
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTrioxatriangulenium (TOTA+) as a robust carbon-based Lewis acid in frustrated Lewis pair chemistry†
Aslam C.
Shaikh
 ,
José M.
Veleta
,
Jules
Moutet
and
Thomas L.
Gianetti
*
,
José M.
Veleta
,
Jules
Moutet
and
Thomas L.
Gianetti
*
University of Arizona, Department of Chemistry and Biochemistry, Tucson, AZ, USA. E-mail: tgianetti@arizona.edu
First published on 8th February 2021
Abstract
We report the reactivity between the water stable Lewis acidic trioxatriangulenium ion (TOTA+) and a series of Lewis bases such as phosphines and N-heterocyclic carbene (NHC). The nature of the Lewis acid–base interaction was analyzed via variable temperature (VT) NMR spectroscopy, single-crystal X-ray diffraction, UV-visible spectroscopy, and DFT calculations. While small and strongly nucleophilic phosphines, such as PMe3, led to the formation of a Lewis acid–base adduct, frustrated Lewis pairs (FLPs) were observed for sterically hindered bases such as P(tBu)3. The TOTA+–P(tBu)3 FLP was characterized as an encounter complex, and found to promote the heterolytic cleavage of disulfide bonds, formaldehyde fixation, dehydrogenation of 1,4-cyclohexadiene, heterolytic cleavage of the C–Br bonds, and interception of Staudinger reaction intermediates. Moreover, TOTA+ and NHC were found to first undergo single-electron transfer (SET) to form [TOTA]·[NHC]˙+, which was confirmed via electron paramagnetic resonance (EPR) spectroscopy, and subsequently form a [TOTA–NHC]+ adduct or a mixture of products depending the reaction conditions used.
Introduction
Frustrated Lewis pairs (FLPs) arise from the combination of Lewis acids (LAs) and bases (LBs) with un-neutralized reactivity due to the steric repulsion between these units.1 Since their discovery, FLPs have received considerable attention for their potential to activate small molecules and applications in catalysis in the absence of transition-metals.2 While significant exploration of the tunability of FLPs has been done, it is interesting to note that the main advances are focused on the nature of the LBs. Common bases for FLPs found in the literature include a wide range of phosphines, amines, pyridines, N-heterocyclic carbenes (NHCs), or carbodiphosphoranes, which show compatibility with various solvents and functional groups during transformations. In contrast, the LA counterparts in FLPs are less versatile and mostly limited to perfluorinated alkyl/aryl boranes and alanes.3 Group 13 molecules are often highly oxophilic which limits their use and stability in wet solvents, tolerance to several functional groups, and reaction conditions when applied in catalysis.4 As a result, the development of versatile and stable LAs for FLP chemistry is still highly desirable.In recent years, several cationic or electron-deficient compounds containing heavy elements of group 14 as LAs have been reported to show FLP reactivity, but their use remains underdeveloped.5 Similarly, examples of Lewis acidic carbon centers in FLP chemistry are scarce,6 and limited to trityl4,7 and substituted trityl cations,8 electron-deficient allenes,9N-methylacridinium salts,10 cationic η6-arene-Ru complex11 and C60-fullerenes12 (Fig. 1a). However, similar to borane systems, most of these carbenium ions are air-sensitive, highly oxophilic, and experience side reactivity, which limit their applications. For example, trityl cation is an attractive LA due to its facile synthesis, but its negative pKR+ of −6.7 makes it more oxophilic than most boron-based FLPs.13 However, triaryl-carbenium ions are susceptible to a nucleophilic attack at the para-aryl, or meta-aryl positions, resulting in the formation of either cyclohexadienyl or aryl-phosphonium cations, which hampers the reactivity.14 Heterocyclic fused carbenium analogues, such as acridinium, helicenium, and angulenium ions, are an attractive alternative.15 Their more planar scaffold and reduced steric hindrance facilitates interaction at the central carbon. Furthermore, despite the increased stabilization and electron richness provided by the heteroatoms,16 recent reports have shown that some of these carbocations retain a certain degree of Lewis acidity.17
 | ||
| Fig. 1 (a) Previously reported carbon-based LA and (b) this work. | ||
