
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNS3/4A helicase inhibitory alkaloids from Aptenia cordifolia as HCV target†
Asmaa Abo Elgoud Saida,
Ahmed H. Afifib,
Taha F. S. Alic,
Mamdouh Nabil Samy a,
Usama Ramadan Abdelmohsen*ad,
Mostafa A. Fouada and
Eman Zekry Attiaa
a,
Usama Ramadan Abdelmohsen*ad,
Mostafa A. Fouada and
Eman Zekry Attiaa
aDepartment of Pharmacognosy, Faculty of Pharmacy, Minia University, 61519 Minia, Egypt. E-mail: usama.ramadan@mu.edu.eg; Fax: +20-86-2369075; Tel: +20-86-2347759
bDepartment of Pharmacognosy, Division of Pharmaceutical Industries, National Research Center, Dokki, 12622, Giza, Egypt
cDepartment of Medicinal Chemistry, Faculty of Pharmacy, Minia University, 61519 Minia, Egypt
dDepartment of Pharmacognosy, Faculty of Pharmacy, Deraya University, Universities Zone, 61111 New Minia City, Egypt
First published on 6th October 2021
Abstract
Chemical investigation of Aptenia cordifolia roots extract, using chromatographic and spectroscopic techniques, resulted in isolation and identification of eight known compounds. The basic ethyl acetate fraction (alkaloidal fraction) afforded O-methylsceletenone, epinine, 4-methoxy phenethylamine, and N-methyl tyramine while, the acidic ethyl acetate fraction (non-alkaloidal fraction) afforded only cis-N-coumaroyl tyramine. Moreover, the petroleum ether fraction afforded capric acid, tricosanol, and a mixture of β-sitosterol & stigma sterol. Upon screening of anti HCV activity of these three fractions, only the basic ethyl acetate fraction had high activity against HCV with an IC50 value equal to 2.4 μg mL−1 which provoked us to carry out structure based in silico virtual screening on the drug targets of HCV of isolated alkaloidal compounds as well as the previously dereplicated alkaloids through metabolomics from the antiviral active fraction. The tortuosamine compound exhibited the strongest binding to the active site of NS3/4A helicase with a binding affinity (−7.1 kcal mol−1) which is very close to the native ligand (−7.7 kcal mol−1).
1. Introduction
Hepatitis C virus (HCV) was first identified in 1989.1 It is an enveloped, positive stranded RNA virus belonging to the family Flaviviridae and considered to be a public health concern which infects more than 185 million people worldwide.2,3 HCV is the major etiological agent of non-A non-B hepatitis as the infection occurs principally through blood or blood-derived products.4 This viral infection is the leading cause of cirrhosis and, as a result, liver transplantation around the world.5,6 The conventional HCV treatment has advanced quickly, as several protease inhibitors, such as boceprevir and telaprevir have recently been recommended as hepatitis C therapy. These novel inhibitors, however, have been linked to drug toxicity, the creation of resistance mutants, high cost and efficacy limitation.7 Indeed, the challenge is to develop a potent, inexpensive and widely available antiviral drug.Medicinal plants contain a variety of compounds that have the potential to treat ailments, particularly infectious disorders. Different studies declared beneficial effects of secondary metabolites derived from medicinal plants against viral diseases. A broad variety of these secondary metabolites, such as alkaloids, coumarins, lignans and polyphenolic compounds affect cellular functions and replication of various infectious viruses. Medicinal plants' history dates back to the origin of human civilization on this planet. Inhibitory activity of medicinal plants extracts against replication of several viruses was reported latterly, such as, herpes simplex virus type2 (HSV-2),8 HIV,9,10 hepatitis B virus (HBV),11,12 emerging viral infections associated with poxvirus, and severe acute respiratory syndrome (SARS) virus.13 Recently, various studies have been conducted to investigate the antiviral activity of a variety of plants. As the methanolic extract of both Terminalia bellerica seeds and Enicostemma axillare showed antiviral activity against hepatitis B.14 On the other hand, Stixis scandens Lour leafe extract showed powerful antiviral activity against porcine epidemic diarrhea virus.15 Moreover, silymarin, epigallocatechin gallate, naringenin,16 phenolic compounds of grape seed and aqueous extract of Eclibta alba leaves exhibited anti HCV activity.14 The alcoholic and/or water soluble extracts of medicinal plants were utilized during furthermost of these antiviral studies. Genus Aptenia is endemic to South Africa and includes four species, which are currently recognized as Aptenia cordifolia L.F. Schwantes, Aptenia geniculiflora L. Bittrich ex Gerbaulet, Aptenia haeckeliana A. Berger Bittrich ex Gerbaulet, and Aptenia lancifolia L. Bolus.17 Aptenia cordifolia, a well-known groundcover, was named heart-leaved ice plant and has only one synonym “Mesembryanthemum cordifolium”.18 So as to provide a complementary therapy for previously existing remedies, the antiviral activity of different fractions (petroleum ether, acidic ethyl acetate and basic ethyl acetate) derived from the total ethanol extract of A. cordifolia roots were examined against HCV by in vitro cells culture using luciferase assay and the cytotoxic effect was accessed by MTT assay. Additionally, we aimed to identify which compound/s in the basic ethyl acetate fraction responsible for the anti HCV activity as well as the mechanism of action by undergoing docking studies on the drug targets of HCV. Although there are many HCV targets, our docking study will focus on the most attractive viral proteins required for HCV replication. These targets are NS5B HCV RNA-dependent RNA polymerase, NS3/4A protease, NS3/4A helicase, and a new allosteric pocket in the HCV NS3–NS4A protein located at the interface between the protease and helicase domains. The compounds used for docking studies were the isolated alkaloids from the active fraction (basic ethyl acetate fraction), as well as, the previously nine dereplicated alkaloids from the basic ethyl acetate fraction through metabolomic analysis.19
2. Results and discussion
2.1. Identification of the isolated compounds
The petroleum ether fraction afforded four compounds. Mixture of β-sitosterol & stigma sterol (8 mg) was isolated as white amorphous powder, and was simply identified by comparison with authentic sample. This mixture was previously isolated from the aerial parts of the plant.20Tricosanol alcohol (5 mg) was isolated as white powder and the complete assignment was confirmed by investigation of 1H-NMR, DEPT-Q and ESI-MS analyses. The molecular formula was determined to be C23H48O by ESI-MS that showed a pseudo molecular ion peak at 339 [M–H]−. The compound was identified by comparison of the ESI-MS and NMR data, with the reported data21 and this is the first time for its isolation from family Aizoaceae.
Capric acid was isolated as white powder (4 mg), the molecular formula was confirmed to be C10H20O2 by ESI-MS that showed a molecular ion peak at 172.8 [M]+. The compound was identified by comparison of the ESI-MS and 1H-NMR data with the reported data,22 and it was previously identified by GC-MS in Attalea dubia family Arecaceae, but this is the first time for its isolation from family Aizoaceae.
