
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering Sr-doping for enabling long-term stable FAPb1−xSrxI3 quantum dots with 100% photoluminescence quantum yield†
Andrés F.
Gualdrón-Reyes
 *a,
David F.
Macias-Pinilla
a,
Sofia
Masi
a,
Carlos
Echeverría-Arrondo
a,
Said
Agouram
bc,
Vicente
Muñoz-Sanjosé
bc,
Jhonatan
Rodríguez-Pereira
d,
Jan M.
Macak
de and
Iván
Mora-Seró
*ac
*a,
David F.
Macias-Pinilla
a,
Sofia
Masi
a,
Carlos
Echeverría-Arrondo
a,
Said
Agouram
bc,
Vicente
Muñoz-Sanjosé
bc,
Jhonatan
Rodríguez-Pereira
d,
Jan M.
Macak
de and
Iván
Mora-Seró
*ac
aInstitute of Advanced Materials (INAM), Universitat Jaume I (UJI), Avenida de Vicent Sos Baynat, s/n, 12071 Castelló de la Plana, Castellón, Spain. E-mail: sero@uji.es; gualdron@uji.es
bDepartment of Applied Physics and Electromagnetism, University of Valencia, 46100 Valencia, Spain
cMaterials for Renewable Energy (MAER), Unitat Mixta d’Investigació UV-UJI, 46010 Valencia, Spain
dCenter of Materials and Nanotechnologies, Faculty of Chemical Technology, University of Pardubice, Nam. Cs. Legii 565, 53002 Pardubice, Czech Republic
eCentral European Institute of Technology, Brno University of Technology, Purkyňova 123, 612 00 Brno, Czech Republic
First published on 1st February 2021
Abstract
The Pb substitution in quantum dots (PQDs) with lesser toxic metals has been widely searched to be environmentally friendly, and be of comparable or improved performance compared to the lead-perovskite. However, the chemical nature of the lead substitute influences the incorporation mechanism into PQDs, which has not been explored in depth. In this work, we analyzed Sr-doping-induced changes in FAPbI3 perovskites by studying the optical, structural properties and chemical environment of FAPb1−xSrxI3 PQDs. The substitution of Pb by 7 at% Sr allows us to achieve FAPb1−xSrxI3 PQDs with 100% PLQY, high stability for 8 months under a relative humidity of 40–50%, and T80 = 6.5 months, one of the highest values reported for halide PQDs under air ambient conditions. FAPb0.93Sr0.07I3 PQDs also exhibit photobrightening under UV illumination for 12 h, recovering 100% PLQY at 15 days after synthesis. The suppression of structural defects mediated by Sr-doping decreases the non-radiative recombination mechanism. By attempting to increase the Sr content in PQDs, a mixture of 2D nanoplatelets/3D nanocubes has emerged, caused by a high Pb deficiency during the FAPb1−xSrxI3 synthesis. This contribution gives a novel insight to understand how the suitable/poor Pb substitution achieved through Sr-doping dictates the photophysical properties of PQDs that may be potentially applicable in optoelectronics.
1 Introduction
Considering the outstanding optoelectronic properties of different materials, it is unavoidable to refer halide perovskites, taking into account the research carried out in the last decade. The main features, such as the long diffusion length, strong sunlight harvesting and improved photocarrier mobility have led to the fabrication of efficient perovskite solar cells (PSCs) with the highest photoconversion efficiency at around 25.2% achieved until now.1 In terms of competitiveness, PSCs have surpassed laboratory efficiencies of well-established technologies, such as polycrystalline Si, CIGS and CdTe solar devices.1,2 However, PSCs suffer two main drawbacks: (i) the relatively low long-term stability reported for 3D halide perovskites based on APbI3 (A = cesium, Cs+; methylammonium, MA+ and formamidinium, FA+)3–6 and (ii) the presence of toxic Pb. Moreover, the maximum theoretical performance from a 3D lead halide perovskite obtained with the black perovskite α-phase of FAPbI3 stands at an efficiency of 32.3%, which is very close to the theoretical Schottky–Queisser limit.7 Nevertheless, while α-MAPbI3 is degraded under soft thermal conditions,8 CsPbI3 and FAPbI3, which present higher long term stability, stabilize into a non-photoactive crystalline yellow δ-phase at room temperature.3,6,9,10 Nonetheless, the stability of the black FAPbI3 phase can be enhanced by the appropriate use of the surface energy.10 To this extent, the synthesis of nanoconfined materials such as quantum dots can maintain the photoactive black phase structure and ensure the intrinsic optical properties.10,11 Perovskite quantum dots (PQDs) also exhibit a tolerant-to-defect structure,12 very high photoluminescence quantum yield (PLQY),13 multiple exciton generation and a tunable band gap (Eg) through the composition engineering or quantum confinement effect.14–16 These features have allowed the facile processing and manufacturing of PQD colloidal solutions, fabricating highly efficient LEDs with external quantum efficiencies higher than 20%.17 PQDs also show a good dot-to-dot electron transport ability,18 providing a long electron lifetime.19,20 Therefore, PQD solar cells have reached a certified record PCE of 16.6%, retaining 94% photoconversion after 600 h, using black-stabilized Cs1−xFAxPbI3 PQDs.19Although PQD-based devices could be available in future industrialization, the abovementioned toxic Pb is still a main concern to practical applications. In this context, the synthesis of less-toxic PQDs has been implemented through the metal replacement or doping21 with different cations, such as Mn2+,22 Zn2+,23 Sb3+,24 Sn2+,25–27 or Ge2+.25,28–31 Sn and Ge are prone to oxidizing to their corresponding tetravalents Sn4+ and Ge4+ forms, respectively. This causes the formation of metal (M) vacancies in the perovskite structure, and deviations in the M–I octahedra connectivity, which in turn, decrease the defect tolerance and consequently the performance.32 In the case of other elements, in general, the doping with new metals resulted in: (i) the suppression of a high density of non-radiative recombination pathways, increasing the photoluminescence quantum yield (PLQY) and providing suitable optoelectronic properties for LED fabrication,23 and (ii) the stabilization of the photoactive black α-phase by reducing the PQDs size, due to the smaller ionic radius of the metal substitutes [for instance, Zn2+ (0.74 Å), Mn2+ (0.83 Å) Sb3+ (0.92 Å) and Sn2+ (0.93 Å)] compared with Pb2+ (1.19 Å).23–25,33 Specifically, in the case of Zn2+-modification, a maximum PLQY of around 98.5% is achieved after adding 36 at% Zn2+ into the CsPbI3 structure. Here, alloyed CsPb1−xZnxI3 PQDs are generated, indicating that a large amount of the cation is introduced to improve the intrinsic properties of the host.23 Therefore, a more recent strategy to substitute Pb emerges with the Sr2+ doping. This non-toxic divalent cation shows an identical ionic radius (1.18 Å) to that of Pb2+,34 meeting all of the main characteristics for a total ion replacement, keeping the intrinsic properties of the host. By incorporating a Sr2+ content between 3–5 at% into the CsPbI3 PQDs, the black phase stability is prolonged to around 2 months, improving the PLQY up to 95%.35 At the doping level, this improvement surpasses the PLQY provided by the CsPb1−xZnxI3 PQDs (x up to 12 at%) around 60–70%.23 In this context, Sr2+ is a promising dopant for enhancing the photophysical properties of iodide PQDs. However, even though the total Pb substitution could be accessible to synthesize ASrI3, DFT calculations predict a wider band gap in these materials than those of the APbI3 counterparts (3.6 eV for the case of MASrI3).36 These works point out that the chemical mechanisms that control the Pb replacement in PQDs are not fully understood.
In this work, we track the Sr-doping-induced photophysical properties in red-emitting FAPb1−xSrxI3 PQDs by varying the Sr fraction during their synthesis. The 7 at% Sr-doping provided FAPb1−xSrxI3 PQDs with 100% PLQY, which is higher and more stable than that for pure FAPbI3, keeping a PLQY as high as 72% for 8 months after preparation. We concluded that Sr-doping maximized the suppression of the non-radiative recombination mechanism by reducing the density of the structural defects. For a higher Sr fraction, we evidenced the simultaneous emergence of 2D nanoplatelets, as the result of altering the stoichiometry of the precursors by a poor compensation of the Pb deficiency by Sr. According to these findings, the chemical nature and the content of lead substitute impacts on the synthesis of high-quality PQDs with desired photophysical properties. Furthermore, a mechanism for the Pb substitution controlled by Sr-doping can be established.
