
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSoluble, crystalline, and thermally stable alkali CO2− and carbonite (CO22−) clusters supported by cyclic(alkyl)(amino) carbenes†
Lucas A.
Freeman
 a,
Akachukwu D.
Obi
a,
Haleigh R.
Machost
a,
Andrew
Molino
b,
Asa W.
Nichols
a,
Diane A.
Dickie
a,
David J. D.
Wilson
*b,
Charles W.
Machan
*a and
Robert J.
Gilliard
Jr
*a
a,
Akachukwu D.
Obi
a,
Haleigh R.
Machost
a,
Andrew
Molino
b,
Asa W.
Nichols
a,
Diane A.
Dickie
a,
David J. D.
Wilson
*b,
Charles W.
Machan
*a and
Robert J.
Gilliard
Jr
*a
aDepartment of Chemistry, University of Virginia, 409 McCormick Road, PO Box 400319, Charlottesville, Virginia 22904, USA. E-mail: machan@virginia.edu; rjg8s@virginia.edu
bDepartment of Chemistry and Physics, La Trobe Institute for Molecular Science, La Trobe University, Bundoora, Victoria 3086, Australia. E-mail: David.Wilson@latrobe.edu.au
First published on 22nd January 2021
Abstract
The mono- and dianions of CO2 (i.e., CO2− and CO22−) have been studied for decades as both fundamentally important oxycarbanions (anions containing only C and O atoms) and as critical species in CO2 reduction and fixation chemistry. However, CO2 anions are highly unstable and difficult to study. As such, examples of stable compounds containing these ions are extremely limited; the unadulterated alkali salts of CO2 (i.e., MCO2, M2CO2, M = alkali metal) decompose rapidly above 15 K, for example. Herein we report the chemical reduction of a cyclic (alkyl)(amino) carbene (CAAC) adduct of CO2 at room temperature by alkali metals, which results in the formation of CAAC-stabilized alkali CO2− and CO22− clusters. One-electron reduction of CAAC–CO2 adduct (1) with lithium, sodium or potassium metal yields stable monoanionic radicals [M(CAAC–CO2)]n (M = Li, Na, K, 2–4) analogous to the alkali CO2− radical, and two-electron alkali metal reduction affords dianionic clusters of the general formula [M2(CAAC–CO2)]n (5–8) with reduced CO2 units which are structurally analogous to the carbonite anion CO22−. It is notable that crystalline clusters of these alkali–CO2 salts may also be isolated via the “one-pot” reaction of free CO2 with free CAAC followed by the addition of alkali metals – a process which does not occur in the absence of carbene. Each of the products 2–8 was investigated using a combination of experimental and theoretical methods.
Introduction
Oxycarbanions, polyatomic ions of the general formula [CxOy]z−, are some of the most fundamentally important chemical entities in chemistry, with applications ranging from organic synthesis to battery technologies.1 Among the most important oxycarbanions are the anions of carbon dioxide, CO2− and CO22− (i.e., carbonite), due to their critical relevance in CO2 conversion chemistry and chemical synthesis. Due the high instability of CO2− and CO22−, stable compounds featuring these moieties are exceedingly rare, and typically involve metalation of the reactive anion to stabilize the electron-rich CO2 fragment.2 In contrast to the extremely stable alkali carbonates (M2CO3 M = alkali metal), alkali salts of carbon dioxide (i.e. MCO2 and M2CO2) formed via the reduction of CO2(g) by alkali metals are highly reactive species that have only been detected under specialized conditions. The Li, Na, K, and Cs salts of CO2− and CO22− were previously detected using matrix isolation IR spectroscopy, where they were observed to decompose to more stable alkali oxalates (M2C2O4) at temperatures above 15 K (Fig. 1, top).3 We hypothesized that to synthesize isolable alkali–CO2 compounds containing CO2− and CO22−, the metalloxycarbene resonance forms of the anions could be stabilized using a singlet carbene (Fig. 1, bottom).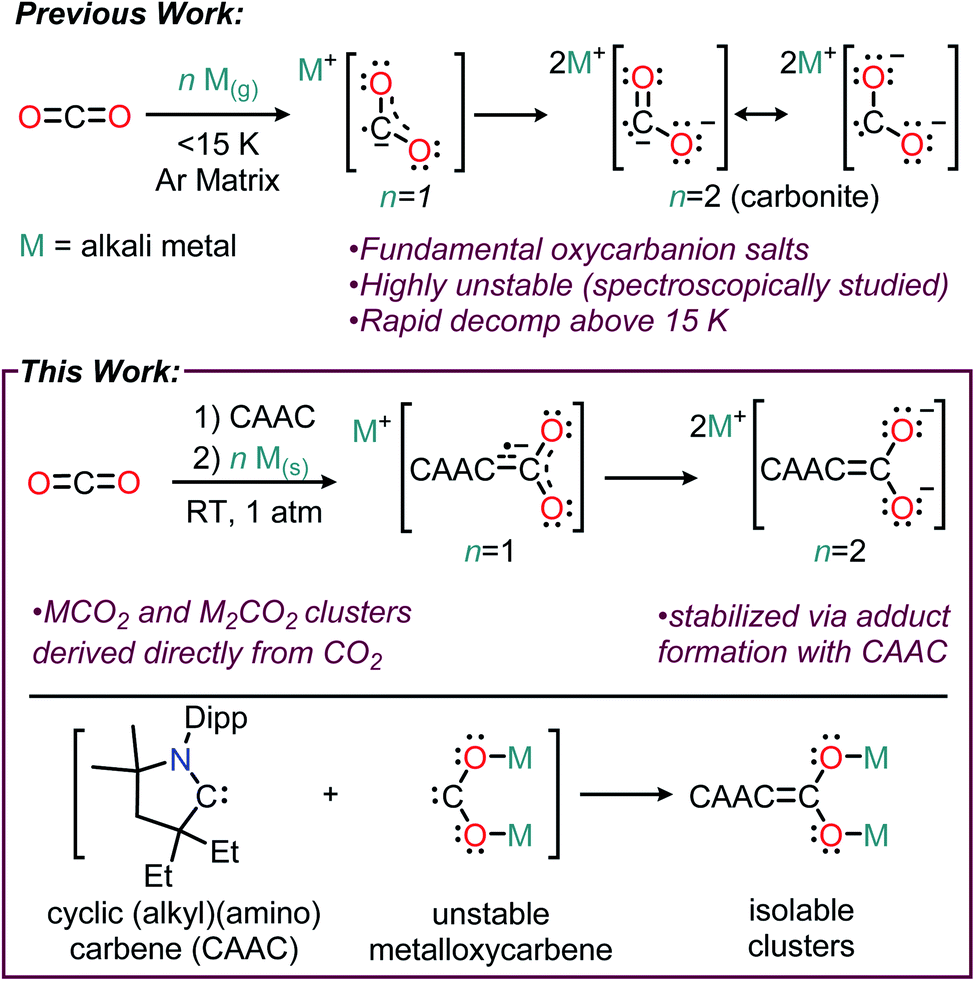 | ||
| Fig. 1 Previous methods used to study highly reactive alkali–CO2 salts (top). Method of synthesizing stable alkali CO2 salts utilizing carbene stabilization reported herein (bottom); Dipp = 2,6-diisopropylphenyl. | ||
Since the isolation of stable N-heterocyclic carbenes (NHCs) by Arduengo,4 and cyclic (alkyl)(amino)carbenes (CAACs) by Bertrand,5 these ligands have had a profound impact on synthetic chemistry.6 Notably, the ambiphilic nature of stable singlet carbenes has allowed the development of a rich body of organic chemistry which is distinct from their utility in metal-based systems.7 In such transformations, a common reaction step involves the carbene functioning as a neutral carbon nucleophile with the desired electrophilic substrate(s). A significant example of this class of reaction is the behavior of free carbenes in the presence of CO2, where the carbene lone pair forms a covalent bond with the electrophilic carbon atom of CO2. This results in the formation of highly stable zwitterionic adducts (i.e. N,N′-disubstituted imidazolium-2-carboxylates or, more generally, carbene-2-carboxylates). It is noteworthy that although CO2 is estimated to be a comparable electrophile to benzaldehyde, similar products of electrophilic reactivity are generally thermodynamically unfavored.8 These adducts have become an important subclass of “masked” carbene due to their high air and moisture stability compared to the free carbene.9 Indeed, a substantial number of imidazolium-2-carboxylate compounds are known, and their reactivity has been studied with organic nucleophiles,7a organic electrophiles,10 and organometallic complexes {e.g. [Rh(COD)Cl]2}.9