Cis-N-coumaroyl tyramine (3),which was isolated as white powder (5 mg), HR-ESI-MS showed a pesudo molecular ion peak at m/z 284.128 [M + H]+, (calculated mass at m/z 284.128) corresponding to the molecular formula C17H18NO3 in addition to, a characteristic fragment peak at m/z 147.057 corresponding to loss of [M–C8H11NO]. It was identified by comparison 1H-NMR data with the literature,23 and this is the first report for its isolation from family Aizoaceae.
4-Methoxyphenethylamine (1) was isolated as white powder (3 mg, purity 97%) HR-ESI-MS of compound 1 showed a pseudo molecular ion peak at m/z 152.1066 [M + H]+ (calculated mas 152.1075), corresponding to the molecular formula C9H14NO. From the HR-ESI-MS, 1H-NMR data and from the reported data in literature24 compound 1 was identified as 4-methoxyphenethylamine which was previously isolated from Coryphantha pectinata25 and this is the first report for its isolation from family Aizoaceae.
N-Methyltyramine (2) was isolated as white powder (4 mg). HR-ESI-MS of compound 2 showed a pseudo molecular ion peak at m/z 152.0697 [M + H]+ (calculated mas 152.1031), corresponding to the molecular formula C9H14NO. From ESI-MS, 1H-NMR data and from the data reported in literature26 compound 2 was identified as N-methyltyramine which was previously isolated from Sawa millet seeds26 and this is the first report for its isolation from family Aizoaceae.
O-methylsceletenone (4) was isolated as reddish brown powder (20 mg, purity 98%), the complete assignment of compound 4 was confirmed by investigation of 1H-NMR, DEPT and HR-ESI-MS. Positive HR-ESI-MS of compound 4 showed a quassi molecular ion peak at m/z 258.1462 [M + H]+, (calculated mass at m/z 258.1494) corresponding to the molecular formula C16H20NO2. From the HR-ESI-MS, NMR data and in comparison with the literature,27 compound 4 was identified as O-methyl sceletenone. It was previously isolated from different organs of the investigated plant.28
Epinine (5) was isolated as yellow powder (4 mg, purity 97%) HR-ESI-MS of compound 5 showed a pseudo molecular ion peak at m/z 168.9802 [M + H]+, (calculated mass at m/z 167.1024) corresponding to the molecular formula C9H14NO2. Comparison of the HR-ESI-MS and 1H-NMR data of compound 5, with the data reported in literature29,30 revealed that compound 5 is epinine alkaloid which was previously isolated from Cytisus scoparius Family Leguminosae31 and this is the first report for its isolation from family Aizoaceae.
