
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExtremely fast triplet formation by charge recombination in a Nile Red/fullerene flexible dyad†
Federica
Faroldi
a,
Brunella
Bardi
 a,
Irene
Tosi
a,
Sandra
Doria
bc,
Jacopo
Isopi
d,
Laura
Baldini
a,
Mariangela
Di Donato
bc,
Massimo
Marcaccio
d,
Francesco
Sansone
a and
Francesca
Terenziani
*a
a,
Irene
Tosi
a,
Sandra
Doria
bc,
Jacopo
Isopi
d,
Laura
Baldini
a,
Mariangela
Di Donato
bc,
Massimo
Marcaccio
d,
Francesco
Sansone
a and
Francesca
Terenziani
*a
aDepartment of Chemistry, Life Sciences and Environmental Sustainability, University of Parma, Parco Area delle Scienze 17/a, 43124 Parma, Italy. E-mail: francesca.terenziani@unipr.it
bICCOM-CNR, Via Madonna del Piano 10, 50019 Sesto Fiorentino (FI), Italy
cLENS, Via N. Carrara 1, 50019 Sesto Fiorentino (FI), Italy
dDipartimento di Chimica “Giacomo Ciamician”, Università di Bologna, Via Selmi 2, 40126 Bologna, Italy
First published on 16th July 2021
Abstract
A donor/acceptor dyad was obtained by linking Nile Red and fullerene to a calix[4]arene scaffold. The dyad was spectroscopically characterized, both with steady-state and ultrafast transient absorption experiments, as well as with electrochemical and spectroelectrochemical techniques. We demonstrate extremely fast and efficient formation of a long-lived excited triplet localized on the fullerene moiety in this system, occurring in about 80 ps in toluene and 220 ps in chloroform. The mechanism of this process is investigated and discussed. The spectroscopic and electrochemical characterization suggests the occurrence of electron transfer from Nile Red to fullerene, leading to the formation of a charge-separated state. This state lives very briefly and, because of the small interaction between the electron donor and acceptor, promotes a singlet/triplet state mixing, inducing charge recombination and efficient triplet formation.
Introduction
When an electron donor/acceptor system is photoexcited, an excited singlet exciton forms, which can dissociate via charge separation. The formation of a long-lived charge separated (CS) state is crucial for (artificial) photosynthesis or solar cells, such that charge recombination (CR) is often an unwanted process.1 Nevertheless, CR can lead to alternative paths that can be of interest for other applications. Most often, photogenerated charges recombine back to a singlet state. However, in specific conditions, triplet formation by CR can occur.1–3 This pathway is of particular interest because it allows for the formation of triplet states without the need of the heavy-atom effect to enhance intersystem crossing (ISC).4 Easily accessible triplet excited states can be of paramount importance for many applications such as photoinitiated polymerization,5 photodynamic therapy,6 photovoltaics,1 as well as light emitting diodes exploiting triplet–triplet upconversion7 or thermally-activated delayed fluorescence (TADF).8Triplet formation by CR has been reported for many compact donor/acceptor dyads in which the two moieties are orthogonal to one another, as to be almost decoupled.9–17 In these systems, because of their geometry, CR is accompanied by a change in molecular orbital angular momentum, which can be compensated by a change in the electron spin angular momentum, in the so-called spin–orbit charge transfer ISC (SOCT-ISC) mechanism.1,2
Another mechanism to produce triplet states from CS states is the so-called radical pair intersystem crossing (RP-ISC). In this case, the singlet radical pair forming the CS state is converted into a triplet radical pair which subsequently recombines to a neutral triplet state.1,2 For this pathway to occur, the spatial overlap between the molecular orbitals of the donor and acceptor species must be tiny. In fact, a small exchange interaction reduces the energy gap between the 1CS and 3CS states, facilitating their mixing and hence activating the RP-ISC pathway.4 When the 3CS state is populated via RP-ISC, the spin-selective CR to produce a localized triplet state is significant, so that, typically, short-lived CS states are observed.2 RP-ISC was observed for many electron donor/acceptor polyads with large separation distance between the electron donor and acceptor units.10,12,17–21
Typically, relatively slow rates of triplet CR (<108 s−1) have been reported for the RP-ISC pathway, while substantially faster rates (>109, up to 2.5 × 1010 s−1) have been correlated to the SOCT-ISC mechanism.1 However, distinguishing between the two mechanisms is not easy and would require time-resolved electron paramagnetic resonance (TREPR).2
Moreover, regardless of the mechanism, an important dependence of the triplet formation by CR has been reported not only according to the donor/acceptor strength, distance and orientation, but also to the polarity of the solvent.20,22–24
In this paper, we report the synthesis and characterization of a donor/acceptor dyad (3) where the donor moiety is constituted by Nile Red (NR) and the acceptor by C60, both anchored to the upper rim of a calix[4]arene scaffold (Fig. 1, also showing the reference donor (4) and acceptor (5) compounds).
 | ||
| Fig. 1 Structures of the investigated compounds. | ||
Calix[4]arenes are ideal scaffolds for the preparation of chromophoric dyads designed for the study of energy/charge transfer phenomena.25–29 The orientation, mutual distance and conformational freedom of the dyes can be accurately tuned thanks to (i) the availability of four different configurations (cone, partial cone, 1,2-alternate and 1,3-alternate) of the calixarene scaffold,30 which project the substituents in different directions;31,32 (ii) the possibility of finely modulating the residual flexibility of the scaffold thanks to the lower rim functionalization;31–34 (iii) the possibility of attaching the dyes directly to the aromatic rings of the macrocycle35 or through linkers of different rigidity/flexibility.31,32
In our system the two dyes are linked to a flexible tetraalkylated cailx[4]arene scaffold through short but flexible linkers in order to endow the dyad of enough conformational freedom to allow the chromophores to get in close contact.
While the dyad was designed to perform charge transfer, we observed an even more interesting behavior upon selective photoexcitation of Nile Red, leading to the very fast formation of a triplet state localized on C60. Other covalent or supramolecular fully organic donor/acceptor dyads comprising fullerene were reported in the literature,20,21,23,36–41 and some of them were able to form the fullerene triplet after photoexcitation of the donor.20,21,23,40,41 To the best of our knowledge, however, our system is peculiar in terms of the impressive speed of formation of the triplet (∼1010 s−1), as well as of the little importance of the solvent polarity on the dynamics of the process.
Experimental
General methods
Dry solvents were prepared according to standard procedures, distilled before use and stored over 3 or 4 Å molecular sieves. Most of the solvents and reagents were obtained from commercial sources and used without further purification. Analytical TLC was performed using prepared plates of silica gel (Merck 60 F-254 on aluminum). Merck silica gel 60 (70–230 mesh) was used for flash chromatography and for preparative TLC plates. 1H and 13C NMR spectra were recorded on a Bruker AV400 spectrometer. All chemical shifts are reported in part per million (ppm) using the residual peak of the deuterated solvent, whose values are referred to tetramethylsilane (TMS, δTMS = 0), as internal standard. All 13C NMR spectra were performed with proton decoupling. Mass spectra were recorded in ESI mode on a single quadrupole instrument SQ Detector, Waters (capillary voltage 3.7 kV, cone voltage 30–160 eV, extractor voltage 3 eV, source block temperature 80 °C, desolvation temperature 150 °C, cone and desolvation gas (N2) flow rates 1.6 and 8 L min−1, respectively).2-(2-Aminoethoxy)-9-diethylamino-5H-benzo[α]phenoxazin-5-one (NR-NH2),32 25,26,27,28-tetra-n-propoxycalix[4]arene-5,17-dicarboxylic acid (1)42 and compound 432 were synthesized according to a literature procedure.
Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2) to give a purple solid in 23% yield (40 mg, 0.022 mmol). M.p.: > 300 °C. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.29 (d, J = 8.7 Hz, 1H, ArH-NR), 8.15 (d, J = 2.6 Hz, 1H, ArH-NR), 7.63 (d, J = 9.1 Hz, 1H, ArH-NR), 7.25 (d, J = 2.6 Hz, 1H, ArH-NR), 7.17 (s, 1H, ArHCO), 7.05 (s, 1H, ArHCO), 6.98 (s, 2H, ArHCO) 6.82 (br s, 1H, NHCH2CH), 6.73 (br s, 1H, ArH), 6.57–6.49 (m, 5H, 4xArH and ArH-NR), 6.53 (t, J = 7.4 Hz, 1H, ArH), 6.48 (d, J = 2.7 Hz, 1H, ArH-NR), 6.37 (t, J = 5.5 Hz, 1H, NHCH2CH2), 6.34 (s, 1H, ArH-NR), 4.94 (d, J = 9.5 Hz, 1H, CH3NCHH), 4.69 (d, J = 12.8, 6.7 Hz, 1H, NHCHH), 4.52–4.40 (m, 4H, ArCHHaxAr), 4.39–4.30 (m, 2H, CH2), 4.23 (d, J = 9.5 Hz, 1H, CH3NCHH), 4.12 (br s, 1H, NHCHH), 4.09 (br s, 1H, NHCH2CH), 3.96 (br s, 1H, CH2), 3.90–3.79 (m, 9H, OCH2 and CH2’), 3.49 (q, J = 7.1 Hz, 4H, NCH2CH3), 3.29–3.12 (m, 4H, ArCHHeqAr), 3.02 (s, 3H, NCH3), 1.97–1.84 (m, 8H, OCH2CH2CH3), 1.28 (t, J = 7.0 Hz, 6H, NCH2CH3), 1.04–0.96 (m, 12H, OCH2CH2CH3). 13C NMR (100 MHz, CDCl3) δ (ppm): 183.2, 167.2, 161.4, 159.5, 156.5, 155.9, 154.3, 153.3, 152.7, 150.8, 147.3, 146.3, 146.2, 146.1, 145.9, 145.6, 145.5, 145.4, 145.26, 145.25, 145.20, 145.1, 144.64, 144.60, 144.4, 144.3, 143.1, 142.6, 142.5, 142.15, 142.10, 142.07, 141.9, 141.7, 141.6, 140.2, 140.1, 139.9, 139.8, 136.0, 135.7, 135.4, 135.3, 135.1, 134.7, 134.1, 131.2, 128.7, 128.5, 128.1, 127.8, 127.4, 127.3, 127.2, 126.9, 126.6, 125.9, 124.8, 122.5, 122.3, 118.3, 109.6, 106.9, 105.3, 96.3, 73.9, 70.0, 69.4, 67.5, 45.1, 39.5, 39.4, 37.5, 31.1, 31.0, 29.7, 23.3, 23.2, 23.1, 12.6, 10.39, 10.36, 10.2, 7.1. ESI-MS: m/z calcd for C128H78N5O9 [(3 + H)+] 1828.6, found 1830.2 (40%); calcd for C128H79N5O9 [(3 + 2H)2+] 914.8, found 915.6 (100%).
