
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelective hydroxylation of aryl iodides to produce phenols under mild conditions using a supported copper catalyst†
Leiduan Hao a,
Anika Aunia,
Guodong Ding*a,
Xiaoyu Lib,
Haiping Xuc,
Tao Li*cd and
Qiang Zhang*ab
a,
Anika Aunia,
Guodong Ding*a,
Xiaoyu Lib,
Haiping Xuc,
Tao Li*cd and
Qiang Zhang*ab
aDepartment of Chemistry, Washington State University, Pullman, Washington 99164, USA. E-mail: guodong.ding@wsu.edu; q.zhang@wsu.edu
bMaterials Science and Engineering Program, Washington State University, Pullman, Washington 99164, USA
cDepartment of Chemistry and Biochemistry, Northern Illinois University, DeKalb, IL 60115, USA. E-mail: tli4@niu.edu
dX-ray Science Division, Argonne National Laboratory, Argonne, IL 60439, USA
First published on 21st July 2021
Abstract
Owing to the high activity and low-cost, copper-based catalysts are promising candidates for transforming aromatic halides to yield phenols. In this work, we report the selective hydroxylation of aromatic iodides to produce phenols using an atomically dispersed copper catalyst (Cu–ZnO–ZrO2) under mild reaction conditions. The reactions were conducted without the use of additional organic ligands, and the protection of an inert atmosphere environment is not required. The catalyst can be easily prepared, scalable, and is very efficient for a wide range of substrates. The catalytic reactions can be carried out with only 1.24 mol% Cu loading, which shows great potential in mass production.
Introduction
Phenol derivatives are highly valuable structural scaffolds widely present in natural and pharmaceutical compounds,1,2 and are also employed as versatile intermediates in synthetic organic chemistry. The development of effective protocols for the preparation of functionalized phenols significantly impacts modern chemical science. Although various approaches have been developed starting from assorted molecules, such as the aryl halides,3 benzyne,4 aromatic boronic acids,5 through C–H activation,6–8 and diazonium salts,9 there is still a challenge to make this process more efficient.10–12 Among these methods, due to the simplicity and availability of aryl halides, metal-catalyzed hydroxylation of aryl halides is the most favorable reaction for the synthesis of phenol derivatives. Palladium,3,13–19 and copper20–37-based catalysts have attracted extensive attention owing to their high activity and great selectivity (Scheme 1a and b). The iron-based catalytic system was also studied at high temperatures.38 However, most of the systems are homogeneous with high catalyst loadings and complicated ligands are indispensable, which makes these methods unacceptable in industry processes. Therefore, designing new, highly efficient catalysts to reduce the cost is very important from both scientific and industrial viewpoints.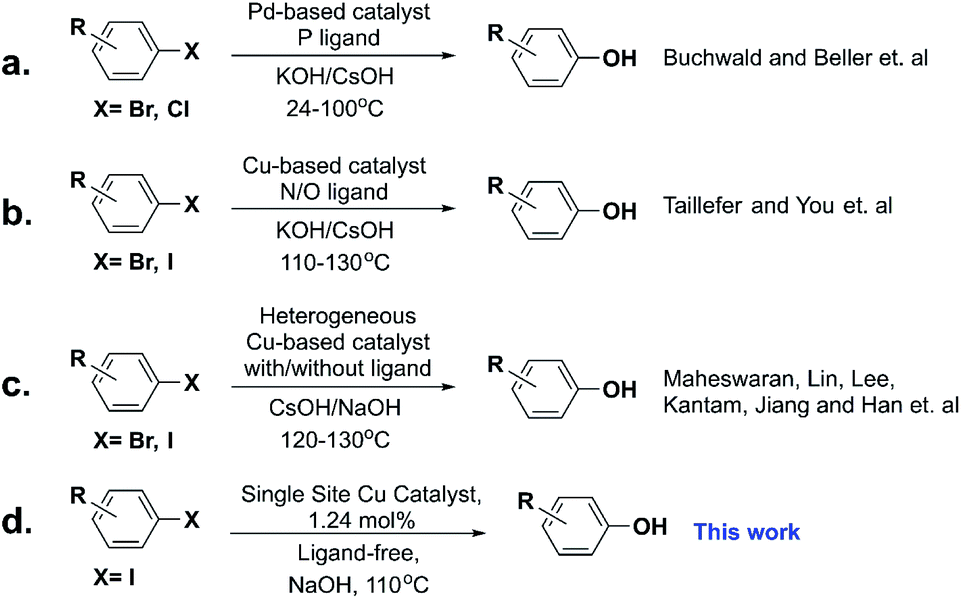 | ||
| Scheme 1 Different catalytic systems in the hydroxylation of aryl halides. | ||
Heterogeneous catalysis has attracted increasing attention due to its high catalyst recyclability and easy product separation and purification. However, heterogeneous catalytic systems for the hydroxylation reaction of aryl halides have not been extensively studied (Scheme 1c). A sulfonic acid resin (INDION-770) in combination with copper salts39 and CuO supported on mesoporous silica40 were reported for the hydroxylation of the aryl halides, but expensive CsOH was used. Cu-MOF was also found to be active in the hydroxylation of aryl iodides; unfortunately, the Cu-MOF was decomposed after the reaction and cannot be recycled.41 The ligand-free heterogeneous copper catalyst, Cu–g–C3N4, was studied for the hydroxylation of aryl iodides to synthesize phenols using low-cost bases, which shows great potential in the synthesis of substituted phenols.42 Therefore, developing heterogeneous, low-cost, and highly efficient catalysts for hydroxylation reactions is highly desirable yet challenging.