2.2. Antiviral activity and molecular docking simulation
Upon screening the HCV antiviral activity of different fractions of A. cordifolia roots extract, only the basic ethyl acetate fraction (alkaloidal fraction) displayed high potential against HCV with IC50 value equal to 2.4 μg mL−1. According to the HCV genome, there are four structural (core, E1, E2, and p7) and six non-structural (NS2, NS3, NS4A, NS4B, NS5A, and NS5B) proteins.32,33 The non-structural proteins that accounted for viral production and replication are the primary targets for the currently FDA-approved drugs designed against HCV.34 In addition, there is a newly discovered binding pocket between the protease and helicase domains, also considered a druggable target. Of the six non-structural viral proteins, NS2/3 autoprotease, which mediates a single cleavage in the polyprotein, has been less well explored. Moreover, the NS5A protein, involved in both RNA replication and infectious virus assembly, is not a promising target because it has no known enzymatic activity.35 Therefore, our main target is in silico prediction of the binding affinities of the four alkaloids isolated from the alkaloidal fraction of the A. cordifolia roots named, 4-methoxyphenethylamine (1), N-methyl tyramine (2), O-methylsceletenone (4) and epinine (5) (Fig. 1) as well as previously reported nine alkaloids dereplicated from alkaloidal fraction through metabolomic analysis (3 and 6–12) (Fig. 2)19 against the four non-structural HCV antiviral targets: namely NS5B HCV RNA-dependent RNA polymerase, NS3/4A protease, NS3/4A helicase, and a new allosteric pocket in the HCV NS3–NS4A protein located at the interface between the protease and helicase domains.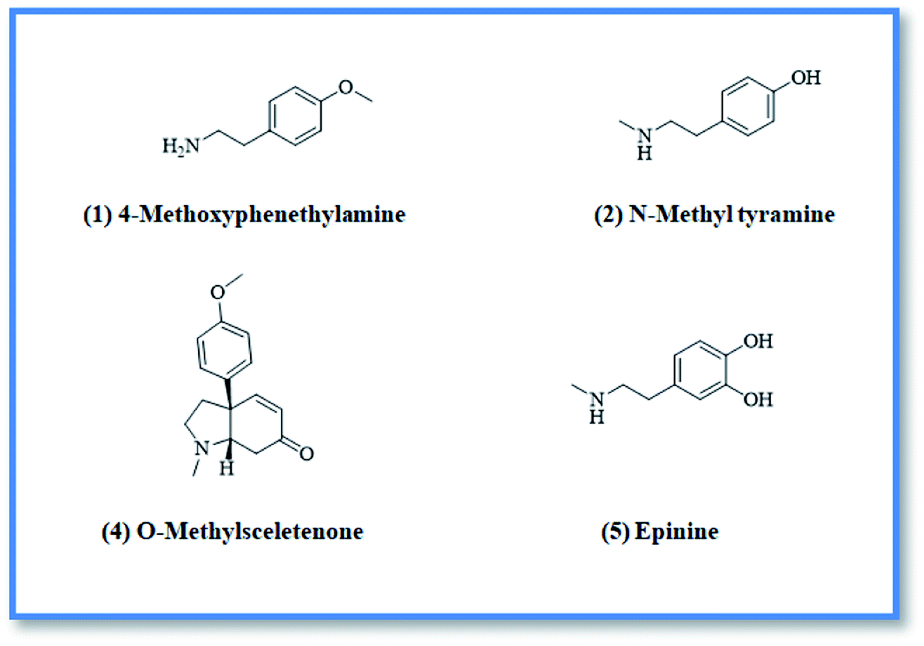 | ||
| Fig. 1 Isolated compounds from alkaloidal fraction. | ||
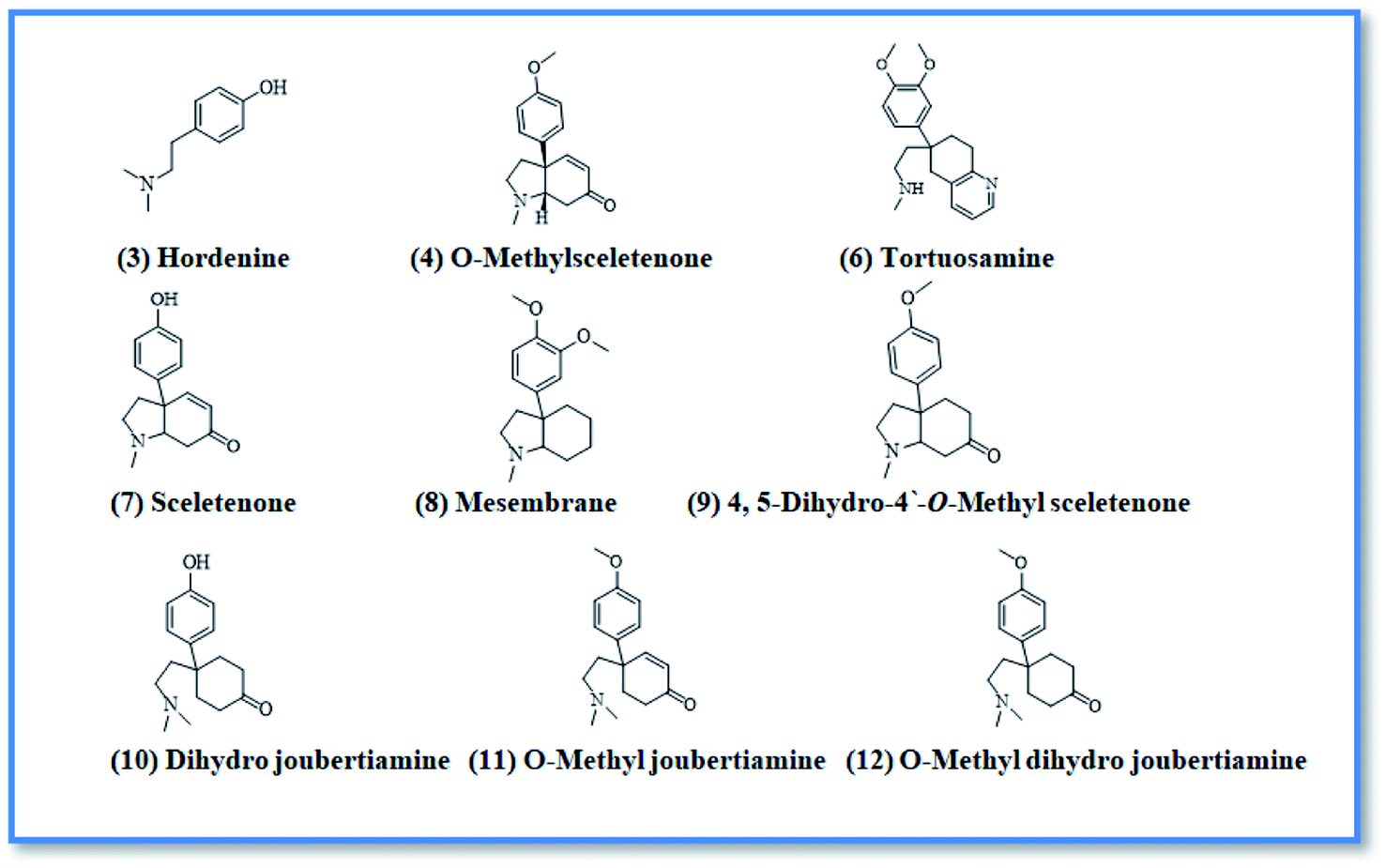 | ||
| Fig. 2 Dereplicated compounds from alkaloidal fraction. | ||
