
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Coinage metal aluminyl complexes: probing regiochemistry and mechanism in the insertion and reduction of carbon dioxide†
Caitilín
McManus
a,
Jamie
Hicks
 a,
Xianlu
Cui
b,
Lili
Zhao
*b,
Gernot
Frenking
*c,
Jose M.
Goicoechea
*a and
Simon
Aldridge
*a
a,
Xianlu
Cui
b,
Lili
Zhao
*b,
Gernot
Frenking
*c,
Jose M.
Goicoechea
*a and
Simon
Aldridge
*a
aInorganic Chemistry Laboratory, Department of Chemistry, University of Oxford, South Parks Road, Oxford, OX1 3QR, UK. E-mail: simon.aldridge@chem.ox.ac.uk
bInstitute of Advanced Synthesis, School of Chemistry and Molecular Engineering, Jiangsu National Synergetic Innovation Center for Advanced Materials, Nanjing Tech University, Nanjing 211816, P. R. China
cFachbereich Chemie, Philipps-Universität, Marburg, D-35043 Marburg, Germany
First published on 16th September 2021
Abstract
The synthesis of coinage metal aluminyl complexes, featuring M–Al covalent bonds, is reported via a salt metathesis approach employing an anionic Al(I) (‘aluminyl’) nucleophile and group 11 electrophiles. This approach allows access to both bimetallic (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) systems of the type (tBu3P)MAl(NON) (M = Cu, Ag, Au; NON = 4,5-bis(2,6-diisopropylanilido)-2,7-di-tert-butyl-9,9-dimethylxanthene) and a 2:1 di(aluminyl)cuprate system, K[Cu{Al(NON)}2]. The bimetallic complexes readily insert heteroallenes (CO2, carbodiimides) into the unsupported M–Al bonds to give systems containing a M(CE2)Al bridging unit (E = O, NR), with the μ-κ1(C):κ2(E,E′) mode of heteroallene binding being demonstrated crystallographically for carbodiimide insertion in the cases of all three metals, Cu, Ag and Au. The regiochemistry of these processes, leading to the formation of M–C bonds, is rationalized computationally, and is consistent with addition of CO2 across the M–Al covalent bond with the group 11 metal acting as the nucleophilic partner and Al as the electrophile. While the products of carbodiimide insertion are stable to further reaction, their CO2 analogues have the potential to react further, depending on the identity of the group 11 metal. (tBu3P)Au(CO)2Al(NON) is inert to further reaction, but its silver counterpart reacts slowly with CO2 to give the corresponding carbonate complex (and CO), and the copper system proceeds rapidly to the carbonate even at low temperatures. Experimental and quantum chemical investigations of the mechanism of the CO2 to CO/carbonate transformation are consistent with rate-determining extrusion of CO from the initially-formed M(CO)2Al fragment to give a bimetallic oxide that rapidly assimilates a second molecule of CO2. The calculated energetic barriers for the most feasible CO extrusion step (ΔG‡ = 26.6, 33.1, 44.5 kcal mol−1 for M = Cu, Ag and Au, respectively) are consistent not only with the observed experimental labilities of the respective M(CO)2Al motifs, but also with the opposing trends in M–C (increasing) and M–O bond strengths (decreasing) on transitioning from Cu to Au.
:1) systems of the type (tBu3P)MAl(NON) (M = Cu, Ag, Au; NON = 4,5-bis(2,6-diisopropylanilido)-2,7-di-tert-butyl-9,9-dimethylxanthene) and a 2:1 di(aluminyl)cuprate system, K[Cu{Al(NON)}2]. The bimetallic complexes readily insert heteroallenes (CO2, carbodiimides) into the unsupported M–Al bonds to give systems containing a M(CE2)Al bridging unit (E = O, NR), with the μ-κ1(C):κ2(E,E′) mode of heteroallene binding being demonstrated crystallographically for carbodiimide insertion in the cases of all three metals, Cu, Ag and Au. The regiochemistry of these processes, leading to the formation of M–C bonds, is rationalized computationally, and is consistent with addition of CO2 across the M–Al covalent bond with the group 11 metal acting as the nucleophilic partner and Al as the electrophile. While the products of carbodiimide insertion are stable to further reaction, their CO2 analogues have the potential to react further, depending on the identity of the group 11 metal. (tBu3P)Au(CO)2Al(NON) is inert to further reaction, but its silver counterpart reacts slowly with CO2 to give the corresponding carbonate complex (and CO), and the copper system proceeds rapidly to the carbonate even at low temperatures. Experimental and quantum chemical investigations of the mechanism of the CO2 to CO/carbonate transformation are consistent with rate-determining extrusion of CO from the initially-formed M(CO)2Al fragment to give a bimetallic oxide that rapidly assimilates a second molecule of CO2. The calculated energetic barriers for the most feasible CO extrusion step (ΔG‡ = 26.6, 33.1, 44.5 kcal mol−1 for M = Cu, Ag and Au, respectively) are consistent not only with the observed experimental labilities of the respective M(CO)2Al motifs, but also with the opposing trends in M–C (increasing) and M–O bond strengths (decreasing) on transitioning from Cu to Au.
Introduction
Combinations of metals – either in discrete complexes or in extended materials – have been shown to facilitate patterns of reactivity inaccessible to individual metals in isolation.1 Molecular heterobimetallic systems, for example, often possess reactivity distinct from complexes containing a single metal centre.2 Such attributes have been exploited to enable stoichiometric and catalytic transformations of kinetically challenging substrates, and to effect selectivity patterns distinct from monometallic systems.3 In the case of directly bonded bimetallics, the combination of two very different metals can lead to highly polarised bonds (and sites of differential Lewis acidity/basicity), which can produce synergistic effects in reactivity towards small molecules.4 Such systems also offer potential as single source precursors for metal alloys and functional materials.5–8Of particular interest are binuclear systems featuring elements from group 13 (Chart 1). The presence of a strongly Lewis acidic centre in close proximity to a d-block metal can assist in substrate activation, and has proven effective, for example, in mixed transition metal/aluminium systems for alkene polymerisation.9 In the case of directly bonded systems, the relative electropositivity of the group 13 elements typically means that ligands derived from them are very good σ-donors towards d-block metals.10 As such, the intermediacy of transition metal boryl complexes in metal catalysed C–H borylation chemistry, for example, owes much to the strong trans influence of this ligand class. This feature, combined with the availability of a formally vacant B-centred p-orbital, are key to both C–H bond breaking and subsequent C–B bond formation steps.11,12 More generally, the boryl ligand family has found extensive use across the Periodic Table with its strong donor properties facilitating a range of chemistries, including E–H activation13 and CO2 reduction.14 While a wide range of M–Ga bonded species have been synthesised using nucleophilic Ga(I) heterocycles,15 the chemistry of bimetallic aluminium-containing systems is less well studied.
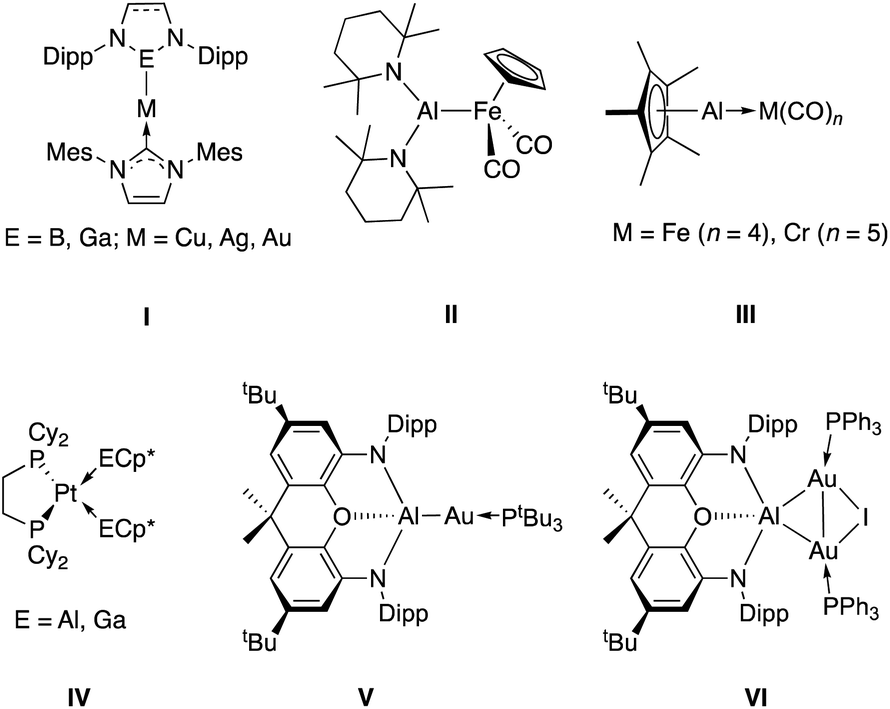 | ||
| Chart 1 Selected examples of transition metal – group 13 complexes: coinage metal complexes made by metathesis using boryl or gallyl nucleophiles (I);17,18 an Fe–Al bond synthesized from [CpFe(CO)2]− (II);19 transition metal complexes of AlCp* and GaCp* (II and III);20,21 previously communicated aluminium–gold complexes formed from an aluminyl nucleophile and Au(I) electrophiles (V and VI).22 | ||
A significant body of work has been carried out by Schnöckel and Fischer, using neutral Al(I) systems such as AlCp* to construct bonds typically to electron-rich transition metal carbonyl and olefin complexes.16 The resulting donor/acceptor M–Al bonds feature the AlCp* moiety acting as an L type ligand (isolobal with CO and PR3) and are relatively labile, rendering studies of their further chemistry challenging. As regards covalent M–Al bonds, a long-standing synthetic approach involves the metathesis reaction between an aluminium electrophile and a transition metal nucleophile, exemplified by the reaction between AlBr(tmp)2 and Na[CpFe(CO)2] to give CpFe(CO)2Al(tmp)2 reported by Nöth and co-workers.19 A shortcoming of this approach, however, is the relative scarcity of suitable electron-rich transition metal nucleophiles, which are typically limited to strongly pi accepting ancillary ligand sets (often carbonyls).