Trioxatriangulenium ion (TOTA+, Fig. 1b), first reported by Smith et al. in 1964 (ref. 18) and with more recent applications in bioimaging and nucleic acid interaction,19 is a planar heterocyclic fused carbenium ion with a pKR+ of 9, a positive charge delocalized across the π-system, yet the central carbon contributing 33% of the LUMO's character, and with an acceptor number (AN) of 23.1.20 As a result, TOTA+ is water stable, resistant to para-aryl nucleophilic attack, and has shown preferential reactivities at the central carbon, with a variety of strong nucleophiles, such as hydride, sulfoxide, and organolithium or Grignard reagents, to form a cup-like molecule with an sp3-hybridized central carbon, known as trioxatricornan.21 While TOTA+ is significantly less acidic than common Lewis acids such as B(C6F5)3 (pKa of 8.4 and AN of 80.1), the reported reactivity of TOTA+ with small and hard nucleophiles,22 led our group to investigate the nature of the interactions between TOTA+ and softer LBs, such as phosphines.
Herein we describe the use of a trioxatriangulene (TOTA+) as water stable LA for FLP chemistry, and its reactivity with different phosphines as LBs. The reactivity of the obtained FLPs was probed via the heterolytic disulfide bond cleavage, fixation of formaldehyde, C(sp3)–H activation of 1,4-cyclohexadiene, heterolytic cleavage of a C–Br bond, and interception of Staudinger reaction intermediate. The interaction and reactivity of these FLPs are best described as encounter complexes. Furthermore, formation of Lewis acid–base adducts via single-electron transfer was observed between TOTA+ and the N-heterocyclic carbene 1,3-di-tert-butylimidazol-2-ylidene (ItBu). The formation of these radical species is supported by UV-vis and EPR spectroscopies.
Results and discussion
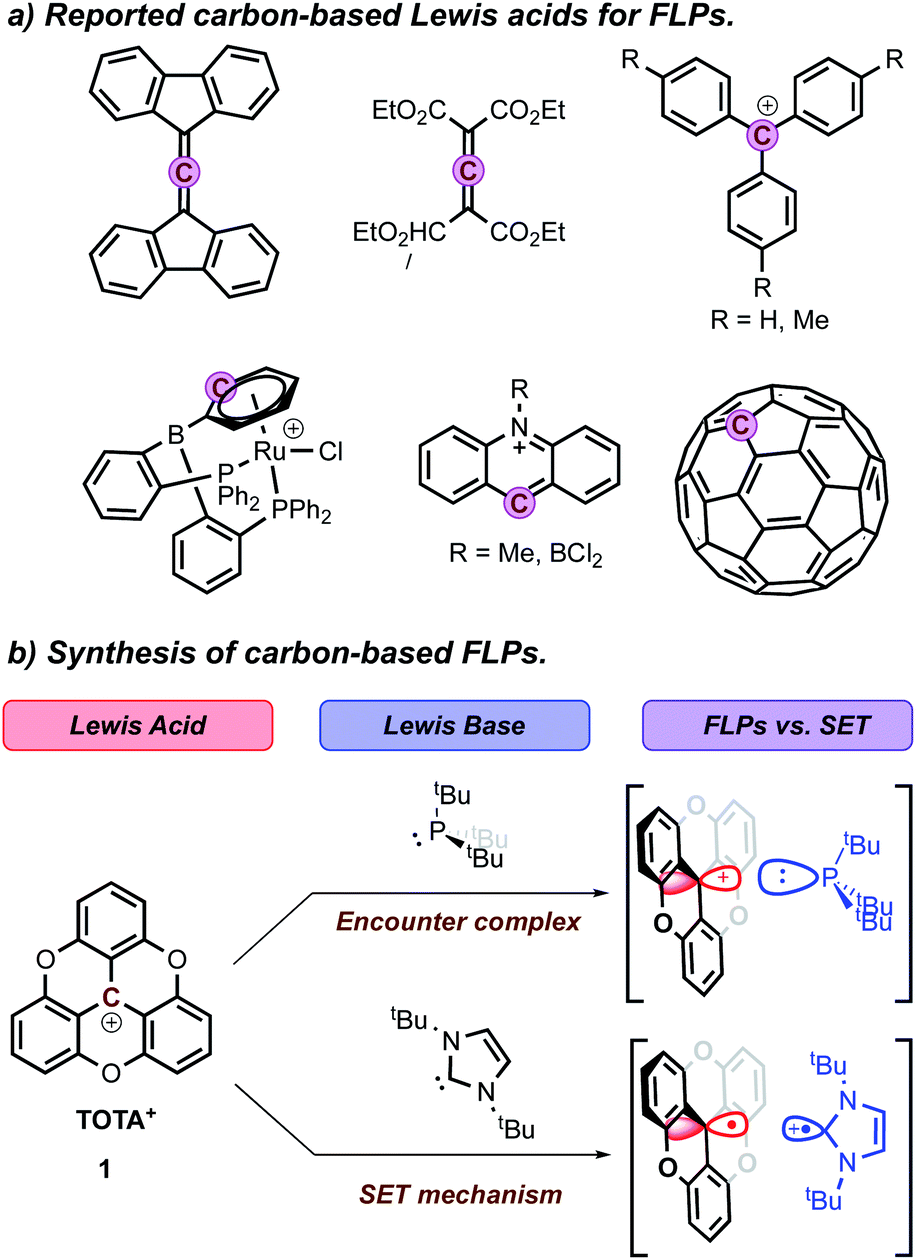
A variety of alkyl, alkyl–aryl, and aryl phosphines were selected to probe the interaction between TOTA+ and phosphines (Scheme 1a). Reacting the less sterical phosphine, PMe3, with TOTA+ resulted in the formation of a classical Lewis acid–base covalent adduct. This was observed by the instantaneous change in color upon the room temperature addition of PMe3, changing from a dark yellow solution, characteristic of TOTA+ (1) in CH3CN, to a colorless solution. Such color change is consistent with the loss of conjugation in the π-system, due to a change in hybridization from sp2 to sp3 in the central carbon. The TOTA–(PMe3)+ adduct (3a) was isolated as a white solid in 96% yield (see Scheme 1 and ESI†). The 31P NMR spectrum indicates the exclusive formation of a phosphonium species with a chemical shift of 39.7 ppm (Fig. S7†). The 1H NMR spectrum of 3a shows two sets of protons at δ = 7.53 (td) and 7.12 (dd) ppm, consistent with the presence of a C3 rotation axis, suggesting that the phosphorous is bound to the central carbon atom in TOTA. Slow CH2Cl2/hexane layering afforded colorless crystals of 3a suitable for single-crystal X-ray diffractometry, that confirmed the formation of the covalent adduct at the central carbon (Scheme 1b and ESI†). The C1–P1 distance (1.864(1) Å) is comparable to typical C–P bonds (ca. 1.87 Å), yet shorter than the P–C bond in [trityl-PMe3]+ (1.887(4) Å)14 and the P–B bond in tris(pentafluorophenyl)borane-phenylphosphane (2.039(4) Å).23 These observations suggest that the planarity of the TOTA+ scaffold promotes the formation of shorter bond in solid-state compared to the more sterically hindered trityl and borane structures. Within the asymmetric unit, the counter-anion BF4− is far from both the carbon and phosphorous centers in 3a (B–C1 = 5.355 Å, B–P = 4.821 Å, or F–C1 = 4.573 Å and F–P = 3.891 Å respectively, see Fig. S110†).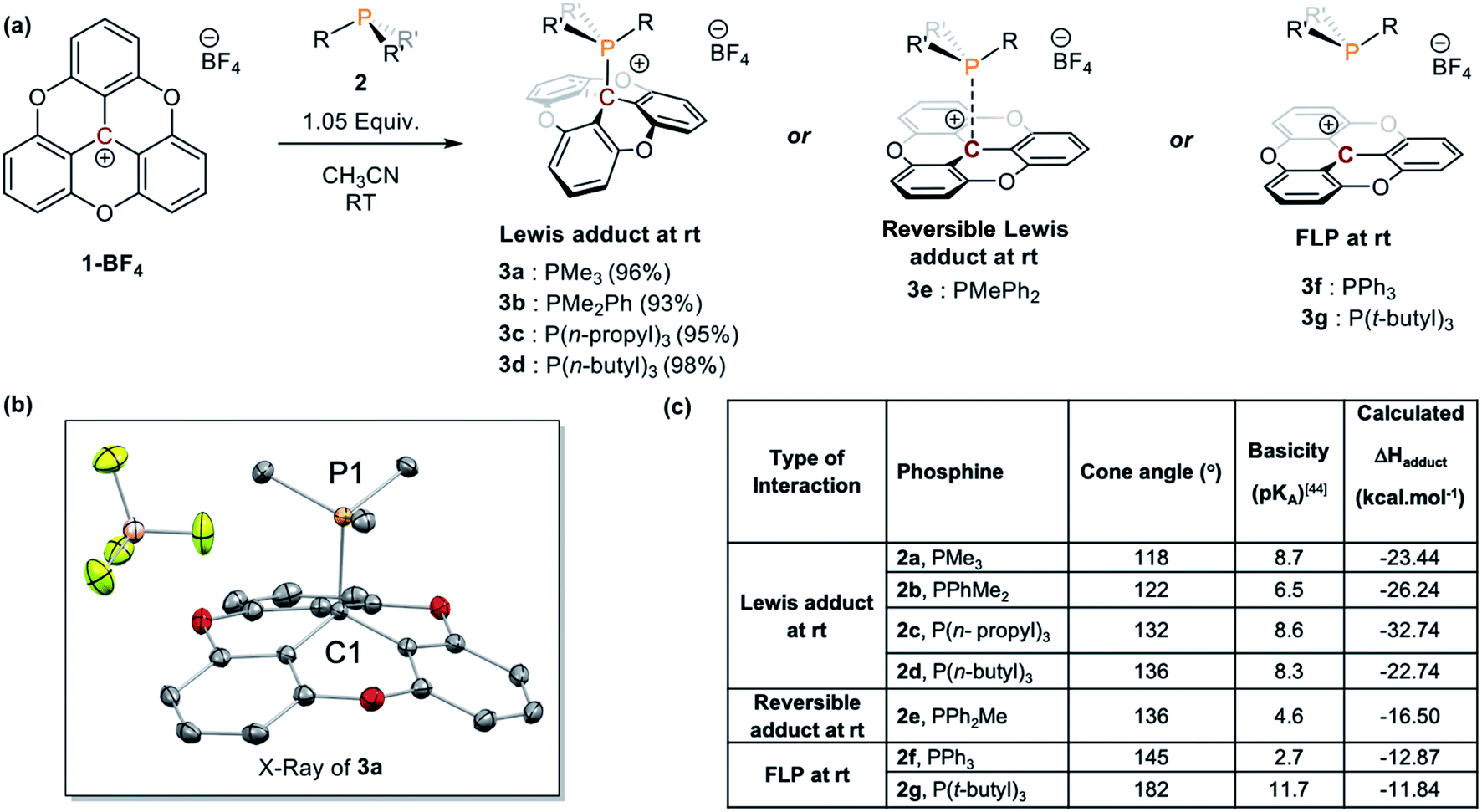 | ||
| Scheme 1 (a) Reactivity of phosphines (2a–2g) towards TOTA+. (b) ORTEP of the X-ray structure of 3a. Hydrogen atoms and counter ion are omitted for clarity. (c) Summary of cone angle, Lewis basicity, and DFT calculated ΔHadduct (enthalpy of adduct formation relative to free TOTA and phosphine) for phosphines 2a–2g. | ||
Other phosphines with small cone angles, such as PPhMe2, P(nPr)3, and P(nBu)3, also formed covalent Lewis adducts at room temperature, which were isolated in excellent yields (Scheme 1, 3a, 3b, 3c, and 3d, respectively).24 Interestingly, when the TOTA–P(nBu)3+ adduct (3d) was warmed up to 353 K in CD3CN, a broadening of the 1H NMR resonances are observed (Fig. S19†), suggesting the presence of a thermodynamic equilibrium. Using PPh2Me, a phosphine of similar cone angle as P(nBu)3 yet with lower basicity, broad and unresolved peaks in the 1H NMR spectrum were observed at room temperature, suggesting that the slow exchange regime of the equilibrium is observable on the NMR time scale at room temperature (Scheme 1a, 3e). Variable temperature 1H NMR analysis, along with the thermochromic change of the solution, confirmed the presence of a reversible adduct in which the Lewis adduct is the major species at low temperatures (see Fig. 2 and ESI†).
