overall animal survival.
To increase CRISPR/Cas9 gene editing specificity to target cells and reduce potential off-target effects Chung et al. formulated oligoplexes consisting of adipose tissue targeting sequence (ATS) peptide (CKGGRAKDC) conjugated to a 9-mer of D-arginine (9R),28 previously reported to specifically bind Prohibitin protein expressed on the membrane of adipose tissues.138 CRISPR plasmid for dead Cas9 (dCas9) and sgRNA against fatty acid binding protein 4 (Fabp4) were complexed with ATS-9R to form nanocomplexes as a treatment for obesity induced metabolic syndrome. Peritoneal injection of these nanocomplexes in high fat diet (HFD) mouse models resulted in reduced inflammation in adipose tissues as well as reduction in blood glucose levels and body weight.
Owing to the amphiphilic nature, R7L10 peptide (NH2-RRRRRRRRLLLLLLLLLL-COOH) has been widely used to encapsulate hydrophobic drugs into the core of self-assembling peptide-micelles.139 R7L10 micelles possess a highly cationic surface charge making them good candidates for nucleic acid encapsulation and delivery. Park et al. used R7L10 peptide to formulate nanocomplexes carrying RNP to target beta-secretase 1 (Bace1) and tyrosine hydroxylase (Th) in post-mitotic neurons.109 The complexation of R7L10 peptide and the RNP was facilitated by electrostatic interactions between the negatively charged RNP and cationic R7L10. As reported in Table 4, gene knockout in the brain was confirmed and was able to induce measurable therapeutic improvement in Alzheimer's disease mouse models such as significant plaque reductions and improved spatial working memory.
5.4 Gold-based nanoparticles
Gold nanoparticles (GNP) are inorganic vectors which due to their tuneable size, shape and large surface area can be utilized to load different cargos via ionic or covalent coupling, electrostatic interactions or physical adsorption.140 Notably GNP benefit from surface plasmon resonance properties making them suitable for thermal and photodynamic therapeutic applications.141In the context of in vivo CRISPR delivery, GNP were utilized to deliver large cargoes such plasmids and RNP. Following the loading of the CRISPR cargoes onto the GNP, these were functionalized through different approaches to enhance their cellular and nuclear internalization. Based on previous reports suggesting that HIV-1-transactivator of transcription (TAT) peptide can promote nuclear localization of GNs and improve pDNA in vitro transfection,142 Jiang group loaded TAT peptide onto gold nanoclusters (GNs) generating positively charged TAT-GNs.86 This binary complex was then electrostatically complexed with Cas9 protein and sgPlk-1 plasmid. To protect the cargoes from enzymatic degradation and facilitate the circulation of the nanoclusters in the bloodstream, TAT-GNs-Cas9-sgPlk-1 complexes were coated with a cationic lipid shell consisting of DOTAP, DOPE and cholesterol and a PEG layer (DSPE-PEG). This vehicle could induce significant reduction in Plk-1 expression as well as ensuring significant suppression of melanoma tumours following a course of multiple intratumoural injections.
To enhance membrane fusion of GNPs and facilitate translocation of the RNP directly into the cytosol bypassing endosomes, Lee et al. formulated arginine coated self-assembling gold nanocomplexes.108 Following systemic administration of these nanocomplexes, significant accumulation of gold was observed in macrophages of liver and spleen, which resulted in efficient gene editing of phosphatase and tensin homolog (PTEN) gene. The authors speculate that the preferential accumulation of the nanocomplexes in macrophages might be due to the large particles size (285 nm) and the absence of PEGylation. These two factors can facilitate the passive targeting of macrophages as described by Chan's group in their studies investigating the macrophages’ uptake of GNs in correlations with the nanoparticles size, PEGylation and protein corona adsorption.143
Lee et al. designed CRISPR-gold nanoparticles which showed efficient transfection in different cell lines.29,30,57 The carrier utilized in this study was composed of an internal gold core which was firstly coated with a 5′ thiol modified single stranded oligonucleotide, containing a region complementary to the donor DNA. Then, the donor DNA was hybridized with DNA-SH, followed by adsorption of RNP, forming a CRISPR-gold complex. Next, the CRISPR-gold complex was coated with a cationic endosomal disruptive polymer Poly-N-[N-(2-aminoethyl)-2-aminoethyl] aspartamide (PAsp(DET)). Pasp(DET) features selective membrane destabilizing properties within the acidic endosomal compartment enabling efficient endosomal escape of the cargoes.144,145 Once in the cytoplasm, glutathione cleaves the thiol linker of DNA-SH allowing the release of the donor DNA and RNP. The cationic charge of the polymer triggered endosomal disruption causing the release of the CRISPR components. Lee et al. utilized these nanoparticles for the correction of a point mutation in the dystrophin gene for the treatment of Duchenne Muscular Dystrophy (DMD).29 Intramuscular injection of the designed nanoparticles could ensure 5.4% editing of the dystrophin gene which was reflected in improved strength and agility of the treated animals. These nanoparticles were then utilized by the same group in another study to deliver RNP targeting the metabotropic glutamate receptor 5 (mGluR5) gene.30 The study showed that intracranial injection of the nanocomplex induced successful mGluR5 knockout and subsequently reduced phenotypic repetitive behaviours associated with Fragile X Syndrome. In vitro biocompatibility testing also concluded that CRISPR-gold showed no adverse effects on neuronal membrane health or neuronal excitability. However, in vivo testing only administered very small quantities (picomoles) of gold to mice brain tissues. Furthermore, the study acknowledged that with multiple injections of CRISPR–Gold, the gold nanoparticle core could potentially accumulate in tissues of the brain and cause toxicity. This is a significant limitation when it comes to clinical translation.
5.5 Hybrid nanoparticles
Hybrid nanoparticles (HNP) are formulated combining multiple types of materials, with the aim of enhancing the properties or creating synergy between the individual components. HNP, with different compositions, were utilized in four studies for the delivery of CRISPR plasmid and RNP.Wang et al. generated hybrid nanoparticles combining gold, targeting peptides and lipids.32 Plk-1 targeting plasmids were electrostatically complexed with the TAT-GNs and coated with a combination of lipids (DOTAP, DOPE, cholesterol, PEG2000-DSPE). The therapeutic effect of TAT-GNs mediated Plk-1 gene editing of subcutaneous melanoma tumour model was further enhanced by concomitant application of irradiation, resulting in greater tumour eradication.
Li et al. developed hybrid nanoparticles featuring a polymeric fluorinated core encapsulating CRISPR plasmid, which was then coated with a multifunctional shell composed of PEG functionalized hyaluronan (HA) and linked to RGD peptide octa-arginine conjugates (RGD-R8).85 The authors hypothesized that the incorporation of RGD-R8 tandem peptide, into the RGD-R8-PEG-HA core–shell resulted in an artificial virus-like structure (RRPHC) that could guarantee specific targeting to integrin αvβ3 often overexpressed on tumour vascular endothelial cells. Once in the tumour, HA could be partially degraded by the hyaluronidase overexpressed in the intracellular compartments of the cells, releasing the plasmid complex. RRPHC, injected intraperitoneally, were able to successfully knockout the MTH1 gene of human ovarian cancer in a metastatic model, which resulted in a significant reduction of tumour nodules compared to control groups.