The recent development of nucleophilic aluminium ‘aluminyl’ anions allows access to a potential metathesis process in the opposite sense;23i.e. by the combination of a nucleophilic aluminium source with a readily available transition metal electrophile.22,24,25 We recently reported the synthesis of two aluminium–gold complexes (V and VI) prepared by salt metathesis between the potassium aluminyl complex, [K{Al(NON)}]2 (1, where NON = 4,5-bis(2,6-diiso-propylanilido)-2,7-di-tert-butyl-9,9-dimethyl-xanthene) and phosphine-supported Au(I) halides.22 These compounds represented the first examples of aluminium–gold covalent bonds, and bimetallic system V is particularly unusual in featuring a gold centre with nucleophilic character. V undergoes insertion of CO2 into the Al–Au bond to form the μ-κ1(C):κ2(O,O′) CO2 complex (tBu3P)Au(CO2)Al(NON), which can also be thought of as a dioxocarbene complex of gold(I). Insertions of this kind are extremely rare; only a handful of such systems have been reported,25,26 and (tBu3P)Au(CO2)Al(NON) represented the first structurally characterized example. Heterobimetallic CO2-bridged complexes of this nature had been prepared previously only via reactions of pre-formed metallacarboxylate nucleophiles with external metal electrophiles.27
From a broader perspective this unusual binding mode is interesting from the perspective of CO2 reduction chemistry, where the initial mode of binding to the metal(s) often determines the nature of the product formed e.g. CO or formate.28 CO2 produced by fossil fuels represents a large component of greenhouse gases; its utilisation as a chemical feedstock is therefore of increasing environmental importance, and understanding its potential modes of interaction with metal centres is therefore highly topical.
Given the unprecedented results seen with gold aluminyl complex V,22 we targeted (i) the synthesis of related compounds of the lighter group 11 elements via reactions of copper and silver electrophiles with the aluminium nucleophile 1; and (ii) investigation of the reactivity of Cu–Al and Ag–Al bonds towards CO2 and other heteroallenes. No covalently bonded silver–aluminium complexes have been synthesised to date, and copper analogues have only been described very recently.25 It was anticipated that such complexes – if accessible – might display differing reactivity towards CO2 compared to V, owing to the variation in electronegativity of the metals on descending group 11.
Results and discussion
Synthesis of aluminium–coinage metal complexes
Our preliminary studies revealed that the reactions of the potassium aluminyl dimer 1 with the gold(I) halides (tBu3P)AuI and (Ph3P)AuI lead to the formation of Au–Al covalent bonds via halide metathesis, and the isolation of the bi- and trimetallic systems V and VI respectively (Chart 1).22 A similar approach was therefore adopted for an initial exploration of the synthesis of copper and silver aluminyl complexes.In contrast to the related gold chemistry, the reaction of dimeric 1 with one equivalent of (Ph3P)CuI generates the bisaluminyl cuprate complex K[Cu{Al(NON)}2] (2; Scheme 1). Dissociation of the phosphine ligand during the reaction is signalled by the appearance of a broad resonance at −6.0 ppm in the in situ31P NMR spectrum. 2 can subsequently be obtained by recrystallization from hexane in ca. 50% yield, characterized by standard spectroscopic and analytical methods, and its structure in the solid state determined by X-ray crystallography. The 1H NMR spectrum of the isolated product in d6-benzene features a pattern of resonances consistent with lower molecular symmetry for the (NON)Al fragment in solution compared to 1. For example, whereas 1 is characterized by only one resonance for the Dipp iPr methine protons, 2 gives rise to four such signals. This lower symmetry is consistent with the structure determined crystallographically for 2 in the solid state which features a potassium cation sandwiched between the π systems of the flanking Dipp substituents on one side of the Al–Cu–Al axis (Fig. 1). Retention of this motif in benzene-d6 solution on the NMR timescale (as has been observed for the K+ cations in 1 itself)23a accounts for the observed lower molecular symmetry. The K+ ion is held in place between two Dipp groups of opposing (NON) ligand frameworks, and the K⋯C distances (3.089(3)–3.296(3) Å) are slightly shorter than the equivalent contacts in 1 (3.226(3)–3.474(3) Å),23a suggesting – if anything – a marginally stronger arene–potassium interaction.
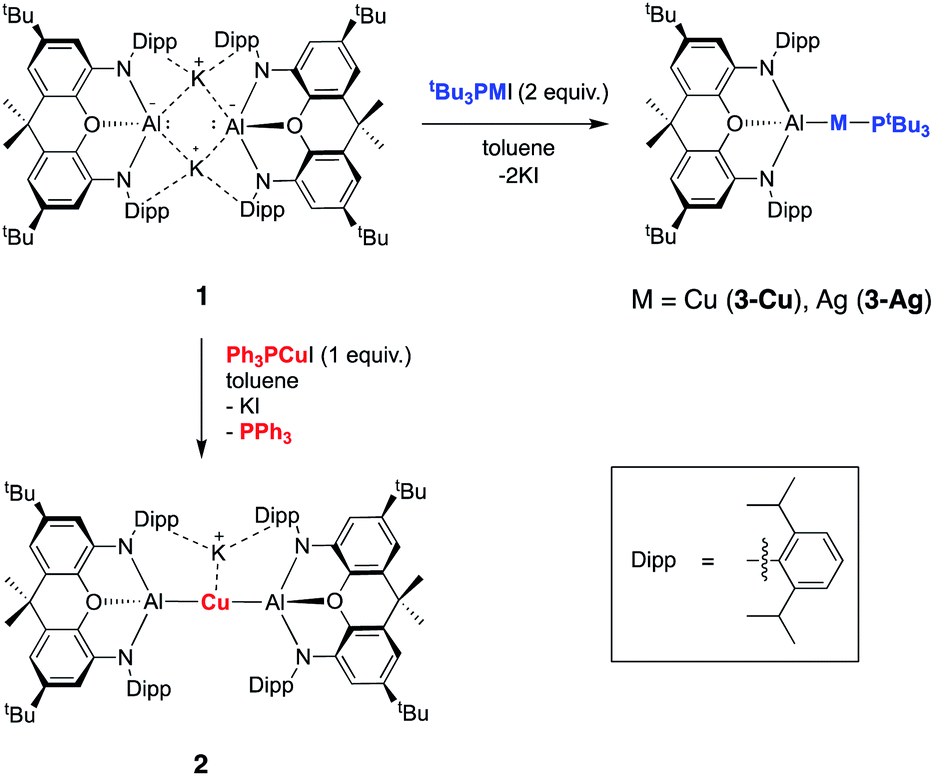 | ||
| Scheme 1 Synthesis of aluminium–silver and aluminium–copper complexes 2, 3-Cu and 3-Ag from potassium aluminyl complex 1via salt metathesis. | ||
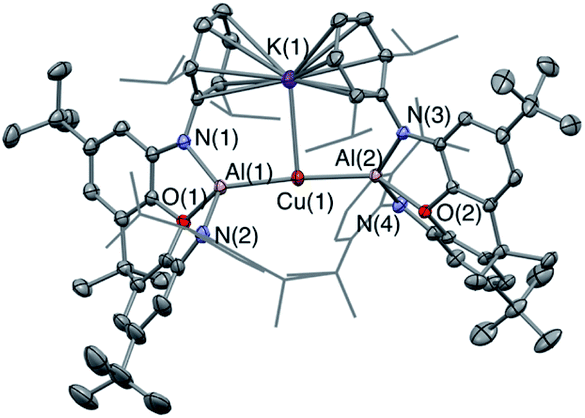 | ||
| Fig. 1 Molecular structure of 2 in the solid state as determined by X-ray crystallography. Thermal ellipsoids set at the 50% probability level. Hydrogen atoms omitted and iPr groups shown in wireframe for clarity. Key bond lengths (Å) and angles (°): Al(1)–O(1) 2.0428(15), Cu⋯K 3.0669(6), Cu(1)–Al(1) 2.4076(5), Cu(2)–Al(1) 2.4075(5), Al(1)–Cu(1)–Al(2) 174.88(2). | ||
From the perspective of coordination chemistry, the solid-state structure of 2 features a near-linear Al–Cu–Al fragment [∠(Al(1)–Cu(1)–Al(2)) = 174.88(2)°], in common with other sterically encumbered [CuX2]− systems.29 The Cu–Al bond lengths (2.4076(5) and 2.4075(5) Å) are shorter than the sum of the respective covalent radii (2.53 Å) and are towards the shorter end of the range observed for solid state Cu/Al clusters, such as Cu9Al4, (2.4027(14) to 2.7189(14) Å).30 They are also similar to those reported very recently by Hill and co-workers for a cyclic (amino)alkyl carbene (cAAC)-supported copper aluminyl complex (2.4028(7) Å),25 but somewhat longer than the dispersion enhanced donor–acceptor bond measured for {HC(MeCMesN)2}CuAl{(NDippCMe)2CH} (2.3010(6) Å).31 The latter observation potentially reflects the mutually trans disposition of the (strongly donating) aluminyl ligands, and the fact that the positioning of the K+ counterion between the arene rings in 2 potentially constrains the approach of the (NON)Al units to the Cu(I) centre.
The reaction of 1 with two equivalents of (Ph3P)CuI (i.e. a 1:1 ratio Al:Cu) does not yield a compound containing a Cu–Al bond, but instead generates the mixed valence Cu(I)/Cu(0) cluster (Ph3P)4Cu4I2 (see ESI†). This chemistry contrasts with the corresponding reactivity of 1 towards (Ph3P)AuI, which yields the trimetallic system (NON)Al{Au(PPh3)}2I (VI; Chart 1), featuring an aluminium fragment bridging two formally Au(0) centres, and presumably reflects the greater electronegativity of gold vs. copper.32
With a view to generating 1:1 bimetallic systems, with retention of the M–P linkage, alternative ancillary phosphine ligands were investigated. In the case of the stronger σ-donor tBu3P, compounds of the type (tBu3P)MAl(NON) (M = Cu (3-Cu) or Ag (3-Ag)) can be synthesized from the reactions of dimeric 1 with two equivalents of (tBu3P)CuI or (tBu3P)AgI, respectively. 3-Cu and 3-Ag have been characterised by 1H, 31P and 13C NMR spectroscopy and elemental microanalysis. The 1H spectra of both compounds feature a similar pattern of resonances to the (structurally characterized) gold analogue (tBu3P)AuAl(NON) (V); the signals corresponding to the phosphine tBu groups and the NON ligand backbone confirm a ratio of one phosphine ligand per NON unit. The 31P NMR resonances for the three compounds are measured at +38.3, +58.9 and +75 ppm for 3-Cu, 3-Ag and 3-Au, respectively, in line with other systems of the type (tBu3P)MX.33 However, the single crystals of both compounds which could be obtained were too small for diffraction studies (even using synchrotron radiation). That said, the NMR and microanalytical data, together with the structure of the gallyl analogue (3′-Ag, see below) and the patterns of reactivity observed for 3-Cu and 3-Ag (see below), suggest strongly that these two compounds are isostructural with the gold system 3-Au.