 | ||
| Fig. 2 (a) Reversible Lewis adduct formation between TOTA+ and PPh2Me. (b) Variable temperature (VT) 500 MHz 1H NMR spectra of TOTA+ (12 mg, 0.01 mmol) and PPh2Me (4.2 mg, 0.02 mmol) in CD2Cl2 indicating formation Lewis adduct at −60 °C. | ||
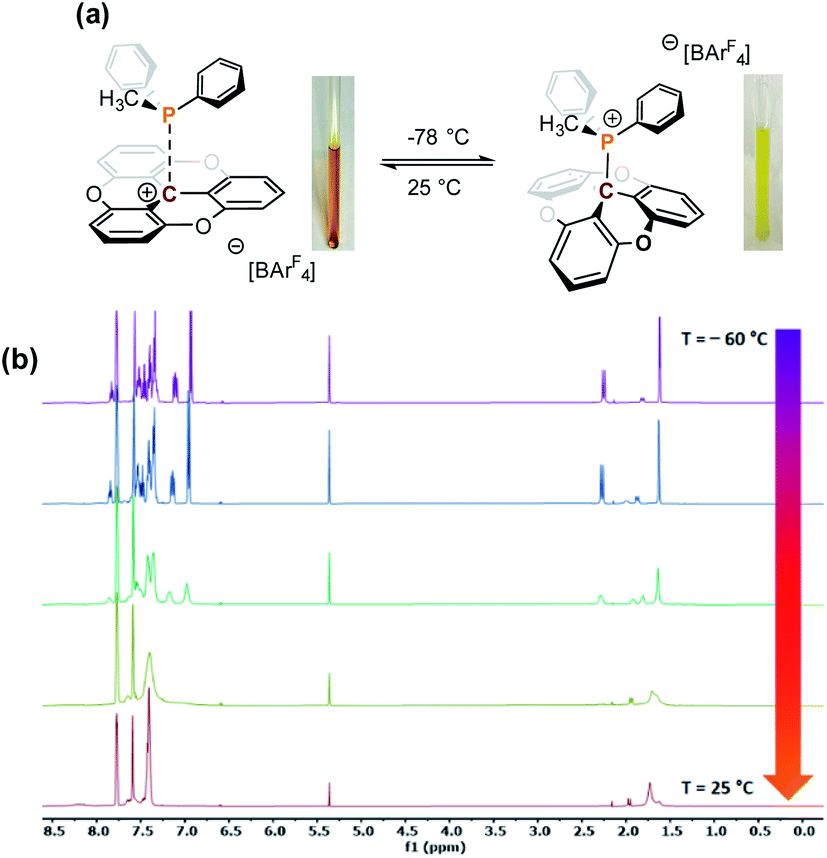
Continuing with the trend, when TOTA+ was mixed with a weakly basic phosphine such as PPh3 (Scheme 1a, 3f), or a sterically hindered phosphines, such as P(tBu)3 (Scheme 1a, 3g), no Lewis adduct was observed by NMR spectroscopy at either room or low temperatures. Neither the appearance of a new peak in 31P NMR spectroscopy nor an upfield shift of the aromatic region in TOTA+ protons, characteristic of the trioxatricornan scaffold, was observed (see ESI†). However, we noted that, in both CD2Cl2 or CD3CN solvents, the aromatic resonances of TOTA+ appear as broad signals at room temperature, and sharpened at lower temperatures (Fig. S27–S30 and S36–S40†). Furthermore, when lowering the temperature below 230 K, a broadening of the tert-butyl and phenyl signals were observed in the 1H NMR spectra (Fig. S27 and S37†). The broadening and coalescences of the TOTA+ aromatic peaks and the phosphine substituents indicate that only a weak van der Waals adduct is formed, as observed in the case of FLPs derived from B(C6F5)3 and bulky phosphines, and that these aggregates have random relative orientations (see DFT calculations vide infra).8,25 Diffusion ordered spectroscopy (DOSY) NMR experiments were performed and further supported the formation of such aggregates (see ESI, Fig. S31–S34 and S41 and S42†).26 For example, the diffusion coefficients (D) for 1-BArF4 and P(tBu3) alone are 1.41 × 10−9 m2 s −1 and 1.88 × 10−9 m2 s−1 respectively. However, upon mixing, diffusion coefficients of 1.80 × 10−9 m2 s −1 (1-BArF4) and 1.74 × 10−9 m2 s−1 (P(tBu3)) were recorded.27 Finally, we noted that upon mixing TOTA+ and P(tBu)3 a slightly darker yellow color, compared to that of TOTA+, was observed. This was further analyzed by mixing an equimolar amounts of each species in CH2Cl2 and analyzing the solution via UV-vis absorption spectroscopy. We observed the appearance of weak absorption bands at λmax of 576 and 626 nm, in addition to the main absorption bands for TOTA+ (458 and 483 nm) (see Fig. 3). These data indicate the presence of an encounter complex in solution.28 We postulate that the increased steric bulk of the phosphine inhibits the formation of a Lewis adduct, resulting in a dynamic equilibrium between free TOTA+/P(tBu)3 and its encounter complex.
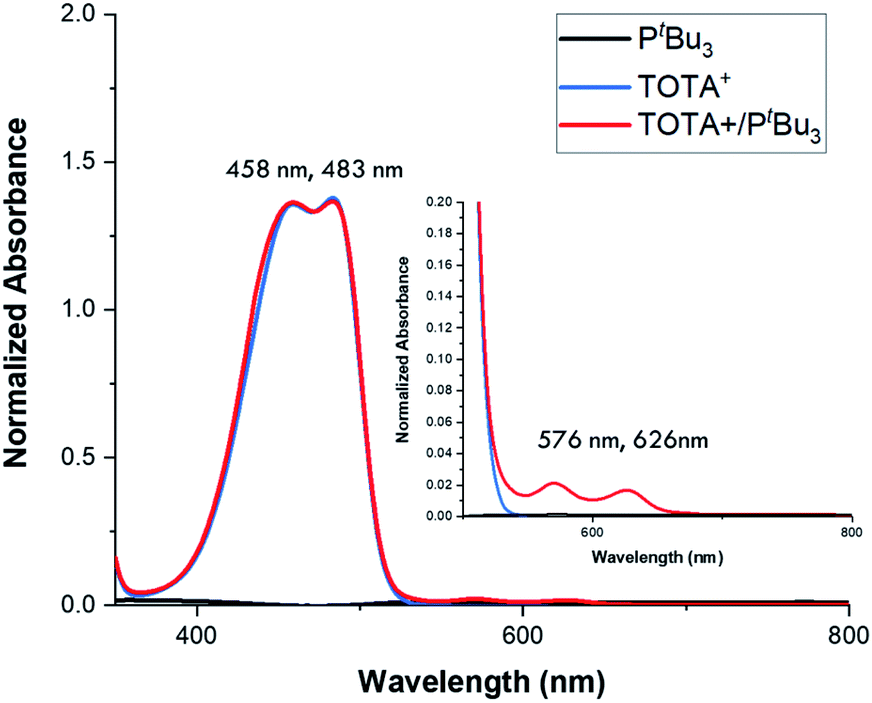 | ||
Fig. 3 Normalized UV-vis spectra of TOTA+ (blue trace, 1 × 10−5 M in CH2Cl2), PtBu3 (black trace, 1 × 10−5 M in CH2Cl2), and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of PtBu3/TOTA+ (red trace, both substrates 1 × 10−5 M in CH2Cl2). :1 mixture of PtBu3/TOTA+ (red trace, both substrates 1 × 10−5 M in CH2Cl2). | ||
Density functional theory (DFT) calculations were used to further understand the energetic and steric factors between TOTA+ and the different phosphines (Scheme 1c and ESI†). As expected, the strength of the LA–LB interaction, quantified via both the [P–CTOTA]+ bond length and stabilization energy, depends on the phosphine's cone angle and basicity. For example, the TOTA+-adducts with phosphines of comparable LB character, such as PMe3, P(nBu)3, and P(tBu)3, show a correlation between increasing cone angle and increasing [P–CTOTA]+ bond length. The [TOTA–PMe3]+ adduct had the shortest bond compared to the [TOTA–P(tBu)3]+ (1.88 vs. 2.06 Å, Table S11†). Similarly, the stabilization energy for these adducts was inversely proportional to the cone angle of the phosphine used, with decreasing energies of −23.4, −16.5, and −11.8 kcal mol−1 for increasing cone angles (Me < nBu < tBu, see ESI†). Looking at phosphines with comparable cone angles, such as PPh2Me and P(nBu)3 (cone angle of 136°), a shorter [P–CTOTA]+ bond length by 0.02 Å and larger stabilization energy (−22.7 vs. −16.5 kcal mol−1) was observed for the more Lewis basic P(nBu)3.
DFT calculations also provided insight on the structure of the FLPs aggregates. For both FLPs, several conformers of close energy were found (see ESI† and Fig. 4), with the most thermodynamic stable aggregate, 3f-I and 3g-I, being more favorable than the free components by ∼12 kcal mol−1 (see Fig. 5). For [TOTA–PPh3]+, the most thermodynamically stable conformer 3f-I contains a π–π stacking interaction between the planar TOTA+ and the arylphosphine, where the phosphorus atom is pointing away from the carbocation scaffold, while 3f-II shows a P–C interaction characteristic of a Lewis adduct (Fig. 4a). With [TOTA–P(tBu)3]+, the thermodynamically favorable conformation 3g-I has the phosphorus atom directly in front of central carbon atom of TOTA+, while in 3g-II the P atom is pointing in the opposite direction (Fig. 4b). These calculations support the NMR spectra obtained for these FLPs (vide supra).