Before docking of the screened compounds, we assessed our MOE induced docking protocol by re-docking the co-crystallized ligand within the active site of NS5B HCV RNA-dependent RNA polymerase (PDB code: 3H2L), NS3/4A protease (PDB code: 6NZT), NS3/4A helicase (PDB code: 4OKS), and a new allosteric pocket in the HCV NS3–NS4A protein located at the interface between the protease and helicase domains (PDB code: 4B73).
As seen in Fig. 3–6, MOE successfully re-docked the native ligand in its original pose with acceptable RMSD values of 0.28, 0.62, 0.31, and 0.16 Å respectively. The binding free energies of the re-docked ligand, as well as the screened compounds, were summarized in Table 1.
 | ||
| Fig. 3 The top-scoring docked pose of compound 6 to the HCV NS5B polymerase active site (PDB code: 3H2L) as predicted by MOE 2019.01. (A) An overview of the HCV NS5B polymerase active site (B) comparison of modeled binding mode of the co-crystallized ligand (a novel bicyclic dihydro-pyridinone inhibitor) (magenta stick model) and its superposed docking conformation (blue stick model). (C) Detailed binding mode of compound 6 (magenta stick model) displaying hydrogen bonds (black dashed line) and H–π interactions (red dashed line) with the key amino acid residue (blue stick model). (D) 2D depiction of compound 6 binding interactions with the critical amino acid residue. | ||
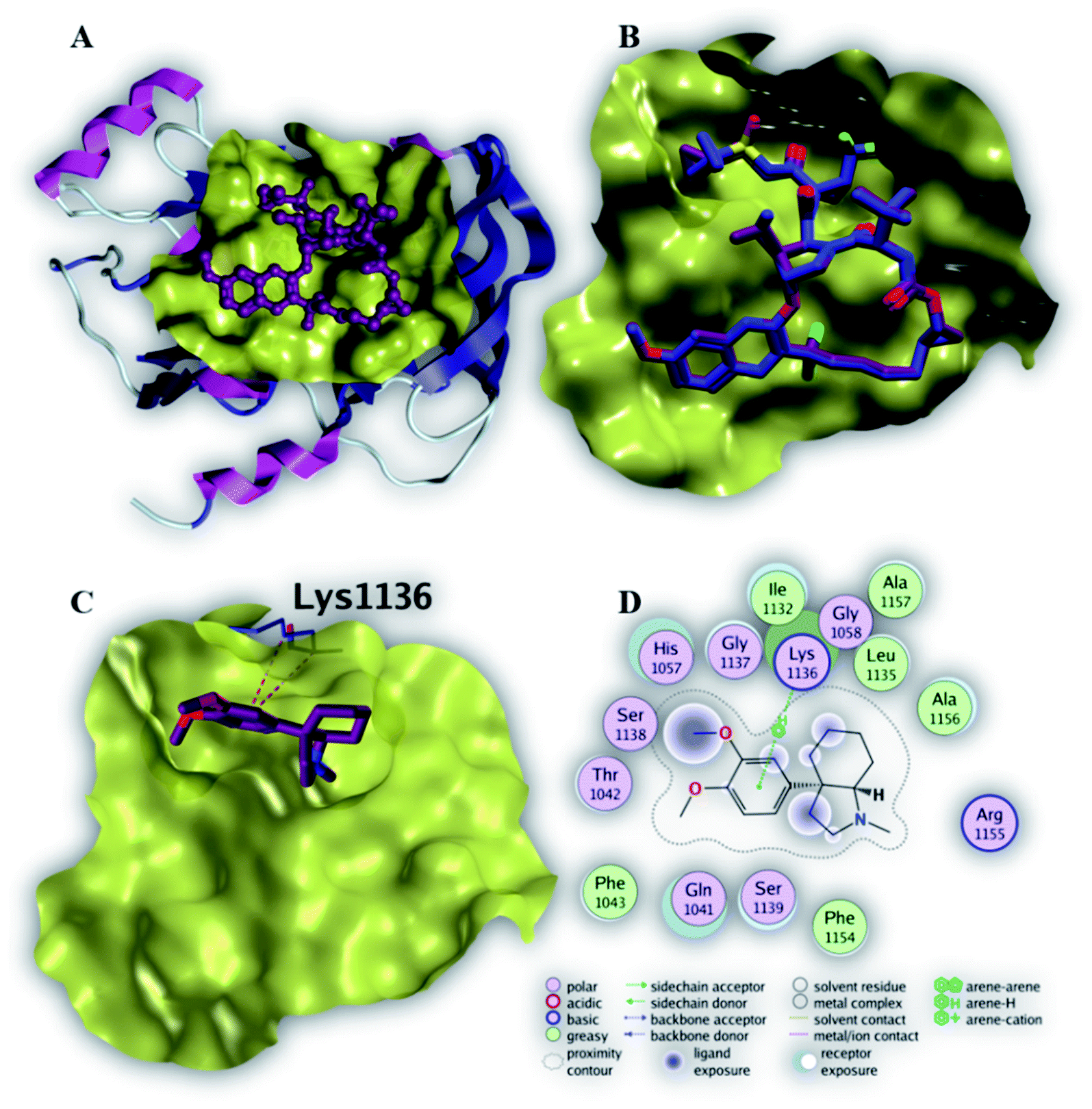 | ||
| Fig. 4 The top-scoring docked pose of compound 8 to the HCV NS3/4A protease active site (PDB code 6NZT) as predicted by MOE 2019.01. (A) An overview of the HCV NS3/4A protease active site (B) comparison of modeled binding mode of the co-crystallized ligand Voxilaprevir (magenta stick model) and its superposed docking conformation (cyan stick model). (C) Detailed binding mode of compound 8 (green stick model) displaying H–π bond (red dashed line) with the key amino acid residue (cyan stick model). (D) 2D depiction of compound 8 binding interactions with the key amino acid residue. | ||
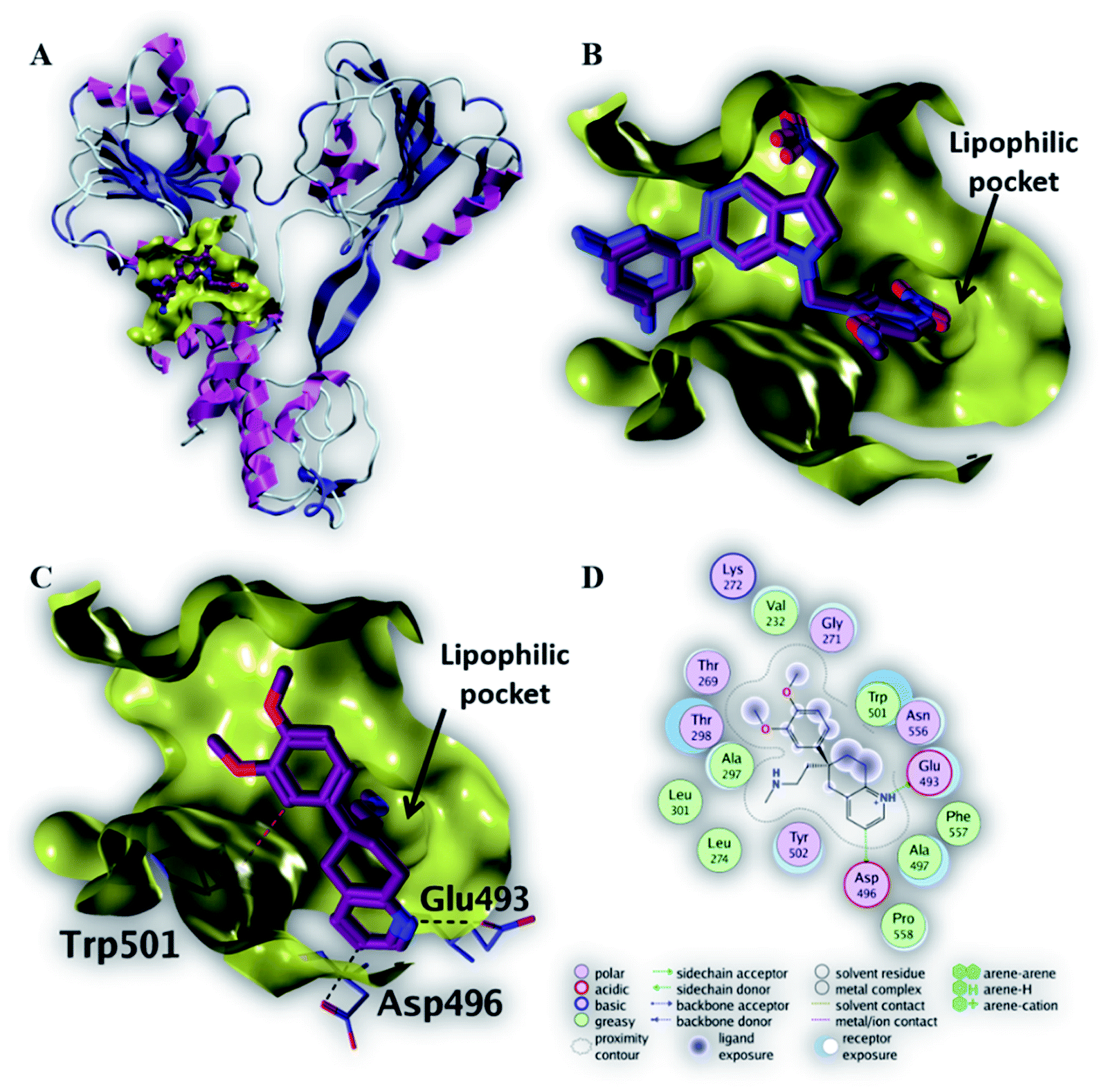 | ||
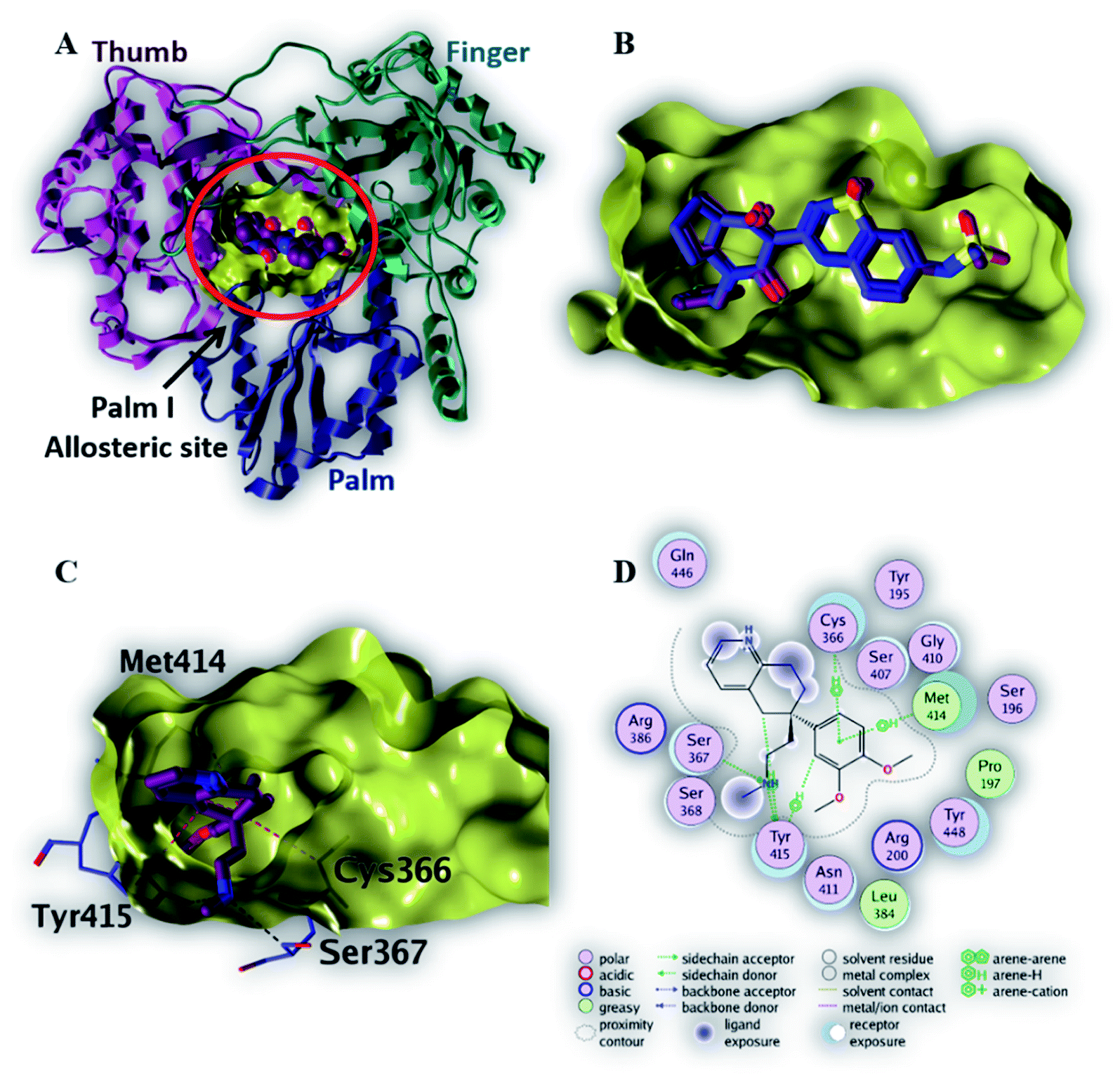
| Fig. 5 The top-scoring docked pose of compound 6 to the HCV NS3 helicase active site (PDB code 4OKS) as predicted by MOE 2019.01. (A) An overview of the HCV NS3 helicase active site (B) comparison of modeled binding mode of the co-crystallized ligand (magenta stick model) and its superposed docking conformation (blue stick model). (C) Detailed binding mode of compound 6 (magenta stick model) displaying hydrogen bonds (black dashed line) with the key amino acid residue (blue stick model). (D) 2D depiction of compound 6 binding interactions with the key amino acid residues. | ||