:2) to give a purple solid in 23% yield (40 mg, 0.022 mmol). M.p.: > 300 °C. 1H NMR (400 MHz, CDCl3) δ (ppm): 8.29 (d, J = 8.7 Hz, 1H, ArH-NR), 8.15 (d, J = 2.6 Hz, 1H, ArH-NR), 7.63 (d, J = 9.1 Hz, 1H, ArH-NR), 7.25 (d, J = 2.6 Hz, 1H, ArH-NR), 7.17 (s, 1H, ArHCO), 7.05 (s, 1H, ArHCO), 6.98 (s, 2H, ArHCO) 6.82 (br s, 1H, NHCH2CH), 6.73 (br s, 1H, ArH), 6.57–6.49 (m, 5H, 4xArH and ArH-NR), 6.53 (t, J = 7.4 Hz, 1H, ArH), 6.48 (d, J = 2.7 Hz, 1H, ArH-NR), 6.37 (t, J = 5.5 Hz, 1H, NHCH2CH2), 6.34 (s, 1H, ArH-NR), 4.94 (d, J = 9.5 Hz, 1H, CH3NCHH), 4.69 (d, J = 12.8, 6.7 Hz, 1H, NHCHH), 4.52–4.40 (m, 4H, ArCHHaxAr), 4.39–4.30 (m, 2H, CH2), 4.23 (d, J = 9.5 Hz, 1H, CH3NCHH), 4.12 (br s, 1H, NHCHH), 4.09 (br s, 1H, NHCH2CH), 3.96 (br s, 1H, CH2), 3.90–3.79 (m, 9H, OCH2 and CH2’), 3.49 (q, J = 7.1 Hz, 4H, NCH2CH3), 3.29–3.12 (m, 4H, ArCHHeqAr), 3.02 (s, 3H, NCH3), 1.97–1.84 (m, 8H, OCH2CH2CH3), 1.28 (t, J = 7.0 Hz, 6H, NCH2CH3), 1.04–0.96 (m, 12H, OCH2CH2CH3). 13C NMR (100 MHz, CDCl3) δ (ppm): 183.2, 167.2, 161.4, 159.5, 156.5, 155.9, 154.3, 153.3, 152.7, 150.8, 147.3, 146.3, 146.2, 146.1, 145.9, 145.6, 145.5, 145.4, 145.26, 145.25, 145.20, 145.1, 144.64, 144.60, 144.4, 144.3, 143.1, 142.6, 142.5, 142.15, 142.10, 142.07, 141.9, 141.7, 141.6, 140.2, 140.1, 139.9, 139.8, 136.0, 135.7, 135.4, 135.3, 135.1, 134.7, 134.1, 131.2, 128.7, 128.5, 128.1, 127.8, 127.4, 127.3, 127.2, 126.9, 126.6, 125.9, 124.8, 122.5, 122.3, 118.3, 109.6, 106.9, 105.3, 96.3, 73.9, 70.0, 69.4, 67.5, 45.1, 39.5, 39.4, 37.5, 31.1, 31.0, 29.7, 23.3, 23.2, 23.1, 12.6, 10.39, 10.36, 10.2, 7.1. ESI-MS: m/z calcd for C128H78N5O9 [(3 + H)+] 1828.6, found 1830.2 (40%); calcd for C128H79N5O9 [(3 + 2H)2+] 914.8, found 915.6 (100%).
Electrochemical and spectroelectrochemical measurements
For CV measurements, electrochemical or analytical grade tetrabutylammonium hexafluorophosphate (TBAH) from Sigma-Aldrich was used as received as a supporting electrolyte. Solvent dichloromethane (DCM) from Sigma-Aldrich was purified and dried by refluxing over and successively distilling from B2O3 and activated 4 Å molecular sieves. Afterward, it was stored in specially designed Schlenk flasks over 3 Å activated molecular sieves, protected from light, and kept under vacuum prior to use, as reported elsewhere.43The solvent was distilled via a closed system into a custom designed electrochemical cell (as described below) containing the supporting electrolyte and the species under examination, immediately before performing the experiment.
Electrochemical experiments were carried out in an airtight single-compartment cell using platinum as working and counter electrodes and a silver spiral as a quasi-reference electrode. The drift of the quasi-reference electrode was negligible during the time required for an experiment. All the E1/2 potentials have been directly obtained from cyclic voltammetric curves as averages of the cathodic and anodic peak potentials and by digital simulation in the case of not Nernstian or overlapping processes. The E1/2 values have been determined by adding ferrocene (Fc), at the end of each experiment, as an internal standard and measuring them with respect to the ferrocinium/ferrocene couple (Fc+/0) standard potential (at 298 K it is +0.42 V vs. SCE, i.e., the aqueous Saturated Calomel Electrode).44
The cell containing the supporting electrolyte and the electroactive compound was dried under vacuum at 100–110 °C for at least 48 hours before each experiment. The pressure measured in the electrochemical cell prior to perform the trap-to-trap distillation of the solvent was typically 1 × 10−5 mbar. Voltammograms were recorded with a custom made fast and low current potentiostat45 controlled by an AMEL Mod. 568 programmable function generator. The potentiostat was interfaced to a Nicolet Mod. 3091 digital oscilloscope and the data transferred to a personal computer by the program Antigona.46 Minimization of the uncompensated resistance effect in the voltammetric measurements was achieved by the positive-feedback circuit of the potentiostat. Digital simulations of the cyclic voltammetric curves were carried out either by Antigona or DigiElch 7, utilizing a best fitting procedure of the experimental curves recorded at different scan rates spanning over, at least, two orders of magnitude.
The spectroelectrochemical experiments have been carried out using a quartz OTTLE cell with a 0.03 cm path length. Temperature control was achieved by a special cell holder with quartz windows, in which two nitrogen fluxes (one at room temperature and the other at low temperature) were regulated by needle valves. The spectra have been recorded by a Cary 5 (Varian) and all the experimental details for the spectroelectrochemical setup have been reported elsewhere.47
Steady-state spectroscopy
Steady-state absorption and emission measurements were performed on freshly prepared air-equilibrated solutions. Solvents (toluene and chloroform) were spectrophotometric grade and were used as received. Absorption spectra were collected with a PerkinElmer Lambda650 double-beam UV/vis spectrophotometer. Molar extinction coefficients were estimated from the absorbances of four different solutions with known concentration, and the Lambert–Beer law was verified over the investigated concentration range (10−5–10−6 M).Fluorescence measurements were performed at room temperature on dilute solutions (optical density <0.1 at the excitation wavelength). Fully corrected emission spectra were recorded with a Fluoromax-3 Horiba-Jobin Yvon fluorometer. Fluorescence quantum yields were estimated using a dilute solution of fluorescein in NaOH 0.1 M as the standard (QY = 90%, λexc = 460 nm).
Singlet oxygen production was assessed under ambient conditions exploiting 1O2 phosphorescence in the near-IR. 1O2 emission spectra were collected with an Edinburgh FLS1000 spectrofluorometer equipped with a Xe excitation source and near-IR PMT detector in liquid nitrogen cooled housing. Singlet oxygen quantum yields were estimated as detailed in ESI.† Phosphorescence lifetimes were obtained with the multi-channel scaling (MCS) technique, following the decay at 1273 nm after photoexcitation with a microsecond flashlamp. Lifetimes were extracted by reconvolution fit or tail fit of the experimental decay traces and the goodness of the fit was judged by the chi-squared test.