2 Analysis and discussion
The synthesis of the FAPbI3 and FAPb1−xSrxI3 PQDs was carried out by varying the precursor Sr fraction. The procedure is the same for each concentration, keeping the total metal amount the same by properly changing the Pb![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Sr ratio (see Experimental section). The final Sr fraction incorporated into the synthesized FAPb1−xSrxI3 PQDs has been determined by Energy-dispersive X-ray spectroscopy (EDS) to be 4, 7, 12 and 6 at%, corresponding to a nominal precursor composition of 30, 50, 60 and 70 at%, respectively, see Table 1. Samples were named, FAPb1−xSrxI3, considering x as the measured EDS composition, if nothing else is commented. Consequently, the amount of Sr incorporated into the PQD is significantly lower than the precursor ratio. The fact that the estimated Sr fraction is in the doping-range of 1–10 at%, compared with the nominal value, is strong evidence of the tough Pb substitution into the PQDs by Sr. A nanocube shape of the PQDs is evidenced for all the compositions, as shown in Fig. 1a–e, with an average particle size from 15 to 8.4 nm as a function of the nominal Sr content, in which higher Sr-doping entailed lower particle size. The size distribution for each composition is shown in Fig. S1a–e (ESI†). On the other hand, we determined the characteristic interplanar spacing of the analyzed PQDs as 0.64, 0.32, 0.29 and 0.23 nm (Fig. S2a–e, ESI†) through selected area electron diffraction (SAED). These values correspond to the (100), (200) (210) and (220) planes of the perovskite cubic crystal phase,37,38 which are in good agreement with the XRD patterns obtained for the PQDs (Fig. 2). Previous reports in the literature have suggested that the reduction of the PQDs size at a higher fraction of metal halide is caused by the high consumption of iodide ions from the mixture reaction used for their stabilization.23,35,39
:Sr ratio (see Experimental section). The final Sr fraction incorporated into the synthesized FAPb1−xSrxI3 PQDs has been determined by Energy-dispersive X-ray spectroscopy (EDS) to be 4, 7, 12 and 6 at%, corresponding to a nominal precursor composition of 30, 50, 60 and 70 at%, respectively, see Table 1. Samples were named, FAPb1−xSrxI3, considering x as the measured EDS composition, if nothing else is commented. Consequently, the amount of Sr incorporated into the PQD is significantly lower than the precursor ratio. The fact that the estimated Sr fraction is in the doping-range of 1–10 at%, compared with the nominal value, is strong evidence of the tough Pb substitution into the PQDs by Sr. A nanocube shape of the PQDs is evidenced for all the compositions, as shown in Fig. 1a–e, with an average particle size from 15 to 8.4 nm as a function of the nominal Sr content, in which higher Sr-doping entailed lower particle size. The size distribution for each composition is shown in Fig. S1a–e (ESI†). On the other hand, we determined the characteristic interplanar spacing of the analyzed PQDs as 0.64, 0.32, 0.29 and 0.23 nm (Fig. S2a–e, ESI†) through selected area electron diffraction (SAED). These values correspond to the (100), (200) (210) and (220) planes of the perovskite cubic crystal phase,37,38 which are in good agreement with the XRD patterns obtained for the PQDs (Fig. 2). Previous reports in the literature have suggested that the reduction of the PQDs size at a higher fraction of metal halide is caused by the high consumption of iodide ions from the mixture reaction used for their stabilization.23,35,39
| Nominal composition | Nominal (x) [Sr/(Sr + Pb)] | Percentage of elements calculated by EDS | Calculated (x) [Sr/(Sr + Pb)] | Calculated I/(Pb + Sr) ratio | ||
|---|---|---|---|---|---|---|
| I | Pb | Sr | ||||
| FAPbI3 | — | 74.9 | 25.1 | — | — | 2.98 |
| x = 0.3 | 0.3 | 76.3 | 22.6 | 1.1 | 0.04 | 3.22 |
| x = 0.5 | 0.5 | 79.0 | 19.5 | 1.5 | 0.07 | 3.76 |
| x = 0.6 | 0.6 | 73.7 | 23.0 | 3.3 | 0.12 | 2.80 |
| x = 0.7 | 0.7 | 73.6 | 24.9 | 1.5 | 0.06 | 2.78 |
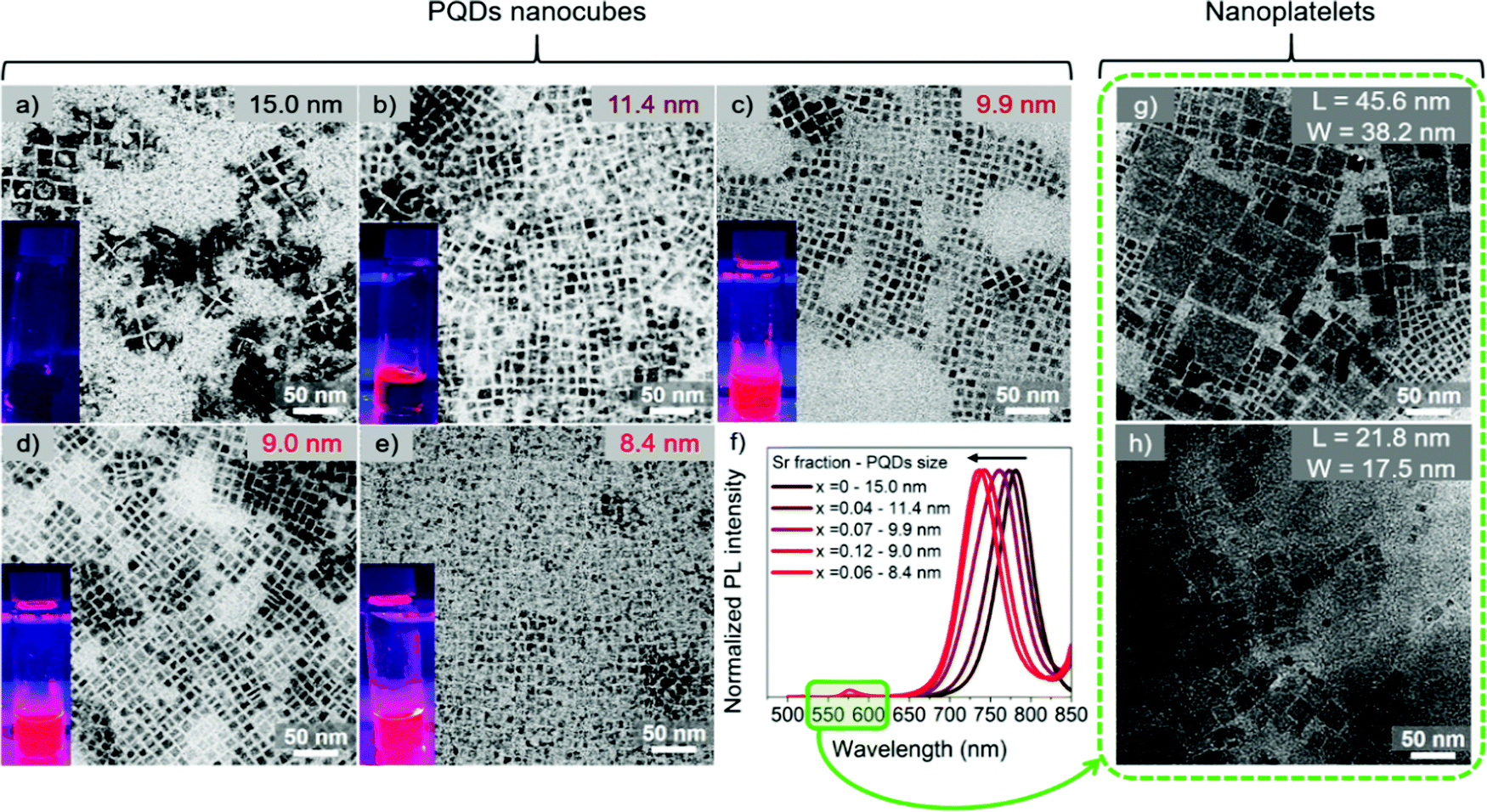 | ||
| Fig. 1 TEM images of (a) FAPbI3 and FAPb1−xSrxI3 perovskite quantum dot (PQDs) colloidal solutions with (b) 4 at%, Sr, (c) 7 at% Sr, (d) 12 at% Sr, and (e) 6 at% Sr. (f) Photoluminescence (PL) spectra of the FAPb1−xSrxI3 PQD colloidal solutions by varying the Sr fraction-particle size. TEM images of the (g) FAPb0.88Sr0.12I3 and (h) FAPb0.94Sr0.06I3 nanoplatelets (NPLs) from the redispersed precipitate by varying their corresponding lateral dimensions (L = length, W = width). | ||
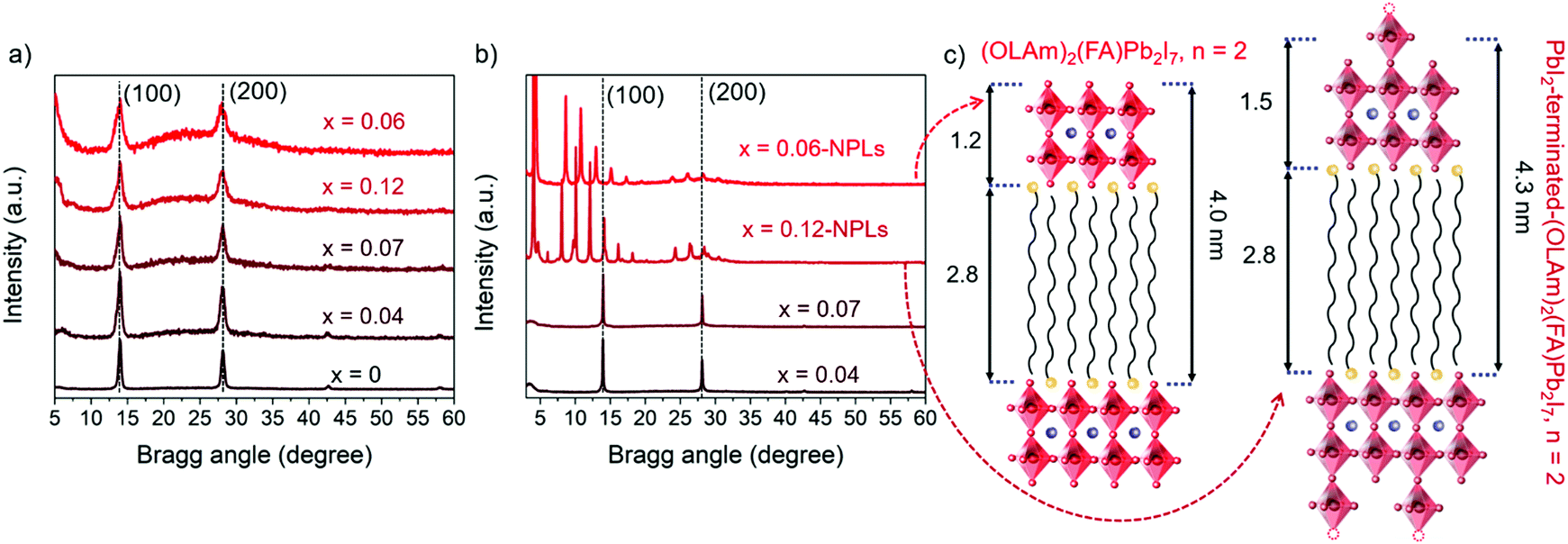 | ||
| Fig. 2 XRD patterns of the (a) FAPb1−xSrxI3 PQDs colloidal solutions, and (b) their corresponding redispersed precipitates. (c) Schematic representations of the layered perovskite spacings and relevant distances for FAPb0.88Sr0.12I3 and FAPb0.94Sr0.06I3 NPLs. | ||
To give a clear understanding about the role of iodide anions on the PQDs growth and stabilization, we consider some characteristics from the surface chemistry of PQDs. First, the high surface-to-volume ratio of small PQDs generates a substantial density of halide vacancies on their surface (undercoordinated Pb).40 These structural defects are quickly passivated by the available halide ions from the mixture reaction. Simultaneously, oleylammonium (OLAm) cations bounded to halides can substitute and fill some A-cation positions into the cuboctahedral place of perovskite,41 compensating the negative charge and decreasing the surface energy.40 The replacement of a high fraction of A-cation sites by the long alkylammonium chain is known as instant capping, and restricts the PQDs dimensions.41 This phenomenon justifies the formation of stable FAPb1−xSrxI3 PQDs with smaller size since the OLA/Pb molar ratio increases as the Pb content decreases (Pb deficiency) by Sr addition (see Table 2). According to this depiction, we suggest that the synthesis of stabilized PQDs requires the use of both SrI2 and PbI2 to keep a high iodide fraction in the mixture reaction to reduce defect formation, even though the Pb deficiency is not fully compensated by Sr incorporation, as we analyze below. To verify this hypothesis, we attempted to grow FAPb0.93Sr0.07I3 PQDs in the absence of the nominal amount of SrI2 added in the mixture reaction, keeping the amount of PbI2 unchanged (Fig. S3, ESI†). As expected, we were not able to synthesize the desired material. Therefore, we assume that the amount of iodide anions in the mixture reaction is constant, and the I/Pb molar ratio increases with the amount of nominal Sr (see Table 2).