More recently, atomically dispersed copper catalysts have emerged as a new frontier in catalysis science and have attracted increasing attention in both academia and industry.43–49 However, till now there is no reported research on hydroxylation of aryl halides catalyzed by atomically dispersed copper catalysts. Also, copper is one of the most abundant elements with relatively low cost. It is urgent and necessary to extend the application of atomically dispersed copper catalysts to a broader scope of chemical transformation reactions.
Herein, we report the first atomically dispersed Cu catalyst for highly efficient hydroxylation of aryl iodides under mild conditions (Scheme 1d). The material can be easily fabricated through an in situ coprecipitation method on large scales. This new Cu catalyst was fully characterized systematically, and TEM and EXAFS characterization tests indicated that the copper atoms are atomically dispersed throughout the sample. The catalytic system tolerates a broad substrate scope with good to excellent isolated yields of phenols, with only 1.24 mol% Cu catalyst loading.
Experimental
General information
All chemicals were used as purchased without further purification. All reactions were carried out in the air. Cu(NO3)2·3H2O, sodium hydroxide, dimethyl sulfoxide, ethanol, methanol, petroleum ether, ethyl acetate, Zn(NO3)2·6H2O, ZrO(NO3)2·xH2O, and (NH4)2CO3 were purchased from Sigma-Aldrich, Alfa Aesar, and Fisher Scientific Co. Ltd. The 1H NMR and 13C NMR spectra were recorded on an Agilent DD2 600 MHz or Agilent Varian 400 MR NMR spectrometer in CDCl3 with TMS as an internal standard. Chemical shifts for protons were referenced to the residual solvent peak (CDCl3, 1H NMR: 7.26 ppm; CD3OD: 1H NMR: 4.87, 3.51; DMSO: 1H NMR: 2.50), and chemical shifts for carbons were referenced to the residual solvent peaks (CDCl3, 13C NMR: 77.16 ppm; CD3OD: 13C NMR: 49.15; DMSO: 13C NMR: 39.52). PXRD patterns were collected on a Rigaku Miniflex 600 and a Bruker D2 Phaser using Cu Kα radiation. N2 adsorption data were collected on a Micrometrics ASAP 2020Plus accelerated surface area and porosimetry system at 77 K. Samples were activated under vacuum at 150 °C for 12 hours with the activation port equipped on ASAP 2020Plus. The morphologies of the catalysts were characterized by aberration-corrected scanning transmission electron microscopy equipped with an energy-dispersive X-ray spectrometer on FEI Titan Cubed Themis G2 300 with a probe corrector. Before microscopy examination, samples were suspended in ethanol with an ultrasonic dispersion for 30 min, and then a drop of the resulting solution was placed onto a porous carbon film on a TEM copper grid. Gas chromatography was operated on Shimadzu GC-MS-QP2010 SE equipped with an FID detector and a DB-5 ms column. XPS was obtained with an ESCALab220i-XL electron spectrometer from VG Scientific using 300 W AlKα radiation. The base pressure was about 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) × 10−9 mbar. The binding energies were referenced to the C 1s line at 284.8 eV from adventitious carbon. X-ray absorption measurements (XAS), including XANES and EXAFS spectroscopy, were performed at 12 BM and 20-BM of the Advanced Photon Source (APS) at Argonne National Lab to investigate the local environment around the Cu atoms of Cu–ZnO–ZrO2 samples. The XANES spectroscopy was performed in fluorescence mode due to the low loading of Cu at the Cu K-edge (8970 eV). The Cu foil EXAFS was measured with the aid of a reference ion chamber for energy calibration for each scan of the samples. Several scans were taken and averaged for each sample to gain a better signal-to-noise ratio. The normalized, energy-calibrated Cu K-edge XANES spectra were obtained using standard data reduction techniques with Athena and Artemis software. The EXAFS oscillations χ(k) as a function of photoelectron wave number k was extracted by following standard procedures.
× 10−9 mbar. The binding energies were referenced to the C 1s line at 284.8 eV from adventitious carbon. X-ray absorption measurements (XAS), including XANES and EXAFS spectroscopy, were performed at 12 BM and 20-BM of the Advanced Photon Source (APS) at Argonne National Lab to investigate the local environment around the Cu atoms of Cu–ZnO–ZrO2 samples. The XANES spectroscopy was performed in fluorescence mode due to the low loading of Cu at the Cu K-edge (8970 eV). The Cu foil EXAFS was measured with the aid of a reference ion chamber for energy calibration for each scan of the samples. Several scans were taken and averaged for each sample to gain a better signal-to-noise ratio. The normalized, energy-calibrated Cu K-edge XANES spectra were obtained using standard data reduction techniques with Athena and Artemis software. The EXAFS oscillations χ(k) as a function of photoelectron wave number k was extracted by following standard procedures.