Carbene-carboxylates have a distinct predisposition toward energy-relevant applications in CO2 reduction and conversion chemistry, and a number of reported studies have focused on functionalizing or selectively transferring the carboxylate motif.7a,11 However, the ability of carbene-carboxylates to participate in direct electron transfer reactions with reducing metals (e.g., the alkali elements) in order to access stable compounds with CO2 anions is hitherto unknown. To begin studying the potential redox chemistry of CAAC–CO2 adducts, we published the first evidence that carbene–CO2 adducts display redox activity under electrochemically reducing conditions.11d In this initial report, we studied the electrochemical reduction of a CAAC–CO2 adduct under both argon and CO2 saturation conditions, and found spectroscopic evidence of reductive chemistry centered around the CAAC–CO2 species at potentials where free CO2 is inert. Most importantly, reduction of CAAC–CO2 under argon saturation showed that the CAAC–CO2 adduct is not only able to readily accept electrons, but that this electron transfer is a reversible redox couple on the CV timescale at −2.15 V (vs. Fc+/Fc). This promising result led us to investigate the ability of carbenes to induce the reaction of CO2 with main-group elements under mild conditions.
Herein we report that CAAC adducts of CO2 can undergo a facile reaction with alkali metals at ambient temperature and pressure. A diverse array of both singly-reduced [(THF)3Li2(CAAC–CO2)2 (2), (THF)4Na4(CAAC–CO2)4 (3), and (THF)4K4(CAAC–CO2)4 (4)] and doubly-reduced [(THF)2Li6(CAAC–CO2)3 (5), Li12(CAAC–CO2)6 (6), Na12(CAAC–CO2)6 (7), and K10(CAAC–CO2)5 (8)] CAAC–CO2 complexes have been synthesized. These compounds may also be prepared via the one-pot reaction of free CO2, alkali metal, and free CAAC. Compounds 2–8 were isolated as structurally diverse organoalkali metal clusters, which exhibited high stability and solubility at room temperature in both polar and non-polar solvents. The reaction sequence consists of the carboxylation of a carbene at room temperature and 1 atm of pressure, followed by the stepwise reduction of CAAC-carboxylate to a CAAC-diolate dianion using two electrons from alkali metals. In contrast to established CO2 reduction reactions which require anionic organic nucleophiles and alkali elements to reduce CO2,12 the reactions reported herein are the first examples of a reductive synthetic protocol which results in the cleavage of CO2 π-bonds at atmospheric pressures and room temperature by elemental alkali metals without using carbanions, organometallic reagents, or catalysts.
Results and discussion
We selected the (diethylCAAC)-2-carboxylate zwitterion, 1, as the subject of this synthetic investigation due to the improved solubility and ease of preparation compared to the spirocyclic (cyclohexylCAAC)-2-carboxylate we previously reported.11d Compound 1 possesses an isostructural CAAC-carboxylate core geometry in the solid-state molecular structure (Fig. S1†) along with nearly identical electronic properties.Our initial studies focused on the isolation and structural characterization of the chemical reduction products resulting from the reaction of 1 with Li, Na, and K. Upon the addition of one equivalent of metal shavings (or KC8 in the case of 4) to a vigorously stirring solution of 1, an immediate coloring of the mixture was observed (Li, Na, or K reducing agent yielded bright red, orange, or red colors, respectively). With vigorous stirring under inert conditions, the time necessary for complete consumption of the metal shavings scaled with the relative activity of the alkali metals (Li: 36 h; Na: 16 h; K: 6 h). The isolation of the products of these reactions via recrystallization from saturated THF or THF/hexane solutions at −39 °C yielded single crystals suitable for X-ray diffraction studies. The crystallographically determined structures of the monoanionic products (THF)3Li2(CAAC–CO2)2 (2), (THF)4Na4(CAAC–CO2)4 (3), and (THF)4K4(CAAC–CO2)4 (4) (Scheme 1, Dipp = 2,6-diisopropylphenyl) revealed a series of ion-contacted clusters incorporating the respective alkali metal cations in a CAAC–CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :M ratio of 1:1 (M = Li, Na, or K).
:M ratio of 1:1 (M = Li, Na, or K).