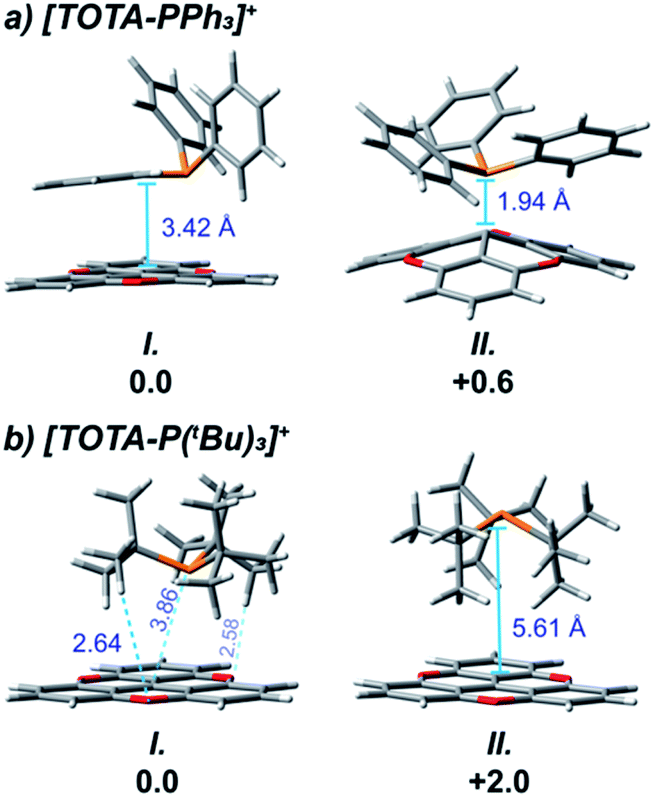 | ||
| Fig. 4 DFT geometry optimized structures of TOTA+ adducts with (a) PPh3 and (b) P(tBu)3 using the CAM-B3LYP functional and 6-311G(d,p) basis set with PCM for acetonitrile (relative energy in kcal mol−1). | ||
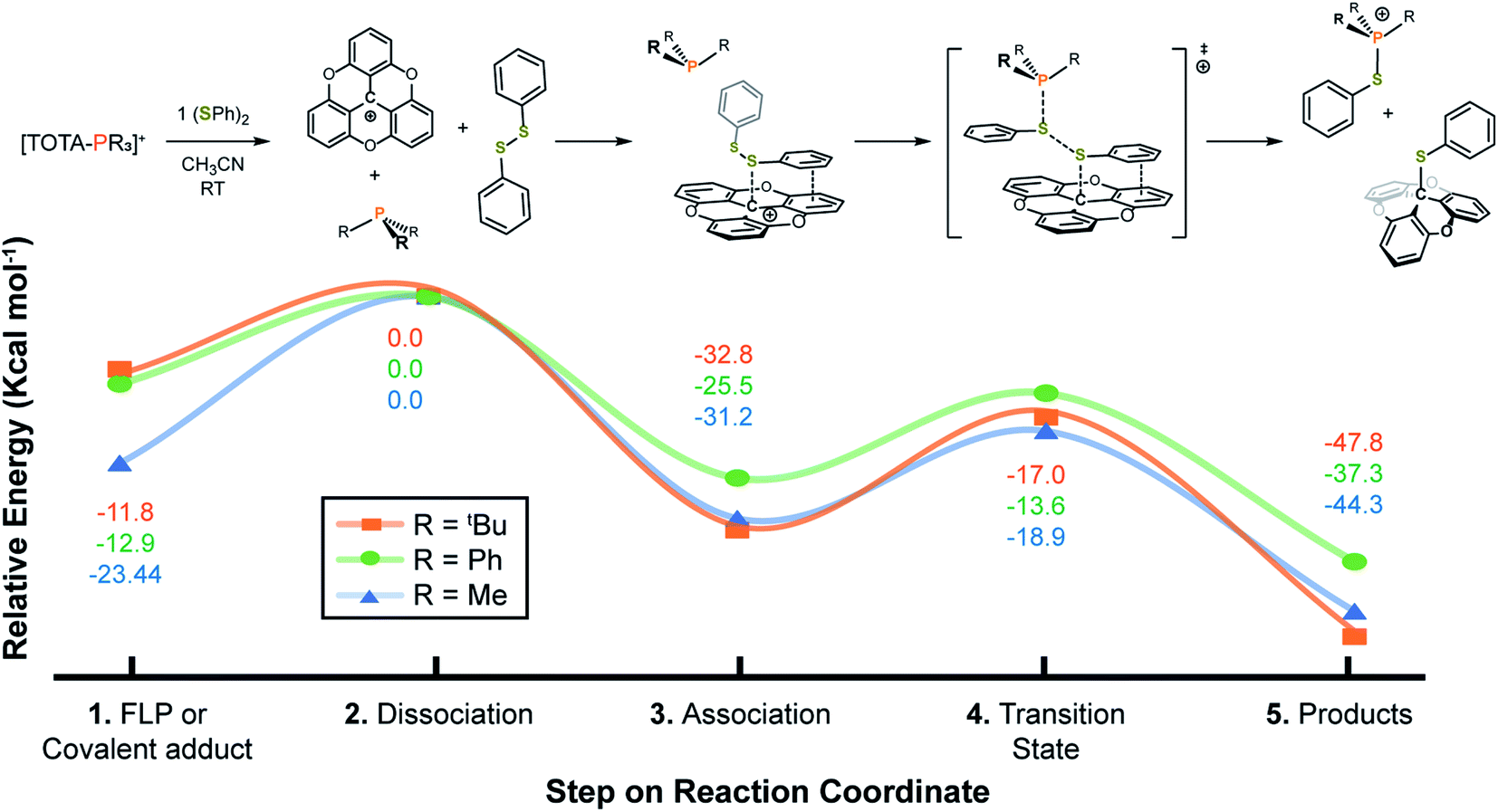 | ||
| Fig. 5 Reaction coordinate for the activation of (SPh)2 using [TOTA–PR3]+ (R = tBu, Ph, or Me) adducts. All models were calculated using the CAM-B3LYP functional, 6-311G(d,p) basis set, SCRF PCM acetonitrile solvation model, and Grimme D3 empirical dispersion damping functions. Transition states were found with a single imaginary frequency and as local maxima by IRC calculations. | ||
We then tested the reactivity of our Lewis pairs for the activation of non-polar covalent bonds, such as those present in disulfides. Using diphenyl sulfide, we observed full conversion in good yields for the desired S–S bond cleavage products at room temperature for the FLP encounter complexes 3f and 3g.
In the case of the reaction with 3g, the initially dark yellow solution gradually turned colorless after 10 min at room temperature. The crude mixture was thoroughly washed with pentane, and crystallization at −20 °C of the pentane washes resulted in TOTA–SPh (4a) as colorless crystals. The formation of 4a was supported by the conservation of the C3 rotation axis and two set of protons for the TOTA+ moieties in the 1H NMR spectrum, as well as the presence of a new quaternary carbon signal at 40.9 ppm in the 13C{1H} NMR spectrum. Furthermore, the structure of 4a was unambiguously confirmed by X-ray diffraction crystallography which attests to the heteroleptic cleavage of the disulfide bond of (SPh)2 (Scheme 2b). Notably, the C1–S1 bond of 1.902(2) Å is slightly longer than the typical value for single C–S bond length (∼1.83 Å), but is consistent with the covalent C–S distance found in the sulfide(trityl) bond (C–S = 1.90 Å).29 Moreover, the solid residue from the washes was dried under vacuum for 12 h, and the yellow compound was crystallized from slow diffusion of pentane into a concentrated solution in CH2Cl2 at −20 °C. The desired thio-phosphonium salt (5a) was isolated in pure form and characterized by NMR spectroscopy (Scheme 2a and ESI†). We further tested the ability of 3g to cleave the S–S bond of a variety of disulfides RS–SR (R = p-tolyl, 4-Cl-C6H4, –Bn) which gave the desired products 4b–4d and 5b–5d in good yields. Similar reactivity was observed when the FLP-3f was used. The desired S–S bond cleavage products were isolated in excellent yields (Scheme 2a and see ESI†).