In the case of 3-Ag, the 31P signal is a doublet arising from coupling to the spin-active silver nuclei. Unlike the starting material (tBu3P)AgI, for example, distinct coupling to 107Ag and 109Ag cannot be resolved, presumably due to the influence of quadrupolar 27Al nucleus (I = 5/2). The mean 1JPAg value (152 Hz), however, is very small indeed. By means of comparison, those measured for related Ag(I) compounds featuring mutually trans tertiary phosphine and N-heterocyclic carbene ligands (e.g.1JPAg = 463.6, 536.0 Hz for [Ag(SIPr)(PCy3)][PF6]) are markedly larger,33 reflecting the very high trans influence of the strongly σ-donating aluminyl ligand (even compared to an NHC).
The corresponding gallyl complex 3′-Ag was prepared via the analogous reaction of the gallyl dimer K2[(NON)Ga]2 (ref. 23a) with (tBu3P)AgI, and in this case crystals could be grown from toluene solution which were suitable for X-ray crystallography (Fig. 2). 3′-Ag shows the expected linear Ga–Ag–P unit (∠(Al(1)–Cu(1)–Al(2)) = 175.00(2)°) analogous to the Al–Au–P motif found in V (∠(Al–Au–P) = 167.47(10)°). The 31P NMR spectrum of 3′-Ag shows coupling to both Ag isotopes, with the signal at 65.4 ppm featuring larger coupling constants (1JPAg = 278, 322 Hz) compared to that measured for 3-Ag. This observation reflects the reduced trans influence of the gallyl ligand compared to its aluminyl counterpart, and (in turn) the higher electronegativity of gallium over aluminium.32
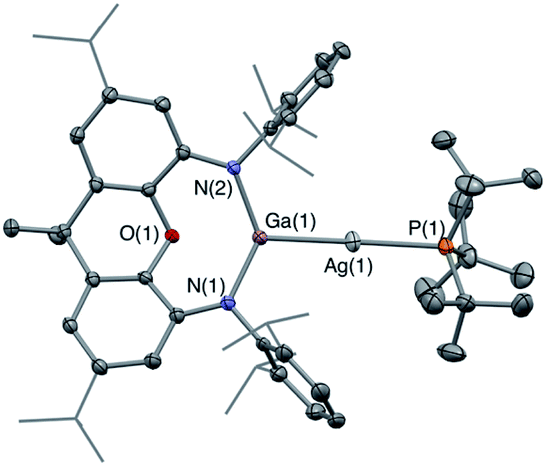 | ||
| Fig. 2 Molecular structure of 3′-Ag in the solid state as determined by X-ray crystallography. Thermal ellipsoids set at the 50% probability level. Hydrogen atoms omitted and iPr groups shown in wireframe for clarity. Key bond lengths (Å) and angles (°): Ga(1)–O(1) 2.3834(14), Ga(1)–Ag(1) 2.4548(3), Ag(1)–P(1) 2.4355(6), Ga(1)–Ag(1)–P(1) 175.00(2). | ||
Experimental studies of the reactivity of copper- and silver aluminyl compounds towards hetero-allenes
In a preliminary communication we showed that 3-Au reacts with carbon dioxide and diisopropylcarbodiimide to yield the respective insertion products featuring Au–C and Al–O or Al–N bonds (Scheme 2).22 The regiochemistry of the insertion process in each case is consistent with polarization of the metal–metal bond in the sense Au(δ−)–Al(δ+), and with the gold centre formally acting as the nucleophilic partner.25,35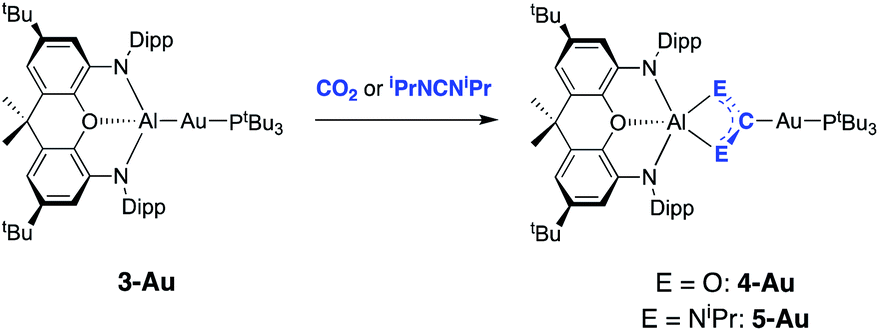 | ||
| Scheme 2 Insertion chemistry of CO2 and iPrNCNiPr with 3-Au.22 | ||
The potential for the lighter group 11 metal centres to act in a similar manner was therefore investigated. Silver aluminyl complex 3-Ag was exposed to a CO2 atmosphere and an immediate reaction was observed. The 1H spectrum of the reaction mixture features a 1:1 ratio of signals corresponding to the (NON) and PtBu3 fragments. The 31P NMR spectrum shows quantitative conversion to one species, with the signal at 76.0 ppm showing two sets of silver satellites, due to coupling to both 107Ag and 109Ag nuclei (1JPAg = 406, 465 Hz). The magnitudes of these couplings are similar to those reported previously for complexes in which silver is bound to both an N-heterocyclic carbene (NHC) and a phosphine ligand.34 Furthermore, the corresponding 31P spectrum obtained from the reaction of 3-Ag with 13CO2 (Fig. 3) shows additional splitting (2JCP = 81 Hz) consistent with the presence of a linear P–Ag–C unit (cf.2JCP = 62 Hz for a related NHC/phosphine complex reported by Braunstein and Danopoulos).34c Consistently, the 13C spectrum of the product features an eight-line multiplet centred at 237.7 ppm due to coupling to 31P and both silver isotopes (1J107AgC = 229 Hz, 1J109AgC = 266 Hz, 2JCP = 81 Hz). The low-field nature of this resonance is characteristic of metal bound carbene systems,36 and is close to that reported for the gold dioxo-carbene complex which is formed from the reaction of V and CO2 (δC = 242.3 ppm). A similar chemical shift of 239.5 ppm has also been reported for an iron bound dioxo-carbene complex.27a The NMR evidence therefore suggests that the product of the reaction of 3-Ag and CO2 is the silver dioxo-carbene complex, 4-Ag, formed by insertion of CO2 into the Al–Ag bond in similar fashion to that seen with 3-Au (Scheme 3).
 | ||
| Fig. 3 (Upper) 31P spectrum of 4-Ag; (lower) 31P spectrum of 4-Ag-13C prepared from 3-Ag and 13CO2, showing additional two-bond coupling to 13C. | ||
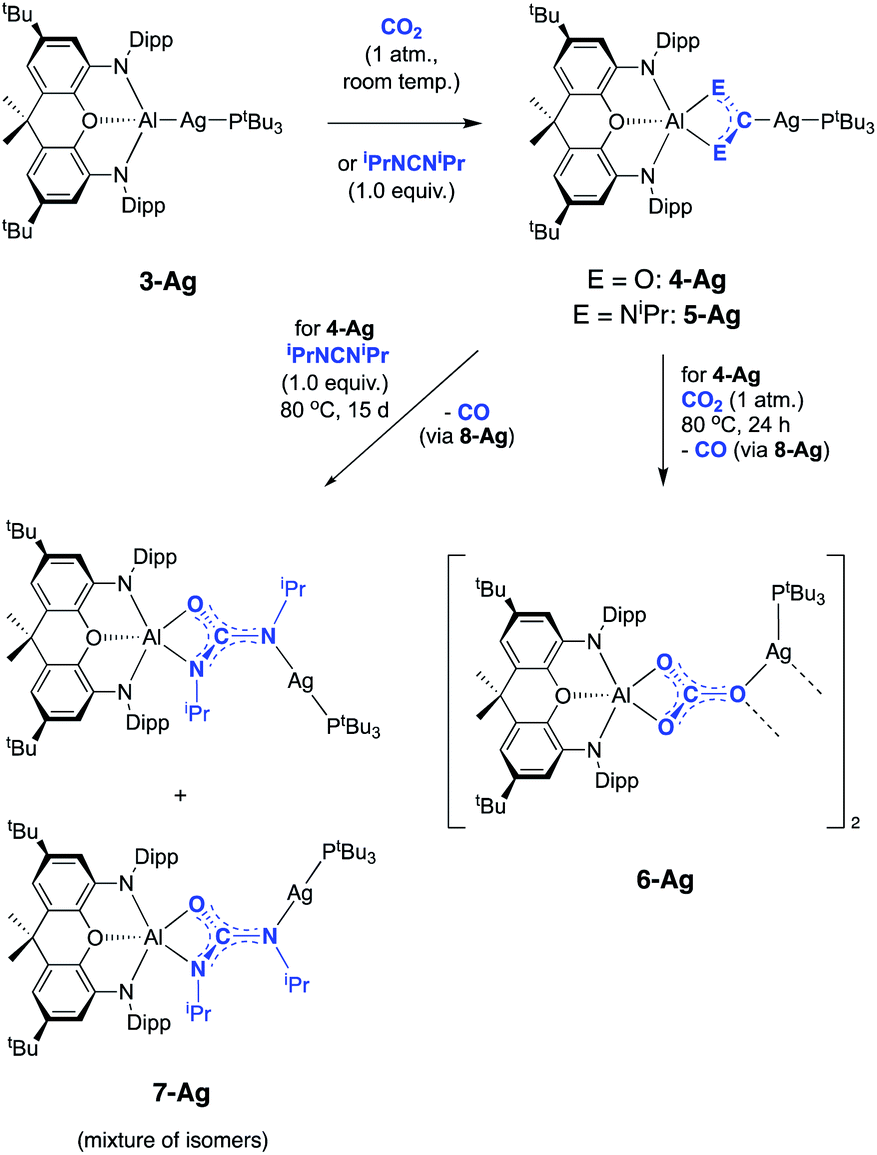 | ||
| Scheme 3 Reactions of silver–aluminium compound 3-Ag with CO2 and iPrNCNiPr. | ||
Although 4-Ag could not be obtained as single crystals suitable for X-ray crystallography, the corresponding chemistry with carbodiimide substrates proved more amenable. The reaction of 3-Ag with diisopropylcarbodiimide proceeds via a similar route, involving insertion into the Al–Ag bond to give 5-Ag (Scheme 3). Spectroscopic evidence for this insertion process (and its regiochemistry) comes from (i) an increase in the number of resonances relating to the carbodiimide isopropyl substituents in the 1H NMR spectrum; (ii) the appearance of a signal in the 31P spectrum at 73.4 ppm showing two sets of silver satellites, with similar coupling constants to those measured for 4-Ag (1JPAg = 352, 408 Hz); and (iii) a 13C spectrum which shows an eight-line multiplet for the carbene ligand carbon (δC = 220.2 ppm; 1J107AgC = 182 Hz, 1J109AgC = 210 Hz, 2JCP = 63 Hz) which is strikingly similar to those seen for both 4-Ag and the product of the reaction of 3-Au with iPrNCNiPr (i.e.5-Au). These data are consistent with the formation of 5-Ag by insertion of the diimide into the Al–Ag bond – an assertion which in this case could be confirmed by X-ray crystallography (Fig. 4).