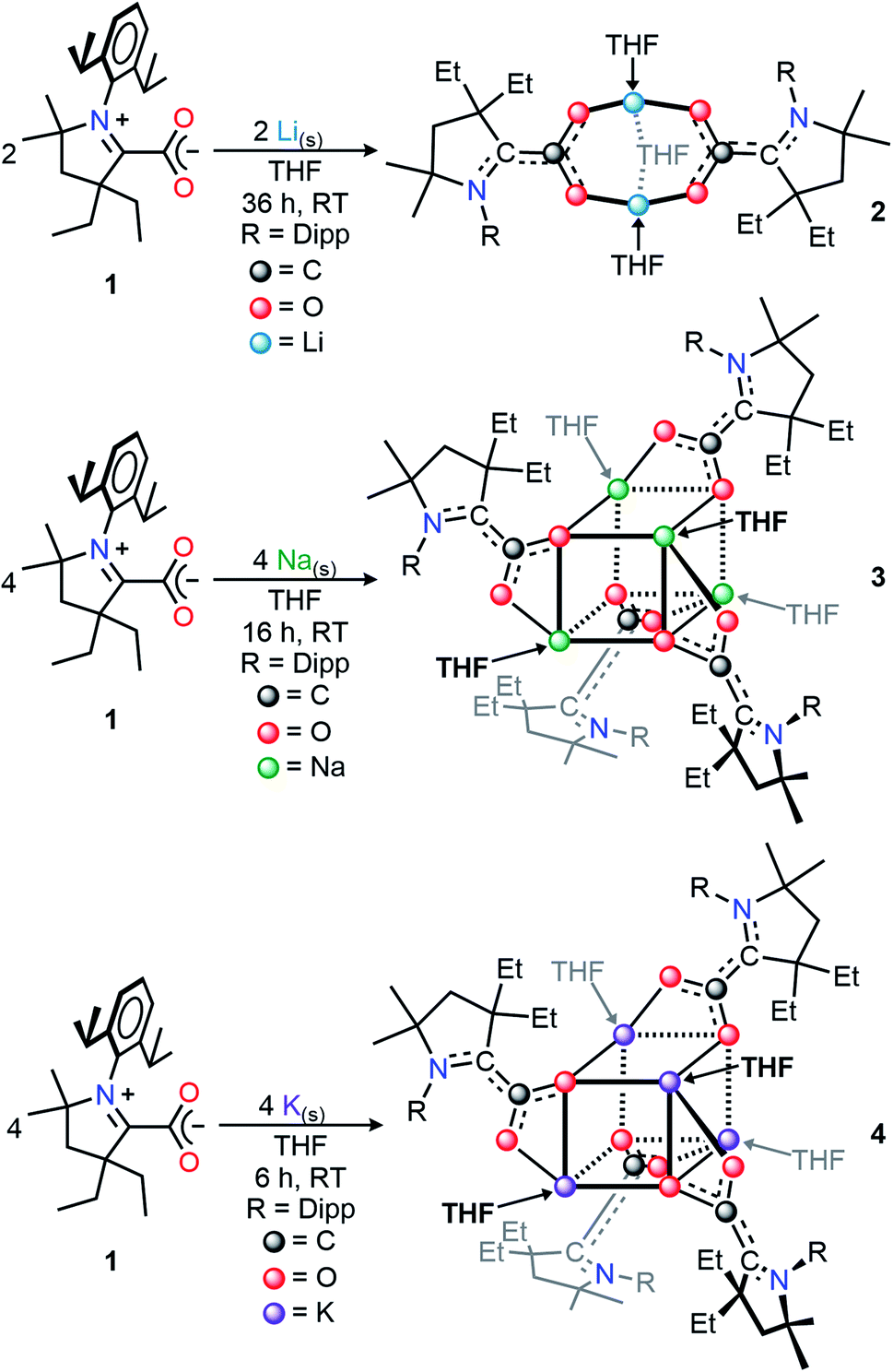 | ||
| Scheme 1 Synthesis of singly-reduced CAAC–CO2 compounds 2–4. | ||
In all three cases, precise control of reaction stoichiometry and times were critical, as reactions were frequently complicated by the presence of either residual CAAC–CO2 starting material which co-crystallizes with the desired products, or overreduction to a doubly-reduced complex.
Single crystals of compounds 2–4 suitable for X-ray diffraction studies were grown from saturated solutions of the complexes in either THF (3) or saturated THF/hexane mixtures (2, 4). Interestingly, the CAAC–CO2 core is completely planar in each singly-reduced species, which matches the predictions of our previously reported DFT calculations.11d The N2–C2 bonds [1.3802(12), 1.385(3), and 1.384(6) Å for 2–4 respectively] and C1–O bonds [1.2732(12) Å, 2; 1.286(3) Å, 3; 1.276(6), 4] in each species are significantly longer than those of the neutral zwitterion [C1–N2 = 1.282(5) Å, C1–O1 = 1.236(5) Å]. Concomitantly, there is a substantial contraction of the C1–C2 bond from 1.516(5) Å in 1 to 1.4533(13) Å, 1.458(3) Å, and 1.451(7) Å in 2–4, respectively. These changes are consistent with the addition of one electron from the alkali metal into a π-symmetric molecular orbital, increasing the bonding character between C1 and C2, and decreasing the bond order of the C2–N1, C1–O1, and C1–O2 bonds. Notably, the CAAC nitrogen atom remains planar in its geometry, indicating that the Nnon-bonding electrons are still involved in a π-symmetric interaction with the C2 center. The cumulative effects of these bonding metrics explain the observed planarity across the CAAC–CO2 core. It is worth noting that the solid-state structures of 2–4 exhibit similar geometries and binding modes to those known for isoelectronic metal-carbamate compounds (R2NCO2M2)n.13 For example, in the case of the lithium carbamate [(TMPCO2)Li·TMEDA]2 (TMP = 2,2,6,6-tetramethylpiperidide), similar binding and C–O bond lengths [1.261(2) and 1.255(2) Å]14 were observed compared to those in 2 [1.2732(12) and 1.2778(12) Å] (Fig. 2).
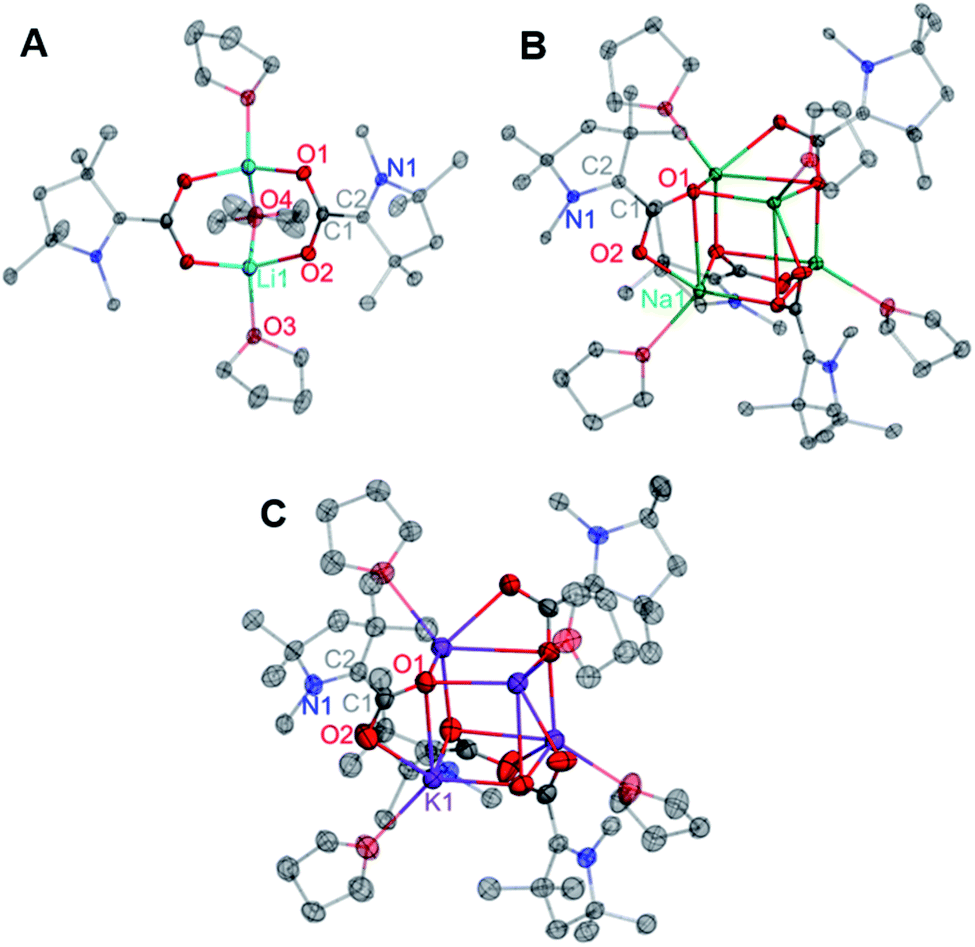 | ||
| Fig. 2 Solid state molecular structures of 2 (A), 3 (B), and 4 (C). All H atoms and carbon atoms not directly attached to the CAAC core ring are omitted for clarity. Selected bond distances (Å) and angles (degree): 2: O1–C1: 1.2732(12); O2–C1: 1.2778(12); C1–C2: 1.4533(13); N1–C2: 1.3802(12); O1–C1–O2: 123.71(9); O1–C1–C2: 119.84(9); O2–C1–C2: 116.45(9); N1–C2–C1: 124.85(9); N1–C2–C3: 110.81(8). 3: O1–C1: 1.286(3); O2–C1: 1.266(3); C1–C2: 1.458(3); N1–C2: 1.385(3); O2–C1–O1: 122.9(2); O2–C1–C2: 118.7(2); O1–C1–C2: 118.3(2); N1–C2–C1: 124.7(2); N1–C2–C3: 109.7(2). 4: O1–C1: 1.276(6); O2–C1: 1.265(7); C1–C2: 1.451(7); N1–C2: 1.384(6); O2–C1–O1: 123.0(5); O2–C1–C2: 119.1(5); O1–C1–C2: 117.9(5); N1–C2–C1: 123.3(5); N1–C2–C3: 110.2(4). | ||
In order to further probe the electronic structure of the reduced CAAC–CO2 species, CW X-band EPR data were collected for compounds 2–4 in toluene solutions at RT (Fig. 3). In spite of the differences in their solid-state molecular structures, the EPR spectra were all nearly identical exhibiting weak splitting by two 13C nuclei, with little to no observable 14N interactions.