Synthesis of catalyst Cu–ZnO–ZrO2
Initially, 0.6 g of Zn(NO3)2·6H2O and 5.8 g of ZrO(NO3)2·xH2O were dissolved in 100 mL of deionized water at 70 °C in a 250 mL round bottle, and 1.0 mmol of Cu(NO3)2·3H2O was added. Next, 3.0 g of (NH4)2CO3 was dissolved in 100 mL DI water and was added dropwise to the aforementioned solution under vigorous stirring at 70 °C to form a precipitate. The suspension was continuously stirred for additional 2 hours at 70 °C, followed by filtering and washing, three times with DI water. The filtered sample was dried at 110 °C for 4 hours and calcined at 500 °C in static air for 3 hours (2.395 g). After which, the sample was reduced in pure H2 atmosphere at 300 °C for 3 h. This was followed by cooling the sample naturally to room temperature with nitrogen gas. The catalyst was finally obtained and named Cu–ZnO–ZrO2 (2.320 g, Cu content: 4.13 × 10−4 mmol mg−1).Procedures for the optimization of the reaction conditions based on iodobenzene
In the experiment, iodobenzene (1.0 mmol), suitable amounts of DMSO, water, base, and Cu–ZnO–ZrO2 were loaded into the reactor. The reactor was placed in an oil bath at the desired temperature. The reaction mixture was cooled to room temperature after the desired reaction time. The reaction mixture was carefully acidified with dilute aqueous HCl, and then 10 mL ethyl acetate was added. After centrifugation, the reaction mother liquid was analyzed by gas chromatography on Shimadzu GC-MS-QP2010 SE equipped with an FID detector and a DB-5 column. An internal standard, tetradecane, was used to quantify the generated phenol.General procedures for the synthesis of phenols from aryl iodides
A mixture of aryl iodides (1.0 mmol), Cu–ZnO–ZrO2 (30 mg, 1.24 mol% Cu) and NaOH (3.0 mmol) were added into the DMSO aqueous solutions. The reaction mixture was stirred at 110 °C for 24 h. After completion of the reaction, 15 mL of ethyl acetate and dilute HCl solution were added to the reaction mixture. The mixture was filtered and washed with 5 mL of ethyl acetate three times. The catalyst was separated and washed with ethanol, methanol, and water. The combined organic phases were washed with water three times. The organic layer was dried over MgSO4 and then evaporated under reduced pressure. The crude residue was purified by column chromatography on silica gel using n-hexane/EtOAc (80/20), to afford the desired phenol. The reaction products were characterized by 1H NMR and 13C NMR.Result and discussion
The catalytic materials were fabricated through a coprecipitation method.50 Cu(NO3)2·3H2O was used as the Cu precursor. The synthesized catalyst was characterized systematically. The powder X-ray diffraction (PXRD) pattern of the Cu–ZnO–ZrO2 between 20 and 80 degrees resembled the pristine bimetal oxide support ZnO–ZrO2 (Fig. 1a).51 Due to the high dispersion of Cu atoms and the low Cu content, no diffraction peaks of copper species were observed. The existence of Cu species was confirmed by XPS analysis (ESI Fig. S1†). The high-resolution Cu 2p XPS spectrum of the sample