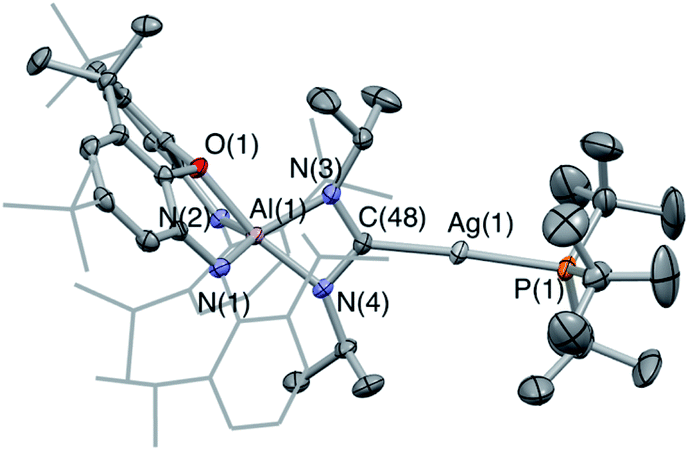 | ||
| Fig. 4 Molecular structure of 5-Ag in the solid state as determined by X-ray crystallography. Thermal ellipsoids set at the 50% probability level. Hydrogen atoms omitted and iPr/tBu shown in wireframe for clarity. Key bond lengths (Å) and angles (°): Ag(1)–C(48): 2.118(2), Ag(1)–P(1): 2.3851(6), N(3)–C(48): 1.346(3), N(4)–C(48): 1.345(3). | ||
The molecular structure of 5-Ag confirms the connectivity implied by NMR measurements and shows that the two carbodiimide nitrogen atoms are bound to aluminium and the central carbon atom is coordinated to silver. As such, carbodiimide insertion into the Al–Ag bond is confirmed, with the same regioselectivity as with 5-Au, i.e. with the silver centre of 3-Ag formally acting as a nucleophile in attacking the electrophilic carbon centre of iPrNCNiPr. The C–N bond lengths (1.346(3) and 1.345(3) Å) lie between the typical values for single and double C–N bonds,37 implying that the reaction of 3-Ag with iPrNCNiPr proceeds via a two-electron reduction of the iPrNCNiPr unit. Consistently, 5-Ag can also be viewed as a Ag(I) complex of the bent (aluminate backbone supported) diisopropylamino carbene formed by the addition of two electrons to the iPrNCNiPr moiety. By extension, the reaction of 3-Ag with CO2 generates (in 4-Ag) the related silver dioxocarbene complex. The lower 13C chemical shift measured for the diamino carbene unit in 5-Ag as compared to the dioxo species formed from CO2 insertion (220.2 vs. 237.7 ppm) finds precedent in the analogous gold compounds (219.9 vs. 242.3 ppm for 5-Au and 4-Au) and is consistent with increased π electron donation from nitrogen compared to oxygen.38 Although there is little further precedent for such silver dioxocarbene species, the coupling constants in the case of 5-Ag align very closely with those reported by Braunstein for the NHC–Ag-phosphine [Ag2(LtBu)2][BF4]2 (LtBu = 3-butyl-1-(3-((di-tert-butyl-phosphino)methyl)phenyl)-imidazol-2-ylidene): 1J107AgC = 190 Hz, 1J109AgC = 219 Hz, 2JCP = 62 Hz cf.1J107AgC = 182 Hz, 1J109AgC = 210 Hz, 2JCP = 63 Hz in 5-Ag.34c
While 4-Ag is stable in solution at room temperature, it undergoes conversion to the corresponding carbonate complex 6-Ag on heating to 80 °C for 24 h under a CO2 atmosphere (Scheme 3). This transformation occurs very slowly at room temperature, with no perceptible conversion observed by 1H NMR spectroscopy after 1 week. Spectroscopically, the 13C NMR spectrum of 6-Ag is most diagnostic, with the carbonate carbon appearing as a singlet at 167.2 ppm. This shift is similar to that of the aluminium carbonate, formed from the reaction of 1 with CO2 (164.5 ppm).23a Interestingly, the 31P NMR spectrum of 6-Ag in d8-toluene at room temperature features a broad resonance at 74.0 ppm, which sharpens into a well-defined doublet (1JPAg = 486 Hz) only on cooling to 203 K, suggesting the possibility that phosphine coordination might be labile at higher temperatures.
6-Ag can be obtained as single crystals by recrystallization from benzene, and X-ray crystallography reveals its structure to be a centrosymmetric dimer in the solid state, with two silver and two oxygen atoms constituting a planar diamond-shaped core (Fig. 5). The C–O distances are indicative of greater double bond character in the C–O bond projected towards the silver centres (d(O(1)–C(61)) = 1.266(6) Å, cf. d(O(3)–C(61)) = 1.307(6) and d(O(2)–C(61)) = 1.297(5) Å), consistent with the idea of greater localization of the negative charge in the [CO3]2− moiety on the oxygen atoms bonded to the harder aluminium centre (i.e. O(2) and O(3)).
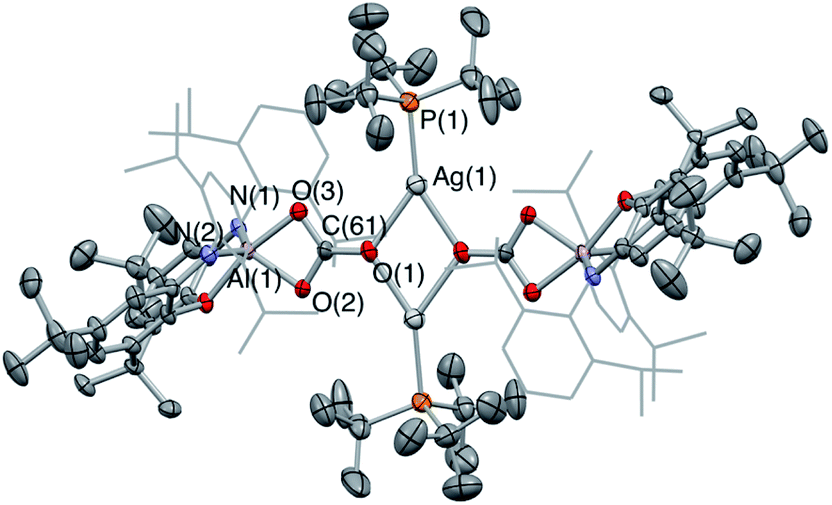 | ||
| Fig. 5 Molecular structure of 6-Ag in the solid state as determined by X-ray crystallography. Thermal ellipsoids shown at 50%. Hydrogen atoms and solvent molecules omitted and Dipp groups shown in wireframe for clarity. Key bond lengths (Å) and angles (°); the corresponding parameters obtained by quantum chemical calculations are given in square parentheses (see below): Ag(1)–O(1): 2.249(4) [2.275(4)], O(3)–C(61): 1.307(6) [1.319(8)], O(2)–C(61): 1.297(5) [1.316(5)], O(1)–C(61): 1.266(6) [1.271(4)]. | ||
Copper aluminyl complex 3-Cu was also reacted with both CO2 and carbodiimide reagents (Scheme 4). In the case of the latter, CyNCNCy was preferred over its isopropyl analogue (due to enhanced crystallinity of the product), and a single new resonance appears in the 31P spectrum of the reaction mixture at 59.6 ppm, accompanied by loss of the signal associated with 3-Cu. The 13C NMR spectrum shows a doublet at 215.5 ppm (2JCP = 57.5 Hz), i.e. at a chemical shift very similar to those measured for the silver and gold carbodiimide insertion products 5-Ag and 5-Au (220.2 and 219.9 ppm, respectively). In addition, single crystals of 5-Cu could be grown from benzene solution, and X-ray diffraction confirms that CyNCNCy undergoes insertion into the Al–Cu bond of 3-Cu in similar manner to its heavier congeners (Fig. 6). Similar reactivity has recently been reported by Hill and co-workers for a cAAC-supported Cu aluminyl complex, although a related NHC-ligated system displays alternative selectivity for the insertion process.25
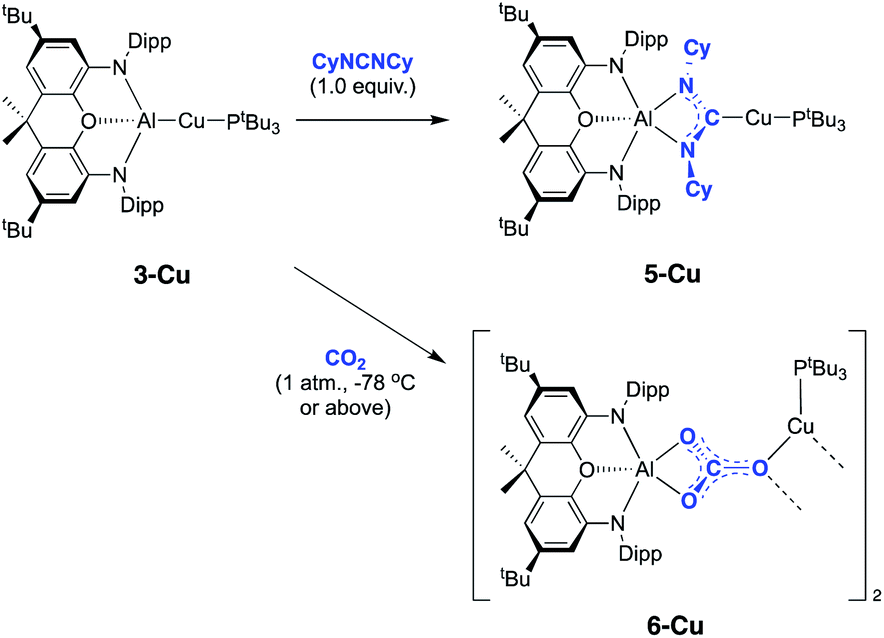 | ||
| Scheme 4 Reactions of copper-aluminium compound 3-Cu with CO2 and CyNCNCy. | ||
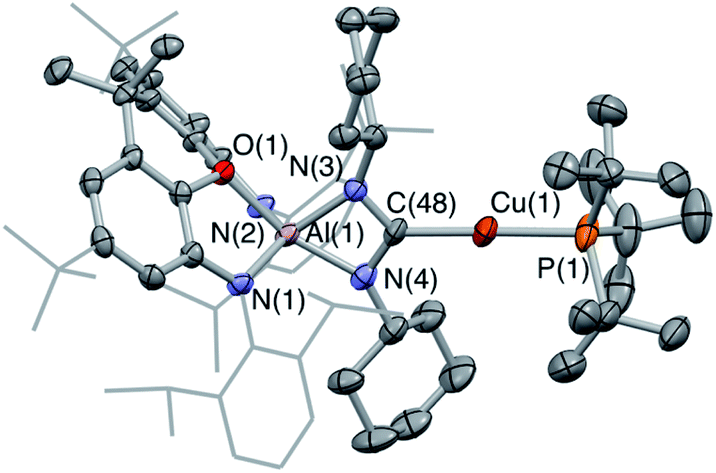 | ||