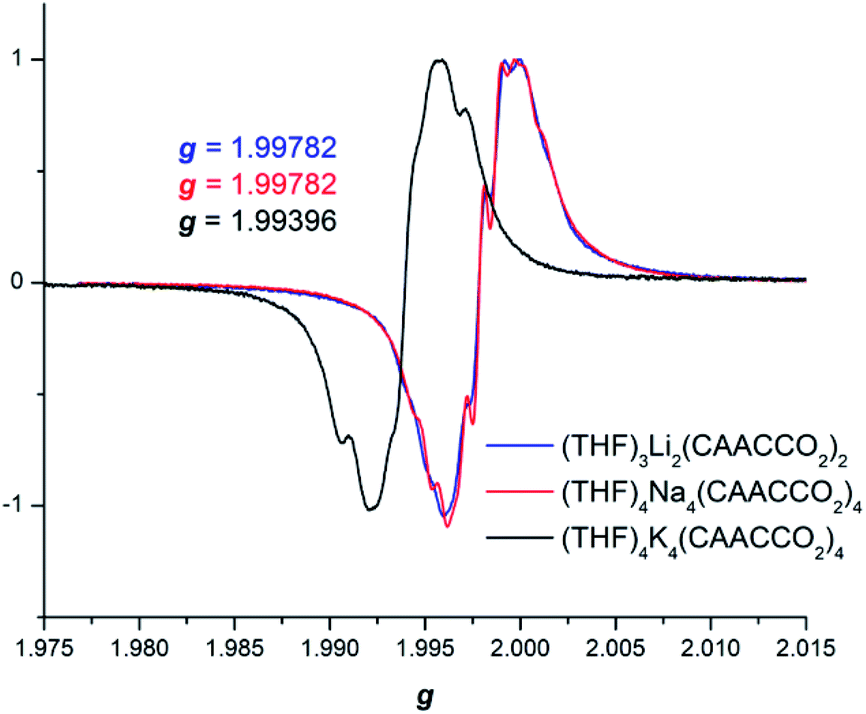 | ||
| Fig. 3 Continuous wave X-band EPR spectra of 2–4 taken on a 250 μM toluene solution at RT. | ||
Theoretical calculations were carried out for compounds 2–4, with geometries optimized using a QM/MM approach including a solvent model for THF (see ESI†). Key bond distances and angles of the QM domain are consistent with SC-XRD studies (C1–C2 1.447, 1.469, 1.471 Å for 2–4, respectively). The CAAC–CO2 core is planar, with optimized N1–C2–C1–O1 dihedral angles of 5.4°, 3.6°, and 2.6° for compounds 2–4, respectively, with NCAAC being planar in all three singly-reduced compounds.
B3LYP-D3(BJ)/def2-SVP (THF solvent) calculations were carried out at the QM/MM optimized geometries for the full complexes. Molecular orbital (MO) plots of 2–4 indicate that the singly-occupied MO (SOMO) largely resides on the carbon atoms that bond the CO2 and carbene units while the LUMO is concentrated on the Dipp substituents of CAAC (Fig. 4). The calculated spin density (see ESI Table S3, Fig. S24†) is mostly located on CCAAC, with smaller contributions from CCO2, NCAAC, and OCO2. X-band EPR simulations of 2–4 were found to be in good agreement with experiment (see ESI†). Calculated hyperfine coupling constants (aiso) were notably weaker for NCAAC relative to CCAAC (2 NCAACaiso = 5.55 MHz, CCAACaiso = 26.36 MHz), indicating that spin density is localized to the CCAAC.
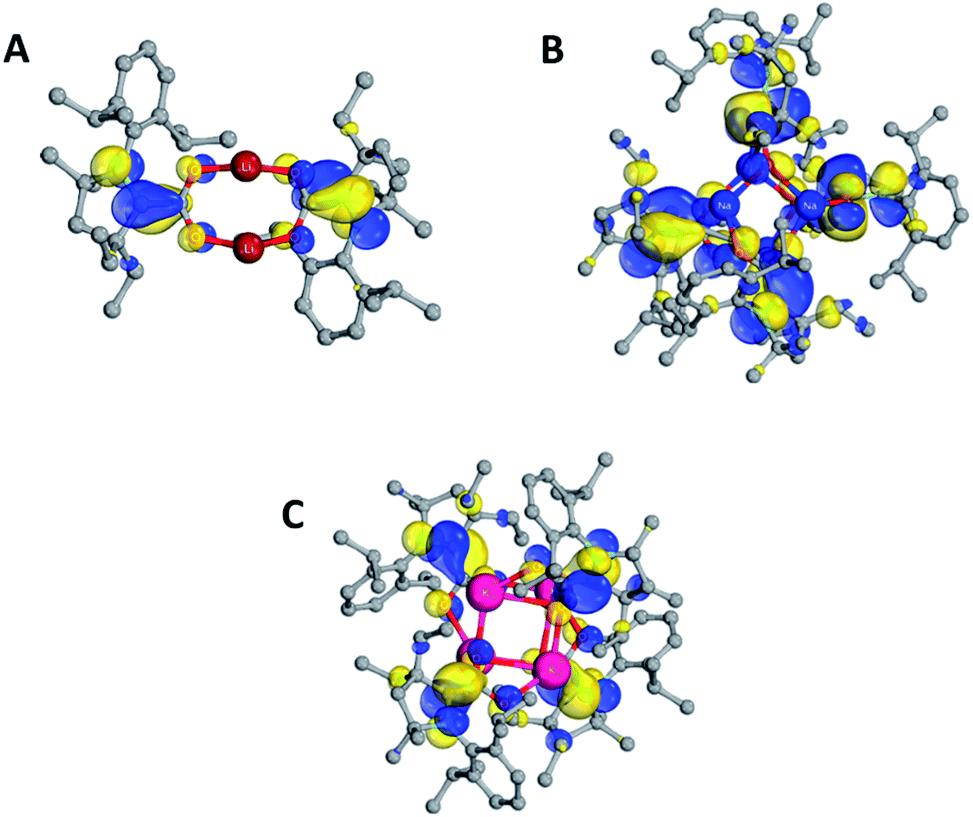 | ||
| Fig. 4 Plots of the SOMOs of 2 (A), 3 (B), and 4 (C). H atoms and THF are omitted for clarity. | ||
The addition of two or more equivalents of metal to a THF suspension of 1 gave gradual conversion (Li: 3 d, Na: 16 h, K: 6 h) to deeply colored, strongly absorbing (i.e., relatively high absorptivity values), homogeneous mixtures containing new reduced species. Completely evaporating these solutions under reduced pressure, followed by extraction and recrystallization in hexanes produced single crystals suitable for X-ray diffraction studies. These revealed the respective solid-state structures of the doubly-reduced complexes (THF)2Li6(CAAC–CO2)3 (5), Li12(CAAC–CO2)6 (6), Na12(CAAC–CO2)6 (7), K10(CAAC–CO2)5 (8) in high yields (Li: 70%;15 Na: 86%; K: 