Guo et al. formulated a tumour targeted non-cationic, deformable nanolipogel (tNLG) to encapsulate CRISPR plasmids.93 The nanolipogel was based on a combination of non-cationic lipid and polymeric components. The use of non-cationic components bypassed reliance on electrostatic interactions to form the complex, whilst also avoiding cationic charge-induced toxicities. The lipid bilayer of tNLG was comprised of two lipids and a naturally occurring biopolymer (alginate). This created a polysaccharide network between the alginate hydrogel and lipid bilayers, which ensured encapsulation and retention of the CRISPR plasmids within the nanoparticles. Intracellular Adhesion Molecule 1 (ICAM-1) antibodies were then conjugated to the surface of the nanoparticles to allow for tissue-specific targeting of triple-negative breast cancer (TNBC) tissues. ICAM-1 tumour specificity was confirmed both in vitro and in vivo. Higher uptake of ICAM-1 tNLG was observed in orthotopic TNBC model compared to TNLG alone. Systemic delivery of these hybrid nanoparticles ensured high Lcn2 gene knockout, resulting in significant tumour growth reduction.
Liu et al. created a virus like nanoparticles (VLN) for the delivery of RNP in a melanoma disease model. Co-delivery of RNP with the anticancer drug Axitinib was also investigated.114 The core of VLN consisted of surface-thiolated mesoporous silica nanoparticles (MSN). For co-delivery studies, Axitinib, a small molecule inhibitor of tyrosine kinase, was loaded into the pores of the nanoparticles. The pores were then ‘locked’ by conjugation with the RNP via disulphide bonds. The complex was coated with a lipid layer in order to protect RNP from enzymatic degradation. The mechanism of RNP release from the VLN core was based on the glutathione cleavage of the disulphide bonds between RNP and the MSN core. Since glutathione tends to be aberrantly overexpressed by tumour cells, the authors hypothesized that this approach could guarantee a tumour-specific release of the RNP. This phenomenon was confirmed by in vitro Förster resonance energy transfer (FRET) analysis, which demonstrated that RNP detachment from VLN occurred exclusively in the presence of glutathione. In vivo co-treatment with RNP and Axitinib showed the most efficient inhibition of tumour growth compared to mono treatment controls.
6. Discussion
The CRISPR/Cas9 system sparked a revolution in the field of gene editing, representing a powerful tool to be exploited in different disciplines from basic science to disease modelling and development of clinical and pre-clinical therapeutic applications. The sharp increase in popularity of this technology relies on the simple design, versatility in potentially targeting any gene, and its relatively low cost. Though promising, several factors must be considered before the CRISPR/Cas9 technology can be adopted for clinical applications.Whilst it is relatively simple to perform in vitro gene editing, the in vivo translation is limited by a multitude of factors. Firstly, a key obstacle is the inability to administer any of the naked CRISPR formats directly into the body. Once injected, CRISPR cargos are quickly degraded by endogenous endonucleases and proteases found in the bloodstream. Secondly, due to their large size, anionic charge and hydrophilic nature, CRISPR formats have limited ability to cross the cell membrane and traffic into the intracellular compartments, thus impeding specific and efficient gene editing. The delivery of naked genetic material (pDNA, RNA) can rapidly induce an innate immune response via the pattern recognition receptors (e.g. TLR3, TLR7/8), which can then result in the acquisition of adaptive immunity.146 Adaptive immunity could also be elicited by the delivery of Cas9 protein which is commonly derived from Streptococcus pyogenes, and Staphylococcus aureus, bacteria that frequently infect humans.65 There is, therefore, the need for a delivery system that could guarantee protection of the CRISPR/Cas9 components, efficiently deliver it to the target cells, and attenuate its immunogenic properties. While viral vectors have been widely used for in vivo CRISPR/Cas9 gene editing, their human translation is intrinsically limited by genotoxicity, mutagenesis, and immunogenicity concerns.
Different organic and inorganic materials have been employed to formulate nanoparticles with controlled size, shape and surface charge enabling efficient encapsulation of the different CRISPR/Cas9 cargos (Fig. 4). Among these, lipid-based NPs represent the major class of non-viral vectors that have been utilized to overcome the main challenges of CRISPR/Cas9 in vivo application. Cationic lipids can electrostatically complex with the anionic nucleic acids and RNP forming nanoparticles characterized by an external phospholipidic bilayer and an aqueous core in which the genetic material lies, thus protecting the payloads from enzymatic degradation.
 | ||
| Fig. 4 Examples of different delivery systems and surface modifications adopted for CRISPR/Cas9 in vivo gene editing. Representative schematic illustration of polymeric nanoparticles (PNP), lipid nanoparticles (LNP), hybrid nanoparticles (HNP), gold nanoparticles (GNP) and cell penetrating peptides (CPP) utilized for the delivery of CRISPR/Cas9 cargos. Surface modification of these nanoparticles includes incorporation of antibodies, peptides, aptamers, and PEGylation. Created with BioRender.com. | ||
Nanoparticles, once injected, can be easily opsonized, and cleared through the reticuloendothelial system (RES), hindering the delivery of the cargos to the target cells. Functionalization of the nanoparticles with hydrophilic polymers, such as PEG, is often implemented to reduce the adsorption of serum proteins onto the NPs. This can prolong the NPs stability in the physiological fluids as well as reducing its clearance by the RES.
Once the target site has been reached, the NPs must guarantee the internalization of the cargos into the intracellular compartments. Due to their fusogenic properties, lipids are often incorporated not only in lipid-based nanocarriers but also as a coating for other types of carriers such as polymeric and gold nanoparticles, to facilitate the endocytosis of the NPs into the intracellular compartments.
Once internalized by the endosome, the NPs must remain intact and facilitate endosomal escape prior to lysosomal digestion, which otherwise will eventually degrade the genetic material. Depending on the composition of the NPs, endosomal escape could be achieved through different approaches. In the acidic environment of the endosome, pH-responsive lipids protonate and fuse with the endosomal membrane destabilizing it, enabling the release of the cargos into the cytosol.147 Cationic polymers like PAMAM and PEI, can also induce endosomal escape via the proton sponge effect.148,149 Cell-penetrating peptides can be utilized alone or to coat NPs to enhance endosomal membrane disruption or facilitate the formation of micropores that enables the release of the cargos.150
NPs are often conjugated with peptides or antibodies that can specifically bind to receptors overexpressed on the target cells (i.e., EGFR or αvβ3 on cancer cells). This approach can enhance the uptake of the nanoparticles in the organ of interest and limit unwanted off-target effects on healthy tissues.
As highlighted in this review, several types of non-viral vectors have been successfully developed to mediate the delivery of CRISRP/Cas9 in vivo with therapeutic efficacy. While promising, each technology holds specific limitations that should be accurately addressed to increase its efficacy, safety and suitability for clinical translation. A noteworthy Phase I clinical study sponsored by Intellia Therapeutics, has been evaluating the safety and pharmacodynamic of NTLA-2001, a lipid-based nanoparticle system encapsulating Cas9 mRNA and sgRNA targeting TTR protein in patients affected by hereditary ATTR amyloidosis with polyneuropathy.151 A dose-dependent response was observed 28 days post single administration, with maximal reduction in TTR expression (87%) in the group that received a dose of 0.3 mg per kilogram. This study represents the first proof of concept of the feasibility of in vivo non-viral CRISPR/Cas9 mediated gene editing in humans. While further examination is still required, these early positive outcomes are highly encouraging, further incentivising pre-clinical and clinical research for a safer and efficient in vivo non-viral CRISPR/Cas9 gene editing.