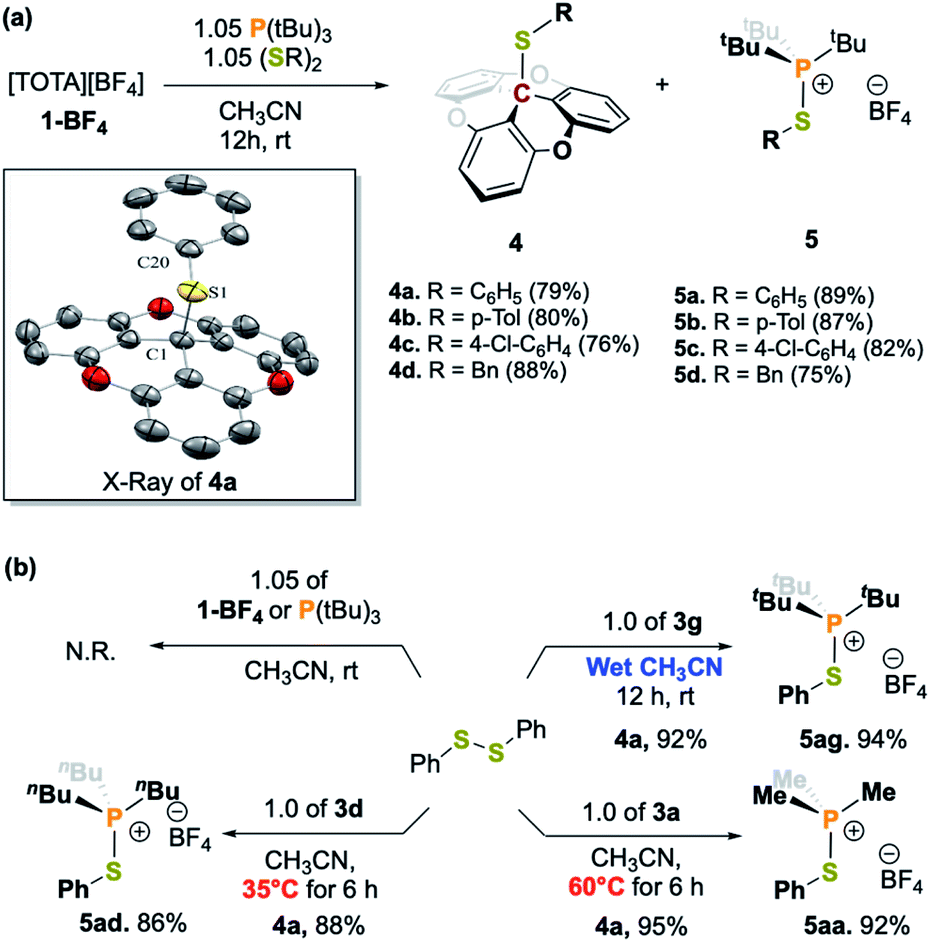 | ||
| Scheme 2 (a) FLP-mediated activation of disulfide bonds and crystal structures of 4a (thermal ellipsoids shown at 50% probability, hydrogen atoms and counter ions are omitted for clarity. C grey, O red, S yellow). (b) Control experiments, Lewis adduct mediated S–S bond cleavage with 3a and 3d, as well as 3g in wet solvent. | ||
It is noted that TOTA+ or phosphine alone were inert towards the disulfide bond cleavage (Scheme 2b). Excitingly, when the disulfide cleavage was performed in wet acetonitrile, the desired products were obtained in high yields (Scheme 2b). This result highlights the robustness and water tolerance of the system. Interestingly, the covalent adducts 3a and 3d also resulted in S–S bond cleavage but required temperatures of 60 °C and 35 °C respectively (see Scheme 2b and ESI†). Furthermore, premixing the PMe3 with disulfide, followed by addition of TOTA+, resulted in a mixture of the desired S–S bond cleavage product along with TOTA–PMe3 adduct 3a. These observations suggest that LA–LB dissociation is needed to promote disulfide activation.30
The reaction coordinate for the activation of disulfide bonds was studied using DFT (Fig. 5). Grimme D3 empirical dispersion damping forces were applied to better understand non-covalent interactions between the TOTA+, (SPh)2, and PR3 (R = Me, Ph, tBu) fragments. Starting from the FLP or covalent adduct (step 1, Fig. 5), a dissociation step in the presence of (SPh)2 is considered (step 2, Fig. 5). As observed experimentally, the dissociation energies for the TOTA+ adducts with PPh3 and P(tBu)3 is thermodynamically more accessible than for PMe3, with a dissociation barrier ducts ca. 10 kcal mol−1 more accessible for PPh3 and P(tBu)3 than for the PMe3 adduct. In the following step, a TOTA–SPh intermediate stabilized by π-stacking interactions between the aryl groups in TOTA and the disulfide substrate is formed (step 3, Fig. 5). The π-complex is then attacked by the phosphine via a concerted transition state (step 4, Fig. 5). The overall reaction coordinate for the activation of (SPh)2 is representative of an exothermic process, with the formation of TOTA–SPh and [R3P–SPh]+ adducts ca. 40 kcal mol−1 lower in energy than the correspondent reactants (FLP or adducts, step 5, Fig. 5).
We further examined the utility of the FLP 3g towards bond activation using 1,4 cyclohexadiene,7a and tbutyl bromide (Scheme 3). Addition of 1,4-cyclohexadiene to a solution of 3g in acetonitrile resulted in the dehydrogenation of hexadiene to form benzene, the formation of the phosphonium salt 7 in 86% yield and trioxatricornan TOTA–H (6) in 64% yield. The phosphonium salt shows a characteristic P–H signal at 6.25 ppm in the 1H NMR spectrum and a doublet at 50.52 ppm in the 31P NMR spectrum, with a 1JP–H coupling constant of 469.8 Hz (see ESI†). The reactivity of 3g towards tbutyl bromide resulted in the formation of phosphonium salt 7 and [TOTA][Br] in quantitative yields, with the expected isobutylene side-product as reported by Wass et al.31 Importantly, no reaction was observed upon exposure of 3g to H2 gas, at either atmospheric pressure or 4 bar, with or without saturated substrates such as alkyne.
Next, we investigated the reactivity of FLP 3g toward unsaturated substrates such as formaldehyde and benzyl azide (Scheme 3). When a 1.0 molar equivalent of paraformaldehyde was added to a solution of 3g–BF4 in CH3CN, we observed the formation of a new product by NMR spectroscopy, assigned to a formaldehyde adduct. However, the new species slowly returns to the starting material suggesting that the strongly coordinating acetonitrile was not suited to isolate the adduct formed. To improve the stability of the adduct formed we turned to the non-coordinating solvent CH2Cl2. Since [TOTA][BF4] is sparingly soluble in this solvent, we synthesized the more soluble [TOTA][BArF4] analog ([BArF4]− = [{3,5-(CF3)2C6H3}4B]−) following previously reported literature.17a An equimolar solution of 3g–BArF4 and paraformaldehyde were stirred for 24 h at room temperature in CH2Cl2. The resultant colorless turbid solution was dried under vacuum, and the residue was washed with pentane affording the formaldehyde adduct 8. Slow diffusion of pentane into a CH2Cl2 concentrated solution at −20 °C led to the formation of colorless single crystals of 8. Analysis via31P and 1H NMR spectroscopies revealed the presence of resonances that support the complexation of formaldehyde in between the FLP 3g (δ31P = 44.2 ppm, δ13C = 50.27 (d, J = 55.5 Hz), and δ1H = 4.63 ppm (d, J = 5.4 Hz, CH2)). The structure of 8 was unambiguously confirmed by single crystal X-ray diffraction (Scheme 3), showing the formation of a single bond between the carbon center of TOTA and the oxygen of paraformaldehyde with a C1–O4 of 1.513(4), longer than a classic C–O bond (ca. 1.43 Å). The C20–P1 bond length of 1.825(4) Å is consistent with a single carbon–phosphorus bond. We also noted that the elongated C20–O4 bond (1.410(5) Å vs. 1.2 Å in classic C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond) support a single bond and the reduction of the paraformaldehyde to an alcoholate (see ESI†).
O bond) support a single bond and the reduction of the paraformaldehyde to an alcoholate (see ESI†).