96%), which were observed to be highly stable, crystalline, analytically pure solids at room temperature under inert atmosphere (Scheme 2). Interestingly, as was observed in compounds 2–4, the identity of the alkali metal used for reduction had a dramatic effect on reaction time, color, and solid-state structure of the products. This behavior corroborates the observations reported in other investigations of alkali metal reductions which have recently been reviewed.16
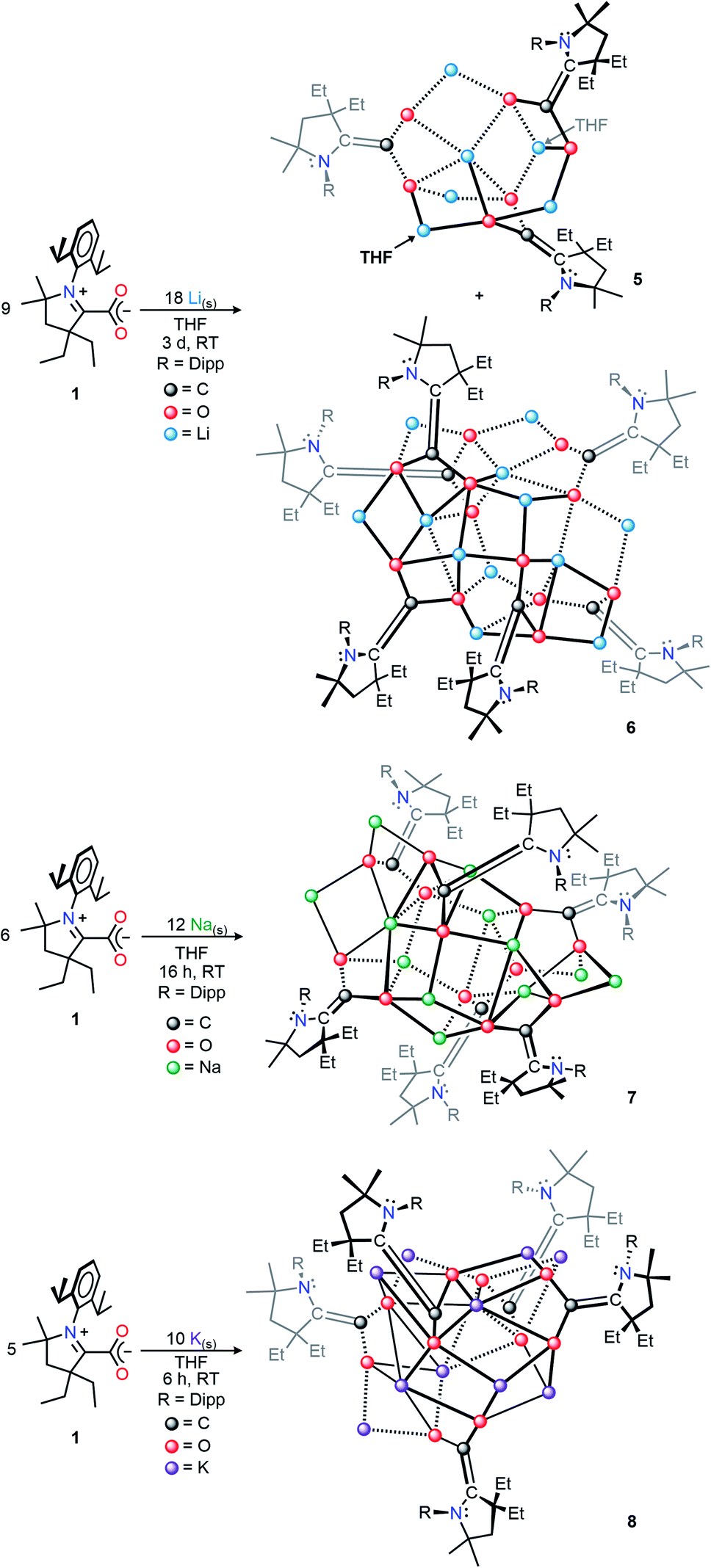 | ||
| Scheme 2 Synthesis of doubly-reduced CAAC–CO2 compounds 5–8. | ||
Notably, when lithium metal is used, the complete removal of THF from the crude reaction residue proved difficult. Even after multiple triturations with hexanes and prolonged drying under reduced pressure, a sticky red-orange semi-solid was obtained. Upon recrystallization of the highly soluble, red-orange bulk material from hexanes, two distinguishable crystalline species – one dark red and one bright yellow – were observed which crystallized from the same solution (Fig. 5A). Surprisingly, similar behavior was not observed when Na or K was used, for which only one crystalline product was obtained in multiple trials [Fig. 5B (Na) and Fig. 5C (K)].
 | ||
| Fig. 5 Digital image of the mixture of compounds 5 (red crystals) and 6 (yellow crystals) (A), 7 (B), and 8 (C) viewed at 40× magnification on an optical microscope. | ||
From the mixture of crystals obtained from the Li reduction, two distinct dilithiated CAAC–CO2 clusters (5, Fig. 6A; 6, Fig. 6B) were structurally characterized. Despite exhibiting drastically different crystal habits and colors, both compounds contained an equal ratio of lithium ions to CAAC–CO2 units (2:1). However, the size of the cluster varies. Compound 5 can be described as a hexanuclear cluster in the solid state, while compound 6 crystallizes as a dodecanuclear cluster. Also, compound 5 is the only doubly-reduced species observed in this study which exhibited THF coordination in the solid-state. Combustion microanalysis performed on a bulk sample of this mixture more closely matched the calculated CHN content for 5, suggesting this is the major species present in the bulk material.