| Nominal composition | Calculated composition/doping, EDS | Amount of added PbI2a|SrI2 (Pb2+|Sr2+) in the synthesis (mmol) | OLAb/Pb molar ratio | Ic/Pb molar ratio |
|---|---|---|---|---|
| a Amount of PbI2 (Pb2+) in the synthesis. b Amount of OLA into the synthesis (mmol): 9.12. c Amount of I− into the synthesis (mmol): 1.48. | ||||
| FAPbI3 | FAPbI3 | 0.74 | 0 | 12.32 | 2 |
| x = 0.3 | x = 0.04 | 0.52 | 0.22 | 17.53 | 2.84 |
| x = 0.5 | x = 0.07 | 0.37 | 0.37 | 24.64 | 4 |
| x = 0.6 | x = 0.12 | 0.30 | 0.44 | 30.40 | 4.93 |
| x = 0.7 | x = 0.06 | 0.22 | 0.52 | 41.45 | 6.73 |
The change in size is correlated with a change in the optical properties of FAPb1−xSrxI3 PQDs colloidal solutions. Sr-doping blueshifts the photoluminescence (PL) emission of pure FAPbI3 progressively, as shown in Fig. 1f and the inset pictures in Fig. 1a–e. This behavior is also reflected in the absorbance spectra of FAPb1−xSrxI3, as shown in Fig. S1f (ESI†). The corresponding optical features of the PQDs are summarized in Table S1 (ESI†). It should be noted that Sr-doping induced a smaller particle size than the Bohr diameter of FAPbI3 (∼ 12.7 nm).42 Consequently, a blueshift is expected due to the quantum confinement effect.23,35 Moreover, two more emission peaks/absorption edges were observed at lower wavelengths at around 511–524 nm and 567–577 nm, respectively, as shown in Fig. S4 (ESI†), when the Sr-doping level surpassed 7 at%. The existence of new PL emission signals suggested the co-existence of diverse structures in the dispersion. To give a suitable explanation for this observation, we first examined the appearance of both PQD colloidal solutions and precipitate materials, which was simultaneously obtained for each FAPb1−xSrxI3 material after the synthesis. It was noted that a precipitate appeared after the PQD synthesis. Precipitates from the FAPb0.96Sr0.04I3 and FAPb0.93Sr0.07I3 syntheses exhibited the same dark coloration as the precipitate from the FAPbI3 synthesis, while precipitates from FAPb0.88Sr0.12I3 and FAPb0.94Sr0.06I3 were more reddish than their corresponding colloidal solutions. Precipitates were separated from the colloidal solutions and redispersed in hexane, as shown in Fig. S5a (ESI†), for further analysis. By conducting TEM images on the FAPb0.88Sr0.12I3 and FAPb0.94Sr0.06I3 precipitates, we were able to find evidence of a mixture of nanoparticle morphology of both nanoplatelets (NPLs) and PQDs nanocubes, as shown in Fig. 1g and h. The predominance of the 2D structure over the 3D one is clear. The dimensions of the FAPb0.88Sr0.12I3 (FAPb0.94Sr0.06I3) NPLs are 45.6 (21.8) nm length, and 38.2 (17.5) nm width, respectively. The distribution of the dimensions for these two materials are displayed in Fig. S5b, c and S5b′, c′ (ESI†), respectively. Additionally, the UV-Vis and PL features of the bare FAPb0.88Sr0.12I3 (FAPb0.94Sr0.06I3) precipitates (Fig. S5d and e, ESI†) exhibit absorption edges/emission peaks centered at 589 (590) nm and 583 (586) and at 530 nm, respectively, pointing out that the origin of the extra peaks in the PQDs colloidal solution comes from the NPLs. This evidence is in accordance with the layered-2D Ruddlesden–Popper structures, showing formula (LA)2(A)n−1PbnI3n+1, n = 1 and n = 2, where A = FA+ and LA is referred to a long alkylammonium chain, and n is the number of the layers of octahedra in the perovskite-like stack, as reported in the literature43–46 For our case, OLAm is the capping ligand occupying the LA positions in the 2D perovskites. At this stage, we deduced that the incorporation of 7 at% Sr (nominal precursor ratio of 0.5, see Table 1) is the maximum Sr-doping that promotes the synthesis of just PQDs without altering their shape and optical properties. By surpassing this nominal Sr precursor ratio of 0.5, the PQDs synthesis is altered, causing the emergence of 2D NPLs.
As shown in Fig. 2a, the FAPbI3 and FAPb1−xSrxI3 PQDs extracted from the colloidal solutions present two main peaks at 14° and 28°, respectively. These signals are associated with the (100) and (200) planes of a typical perovskite lattice FAPbI3 crystallizing in the simple cubic black phase (PDF 54-0752).14,37 Moreover, after Sr-doping, the XRD peaks do not exhibit any shift,34,47,48 which is supportive of the non-detected lattice distortion.34 However, the FAPb1−xSrxI3 PQDs XRD peaks suffer a clear broadening, which can be associated with the PQD size decrease, although an inhomogeneous strain or microstrain cannot be ruled out.34 The low Sr fraction incorporated into the FAPbI3 PQDs structure preserved the crystal structure until a doping level of 7 at%. Sr was achieved. Phung et al.34 revealed that the Sr-MAPbI3 bulk films can incorporate up to 0.2 at% Sr, promoting Sr2+ segregation to the perovskite surface at a higher content. We believe that the presence of a higher Sr fraction into the PQDs than the estimated one in bulk films can be attributed to the facile dissolution of the Sr-precursor by the presence of capping ligands, releasing more Sr2+ cations during the synthesis to occupy Pb2+ positions. Thus, the emergence of the 2D structures is related to the different processability of Sr- and Pb-precursors to define the PQDs growth. The cubic phase is preserved in the PQDs by introducing Sr up to 7 at%, as shown in Fig. 2b, while well-defined peaks at low angles appear in the XRD profiles of the redispersed precipitates, for a nominal Sr precursor ratio higher than 0.5, confirming the NPL crystalline nature observed by TEM.43,49
To determinate the number of layers contained in the NPLs, the n value from the Ruddlesden–Popper structures was calculated. The XRD peaks of each sample follow periodic small angle intervals, associated with nanoplatelet stacking.43,50 The diffraction peaks of FAPb0.88Sr0.12I3 and FAPb0.94Sr0.06I3 NPLs display a regular 2θ increment of around 2.04° and 2.25°, respectively, as shown in Fig. S6 (ESI†). By using the Bragg equation, the periodic spacing (d) forming these periodicities is estimated to be around 4.3 nm and 4.0 nm, respectively. As shown in Fig. 2c, these values are associated with the distance between the [PbX6]4− octahedra layers, separated by OLAm cations.43,50 Consequently, the thickness of the inorganic framework, excluding the long organic chains of the OLAm, of the FAPb0.88Sr0.12I3 and FAPb0.94Sr0.06I3 NPLs is calculated as 1.5 nm and 1.2 nm thick, respectively. The FAPb0.94Sr0.06I3 NPLs structure is the same as that for the Ruddlesden–Popper (OLAm)2(FA)Pb2I7 perovskite (n = 2),49,51 even if we do not discard the mixture of 2D structures with different “n”, since the optical features of the colloidal solutions and precipitates at least suggest the formation of two layered structures (n = 1, n = 2). Regarding the FAPb0.88Sr0.12I3 NPLs, the disparity in thickness compared with the benchmark value for a double metal halide octahedra layer (1.2 nm),51 matches well with the presence of undercoordinated Pb positions in the form of distorted [PbI5]3− units on the NPLs surface, as shown in Fig. 2c. In this context, the FAPb0.88Sr0.12I3 NPLs present a PbI2-terminated-(OLAm)2(FA)Pb2I7 structure (n = 2). Thus, the co-existence of 2D/3D structures is the result of the poor compensation of Pb positions by Sr into PQDs, associated with structural defects, like undercoordinated Pb.
The increase of the nominal Sr precursor fraction significantly impacts the chemical environment of FAPb1−xSrxI3, as it has been analyzed by X-ray photoelectron spectroscopy (XPS) (Fig. S7a and b, ESI†). It has identified the existence of C, N, O, Pb and I in PQDs and NPLs, while the presence of Sr is detected only in the PQDs. The corresponding chemical composition of PQDs and NPLs are summarized in Tables S2 and S3 (ESI†), respectively. In the high resolution (HR) XPS Sr 3p3/2 spectra of FAPb1−xSrxI3 PQDs, as shown in Fig. S7c and d (ESI†), a weak and noisy signal was attained. This response is ascribed to the presence of substitutional Sr2+ in the perovskite structure,34,52 replacing Pb2+. The low signal-to-noise ratio, the difference in intensity, and the absence of the main signal of substitutional Sr2+ in the Sr 3p3/2 spectra of some samples points to an inhomogeneity of Sr incorporation into PQDs.52 On the other hand, Fig. 3a and b show the HR-XPS Pb 4f spectra obtained for PQDs and NPLs, where the main Pb4f7/2 and Pb4f5/2 peaks are decomposed into two doublets. The first one is located at 138/143 eV, associated with the presence of Pb2+ from the perovskite lattice, while the second doublet at 136/141 eV corresponds to metallic lead, Pb0. The existence of Pb0 is closely related with the presence of iodide vacancies in the materials.14 According to the low complexation affinity between Pb2+ and I−, and the dynamic nature of the capping ligands on the PQDs surface, iodide loss can be facilitated,53 leaving undercoordinated Pb or releasing Pb0 to the surface. This fact provides a more defective material.14 Therefore, in a Pb-deficient iodide perovskite, halide defects are formed with the concomitant generation of Pb2+ vacancies.34 The absence of one neighboring Pb2+ causes the formation of two I− vacancies in order to keep the electroneutrality in the perovskite lattice, creating Schottky defects.34,54 Thus, it is expected that more Schottky defects (higher Pb0 fraction) are formed by intensifying the Pb deficiency, as the amount of Pb precursor is decreased in the PQD synthesis. However, our findings show that incorporated Sr (Fig. 3c) can decrease the Pb2+ vacancies, and consequently, the defect density in PQDs under certain circumstances.