Transient spectroscopy
The system used to record transient absorption spectra (TAS) has been previously described.48 Briefly, the setup is composed of an integrated Ti:sapphire oscillator-regenerative amplifier system (Spectra Physics Tsunami-BMI Alpha 1000) delivering 100 fs pulses centred at 800 nm. A small portion of the fundamental laser radiation is used to produce the white light probe beam, obtained by focusing the 800 nm light on a 3 mm thick CF2 window, kept under continuous rotation to avoid damage. The generated white light is then divided into a probe and reference beam through a 50% beamsplitter. The visible pump pulse has been set at 530 nm for excitation of 3 and 4 while at 400 nm for exciting 5. The 530 nm excitation beam was generated by pumping a home-made non collinear optical parametric amplifier (NOPA) with a portion of the fundamental 800 nm light. Excitation light at 400 nm was obtained through second harmonic generation of the fundamental laser output using a 1 mm thick BBO crystal. Excitation powers were about 50–100 nJ. The pump beam polarization has been set to magic angle with respect to the probe beam by rotating a λ/2 plate, to exclude rotational contributions. Before the generation of the probe light, the portion of fundamental radiation directed on the probe/reference pathway was sent through a motorized delay stage, which allows to vary the relative time of arrival of pump and probe beams and to collect transient spectra in a temporal interval going up to 1.5 ns. Both pump and probe were overlapped at the sample position through a 150 mm spherical mirror. After passing through the sample, the white light probe and reference pulses were both directed to a flat field monochromator coupled to a home-made CCD detector. The sample was contained in a 2 mm quartz cuvette, mounted on a motorized stage in order to minimize photodegradation. Transient data have been analysed using a global analysis procedure, which consists of the simultaneous fit at all the acquired frequencies with sums or combination of exponential decay functions.49 Global analysis was performed using the GLOTARAN package (http://glotaran.org), employing a linear unidirectional “sequential” model.50,51 The number of kinetic components to be used in the global fit is determined by a preliminary singular values decomposition (SVD) analysis performed with the same software.52Computational details
A preliminary conformational search for dyad 3 was performed in vacuum with molecular mechanics. In order to restrict the number of possible structures, as suggested by a previous work,32 we performed a scan along each dihedral angle of the two spacers and, for each point of the scan, we optimized the geometry with MMFF94 force field53–57 (PCmodel software, v. 10.0).58 Among the structures characterized by an open conformation of the calixarene macrocycle (suggested by NMR data), the most stable were then reoptimized with MMFF94 and were used as starting point for DFT calculations. DFT optimizations were performed in gas phase with Gaussian16 package,59 adopting the long-range-corrected hybrid ωB97XD functional60 and 6-31G basis set. Stationary points were characterized by frequency analysis. The geometry of the most stable conformer (open) was reoptimized in solution (toluene), treating the solvent according to the polarizable continuum model (PCM) formalism.61Results and discussion
Synthesis
The synthesis of Nile Red-C60 calix[4]arene 3 is reported in Scheme 1.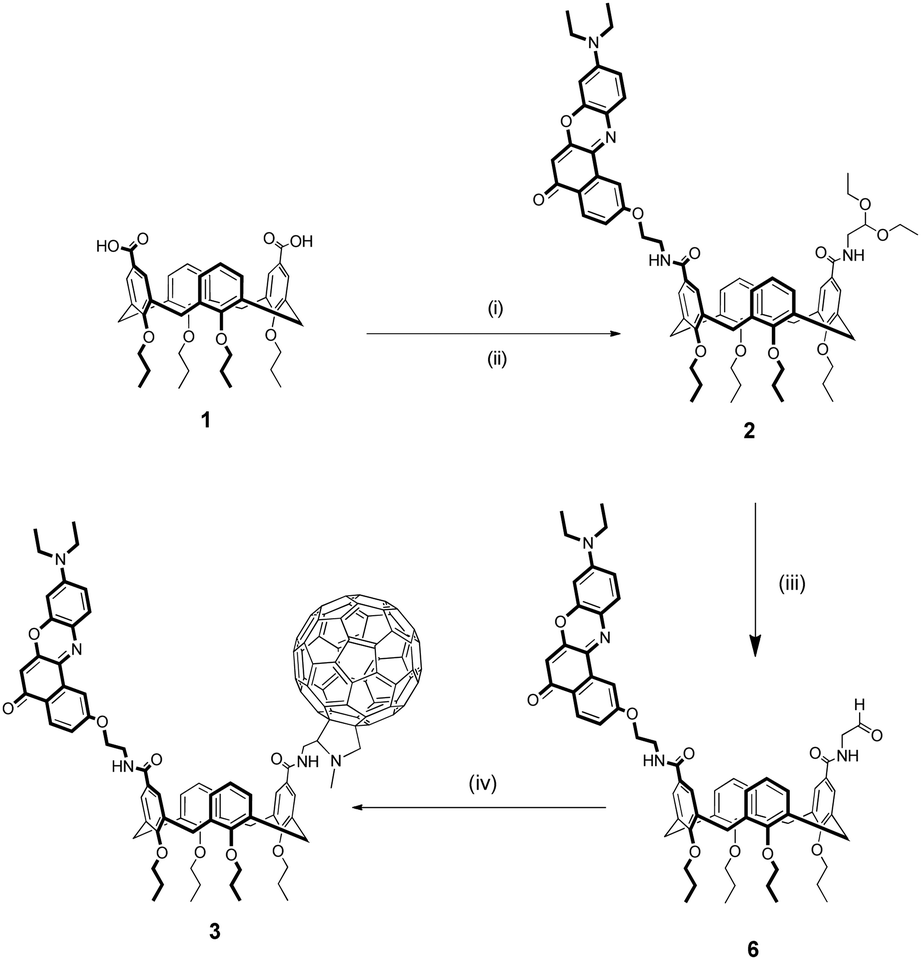 | ||
| Scheme 1 Synthesis of Nile Red-C60 calix[4]arene 3: (i) (COCl)2, DMF, DCM, 5 h, r.t., quantitative; (ii) 2,2-diethoxyethanamine, NR-NH2, DIPEA, DCM, 12 h, r.t., 47%; (iii) HCl (1M), dioxane, 4 h, r.t., 77%; (iv) C60, sarcosine, toluene, reflux, 3 h, 23%. | ||
The dicarboxylic acid tetrapropoxycalix[4]arene 142 was first activated as acyl chloride and immediately reacted with an equimolar mixture of NR-NH2 and 2,2-diethoxyethanamine. This mixed-condensation strategy allowed to obtain the heterodifunctionalized calixarene 2 in a single step after chromatography separation from the homodifunctionalized byproducts. After removal of the acetal protecting group, the resulting aldeyhde was subjected to the Prato reaction62 with C60 and sarcosine in toluene to afford product 3 as a dark purple solid in 23% yield.
Reference monochromophoric compound 5 was synthesized with a similar strategy starting from monocarboxylic acid tetrapropoxycalix[4]arene (see ESI†).
Conformational properties
C60-Nile Red calixarene 3 was characterized in CDCl3 solution by NMR. 2D COSY and ROESY spectra (Fig. S2 and S3, ESI†) were required to fully assign all the 1H NMR signals, due to the asymmetric nature of the molecule. Importantly, besides confirming the identity of the compound, 1H NMR spectroscopy allowed to investigate the conformational properties of the calixarene scaffold.In general, the most stable conformation of a calix[4]arene in the cone geometry tetra-alkylated at the lower rim is the so-called pinched cone structure (Fig. 2a), where two opposite aromatic rings point slightly inwards and the other two are tilted outwards.33 Calix[4]arenes with identical substituents on the four rings rapidly interconvert between the two opposite pinched cone structures and their 1H NMR spectrum, which reflects the time-averaged conformation, is indicative of a regular cone structure. On the contrary, for calixarenes like 3, with two substituents on the distal positions of the upper rim, the stabilities of the two conformations are no longer equal, depending on the interactions between the substituents (attractive or repulsive). In cases like this, 1H NMR spectroscopy allows to distinguish if the preferred conformation is the open (with the functionalized rings pointing outwards) or the closed (functionalized rings pointing inwards) pinched cone (Fig. 2b) from the relative position of the signals of the aromatic protons: the two rings pointing inwards experience the magnetic shielding cone of the other two rings and their resonances are shifted to higher fields.
 | ||
| Fig. 2 (a) Pinched cone conformation of a calix[4]arene; (b) equilibrium between the open and closed conformations of a cone calix[4]arene with two substituents on the distal positions of the upper rim. | ||
For compound 3, the aromatic signals (singlets at 7.17, 7.05 and 6.98 ppm for the protons ortho to the amide group, which resonate at lower fields than the multiplet between 6.57–6.49 ppm and the triplet at 6.53 ppm of the protons on the unsubstituted aromatic rings) suggest an open conformation for the calixarene scaffold. This finding was not obvious, according to our experience. In previous works some of us have studied calix[4]arenes similar to 3, functionalized at the upper rim with pairs of different chromophores linked to the calixarene through an analogous amide linkage.28,32 These compounds were found to adopt a closed structure thanks to the formation of an intramolecular hydrogen bond between the NH and CO groups of the amide moieties. In case of compound 3, most likely the steric hindrance of the fullerene prevents the formation of the intramolecular CO⋯HN hydrogen bond. Even in presence of a prevalent open conformation of the macrocycle, however, the flexibility of the linkers should guarantee the possibility for the dyes to get in close contact.
The conformational properties of the dyad were also investigated computationally. The four structures reported in Fig. S4 (ESI†) were singled out from conformational analysis in gas phase: the open conformer, where the two chromophores point in two different directions out of the calixarene; the open_NR and open_C60 conformers, where the Nile Red and the C60 moiety, respectively, point toward the center of the calixarene; the open_NR_C60 conformer, where both Nile Red and C60 point toward the center of the calixarene. Total ground state free energies of the conformers are summarized in Table S1 (ESI†). The open conformation is the most stable, the open_NR is only slightly more energetic, the open_NR_C60 and open_C60 have significantly higher energies but are still accessible at room temperature. The interchromophoric distance, taken as the distance between the centroids of Nile Red and C60, is also reported in Table S1 (ESI†), ranging from 8 to 14 Å according to the conformer. The geometry of the most stable conformer was also optimized in toluene, giving a very similar structure as in gas phase (compare the interchromophoric distance in Table S1, ESI†). We underline that the discussed conformers are just four out of a plethora of possible conformations allowed by the flexibility of the linkers: the data on these selected conformers should just be used to have an idea of the mean distance between the two donor/acceptor moieties and about the frontier orbitals (reported in Fig. S5–S9, ESI†).