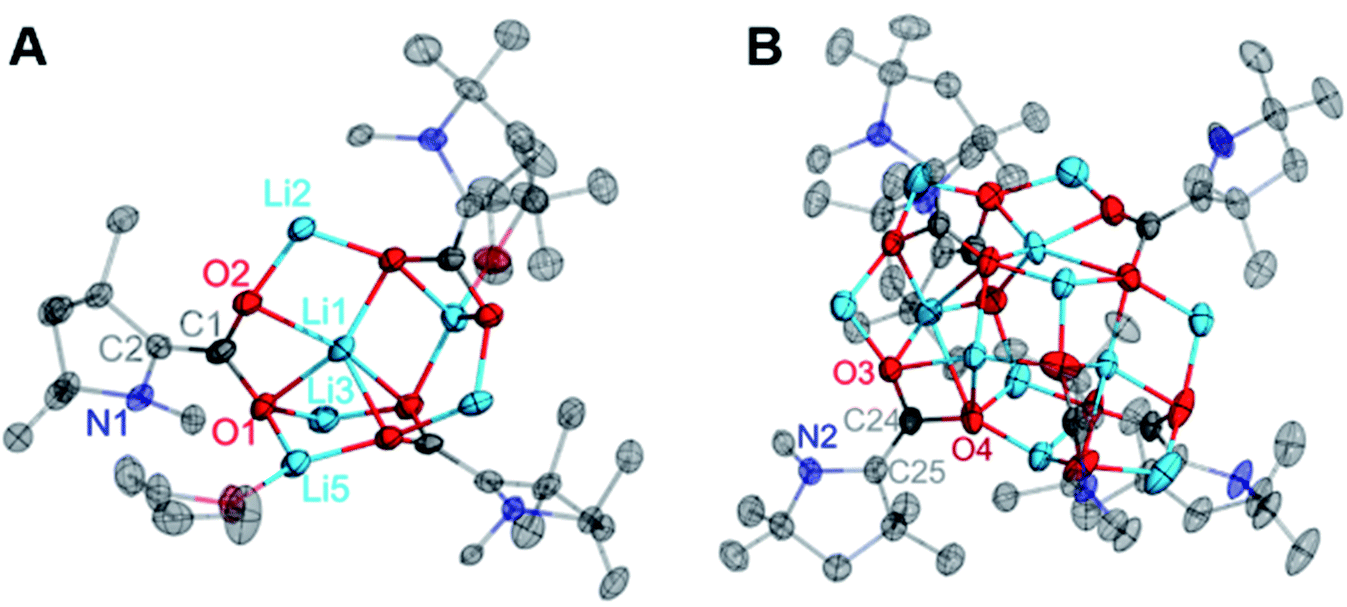 | ||
| Fig. 6 Solid state molecular structure of 5 (A) and 6 (B). All H atoms and carbon atoms not directly attached to the CAAC core ring are omitted for clarity. Selected bond distances (Å) and angles (degree): 5: O1–C1: 1.354(7); O2–C1: 1.332(7); C1–C2: 1.379(8); N1–C2: 1.463(7); O2–C1–O1: 114.1(5); O1–C1–C2: 121.2(5); O2–C1–C2: 124.6(5); C1–C2–N1: 120.0(5); N1–C2–C3: 109.6(5). 6: O1–C1: 1.365(2); O2–C1: 1.360(2); C1–C2: 1.343(3); N1–C2: 1.452(2); O2–C1–O1: 114.20(15); C2–C1–O1: 120.63(16); C2–C1–O2: 125.16(16); C1–C2–N1: 121.20(16); N1–C2–C3: 110.58(15). | ||
As in the cases of the singly-reduced species, the structural features of the CAAC–CO2 core yield information about the extent of reduction. Perhaps the most notable structural change between 2 and 5/6 is the pyramidal geometry of the CAAC nitrogen atom in the doubly-reduced complexes. This geometric change, along with a shortening of the C1–C2 bonds from 1.4533(13) Å in 2 to 1.379(8) Å (5) and 1.343(3) Å (6) clearly supports the existence of a nonbonding lone pair on the NCAAC atom and a formal π-bond between C1 and C2. Moreover, the C1–O1 and C1–O2 bond distances in 5 [1.354(7) Å, 1.332(7) Å] and 6 [1.365(2) Å, 1.360(2) Å] indicate C–O bond orders of 1. The only major structural differences between 5 and 6 are in the nature of the non-covalent ion contacts between the electron-rich π-system of the CAAC–CO2 core and nearby lithium cations. Perturbations in the π–π* energy gap by these nearby charges presumably lead to the observed differences in the absorption of visible light by complexes 5 and 6 (UV-Vis spectroscopic data for the bulk mixture are given in the ESI†).
Similar reactions conducted with sodium and potassium yielded highly pure single crystals suitable for X-ray diffraction studies from a saturated hexane extract of the crude product mixture. SC-XRD data collected on crystals of the Na and K products revealed the structures shown in Fig. 7A and B.
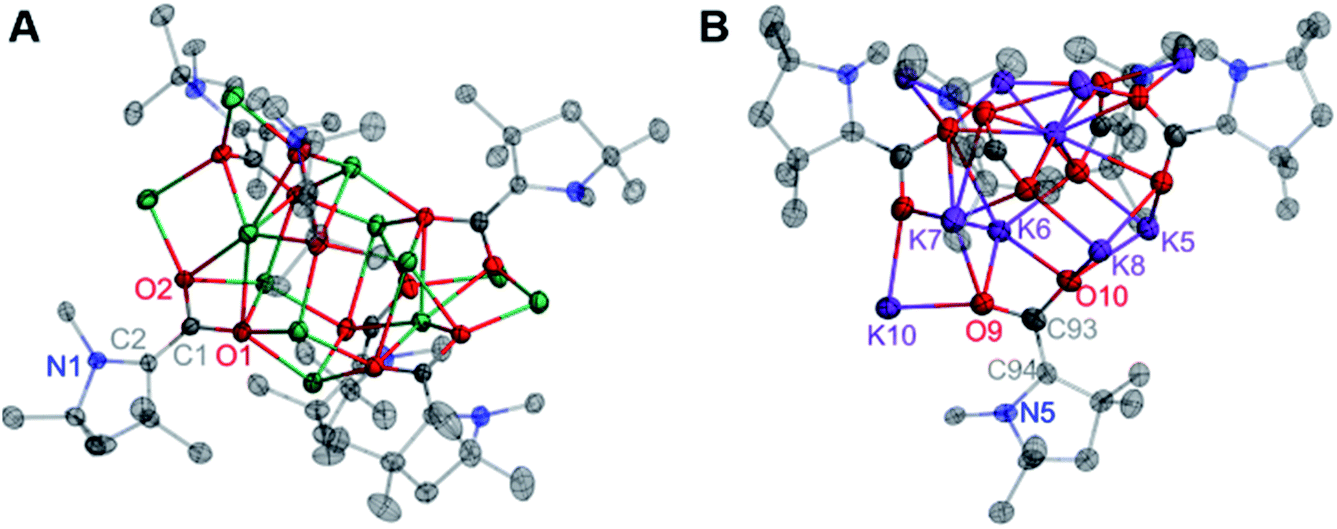 | ||
| Fig. 7 Solid state molecular structures of 7 (A) and 8 (B). All H atoms and all carbon atoms not directly attached to the CAAC core ring are omitted for clarity. All H atoms and carbon atoms not directly attached to the CAAC core ring are omitted for clarity. Selected bond distances (Å) and angles (degree): 7: O1–C1: 1.364(4); O2–C1: 1.336(4); C1–C2: 1.365(5); N1–C2: 1.452(4); O2–C1–O1: 113.4(3); O1–C1–C2: 124.0(3); C1–C2–N1: 122.9(3); N1–C2–C3: 107.9(3). 8: O9–C93: 1.342(10); O10–C93: 1.304(9); C93–C94: 1.385(11); N5–C94: 1.455(9); O10–C93–O9: 115.8(7); O9–C93–C94: 120.0(7); C93–C94–N5: 119.5(6); N5–C94–C95: 108.7(6). | ||
The solid-state molecular structure of 7 shows an oblong Na12(CO2)6 core “capped” by six bound CAACs, which is pseudo-S4 symmetric (S4 axis containing Na2 and Na9). The ratio of Na ions to CAAC–CO2 units (2:1) and the bond distances across the CAAC–CO2 core [N1–C2: 1.452(4) Å; C1–C2: 1.365(5) Å; O1–C1: 1.364(4) Å; O2–C1: 1.336(4) Å] indicate that each unit is doubly-reduced, analogous to 5 and 6. This description is further supported by pyramidal geometries exhibited by all NCAAC atoms in the cluster. In the solid-state structure of 8, a C1-symmetric K10(CO2)5 core was observed. All of the core structural features noted for complexes 5–7 were also observed in 8, however all five Dipp substituents in this complex exhibited substantial K–(η6-Dipp) interactions with externally oriented K ions. These interactions were present in 7 but weak, and not observed in 5 or 6. Notably, the products 5–8 were observed to be NMR silent, and gave no signal in a parallel mode EPR experiment (which only detects species with S = 1/2, 3/2, …, etc.). These experimental data are consistent with the possibility of a significant population of an S = 1 (i.e. non-Kramers') paramagnetic electronic state for all doubly-reduced compounds, which is supported by computational analysis (vide infra) and indicates some degree of chemical fluxionality at room temperature.17