Conflicts of interest
The authors have declared that no competing interest exists.Acknowledgements
Authors would like to thank Dr Adam A Waters for proof reading the manuscript. Khuloud T. Al-Jamal acknowledges funding from the Brain Tumour Charity (GN-000398).References
- J. A. Kulkarni, D. Witzigmann, S. B. Thomson, S. Chen, B. R. Leavitt and P. R. Cullis, et al., The current landscape of nucleic acid therapeutics, Nat. Nanotechnol., 2021, 16(6), 630–643 CrossRef CAS PubMed.
- P. Hair, F. Cameron and K. McKeage, Mipomersen sodium: first global approval, Drugs, 2013, 73(5), 487–493 CrossRef CAS PubMed.
- U. Patel, M. Boucher, L. de Léséleuc and S. Visintini, in Voretigene Neparvovec: An Emerging Gene Therapy for the Treatment of Inherited Blindness, Canadian Agency for Drugs and Technologies in Health, Ottawa (ON), 2016 Search PubMed.
- M. Caillaud, M. El Madani and L. Massaad-Massade, Small interfering RNA from the lab discovery to patients’ recovery, J. Controlled Release, 2020, 321, 616–628 CrossRef CAS PubMed.
- F. P. Polack, S. J. Thomas, N. Kitchin, J. Absalon, A. Gurtman and S. Lockhart, et al., Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine, N. Engl. J. Med., 2020, 383(27), 2603–2615 CrossRef CAS PubMed.
- L. R. Baden, H. M. El Sahly, B. Essink, K. Kotloff, S. Frey and R. Novak, et al., Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine, N. Engl. J. Med., 2021, 384(5), 403–416 CrossRef CAS PubMed.
- N. Lurie, M. Saville, R. Hatchett and J. Halton, Developing Covid-19 Vaccines at Pandemic Speed, N. Engl. J. Med., 2020, 382(21), 1969–1973 CrossRef CAS PubMed.
- J. Sadoff, G. Gray, A. Vandebosch, V. Cárdenas, G. Shukarev and B. Grinsztejn, et al., Safety and Efficacy of Single-Dose Ad26.COV2.S Vaccine against Covid-19, N. Engl. J. Med., 2021, 384(23), 2187–2201 CrossRef CAS PubMed.
- J. C. Miller, M. C. Holmes, J. Wang, D. Y. Guschin, Y.-L. Lee and I. Rupniewski, et al., An improved zinc-finger nuclease architecture for highly specific genome editing, Nat. Biotechnol., 2007, 25(7), 778–785 CrossRef CAS PubMed.
- J. P. Guilinger, V. Pattanayak, D. Reyon, S. Q. Tsai, J. D. Sander and J. K. Joung, et al., Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity, Nat. Methods, 2014, 11(4), 429–435 CrossRef CAS PubMed.
- S. Chandrasegaran and D. Carroll, Origins of Programmable Nucleases for Genome Engineering, J. Mol. Biol., 2016, 428(5 Pt B), 963–989 CrossRef CAS PubMed.
- F. A. Ran, P. D. Hsu, J. Wright, V. Agarwala, D. A. Scott and F. Zhang, Genome engineering using the CRISPR-Cas9 system, Nat. Protoc., 2013, 8(11), 2281–2308 CrossRef CAS PubMed.
- L. Cong, F. A. Ran, D. Cox, S. Lin, R. Barretto and N. Habib, et al., Multiplex genome engineering using CRISPR/Cas systems, Science, 2013, 339(6121), 819–823 CrossRef CAS PubMed.
- A. E. Friedland, Y. B. Tzur, K. M. Esvelt, M. P. Colaiácovo, G. M. Church and J. A. Calarco, Heritable genome editing in C. elegans via a CRISPR-Cas9 system, Nat. Methods, 2013, 10(8), 741–743 CrossRef CAS PubMed.
- N. Chang, C. Sun, L. Gao, D. Zhu, X. Xu and X. Zhu, et al., Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos, Cell Res., 2013, 23(4), 465–472 CrossRef CAS PubMed.
- H. Wang, H. Yang, C. S. Shivalila, M. M. Dawlaty, A. W. Cheng and F. Zhang, et al., One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering, Cell, 2013, 153(4), 910–918 CrossRef CAS PubMed.
- C. Q. Song, Y. Li, H. Mou, J. Moore, A. Park and Y. Pomyen, et al., Genome-Wide CRISPR Screen Identifies Regulators of Mitogen-Activated Protein Kinase as Suppressors of Liver Tumors in Mice, Gastroenterology, 2017, 152(5), 1161–1173 CrossRef CAS PubMed.
- F. J. Sánchez-Rivera and T. Jacks, Applications of the CRISPR–Cas9 system in cancer biology, Nat. Rev. Cancer, 2015, 15(7), 387–393 CrossRef PubMed.
- T. Wan, D. Niu, C. Wu, F.-J. Xu, G. Church and Y. Ping, Material solutions for delivery of CRISPR/Cas-based genome editing tools: Current status and future outlook, Mater. Today, 2019, 26, 40–66 CrossRef CAS.
- Y. Li, Z. Glass, M. Huang, Z.-Y. Chen and Q. Xu, Ex vivo cell-based CRISPR/Cas9 genome editing for therapeutic applications, Biomaterials, 2020, 234, 119711 CrossRef CAS PubMed.
- X. Ou, Q. Ma, W. Yin, X. Ma and Z. He, CRISPR/Cas9 Gene-Editing in Cancer Immunotherapy: Promoting the Present Revolution in Cancer Therapy and Exploring More, Front. Cell Dev. Biol., 2021, 9, 674467 CrossRef PubMed.
- K. Khalaf, K. Janowicz, M. Dyszkiewicz-Konwińska, G. Hutchings, C. Dompe and L. Moncrieff, et al., CRISPR/Cas9 in Cancer Immunotherapy: Animal Models and Human Clinical Trials, Genes, 2020, 11(8), 921 CrossRef CAS PubMed.
- H. Frangoul, D. Altshuler, M. D. Cappellini, Y.-S. Chen, J. Domm and B. K. Eustace, et al., CRISPR-Cas9 gene editing for sickle cell disease and β-thalassemia, N. Engl. J. Med., 2021, 384(3), 252–260 CrossRef CAS PubMed.
- C. E. Nelson, C. H. Hakim, D. G. Ousterout, P. I. Thakore, E. A. Moreb and R. M. C. Rivera, et al., In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy, Science, 2016, 351(6271), 403–407 CrossRef CAS PubMed.
- X. Wang, A. Raghavan, T. Chen, L. Qiao, Y. Zhang and Q. Ding, et al., CRISPR-Cas9 Targeting of PCSK9 in Human Hepatocytes In Vivo-Brief Report, Arterioscler., Thromb., Vasc. Biol., 2016, 36(5), 783–786 CrossRef CAS PubMed.