Steady-state spectroscopic characterization
Dyad 3 and reference compounds 4 and 5 were characterized by steady-state spectroscopy in toluene and chloroform air-equilibrated solutions. Absorption and emission spectra are shown in Fig. 3, while main spectroscopic properties are listed in Table 1.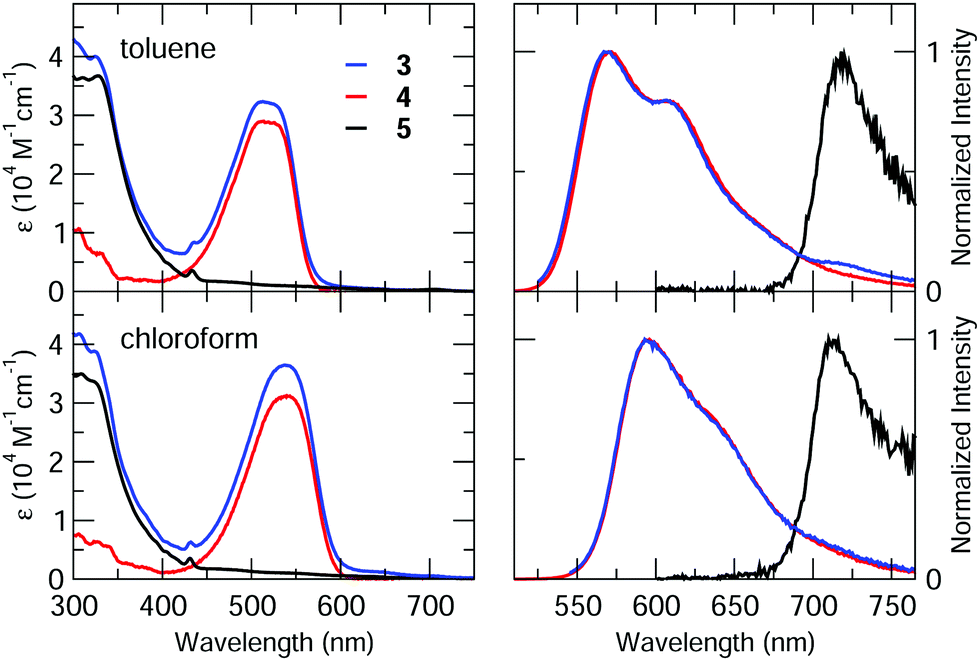 | ||
| Fig. 3 Steady-state absorption (left) and normalized emission (right) spectra of the investigated compounds in toluene (top panels) and chloroform (bottom panels). Emission spectra of 3 were collected for excitation at λ = 515 nm in toluene and λ = 540 nm in chloroform. | ||
Spectra of 4 feature the characteristic absorption band of Nile Red between 450 and 600 nm, and an intense emission signal peaking at λ > 550 nm. Fluorescence quantum yields are high (≈80%) in both the solvents, with a radiative rate constant amounting to 1.7 × 108 s−1 quite independently of the solvent.32 Absorption and emission bands of 4 retain the positive solvatochromism of Nile Red, suggesting that the presence of the calixarene scaffold does not perturb the electronic properties of the chromophore.63
Similarly, compound 5 displays the main spectroscopic properties of C60. Indeed, its absorption spectrum covers most of the UV/visible range, with small molar extinction coefficient in the 400–750 nm spectral window (ε < 103 M−1 cm−1) and intense bands (ε > 104 M−1 cm−1) at shorter wavelengths.64 The peak at λ ≈ 430 nm is instead related to C60 substitution, and is usually found in fulleropyrrolidine derivatives.23,65–67 The weak fluorescence of 5, which is independent of solvent polarity and excitation wavelength, was found beyond 700 nm as typical of C60.68
Absorption spectra of the dyad 3 are roughly the sum of the corresponding spectra of 4 and 5, ruling out the occurrence of strong interchromophoric interactions in the ground state. Emission spectra of 3 can also be qualitatively viewed as the sum of the contributions from the two chromophoric units, however, because of the sizeable difference between the fluorescence efficiencies of the two dyes, emission of 3 is dominated by the Nile Red contribution, while fluorescence of C60 can be recognized only in the long-wavelength tail of the spectrum. Nevertheless, fluorescence of Nile Red is strongly quenched in 3, being approximately 40 and 30 times less intense than in 4 in toluene and chloroform, respectively (see Table 1). This strong quenching suggests the activation of new efficient decay channels (energy and/or charge transfer towards C60).
Distinguishing energy and charge transfer processes through the analysis of steady-state absorption and emission spectra is not an easy task, most of all when donor and acceptor moieties are characterized by very different emission quantum yields, as in this case. In fact, the emission spectrum of 3 is dominated by the fluorescence of Nile Red despite the strong quenching that it undergoes, so that deconvolving the emission of the C60 moiety with a good confidence is not possible. We then decided to investigate the fate of the excited state of Nile Red in the dyad via the evaluation of the ability of 3 to produce singlet oxygen.
Fullerenes are good singlet molecular oxygen sensitizers.69,70 Indeed, photoexcitation of 5 in the UV (370 nm) leads to the appearance of the characteristic 1O2 photoluminescence at 1273 nm in both the analysed solvents. Values estimated in chloroform amount to ≈50% with respect to those retrieved in toluene (Table S3, ESI†). Singlet oxygen production was detected also for dyad 3 for excitation within the Nile Red absorption band (Fig. S10, ESI†), while was not found in reference compound 4 under the same experimental conditions (Fig. S11, ESI†). The comparison between the 1O2 emission intensities for 3 and 5 allowed for the estimate of their relative singlet oxygen quantum yield (see ESI†). Noticeably, in both solvents, the singlet oxygen quantum yield of the dyad 3, obtained for (almost) selective excitation of Nile Red (λexc = 530 and 540 nm in toluene and chloroform, respectively) is not reduced, within the experimental error, compared to reference compound 5. This suggests that, in the dyad, the singlet excited state localized on Nile Red quantitatively converts into an excitation localized on C60.
The quantitative conversion of excitation energy from Nile Red to C60 in 3 deserves more investigation. In fact, the formation of the C60 triplet (responsible for the formation of singlet oxygen) can be due to different mechanisms, namely: (i) FRET from Nile Red to C60, creating the singlet excited state of C60, followed by intersystem crossing (ISC) to form the C60 triplet; (ii) electron transfer from the singlet excited state of Nile Red towards an empty MO of C60, creating a biradical charge-separated state, followed by triplet formation by charge recombination.2,4
Electrochemical and spectroelectrochemical characterization
The redox behaviour of the compounds has been investigated by cyclic voltammetry in highly dry dichloromethane (DCM) solution.The voltammetric curves of the dyad 3 and those of the different constituent moieties, namely compound 5, a reference C60-Ph compound (see ESI†), and compound 4, are reported in Fig. 4. For all the compounds, the redox processes studied at room temperature occur within the potential window comprised between about +1.2 V and −2.3 V, with a particular rich voltammetric pattern in the negative potentials region for the species containing fullerene. The E1/2 potentials of the various processes are collected in Table 2.
 | ||
| Fig. 4 Cyclic voltammetric curves of (a) 0.6 mM species 3 (three scans at different inversion potential); (b) 0.7 mM compound C60-Ph (blue line) and 5 (red line); (c) 0.7 mM species 4 recorded in TBAH/DCM at 298 K. Scan rate = 1 V s−1; working electrode: platinum disk (diameter 125 μm); reference electrode: SCE. | ||
| Species | E 1/2/V (oxidation) | E 1/2/V – (reduction) | |||
|---|---|---|---|---|---|
| I | II | III | IV | ||
| a Irreversible process. b E 1/2 potential determined by digital simulation of the voltammetric curve. c Peak potential. | |||||
| 3 | 0.96ac | −0.73 | −1.09b | −1.62 | −2.15ab |
| −1.14b | |||||
| 5 | — | −0.72 | −1.11 | −1.68ac | — |
| C60-Ph | — | −0.73 | −1.11 | −1.62 | — |
| 4 | 0.93b (0.97)ac | −1.10 | — | — | — |
Dyad 3 (Fig. 4a) shows globally an oxidation process at positive potentials and four voltammetric waves at negative potential values. The oxidation is a completely irreversible process at a scan rate of 1 V s−1 and the voltammetric peak morphology indicates a sluggish electron transfer. Moreover, a repetitive potential scan evidences a progressive passivation of the electrode surface, due to some product of the irreversibility.
Examination of Fig. 4a and c shows that the oxidation of the dyad can be easily attributed to the Nile Red moiety, as the first oxidation of fullerene is at 1.69 V (vs. SCE)71 and the calixarene does not show any redox process neither at the positive nor at the negative potential range explored in this work.
Concerning the reductions of 3, the first peak at negative potentials, centred at about −0.75 V, is completely reversible and can be attributed to the C60 unit. This is supported by comparing the first reduction of the cyclic voltammetric curves of 3 (Fig. 4a) and those of 5 and C60-Ph (Fig. 4b, blue and red lines, respectively), that occur at the same potential.
The second reduction voltammetric wave of dyad 3 is reversible and is due to two closely spaced reduction processes: one attributed to the C60 and the other to the Nile Red moiety, as it can be easily inferred by comparing the corresponding potential values in Table 2 and the voltammetric curves in Fig. 4b and 4c. It is worth noticing that, despite the cathodic peak potential of the sluggish reduction process of the Nile Red species (see Fig. 4c) is at −1.30 V, it does not give rise to any shoulder at the more negative potential side of the second voltammetric wave. In fact, the E1/2 potential of the Nile Red reduction is at −1.10 V, that is very close to the second reduction potential of C60 and thus a catalytic reduction of the Nile Red moiety by the more reversible C602− is observed. This suggests some kind of intramolecular interaction between the units C60 and Nile Red in the dyad.
The third reduction peak of 3 (Fig. 4a) is attributed to the third reduction of fullerene, on the basis of similar arguments as above. The fourth irreversible peak is also centred on the C60, as reported in literature.71
The redox results are in agreement with the scenario given by the calculated frontier molecular orbitals, for all the four selected conformers (Fig. S5–S9, ESI†).