Computationally optimized geometries of 5–8 follow the trends observed of the crystal structures, with the optimized bond distances in general slightly greater than in the crystal structures. In all doubly-reduced species, the NCAAC is pyramidal and the C1–C2 bond distance is shorter than in the singly-reduced analogue. The pyramidal geometry of NCAAC atoms is suggestive of a non-bonding lone-pair on the nitrogen atom and a C1–C2 π-bond, which is supported by intrinsic bonding orbital (IBO) results (Fig. 8). The NCAAC atoms of the doubly-reduced species each possess a π lone pair and a N1–C2 σ bond. There is no N1–C2 π-bond observed in 5–8, unlike the singly-reduced species 2–4 (Fig. 8).
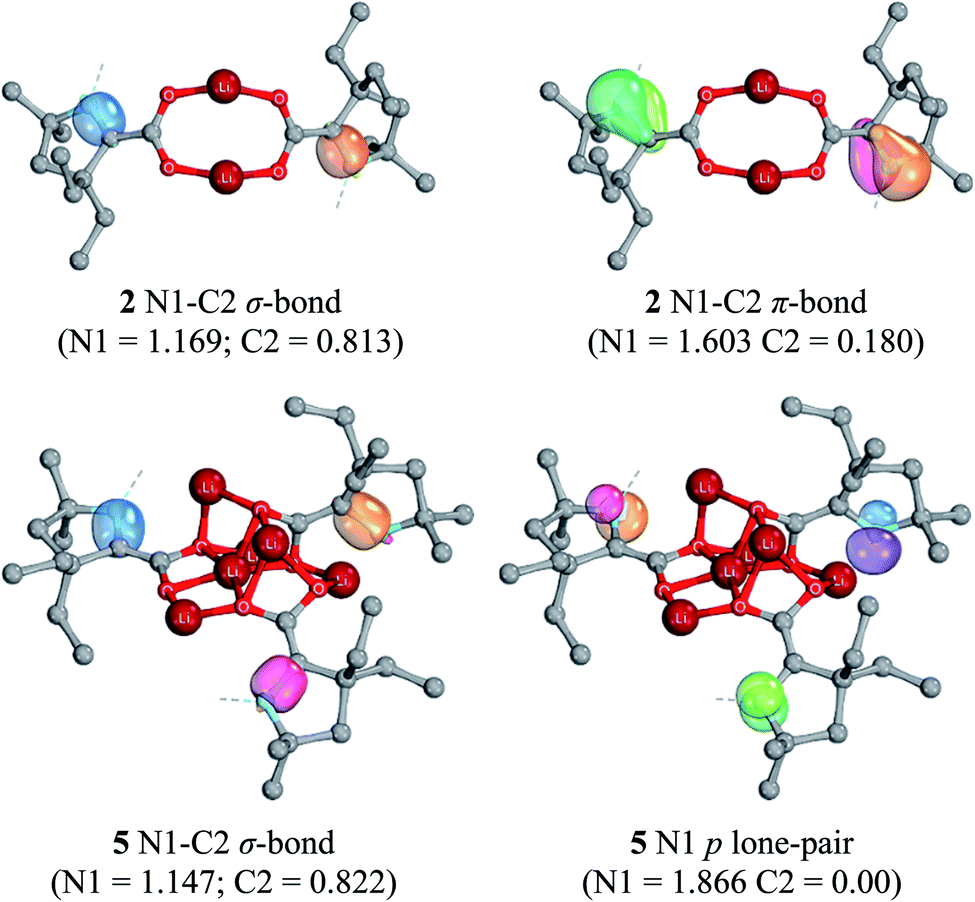 | ||
| Fig. 8 B3LYP-D3(BJ)/def2-SVP intrinsic bonding orbitals (IBOs) of 2 (top) and 5 (bottom). Numbers in parentheses indicate the partial charge distribution of the IBO. Orbital iso-surfaces enclose 80% of the integrated electron densities of the orbital. H atoms, Dipp and THF substituents are omitted for clarity. | ||
The synthetic protocols reported herein result in the carboxylation of CAAC at RT and 1 atm of pressure, followed by the stepwise one- and two-electron alkali metal reduction of CAAC-carboxylate to monoanionic radical clusters and dianionic diolate clusters. The reduced products are isolable as soluble, crystalline products with diverse electronic properties dependent on the nature of the cation and cluster topology. Notably, this represents the first example of the chemical reduction of a carbene–CO2 adduct, and provides a facile method for the cleavage of CO2 π-bonds by alkali metals. Moreover, this work highlights the ability of CAAC to mediate a reductive process between base alkali metals and CO2 – a reaction that does not occur in the absence of carbene. The newfound redox behavior of carbene-carboxylates will facilitate the development of new chemistries using these compounds as simple and efficient electron-transfer platforms in both stoichiometric and catalytic CO2 conversion chemistry.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge the University of Virginia for support of this work. L. A. F. and A. D. O. also thank the Jefferson Scholars Foundation at the University of Virginia for support of this research through the Mary Anderson Harrison and Melville Foundation Graduate Fellowships, respectively. We also thank Sarah Nyenhuis from the Cafiso Group at UVA for her assistance with the collection of EPR data. Generous allocation of computing resources from National Computational Infrastructure (NCI) and La Trobe University are acknowledged.References
- (a) R. A. Moss, M. Platz and M. Jones, Reactive intermediate chemistry, Wiley Online Library, 2004 Search PubMed; (b) H. Chen, M. Armand, M. Courty, M. Jiang, C. P. Grey, F. Dolhem, J.-M. Tarascon and P. Poizot, J. Am. Chem. Soc., 2009, 131, 8984–8988 CrossRef CAS; (c) H. F. Cordes, H. P. Richter and C. A. Heller, J. Am. Chem. Soc., 1969, 91, 7209 CrossRef CAS; (d) K. S. Lackner, C. H. Wendt, D. P. Butt, E. L. Joyce and D. H. Sharp, Energy, 1995, 20, 1153–1170 CrossRef CAS.