| Fig. 6 Molecular structure of 5-Cu in the solid state as determined by X-ray crystallography. Thermal ellipsoids set at the 50% probability level. Hydrogen atoms omitted and Dipp groups shown in wireframe for clarity. Key bond lengths (Å) and angles (°): Cu(1)–C(48): 1.952(4), Al(1)–N(4): 1.908(3), Al(1)–N(3): 1.921(3). | ||
Comparisons of the products of carbodiimide insertion into the respective M–Al bonds (i.e.5-Cu, 5-Ag and 5-Au), reveal properties consistent with descriptions of the three compounds as M(I) complexes featuring mutually trans phosphine and carbene ligands.36 As such, the linear geometries at M, the M–P distances and the 13C chemical shifts (Table 1) find precedent in the corresponding parameters reported for [(R3P)M(IDipp)]+ (M = Cu, Ag, Au; IDipp = 1,3-(diisopropylphenyl)imidazolylidene; R = Cy/tBu).39 The close similarities presumably reflect the fact that the angles at the carbenic carbon defined by the two α-N substituents (108.5(3) to 109.8(9)°) are relatively wide compared to other carbene ligands featuring a four-membered heterocycle,40 being more in line with those measured for imidazolylidene systems. This in turn presumably reflects the relatively large size of the aluminium atom in the heterocycle backbone and the polar nature of the Al–N bonds.
| M | d(M–C) (Å) | d(M–P) (Å) | ∠(C–M–P) (°) | δ C (ppm) | δ P (ppm) |
|---|---|---|---|---|---|
| Cu | 1.952(4) | 2.2163(13) | 179.04(13) | 215.5 | 59.6 |
| Ag | 2.118(2) | 2.3851(6) | 176.76(8) | 220.2 | 73.4 |
| Au | 2.158(10) | 2.316(3) | 175.7(3) | 219.9 | 90.6 |
In the reaction of 3-Cu with CO2, the 31P NMR spectrum of the reaction mixture shows immediate loss of the signal associated with the starting material, and the appearance of a single resonance at δP = 62.5 ppm. The 13C NMR spectrum, however, shows no highly deshielded doublet diagnostic of a carbene-like moiety. Instead, a singlet at δC = 170 ppm implies that the product of this reaction, even under mild conditions, features a carbonate group. This hypothesis can be confirmed by X-ray crystallography, which shows that the carbonate product formed (6-Cu), is isostructural with its silver analogue 6-Ag (Fig. 7). The reaction was also studied in situ using low temperature multinuclear NMR spectroscopy. However, it was found that full conversion to 6-Cu is observed even at −78 °C and no carbene-like intermediate could be observed even under such conditions.
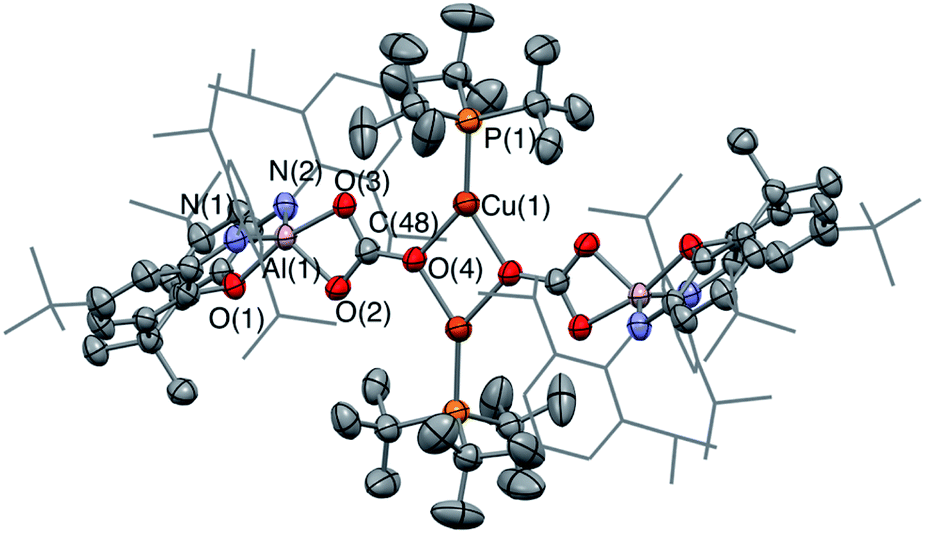 | ||
| Fig. 7 Molecular structure of 6-Cu in the solid state as determined by X-ray crystallography. Thermal ellipsoids set at the 50% probability level. Hydrogen atoms omitted and Dipp groups shown in wireframe format for clarity. Key bond lengths (Å) and angles (°): Cu(1)–O(4): 2.062(3), C(48)–O(3): 1.874(3), C(48)–O(4): 1.268(6), C(48)–O(2): 1.296(6). | ||
A number of general observations can be made concerning the reactivity of the copper, silver and gold complexes towards CO2 and carbodiimides. In the latter case, all three systems, 3-Cu, 3-Ag and 3-Au, can be shown by X-ray crystallography to undergo insertion of the heteroallene via a process which implies nucleophilic character at the group 11 metal centre. In none of these cases is any further reactivity observed with excess carbodiimide. In the case of CO2, however, onward reactivity appears to be possible (to an extent depending on the identity of the group 11 metal), proceeding from the analogous first-formed dioxocarbene species to yield a carbonate. In the case of gold, the carbene-like species 4-Au shows no hint of onward reactivity with excess CO2, even under forcing conditions. In the silver case, further reaction with CO2 is possible to give 6-Ag, but this reaction proceeds slowly and only at elevated temperatures. For the copper system, conversion to the carbonate species 6-Cuvia reaction with two equivalents of CO2 occurs so rapidly even at low temperatures that the initial insertion product, 4-Cu, cannot be directly observed. Superficially, this trend reflects the respective oxo- and carbophilicities of the group 11 elements, with M–O bond strengths decreasing and M-C bonds becoming stronger on going from copper to gold.41
Probes of the mechanism of CO2 insertion and transformation
:1 ratio at 78.8 and 80.1 ppm (both with 1J107AgP = 540, 1J109AgP = 623 Hz). The 13C spectrum shows two signals also in an approximately 2:1 ratio at 167.8 and 167.2 ppm (i.e. at chemical shifts similar to the carbonate resonances in 6-Ag and 6-Cu), and also shows a signal for free carbon monoxide (at δC = 184.5 ppm). X-ray crystallography reveals that the silver-containing product of this reaction is the ureate compound 7-Ag (Fig. 8 and Scheme 3) formed as a result of net CO extrusion from 4-Ag and uptake of the carbodiimide. In the solid state, the product is characterized by a short central C–N bond (d(N(1)–C(51)) = 1.304(6) Å) featuring a trans arrangement of the O and Ag(PtBu3) substituents. Restricted rotation about the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N bond in solution then provides a rationale for the two products observed by NMR (i.e. with the second species featuring the alternative cis alignment of the O and Ag(PtBu3) functions).
N bond in solution then provides a rationale for the two products observed by NMR (i.e. with the second species featuring the alternative cis alignment of the O and Ag(PtBu3) functions).