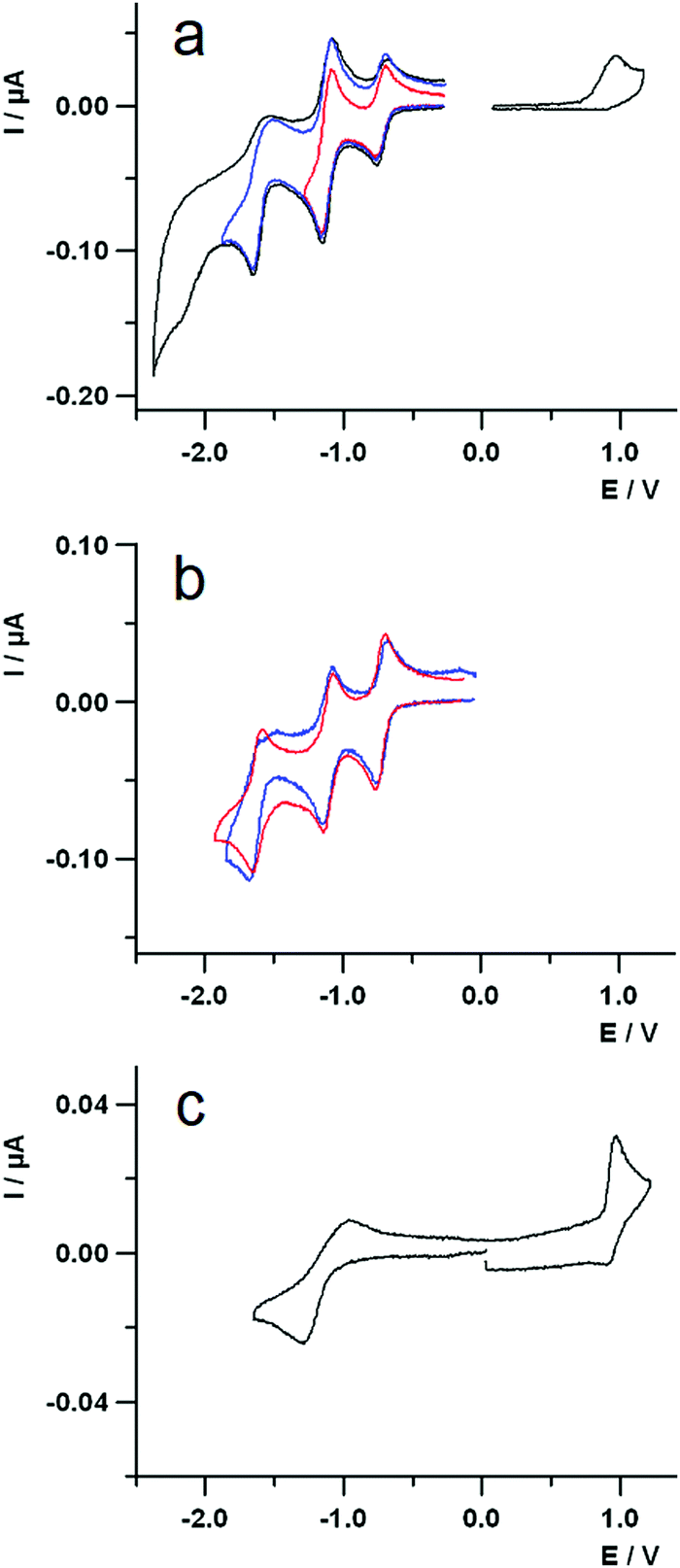
The UV-Vis-NIR spectroelectrochemical study of the dyad has been performed to ascertain both the attribution of the first redox processes as well as to gain further insights on the photoinduced processes. Fig. 5a shows the spectra of the pristine compound (black line), that of the one-electron reduced species (red line) and all those representing the spectral evolution from the initial to the final reduced species (thin black lines). The difference spectrum, representing the change upon reduction, is reported in Fig. 5b, showing a sharp negative band at about 270 nm, due to the injection of the first electron into the fullerene,72 another negative, larger band at about 540 nm, and a positive band forming at longer wavelengths. The peak at about 540 nm is a transition occurring on the Nile Red moiety that, unexpectedly, is involved by the first reduction centred on C60. This suggests that some interaction exists in dyad 3 between the one-electron reduced fullerene and Nile Red, thus perturbing the electronic transitions of the Nile Red itself.73
 | ||
| Fig. 5 (a) UV-Vis-NIR spectra of 0.6 mM dyad 3 in 0.08 M TBAH/DCM solution; working electrode, Pt gauze; temperature = 0 °C. Spectra of the pristine compound (black line) and one-electron reduced species (red line). (b) Difference spectrum of the reduced state (red trace; spectrum of the pristine compound subtracted). (c) UV-Vis-NIR difference spectrum of the oxidized state of dyad 3 0.6 mM in 0.08 M TBAH/DCM solution; working electrode, Pt gauze; temperature = 0 °C. | ||
A spectroelectrochemical investigation about the oxidation of 3 has been tentatively performed with less clear evidences, due to the chemically irreversible electron transfer, as discussed in the electrochemical characterization. The difference spectrum for the oxidation is plotted in Fig. 5c, showing a bleaching of the band at about 540 nm and the increase of some bands both in the 380–450 nm spectral range and on the red side of the spectrum, from about 600 nm up to the near IR region (highlighted by a grey box in Fig. 5c), with a peak at about 1000 nm.
Transient spectroscopy
To better investigate the mechanism and dynamics of energy/charge transfer upon excitation of Nile Red in dyad 3, we performed transient absorption experiments with sub-picosecond time resolution, comparing the results for the dyad with those for the single-chromophore reference compounds 4 and 5.In particular, compounds 3 and 4 were excited at 530 nm, and compound 5 was excited at 400 nm, and the excited state dynamics was recorded in the visible spectral region (400–750 nm). Measurements were performed both in toluene and chloroform.
Fig. 6a reports the experimental transient spectra of dyad 3 dissolved in toluene (the transient spectra and EADS of all compounds measured in chloroform are reported in ESI,† Fig. S12). The negative bands centered around 540 nm and 615 nm are respectively associated with ground state bleaching (GSB) and stimulated emission (SE), while the positive bands around 470 and 545 nm are excited state absorption (ESA) of the Nile Red unit. Indeed, all these spectral features are also visible in the transient spectra of compound 4 (Fig. 6b). In both solvents, the GSB and SE signals of dyad 3 recover on a faster timescale as compared to the isolated compound 4. Furthermore, on a longer timescale, the SE signal of dyad 3 is replaced by a positive broad signal peaking at about 680–700 nm. The latter band is also recognized in the transient spectra of the reference compound 5 (Fig. 6c), allowing to assign this feature to the C60 unit.
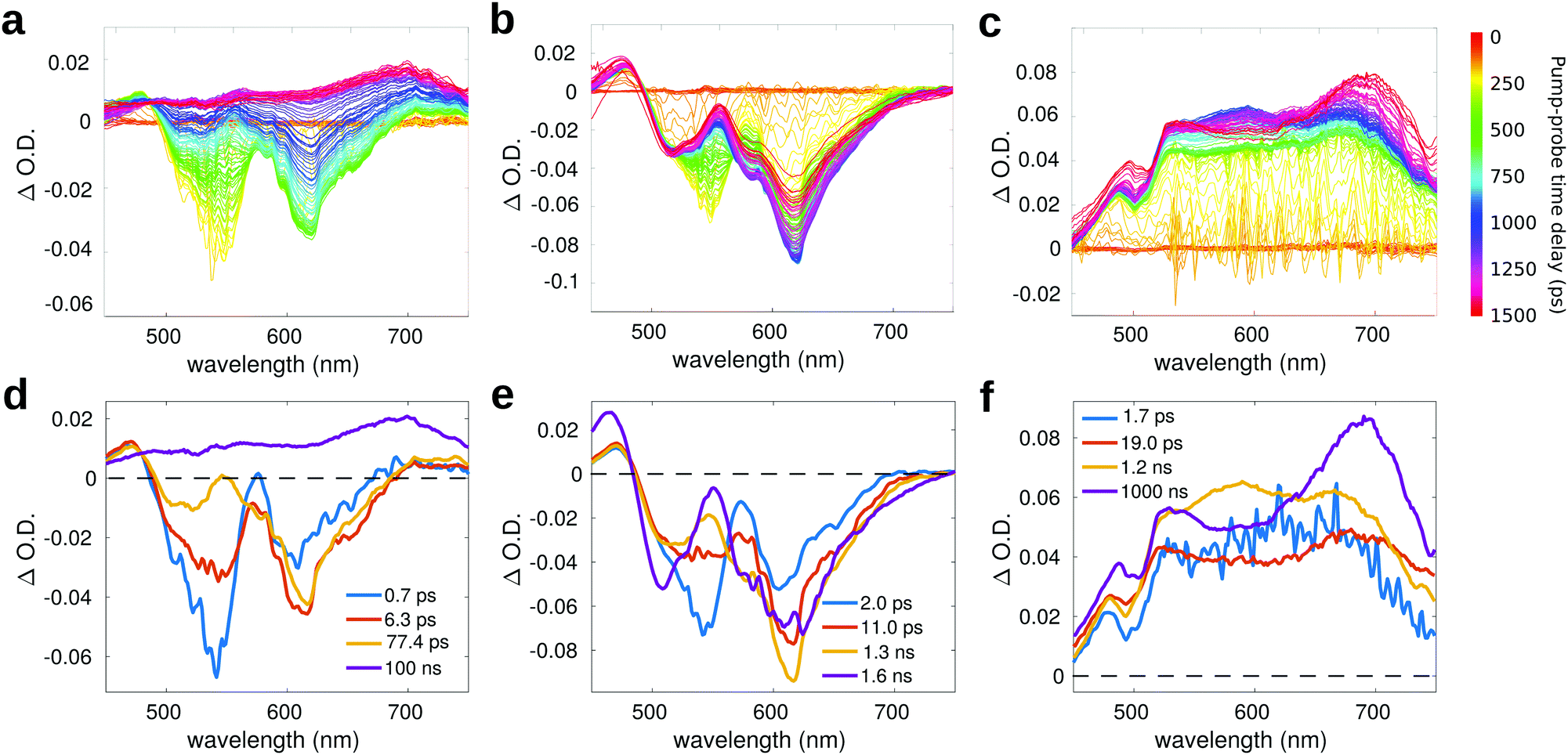 | ||
| Fig. 6 Transient spectra of: (a) compound 3 (λexc = 530 nm), (b) compound 4 (λexc = 530 nm) and (c) compound 5 (λexc = 400 nm) in toluene. Panels (d–f) report the respective EADS obtained from global analysis. | ||
For compound 5 (Fig. 6c), the GSB is not observed in the probed spectral region, because the most intense absorption of compound 5 is located at shorter wavelengths (the excitation wavelength in this case is at 400 nm). In the visible region, the absorption of compound 5 has a very low intensity, so that the GSB is compensated by a broad ESA band, which covers all the probed spectral range. This broad ESA appears immediately after excitation, and does not show significant spectral changes within the first nanosecond. On a timescale of about 1 ns, we observe the rise of a positive signal peaked at around 700 nm, that has been ascribed in the literature to the absorption of the fullerene triplet.74
Global analysis has been performed to extract the kinetic constants associated to the excited state relaxation of dyad 3 and of the reference compounds in the two solvents. The Evolution Associated Difference Spectra (EADS) are obtained under the assumption that the excited state population evolves following a sequential kinetic scheme described by a multi-exponential trend: each EADS represents the spectral component associated with a decay time constant. In our case, the results of global analysis show that the excited state dynamics can be well described using four time constants in all the studied samples.