- X. Song, C. Liu, N. Wang, H. Huang, S. He and C. Gong, et al., Delivery of CRISPR/Cas systems for cancer gene therapy and immunotherapy, Adv. Drug Delivery Rev., 2021, 168, 158–180 CrossRef CAS PubMed.
- S. J. Chou, P. Yang, Q. Ban, Y. P. Yang, M. L. Wang and C. S. Chien, et al., Dual Supramolecular Nanoparticle Vectors Enable CRISPR/Cas9-Mediated Knockin of Retinoschisin 1 Gene-A Potential Nonviral Therapeutic Solution for X-Linked Juvenile Retinoschisis, Adv. Sci., 2020, 7(10), 1903432 CrossRef CAS PubMed.
- J. Y. Chung, Q. U. Ain, Y. Song, S. B. Yong and Y. H. Kim, Targeted delivery of CRISPR interference system against Fabp4 to white adipocytes ameliorates obesity, inflammation, hepatic steatosis, and insulin resistance, Genome Res., 2019, 29(9), 1442–1452 CrossRef CAS PubMed.
- K. Lee, M. Conboy, H. M. Park, F. Jiang, H. J. Kim and M. A. Dewitt, et al., Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair, Nat. Biomed. Eng., 2017, 1, 889–901 CrossRef CAS PubMed.
- B. Lee, K. Lee, S. Panda, R. Gonzales-Rojas, A. Chong and V. Bugay, et al., Nanoparticle delivery of CRISPR into the brain rescues a mouse model of fragile X syndrome from exaggerated repetitive behaviours, Nat. Biomed. Eng., 2018, 2(7), 497–507 CrossRef CAS PubMed.
- Q. Cheng, T. Wei, L. Farbiak, L. T. Johnson, S. A. Dilliard and D. J. Siegwart, Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing, Nat. Nanotechnol., 2020, 15(4), 313–320 CrossRef CAS PubMed.
- P. Wang, L. Zhang, W. Zheng, L. Cong, Z. Guo and Y. Xie, et al., Thermo-triggered Release of CRISPR-Cas9 System by Lipid-Encapsulated Gold Nanoparticles for Tumor Therapy, Angew. Chem., Int. Ed., 2018, 57(6), 1491–1496 CrossRef CAS PubMed.
- D. Rosenblum, A. Gutkin, R. Kedmi, S. Ramishetti, N. Veiga and A. M. Jacobi, et al., CRISPR-Cas9 genome editing using targeted lipid nanoparticles for cancer therapy, Sci. Adv., 2020, 6(47), eabc9450 CrossRef CAS PubMed.
- Y. Ishino, H. Shinagawa, K. Makino, M. Amemura and A. Nakata, Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isozyme conversion in Escherichia coli, and identification of the gene product, J. Bacteriol., 1987, 169(12), 5429–5433 CrossRef CAS PubMed.
- Y. Ishino, M. Krupovic and P. Forterre, History of CRISPR-Cas from Encounter with a Mysterious Repeated Sequence to Genome Editing Technology, J. Bacteriol., 2018, 200(7), e00580-17 CrossRef PubMed.
- R. Barrangou, C. Fremaux, H. Deveau, M. Richards, P. Boyaval and S. Moineau, et al., CRISPR provides acquired resistance against viruses in prokaryotes, Science, 2007, 315(5819), 1709–1712 CrossRef CAS PubMed.
- K. S. Makarova and E. V. Koonin, Annotation and Classification of CRISPR-Cas Systems, Methods Mol. Biol., 2015, 1311, 47–75 CrossRef PubMed.
- A. V. Anzalone, L. W. Koblan and D. R. Liu, Genome editing with CRISPR-Cas nucleases, base editors, transposases and prime editors, Nat. Biotechnol., 2020, 38(7), 824–844 CrossRef CAS PubMed.
- F. Jiang and J. A. Doudna, CRISPR-Cas9 Structures and Mechanisms, Annu. Rev. Biophys., 2017, 46, 505–529 CrossRef CAS PubMed.
- B. Wiedenheft, S. H. Sternberg and J. A. Doudna, RNA-guided genetic silencing systems in bacteria and archaea, Nature, 2012, 482(7385), 331–338 CrossRef CAS PubMed.
- P. D. Hsu, E. S. Lander and F. Zhang, Development and applications of CRISPR-Cas9 for genome engineering, Cell, 2014, 157(6), 1262–1278 CrossRef CAS PubMed.
- D. Gleditzsch, P. Pausch, H. Muller-Esparza, A. Ozcan, X. Guo and G. Bange, et al., PAM identification by CRISPR-Cas effector complexes: diversified mechanisms and structures, RNA Biol., 2019, 16(4), 504–517 CrossRef PubMed.
- K. S. Makarova, Y. I. Wolf and E. V. Koonin, Classification and Nomenclature of CRISPR-Cas Systems: Where from Here?, CRISPR J., 2018, 1(5), 325–336 CrossRef PubMed.
- S. Shmakov, A. Smargon, D. Scott, D. Cox, N. Pyzocha and W. Yan, et al., Diversity and evolution of class 2 CRISPR-Cas systems, Nat. Rev. Microbiol., 2017, 15(3), 169–182 CrossRef CAS PubMed.
- K. Chylinski, A. Le Rhun and E. Charpentier, The tracrRNA and Cas9 families of type II CRISPR-Cas immunity systems, RNA Biol., 2013, 10(5), 726–737 CrossRef CAS PubMed.
- A. Mir, J. F. Alterman, M. R. Hassler, A. J. Debacker, E. Hudgens and D. Echeverria, et al., Heavily and fully modified RNAs guide efficient SpyCas9-mediated genome editing, Nat. Commun., 2018, 9(1), 2641 CrossRef PubMed.
- T. Gaj, C. A. Gersbach and C. F. Barbas 3rd., ZFN, TALEN, and CRISPR/Cas-based methods for genome engineering, Trends Biotechnol., 2013, 31(7), 397–405 CrossRef CAS PubMed.
- Z. Glass, M. Lee, Y. Li and Q. Xu, Engineering the Delivery System for CRISPR-Based Genome Editing, Trends Biotechnol., 2018, 36(2), 173–185 CrossRef CAS PubMed.
- Y. Fu, J. A. Foden, C. Khayter, M. L. Maeder, D. Reyon and J. K. Joung, et al., High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells, Nat. Biotechnol., 2013, 31(9), 822–826 CrossRef CAS PubMed.
- N. Rivera-Torres, K. Banas, P. Bialk, K. M. Bloh and E. B. Kmiec, Insertional Mutagenesis by CRISPR/Cas9 Ribonucleoprotein Gene Editing in Cells Targeted for Point Mutation Repair Directed by Short Single-Stranded DNA Oligonucleotides, PLoS One, 2017, 12(1), e0169350 CrossRef PubMed.
- A. K. Fajrial, Q. Q. He, N. I. Wirusanti, J. E. Slansky and X. Ding, A review of emerging physical transfection methods for CRISPR/Cas9-mediated gene editing, Theranostics, 2020, 10(12), 5532–5549 CrossRef CAS PubMed.