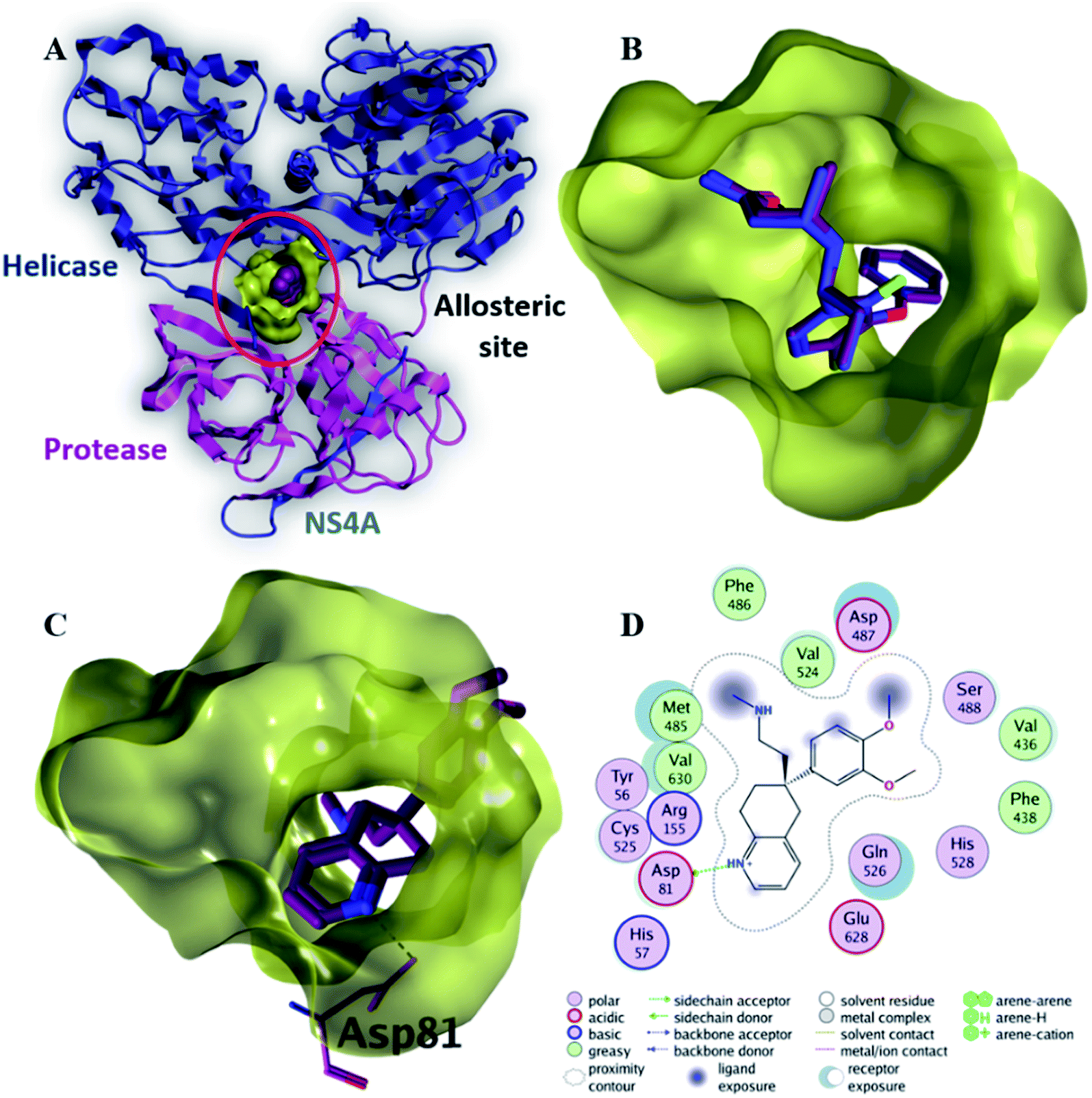 | ||
| Fig. 6 The top-scoring docked pose of compound 6 to the allosteric site on the HCV NS3–NS4A protein, located between the protease and helicase domains of the HCV NS3 protein (PDB code: 4B73) as predicted by MOE 2019.01. (A) An overview of the allosteric site on the HCV NS3–NS4A protein (B) comparison of modeled binding mode of the co-crystallized ligand (magenta stick model) and its superposed docking conformation (blue stick model). (C) Detailed binding mode of compound 6 (green stick model) displaying hydrogen and ionic bonds (black dashed line) with the key amino acid residue (blue stick model). (D) 2D depiction of compound 6 binding interactions with the key amino acid residue. | ||
| Ligand | Binding affinity ΔG (kcal mol−1) | |||
|---|---|---|---|---|
| Polymerase | Protease | Helicase | Protease–helicase allosteric | |
| Native ligand (RMSD) | −9.3 (0.28) | −11.3 (0.62) | −7.7 (0.31) | −9.4 (0.16) |
| 1 | −5.1 | −4.8 | −4.7 | −5.5 |
| 2 | −5.1 | −4.8 | −4.9 | −5.7 |
| 3 | −5.3 | −5.0 | −5.1 | −5.9 |
| 4 | −6.6 | −5.9 | −6.5 | −7.0 |
| 5 | −5.2 | −4.8 | −5.3 | −5.7 |
| 6 | −7.4 | −6.7 | −7.1 | −7.9 |
| 7 | −6.1 | −5.6 | −5.7 | −6.7 |
| 8 | −7.1 | −6.9 | −6.0 | −7.4 |
| 9 | −6.3 | −6.4 | −5.7 | −7.3 |
| 10 | −6.4 | −5.9 | −6.0 | −7.2 |
| 11 | −7.1 | −6.4 | −6.4 | −7.1 |
| 12 | −6.8 | −6.9 | −6.4 | −7.3 |
Among the screened compounds, compound 6 showed the highest binding affinity (7.4 kcal mol−1) to the active site of NS5B HCV RNA-dependent RNA polymerase. Fig. 3C demonstrated that compound 6 binds within the palm I region, where the phenyl ring makes three H–π interactions with three important amino acid residues (Tyr 415, Met 414 and Cys 366) in addition to two important hydrogen bonds made by NH atom to two key amino acid residues (Tyr 415 and Ser 367). These binding interactions explain the strong binding affinity of compound 6 to the active site of NS5B HCV RNA-dependent RNA polymerase (Table 2).