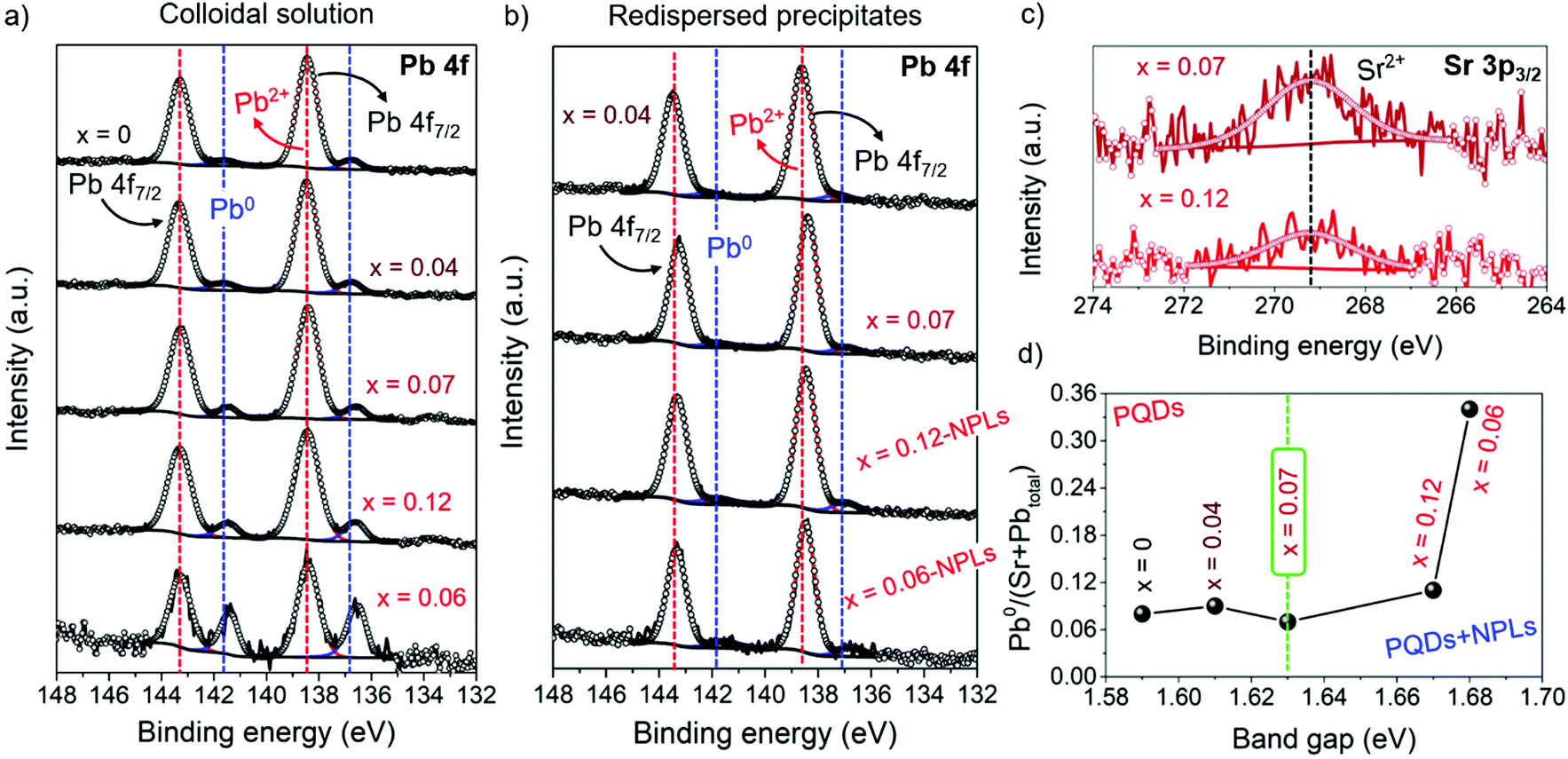 | ||
| Fig. 3 High-resolution (HR) XPS spectra of (a and b) Pb 4f, (c) XPS Sr 3p3/2 and (d) calculated metallic Pb-to-(Sr + Pbtotal) ratio for (a, c and d) FAPb1−xSrxI3 PQDs colloidal solutions, and (b) their corresponding redispersed precipitates. | ||
A good parameter to evaluate the fraction of defects is the metallic lead-to-total metal ratio denoted as Pb0/(Sr + Pbtotal) (see Table S2, ESI† and Fig. 3d). This parameter has been calculated in order to analyze the Sr-doping-induced variation of the Pb-/iodide-vacancies fraction contained in the FAPb1−xSrxI3 PQDs. As seen in Fig. 3d, the lowest Pb0/(Sr + Pbtotal) ratio is obtained for FAPb0.93Sr0.07I3 PQDs (see Table S2, ESI†). In this case, the incorporation of Sr2+ maximizes the reduction of Pb0, and consequently reduces the formation of Pb2+ vacancies and Schottky defects.
Regarding iodine, its corresponding HR-XPS spectra exhibit the I 3d doublet centered at 619/631 eV, as shown in Fig. S8a and b (ESI†). This doublet is attributed to the presence of iodide anions from the [PbI6] octahedra that make up the PQDs and NPLs structure.14 Even though the amount of Pb decreases due to the Sr incorporation, as shown in Table 2, the addition of stoichiometric amounts of PbI2 and SrI2 to the reaction mixture allows us to assume that the iodide fraction is kept constant. In this context, a lower amount of iodide anions from the total available amount will react with a lesser amount of Pb to synthesize PQDs, increasing the amount of free iodide anions. This is the source of iodide needed to support the Schottky defect reduction, facilitating the Sr2+-doping. This fact can also explain the rise of the I/(Pb + Sr) ratio determined in the FAPb1−xSrxI3 colloidal solutions through EDS (Table 1), and the gradual increase of the I/Pb2+ atomic ratio in the surface chemical atomic composition of these materials, as shown in Table S2 (ESI†), to reach the maximum point for the FAPb0.93Sr0.07I3 PQDs. However, when the Pb deficiency is intensified after adding a large amount of Sr precursor to the mixture reaction, there is not enough available iodide species (as it has been employed in the formation of NPLs, see below) to restrain the formation of Schottky defects (see Table S2 (ESI†), Fig. 3d). Consequently, a lower Sr fraction is incorporated to replace the Pb2+ positions, as we can observe in Table 1.
The existence of Schottky defects induced by the distribution of free iodide anions can be also corroborated through the rise of Pb0 density estimated for the FAPb1−xSrxI3 redispersed samples with x = 0.12 and x = 0.06 (Table S3, ESI†). Without a doubt, the distribution of building blocks to obtain both PQDs and NPLs does not satisfy their own chemical composition, contributing to the growth of high-defective perovskites. The Sr-doping is also appreciated in the shift of the Pb0 doublets, as shown in Fig. 3a, from the FAPb1−xSrxI3 PQDs to the lower binding energies (BEs), as a result of the lower electronegativity of Sr compared with Pb.34 On the other hand, by attempting to introduce a higher Sr fraction to reduce the Pb fraction, the Pb0/(Sr + Pbtotal) markedly increases. Hence, we can claim that the incorporation of 7 at% Sr into the FAPb1−xSrxI3 PQDs generates a suitable compensation of Pb deficiency, preserving the PQDs structure and producing less-defective materials.
These results also show the problem of using Sr to substitute Pb in higher ratios, despite their similarities, as the Sr incorporation in PQDs is significantly below that of the precursor ratio. According to these features, Sr-doping is preferred instead of the total Pb replacement. Differences in the chemistry of the two metals justify the difficulty of Sr incorporation, as shown in the sketch in Fig. 4a. First of all, PbI2 is known to crystallize in a rhombohedral structure,36 where the distorted PbI6 octahedra are face-sharing one another into layers connected by weak van der Waals forces. In the presence of OA and OLA in stoichiometric amounts, two simultaneous reactions happen: PbI2 rapidly dissolves with OA to produce hydroiodic acid (HI); then, this strong acid reacts with OLA, yielding the OLAm-I species in the mixture reaction (reactions 1 and 2):55,56
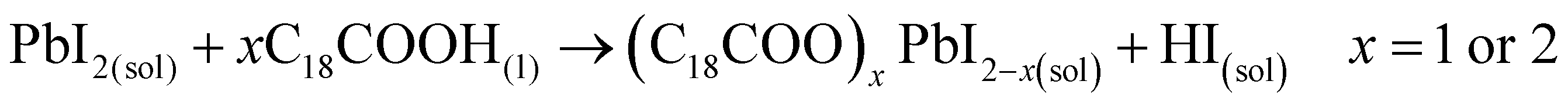 | (1) |
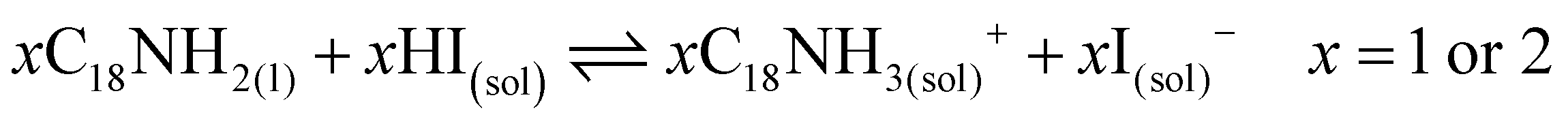 | (2) |
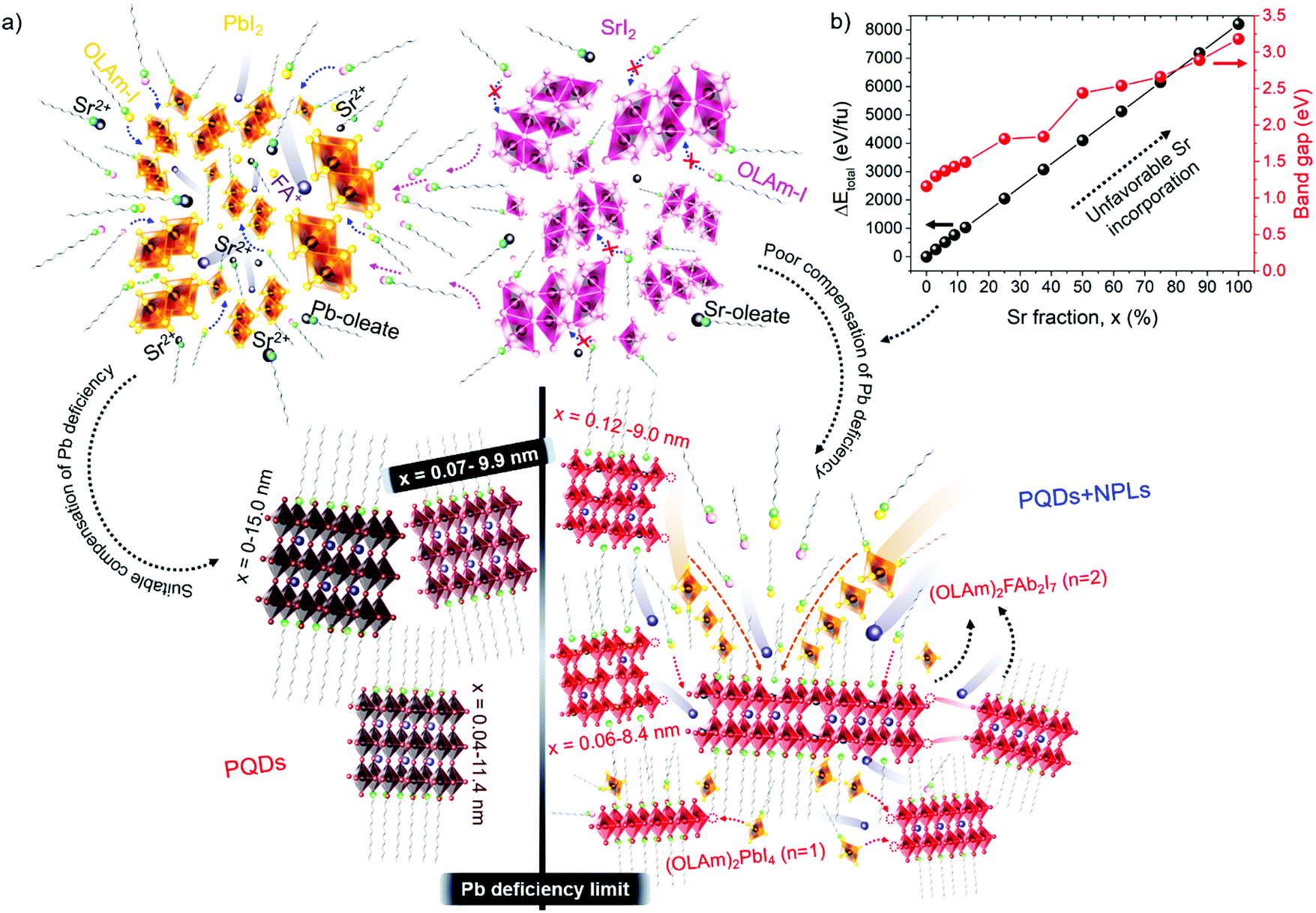 | ||
| Fig. 4 (a) Schematic representation of the emergence of PQDs and NPLs caused by the Pb deficiency. A suitable Sr fraction compensating Pb positions provides Sr-modified nanocubes with a low density of structural defects. The poor compensation of Pb deficiency by Sr produces a mixture of defective 2D/3D structures. (b) DFT total energy of bulk cubic FAPb1−xSrxI3 as a function of the Sr fraction and corresponding band gap, where “fu” in the y-axis refers to formula unit. | ||
A suitable amount of OLAm-I is now available to be intercalated into the lead octahedra frameworks, transforming from a face-sharing to a corner-sharing structure. Then, the coordinated PbI3− structures are achieved (reaction 3):55
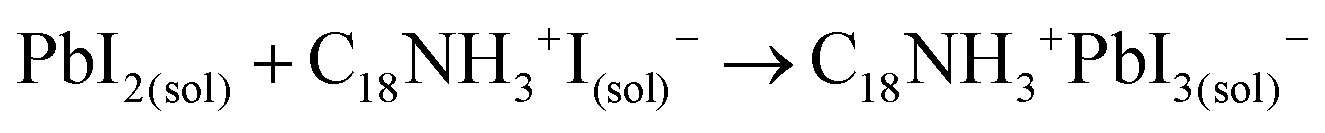 | (3) |
The coordinated PbI3− react with FA+ cations during hot-injection to provide the FAPbI3 structure.