- S. Vaidyanathan, K. T. Azizian, A. Haque, J. M. Henderson, A. Hendel and S. Shore, et al., Uridine Depletion and Chemical Modification Increase Cas9 mRNA Activity and Reduce Immunogenicity without HPLC Purification, Mol. Ther.–Nucleic Acids, 2018, 12, 530–542 CrossRef CAS PubMed.
- H. Zhang and X. Xia, RNA cancer vaccines: developing mRNA nanovaccine with self-adjuvant property for cancer immunotherapy, Hum. Vaccines Immunother., 2021, 1–4 Search PubMed.
- J. Devoldere, H. Dewitte, S. C. De Smedt and K. Remaut, Evading innate immunity in nonviral mRNA delivery: don't shoot the messenger, Drug Discovery Today, 2016, 21(1), 11–25 CrossRef CAS PubMed.
- G. Mullally, K. van Aelst, M. M. Naqvi, F. M. Diffin, T. Karvelis and G. Gasiunas, et al., 5′ modifications to CRISPR-Cas9 gRNA can change the dynamics and size of R-loops and inhibit DNA cleavage, Nucleic Acids Res., 2020, 48(12), 6811–6823 CrossRef CAS PubMed.
- J. Filippova, A. Matveeva, E. Zhuravlev and G. Stepanov, Guide RNA modification as a way to improve CRISPR/Cas9-based genome-editing systems, Biochimie, 2019, 167, 49–60 CrossRef CAS PubMed.
- S. Aghamiri, S. Talaei, A. A. Ghavidel, F. Zandsalimi, S. Masoumi and N. H. Hafshejani, et al., Nanoparticles-mediated CRISPR/Cas9 delivery: Recent advances in cancer treatment, J. Drug Delivery Sci. Technol., 2020, 56, 101533 CrossRef CAS.
- M. L. Guevara, F. Persano and S. Persano, Advances in Lipid Nanoparticles for mRNA-Based Cancer Immunotherapy, Front. Chem., 2020, 8, 589959 CrossRef CAS PubMed.
- C.-F. Xu, G.-J. Chen, Y.-L. Luo, Y. Zhang, G. Zhao and Z.-D. Lu, et al., Rational designs of in vivo CRISPR-Cas delivery systems, Adv. Drug Delivery Rev., 2021, 168, 3–29 CrossRef CAS PubMed.
- B. Sun, H. Chen and X. Gao, Versatile modification of the CRISPR/Cas9 ribonucleoprotein system to facilitate in vivo application, J. Controlled Release, 2021, 337, 698–717 CrossRef CAS PubMed.
- W. L. Chew, M. Tabebordbar, J. K. Cheng, P. Mali, E. Y. Wu and A. H. Ng, et al., A multifunctional AAV-CRISPR-Cas9 and its host response, Nat. Methods, 2016, 13(10), 868–874 CrossRef CAS PubMed.
- A. Li, M. R. Tanner, C. M. Lee, A. E. Hurley, M. De Giorgi and K. E. Jarrett, et al., AAV-CRISPR Gene Editing Is Negated by Pre-existing Immunity to Cas9, Mol. Ther., 2020, 28(6), 1432–1441 CrossRef CAS PubMed.
- J. M. Crudele and J. S. Chamberlain, Cas9 immunity creates challenges for CRISPR gene editing therapies, Nat. Commun., 2018, 9(1), 3497 CrossRef PubMed.
- C. T. Charlesworth, P. S. Deshpande, D. P. Dever, J. Camarena, V. T. Lemgart and M. K. Cromer, et al., Identification of preexisting adaptive immunity to Cas9 proteins in humans, Nat. Med., 2019, 25(2), 249–254 CrossRef CAS PubMed.
- D. L. Wagner, L. Amini, D. J. Wendering, L.-M. Burkhardt, L. Akyüz and P. Reinke, et al., High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population, Nat. Med., 2019, 25(2), 242–248 CrossRef CAS PubMed.
- C. Liu, L. Zhang, H. Liu and K. Cheng, Delivery strategies of the CRISPR-Cas9 gene-editing system for therapeutic applications, J. Controlled Release, 2017, 266, 17–26 CrossRef CAS PubMed.
- M. Hashimoto and T. Takemoto, Electroporation enables the efficient mRNA delivery into the mouse zygotes and facilitates CRISPR/Cas9-based genome editing, Sci. Rep., 2015, 5, 11315 CrossRef CAS PubMed.
- C. L. Xu, M. Z. C. Ruan, V. B. Mahajan and S. H. Tsang, Viral Delivery Systems for CRISPR, Viruses, 2019, 11(1), 28 CrossRef CAS PubMed.
- Y. Yang, L. Wang, P. Bell, D. McMenamin, Z. He and J. White, et al., A dual AAV system enables the Cas9-mediated correction of a metabolic liver disease in newborn mice, Nat. Biotechnol., 2016, 34(3), 334–338 CrossRef CAS PubMed.
- L. B. Ryø, E. A. Thomsen and J. G. Mikkelsen, Production and Validation of Lentiviral Vectors for CRISPR/Cas9 Delivery, Methods Mol. Biol., 2019, 1961, 93–109 CrossRef PubMed.
- C. A. Lino, J. C. Harper, J. P. Carney and J. A. Timlin, Delivering CRISPR: a review of the challenges and approaches, Drug Delivery, 2018, 25(1), 1234–1257 CrossRef CAS PubMed.
- H. Yin, W. Xue, S. Chen, R. L. Bogorad, E. Benedetti and M. Grompe, et al., Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype, Nat. Biotechnol., 2014, 32(6), 551–553 CrossRef CAS PubMed.
- M. J. Kim and N. Ahituv, The hydrodynamic tail vein assay as a tool for the study of liver promoters and enhancers, Methods Mol. Biol., 2013, 1015, 279–289 CrossRef CAS PubMed.
- B. Bonamassa, L. Hai and D. Liu, Hydrodynamic Gene Delivery and Its Applications in Pharmaceutical Research, Pharm. Res., 2011, 28(4), 694–701 CrossRef CAS PubMed.
- J. E. Hagstrom, J. Hegge, G. Zhang, M. Noble, V. Budker and D. L. Lewis, et al., A Facile Nonviral Method for Delivering Genes and siRNAs to Skeletal Muscle of Mammalian Limbs, Mol. Ther., 2004, 10(2), 386–398 CrossRef CAS PubMed.
- J. O. Hegge, C. I. Wooddell, G. Zhang, J. E. Hagstrom, S. Braun and T. Huss, et al., Evaluation of hydrodynamic limb vein injections in nonhuman primates, Hum. Gene Ther., 2010, 21(7), 829–842 CrossRef CAS PubMed.
- H. Herweijer and J. A. Wolff, Gene therapy progress and prospects: Hydrodynamic gene delivery, Gene Ther., 2007, 14(2), 99–107 CrossRef CAS PubMed.
- J. E. Rittiner, M. Moncalvo, O. Chiba-Falek and B. Kantor, Gene-Editing Technologies Paired With Viral Vectors for Translational Research Into Neurodegenerative Diseases, Front. Mol. Neurosci., 2020, 13, 148 CrossRef CAS PubMed.