in Fig. 1b shows two main peaks located at about 952.3 and 932.5 eV corresponding to Cu 2p1/2 and Cu 2p3/2,52,53 which confirms the existence of Cu(0) and Cu(I) species on the surfaces of the catalyst. These results indicated that the Cu(II) was reduced during the H2 thermal process. The color of the material also changed from greenish gray to black (Fig. S2†), which also indicates the successful reduction of Cu(II) to Cu(0). The UV-vis spectrum and the Tauc plot were also consistent with the color of the material, Fig. S6.† Moreover, the existence of Cu species was further confirmed by aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC HAADF-STEM) and corresponding energy-dispersive X-ray spectroscopy (EDX) element mapping of the Cu–ZnO–ZrO2 catalyst (Fig. 2c). The FTIR spectra of the material before and after the incorporation of Cu species do not show obvious differences (Fig. S4†).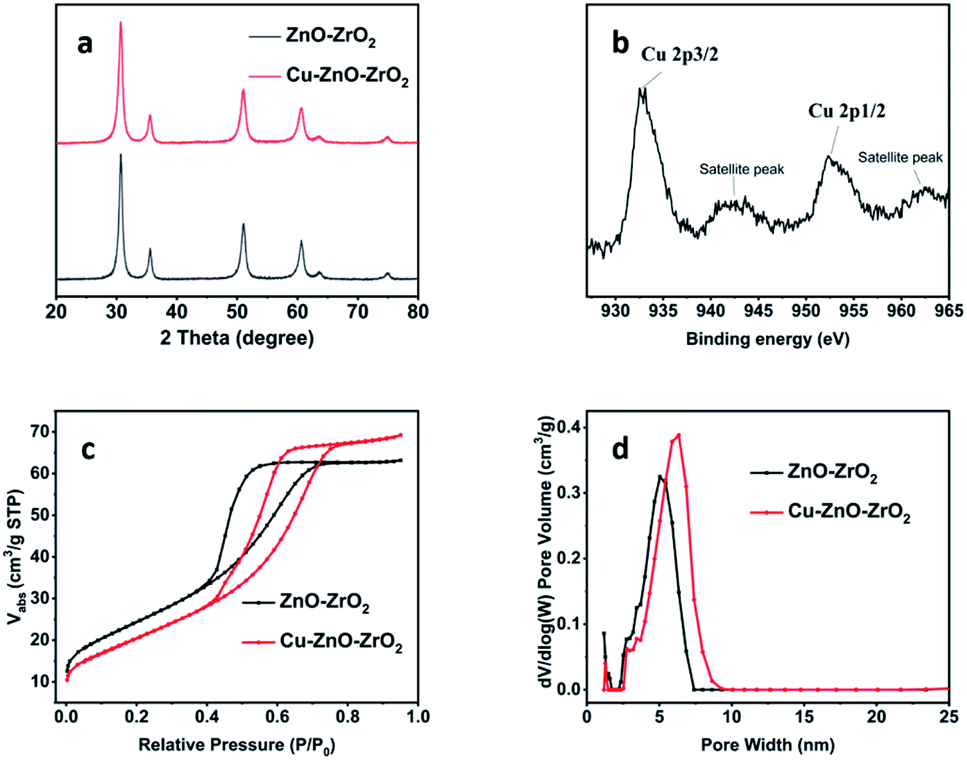 | ||
| Fig. 1 PXRD patterns (a), high-resolution XPS of Cu element (b), nitrogen isotherms (c), and pore size distribution (d) for the catalyst Cu–ZnO–ZrO2 and the pristine bimetal oxide support ZnO–ZrO2. | ||
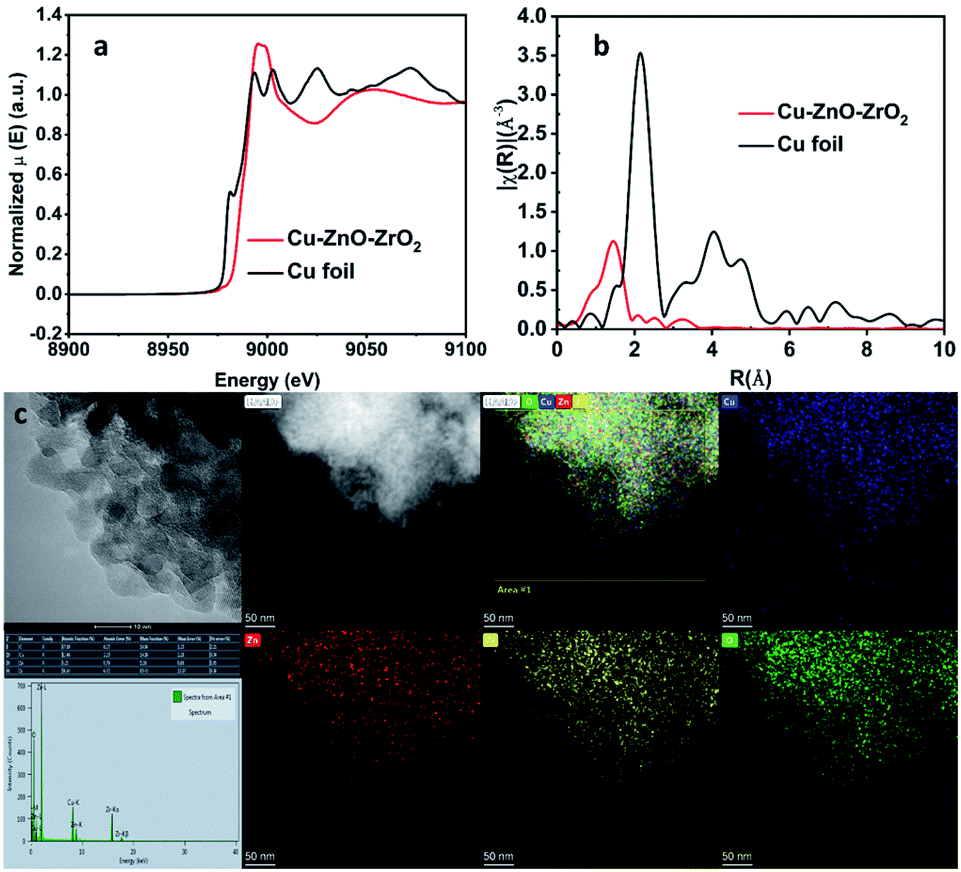 | ||
| Fig. 2 Characterizations of the catalyst Cu–ZnO–ZrO2. XANES (a), EXAFS (b), and AC-HAADF (c) with EDS mapping. Cu (blue), Zn (red), Zr (yellow), and O (green). | ||
N2 adsorption–desorption tests show type II hysteresis loops for both ZnO–ZrO2 and Cu–ZnO–ZrO2, revealing the existence of mesopores (Fig. 1c). The Brunauer–Emmett–Teller (BET) surface area of Cu–ZnO–ZrO2 is 77.5 m2 g−1, slightly decreased from that of the pristine ZnO–ZrO2.50 The internal surface area is 7.0 m2 g−1, and the external surface area is about 70.4 m2 g−1, Fig. S8.† For pore size distributions, the Cu–ZnO–ZrO2 show a much broader range (centered around 6.32 nm) than that of the pristine support ZnO–ZrO2 (centered at 5.04 nm) (Fig. 1d), which is beneficial for higher mass transfer in catalysis.54
To probe the detailed structural information of Cu species in Cu–ZnO–ZrO2, synchrotron extended X-ray absorption fine structure (EXAFS) experiment was conducted. As shown in Fig. 2b, there is only a main peak located at 1.46 Å, which can be attributed to Cu–O coordination, whereas the Cu–Cu peak (2.15 Å) was not detected in the catalyst Cu–ZnO–ZrO2, indicating the isolated copper atoms were dispersed discretely on the support. This is in a good agreement with the X-ray diffraction result, which shows no Cu nanoparticle diffraction peaks (Fig. 1a). X-ray absorption near-edge structure spectroscopy shows an absorption edge energy located at 8975.8 eV which is higher than that of the Cu foil (8973.6 eV), further indicating that the Cu species were partially positively charged (Cuδ+). This observation agrees well with the XPS results (Fig. 1b). The single-atom Cu structure was further demonstrated by the HAADF-STEM image and corresponding EDX element mappings, as shown in Fig. 2c. These results corroborated the atomic distribution of Cu dispersed evenly on the support.