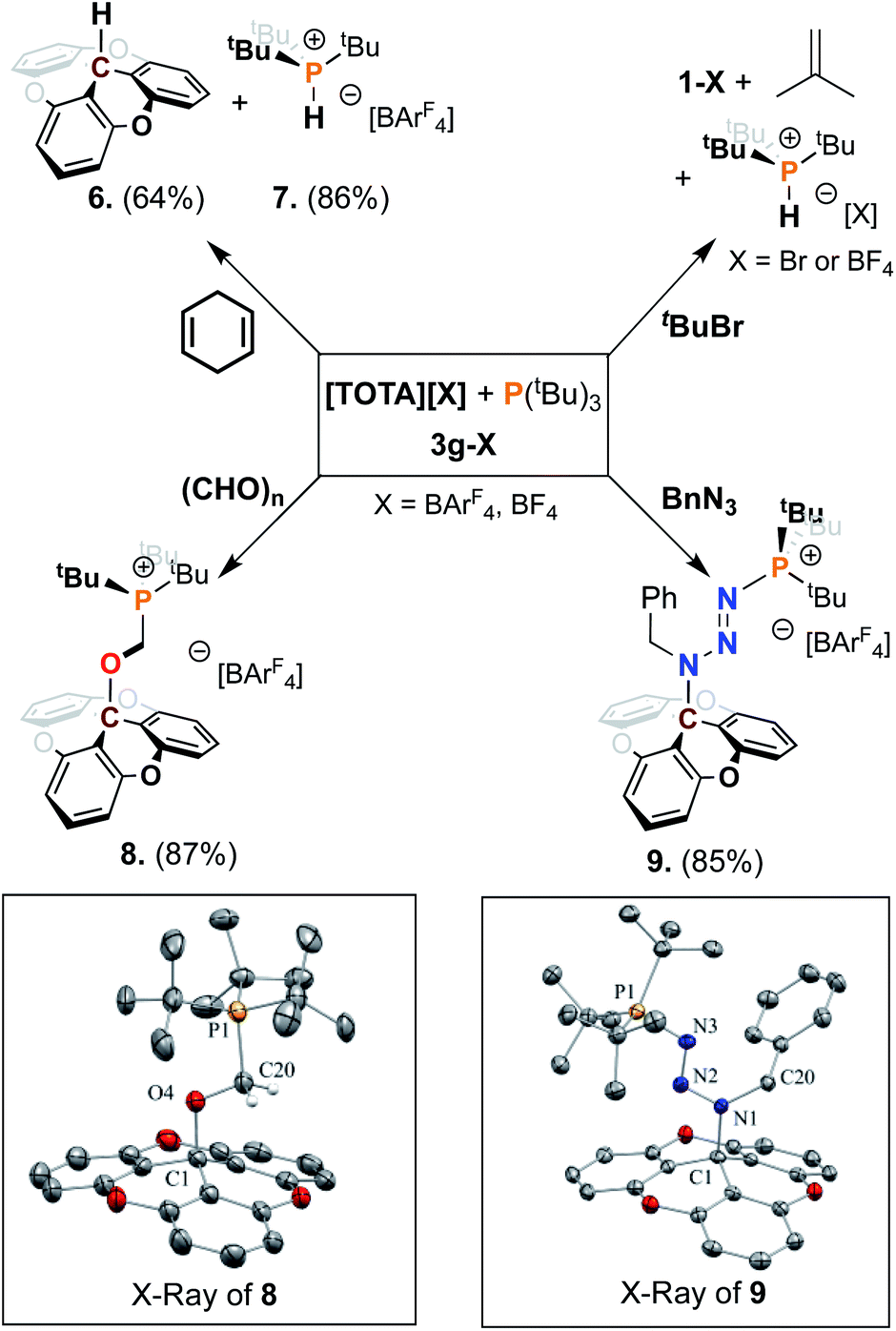 | ||
| Scheme 3 FLPs mediated activation of small molecules. Unless otherwise noted all reactions were carried out on a 0.05 mmol scale using [1+], 0.05 mmol P(tBu)3, 4 ml solvent, at room temperature. Isolated yields are given in parentheses (BArF4 = tetrakis(3,5-bis(trifluoromethyl)phenyl)borate). Crystal structures of 8 and 9; (thermal ellipsoids shown at 50% probability, hydrogen atoms and counter ion are omitted for clarity. P orange, N blue, C grey, O red). | ||
Finally, an organic azide was employed which led to the isolation of a Staudinger-type reaction intermediate from the reaction between P(tBu)3, TOTA+, and benzyl azide. The combination of 3g and benzyl azide in CH2Cl2 afforded adduct 9 in 85% yield after 12 h at room temperature (Scheme 3). NMR and crystallographic analysis of 9 revealed it to be the addition product of BnN3 between TOTA+ and P(tBu)3 (Scheme 3). Suitable crystals for X-ray diffraction studies of 9 were obtained by slow diffusion of pentane into a concentrated CH2Cl2 solution, resulting in pale-yellow plates. The trapping of Staudinger-type intermediate by 3g is characterized by the creation of a single C1–N1 bond (benzyl nitrogen from the substrate) length of 1.524(4) Å longer than the classic C–N bond (∼1.47 Å). At the same time, an N3–P1 bond is formed with the terminal nitrogen (1.680(3) Å) and the P(tBu)3 having a typical length of a phosphorus–azide bond (1.658(3) Å).27 It was further noted that the distances N1–N2 and N2–N3 (respectively 1.310(3) and 1.299(3) Å) retain a double bond character while the length of the N1–C20 bond (1.467(3) Å) of the benzyl group is also unaffected. This FLP addition reaction is consistent and can be compared to literature examples for a range of aryl azides and trityl-boron or phosphorous–boron FLPs.7a,27,32
To test the compatibility of TOTA+ with other Lewis bases, we used an NHC, 1,3-di-tert-butylimidazol-2-ylidene (ItBu, 10), as a possible LB (see Scheme 4 and Table S2†). Upon addition of 10 to a solution of [TOTA][BArF4] in toluene at room temperature, an immediate light green solution was observed which quickly turned to pale yellow. Analysis via1H NMR spectroscopy of the crude reaction mixture shows the formation of C4-adduct (11, 79%), and C2-adduct (11′, 10%), and a minor byproduct assigned to the imidazole salt (12, 10%). When the reaction was performed at −78 °C, the C4/C2 selectivity was reversed, and the predominant formation of the C2 adduct 11′ was observed, along with a trace of imidazole salt. The two isomers are easily differentiated by 1H NMR spectroscopy, specifically with the signals corresponding to the tBu groups. In the C4-adduct 11, the tBu groups are non-equivalent (1.36 and 1.34 ppm), while the C2-adduct 11′ conserves a C2 rotation axis rendering both tBu groups equivalent (1.53 ppm). Furthermore, crystals of 11, obtained by slow diffusion of pentane into CH2Cl2 at −20 °C, were subjected to X-ray diffraction analysis which confirmed its structure as a C4–NHC adduct (Scheme 4). As observed in NMR, the formation of the thermodynamically favorable compound (“C4” adduct) is characterized by the formation of the single bond C1–C20 (1.565(3) Å) corresponds to the homolytic coupling product following the SET process. This bond, although slightly longer than a standard C–C ((∼1.54) Å, +0.02 Å) has a covalent character that is only weakened by the steric hindrance imposed by the tBu groups. The presence of a CC double bond is also noted between the C20–C21 atoms (1.361(3) Å) as postulated by 1H NMR spectroscopy.
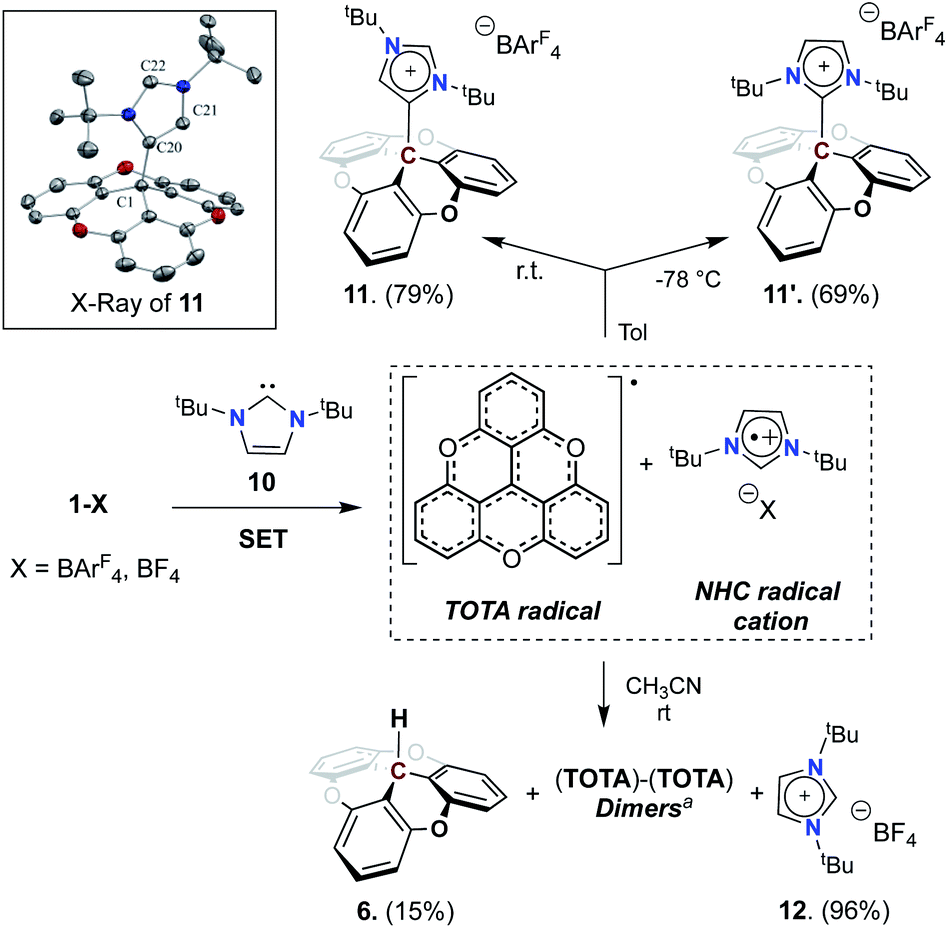 | ||
| Scheme 4 Reactivity of TOTA+ with NHC carbene. aUnless otherwise noted all reactions were carried out on a 0.05 mmol scale using [1+], 0.05 mmol ItBu (10), 4 ml solvent, rt. aSee ESI† for dimer structures. | ||
Interestingly, when TOTA+ and ItBu were mixed in CH3CN at room temperature a deep-green solution was formed, which immediately turned to brown with a purple precipitate. From the reaction mixture, the imidazolium salt (12), TOTA–H (7), TOTA-dimers were isolated, with only traces of the C4-adduct (11) (see Scheme 4 and ESI†). The TOTA-dimers formation can be rationalized by the dimerization of TOTA˙ radicals.33 However, consistent with previous report of such radical,34 monitoring the CH3CN reaction mixture by UV-Vis spectroscopy and EPR spectroscopy did not allow the observation of TOTA˙ radical (see Fig. S107†).35 Trapping experiments with TEMPO and benzoyl peroxide were also unsuccessful (see ESI†). These observations suggest that a single electron transfer (SET)36 between TOTA+ and the NHC occurred, and then followed by radical recombination to form 11 or 11′ in non-polar solvent such as toluene, or a mixture of products in dissociative solvent such as acetonitrile. The calculated electron affinity and ionization potential of TOTA+ (−7.41 eV) and ItBu (+5.59 eV) suggest that SET process in the ground state is accessible (see Table S15†).37
The TOTA–NHC adducts 11 and 11′ were studied using computational methods (Fig. 6). As observed experimentally, the ground state configuration of the adduct corresponds to 11 (C4–NHC adduct); being lower in energy than its conformational isomer 11′ by 37.8 kcal mol−1. The difference in energy is also reflected in the elongation of the C(TOTA)–C(NHC) bond distances, with a 0.06 Å difference between the C4 and C2 adduct. These differences in energy and bond distances are attributed to the increased steric hindrance of the C2 adduct, where both tBu groups are in close proximity to TOTA, compared to the C4 adduct, where one of the tBu groups is pointing away of TOTA.