- R. Goswami, G. Subramanian, L. Silayeva, I. Newkirk, D. Doctor and K. Chawla, et al., Gene Therapy Leaves a Vicious Cycle, Front. Oncol., 2019, 9, 297 CrossRef PubMed.
- X. Chen and M. A. Goncalves, Engineered Viruses as Genome Editing Devices, Mol. Ther., 2016, 24(3), 447–457 CrossRef CAS PubMed.
- G. Ronzitti, D.-A. Gross and F. Mingozzi, Human Immune Responses to Adeno-Associated Virus (AAV) Vectors, Front. Immunol., 2020, 11, 670 CrossRef CAS PubMed.
- G. J. Xu, T. Kula, Q. Xu, M. Z. Li, S. D. Vernon and T. Ndung'u, et al., Viral immunology. Comprehensive serological profiling of human populations using a synthetic human virome, Science, 2015, 348(6239), aaa0698 CrossRef PubMed.
- K. A. Hajj and K. A. Whitehead, Tools for translation: non-viral materials for therapeutic mRNA delivery, Nat. Rev. Mater., 2017, 2(10), 17056 CrossRef CAS.
- C. Liang, F. Li, L. Wang, Z.-K. Zhang, C. Wang and B. He, et al., Tumor cell-targeted delivery of CRISPR/Cas9 by aptamer-functionalized lipopolymer for therapeutic genome editing of VEGFA in osteosarcoma, Biomaterials, 2017, 147, 68–85 CrossRef CAS PubMed.
- L. Li, L. Song, X. Liu, X. Yang, X. Li and T. He, et al., Artificial Virus Delivers CRISPR-Cas9 System for Genome Editing of Cells in Mice, ACS Nano, 2017, 11(1), 95–111 CrossRef CAS PubMed.
- P. Wang, L. Zhang, Y. Xie, N. Wang, R. Tang, W. Zheng and X. Jiang, Genome editing for cancer therapy: delivery of Cas9 protein/sgRNA plasmid via a gold nanocluster/lipid core–shell nanocarrier, Adv. Sci., 2017, 4(11), 1700175 CrossRef PubMed.
- L. Zhang, P. Wang, Q. Feng, N. Wang, Z. Chen and Y. Huang, et al., Lipid nanoparticle-mediated efficient delivery of CRISPR/Cas9 for tumor therapy, NPG Asia Mater., 2017, 9(10), e441 CrossRef CAS.
- Y.-L. Luo, C.-F. Xu, H.-J. Li, Z.-T. Cao, J. Liu and J.-L. Wang, et al., Macrophage-specific in vivo gene editing using cationic lipid-assisted polymeric nanoparticles, ACS Nano, 2018, 12(2), 994–1005 CrossRef CAS PubMed.
- H. X. Wang, Z. Song, Y. H. Lao, X. Xu, J. Gong and D. Cheng, et al., Nonviral gene editing via CRISPR/Cas9 delivery by membrane-disruptive and endosomolytic helical polypeptide, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(19), 4903–4908 CrossRef CAS PubMed.
- Y. Liu, Z. T. Cao, C. F. Xu, Z. D. Lu, Y. L. Luo and J. Wang, Optimization of lipid-assisted nanoparticle for disturbing neutrophils-related inflammation, Biomaterials, 2018, 172, 92–104 CrossRef CAS PubMed.
- Y. Liu, G. Zhao, C.-F. Xu, Y.-L. Luo, Z.-D. Lu and J. Wang, Systemic delivery of CRISPR/Cas9 with PEG-PLGA nanoparticles for chronic myeloid leukemia targeted therapy, Biomater. Sci., 2018, 6(6), 1592–1603 RSC.
- M. Li, H. Xie, Y. Liu, C. Xia, X. Cun and Y. Long, et al., Knockdown of hypoxia-inducible factor-1 alpha by tumor targeted delivery of CRISPR/Cas9 system suppressed the metastasis of pancreatic cancer, J. Controlled Release, 2019, 304, 204–215 CrossRef CAS PubMed.
- P. Guo, J. Yang, J. Huang, D. T. Auguste and M. A. Moses, Therapeutic genome editing of triple-negative breast tumors using a noncationic and deformable nanolipogel, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(37), 18295–18303 CrossRef CAS PubMed.
- Y. Qi, Y. Liu, B. Yu, Y. Hu, N. Zhang and Y. Zheng, et al., A Lactose-Derived CRISPR/Cas9 Delivery System for Efficient Genome Editing In Vivo to Treat Orthotopic Hepatocellular Carcinoma, Adv. Sci., 2020, 7(17), 2001424 CrossRef CAS PubMed.
- Y.-L. Luo, L.-F. Liang, Y.-J. Gan, J. Liu, Y. Zhang and Y.-N. Fan, et al., An All-in-One Nanomedicine Consisting of CRISPR-Cas9 and an Autoantigen Peptide for Restoring Specific Immune Tolerance, ACS Appl. Mater. Interfaces, 2020, 12(43), 48259–48271 CrossRef CAS PubMed.
- K. Tu, H. Deng, L. Kong, Y. Wang, T. Yang and Q. Hu, et al., Reshaping Tumor Immune Microenvironment through Acidity-Responsive Nanoparticles Featured with CRISPR/Cas9-Mediated Programmed Death-Ligand 1 Attenuation and Chemotherapeutics-Induced Immunogenic Cell Death, ACS Appl. Mater. Interfaces, 2020, 12(14), 16018–16030 CrossRef CAS PubMed.
- H. Deng, S. Tan, X. Gao, C. Zou, C. Xu and K. Tu, et al., Cdk5 knocking out mediated by CRISPR-Cas9 genome editing for PD-L1 attenuation and enhanced antitumor immunity, Acta Pharm. Sin. B, 2020, 10(2), 358–373 CrossRef CAS PubMed.
- H. Yin, X. Yuan, L. Luo, Y. Lu, B. Qin and J. Zhang, et al., Appropriate Delivery of the CRISPR/Cas9 System through the Nonlysosomal Route: Application for Therapeutic Gene Editing, Adv. Sci., 2020, 7(14), 1903381 CAS.
- X. Gao, Z. Jin, X. Tan, C. Zhang, C. Zou and W. Zhang, et al., Hyperbranched poly(β-amino ester) based polyplex nanopaticles for delivery of CRISPR/Cas9 system and treatment of HPV infection associated cervical cancer, J. Controlled Release, 2020, 321, 654–668 CrossRef CAS PubMed.
- C. Jiang, M. Mei, B. Li, X. Zhu, W. Zu and Y. Tian, et al., A non-viral CRISPR/Cas9 delivery system for therapeutically targeting HBV DNA and pcsk9 in vivo, Cell Res., 2017, 27(3), 440–443 CrossRef CAS PubMed.
- H. Yin, C. Q. Song, S. Suresh, Q. Wu, S. Walsh and L. H. Rhym, et al., Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing, Nat. Biotechnol., 2017, 35(12), 1179–1187 CrossRef CAS PubMed.