The catalytic performance of Cu–ZnO–ZrO2 was evaluated in the hydroxylation cross-coupling between aryl iodides and sodium hydroxide. Iodobenzene was selected as the model substrate to optimize the reaction conditions (Table 1). Initially, the hydroxylation reaction was performed in DMSO/water solution using 50 mg of Cu–ZnO–ZrO2 at 120 °C, with sodium hydroxide as the nucleophile (Table 1, entry 1). The product was obtained in 75% yield after 15 hours. Encouraged by the results, the effect of reaction temperature, amounts of NaOH, and reaction solvents were further investigated. Surprisingly, the yield of phenol reached 99% at 110 °C after 24 h (Table 1, entry 2). When the temperature was further lowered to 100 °C, the yield of phenol was dramatically decreased (Table 1, entry 3). Settled with the ideal temperature, we sought to alter the amount of nucleophiles. When three equivalents of NaOH were used, quantitative conversion was realized without any detectable byproducts (Table 1, entries 4–6). For a catalytic reaction, the solvent usually possesses a remarkable impact on the reactivity of the catalyst.55 The ethanol/water mixture or water only solvent systems did not produce any products (Table 1, entries 7 and 9). Only a 0.1% yield of phenol was formed when DMSO was used (Table 1, entry 8). The DMSO/water mixture is ideal for the hydroxylation reaction of iodobenzene (Table 1, entries 2, 4, and 10). This can be attributed to the high solubility of iodobenzene and NaOH in DMSO and water, respectively. Given the increased activity of single-atom catalysts, a low amount of the catalyst was used to test the activity. When the amount of the catalyst was lowered from 50 mg to 30 mg, the reaction yield was not affected (Table 1, entry 10). However, when the amount of catalyst was further decreased to 17 mg, the product yield was only 48% after 24 h (Table 1, entry 11). To confirm the crucial role of the Cu species and NaOH, control experiments were also conducted, and no reaction took place in the absence of the catalyst and NaOH (Table 1, entries 12–14). Finally, the optimized reaction condition was selected with 30 mg Cu–ZnO–ZrO2, three equivalent of NaOH (3 equiv.) in DMSO/water mixture solution at 110 °C in the air for 24 h (Table 1, entry 10). It is worth noting that Cu–ZnO and Cu–ZrO2 also possess high activity for the hydroxylation of aryl iodides (93% and 95% yields, respectively). However, aggregation of Cu to form nanoparticles was observed in those systems. Therefore, those two catalysts were not investigated further. The recycling of the catalyst was also conducted, and the result shows that the catalyst could be used for two cycles with very high product yields, Fig. S9.† Further cycling experiments were not conducted due to the loss of catalyst during the reaction. This could be attributed to the decreased particle size of the material, which is supported by the broadening of the XRD pattern after the reaction, Fig. S3.†
| Entry | Base/equiv. | T/°C | Solventsb | Catalyst | t/h | Yieldc/% |
|---|---|---|---|---|---|---|
| a All reactions were performed using 1.0 mmol of iodobenzene.b 4 mL solvent was used. Volume ratio of DMSO/H2O or EtOH/H2O was 1/1).c Yield was determined by GC with tetradecane as the internal standard based on the added iodobenzene. | ||||||
| 1 | 4 | 120 | DMSO/H2O | 50 mg | 15 | 75 |
| 2 | 4 | 110 | DMSO/H2O | 50 mg | 24 | 99 |
| 3 | 4 | 100 | DMSO/H2O | 50 mg | 24 | 5 |
| 4 | 3 | 110 | DMSO/H2O | 50 mg | 24 | 99 |
| 5 | 2 | 110 | DMSO/H2O | 50 mg | 24 | 79 |
| 6 | 1 | 110 | DMSO/H2O | 50 mg | 24 | 2 |
| 7 | 4 | 120 | EtOH/H2O | 50 mg | 15 | — |
| 8 | 3 | 110 | DMSO | 30 mg | 24 | 0.1 |
| 9 | 3 | 110 | H2O | 30 mg | 24 | — |
| 10 | 3 | 110 | DMSO/H2O | 30 mg | 24 | 99 |
| 11 | 3 | 110 | DMSO/H2O | 17 mg | 24 | 48 |
| 12 | 3 | 110 | DMSO/H2O | — | 24 | — |
| 13 | 3 | 110 | DMSO/H2O | ZnO–ZrO2 | 24 | — |
| 14 | — | 110 | DMSO/H2O | 50 mg | 24 | — |
With the optimized reaction conditions determined, the reaction was then investigated with a wider scope of aryl iodides as the substrates and NaOH as the base. The results are listed in Table 2. As shown in Table 2, the hydroxylation reactions of aryl iodides with NaOH proceeded efficiently at 110 °C in the air to afford the corresponding products. Various substrates with electron-donating and electron-withdrawing groups such as –CH3, –OCH3, –F, –Cl, –Br, –NO2, and –CN were well tolerated to yield the desired phenols in good to excellent yields. Since the electronic effect did not show appreciable difference in the catalytic reactions, similar isolated product yields were obtained (Table 2, entries 2–6, and 12). For substrates with bulk functional groups, a potent steric effect was observed. For example, a slightly lower reaction yield was obtained when 2-methyl-iodobenzene was used (Table 2, entry 3). Interestingly, compared with 3-methoxyl-iodobenzene and 4-methoxyl-iodobenzene, when 2-methoxyl-iodobenzene was employed as the substrate, no desired product was detected under the same reaction conditions (Table 2, entries 5–7). It should be noted that, besides the steric effect, the interaction between oxygen from the –OCH3 group and the catalytic Cu species might play an more important role. Remarkably, we can see from Table 2 that the reaction only happens to the iodide functional group, when 1-fluoro-4-iodobenzene, 1-chloro-4-iodobenzene, and 1-bromo-4-iodobenzene were used, –F, –Cl, and –Br functionalize phenols were produced (Table 2, entries 8–10). It has been reported that when excess amount of potassium iodide was added, the substrates with –Cl and –Br groups can be converted to phenols to some degree.42 However, in the present catalytic systems, –Cl and –Br groups are still inert even at high temperatures with longer reaction times. Thus our catalytic system provides a highly selective cross-coupling reaction of aryl iodides with NaOH under practical, mild conditions (Table 2, entries 15 and 16). The high selectivity of the reaction to only the iodide functional group makes it the ideal process to synthesize fluoro-, chloro-, or bromo-phenols, which can be further modified through subsequent cross-coupling strategies.56–58 The conversion of 4,4′-diiodo-1,1′-biphenyl to [1,1′-biphenyl]-4,4′-diol could be realized in 85% yield (entry 11), indicating the catalyst is capable of converting multiple iodide groups. When 2-iodo-1,3,5-trimethylbenzene was used as the starting material, 2, 4, 6-trimethylphenol could be produced in 90% yield (entry 12), illustrating the fact that the reaction is not affected very much by the steric hindrance of methyl groups on the ortho positions. Entry 13 indicated that the nitro functional group would not affect the reaction. Interestingly, when 4-iodobenzonitrile was used as the starting material, 4-hydroxybenzoic acid was produced with a low yield (entry 14). To reveal the reaction mechanisms, 4-iodobenzoic acid was selected as the starting material, and no 4-hydroxybenzoic acid could be detected in the reaction mixture after 24 hours. The result indicated that the carboxylate group hinders the conversion of the iodo group to phenol, which explains the low yield of the reaction when 4-iodobenzonitrile was used as the starting material.