| Ligand | Binding affinity (ΔG in kcal mol−1) | Interaction parameters | |||
|---|---|---|---|---|---|
| Interaction | AA residue | δ (Å) | E (kcal mol−1) | ||
| Native ligand | −9.3 | H-Donor | Asp 318 (side chain) | 2.71 | −7.2 |
| H-Acceptor | Ser 556 (side chain) | 3.11 | −1.0 | ||
| H-Acceptor | Asn 291 (side chain) | 2.88 | −3.5 | ||
| H–π | Met 414 (side chain) | 4.77 | −0.5 | ||
| 6 | −7.4 | H-Donor | Tyr 415 (side chain) | 3.10 | −0.2 |
| H-Acceptor | Ser 367 (side chain) | 3.40 | −0.2 | ||
| H–π | Tyr 415 (side chain) | 4.59 | −0.6 | ||
| H–π | Met 414 (side chain) | 4.81 | −0.3 | ||
| H–π | Cys 366 (side chain) | 4.23 | −0.2 | ||
Compounds 6, 8 and 12 demonstrated the best binding affinities (−6.7 and −6.9 and −6.9 kcal mol−1, respectively), among the screened compounds, to the active site of NS3/4A protease. Fig. 4C revealed that the phenyl ring of compound 8 binds within protease active site via a single H–π interaction with amino acid residue (Lys 1136). This binding interaction elucidates the weak binding affinity of compound 8 to the active site of NS3/4A protease (Table 3). Therefore, NS3/4A HCV protease protein is less likely to be a considerable target for our screened compounds.
| Ligand | Binding affinity (ΔG in kcal mol−1) | Interaction parameters | |||
|---|---|---|---|---|---|
| Interaction | AA residue | δ (Å) | E (kcal mol−1) | ||
| Native ligand | −11.3 | H-Donor | Arg 1155 (backbone) | 2.82 | −4.6 |
| H-Donor | His 1057 (side chain) | 3.01 | −7.5 | ||
| H-Donor | Ala 1157 (backbone) | 2.88 | −4.3 | ||
| H-Acceptor | Gly 1137 (backbone) | 3.00 | −1.1 | ||
| H-Acceptor | Gly 1137 (backbone) | 3.02 | −2.8 | ||
| H-Acceptor | Ala 1157 (backbone) | 2.95 | −3.7 | ||
| H–π | His 1057 (side chain) | 3.68 | −1.6 | ||
| 8 | −6.9 | H–π | Lys 1136 (side chain) | 4.36 | −0.6 |
As depicted in Fig. 5C, compound 6 binds strongly to the active site of NS3/4A helicase with binding affinity (−7.1 kcal mol−1) very close to the native ligand (−7.7 kcal mol−1). This strong binding affinity could be explained by two reasons. Firstly, compound 6 binds mainly through two strong hydrogen bonds to the key amino acid residues (Asp 496 and Glu 493) as well as a strong ionic bond to Glu 493 (Table 4). Secondly, the alkylamine side chain of compound 6 extended deeply in the lipophilic pocket of helicase active site which also increases the binding affinity of this compound to the target helicase protein. Consequently, NS3/4A helicase protein is more likely to be a potential target for compound 6, among our screened compounds.
| Ligand | Binding affinity (ΔG in kcal mol−1) | Interaction parameters | |||
|---|---|---|---|---|---|
| Interaction | AA residue | δ (Å) | E (kcal mol−1) | ||
| Native ligand | −7.7 | H-Donor | Trp 501 (backbone) | 2.81 | −2.3 |
| H-Acceptor | Gly 255 (backbone) | 2.91 | −3.5 | ||
| H-Acceptor | Gly 255 (backbone) | 2.92 | −4.6 | ||
| H-Acceptor | Thr 269 (side chain) | 3.52 | −1.9 | ||
| 6 | −7.1 | H-Donor | Asp 496 (side chain) | 3.35 | −0.7 |
| H-Donor | Glu 493 (side chain) | 2.91 | −13.1 | ||
| Ionic | Glu 493 (side chain) | 2.91 | −5.1 | ||
Compound 6 displayed the highest binding affinity (−7.9 kcal mol−1) to the allosteric site of the HCV NS3–NS4A protein. As summarized in Table 5, compound 6 binds to the curial amino acid residue Asp 81 forms two bonds, one hydrogen bond and one ionic bond. Fig. 6C and D showed the 3D and 2D depiction of compound 6 within the allosteric site of the HCV NS3–NS4A protein displaying hydrogen and ionic bonds with the key amino acid residue (Asp 81). Subsequently, the new allosteric site of the HCV NS3–NS4A protein is considered as a plausible target for compound 6.
| Ligand | Binding affinity (ΔG in kcal mol−1) | Interaction parameters | |||
|---|---|---|---|---|---|
| Interaction | AA residue | δ (Å) | E (kcal mol−1) | ||
| Native ligand | −9.4 | H-Donor | Asp 79 (backbone) | 3.46 | −0.7 |
| H-Donor | Cys 525 (backbone) | 3.00 | −4.2 | ||
| H-Donor | H2O (glu 628) | 3.04 | −5.3 | ||
| H-Donor | Leu 517 (backbone) | 2.99 | −1.1 | ||
| H-Acceptor | Cys 525 (backbone) | 2.97 | −4.8 | ||
| 6 | −7.9 | H-Donor | Asp 81 (side chain) | 2.97 | −4.9 |
| Ionic | Asp 81 (side chain) | 3.47 | −2.0 | ||
According to our molecular docking simulation study, compound 6 was the most promising anti-HCV candidate among the twelve members of the alkaloidal fraction of A. cordifolia roots as it gives an excellent binding affinity; specifically against NS3/4A helicase and a new allosteric pocket in the HCV NS3–NS4a protein located at the interface between the protease and helicase domains with a relative good binding energy and distance values from the amino acids of the active binding site compared with the native ligands in these proteins.