Lastly, the combination of OA and OLA into the mixture reaction produces oleylammonium oleate (reaction 4).57,58 This species is ionically coordinated to the PQDs surface, providing the material stabilization.
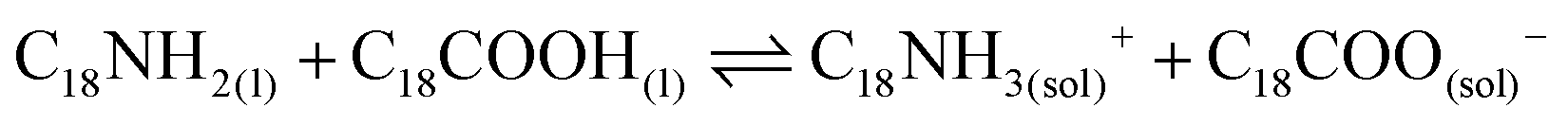 | (4) |
Unlike the Pb precursor, SrI2 shows an orthorhombic crystal lattice, where Sr is coordinated to 7 iodine atoms, and prefers the formation of a [SrI7]5− pentagonal bipyramidal structure.36 This system does not present a face-sharing structure as PbI2, but each bipyramidal unit is interconnected to each other. Based on the low Sr fraction found in the FAPb1−xSrxI3 PQDs, we propose that the availability of Sr2+ cations to dope the PQDs is promoted by the dissolution of some bipyramidal units of SrI2 in the presence OLA and OA, similar to PbI2 (reactions 5 and 6):
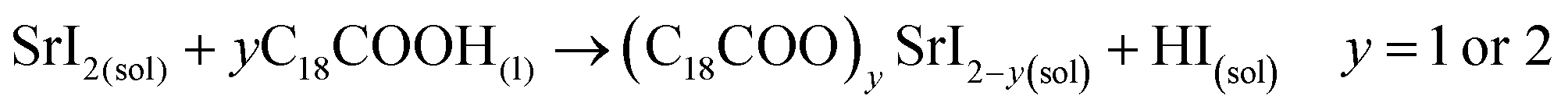 | (5) |
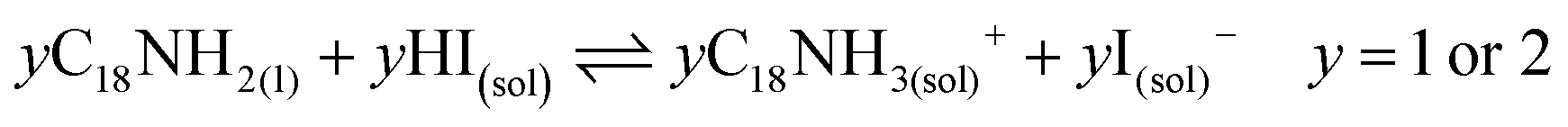 | (6) |
Nevertheless, the poor compensation of the Pb deficiency by Sr incorporation allows us to deduce that the OLAm-I product formed in reaction 6 is not able to modify the interconnected [SrI7]5− bipyramidal units, unlike PbI2, restraining the reaction with FA+ cations. This fact can explain why it is necessary to add a high Sr-precursor content in the mixture reaction to reach the Sr2+-doping level, as we detected by EDS. On the other hand, the non-incorporated Sr2+ can also react with oleate anions to produce Sr-oleate in the mixture reaction. However, attending to reactions 1, 2, 5 and 6, Sr-oleate can be formed without altering the acid–base balance and the subsequent formation of the OLAm+ cations.
To understand the difficulty of the Sr incorporation, we have calculated by density functional theory (DFT) methods the total energy of cubic FAPb1−xSrxI3 in the bulk (0 ≤ x ≤ 100%), as shown in Fig. 4b. It is apparent that small fractions of Pb up to 7% can be substituted by Sr atoms in the lattice without significantly altering the cubic symmetry of FAPbI3. However, the total energy continuously increases with a higher Sr fraction, confirming the eventual destabilization of the cubic geometry.35 The rise of the total energy was accompanied by a band gap broadening in agreement with the experimental trend, as shown in Table S1 (ESI†). Moreover, we have calculated the formation energies of the bulk compounds FAPbI3 and FASrI3 in the absence of defects. They are −0.072 Ry (−94.52 kJ mol−1) and −0.069 Ry (−90.58 kJ mol−1), respectively, and more details can be found in the ESI.† The smaller stability of the pure FASrI3 perovskite is consistent with the proven difficulty to convert one compound into the other by Sr incorporation. These results prove that a greater reorganization of the SrI2 structure is required to synthesize FAPb1−xSrxI3 PQDs with stoichiometric fractions of SrI2 and PbI2, which is not possible at least under our synthetic conditions. On the other hand, DFT also predicts that the incorporation of Sr produces a widening of the bandgap, as shown in Fig. 4b. This fact allows us to explain the observed blueshift in the PL emission, shown in Fig. 1f, despite the increase of the quantum confinement regime, shown in Fig. 1a–e.
Evidently, if the Pb deficiency is poorly compensated by Sr during synthesis, the Sr-doping-induced modification in the stoichiometry of the precursors determines the growth of different perovskite structures from 3D. In this scenario, the OLA/Pb molar ratio is increased, while the mixture reaction shows an iodide-rich environment (higher I/Pb molar ratio). Under this consideration, the formation of OLAm-I is accelerated to produce highly coordinated iodoplumbates species, schematized in reaction 7:55
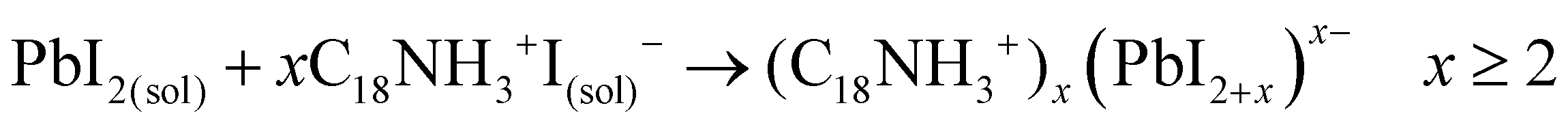 | (7) |
The number of [PbI4]2− species counterbalanced with OLAm cations are the basic building blocks for establishing the structure of the layered 2D perovskites with formula [(RNH3)2(PbI4)]n.55 On the other hand, the thickness of a 2D perovskite depends on the competition between the A-site cations, trying to expand the perovskite growth from the planar layer, and the alkylammonium cations attempting to compress the growth into the planar layer.51 In our context, the small thickness of the obtained NPLs implies that a lower density of FA+ cannot balance the effect of OLAm. In other words, a high density of the OLAm species would occupy the FA+ positions, confining its thickness to two [PbI6]4- layers (∼1.2 nm). This argument can explain the concurrent formation of PQDs and NPLs in our samples, as a direct reflection that the fraction of FA+ cations is distributed to stabilize both 2D and 3D structures. At the same time, the excess of OLAm-I needed to promote the NPLs growth according to the reaction 7 (higher coverage of OLAm/surface area, as shown in Table S4 (ESI†), can also mediate the growth of PQDs (reaction 3). This fact indicates that not only iodide vacancies are formed due to the free iodide distribution in both kinds of structures, generating consequently Schottky defects. Hence, if the free iodide fraction from the mixture reaction is consumed by the NPLs formation as a consequence of a marked Pb deficiency, it is expected that a lower Sr fraction can dope PQDs, reducing the likelihood to suppress Schottky defects. This would explain why, upon adding more Sr from 60 to 70 at% nominal Sr content, there is a decrease in the found Sr into PQDs from 12 to 6 at%, respectively. However, the co-existence of PQDs with NPLs indicates that there is enough OLM to stabilize both morphologies (higher OLA/Pb), facilitating the instant capping of PQDs, which is reflected in the smaller particle size observed by TEM. At this stage, we concluded that the poor compensation of the Pb deficiency by Sr substitution lies in the difficulty to modify the crystal structure of the Sr precursor compared with its lead analogue during PQDs synthesis. This fact is the critical point to trigger the alteration in the stoichiometry of the precursors to favor the growth of defective PQDs and NPLs.
Moreover, it has been established that the halide vacancies are the Achilles’ heel of perovskite stability, providing a facile way for the oxygen incorporation and, in turn, causing its self-degradation.14 After introducing a controlled amount of Sr into the FAPbI3 PQDs, we significantly reduce the structural defects fraction. Thus, we could expect a Sr-doping-induced improvement in their long-term stability, and also of the optical properties in comparison with FAPbI3 PQDs.