 | ||
| Fig. 6 Relative energy and bond TOTA–NHC bond distances from the calculated DFT models of 11 and 11′. | ||
Conclusions
We have demonstrated that trioxatrianguleniums (TOTA+) and sterically hindered phosphines, such as P(tBu)3, can act as a frustrated Lewis pair. The VT-NMR experiments, single crystal X-ray diffraction crystallography, and UV-visible spectroscopic analysis supports the existence of a FLP encounter complex. These newly identified FLPs mimic the reactivity of main group FLPs in reactions such as heterolytic S–S bond cleavage, dehydrogenation of 1,4-cyclohexadiene, fixation of formaldehyde, C–Br bond cleavage in tert-butyl bromide, and interception of a Staudinger reaction intermediate. DFT calculations have demonstrated the possible reactivity mode of the FLP system for disulfide cleavage. Furthermore, the reactivity of TOTA+ with other Lewis bases such as NHC was investigated. Control experiments and EPR studies revealed that this pair undergoes the SET step for the formation Lewis acid–base adduct. It is noteworthy that this carbon-based Lewis acid is significantly less oxophilic than boron, aluminum, and silicon-based Lewis acids, making it both air and moisture stable. Considering the efficiency of this FLPs system, it could be useful for other organic transformation in the future. Extending this family of carbon Lewis acids by developing FLPs containing other trioxatrianguleniums salts with and different Lewis bases are ongoing in our lab.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful for financial support from the University of Arizona for this work. We thank Dr Andrei Astashkin (EPR and X-Ray facility of the University of Arizona) for performing the EPR measurements and helping with the analysis of the EPR spectra, as well as providing help for the XRD data collection and analysis. All NMR data were collected in the NMR facility of the Department of Chemistry and Biochemistry at the University of Arizona, and we thank Dr Jixun Dai for his help with 2D-NMR analysis. The purchase of the Bruker NEO 500 MHz spectrometer were supported by the National Science Foundation under Grant Number 1920234, and the University of Arizona. J. M. V. thanks the diversity and inclusion efforts from individuals, groups, and institutions towards a more united science community.Notes and references
- (a) D. W. Stephan and G. Erker, Angew. Chem. Int. Ed., 2015, 54, 6400–6441 ( Angew. Chem. , 2015 , 127 , 6498–6541 ) CrossRef CAS; (b) D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 10018–10032 CrossRef CAS; (c) G. Erker and D. W. Stephan, Frustrated Lewis Pairs I: Uncovering and Understanding, Springer, New York, 2013 Search PubMed; (d) G. Erker and D. W. Stephan, Frustrated Lewis Pairs II: Expanding the Scope, Springer, New York, 2013 Search PubMed; (e) J. S. J. McCahill, G. C. Welch and D. W. Stephan, Angew. Chem. Int. Ed., 2007, 46, 4968–4971 ( Angew. Chem. , 2007 , 119 , 5056–5059 ) CrossRef CAS.
- (a) L. Liu, B. Lukose, P. Jaque and B. Ensing, Green Energy Environ., 2019, 4, 20–28 CrossRef; (b) S. Mukherjee and P. Thilagar, J. Chem. Sci., 2015, 127, 241–255 CrossRef CAS; (c) D. W. Stephan, Acc. Chem. Res., 2015, 48, 306–316 CrossRef CAS; (d) D. W. Stephan, Org. Biomol. Chem., 2008, 6, 1535–1539 RSC.
- (a) L. C. Wilkins and R. L. Melen, Encycl. Inorg. Bioinorg. Chem., 2017, 1, 1–24 Search PubMed; (b) D. W. Stephan, Science, 2016, 354, aaf7229 CrossRef.
- S. A. Weicker and D. W. Stephan, Bull. Chem. Soc. Jpn., 2015, 88, 1003–1016 CrossRef CAS.
- (a) T. A. Kinder, R. Pior, S. Blomeyer, B. Neumann, H. G. Stammler and N. W. Mitzel, Chem.–Eur. J., 2019, 25, 5899–5903 CrossRef CAS; (b) D. J. Scott, N. A. Phillips, J. S. Sapsford, A. C. Deacy, M. J. Fuchter and A. E. Ashley, Angew. Chem., Int. Ed., 2016, 55, 14738–14742 ( Angew. Chem. , 2016 , 128 , 14958–14962 ) CrossRef CAS; (c) T. J. Herrington, B. J. Ward, L. R. Doyle, J. McDermott, A. J. P. White, P. A. Hunt and A. E. Ashley, Chem. Commun., 2014, 50, 12753–12756 RSC; (d) M. Reißmann, A. Schäfer, S. Jung and T. Müller, Organometallics, 2013, 32, 6736–6744 CrossRef.
- M. Alcarazo, Dalton Trans., 2011, 40, 1839–1845 RSC.
- (a) J. Zhou, L. L. Cao, L. Liu and D. W. Stephan, Dalton Trans., 2017, 46, 9334–9338 RSC; (b) M. H. Holthausen, T. Mahdi, C. Schlepphorst, L. J. Hounjet, J. J. Weigand and D. W. Stephan, Chem. Commun., 2014, 50, 10038–10040 RSC; (c) J. W. Runyon, O. Steinhof, H. V. R. Dias, J. C. Calabrese, W. J. Marshall and A. J. Arduengo III, Aust. J. Chem., 2011, 64, 1165–1172 CrossRef CAS; (d) P. A. Chase, A. L. Gille, T. M. Gilbert and D. W. Stephan, Dalton Trans., 2009, 7179–7188 RSC.
- E. Follet, P. Mayer, D. S. Stephenson, A. R. Ofial and G. Berionni, Chem.–Eur. J., 2017, 23, 7422–7427 CrossRef CAS.
- (a) D. Palomas, S. Holle, B. Inés, H. Bruns, R. Goddard and M. Alcarazo, Dalton Trans., 2012, 41, 9073–9082 RSC; (b) B. Inés, D. Palomas, S. Holle, S. Steinberg, J. A. Nicasio and M. Alcarazo, Angew. Chem., Int. Ed., 2012, 51, 12367–12369 ( Angew. Chem. , 2012 , 124 , 12533–12536 ) CrossRef; (c) B. Inés, S. Holle, R. Goddard and M. Alcarazo, Angew. Chem., Int. Ed., 2010, 49, 8389–8391 ( Angew. Chem. , 2010 , 122 , 8567–8569 ) CrossRef.
- (a) E. J. Lawrence, E. R. Clark, L. D. Curless, J. M. Courtney, R. J. Blagg, M. J. Ingleson and G. G. Wildgoose, Chem. Sci., 2016, 7, 2537–2543 RSC; (b) E. R. Clark and M. J. Ingleson, Angew. Chem., Int. Ed., 2014, 53, 11306–11309 ( Angew. Chem. , 2014 , 126 , 11488–11491 ) CrossRef CAS; (c) E. R. Clark and M. J. Ingleson, Organometallics, 2013, 32, 6712–6717 CrossRef CAS.
- M. P. Boone and D. W. Stephan, J. Am. Chem. Soc., 2013, 135, 8508–8511 CrossRef CAS.
- (a) J. Iglesias-Sigüenza and M. Alcarazo, Angew. Chem., Int. Ed., 2012, 51, 1523–1524 ( Angew. Chem. , 2012 , 124 , 1553–1555 ) CrossRef; (b) H. Li, C. Risko, J. H. Seo, C. Campbell, G. Wu, J. L. Brédas and G. C. Bazan, J. Am. Chem. Soc., 2011, 133, 12410–12413 CrossRef CAS.
- V. R. Naidu, S. Ni and J. Franzén, ChemCatChem, 2015, 7, 1896–1905 CrossRef CAS.