| Entry | Aryl halides | t/h | Product | Yieldb/% |
|---|---|---|---|---|
| a Reactions were carried out with 1.0 mmol of aryl halides, 30 mg of Cu, 3 eq. NaOH, 2.0 mL DMSO and 2.0 mL H2O at 110 °C under air conditions.b Isolated yields based on the added aryl iodides.c Data in parentheses indicate 2.0 equiv. KI was added, 150 °C. | ||||
| 1 |  |
24 | 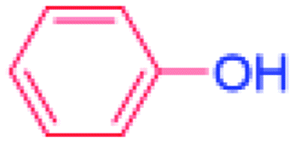 |
91 |
| 2 |  |
24 | 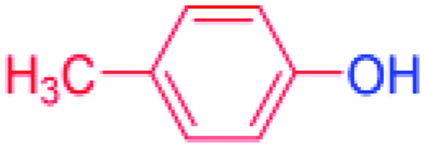 |
88 |
| 3 | 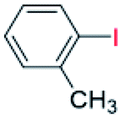 |
24 |  |
80 |
| 4 | 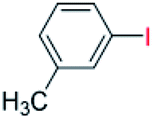 |
24 | 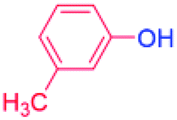 |
88 |
| 5 | 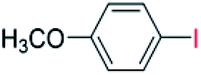 |
24 | 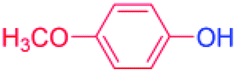 |
98 |
| 6 | 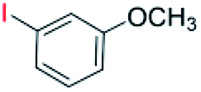 |
24 | 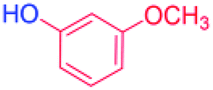 |
93 |
| 7 | 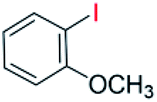 |
24 | 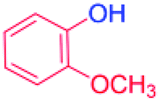 |
0 |
| 8 | 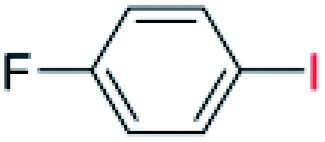 |
24 | 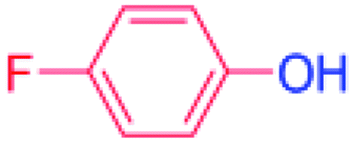 |
87 |
| 9 |  |
24 | 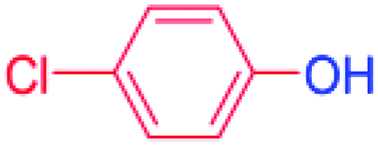 |
92 |
| 10 | 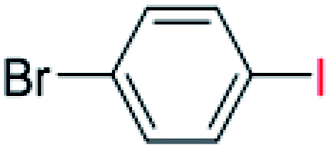 |
24 | 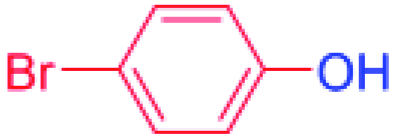 |
85 |
| 11 | 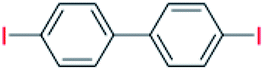 |
24 | 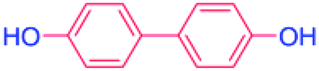 |
85 |
| 12 |  |
24 | 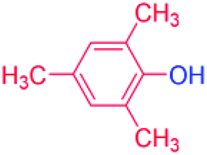 |
90 |
| 13 | 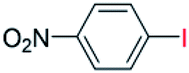 |
24 | 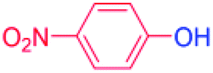 |
94 |
| 14 | 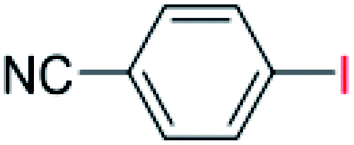 |
24 | 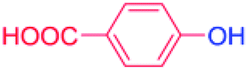 |
40 |
| 15 |  |
36 | 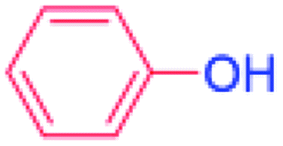 |
0 (0)c |
| 16 | 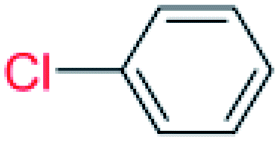 |
36 | 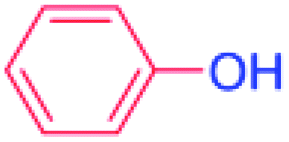 |
0 (0)c |
Conclusions
In summary, we have developed a new atomically dispersed copper catalyst (Cu–ZnO–ZrO2). The new Cu catalyst was characterized systematically. The cross-coupling reactions of various aryl iodides with NaOH proceeded very effectively with low loading of Cu catalyst at 110 °C in the air to afford the corresponding products in good to excellent yields. The catalyst can be easily prepared with low-cost and scalable, and is air and moisture-tolerant. The high selectivity of the reaction, targeting only the iodide functional group, makes the synthesis of halogenated phenols much more accessible at a low cost.Author contributions
GD and QZ conceived the idea, LH, AA, and GD conducted the experiment with the help of XL under the supervision of QZ. HX helped with the XANES and EXAFS data collection and data analysis under the supervision of TL.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank the Department of Chemistry and the College of Arts and Sciences at Washington State University for their financial support. The Bruker D2 Phaser diffractometer at Washington State University was purchased with the support from the M. J. Murdock Charitable Trust. The characterization used resources of the Advanced Photon Source and the Center for Nanoscale Materials, Office of Science user facilities, supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under contract no. DE-AC02-06CH11357.Notes and references
- J. H. P. Tyman, Synthetic and Natural Phenols, Elsevier Science, 1996 Search PubMed.