From this perspective, we studied the photoluminescence quantum yield (PLQY) for all of the FAPb1−xSrxI3 PQDs. It is worth noting that 100% PLQY corresponding to a Sr fraction of 7 at% was obtained, shown in Fig. 5a, associating this outstanding optical behavior to the maximum suppression of halide defects, shown in Fig. 3d. Here, the non-radiative carrier recombination channel is suppressed. However, when the Sr fraction is raised, the PLQY value rapidly decreased, as shown in Fig. 5a, indicating the appearance of non-radiative recombination channels. To get a closer look at the recombination kinetics of the PQDs samples, we carried out time-resolved PL measurements, as shown in Fig. S9 (ESI†), collecting their corresponding average electron lifetimes, τavg, by fitting the PL decays through a bi-exponential equation,y = y0 + A1e−x/τ1 + A2e−x/τ2. As seen in Fig. 5a, the higher the band gap of FAPb1−xSrxI3 PQDs, the lower the τavg (see the discussion below). An Excel file with the raw data of the PLQY measurements is included as ESI.†
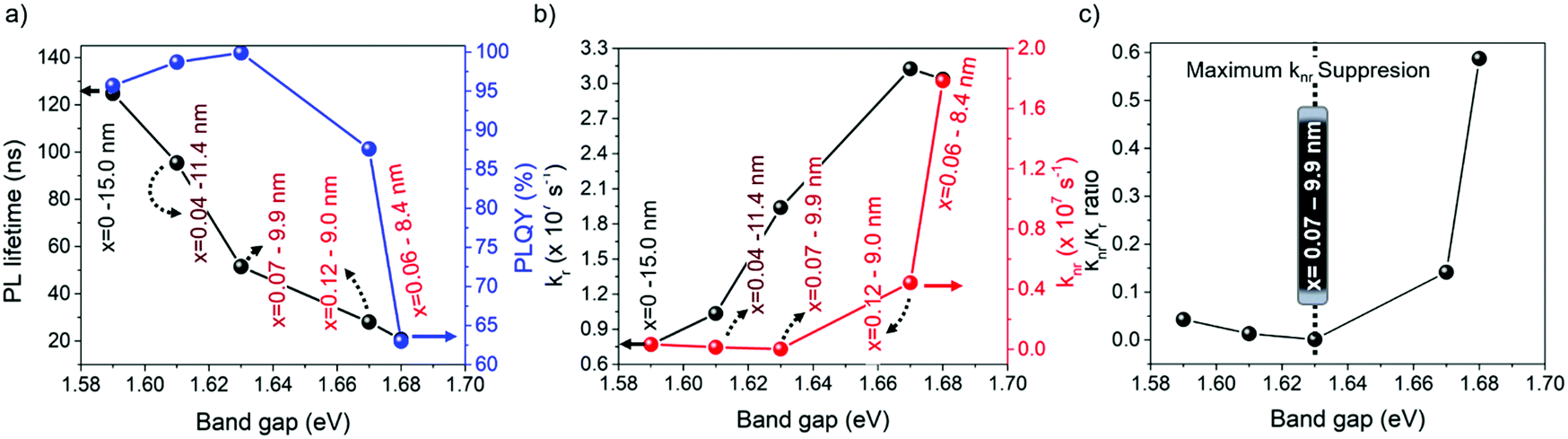 | ||
| Fig. 5 (a) Behavior of the PL lifetime and PLQY, (b) radiative (kr) and non-radiative recombination (knr) decay rate constants, and (c) their corresponding knr/kr ratio for the FAPb1−xSrxI3 PQDs colloidal solutions by varying the Sr fraction. | ||
Taking into account the PLQY, it is possible to determine the radiative (kr) and non-radiative recombination (knr) decay rate constants (Table S5, ESI†).59 Interestingly, knr, shown in Fig. 5b, shows a similar behavior to that of the Pb0/(Sr + Pbtotal) ratio estimated by XPS, shown in Fig. 3d, confirming that the suppression of iodide vacancies (Schottky defects) by adding Sr up to 7 at% into the FAPb1−xSrxI3 PQDs quenches the non-radiative channel. At this stage, we expected to obtain a single-exponential behavior to describe the longer PL decays. Nevertheless, a bi-exponential behavior depicts the PL dynamics better. The PL decay profile of 100% PLQYs does not exhibit single-exponential decay behavior, a fact that could be explained by the trapping detrapping-processes with shallow defects in the perovskite conduction band, as it has been previously reported.60 Accordingly, we suggest that Sr modification also plays a main role in the recombination kinetics of PQDs. Yao et al. reported that Sr2+ can provide radiative energy levels near or into the conduction band (CB) states from CsPbI3 QDs. Then, these states induce a radiative recombination, which is the key point to increasing the PLQY.35
According to this fact, Sr not only mitigates the formation of carrier traps provided by the existence of iodide vacancies, but also introduces radiative recombination sites, which can explain the formation of high-quality FAPb1−xSrxI3 PQDs with 100% PLQY. Nevertheless, the reduction of the PL lifetime is ascribed to the growth of smaller PQDs, a consequence of the poor compensation of Pb deficiency (instant capping). In this context, a stronger quantum confinement is achieved, increasing the exciton binding energy and making the carrier recombination faster. This trend has been also reported for PQDs with near-unity PLQY, where the metal-doping is chosen as an alternative to improve the photophysical properties of the perovskites.23,35 On the other hand, the rapid raise of knr, as shown in Fig. 5b, at higher Sr fraction causes an increase of the knr/kr rate, as shown in Fig. 5c. In this case, the difficult incorporation of Sr into the FAPbI3 PQDs causes Sr to not completely compensate the higher Pb deficiency, and the use of free iodide anions from the mixture reaction in the formation of NPLs avoids the suppression of Schottky defects. According to the fact that these defects act like non-recombination sites for carriers, the PLQY of the PQDs is reduced.
The PLQY of the synthesized samples was followed for 30 days in the dark under ambient conditions, as shown in Fig. 6a, and exposed to a high relative ambient humidity between 40–50%. The PLQY values of the PQDs obtained during measurements as a function of time are summarized in Table S6 (ESI†). As shown in Fig. 6b, the PLQY of the FAPb0.93Sr0.07I3 PQDs continuously decreases after being practically null after 15 days. However, for samples containing Sr, the PLQY stabilizes after 3–6 days, preserving a value as high as 78% FAPb0.93Sr0.07I3 PQDs after 30 days. The stability of the different PQDs under continuous UV illumination has also been analyzed, as shown in Fig. 6c. The PLQY of samples aged for 15 days at room conditions was tracked during 12 h under continuous UV illumination. We observed that the relative PL intensity increased for all Sr-modified PQDs, associated with a phenomenon known as photobrightening.2 Although the reasons to describe this effect are still under debate, the passivation of halide defects by the presence of oxygen/superoxide anions has been considered as the most accepted explanation.61 In this context, we suggest that the suppression of a high density of Schottky defects by the Sr inclusion facilitates the passivation of residual electron traps by oxygen species. Interestingly, FAPb0.93Sr0.07I3 PQDs aged for 15 days recovered the 100% PLQY after UV illumination. Nonetheless, an excess of adsorbed oxygen also instigates the self-degradation of the perovskites, origin of the immediate PL quenching of FAPbI3, and the hampered photobrightening of the FAPb0.94Sr0.06I3 PQDs.
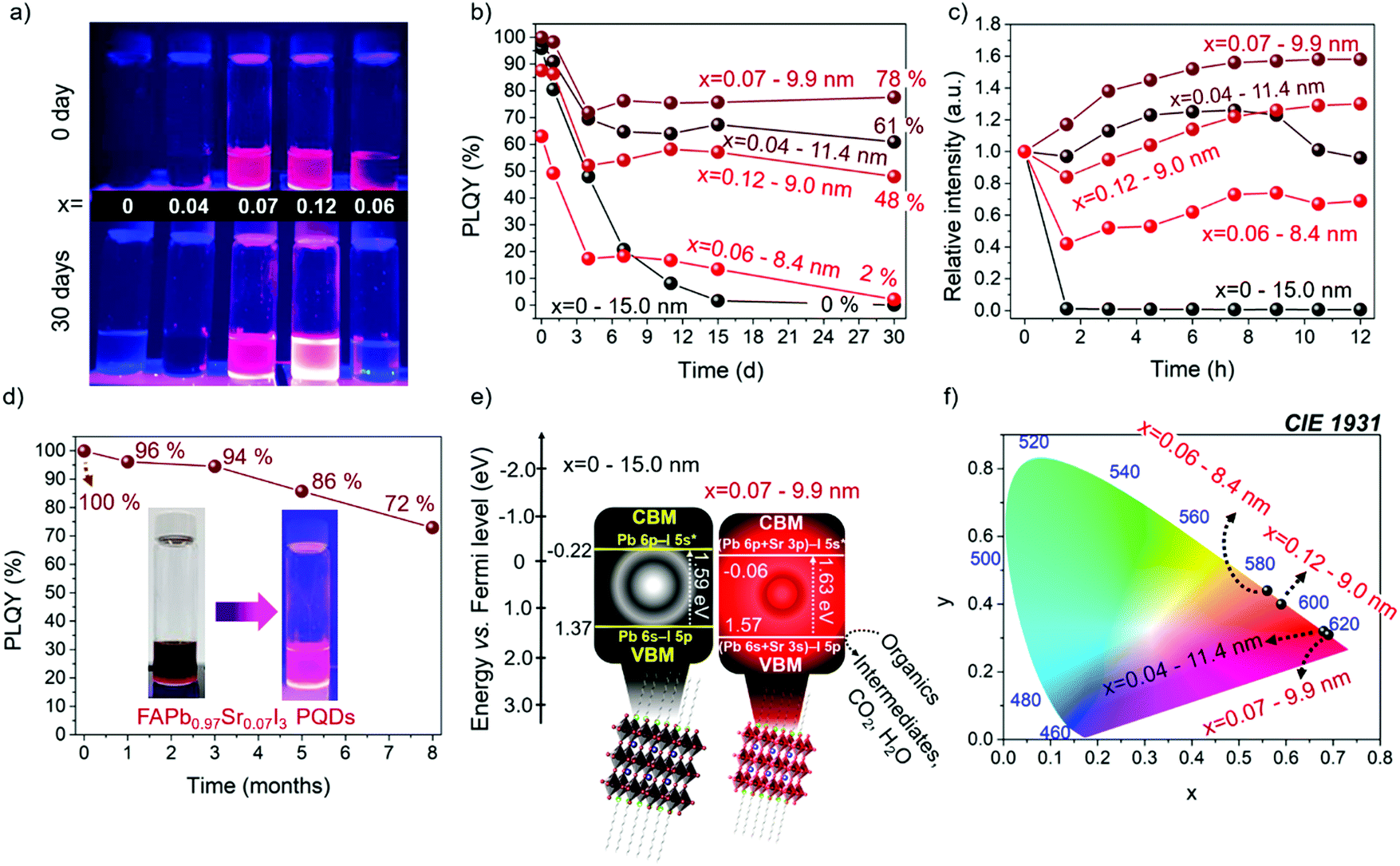 | ||
| Fig. 6 (a) Photographs of fresh and aged (after 30 days) FAPb1−xSrxI3 PQDs colloidal solutions under UV-light irradiation. (b) PLQY values as a function of aged days for PQDs colloidal solutions. (c) Photostability of 15 days-aged PQDs exposed to UV light irradiation for 15 h. (d) PLQY stability of FAPb0.93Sr0.07I3 PQDs colloidal solutions for 8 months, with T80 = 6.5 months. (e) Band structure of FAPbI3 and FAPb0.93Sr0.07I3 PQDs obtained from XPS VB spectra through the extrapolation method. (f) CIE chromaticity of Sr-substituted PQDs. | ||
It is worth highlighting that FAPb0.93Sr0.07I3 sample stored in the fridge (under dark conditions and 4 °C) is stable for at least 8 months preserving a significant PLQY of 72%, as shown in Fig. 6d and Table S7 (ESI†), with T80 = 6.5 months, where T80 is the time for which the initial PLQY is reduced by 20%. Accordingly, the red-emitting FAPb1−xSrxI3 PQDs synthetized in this work shows higher PLQY and stability than other modified FA-based iodide perovskite PQDs, such as FAPb1−xSnxI3, (PLQY = 63% for 2 months),25 established red-emitting Zn- (PLQY = 98.5% for 2.3 months)23 and Sr-doped CsPbI3 PQDs (PLQY = 95% for 2 months),35 and competitive optical properties comparable with the longest-term stable FAPbI3 QDs reported until now: Ding et al.38 (PLQY = 100% for 4 months), Protesescu et al.45 (PLQY = 70% for 6 months).