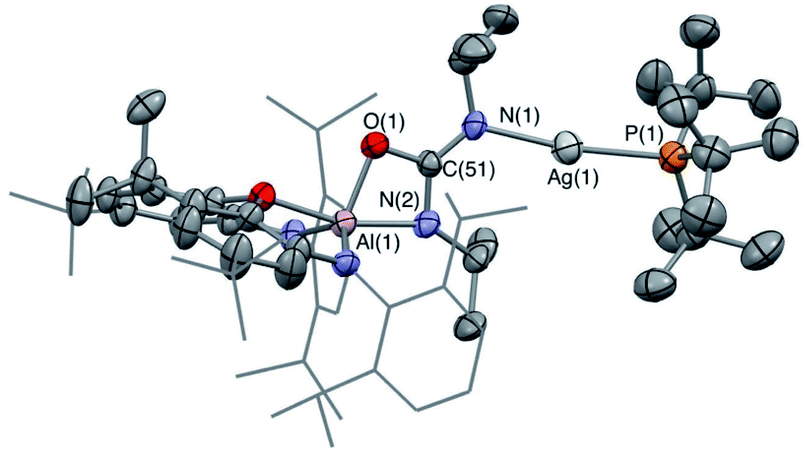 | ||
| Fig. 8 Molecular structure of 7-Ag as determined by X-ray crystallography. Thermal ellipsoids set at the 50% probability level. Hydrogen atoms omitted and Dipp groups shown in wireframe format for clarity. Key bond lengths (Å) and angles (°): Ag(1)–P(1): 2.3572(14), Ag(1)–N(1): 2.130(4), N(1)–C(51): 1.304(6), N(2)–C(51): 1.339(6), Al(1)–O(1): 1.842(3), O(1)–C(51): 1.344(5). | ||
While the formation of 7-Ag from 4-Ag and iPrNCNiPr implies that CO is lost from the dioxocarbene compound, in an overall process also involving uptake of the carbodiimide, we sought to obtain experimental evidence for similar steps in the formation of the carbonate compound 6-Ag. An in situ13C NMR spectrum of the reaction mixture comprised of 4-Ag and CO2 at 80 °C is consistent with the evolution of carbon monoxide. In addition, details of the CO extrusion process and uptake of a second CO2 molecule were probed using 13CO2 labelling studies (Fig. 9). These involved two separate experiments: (a) exposure of a sample of 4-Ag to 13CO2 at 80 °C and (b) exposure of a sample of 4-Ag-13C (itself prepared from 3-Ag and 13CO2) to unlabelled CO2 under similar conditions. The resulting 13C NMR spectra clearly show that the extruded CO originates from the first CO2 unit (i.e. from 4-Ag or 4-Ag-13C), and that the carbon incorporated into the resulting carbonate complex arises from the second molecule of CO2.
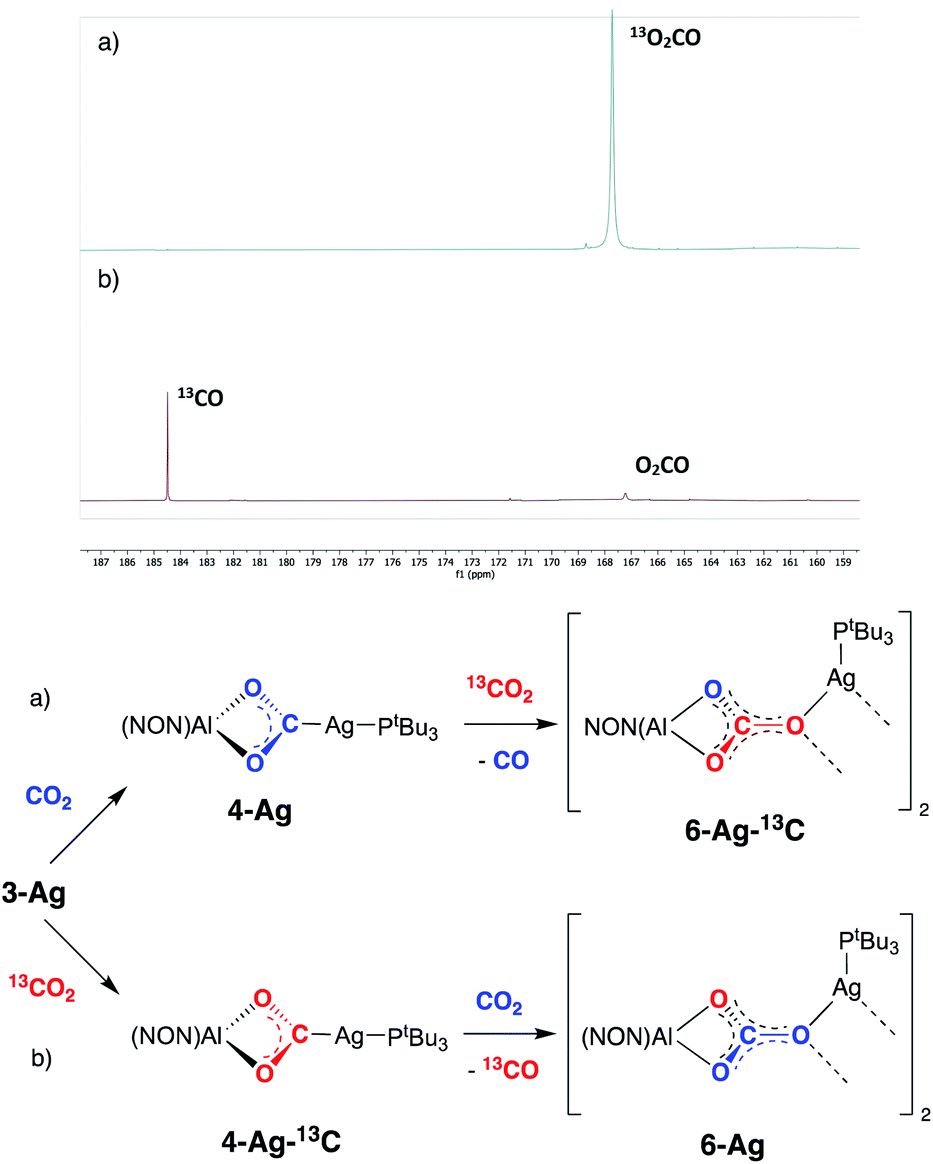 | ||
| Fig. 9 13CO2 ‘crossover’ labelling experiments (and their associated spectra) carried out with 4-Ag and 4-Ag-13C: (a) addition of 13CO2 to 4-Ag to yield 6-Ag-13C and CO; (b) addition of unlabelled CO2 to 4-Ag-13C to yield 6-Ag and 13CO. | ||
On this basis, a potential intermediate in the conversion of 4-Ag to 6-Ag is a bimetallic species featuring a bridging oxide between the aluminium and silver centres. Such a species would be formed via CO extrusion from 4-Ag, and then assimilate the second molecule of CO2 to give the carbonate product 6-Ag (Scheme 5).42 A similar mechanism for CO2 reduction to CO by a digermyne complex has been proposed by Jones and Frenking.26b
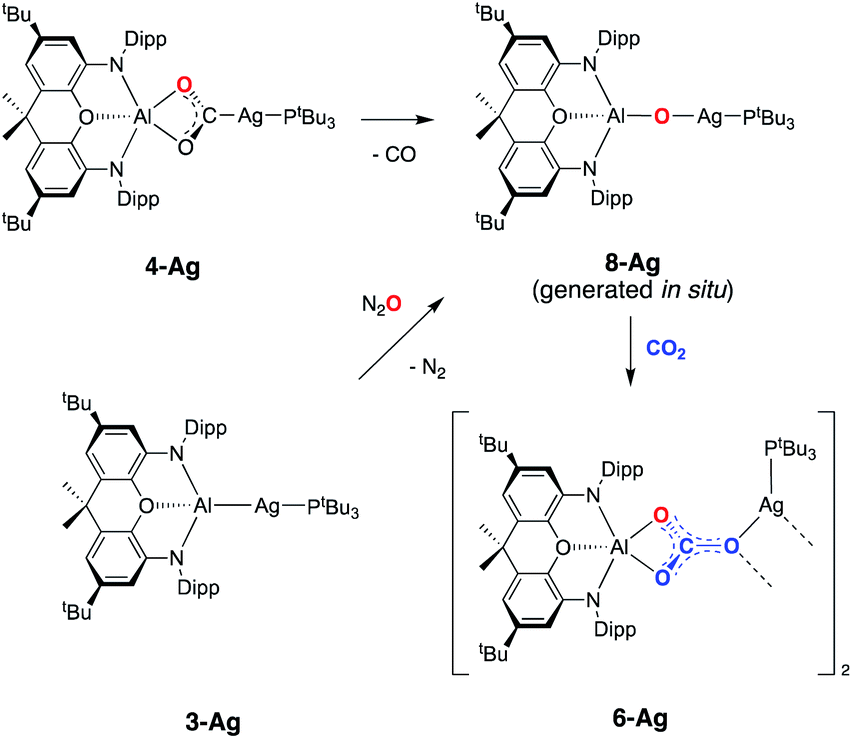 | ||
| Scheme 5 Potential pathway for the formation of carbonate complex 6-Agvia an oxide intermediate. | ||
To obtain experimental evidence for this proposal, the proposed intermediate (tBu3P)AgOAl(NON) (8-Ag) was independently synthesised by exposing 3-Ag to an N2O atmosphere at −78 °C (Scheme 5). The reaction was followed in situ by 1H NMR spectroscopy, which shows conversion under such conditions to a single species which features a 1:1 ratio of NON and PtBu3 ligands. The corresponding 31P spectrum features a single signal at 82.0 ppm displaying silver satellites (1J107AgP = 551, 1J109AgP = 640 Hz). The greatly increased magnitudes of the 1JPAg coupling constants (compared to 3-Ag) implies that the aluminyl ligand is no longer directly bonded to silver, and that it has been replaced by an appreciably poorer σ-donor. Although 8-Ag proved to be too thermally fragile to be isolated in bulk quantities, it could be generated in situ for reactivity studies by degassing the reaction mixture to remove excess N2O. At this point, exposure to an atmosphere of CO2 led to an immediate reaction and quantitative formation of carbonate compound 6-Ag, as evidenced by 1H and 31P NMR spectroscopy, and X-ray crystallography (Scheme 5). In similar fashion, the reaction of 8-Ag with diisopropylcarbodiimide at room temperature rapidly forms ureate complex 7-Ag, with the same apparent selectivity for the two isomers as seen in the reaction with 4-Ag.