- S. Quideau, D. Deffieux, C. Douat-Casassus and L. Pouységu, Angew. Chem., Int. Ed., 2011, 50, 586–621 CrossRef CAS PubMed.
- K. W. Anderson, T. Ikawa, R. E. Tundel and S. L. Buchwald, J. Am. Chem. Soc., 2006, 128(33), 10694–10695 CrossRef CAS PubMed.
- T. George and R. Mabon, J. Chem. Soc., Perkin Trans. 1, 2000, 1, 2529 RSC.
- L. D. Hao, G. D. Ding, D. A. Deming and Q. Zhang, Eur. J. Org. Chem., 2019, 2019, 7307–7321 CrossRef CAS.
- R. E. Maleczka, F. Shi, D. Holmes and M. R. Smith, J. Am. Chem. Soc., 2003, 125, 7792–7793 CrossRef CAS PubMed.
- Y.-H. Zhang and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 14654–14655 CrossRef CAS PubMed.
- J. Börgel, L. Tanwar, F. Berger and T. Ritter, J. Am. Chem. Soc., 2018, 140, 16026–16031 CrossRef PubMed.
- T. Cohen, A. G. Dietz Jr and J. R. Miser, J. Org. Chem., 1977, 42, 2053–2058 CrossRef CAS.
- D. A. Alonso, C. Najera, I. M. Pastor and M. Yus, Chemistry, 2010, 16, 5274–5284 CrossRef CAS PubMed.
- Y. Liu, S. Liu and Y. Xiao, Beilstein J. Org. Chem., 2017, 13, 589–611 CrossRef CAS PubMed.
- P. Amal Joseph and S. Priyadarshini, Org. Process Res. Dev., 2017, 21, 1889–1924 CrossRef CAS.
- T. Schulz, C. Torborg, B. Schäffner, J. Huang, A. Zapf, R. Kadyrov, A. Börner and M. Beller, Angew. Chem., 2009, 121, 936–939 CrossRef.
- A. G. Sergeev, T. Schulz, C. Torborg, A. Spannenberg, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 7595–7599 CrossRef CAS PubMed.
- A. Dumrath, X.-F. Wu, H. Neumann, A. Spannenberg, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 8988–8992 CrossRef CAS PubMed.
- C.-W. Yu, G. S. Chen, C.-W. Huang and J.-W. Chern, Org. Lett., 2012, 14, 3688–3691 CrossRef CAS PubMed.
- C. B. Lavery, N. L. Rotta-Loria, R. McDonald and M. Stradiotto, Adv. Synth. Catal., 2013, 355, 981–987 CrossRef CAS.
- A. Buitrago Santanilla, M. Christensen, L.-C. Campeau, I. W. Davies and S. D. Dreher, Org. Lett., 2015, 17, 3370–3373 CrossRef CAS PubMed.
- P. S. Fier and K. M. Maloney, Angew. Chem., 2017, 129, 4549–4553 CrossRef.
- A. Tlili, N. Xia, F. Monnier and M. Taillefer, Angew. Chem., Int. Ed., 2009, 48, 8725–8728 CrossRef CAS PubMed.
- D. Zhao, N. Wu, S. Zhang, P. Xi, X. Su, J. Lan and J. You, Angew. Chem., Int. Ed., 2009, 48, 8729–8732 CrossRef CAS PubMed.
- L. Jing, J. Wei, L. Zhou, Z. Huang, Z. Li and X. Zhou, Chem. Commun., 2010, 46, 4767–4769 RSC.
- S. Maurer, W. Liu, X. Zhang, Y. Jiang and D. Ma, Synlett, 2010, 2010, 976–978 CrossRef.
- R. Paul, M. A. Ali and T. Punniyamurthy, Synthesis, 2010, 2010, 4268–4272 CrossRef.
- D. Yang and H. Fu, Chem.–Eur. J., 2010, 16, 2366–2370 CrossRef CAS PubMed.
- K. G. Thakur and G. Sekar, Chem. Commun., 2011, 47, 6692–6694 RSC.
- K. Yang, Z. Li, Z. Wang, Z. Yao and S. Jiang, Org. Lett., 2011, 13, 4340–4343 CrossRef CAS PubMed.
- H.-J. Xu, Y.-F. Liang, Z.-Y. Cai, H.-X. Qi, C.-Y. Yang and Y.-S. Feng, J. Org. Chem., 2011, 76, 2296–2300 CrossRef CAS PubMed.
- Y. Xiao, Y. Xu, H.-S. Cheon and J. Chae, J. Org. Chem., 2013, 78, 5804–5809 CrossRef CAS PubMed.
- J. Kim, O. Battsengel, Y. Liu and J. Chae, Bull. Korean Chem. Soc., 2015, 36, 2833–2840 CrossRef CAS.
- Y. Wang, C. Zhou and R. Wang, Green Chem., 2015, 17, 3910–3915 RSC.