For dyad 3 in toluene (Fig. 6d), the initial EADS (blue line) forms almost instantaneously after excitation and decays with a lifetime of 0.7 ps. This timescale can be associated to a fast relaxation of the excited state. In the second EADS (orange line), it is possible to observe a partial decay of the GSB band at 540 nm and the increase of the negative SE signal at 615 nm, due to increased emission from a relaxed excited state. The second EADS evolves into the third spectral component (yellow line) with a time constant of (6.3 ps)−1: on this timescale, the negative bleaching signal (540 nm) keeps decaying and a small ESA signal grows at around 550 nm. The final spectral evolution, occurring on a timescale of 77.4 ps, is associated to the formation of the fullerene triplet. The final very long lifetime has been arbitrarily fixed at 100 ns, which has to be considered only as an indicative value. The lifetime of the triplet state, which is probably longer than 100 ns, is well beyond the time window probed by our measurement, which allows to record transient spectra with a maximum pump–probe delay of 1.5 ns. The spectral shape of the final EADS of dyad 3 is very well comparable to the long-living EADS retrieved for compound 5. A similar excited state evolution can be observed in case of chloroform (Fig. S12, ESI†), with slightly different kinetic constants.
Fig. 6e reports the EADS obtained by global analysis for compound 4 in toluene. Although the spectral shape of the transient signal is very similar to that observed for dyad 3 (with the exclusion of the long-living spectral component), the dynamic evolution of 4 is quite different: both GSB and SE bands do not recover on the investigated timescale, indicating that the excited state lifetime of 4 is >1.5 ns, in agreement with the high fluorescence quantum yield measured for this compound.32
To better highlight the contribution of the isolated moieties to the spectra of dyad 3, and to evidence the occurrence of energy/charge transfer processes, in Fig. 7 we compare the transient spectra of compounds 3 and 4 at selected pump–probe delay times.
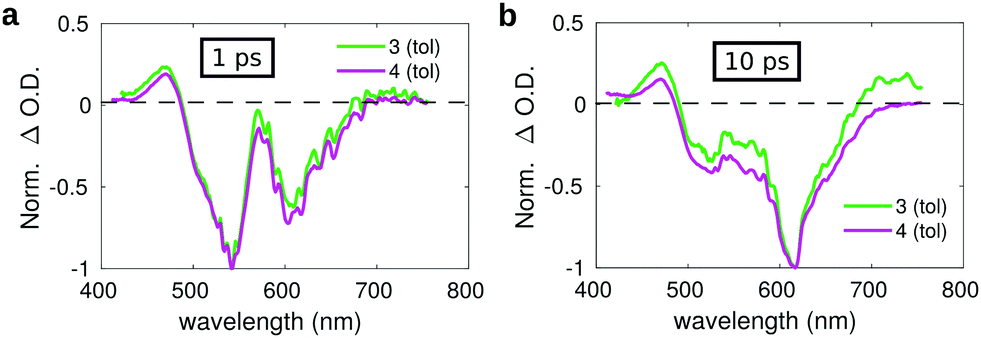 | ||
| Fig. 7 Comparison between the transient spectra of compound 3 (green lines) and 4 (violet lines) in toluene registered at a pump–probe delay of: (a) 1 ps and (b) 10 ps. | ||
The spectrum measured on a short delay (1 ps, Fig. 7a) for dyad 3 is almost superimposed to that of compound 4 upon normalization of their respective intensities. However, the comparison of the transient spectra of compounds 3 and 4 registered at a pump–probe delay of 10 ps evidences slight differences in the respective spectral shapes. Normalizing the spectra on the intensity of the SE band, an overall increase of the positive signals is observed for dyad 3 as compared to compound 4, both at the blue side of the probed spectral region (400–450 nm) and on the red side (650–750 nm). The small overall positive intensity of the spectrum of dyad 3 as compared to compound 4 could indicate a contribution of the C60 moiety, whose transient spectrum is characterized by a broad positive band extending over the full investigated spectral window on the short timescale, suggesting the occurrence of fast energy transfer. The excited state evolution of compound 3 and 4 is markedly different on a longer timescale, as evidenced by comparing the kinetic traces recorded on the maximum of the SE band (615 nm) (Fig. S15, ESI†), showing that the band recovery of dyad 3 is much faster as compared to compound 4. This suggests the occurrence of efficient energy/charge transfer in the dyad.
As previously mentioned, starting from a timescale of about 100 ps up to the maximum pump–probe delay probed with our measurements, the transient spectra of dyad 3 are dominated by a positive band which closely reminds that observed in the isolated fullerene. Indeed the comparison of the long-living EADS of compounds 3 and 5 (Fig. 8a), shows that the transient spectra of the two compounds are almost superimposable and characterized by the presence of the intense ESA band peaked at about 700 nm, which has been previously ascribed to the C60 triplet.74
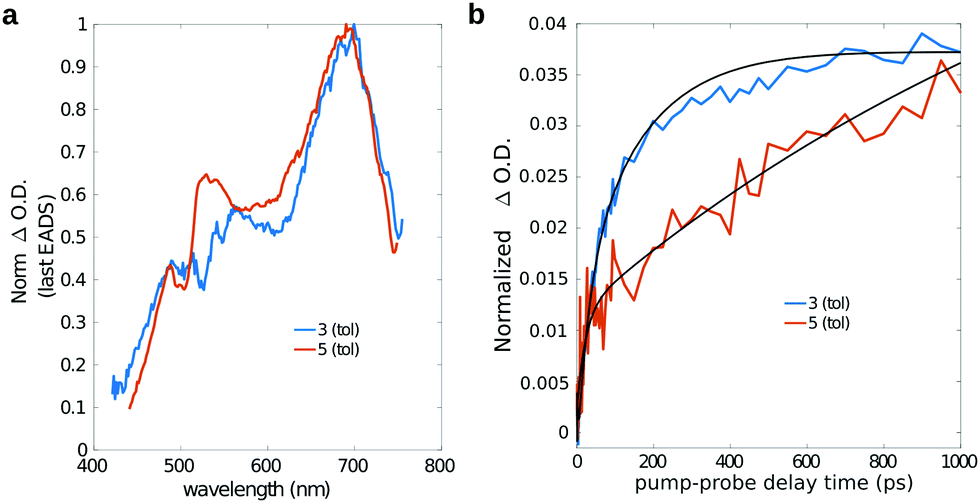 | ||
| Fig. 8 (a) Long-lived EADS and (b) single-wavelength kinetics at 680 nm for compound 3 (blue lines) and 5 (orange lines) in toluene. For a better comparison, a constant offset has been added/subtracted from the time traces of both compound 3 and 5 to shift their intensity to zero at zero pump–probe delay time. | ||
This observation confirms that, in the dyad, the initially formed singlet localized on Nile Red efficiently converts into the triplet excited state of fullerene. The comparison between the single-wavelength kinetic traces recorded at the maximum of the positive band associated to the fullerene triplet (Fig. 8b, other single-wavelength kinetic traces are shown and discussed in ESI†), indicates that the growth of this peak is highly accelerated in dyad 3 (about 80 ps in toluene and 220 ps in chloroform) with respect to the reference compound 5 (∼1 ns). This result implies that the main formation of the fullerene triplet in dyad 3 does not step through FRET followed by ISC, since in that case the kinetics would not be accelerated with respect to 5. Indeed, previous literature shows that when Förster-type energy transfer between a donor and fullerene occurs, triplet formation is not usually observed below the nanosecond timescale.1,75–80 Our results suggest that the mechanism of formation of the fullerene triplet in dyad 3 is related to fast electron transfer (charge separation) and back electron transfer (recombination) involving the frontier orbitals of the Nile Red and C60 moieties. Charge recombination in dyad 3 seems to rapidly produce the fullerene triplet in a preferential way with respect to the fullerene singlet. The mechanism that we envisage is the so-called radical-pair intersystem crossing (RP-ISC),2,4i.e. a long-distance charge separation, followed by a singlet/triplet state mixing and, eventually, a charge recombination, as schematically resumed in Scheme 2. The Rehm–Weller analysis performed on this donor/acceptor dyad confirmed that the charge separation process is thermodynamically allowed for three of the four conformers singled out by the computational screening (see ESI†).