- (a) J. Bosson, G. M. Labrador, C. Besnard, D. Jacquemin and J. Lacour, Angew. Chem., Int. Ed., 2021 DOI:10.1002/anie.202016643; (b) A. C. Shaikh, J. M. Veleta, J. Bloch, H. J. Goodman and T. L. Gianetti, Eur. J. Org. Chem., 2020, 17, 2553–2559 CrossRef; (c) K. Chansaenpak, M. Yang and F. P. Gabbaï, Philos. Trans. R. Soc. London, Ser. A, 2017, 375, 2017000 CrossRef; (d) R. Vanel, F.-A. Miannay, E. Vauthey and J. Lacour, Chem. Commun., 2014, 50, 12169–12172 RSC; (e) L. Cabrera, G. C. Welch, J. D. Masuda, P. Wei and D. W. Stephan, Inorg. Chim. Acta, 2006, 359, 3066–3071 CrossRef CAS.
- J. Bosson, J. Gouin and J. Lacour, Chem. Soc. Rev., 2014, 43, 2824–2840 RSC.
- (a) M. Marinova, S. Pascal, L. Guénée, C. Besnard, B. Shivachev, K. Kostova, C. Villani, R. Franzini, V. Dimitrov and J. Lacour, J. Org. Chem., 2020, 85, 11908–11923 CrossRef CAS; (b) I. H. Delgado, S. Pascal, C. Besnard, S. Voci, L. Bouffier, N. Sojic and J. Lacour, Chem.–Eur. J., 2018, 24, 10186–10195 CrossRef CAS; (c) I. Hernandez Delgado, S. Pascal, A. Wallabregue, R. Duwald, C. Besnard, L. Guénée, C. Nançoz, E. Vauthey, R. C. Tovar, J. L. Lunkley, G. Muller and J. Lacour, Chem. Sci., 2016, 7, 4685–4693 RSC.
- (a) W.-C. Liu, Y. Kim and F. P. Gabbai, Chem.–Eur. J., 2021 DOI:10.1002/chem.202100389; (b) G. Park and F. P. Gabbaï, Chem. Sci., 2020, 11, 10107–10112 RSC; (c) A. C. Shaikh, J. Moutet, J. M. Veleta, M. Hossain, J. Bloch, A. V. Astashkin and T. L. Gianetti, Chem. Sci., 2020, 11, 11060–11067 RSC; (d) L. Mei, J. M. Veleta, J. Bloch, H. J. Goodman, D. Pierce-Navarro, A. Villalobos and T. L. Gianetti, Dalton Trans., 2020, 49, 16095–16105 RSC; (e) M. Karimi, R. Borthakur, C. L. Dorsey, C.-H. Chen, S. Lajeune and F. P. Gabbaï, J. Am. Chem. Soc., 2020, 142, 13651–13656 CrossRef CAS; (f) L. Wilkins, Y. Kim, E. Litle and F. P. Gabbaï, Angew. Chem., Int. Ed., 2019, 58, 18266–18270 ( Angew. Chem. , 2019 , 131 , 18434–18438 ) CrossRef CAS.
- J. C. Martin and R. G. Smith, J. Am. Chem. Soc., 1964, 86, 2252–2256 CrossRef CAS.
- (a) B. Qiao, B. E. Hirsch, S. Lee, M. Pink, C.-H. Chen, B. W. Laursen and A. H. Flood, J. Am. Chem. Soc., 2017, 139, 6226–6233 CrossRef CAS; (b) A. Shivalingam, A. Vyšniauskas, T. Albrecht, A. J. P. White, M. K. Kuimova and R. Vilar, Chem.–Eur. J., 2016, 22, 4129–4139 CrossRef CAS; (c) T. J. Sørensen and B. W. Laursen, J. Org. Chem., 2010, 75, 6182–6190 CrossRef; (d) F. Westerlund, C. B. Hildebrandt, T. J. S. ørensen and B. W. Laursen, Chem.–Eur. J., 2010, 16, 2992–2996 CrossRef CAS; (e) A. Pothukuchy, S. Ellapan, K. R. Gopidasb and M. Salazara, Bioorg. Med. Chem. Lett., 2003, 13, 1491–1494 CrossRef CAS; (f) J. Reynisson, R. Wilbrandt, V. Brinck, B. W. Laursen, K. Nørgaard, N. Harrit and A. M. Brouwer, Photochem. Photobiol. Sci., 2002, 1, 763–773 CrossRef CAS; (g) B. W. Laursen and F. C. Krebs, Angew. Chem., Int. Ed., 2000, 39, 3432–3434 ( Angew. Chem. , 2000 , 112 , 3574–3576 ) CrossRef CAS.
- (a) DFT calculations on TOTA+ are reported in the ESI,†; (b) The AN of 1-BArF4 was measured following the Gutmann–Beckett method (see ESI†)..
- (a) R. Löw, T. Rusch, T. Moje, F. Röhricht, O. M. Magnussen and R. Herges, Beilstein J. Org. Chem., 2019, 15, 1815–1821 CrossRef; (b) M. Lofthagen, R. Vernonclark and K. K. Baldridge, J. Org. Chem., 1992, 57, 61–69 CrossRef CAS; (c) P. Huszthy, K. Lempert and G. Simig, J. Chem. Soc., Perkin Trans. 2, 1985, 1351–1354 RSC.
- T. J. Herrington, A. J. W. Thom, A. J. P. White and A. E. Ashley, Dalton Trans., 2012, 41, 9019–9022 RSC.
- J.-M. Denis, H. Forintos, H. Szelke, L. Toupet, T.-N. Pham, P.-J. Madecb and A.-C. Gaumont, Chem. Commun., 2003, 54–55 RSC.
- (a) H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC; (b) C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- L. Rocchigiani, G. Ciancaleoni, C. Zuccaccia and A. Macchioni, J. Am. Chem. Soc., 2014, 136, 112–115 CrossRef CAS.
- (a) L. Fan, A. R. Jupp and D. W. Stephan, J. Am. Chem. Soc., 2018, 140, 8119–8123 CrossRef CAS; (b) O. J. Metters, S. J. K. Forrest, H. A. Sparkes, I. Manners and D. F. Wass, J. Am. Chem. Soc., 2016, 138, 1994–2003 CrossRef CAS.
- 19F–1H HOESY NMR experiments did not reveal the presence of an interaction between TOTA+ and the BArF4 counter ions (Fig. S35 and S43†)..
- (a) A. J. V. Marwitz, J. L. Dutton, L. G. Mercier and W. E. Piers, J. Am. Chem. Soc., 2011, 133, 10026–10029 CrossRef CAS; (b) L. C. Brown, J. M. Hogg, M. Gilmore, L. Moura, S. Imberti, S. Gärtner, H. Q. N. Gunaratne, R. J. O'Donnell, N. Artioli, J. D. Holbrey and M. Swadźba-Kwaśny, Chem. Commun., 2018, 54, 8689–8692 RSC.
- P. Skowronek, J. Ścianowski, A. J. Pacuła and J. Gawroński, RSC Adv., 2015, 5, 69441–69444 RSC.
- T. C. Johnstone, G. N. J. H. Wee and D. W. Stephan, Angew. Chem. Int. Ed., 2018, 57, 5881–5884 ( Angew. Chem. , 2008 , 130 , 5983–5986 ) CrossRef CAS.
- A. M. Chapman, M. F. Haddow and D. F. Wass, J. Am. Chem. Soc., 2011, 133, 18463–18478 CrossRef CAS.
- D. H. A. Boom, A. R. Jupp, M. Nieger, A. W. Ehlers and J. C. Slootweg, Chem.–Eur. J., 2019, 25, 13299–13308 CrossRef CAS.
- M. J. Sabacky, C. S. Johnson, R. G. Smith, H. S. Gutowsky and J. C. Martin, J. Am. Chem. Soc., 1967, 89, 2054–2058 CrossRef CAS.
- E. Müller, A. Moosmayer, A. Rieker and K. Scheffler, Tetrahedron Lett., 1967, 39, 3877–3880 CrossRef.
- S. Dileesh and K. R. Gopidas, Chem. Phys. Lett., 2000, 330, 397–402 CrossRef CAS.
- Selected review: (a) D. Ayan, E. Richards and R. Melen, Angew. Chem., Int. Ed., 2021, 60, 53–65 CrossRefselected examples: (b) Z. Dong, C. Pezzato, A. Sienkiewicz, R. Scopelliti, F. Fadaei-Tirani and K. Severin, Chem. Sci., 2020, 11, 7615–7618 RSC; (c) R. J. Andrews and D. W. Stephan, Chem.–Eur. J., 2020, 26, 7194–7198 CrossRef CAS.
- (a) F. Holtrop, A. R. Jupp, B. J. Kooij, N. P. van Leest, B. de Bruin and J. C. Slootweg, Angew. Chem., Int. Ed., 2020, 59, 22210–22216 CrossRef CAS; (b) A. Merk, H. Großekappenberg, M. Schmidtmann, M.-P. Luecke, C. Lorent, M. Driess, M. Oestreich, H. F. T. Klare and T. Müller, Angew. Chem., Int. Ed., 2018, 57, 15267–15271 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2035921–2035925. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc05893a |
| This journal is © The Royal Society of Chemistry 2021 |