Finally, to give clear insight into the potential application of the FAPb1−xSrxI3 PQDs, we estimated the band structure of the materials by XPS valence band spectrum in the presence and absence of Sr2+ energy levels. The valence band energy (EVB) of the FAPb0.93Sr0.07I3 PQDs is calculated by extrapolation method, as shown in Fig. S10 (ESI†), and then the associated CB energy (ECB) is obtained through the equation EVB = ECB + Eg. By comparing with un-substituted FAPbI3, the VB position of the Sr-substituted PQDs is displaced to more positive energy values (vs. Fermi level) (Fig. 6e). This Sr-doping-induced modification potentially boost the oxidizing power to FAPb0.93Sr0.07I3 PQDs to carry out, for instance, the degradation of organics,14 whether some reaction conditions (electrolyte, irradiation source/time and reaction medium) are well established. On the other hand, the downward shift of the relative position of CB of PQDs with the incorporation of Sr was also shifted to more positive values. This indicated that the presence of Sr2+ can suppress some electron traps near the CB, and favors the radiative recombination pathway, as shown in Fig. 5b. Moreover the CIE chromaticity coordinates (x,y) are calculated from the PL spectra of the FAPb1−xSrxI3 PQDs samples, as shown in Table S8 (ESI†). Interestingly, the chromaticity values of the FAPb0.93Sr0.07I3 PQDs are close to the Rec. 2020 standards for the red color (0.708, 0.292)62 (Fig. 6f). This fact allows us to deduce that the Sr-doping is suitable in this synthetic protocol to obtain PQDs with a purer color. Additionally, features such as high PLQY and stability of the PQDs provide the potential to fabricate competitive red-emitting diodes.
3 Conclusions
In this work, we synthesized FAPb1−xSrxI3 PQDs by using the hot-injection method, with the purpose of enhancing their photophysical properties and extending the long-term stability of the PQDs. These facts that undoubtedly increase the interest of these systems for optoelectronic applications. We elucidated that the difficulty of Sr incorporation to replace Pb during the PQDs synthesis lies in the different crystalline structure of the Sr precursor compared with its Pb analogues, restraining the formation of building blocks for PQDs growth. Thus, metal doping in PQDs is carried out instead of total Pb substitution. In this context, just a relatively low amount of Sr (in the doping range of 4–12 at%) can be incorporated into the PQDs substituting Pb. In addition, the progressive substitution of Pb by Sr increases the total energy of the system, as we have calculated by DFT. This is an indication of the hindered Sr incorporation. We observe that for ratios of Sr/Pb precursors higher than 0.5, the synthesis produces not just quantum dots but also nanoplatelets. Interestingly, we observed that for ratios of Sr/Pb precursors lower than 0.5, both the Sr incorporation and the excess of I into the PQDs decrease the Pb- and I-vacancies to form Schottky defects. Additionally, the energy states provided by Sr-doping induce the radiative recombination into PQDs. Nevertheless, for ratios of Sr/Pb precursors higher than 0.5, the formation of nanoplatelets removes the excess I, and the density of Schottky defects increases. Thus, the Sr-doping-induced change in the optical properties and PQDs long-term stability have been unveiled. Under optimized conditions, FAPb0.93Sr0.07I3 PQDs synthesized with a Sr/Pb precursor ratio of 0.5 eliminates the non-radiative recombination pathways reaching a 100% PLQY. The decrease of defects also improves the long-term stability of FAPb1−xSrxI3 PQDs. The FAPb0.93Sr0.07I3 PQDs present a significant 72% PLQY after 8 months, with T80 = 6.5 months, one of the highest reported for halide perovskite quantum dots, owing to the fact that the PQDS were not stored under an inert atmosphere. FAPb0.93Sr0.07I3 PQDs also exhibit photobrightening under UV illumination for 12 h, recovering the 100% PLQY after aging for 15 days. All of these results point to the significant Sr-doping-induced reduction of defects in the synthesized FAPb0.93Sr0.07I3 PQDs. Consequently, the Sr-doping-induced outstanding photophysical properties makes the FAPb1−xSrxI3 PQDs excellent candidates to improve the performance of optoelectronic devices and to conduct lead solar driven processes more efficiently.Author contributions
A. F. G.-R. and I. M.-S. conceived the project. A. F. G.-R., S. M. and I. M.-S. designed the experiments. A. F. G.-R. synthesized the PQDs and performed the corresponding optical properties. S. A. and V. M.-S. conducted the TEM and EDS measurements of the materials. J. R.-P. and J. M. M. contributed to the XPS measurements and analysis. D. F. M. P. and C. E. A. carried out the DFT calculations for energy formation. A. F. G.-R. coordinated the experimental work. D. F. M. P. and C. E. A. coordinated the theoretical work. I. M.-S. coordinated the whole project. A. F. G.-R., S. M. and I. M.-S. wrote the manuscript. All of the authors contributed to the discussions.Conflicts of interest
The authors declare no competing interests.Acknowledgements
This work was supported by the European Research Council (ERC) via Consolidator Grant (724424–No-LIMIT) and the European Commission via FET Open Grant (862656 – DROP-IT), EU (FEDER) and MINECO under project TEC2017-85912-C2-2, and Generalitat Valenciana via Prometeo Grant Q-Devices (Prometeo/2018/098). We acknowledge the SCSIE from the University of Valencia for providing TEM facilities. CEMNAT project LM2018103 funded by the Ministry of Youth, Education and Sports of the Czech Republic is acknowledged for the financial support of the XPS measurements. We acknowledge SCIC from Jaume I University (UJI) for help with the XRD characterization. We also acknowledge to Juan I. Climente for his assistance in the theoretical work.References
- M. A. Green, E. D. Dunlop, J. Hohl-Ebinger, M. Yoshita, N. Kopidakis and A. W. Y. Ho-Baillie, Prog. Photovoltaics Res. Appl., 2020, 28, 3–15 CrossRef.
- Z. Andaji-Garmaroudi, M. Anaya, A. J. Pearson and S. D. Stranks, Adv. Energy Mater., 2020, 10, 1903109 CrossRef CAS.
- S. Masi, C. Echeverría-Arrondo, K. M. M. Salim, T. T. Ngo, P. F. Mendez, E. López-Fraguas, D. F. Macias-Pinilla, J. Planelles, J. I. Climente and I. Mora-Seró, ACS Energy Lett., 2020, 5, 418–427 CrossRef CAS.
- X. Zheng, C. Wu, S. K. Jha, Z. Li, K. Zhu and S. Priya, ACS Energy Lett., 2016, 1, 1014–1020 CrossRef CAS.
- J. A. Steele, H. Jin, I. Dovgaliuk, R. F. Berger, T. Braeckevelt, H. Yuan, C. Martin, E. Solano, K. Lejaeghere, S. M. J. Rogge, C. Notebaert, W. Vandezande, K. P. F. Janssen, B. Goderis, E. Debroye, Y.-K. Wang, Y. Dong, D. Ma, M. Saidaminov, H. Tan, Z. Lu, V. Dyadkin, D. Chernyshov, V. Van Speybroeck, E. H. Sargent, J. Hofkens and M. B. J. Roeffaers, Science, 2019, 365, 679–684 CrossRef CAS.
- A. Marronnier, G. Roma, S. Boyer-Richard, L. Pedesseau, J.-M. Jancu, Y. Bonnassieux, C. Katan, C. C. Stoumpos, M. G. Kanatzidis and J. Even, ACS Nano, 2018, 12, 3477–3486 CrossRef CAS.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510–519 CrossRef CAS.
- S. Ma, S. H. Kim, B. Jeong, H.-C. Kwon, S.-C. Yun, G. Jang, H. Yang, C. Park, D. Lee and J. Moon, Small, 2019, 15, 1900219 CrossRef.
- Y. Fan, H. Meng, L. Wang and S. Pang, Sol. RRL, 2019, 3, 1900215 CrossRef.
- S. Masi, A. F. Gualdrón-Reyes and I. Mora-Seró, ACS Energy Lett., 2020, 1974–1985, DOI:10.1021/acsenergylett.0c00801.
- J. Xue, J.-W. Lee, Z. Dai, R. Wang, S. Nuryyeva, M. E. Liao, S.-Y. Chang, L. Meng, D. Meng, P. Sun, O. Lin, M. S. Goorsky and Y. Yang, Joule, 2018, 2, 1866–1878 CrossRef CAS.
- A. Swarnkar, V. K. Ravi and A. Nag, ACS Energy Lett., 2017, 2, 1089–1098 CrossRef CAS.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692–3696 CrossRef CAS.
- A. F. Gualdrón-Reyes, J. Rodríguez-Pereira, E. Amado-González, J. Rueda-P, R. Ospina, S. Masi, S. J. Yoon, J. Tirado, F. Jaramillo, S. Agouram, V. Muñoz-Sanjosé, S. Giménez and I. Mora-Seró, ACS Appl. Mater. Interfaces, 2020, 12, 914–924 CrossRef.
- M. Suri, A. Hazarika, B. W. Larson, Q. Zhao, M. Vallés-Pelarda, T. D. Siegler, M. K. Abney, A. J. Ferguson, B. A. Korgel and J. M. Luther, ACS Energy Lett., 2019, 4, 1954–1960 CrossRef CAS.
- M. Que, Z. Dai, H. Yang, H. Zhu, Y. Zong, W. Que, N. P. Padture, Y. Zhou and O. Chen, ACS Energy Lett., 2019, 4, 1970–1975 CrossRef CAS.
- K. Lin, J. Xing, L. N. Quan, F. P. G. de Arquer, X. Gong, J. Lu, L. Xie, W. Zhao, D. Zhang, C. Yan, W. Li, X. Liu, Y. Lu, J. Kirman, E. H. Sargent, Q. Xiong and Z. Wei, Nature, 2018, 562, 245–248 CrossRef CAS.
- Q. Zhao, A. Hazarika, X. Chen, S. P. Harvey, B. W. Larson, G. R. Teeter, J. Liu, T. Song, C. Xiao, L. Shaw, M. Zhang, G. Li, M. C. Beard and J. M. Luther, Nat. Commun., 2019, 10, 2842 CrossRef.