- C. D. Sago, M. P. Lokugamage, K. Paunovska, D. A. Vanover, C. M. Monaco and N. N. Shah, et al., High-throughput in vivo screen of functional mRNA delivery identifies nanoparticles for endothelial cell gene editing, Proc. Natl. Acad. Sci. U. S. A., 2018, 115(42), E9944–E52 CrossRef CAS PubMed.
- C. Xu, Z. Lu, Y. Luo, Y. Liu, Z. Cao and S. Shen, et al., Targeting of NLRP3 inflammasome with gene editing for the amelioration of inflammatory diseases, Nat. Commun., 2018, 9(1), 4092 CrossRef PubMed.
- J. Liu, J. Chang, Y. Jiang, X. Meng, T. Sun and L. Mao, et al., Fast and Efficient CRISPR/Cas9 Genome Editing In Vivo Enabled by Bioreducible Lipid and Messenger RNA Nanoparticles, Adv. Mater., 2019, 31(33), e1902575 CrossRef PubMed.
- Y. Zhang, S. Shen, G. Zhao, C.-F. Xu, H.-B. Zhang and Y.-L. Luo, et al., In situ repurposing of dendritic cells with CRISPR/Cas9-based nanomedicine to induce transplant tolerance, Biomaterials, 2019, 217, 119302 CrossRef CAS PubMed.
- J. D. Finn, A. R. Smith, M. C. Patel, L. Shaw, M. R. Youniss and J. van Heteren, et al., A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing, Cell Rep., 2018, 22(9), 2227–2235 CrossRef CAS PubMed.
- Z. Chen, F. Liu, Y. Chen, J. Liu, X. Wang and A. T. Chen, et al., Targeted Delivery of CRISPR/Cas9-Mediated Cancer Gene Therapy via Liposome-Templated Hydrogel Nanoparticles, Adv. Funct. Mater., 2017, 27(46), 1703036 CrossRef PubMed.
- Y.-W. Lee, R. Mout, D. C. Luther, Y. Liu, L. Castellanos-García and A. S. Burnside, et al., In Vivo Editing of Macrophages through Systemic Delivery of CRISPR-Cas9-Ribonucleoprotein-Nanoparticle Nanoassemblies, Adv. Ther., 2019, 2(10), 1900041 CAS.
- H. Park, J. Oh, G. Shim, B. Cho, Y. Chang and S. Kim, et al., In vivo neuronal gene editing via CRISPR-Cas9 amphiphilic nanocomplexes alleviates deficits in mouse models of Alzheimer's disease, Nat. Neurosci., 2019, 22(4), 524–528 CrossRef CAS PubMed.
- E. Y. Cho, J. Y. Ryu, H. A. R. Lee, S. H. Hong, H. S. Park and K. S. Hong, et al., Lecithin nano-liposomal particle as a CRISPR/Cas9 complex delivery system for treating type 2 diabetes, J. Nanobiotechnol., 2019, 17(1), 19 CrossRef PubMed.
- J. Y. Ryu, E. J. Won, H. A. R. Lee, J. H. Kim, E. Hui and H. P. Kim, et al., Ultrasound-activated particles as CRISPR/Cas9 delivery system for androgenic alopecia therapy, Biomaterials, 2020, 232, 119736 CrossRef CAS PubMed.
- S. Deng, X. Li, S. Liu, J. Chen, M. Li and S. Y. Chew, et al., Codelivery of CRISPR-Cas9 and chlorin e6 for spatially controlled tumor-specific gene editing with synergistic drug effects, Sci. Adv., 2020, 6(29), eabb4005 CrossRef CAS PubMed.
- T. Wei, Q. Cheng, Y. L. Min, E. N. Olson and D. J. Siegwart, Systemic nanoparticle delivery of CRISPR-Cas9 ribonucleoproteins for effective tissue specific genome editing, Nat. Commun., 2020, 11(1), 3232 CrossRef CAS PubMed.
- Q. Liu, C. Wang, Y. Zheng, Y. Zhao, Y. Wang and J. Hao, et al., Virus-like nanoparticle as a co-delivery system to enhance efficacy of CRISPR/Cas9-based cancer immunotherapy, Biomaterials, 2020, 258, 120275 CrossRef CAS PubMed.
- W. Sun, W. Ji, J. M. Hall, Q. Hu, C. Wang and C. L. Beisel, et al., Self-assembled DNA nanoclews for the efficient delivery of CRISPR-Cas9 for genome editing, Angew. Chem., Int. Ed., 2015, 54(41), 12029–12033 CrossRef CAS PubMed.
- Y. Wang, P. K. Shahi, R. Xie, H. Zhang, A. A. Abdeen and N. Yodsanit, et al., A pH-responsive silica–metal–organic framework hybrid nanoparticle for the delivery of hydrophilic drugs, nucleic acids, and CRISPR-Cas9 genome-editing machineries, J. Controlled Release, 2020, 324, 194–203 CrossRef CAS PubMed.
- S. Samimi, N. Maghsoudnia, R. B. Eftekhari and F. Dorkoosh, Chapter 3 - Lipid-Based Nanoparticles for Drug Delivery Systems, in Characterization and Biology of Nanomaterials for Drug Delivery, ed. S.S Mohapatra, S. Ranjan, N. Dasgupta, R.K Mishra and S. Thomas, Elsevier, 2019, pp. 47–76 Search PubMed.
- P. R. Cullis and M. J. Hope, Lipid Nanoparticle Systems for Enabling Gene Therapies, Mol. Ther., 2017, 25(7), 1467–1475 CrossRef CAS PubMed.
- X. Cheng and R. J. Lee, The role of helper lipids in lipid nanoparticles (LNPs) designed for oligonucleotide delivery, Adv. Drug Delivery Rev., 2016, 99, 129–137 CrossRef CAS PubMed.
- X. Ge, L. Chen, B. Zhao and W. Yuan, Rationale and Application of PEGylated Lipid-Based System for Advanced Target Delivery of siRNA, Front. Pharmacol., 2020, 11, 598175 CrossRef CAS PubMed.
- Z.-Y. He, Y.-G. Zhang, Y.-H. Yang, C.-C. Ma, P. Wang and W. Du, et al., In vivo ovarian cancer gene therapy using CRISPR-Cas9, Hum. Gene Ther., 2018, 29(2), 223–233 CrossRef CAS PubMed.
- X.-Z. Yang, S. Dou, T.-M. Sun, C.-Q. Mao, H.-X. Wang and J. Wang, Systemic delivery of siRNA with cationic lipid assisted PEG-PLA nanoparticles for cancer therapy, J. Controlled Release, 2011, 156(2), 203–211 CrossRef CAS PubMed.
- E. Samaridou, J. Heyes and P. Lutwyche, Lipid nanoparticles for nucleic acid delivery: Current perspectives, Adv. Drug Delivery Rev., 2020, 154-155, 37–63 CrossRef CAS PubMed.
- K. Garber, Alnylam launches era of RNAi drugs, Nat. Biotechnol., 2018, 36(9), 777–778 CrossRef CAS PubMed.
- S. Ramishetti, I. Hazan-Halevy, R. Palakuri, S. Chatterjee, S. Naidu Gonna and N. Dammes, et al., A combinatorial library of lipid nanoparticles for RNA delivery to leukocytes, Adv. Mater., 2020, 32(12), 1906128 CrossRef CAS PubMed.