 | ||
| Scheme 2 Schematic representation of the decay pathway of dyad 3 upon photoexcitation of the Nile Red moiety. | ||
The spectral range covered by our transient setup (up to 750 nm) does not allow to check for the presence of the fullerene anion, which is expected to feature a peak at around 1000 nm.81 The results obtained by the spectroelectrochemistry measurements on dyad 3 highlight that the formation of a radical pair NR+-C60− would induce a decreased absorption at about 540 nm and the rise of a positive band peaked at about 600-620 nm, besides a small increased absorption both in the 400–450 nm and in the near IR spectral ranges. Since electron transfer would occur upon photoexcitation of the sample, we do not expect to see, in the transient spectra, variations of intensity in the bleaching region in response to charge separation, while, in principle, all the positive signals observed in the spectroelectrochemistry difference spectra should appear also in the pump–probe difference spectra, at least in a certain time window. Unfortunately, the main positive band associated with charge separation, peaked at about 600 nm, is superimposed with the negative intense stimulated emission signal (see Fig. S16, ESI†). This, together with the likely very short lifetime of the CS state, probably hampers the observation of the CS state itself in the pump–probe spectra. Nevertheless, the observation of triplet formation on a much faster timescale than expected in case of FRET strongly suggests that charge separation and recombination are essential in order to justify the kinetics of triplet formation for the dyad in both toluene and chloroform.
Conclusions
We reported the synthesis and the electrochemical and photophysical characterization of a Nile Red/Calix[4]arene/C60 donor/bridge/acceptor dyad where the donor and acceptor moieties are grafted to the central scaffold through flexible linkers. The properties of the dyad were compared with the reference isolated donor and acceptor. Steady-state optical spectra show a strong quenching of the Nile Red emission and an efficient production of singlet oxygen when selectively exciting Nile Red in the dyad, suggesting a complete conversion of the photoexcited Nile Red singlet into the C60 triplet. The mechanism and dynamics of this conversion was investigated via sub-picosecond transient absorption spectroscopy that, with the support of spectroelectrochemistry measurements, strongly suggests the formation of a CS state on a timescale of about a few tens of ps, followed by extremely fast charge recombination leading to the formation of the triplet localized on the C60 moiety, on a hundred ps timescale. Considering the very fast formation of the C60 triplet in dyad 3 as compared to compound 5, the most probable mechanism for intersystem crossing is the so-called radical pair ISC, which is facilitated when the donor and acceptor groups are quite far apart and weakly interacting. The flexible structure of our dyad and the presence of a quite bulky bridge can favor the decoupling of the radical pair allowing for the mixing of the 1CS and 3CS states. The rate of formation of the triplet state localized on C60 is high and is barely dependent on solvent polarity (on the order of 1010 s−1 in toluene and 5 × 109 s−1 in chloroform).We exclude FRET followed by ISC as a possible mechanism for the formation of the C60 triplet, because of the too fast dynamics. Other mechanisms of triplet formation cannot be excluded, like SOCT-ISC or vibronically assisted ISC,4 but our spectroelectrochemical and transient absorption data, together with the conformation of our dyad, mostly support the RP-ISC mechanism. What is surprising is the speed of formation of the triplet, despite the conformational flexibility, being comparable to the SOCT-ISC mediated mechanism, typically observed in compact and orthogonal dyads.
The fast and efficient formation of the fullerene triplet in easily affordable dyads with high absorption coefficient in the visible range, in solvents of different polarity, could be of paramount importance in many applications, from the production of reactive oxygen species (photodynamic therapy) to photon upconversion.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We dedicate this paper to Concepció Rovira and Jaume Veciana, with gratitude for their contribution in the field of advanced functional materials and for their collaborative and friendly enthusiasm. The work in Parma benefited from the equipment and support of the COMP-HUB Initiative, funded by the “Departments of Excellence” program of the Italian Ministry for Education, University and Research (MIUR, 2018-2022), and from the HPC (High Performance Computing) facility of the University of Parma, Italy. F. T. acknowledges funding from the European Union's Horizon 2020 research and innovation program under Grant Agreement No. 812872 (TADFlife). M. D. D. and S. D. acknowledge European Union's Horizon 2020 research and innovation program under grant agreement n. 871124 Laserlab-Europe. The “Centro Interfacoltà di Misure” (CIM) of the University of Parma is acknowledged for use of their NMR and mass spectrometers. University of Bologna is also gratefully acknowledged.Notes and references
- D. J. Gibbons, A. Farawar, P. Mazzella, S. Leroy-Lhez and R. M. Williams, Photochem. Photobiol. Sci., 2020, 19, 136–158 CrossRef CAS PubMed.
- Y. Hou, X. Zhang, K. Chen, D. Liu, Z. Wang, Q. Liu, J. Zhao and A. Barbon, J. Mater. Chem. C, 2019, 7, 12048–12074 RSC.
- A. Aster, C. Rumble, A.-B. Bornhof, H.-H. Huang, N. Sakai, T. Šolomek, S. Matile and E. Vauthey, Chem. Sci., 2021, 12, 4908–4915 RSC.
- D. Sasikumar, A. T. John, J. Sunny and M. Hariharan, Chem. Soc. Rev., 2020, 49, 6122–6140 RSC.
- J. P. Fouassier and J. Lalevée, Photoinitiators for Polymer Synthesis, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2012 Search PubMed.
- X. Zhao, J. Liu, J. Fan, H. Chao and X. Peng, Chem. Soc. Rev., 2021, 50, 4185–4219 RSC.
- P. Bharmoria, H. Bildirir and K. Moth-Poulsen, Chem. Soc. Rev., 2020, 49, 6529–6554 RSC.
- T. J. Penfold, F. B. Dias and A. P. Monkman, Chem. Commun., 2018, 54, 3926–3935 RSC.
- Z. Wang and J. Zhao, Org. Lett., 2017, 19, 4492–4495 CrossRef CAS PubMed.
- J. Verhoeven, J. Photochem. Photobiol., C, 2006, 7, 40–60 CrossRef CAS.
- Z. E. X. Dance, S. M. Mickley, T. M. Wilson, A. B. Ricks, A. M. Scott, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. A, 2008, 112, 4194–4201 CrossRef CAS PubMed.
- A. Karimata, S. Suzuki, M. Kozaki, K. Kimoto, K. Nozaki, H. Matsushita, N. Ikeda, K. Akiyama, D. Kosumi, H. Hashimoto and K. Okada, J. Phys. Chem. A, 2014, 118, 11262–11271 CrossRef CAS PubMed.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, H. Savoie, K. J. Flanagan, C. Sy, E. Sitte, M. Telitchko, F. Laquai, R. W. Boyle and M. O. Senge, J. Am. Chem. Soc., 2017, 139, 6282–6285 CrossRef CAS PubMed.
- K. Chen, W. Yang, Z. Wang, A. Iagatti, L. Bussotti, P. Foggi, W. Ji, J. Zhao and M. Di Donato, J. Phys. Chem. A, 2017, 121, 7550–7564 CrossRef CAS PubMed.
- Y. Hou, T. Biskup, S. Rein, Z. Wang, L. Bussotti, N. Russo, P. Foggi, J. Zhao, M. Di Donato, G. Mazzone and S. Weber, J. Phys. Chem. C, 2018, 122, 27850–27865 CrossRef CAS.
- D. Liu, A. M. El-Zohry, M. Taddei, C. Matt, L. Bussotti, Z. Wang, J. Zhao, O. F. Mohammed, M. Di Donato and S. Weber, Angew. Chem., 2020, 132, 11688–11696 CrossRef.
- M. T. Colvin, A. B. Ricks, A. M. Scott, D. T. Co and M. R. Wasielewski, J. Phys. Chem. A, 2012, 116, 1923–1930 CrossRef CAS PubMed.
- E. A. Weiss, M. A. Ratner and M. R. Wasielewski, J. Phys. Chem. A, 2003, 107, 3639–3647 CrossRef CAS.
- G. P. Wiederrecht, W. A. Svec, M. R. Wasielewski, T. Galili and H. Levanon, J. Am. Chem. Soc., 2000, 122, 9715–9722 CrossRef CAS.
- C. B. Kc, G. N. Lim, V. N. Nesterov, P. A. Karr and F. D’Souza, Chem. – Eur. J., 2014, 20, 17100–17112 CrossRef CAS PubMed.
- F. Scarel, C. Ehli, D. M. Guldi and A. Mateo-Alonso, Chem. Commun., 2013, 49, 9452 RSC.
- T. Okada, I. Karaki, E. Matsuzawa, N. Mataga, Y. Sakata and S. Misumi, J. Phys. Chem., 1981, 85, 3957–3960 CrossRef CAS.
- R. M. Williams, J. M. Zwier and J. W. Verhoeven, J. Am. Chem. Soc., 1995, 117, 4093–4099 CrossRef CAS.
- M. A. Filatov, S. Karuthedath, P. M. Polestshuk, S. Callaghan, K. J. Flanagan, M. Telitchko, T. Wiesner, F. Laquai and M. O. Senge, Phys. Chem. Chem. Phys., 2018, 20, 8016–8031 RSC.
- C. Hippius, I. H. M. van Stokkum, M. Gsänger, M. M. Groeneveld, R. M. Williams and F. Würthner, J. Phys. Chem. C, 2008, 112, 2476–2486 CrossRef CAS.