- M. Hao, Y. Bai, S. Zeiske, L. Ren, J. Liu, Y. Yuan, N. Zarrabi, N. Cheng, M. Ghasemi, P. Chen, M. Lyu, D. He, J.-H. Yun, Y. Du, Y. Wang, S. Ding, A. Armin, P. Meredith, G. Liu, H.-M. Cheng and L. Wang, Nat. Energy, 2020, 5, 79–88 CrossRef CAS.
- S. Seth, T. Ahmed, A. De and A. Samanta, ACS Energy Lett., 2019, 4, 1610–1618 CrossRef CAS.
- Y. Zhou, J. Chen, O. M. Bakr and H.-T. Sun, Chem. Mater., 2018, 30, 6589–6613 CrossRef CAS.
- Q. A. Akkerman, D. Meggiolaro, Z. Dang, F. De Angelis and L. Manna, ACS Energy Lett., 2017, 2, 2183–2186 CrossRef CAS.
- X. Shen, Y. Zhang, S. V. Kershaw, T. Li, C. Wang, X. Zhang, W. Wang, D. Li, Y. Wang, M. Lu, L. Zhang, C. Sun, D. Zhao, G. Qin, X. Bai, W. W. Yu and A. L. Rogach, Nano Lett., 2019, 19, 1552–1559 CrossRef CAS.
- S. Bera, D. Ghosh, A. Dutta, S. Bhattacharyya, S. Chakraborty and N. Pradhan, ACS Energy Lett., 2019, 4, 1364–1369 CrossRef CAS.
- R. Begum, X. Y. Chin, M. Li, B. Damodaran, T. C. Sum, S. Mhaisalkar and N. Mathews, Chem. Commun., 2019, 55, 5451–5454 RSC.
- F. Liu, C. Ding, Y. Zhang, T. S. Ripolles, T. Kamisaka, T. Toyoda, S. Hayase, T. Minemoto, K. Yoshino, S. Dai, M. Yanagida, H. Noguchi and Q. Shen, J. Am. Chem. Soc., 2017, 139, 16708–16719 CrossRef CAS.
- A. B. F. Vitoreti, S. Agouram, M. Solis de la Fuente, V. Muñoz-Sanjosé, M. A. Schiavon and I. Mora-Seró, J. Phys. Chem. C, 2018, 122, 14222–14231 CrossRef CAS.
- X. Wu, W. Song, Q. Li, X. Zhao, D. He and Z. Quan, Chem. – Asian J., 2018, 13, 1654–1659 CrossRef CAS.
- F. Liu, C. Ding, Y. Zhang, T. Kamisaka, Q. Zhao, J. M. Luther, T. Toyoda, S. Hayase, T. Minemoto, K. Yoshino, B. Zhang, S. Dai, J. Jiang, S. Tao and Q. Shen, Chem. Mater., 2019, 31, 798–807 CrossRef CAS.
- Y. Wang, J. Tu, T. Li, C. Tao, X. Deng and Z. Li, J. Mater. Chem. A, 2019, 7, 7683–7690 RSC.
- F. Liu, Y. Zhang, C. Ding, K. Kawabata, Y. Yoshihara, T. Toyoda, S. Hayase, T. Minemoto, R. Wang and Q. Shen, Chem. Mater., 2020, 32, 1089–1100 CrossRef CAS.
- S. Khalfin and Y. Bekenstein, Nanoscale, 2019, 11, 8665–8679 RSC.
- S. Zhou, Y. Zhu, J. Zhong, F. Tian, H. Huang, J. Chen and D. Chen, Nanoscale, 2019, 11, 12465–12470 RSC.
- N. Phung, R. Félix, D. Meggiolaro, A. Al-Ashouri, G. Sousa e Silva, C. Hartmann, J. Hidalgo, H. Köbler, E. Mosconi, B. Lai, R. Gunder, M. Li, K.-L. Wang, Z.-K. Wang, K. Nie, E. Handick, R. G. Wilks, J. A. Marquez, B. Rech, T. Unold, J.-P. Correa-Baena, S. Albrecht, F. D. Angelis, M. Bär and A. Abate, J. Am. Chem. Soc., 2020, 142, 2364–2374 CrossRef CAS.
- J.-S. Yao, J. Ge, K.-H. Wang, G. Zhang, B.-S. Zhu, C. Chen, Q. Zhang, Y. Luo, S.-H. Yu and H.-B. Yao, J. Am. Chem. Soc., 2019, 141, 2069–2079 CrossRef CAS.
- T. J. Jacobsson, M. Pazoki, A. Hagfeldt and T. Edvinsson, J. Phys. Chem. C, 2015, 119, 25673–25683 CrossRef CAS.
- L.-Q. Xie, L. Chen, Z.-A. Nan, H.-X. Lin, T. Wang, D.-P. Zhan, J.-W. Yan, B.-W. Mao and Z.-Q. Tian, J. Am. Chem. Soc., 2017, 139, 3320–3323 CrossRef CAS.
- C. Ding, F. Liu, Y. Zhang, D. Hirotani, X. Rin, S. Hayase, T. Minemoto, T. Masuda, R. Wang and Q. Shen, Nano Energy, 2020, 67, 104267 CrossRef CAS.
- Y. Dong, T. Qiao, D. Kim, D. Parobek, D. Rossi and D. H. Son, Nano Lett., 2018, 18, 3716–3722 CrossRef CAS.
- Y. Fu, T. Wu, J. Wang, J. Zhai, M. J. Shearer, Y. Zhao, R. J. Hamers, E. Kan, K. Deng, X. Y. Zhu and S. Jin, Nano Lett., 2017, 17, 4405–4414 CrossRef CAS.
- A. Dutta, S. K. Dutta, S. D. Adhikari and N. Pradhan, ACS Energy Lett., 2018, 3, 329–334 CrossRef CAS.
- I. Levchuk, A. Osvet, X. Tang, M. Brandl, J. D. Perea, F. Hoegl, G. J. Matt, R. Hock, M. Batentschuk and C. J. Brabec, Nano Lett., 2017, 17, 2765–2770 CrossRef CAS.
- M. C. Weidman, M. Seitz, S. D. Stranks and W. A. Tisdale, ACS Nano, 2016, 10, 7830–7839 CrossRef CAS.
- J. C. Blancon, H. Tsai, W. Nie, C. C. Stoumpos, L. Pedesseau, C. Katan, M. Kepenekian, C. M. M. Soe, K. Appavoo, M. Y. Sfeir, S. Tretiak, P. M. Ajayan, M. G. Kanatzidis, J. Even, J. J. Crochet and A. D. Mohite, Science, 2017, 355, 1288–1292 CrossRef CAS.
- L. Protesescu, S. Yakunin, S. Kumar, J. Bär, F. Bertolotti, N. Masciocchi, A. Guagliardi, M. Grotevent, I. Shorubalko, M. I. Bodnarchuk, C.-J. Shih and M. V. Kovalenko, ACS Nano, 2017, 11, 3119–3134 CrossRef CAS.
- C. Liang, D. Zhao, Y. Li, X. Li, S. Peng, G. Shao and G. Xing, Energy Environ. Mater., 2018, 1, 221–231 CrossRef CAS.
- S. Adjokatse, S. Kahmann, H. Duim and M. A. Loi, APL Mater., 2019, 7, 031116 CrossRef.
- C. F. J. Lau, M. Zhang, X. Deng, J. Zheng, J. Bing, Q. Ma, J. Kim, L. Hu, M. A. Green, S. Huang and A. Ho-Baillie, ACS Energy Lett., 2017, 2, 2319–2325 CrossRef CAS.
- L. Martínez-Sarti, S. H. Jo, Y.-H. Kim, M. Sessolo, F. Palazon, T.-W. Lee and H. J. Bolink, Nanoscale, 2019, 11, 12793–12797 RSC.
- X. Zhang, C. Wang, Y. Zhang, X. Zhang, S. Wang, M. Lu, H. Cui, S. V. Kershaw, W. W. Yu and A. L. Rogach, ACS Energy Lett., 2018, 4, 242–248 CrossRef.
- D. H. Cao, C. C. Stoumpos, O. K. Farha, J. T. Hupp and M. G. Kanatzidis, J. Am. Chem. Soc., 2015, 137, 7843–7850 CrossRef CAS.
- P. Caprioglio, F. Zu, C. M. Wolff, J. A. Márquez Prieto, M. Stolterfoht, P. Becker, N. Koch, T. Unold, B. Rech, S. Albrecht and D. Neher, Sustainable Energy Fuels, 2019, 3, 550–563 RSC.
- K. Chen, Q. Zhong, W. Chen, B. Sang, Y. Wang, T. Yang, Y. Liu, Y. Zhang and H. Zhang, Adv. Funct. Mater., 2019, 29, 1900991 CrossRef.
- A. Walsh, D. O. Scanlon, S. Chen, X. G. Gong and S.-H. Wei, Angew. Chem., Int. Ed., 2015, 54, 1791–1794 CrossRef CAS.
- R. Grisorio, E. Fanizza, I. Allegretta, D. Altamura, M. Striccoli, R. Terzano, C. Giannini, V. Vergaro, G. Ciccarella, N. Margiotta and G. P. Suranna, Nanoscale, 2020, 12, 623–637 RSC.
- J. De Roo, M. Ibáñez, P. Geiregat, G. Nedelcu, W. Walravens, J. Maes, J. C. Martins, I. Van Driessche, M. V. Kovalenko and Z. Hens, ACS Nano, 2016, 10, 2071–2081 CrossRef CAS.
- G. Almeida, L. Goldoni, Q. Akkerman, Z. Dang, A. H. Khan, S. Marras, I. Moreels and L. Manna, ACS Nano, 2018, 12, 1704–1711 CrossRef CAS.
- E. Hassanabadi, M. Latifi, A. F. Gualdrón-Reyes, S. Masi, S. Y. joon, M. Poyatos, B. Julián-López and I. Mora Seró, Nanoscale, 2020, 12, 14194–14203 RSC.
- V. K. Ravi, R. A. Scheidt, A. Nag, M. Kuno and P. V. Kamat, ACS Energy Lett., 2018, 3, 1049–1055 CrossRef CAS.
- V. S. Chirvony, K. S. Sekerbayev, H. Pashaei Adl, I. Suárez, Y. T. Taurbayev, A. F. Gualdrón-Reyes, I. Mora-Seró and J. P. Martínez-Pastor, J. Lumin., 2020, 221, 117092 CrossRef CAS.
- M. Anaya, J. F. Galisteo-López, M. E. Calvo, J. P. Espinós and H. Míguez, J. Phys. Chem. Lett., 2018, 9, 3891–3896 CrossRef CAS.
- R. Zhu, Z. Luo, H. Chen, Y. Dong and S.-T. Wu, Opt. Express, 2015, 23, 23680 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental setup, histograms, photoimages, SAED measurements, UV-Vis, PL, and XPS spectra, PL decays and tables of the optical properties, surface chemical composition, carrier lifetime and PLQY of the FAPb1-xSrxI3 PQDs colloidal solutions and redispersed precipitates. Excel file with the raw data of PLQY measurements. See DOI: 10.1039/d0tc04625f |
| This journal is © The Royal Society of Chemistry 2021 |