- Y. Dong, K. T. Love, J. R. Dorkin, S. Sirirungruang, Y. Zhang and D. Chen, et al., Lipopeptide nanoparticles for potent and selective siRNA delivery in rodents and nonhuman primates, Proc. Natl. Acad. Sci. U. S. A., 2014, 111(11), 3955–3960 CrossRef CAS PubMed.
- L. Miao, J. Lin, Y. Huang, L. Li, D. Delcassian and Y. Ge, et al., Synergistic lipid compositions for albumin receptor mediated delivery of mRNA to the liver, Nat. Commun., 2020, 11(1), 2424 CrossRef CAS PubMed.
- Y.-N. Zhang, W. Poon, A. J. Tavares, I. D. McGilvray and W. C. W. Chan, Nanoparticle–liver interactions: Cellular uptake and hepatobiliary elimination, J. Controlled Release, 2016, 240, 332–348 CrossRef CAS PubMed.
- R. Rai, S. Alwani and I. Badea, Polymeric Nanoparticles in Gene Therapy: New Avenues of Design and Optimization for Delivery Applications, Polymers, 2019, 11(4), 745 CrossRef CAS PubMed.
- G. Lin, H. Zhang and L. Huang, Smart Polymeric Nanoparticles for Cancer Gene Delivery, Mol. Pharm., 2015, 12(2), 314–321 CrossRef CAS PubMed.
- X. Zhang, C. Xu, S. Gao, P. Li, Y. Kong and T. Li, et al., CRISPR/Cas9 Delivery Mediated with Hydroxyl-Rich Nanosystems for Gene Editing in Aorta, Adv. Sci., 2019, 6(12), 1900386 CrossRef PubMed.
- S. Chesnoy and L. Huang, Structure and function of lipid-DNA complexes for gene delivery, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 27–47 CrossRef CAS PubMed.
- Y. Sun, H. Hu, N. Zhao, T. Xia, B. Yu and C. Shen, et al., Multifunctional polycationic photosensitizer conjugates with rich hydroxyl groups for versatile water-soluble photodynamic therapy nanoplatforms, Biomaterials, 2017, 117, 77–91 CrossRef CAS PubMed.
- J. Chen, C. Wu and D. Oupický, Bioreducible hyperbranched poly(amido amine)s for gene delivery, Biomacromolecules, 2009, 10(10), 2921–2927 CrossRef CAS PubMed.
- Y. Liu, J. Du, J. S. Choi, K. J. Chen, S. Hou and M. Yan, et al., A High-Throughput Platform for Formulating and Screening Multifunctional Nanoparticles Capable of Simultaneous Delivery of Genes and Transcription Factors, Angew. Chem., Int. Ed., 2016, 55(1), 169–173 CrossRef PubMed.
- P. P. Tripathi, H. Arami, I. Banga, J. Gupta and S. Gandhi, Cell penetrating peptides in preclinical and clinical cancer diagnosis and therapy, Oncotarget, 2018, 9(98), 37252–37267 CrossRef PubMed.
- H. Lu, J. Wang, Y. Bai, J. W. Lang, S. Liu, Y. Lin and J. Cheng, Ionic polypeptides with unusual helical stability, Nat. Commun., 2011, 2(1), 1–9 Search PubMed.
- Y.-W. Won, P. P. Adhikary, K. S. Lim, H. J. Kim, J. K. Kim and Y.-H. Kim, Oligopeptide complex for targeted non-viral gene delivery to adipocytes, Nat. Mater., 2014, 13(12), 1157–1164 CrossRef CAS PubMed.
- E. Choi, J. Han, X. Tan, J. Oh, D. Lee and T. Rhim, et al., Combined delivery of temozolomide and the thymidine kinase gene for treatment of glioblastoma, J. Drug Targeting, 2017, 25(2), 156–162 CrossRef CAS PubMed.
- R. Bahadur K.C., B. Thapa and N. Bhattarai, Gold nanoparticle-based gene delivery: promises and challenges, Nanotechnol. Rev., 2013, 3, 269–280 Search PubMed.
- J. K. Patra, G. Das, L. F. Fraceto, E. V. R. Campos, M. D. P. Rodriguez-Torres and L. S. Acosta-Torres, et al., Nano based drug delivery systems: recent developments and future prospects, J. Nanobiotechnol., 2018, 16(1), 71 CrossRef PubMed.
- R. Vankayala, C. L. Kuo, K. Nuthalapati, C. S. Chiang and K. C. Hwang, Nucleus–targeting gold nanoclusters for simultaneous in vivo fluorescence imaging, gene delivery, and NIR–light activated photodynamic therapy, Adv. Funct. Mater., 2015, 25(37), 5934–5945 CrossRef CAS.
- C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. W. Chan, Nanoparticle Size and Surface Chemistry Determine Serum Protein Adsorption and Macrophage Uptake, J. Am. Chem. Soc., 2012, 134(4), 2139–2147 CrossRef CAS PubMed.
- H. J. Kim, A. Ishii, K. Miyata, Y. Lee, S. Wu and M. Oba, et al., Introduction of stearoyl moieties into a biocompatible cationic polyaspartamide derivative, PAsp(DET), with endosomal escaping function for enhanced siRNA-mediated gene knockdown, J. Controlled Release, 2010, 145(2), 141–148 CrossRef CAS PubMed.
- F. Pittella, M. Zhang, Y. Lee, H. J. Kim, T. Tockary and K. Osada, et al., Enhanced endosomal escape of siRNA-incorporating hybrid nanoparticles from calcium phosphate and PEG-block charge-conversional polymer for efficient gene knockdown with negligible cytotoxicity, Biomaterials, 2011, 32(11), 3106–3114 CrossRef CAS PubMed.
- H. Sakurai, K. Kawabata, F. Sakurai, S. Nakagawa and H. Mizuguchi, Innate immune response induced by gene delivery vectors, Int. J. Pharm., 2008, 354(1), 9–15 CrossRef CAS PubMed.
- M. Schlich, R. Palomba, G. Costabile, S. Mizrahy, M. Pannuzzo and D. Peer, et al., Cytosolic delivery of nucleic acids: The case of ionizable lipid nanoparticles, Bioeng. Transl. Med., 2021, e10213 CAS.
- A. K. Varkouhi, M. Scholte, G. Storm and H. J. Haisma, Endosomal escape pathways for delivery of biologicals, J. Controlled Release, 2011, 151(3), 220–228 CAS.
- B. Ashok, N. A. Peppas and M. E. Wechsler, Lipid- and Polymer-Based Nanoparticle Systems for the Delivery of CRISPR/Cas9, J. Drug Delivery Sci. Technol., 2021, 65, 102728 CrossRef CAS PubMed.
- R. E. Taylor and M. Zahid, Cell Penetrating Peptides, Novel Vectors for Gene Therapy, Pharmaceutics, 2020, 12(3), 225 CrossRef CAS PubMed.
- J. D. Gillmore, E. Gane, J. Taubel, J. Kao, M. Fontana and M. L. Maitland, et al., CRISPR-Cas9 In Vivo Gene Editing for Transthyretin Amyloidosis, N. Engl. J. Med., 2021, 385(6), 493–502 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2022 |