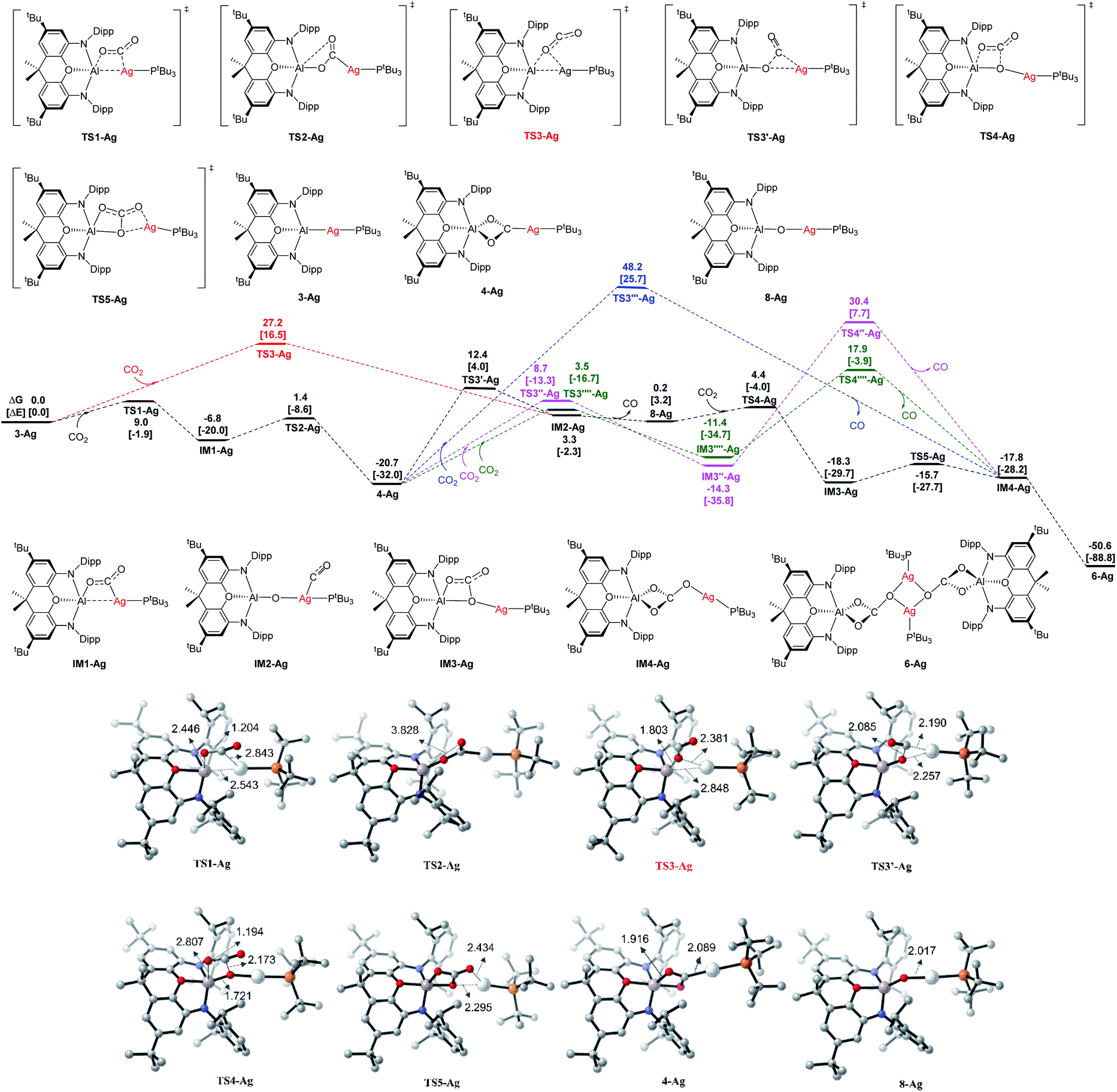 | ||
| Fig. 10 Computed Gibbs energy profiles (in kcal mol−1) for Ag at the BP86+D3(BJ)/def2-TZVPP (SMD, solvent = benzene)//BP86/def2-SVP level, and the electronic energies are given in bracket for reference. Other structures not shown along the reaction pathways are given in Fig. S18.† Key bond distances are given in Å for some optimized key intermediate and transition states. Color code, C: grey, N: blue, O: red, P: orange, Al: pink, Ag: white. | ||
The addition of one molecule of CO2 to 3-Ag takes place with very low activation energies via the intermediate IM1-Ag, giving the dioxocarbene complex 4-Ag as the first product. The calculations suggest that CO2 uptake starts with side-on [2 + 2] addition of one CO bond to the Al–Ag bond (with a regiochemistry reflecting polarization in the sense Ag(δ−)–Al(δ+)),25,35,43 and that the intermediate IM1-Ag then rearranges viaTS2-Ag to give dioxocarbene complex 4-Ag. From this species, the second part of the reaction sequence 4-Ag → 6-Ag proceeds via initial CO extrusion, yielding 8-Ag as an intermediate (featuring an Ag–O–Al unit), preceded by IM2-Ag as a weakly bonded silver–CO complex precursor. Subsequent reaction steps from 8-Ag have low activation barriers, leading first to IM3-Agvia uptake of a second CO2 molecule across the Ag–O bond, followed by isomerization to the monomer Im4-Ag, which then dimerizes to give 6-Ag as the final product.
The highest energy barrier for the overall reaction is the rearrangement/CO extrusion step from 4-Ag to IM2-AgviaTS3′-Ag, which has a value of ΔG‡ = 33.1 kcal mol−1. The alternative one-step pathway 3-Ag → IM2-AgviaTS3-Ag has a lower overall barrier of ΔG‡ = 27.2 kcal mol−1, but the uptake of CO2 in the initial step, leading to 4-Ag is both kinetically and thermodynamically much more favorable. The compound 4-Ag is thus the energy reference species for the CO2 addition. The most favorable reaction pathway is given in black lines in Fig. 10. Energetically less favorable courses are shown in red, blue and green.
We also calculated the analogous reactions of CO2 with the copper and gold aluminyl complexes 3-Cu and 3-Au. The calculated energy profiles are shown in Fig. S19 and S20 of ESI.† The theoretical data suggest that the reaction sequence in the case of the copper species follows the analogous pathway from 3-Cu to 6-Cu as the silver species with a lower activation barrier for the rate determining step (4-Cu → IM2-CuviaTS3-Cu) of ΔG‡ = 27.4 kcal mol−1, which agrees with the experimental finding of much more facile carbonate formation in the case of copper. The calculations for the gold homologues give a similar reaction profile for the initial CO2 uptake leading to 4-Au, but the subsequent rearrangement has a very high barrier of ΔG‡ = 44.5 kcal mol−1 with concomitant loss of CO leading to IM3-Au (Fig. S20†). This finding is also in agreement with the experimental results, i.e. with the fact that 4-Au can be isolated without complications arising from further onward reactivity.
Conclusions
The reactions of group 11 metal aluminyl compounds of the type (tBu3P)MAl(NON) (M = Cu, Ag, Au) with carbon dioxide and with carbodiimides are shown to proceed via insertion into the polar metal-aluminium bonds to yield species featuring μ-κ1(C):κ2(E,E′) bridging units and M–C/Al–E bonds, and which can be thought of as aluminium-functionalized carbene adducts of the respective coinage metals. While the products of carbodiimide insertion, (tBu3P)M{C(NR)2}Al(NON), are stable to further reaction in the presence of excess of the heteroallene (and have been structurally characterized for all three metals), the corresponding CO2 insertion products are labile to an extent dependent on the nature of M. Thus, (tBu3P)Au(CO2)Al(NON) is inert to further reaction under all conditions examined (and can be structurally characterized), while (tBu3P)Ag(CO2)Al(NON) can be generated at room temperature but slowly converts into the corresponding carbonate (tBu3P)Ag(CO3)Al(NON) (and CO), and (tBu3P)Cu(CO2)Al(NON) has been identified only in quantum chemical experiments as part of a facile route to (tBu3P)Cu(CO3)Al(NON). Both experimental and computational studies are consistent with a mechanism for the formation of the carbonate species which proceeds via rate-limiting extrusion of CO from (tBu3P)M(CO2)Al(NON) (M = Cu, Ag) to give a reactive oxide species containing a M–O–Al unit, which then rapidly assimilates further CO2. The comparative labilities of the species (tBu3P)M(CO2) Al(NON) (M = Cu, Ag, Au) can be quantified by activation energies of ΔG‡ = 26.6, 33.1, 44.5 kcal mol−1, respectively, for the most feasible CO extrusion step, and rationalized in turn on the basis of the relative carbophilicities of the coinage metals.41aData availability
See ESI for complete synthetic, quantum chemical and crystallographic details.†Author contributions
C. McManus: carried out the experimental investigation; J. Hicks carried out the experimental investigation; X. Cui: carried out the quantum chemical investigation; L. Zhao: conceptualization and supervision of quantum chemical investigation; G. Frenking: conceptualization and supervision of quantum chemical investigation; J. M. Goicoechea: conceptualization and supervision of experimental investigation; S. Aldridge: conceptualization and supervision of experimental investigation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Leverhulme Trust and the EPSRC for funding various aspects of this work. L. Z. and G. F. acknowledge financial support from Nanjing Tech University (grant numbers 39837123, 39837132) and SICAM Fellowship from Jiangsu National Synergetic Innovation Center for Advanced Materials, National Natural Science Foundation of China (grant number 21973044), and the high performance center of Nanjing Tech University for supporting the computational resources. GF is grateful to the Deutsche Forschungsgemeinschaft for financial support.Notes and references
- P. Buchwalter, J. Rosé and P. Braunstein, Chem. Rev., 2015, 115, 28 CrossRef CAS PubMed.
- N. Wheatley and P. Kalck, Chem. Rev., 2002, 99, 3379–3420 CrossRef PubMed.
- See, for example: (a) M. Delferro and T. J. Marks, Chem. Rev., 2011, 111, 2450–2485 CrossRef CAS PubMed; (b) B. G. Cooper, J. Napoline and C. M. Thomas, Catal. Rev.: Sci. Eng., 2012, 54, 1–40 CrossRef CAS; (c) J. Park and S. Hong, Chem. Soc. Rev., 2012, 41, 6931–6943 RSC; (d) N. P. Mankad, Chem.–Eur. J., 2016, 22, 5822–5829 CrossRef CAS PubMed; (e) D. R. Pye and N. P. Mankad, Chem. Sci., 2017, 8, 1705–1718 RSC; (f) R. C. Cammarota, L. J. Clouston and C. C. Lu, Coord. Chem. Rev., 2017, 334, 100–111 CrossRef CAS; (g) S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332–8405 CrossRef CAS PubMed.
- M. Herberhold and G.-X. Jin, Angew. Chem., Int. Ed. Engl., 1994, 33, 964–966 CrossRef.
- Z. Wei, A. S. Filatov and E. V. Dikarev, J. Am. Chem. Soc., 2013, 135, 12216–12219 CrossRef CAS PubMed.
- A. Navulla, A. A. Tsirlin, A. M. Abakumov, R. V. Shpanchenko, H. Zhang and E. V. Dikarev, J. Am. Chem. Soc., 2011, 133, 692–694 CrossRef CAS PubMed.