The ADME analysis has revealed that all the studied compounds show no violations toward Lipinski's rule and hence prove acceptable drug-likeness and pharmacokeintic properties with no potential toxicity (Tables 6 and 7).
| Molecule | Hydrogen bond donors | Hydrogen bond acceptors | Number of rotatable bonds | M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logP logP |
Molecular weight | Number of violations |
|---|---|---|---|---|---|---|
| 1 | 1 | 2 | 3 | 1.53 | 151.21 | 0 |
| 2 | 2 | 2 | 3 | 1.53 | 151.21 | 0 |
| 3 | 1 | 2 | 3 | 1.83 | 165.23 | 0 |
| 4 | 0 | 3 | 2 | 2.00 | 257.33 | 0 |
| 5 | 3 | 3 | 3 | 0.92 | 167.21 | 0 |
| 6 | 1 | 4 | 6 | 2.27 | 326.44 | 0 |
| 7 | 1 | 3 | 1 | 1.75 | 243.31 | 0 |
| 8 | 0 | 3 | 3 | 2.68 | 275.39 | 0 |
| 9 | 0 | 3 | 2 | 2.08 | 259.35 | 0 |
| 10 | 1 | 3 | 4 | 2.08 | 261.37 | 0 |
| 11 | 0 | 3 | 5 | 2.24 | 273.38 | 0 |
| 12 | 0 | 3 | 5 | 2.33 | 275.39 | 0 |
| Molecule | Bioavilability score | GI absorption | BBB permeation | Pgp substrate | PAINS alerts | Synthetic accessibility |
|---|---|---|---|---|---|---|
| 1 | 0.55 | High | Yes | No | 0 | 1.00 |
| 2 | 0.55 | High | Yes | No | 0 | 1.00 |
| 3 | 0.55 | High | Yes | No | 0 | 1.00 |
| 4 | 0.55 | High | Yes | No | 0 | 3.32 |
| 5 | 0.55 | High | Yes | No | 1, catechol | 1.09 |
| 6 | 0.55 | High | Yes | Yes | 0 | 3.32 |
| 7 | 0.55 | High | Yes | No | 0 | 3.25 |
| 8 | 0.55 | High | Yes | No | 0 | 2.99 |
| 9 | 0.55 | High | Yes | No | 0 | 2.66 |
| 10 | 0.55 | High | Yes | No | 0 | 1.80 |
| 11 | 0.55 | High | Yes | No | 0 | 3.02 |
| 12 | 0.55 | High | Yes | No | 0 | 1.95 |
3. Material and methods
3.1. Chemicals
All chemicals and solvents were acquired from Merck (Germany) and Sigma Chemical Co. (USA), respectively and were of analytical grades.3.2. Extraction and isolation
The roots of A. cordifolia (250 g) were extracted using 95% ethyl alcohol, the dried ethanol extract (21.5 g) was then suspended in the least amount of distilled water, transferred to separating funnel, then defatted with light petroleum ether to afford pet. ether fraction (3 g). The remaining aqueous solution was then acidified and extracted with ethyl acetate to give acidic ethyl acetate fraction (2.8 g) (non-alkaloidal fraction). The mother liquor was then completely basified and extracted again with ethyl acetate to give basic ethyl acetate fraction (1.8 g) (alkaloidal fraction). The petroleum ether soluble fraction was subjected to silica gel column chromatography and eluted with pet. ether:EtOAc gradient elution starting with 100% pet. ether till 70:30 pet. ether:EtOAc to afford four compounds named, capric acid, tricosanol and mixture of β-sitosterol & stigma sterol. Then the acidic EtOAc fraction was subjected to silica gel column chromatography eluted with DCM:MeOH gradient elution to afford five subfractions. Subfraction no. three was further subjected to sephadex LH-20 (Merck, Germany) column chromatography using isocratic elution with DCM:MeOH (50:50) to yield cis-N-coumaroyl tyramine compound. Finally the alkaloidal fraction was subjected to silica gel column chromatography eluted with DCM:MeOH gradient elution to afford seven subfractions. Subfraction no. 3 was further eluted on sephadex LH-20 (Merck, Germany) column chromatography using isocratic elution with MeOH (100%) to yield compound O-methyl sceletenone (4), while 4-methoxyphenethylamine (1), N-methyl tyramine (2) and epinine (5) compounds were afforded by further purification of subfraction no. 4 on semi preparative HPLC (Knauer, Smartline, Germany) using H2O/methanol (95:5) initially for 5 min, followed by a linear gradient to 100% methanol within 40 min and maintained isocratically for 5 min. Separation was achieved on a preparative C18 column (5 μm, 10 × 250 mm, Waters XBridge, Eschborn, Germany), with a flow rate of 2.0 mL min−1 and solvent mixture complemented by 0.05% trifluoroacetic acid.
3.3. Antiviral activity
HCV replicon cells were inoculated at 26 × 104 cells per well in a 48-well plate for 24 h prior to the experiment. Each investigated fraction (petroleum ether, acidic ethyl acetate and basic ethyl acetate) was added to the culture broth at different concentrations (1–200 μg mL−1). After 72 h, the treated cells were harvested and lysed in cell culture lysis reagent. Luciferase activity was evaluated with a luciferase assay system and the resulting luminescence was detected by the luminescence plate reader (PerkinElmer) and corresponded to the expression level of the HCV replicon. MTT assay was used to monitor the cytotoxicity which could be attributed to some compounds in the extracts.3.4. Molecular docking
Molecular operating environment software (MOE 2019.01, Chemical Computing Group, Montreal, QC, Canada) was used to perform all the docking algorithms in this study in addition to ranking and visualization of the generated docking poses. Four co-crystal structures (PDB ID: 3H2L, 6NZT, 4OKS, 4B73) [10.1016/j.bmcl.2009.09.045, 10.1016/j.bmcl.2019.03.037, 10.1021/jm401432c, 10.1038/nchembio.1081], which represent the four non-structural HCV targets, were retrieved from the protein data bank and involved in our molecular docking simulation. All protein structures were prepared by QuickPrep module implemented in MOE before docking. The chemical structures of the screened compounds were drawn and energy minimized by MOE default settings. MOE induced docking protocol was applied, where the co-crystallized ligand was used as a center for the docking simulation grid. All water molecules as well as other non-essential ligands were removed before docking. Other parameters were left in their default value.3.5. In silico drug-likeness, ADME profiling, and toxicity risk assessment
The drug-likeness and pharmacokinetic properties of all identified compounds were predicted using Swiss ADME database.364. Conclusion
The investigation of A. cordifolia roots revealed isolation and structure elucidation of eight compounds. Additionally, the alkaloidal fraction showed potent anti HCV activity with IC50 value of 2.4 μg mL−1 so, we undergo docking studies of compounds isolated from alkaloidal fraction as well as, alkaloidal compounds dereplicated from the same fraction through metabolomics. The structure-based screening of the compounds was elucidated against four non-structural HCV antiviral targets and revealed that tortuosamine compound could be a promising anti-HCV candidate as it showed the strongest binding to the active site of NS3/4A helicase among the others. Accordingly, our in silico study highlights the importance of the alkaloidal fraction of the A. cordifolia roots as a potential complementary remedy for HCV infections after further validation.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication was funded by Minia University.References
- Q.-L. Choo, G. Kuo, A. J. Weiner, L. R. Overby, D. W. Bradley and M. Houghton, Science, 1989, 244, 359–362 CrossRef CAS PubMed.