- N. Vân Anh, F. Schlosser, M. M. Groeneveld, I. H. M. van Stokkum, F. Würthner and R. M. Williams, J. Phys. Chem. C, 2009, 113, 18358–18368 CrossRef.
- I. Tosi, B. Bardi, M. Ambrosetti, E. Domenichini, A. Iagatti, L. Baldini, C. Cappelli, M. Di Donato, F. Sansone, C. Sissa and F. Terenziani, Dyes Pigm., 2019, 171, 107652 CrossRef CAS.
- B. Bardi, I. Tosi, F. Faroldi, L. Baldini, F. Sansone, C. Sissa and F. Terenziani, Chem. Commun., 2019, 55, 8098–8101 RSC.
- A. Issac, R. Hildner, C. Hippius, F. Würthner and J. Köhler, ACS Nano, 2014, 8, 1708–1717 CrossRef CAS PubMed.
- S. Shinkai, Tetrahedron, 1993, 49, 8933–8968 CrossRef CAS.
- G. Pognon, J. A. Wytko, P. D. Harvey and J. Weiss, Chem. – Eur. J., 2009, 15, 524–535 CrossRef CAS PubMed.
- I. Tosi, M. Segado Centellas, E. Campioli, A. Iagatti, A. Lapini, C. Sissa, L. Baldini, C. Cappelli, M. Di Donato, F. Sansone, F. Santoro and F. Terenziani, ChemPhysChem, 2016, 17, 1686–1706 CrossRef CAS PubMed.
- A. Arduini, M. Fabbi, M. Mantovani, L. Mirone, A. Pochini, A. Secchi and R. Ungaro, J. Org. Chem., 1995, 60, 1454–1457 CrossRef CAS.
- C. Hippius, I. H. M. van Stokkum, E. Zangrando, R. M. Williams, M. Wykes, D. Beljonne and F. Würthner, J. Phys. Chem. C, 2008, 112, 14626–14638 CrossRef CAS.
- C. Hippius, I. H. M. van Stokkum, E. Zangrando, R. M. Williams and F. Würthner, J. Phys. Chem. C, 2007, 111, 13988–13996 CrossRef CAS.
- P. S. Baran, R. R. Monaco, A. U. Khan, D. I. Schuster and S. R. Wilson, J. Am. Chem. Soc., 1997, 119, 8363–8364 CrossRef CAS.
- S. Shao, H. B. Gobeze, V. Bandi, C. Funk, B. Heine, M. J. Duffy, V. Nesterov, P. A. Karr and F. D’Souza, ChemPhotoChem, 2020, 4, 68–81 CrossRef CAS.
- A. Iagatti, L. Cupellini, G. Biagiotti, S. Caprasecca, S. Fedeli, A. Lapini, E. Ussano, S. Cicchi, P. Foggi, M. Marcaccio, B. Mennucci and M. Di Donato, J. Phys. Chem. C, 2016, 120, 16526–16536 CrossRef CAS.
- D. Gust, T. A. Moore and A. L. Moore, J. Photochem. Photobiol., B, 2000, 58, 63–71 CrossRef CAS.
- R. Ziessel, B. D. Allen, D. B. Rewinska and A. Harriman, Chem. – Eur. J, 2009, 15, 7382–7393 CrossRef CAS PubMed.
- H. Ünlü and E. Okutan, Dyes Pigm., 2017, 142, 340–349 CrossRef.
- F. Sansone, S. Barboso, A. Casnati, M. Fabbi, A. Pochini, F. Ugozzoli and R. Ungaro, Eur. J. Org. Chem., 1998, 897–905 CrossRef CAS.
- C. Bruno, I. Doubitski, M. Marcaccio, F. Paolucci, D. Paolucci and A. Zaopo, J. Am. Chem. Soc., 2003, 125, 15738–15739 CrossRef CAS PubMed.
- C. Bruno, R. Benassi, A. Passalacqua, F. Paolucci, C. Fontanesi, M. Marcaccio, E. A. Jackson and L. T. Scott, J. Phys. Chem. B, 2009, 113, 1954–1962 CrossRef CAS PubMed.
- C. Amatore and C. Lefrou, J. Electroanal. Chem., 1992, 324, 33–58 CrossRef CAS.
- L. Mottier, Antigona, University of Bologna, Bologna, Italy, 1999 Search PubMed.
- S.-M. Lee, M. Marcaccio, J. A. McCleverty and M. D. Ward, Chem. Mater., 1998, 10, 3272–3274 CrossRef CAS.
- P. L. Gentili, M. Mugnai, L. Bussotti, R. Righini, P. Foggi, S. Cicchi, G. Ghini, S. Viviani and A. Brandi, J. Photochem. Photobiol., A, 2007, 187, 209–221 CrossRef CAS.
- I. H. M. van Stokkum, D. S. Larsen and R. van Grondelle, Biochim. Biophys. Acta, Bioenerg., 2004, 1657, 82–104 CrossRef CAS PubMed.
- J. J. Snellenburg, S. P. Laptenok, R. Seger, K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2012, 49, 1–22 Search PubMed.
- K. M. Mullen and I. H. M. van Stokkum, J. Stat. Softw., 2007, 18, 1–5 CrossRef.
- E. R. Henry and J. Hofrichter, Methods Enzymol., 1992, 210, 129–192 CAS.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 490–519 CrossRef CAS.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 520–552 CrossRef CAS.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 553–586 CrossRef CAS.
- T. A. Halgren and R. B. Nachbar, J. Comput. Chem., 1996, 17, 587–615 CAS.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 616–641 CrossRef CAS.
- PCmodel v. 10.0, http://www.serenasoft.com.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Revision B.01, Wallingfor CT, 2016 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615 RSC.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 CrossRef CAS PubMed.
- M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 1993, 115, 9798–9799 CrossRef CAS.
- J. F. Deye, T. A. Berger and A. G. Anderson, Anal. Chem., 1990, 62, 615–622 CrossRef CAS.
- J. P. Hare, H. W. Kroto and R. Taylor, Chem. Phys. Lett., 2013, 589, 57–60 CrossRef CAS.
- R. K. Dubey, T. Kumpulainen, A. Efimov, N. V. Tkachenko and H. Lemmetyinen, Eur. J. Org. Chem., 2010, 3428–3436 CrossRef CAS.
- R. M. Williams, M. Koeberg, J. M. Lawson, Y.-Z. An, Y. Rubin, M. N. Paddon-Row and J. W. Verhoeven, J. Org. Chem., 1996, 61, 5055–5062 CrossRef CAS.
- D. M. Guldi and M. Prato, Acc. Chem. Res., 2000, 33, 695–703 CrossRef CAS PubMed.
- Y. P. Sun, P. Wang and N. B. Hamilton, J. Am. Chem. Soc., 1993, 115, 6378–6381 CrossRef CAS.
- R. W. Redmond and J. N. Gamlin, Photochem. Photobiol., 1999, 70, 391–475 CrossRef CAS PubMed.
- F. Prat, C. Martí, S. Nonell, X. Zhang, C. S. Foote, R. González Moreno, J. L. Bourdelande and J. Font, Phys. Chem. Chem. Phys., 2001, 3, 1638–1643 RSC.
- C. Bruno, M. Marcaccio, D. Paolucci, C. Castellarin-Cudia, A. Goldoni, A. V. Streletskii, T. Drewello, S. Barison, A. Venturini, F. Zerbetto and F. Paolucci, J. Am. Chem. Soc., 2008, 130, 3788–3796 CrossRef CAS PubMed.
- D. Dubois, K. M. Kadish, S. Flanagan, R. E. Haufler, L. P. F. Chibante and L. J. Wilson, J. Am. Chem. Soc., 1991, 113, 4364–4366 CrossRef CAS.
- G. Valenti, M. Iurlo, R. M. Claramunt, G. Accorsi, F. Paolucci, M. Á. Farrán and M. Marcaccio, ChemElectroChem, 2018, 5, 985–990 CrossRef CAS.
- R. B. Weisman, K. D. Ausman, A. F. Benedetto and D. A. Samuels, Proc. SPIE, 1997, 3142, 26–35 CrossRef CAS.
- J. T. Buck, A. M. Boudreau, A. DeCarmine, R. W. Wilson, J. Hampsey and T. Mani, Chem, 2019, 5, 138–155 CAS.
- P. K. Poddutoori, Y. E. Kandrashkin, C. O. Obondi, F. D’Souza and A. van der Est, Phys. Chem. Chem. Phys., 2018, 20, 28223–28231 RSC.
- I. R. Gould, J. A. Boiani, E. B. Gaillard, J. L. Goodman and S. Farid, J. Phys. Chem. A, 2003, 107, 3515–3524 CrossRef CAS.
- P. C. Y. Chow, S. Albert-Seifried, S. Gélinas and R. H. Friend, Adv. Mater., 2014, 26, 4851–4854 CrossRef CAS PubMed.
- E. G. Petrov, B. Robert, S. H. Lin and L. Valkunas, Biophys. J., 2015, 109, 1735–1745 CrossRef CAS PubMed.
- R. M. Williams, H.-C. Chen, D. Di Nuzzo, S. C. J. Meskers and R. A. J. Janssen, J. Spectrosc., 2017, 1–16 CAS.
- T. Kato, T. Kodama, T. Shida, T. Nakagawa, Y. Matsui, S. Suzuki, H. Shiromaru, K. Yamauchi and Y. Achiba, Chem. Phys. Lett., 1991, 180, 446–450 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis; 1H NMR spectroscopy; computational results; Rehm–Weller analysis; singlet oxygen generation; supplementary transient spectroscopy data. See DOI: 10.1039/d1tc01885j |
| This journal is © The Royal Society of Chemistry 2021 |