- N. Mketo, J. H. Jordaan, A. Jordaan, A. J. Swarts and S. F. Mapolie, Eur. J. Inorg. Chem., 2016, 2016, 3781–3790 CrossRef CAS.
- B. Y.-H. Tan and Y.-C. Teo, Synlett, 2016, 27, 1814–1819 CrossRef.
- S. Xia, L. Gan, K. Wang, Z. Li and D. Ma, J. Am. Chem. Soc., 2016, 138, 13493–13496 CrossRef CAS PubMed.
- G. Li, X. Zhao, K. Fang, J. Li and Y. She, J. Org. Chem., 2017, 82, 8634–8644 CrossRef CAS PubMed.
- V. S. Chan, S. W. Krabbe, C. Li, L. Sun, Y. Liu and A. J. Nett, ChemCatChem, 2019, 11, 5748–5753 CrossRef CAS.
- X. Liang, H. Li, F. Du, Y. Zhang, J. Dong, X. Bao, Y. Wu and G. Chen, Tetrahedron Lett., 2020, 61(33), 152222 CrossRef CAS.
- Y. Ren, L. Cheng, X. Tian, S. Zhao, J. Wang and C. Hou, Tetrahedron Lett., 2010, 51, 43–45 CrossRef CAS.
- P. J. Amal Joseph, S. Priyadarshini, M. Lakshmi Kantam and H. Maheswaran, Catal. Sci. Technol., 2011, 1, 582–585 RSC.
- C.-C. Chan, Y.-W. Chen, C.-S. Su, H.-P. Lin and C.-F. Lee, Eur. J. Org. Chem., 2011, 2011, 7288–7293 CrossRef CAS.
- S. Priyadarshini, P. J. Amal Joseph, M. L. Kantam and B. Sreedhar, Tetrahedron, 2013, 69, 6409–6414 CrossRef CAS.
- G. Ding, H. Han, T. Jiang, T. Wu and B. Han, Chem. Commun., 2014, 50, 9072–9075 RSC.
- Y. Qu, Z. Li, W. Chen, Y. Lin, T. Yuan, Z. Yang, C. Zhao, J. Wang, C. Zhao, X. Wang, F. Zhou, Z. Zhuang, Y. Wu and Y. Li, Nat. Catal., 2018, 1, 781–786 CrossRef CAS.
- A. M. Abdel-Mageed, B. Rungtaweevoranit, M. Parlinska-Wojtan, X. Pei, O. M. Yaghi and R. J. Behm, J. Am. Chem. Soc., 2019, 141, 5201–5210 CrossRef CAS PubMed.
- A. Bakandritsos, R. G. Kadam, P. Kumar, G. Zoppellaro, M. Medved', J. Tuček, T. Montini, O. Tomanec, P. Andrýsková, B. Drahoš, R. S. Varma, M. Otyepka, M. B. Gawande, P. Fornasiero and R. Zbořil, Adv. Mater., 2019, 31, 1900323 CrossRef PubMed.
- F. Huang, Y. Deng, Y. Chen, X. Cai, M. Peng, Z. Jia, J. Xie, D. Xiao, X. Wen, N. Wang, Z. Jiang, H. Liu and D. Ma, Nat. Commun., 2019, 10, 4431 CrossRef PubMed.
- B.-H. Lee, S. Park, M. Kim, A. K. Sinha, S. C. Lee, E. Jung, W. J. Chang, K.-S. Lee, J. H. Kim, S.-P. Cho, H. Kim, K. T. Nam and T. Hyeon, Nat. Mater., 2019, 18, 620–626 CrossRef CAS PubMed.
- H. Yang, Y. Wu, G. Li, Q. Lin, Q. Hu, Q. Zhang, J. Liu and C. He, J. Am. Chem. Soc., 2019, 141, 12717–12723 CrossRef CAS PubMed.
- W. Zang, T. Yang, H. Zou, S. Xi, H. Zhang, X. Liu, Z. Kou, Y. Du, Y. P. Feng, L. Shen, L. Duan, J. Wang and S. J. Pennycook, ACS Catal., 2019, 9, 10166–10173 CrossRef CAS.
- G. Ding, L. Hao, H. Xu, L. Wang, J. Chen, T. Li, X. Tu and Q. Zhang, Commun. Chem., 2020, 3, 43 CrossRef CAS.
- J. Wang, G. Li, Z. Li, C. Tang, Z. Feng, H. An, H. Liu, T. Liu and C. Li, Sci. Adv., 2017, 3, e1701290 CrossRef PubMed.
- A. Mansour, Surf. Sci. Spectra, 1994, 3, 202–210 CrossRef CAS.
- J. Haber, T. Machej, L. Ungier and J. Ziółkowski, J. Solid State Chem., 1978, 25, 207–218 CrossRef CAS.
- P. T. Tanev, M. Chibwe and T. J. Pinnavaia, Nature, 1994, 368, 321 CrossRef CAS PubMed.
- F. Proutiere and F. Schoenebeck, Angew. Chem., Int. Ed., 2011, 50, 8192–8195 CrossRef CAS PubMed.
- U. Sharma, T. Naveen, A. Maji, S. Manna and D. Maiti, Angew. Chem., Int. Ed., 2013, 52, 12669–12673 CrossRef CAS PubMed.
- S. Bhunia, G. G. Pawar, S. V. Kumar, Y. Jiang and D. Ma, Angew. Chem., Int. Ed., 2017, 56, 16136–16179 CrossRef CAS PubMed.
- L. Fu, S. Li, Z. Cai, Y. Ding, X.-Q. Guo, L.-P. Zhou, D. Yuan, Q.-F. Sun and G. Li, Nat. Catal., 2018, 1, 469–478 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1ra04112f |
| This journal is © The Royal Society of Chemistry 2021 |