- (a) A. Paparo, J. S. Silvia, C. E. Kefalidis, T. P. Spaniol, L. Maron, J. Okuda and C. C. Cummins, Angew. Chem., Int. Ed., 2015, 54, 9115–9119 CrossRef CAS; (b) A. Paparo and J. Okuda, J. Organomet. Chem., 2018, 869, 270–274 CrossRef CAS.
- (a) K. von Baczko, Kohlenstoff: Chemisches Verhalten von CO und CO2. Teil C2, Verlag Chemie, 1972 Search PubMed; (b) R. H. Hauge, J. L. Margrave, J. W. Kauffman, N. A. Rao, M. M. Konarski, J. P. Bell and W. E. Billups, J. Chem. Soc., Chem. Commun., 1981, 1258–1260 RSC; (c) Z. H. Kafafi, R. H. Hauge, W. E. Billups and J. L. Margrave, Inorg. Chem., 1984, 23, 177–183 CrossRef CAS.
- A. J. Arduengo, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef CAS.
- V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., 2005, 117, 5851–5855 CrossRef.
- (a) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046–10068 CrossRef CAS; (b) N. Marion and S. P. Nolan, Chem. Soc. Rev., 2008, 37, 1776–1782 RSC; (c) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef; (d) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS; (e) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485 CrossRef CAS; (f) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS; (g) V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS.
- (a) S. N. Riduan, Y. Zhang and J. Y. Ying, Angew. Chem., Int. Ed., 2009, 48, 3322–3325 CrossRef CAS; (b) G. D. Frey, V. Lavallo, B. Donnadieu, W. W. Schoeller and G. Bertrand, Science, 2007, 316, 439 CrossRef CAS; (c) E. M. Phillips, T. E. Reynolds and K. A. Scheidt, J. Am. Chem. Soc., 2008, 130, 2416–2417 CrossRef CAS; (d) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS; (e) V. Regnier, E. A. Romero, F. Molton, R. Jazzar, G. Bertrand and D. Martin, J. Am. Chem. Soc., 2019, 141, 1109–1117 CrossRef CAS; (f) A. Grossmann and D. Enders, Angew. Chem., Int. Ed., 2012, 51, 314–325 CrossRef CAS; (g) J. K. Mahoney, V. Regnier, E. A. Romero, F. Molton, G. Royal, R. Jazzar, D. Martin and G. Bertrand, Org. Chem. Front., 2018, 5, 2073–2078 RSC; (h) S. J. Ryan, L. Candish and D. W. Lupton, Chem. Soc. Rev., 2013, 42, 4906–4917 RSC.
- Z. Li, R. J. Mayer, A. R. Ofial and H. Mayr, J. Am. Chem. Soc., 2020, 142, 8383–8402 CrossRef CAS.
- A. M. Voutchkova, L. N. Appelhans, A. R. Chianese and R. H. Crabtree, J. Am. Chem. Soc., 2005, 127, 17624–17625 CrossRef CAS.
- (a) Y.-B. Wang, D.-S. Sun, H. Zhou, W.-Z. Zhang and X.-B. Lu, Green Chem., 2015, 17, 4009–4015 RSC; (b) H. Zhou, W.-Z. Zhang, C.-H. Liu, J.-P. Qu and X.-B. Lu, J. Org. Chem., 2008, 73, 8039–8044 CrossRef CAS.
- (a) E. Theuergarten, T. Bannenberg, M. D. Walter, D. Holschumacher, M. Freytag, C. G. Daniliuc, P. G. Jones and M. Tamm, Dalton Trans., 2014, 43, 1651–1662 RSC; (b) S. N. Talapaneni, O. Buyukcakir, S. H. Je, S. Srinivasan, Y. Seo, K. Polychronopoulou and A. Coskun, Chem. Mater., 2015, 27, 6818–6826 CrossRef CAS; (c) C. Finn, S. Schnittger, L. J. Yellowlees and J. B. Love, Chem. Commun., 2012, 48, 1392–1399 RSC; (d) L. E. Lieske, L. A. Freeman, G. Wang, D. A. Dickie, R. J. Gilliard Jr and C. W. Machan, Chem.–Eur. J., 2019, 25, 6098–6101 CrossRef CAS; (e) G. Kuchenbeiser, M. Soleilhavoup, B. Donnadieu and G. Bertrand, Chem.–Asian J., 2009, 4, 1745–1750 CrossRef CAS; (f) M. Hans, L. Delaude, J. Rodriguez and Y. Coquerel, J. Org. Chem., 2014, 79, 2758–2764 CrossRef CAS; (g) D. R. Tolentino, S. E. Neale, C. J. Isaac, S. A. Macgregor, M. K. Whittlesey, R. Jazzar and G. Bertrand, J. Am. Chem. Soc., 2019, 141, 9823–9826 CrossRef CAS; (h) A. P. Singh, P. P. Samuel, H. W. Roesky, M. C. Schwarzer, G. Frenking, N. S. Sidhu and B. Dittrich, J. Am. Chem. Soc., 2013, 135, 7324–7329 CrossRef CAS; (i) L. Delaude, Adv. Synth. Catal., 2020, 362, 3259–3310 CrossRef CAS; (j) O. R. Luca, C. C. L. McCrory, N. F. Dalleska and C. A. Koval, J. Electrochem. Soc., 2015, 162, H473–H476 CrossRef CAS; (k) O. R. Luca and A. Q. Fenwick, J. Photochem. Photobiol., B, 2015, 152, 26–42 CrossRef CAS.
- (a) B. J. Wakefield, The chemistry of organolithium compounds, Elsevier, 2013 Search PubMed; (b) E. Kaufmann, S. Sieber and P. v. R. Schleyer, J. Am. Chem. Soc., 1989, 111, 4005–4008 CrossRef CAS.
- G. Bresciani, L. Biancalana, G. Pampaloni and F. Marchetti, Molecules, 2020, 25, 3603 CrossRef CAS.
- A. R. Kennedy, R. E. Mulvey, D. E. Oliver and S. D. Robertson, Dalton Trans., 2010, 39, 6190–6197 RSC.
- The bulk mixture of 5 and 6 was analyzed as one product with a varying amount of THF, as discussed in the ESI.† As such, this yield is given with respect to the chemical formula of the bulk mixture as determined by combustion analysis.
- T. X. Gentner and R. E. Mulvey, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202010963.
- (a) A. Rajca, Chem. Rev., 1994, 94, 871–893 CrossRef CAS; (b) W. Carl Lineberger and W. Thatcher Borden, Phys. Chem. Chem. Phys., 2011, 13, 11792–11813 RSC; (c) L. Salem and C. Rowland, Angew. Chem., Int. Ed. Engl., 1972, 11, 92–111 CrossRef CAS; (d) M. Abe, Chem. Rev., 2013, 113, 7011–7088 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1991606–1991613. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc06851a |
| This journal is © The Royal Society of Chemistry 2021 |