- B. D. Dhanapala, H. N. Munasinghe, L. Suescun and F. A. Rabuffetti, Inorg. Chem., 2017, 56, 13311–13320 CrossRef CAS PubMed.
- K. R. Margulieux, C. Sun, M. T. Kihara, A. C. Colson, L. N. Zakharov, K. H. Whitmire, A. W. Holland and J. J. Pak, Eur. J. Inorg. Chem., 2017, 2068–2077 CrossRef CAS.
- R. A. Collins, A. F. Russell and P. Mountford, Appl. Petrochem. Res., 2015, 5, 153–171 CrossRef CAS.
- See, for example: (a) J. Zhu, Z. Lin and T. B. Marder, Inorg. Chem., 2005, 44, 9384–9390 CrossRef CAS PubMed; (b) H. Niu, R. Mangan, A. V. Protchenko, N. Phillips, W. Unkrig, C. Friedmann, E. Kolychev, R. Tirfoin, J. Hicks and S. Aldridge, Dalton Trans., 2018, 47, 7445–7455 RSC.
- I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890–931 CrossRef CAS PubMed.
- H. Braunschweig, R. D. Dewhurst and A. Schneider, Chem. Rev., 2010, 110, 3924–3957 CrossRef CAS PubMed.
- (a) A. V. Protchenko, K. H. Birjkumar, D. Dange, A. D. Schwarz, D. Vidovic, C. Jones, N. Kaltsoyannis, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2012, 134, 6500–6503 CrossRef CAS PubMed; (b) A. V. Protchenko, J. I. Bates, L. M. A. Saleh, M. P. Blake, A. D. Schwarz, E. L. Kolychev, A. L. Thompson, C. Jones, P. Mountford and S. Aldridge, J. Am. Chem. Soc., 2016, 138, 4555–4564 CrossRef CAS PubMed.
- (a) D. S. Laitar, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2005, 127, 17196–17197 CrossRef CAS PubMed; (b) D. S. Laitar, E. Y. Tsui and J. P. Sadighi, J. Am. Chem. Soc., 2006, 128, 11036–11037 CrossRef CAS PubMed.
- M. Asay, C. Jones and M. Driess, Chem. Rev., 2011, 111, 354–396 CrossRef CAS PubMed.
- S. González-Gallardo, T. Bollermann, R. A. Fischer and R. Murugavel, Chem. Rev., 2012, 112, 3136–3170 CrossRef PubMed.
- Y. Segawa, M. Yamashita and K. Nozaki, Angew. Chem., Int. Ed., 2007, 46, 6710–6713 CrossRef CAS PubMed.
- S. P. Green, C. Jones, D. P. Mills and A. Stasch, Organometallics, 2007, 26, 3424–3430 CrossRef CAS.
- B. N. Anand, I. Krossing and H. Nöth, Inorg. Chem., 1997, 36, 1979–1981 CrossRef CAS PubMed.
- Q. Yu, A. Purath, A. Donchev and H. Schnöckel, J. Organomet. Chem., 1999, 584, 94–97 CrossRef CAS.
- D. Weiss, T. Steinke, M. Winter, R. A. Fischer, N. Fröhlich, J. Uddin and G. Frenking, Organometallics, 2000, 19, 4583–4588 CrossRef CAS.
- J. Hicks, A. Mansikkamäki, P. Vasko, J. M. Goicoechea and S. Aldridge, Nat. Chem., 2019, 11, 237–241 CrossRef CAS PubMed.
- (a) J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Nature, 2018, 557, 92–95 CrossRef CAS PubMed; (b) R. J. Schwamm, M. D. Anker, M. Lein and M. P. Coles, Angew. Chem., Int. Ed., 2019, 58, 1489–1493 CrossRef CAS PubMed; (c) R. J. Schwamm, M. P. Coles, M. S. Hill, M. F. Mahon, C. L. McMullin, N. A. Rajabi and A. S. S. Wilson, Angew. Chem., Int. Ed., 2020, 59, 3928–3932 CrossRef CAS PubMed; (d) J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, J. Am. Chem. Soc., 2019, 141, 11000–11003 CrossRef CAS PubMed; (e) S. Kurumada, S. Takamori and M. Yamashita, Nat. Chem., 2020, 12, 36–39 CrossRef CAS PubMed; (f) K. Koshino and R. Kinjo, J. Am. Chem. Soc., 2020, 142, 9057–9062 CrossRef CAS PubMed; (g) S. Grams, J. Eyselein, J. Langer, C. Färber and S. Harder, Angew. Chem., Int. Ed., 2020, 59, 15982–15986 CrossRef CAS PubMed; (h) J. Hicks, P. Vasko, J. M. Goicoechea and S. Aldridge, Angew. Chem., Int. Ed., 2021, 60, 1702–1713 CrossRef CAS PubMed.
- K. Sugita and M. Yamashita, Chem.–Eur. J., 2020, 26, 4520–4523 CrossRef CAS PubMed.
- H. Y. Liu, R. J. Schwamm, M. S. Hill, M. F. Mahon, C. F. McMullin and N. A. Rajabi, Angew. Chem., Int. Ed., 2021, 60, 14390–14393 CrossRef CAS PubMed.
- (a) J. R. Pinkes, B. D. Steffey, J. C. Vites and A. R. Cutler, Organometallics, 1994, 13, 21–23 CrossRef CAS; (b) J. Li, M. Hermann, G. Frenking and C. Jones, Angew. Chem., Int. Ed., 2012, 51, 8611–8614 CrossRef CAS PubMed.
- (a) M. Hirano, M. Akita, K. Tani, K. Kumagai, N. C. Kasuga, A. Fukuoka and S. Komiya, Organometallics, 1997, 16, 4206–4213 CrossRef CAS; (b) C. Yoo and Y. Lee, Chem. Sci., 2016, 8, 600–605 RSC.
- J. Mascetti, Carbon Dioxide Coordination Chemistry and Reactivity of Coordinated CO2, in Carbon Dioxide as Chemical Feedstock, ed. M. Aresta, Wiley-VCH, Weinheim, 2010, pp. 55–88 Search PubMed.
- See, for example: P. P. Power, K. Ruhlandt-Senge and S. C. Shoner, Inorg. Chem., 1991, 30, 5013–5015 CrossRef CAS.
- L. Arnberg and S. Westman, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1978, 34, 399–404 CrossRef.
- K. L. Mears, C. R. Stennett, E. K. Taskinen, C. E. Knapp, C. J. Carmalt, H. M. Tuononen and P. P. Power, J. Am. Chem. Soc., 2020, 142, 19874–19878 CrossRef CAS PubMed.
- L. Pauling, The Nature of the Chemical Bond, Cornell University Press, Ithaca, NY, 3rd edn, 1960 Search PubMed.
- For example, complexes of the type [(tBu3P)2M]+: (a) (Au, δP = +97.0 ppm) P. Diversi, A. Cuzzola and F. Ghiotto, Eur. J. Inorg. Chem., 2009, 4, 545–553 CrossRef; (b) (Ag, δP = +80.0 ppm) R. P. Goel and P. Pilon, Inorg. Chem., 1978, 17, 2876–2879 CrossRef CAS; (c) (Cu, δP = +58.6 ppm) R. P. Goel and A. L. Beauchamp, Inorg. Chem., 1983, 22, 395–400 CrossRef CAS.
- (a) C. Gibard, K. Fauché, R. Guillot, L. Jouffret, M. Traïkia, A. Gautier and F. Cisnetti, J. Organomet. Chem., 2017, 840, 70–74 CrossRef CAS; (b) X. Liu and P. Braunstein, Inorg. Chem., 2013, 52, 7367–7379 CrossRef CAS PubMed; (c) T. Simler, P. Braunstein and A. A. Danopoulos, Dalton Trans., 2016, 45, 5122 RSC.
- For other recent examples, see: (a) A. Suzuki, X. Guo, Z. Lin and M. Yamashita, Chem. Sci., 2021, 12, 917–928 RSC; (b) L. Escomel, I. Del Rosal, L. Maron, E. Jeanneau, L. Veyre, C. Thieuleux and C. Camp, J. Am. Chem. Soc., 2021, 143, 4844–4856 CrossRef CAS PubMed.
- J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef CAS PubMed.
- B. Cordero, V. Gómez, A. E. Platero-Prats, M. Revés, J. Echeverría, E. Cremades, F. Barragán and S. Alvarez, Dalton Trans., 2008, 2832–2838 RSC.
- D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–92 CrossRef CAS PubMed.
- (a) F. Lazreg, A. M. Z. Slawin and C. S. J. Cazin, Organometallics, 2012, 31, 7969–7975 CrossRef CAS; (b) C. Gibard, K. Fauché, R. Guillot, L. Jouffret, M. Traïkia, A. Gautier and F. Cisnetti, J. Organomet. Chem., 2017, 840, 70–74 CrossRef CAS; (c) S. Gaillard, P. Nun, A. M. Z. Slawin and S. P. Nolan, Organometallics, 2010, 29, 5402–5408 CrossRef CAS.
- E. Despagnet-Ayoub and R. H. Grubbs, J. Am. Chem. Soc., 2004, 126, 10198–10199 CrossRef CAS PubMed.
- (a) C. Boehme and G. Frenking, Organometallics, 1998, 17, 5801–5809 CrossRef CAS; (b) K. A. Moltved and K. P. Kepp, J. Phys. Chem. C, 2019, 123, 18432–18444 CrossRef CAS.
- For a similar series of reactions leading to the formation of CO and [CO3]2− from CO2 using a wholly main group bimetallic system see R. Lalrempuia, A. Stasch and C. Jones, Chem. Sci., 2013, 4, 4383–4388 RSC.
- For a very recent computational study of one of the systems presented here see: D. Sorbelli, L. Belpassi and P. Belanzoni, J. Am. Chem. Soc., 2021 DOI:10.1021/jacs.1c06728 , ASAP. These authors suggest the presence of a Al–Au covalent bond in compound V which is most easily fragmented homolytically (in accordance with findings reported previously in ref. 22), and calculate a potential energy profile for the reaction with CO2 which is very similar to that provided in Fig. S20.†.
Footnote |
| † Electronic supplementary information (ESI) available: Complete synthetic and characterizing data for new compounds; crystallographic data (including CIFs); details of quantum chemical studies. CCDC 2085387–2085394. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1sc04676d |
| This journal is © The Royal Society of Chemistry 2021 |