- R. De Francesco, L. Tomei, S. Altamura, V. Summa and G. Migliaccio, Antiviral Res., 2003, 58, 1–16 CrossRef CAS PubMed.
- S. T. Hassan, K. Berchová-Bímová and J. Petráš, Phytother. Res., 2016, 30, 1487–1492 CrossRef CAS PubMed.
- G. Hussein, H. Miyashiro, N. Nakamura, M. Hattori, N. Kakiuchi and K. Shimotohno, Phytother. Res., 2000, 14, 510–516 CrossRef CAS PubMed.
- A. Petruzziello, S. Marigliano, G. Loquercio, A. Cozzolino and C. Cacciapuoti, World J. Gastroenterol., 2016, 22, 7824 CrossRef PubMed.
- A. A. Youssef, N. Magdy, L. A. Hussein and A. El-Kosasy, J. Chromatogr. Sci., 2019, 57, 636–643 CAS.
- D. L. Wyles and A. F. Luetkemeyer, Top. Antivir. Med., 2017, 25, 103 Search PubMed.
- M. Debiaggi, L. Pagani, P. Cereda, P. Landini and E. Romero, Microbiologica, 1988, 11, 55–61 CAS.
- K. Asres and F. Bucar, Ethiop. Med. J., 2005, 43, 15–20 Search PubMed.
- K. Vermani and S. Garg, J. Ethnopharmacol., 2002, 80, 49–66 CrossRef PubMed.
- K.-L. Huang, Y.-K. Lai, C.-C. Lin and J.-M. Chang, World J. Gastroenterol., 2006, 12, 5721 CrossRef PubMed.
- D. H. Kwon, H. Y. Kwon, H. J. Kim, E. J. Chang, M. B. Kim, S. K. Yoon, E. Y. Song, D. Y. Yoon, Y. H. Lee and I. S. Choi, Phytother. Res., 2005, 19, 355–358 CrossRef PubMed.
- G. J. Kotwal, J. N. Kaczmarek, S. Leivers, Y. T. Ghebremariam, A. P. Kulkarni, G. Bauer, C. De Beer, W. Preiser and A. R. Mohamed, Ann. N. Y. Acad. Sci., 2005, 1056, 293–302 CrossRef PubMed.
- M. Musarra-Pizzo, R. Pennisi, I. Ben-Amor, G. Mandalari and M. T. Sciortino, Viruses, 2021, 13, 828 CrossRef CAS PubMed.
- T. B. N. Trinh, D. H. Le, T. T. K. Nguyen, M. H. Nguyen, M. Muller and T. K. N. Nguyen, Virusdisease, 2021, 1–7 Search PubMed.
- T. S. Wahyuni, A. A. Permatasari, T. Widiandani, A. Fuad, A. Widyawaruyanti, C. Aoki-Utsubo and H. Hotta, Nat. Prod. Commun., 2018, 13, 1579–1582 CrossRef.
- G. Germishuizen and N. Meyer, Plants of southern Africa: an annotated checklist, National Botanical Institute Pretoria, 2003 Search PubMed.
- Mifsud S (2002–2014), MaltaWildPlants.com – an online flora of the Maltese islands, Accessed 20 Nov 2018, http://www.maltawildplants.com.
- A. A. E. Said, T. F. Ali, E. Z. Attia, A.-S. F. Ahmed, A. H. Shehata, U. R. Abdelmohsen and M. A. Fouad, Nat. Prod. Res., 2020, 1–5 Search PubMed.
- D. Elgindi and T. Mabry, Asian J. Chem., 1999, 11, 1525 Search PubMed.
- S. Sultana, M. Ali and S. R. Mir, Int. J. Adv. Pharm. Med. Bioallied Sci., 2017, 5, 217–224 Search PubMed.
- B. Silva Ferreira, L. Pereira Faza and M. Le Hyaric, Sci. World J., 2012, 2012, 1–4 CrossRef PubMed.
- D. K. Kim, J. P. Lim, J. W. Kim, H. W. Park and J. S. Eun, Arch. Pharm. Res, 2005, 28, 39–43 CrossRef CAS PubMed.
- N. Hiroka and D. P. Carew, J. Nat. Prod., 1981, 44, 285–288 CrossRef.
- K. K. Hornemann, J. Neal and J. McLaughlin, J. Pharm. Sci., 1972, 61, 41–45 CrossRef CAS PubMed.
- H. Sato, S. Sakamura and Y. Obata, Agric. Biol. Chem., 1970, 34, 1254–1255 CAS.
- P. W. Jeffs, T. Capps, D. Johnson, J. Karle, N. Martin and B. Rauckman, J. Org. Chem., 1974, 39, 2703–2710 CrossRef CAS.
- C. D. Gaffney, PhD thesis, University of Johannesburg, South Africa, 2008.
- R. J. Borgman, J. J. McPhillips, R. E. Stitzel and I. J. Goodman, J. Med. Chem., 1973, 16, 630–633 CrossRef CAS PubMed.
- C. G. Chavdarian, D. Karashima, N. Castagnoli Jr and H. K. Hundley, J. Med. Chem., 1978, 21, 548–554 CrossRef CAS PubMed.
- T. A. Smith, Phytochemistry, 1977, 16, 9–18 CrossRef CAS.
- P. Halfon and S. Locarnini, J. Hepatol., 2011, 55, 192–206 CrossRef CAS PubMed.
- B. Nutho, W. Khuntawee, C. Rungnim, P. Pongsawasdi, P. Wolschann, A. Karpfen, N. Kungwan and T. Rungrotmongkol, Beilstein J. Org. Chem., 2014, 10, 2789–2799 CrossRef PubMed.
- A. H. Al-Saeedi, M. T. H. Al- Ghafri and M. A. Hossain, Pac. Sci. Rev., 2016, 18, 78–83 Search PubMed.
- C. M. Rice, Top. Antivir. Med., 2011, 19, 117 Search PubMed.
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 1–13 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra06139a |
| This journal is © The Royal Society of Chemistry 2021 |