
Rational design of selective inhibitors of PARP4†
Ilsa T.
Kirby
a,
Ashley
Person
b and
Michael
Cohen
 *c
*c
aInitial Therapeutics, South San Francisco, CA 94080, USA
bEunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, Bethesda, MD 20892, USA
cDepartment of Chemical Physiology and Biochemistry, Oregon Health and Science University, Portland, OR 97239, USA. E-mail: cohenmic@ohsu.edu
First published on 4th October 2021
Abstract
PARPs (PARP1-16 in humans) are a large family of ADP-ribosyltransferases (ARTs) that have diverse roles in cellular physiology and pathophysiology. Most PARP family members mediate mono-ADP-ribosylation (MARylation) of targets. The function of PARP-mediated MARylation in cells is poorly characterized, due in large part to the paucity of selective small molecule inhibitors of the catalytic activity of individual PARP enzymes. Herein we describe the rational design of selective small molecule inhibitors of PARP4 (also known as vPARP). These inhibitors are based on a quinazolin-4(3H)-one scaffold, and contain substituents at the C-8 position designed to exploit a unique threonine (Thr484, human PARP4 numbering) in the PARP4 nicotinamide sub-pocket. Our most potent analog, AEP07, which contains an iodine at the C-8 position, is at least 12-fold selective over other PARP family members. AEP07 will serve as a useful lead compound for the further development of PARP4 inhibitors that can be used to probe the cellular functions of PARP4 catalytic activity.
Introduction
PARPs are a family of 17 enzymes in humans that catalyze the transfer of ADP-ribose from nicotinamide adenine dinucleotide (NAD+) to targets—mostly proteins, but also DNA and RNA.1,2 This process, referred to as ADP-ribosylation, can occur either as the addition of a single monomer of ADP-ribose (MARylation), catalyzed by the majority of PARPs (i.e., PARP3, 4, 6–8, 10–12, 14–16), or the addition of ADP-ribose polymers (PARylation) catalyzed by four well-studied PARPs (i.e., PARP1, 2, 5a, 5b).3 All active PARPs contain an H–Y–X triad motif in the NAD+ binding pocket that is required for their ADP-ribosylating activity: the HYE-PARPs (PARP1, 2, 3, 4, 5a and 5b) and the HYΦ-PARPs (PARP 6–8, 10–12, 14–16).2 The glutamate in the HYE motif is necessary, but not sufficient, for the PARylation activity.4,5 Indeed, PARP3 and PARP4 are HYE-PARPs but have only been shown to catalyze MARylation.3 PARP4 is unique among PARP family members in that its catalytic domain is embedded in the middle of its protein sequence, whereas all other PARPs contain their catalytic domain at their C-terminus.6PARP4 was originally identified in mammalian cells as a protein component of “vaults”—massive ribonucleoprotein (RNP) nanoparticles.7–10 In addition to PARP4, vaults contain major vault protein (MVP), telomerase associated protein (TEP1), and vault RNA (vRNA).11 While the functions of vaults are not well understood, they have been implicated in diverse cellular functions: from DNA repair to immune defense.12–14 The function of PARP4 is equally enigmatic; however, knockout studies in mice suggest that PARP4 may play a role in carcinogen-induced tumorigenesis.12 Although PARP4 can auto-MARylate—a common feature among PARPs—its targets are unknown. To better understand the function of PARP4-mediated MARylation in physiology and disease, selective inhibitors of PARP4 catalytic activity are needed. Although PARP4 is potently inhibited by several clinical PARP1/2 inhibitors, namely olaparib, rucaparib and talazoparib,15–17 no selective inhibitors of PARP4 exist. Indeed, this is part of a general shortage of selective inhibitors for PARPs, particularly the MARylating PARP family members.18 To date, selective MARylating PARP inhibitors exist only for PARP7 (ref. 19) PARP10,20–23 PARP11,24 and PARP14.25 Herein, we describe the rational design of the first selective inhibitors of PARP4 using a structural bioinformatics approach.
Results and discussion
The PARP family is defined by a shared catalytic domain, which binds to its substrate NAD+. The NAD+ binding site is highly conserved across PARPs, making selective inhibition of individual PARP family members challenging. Guided by work in the kinase field26 we previously employed a bump-hole strategy to create engineered PARP—orthogonal inhibitor pairs.27,28 In this strategy we mutated an amino acid (e.g., Lys903 in human PARP1, Leu926 in human PARP10) in the nicotinamide sub-site (a position we refer to as the “ceiling position”) to a smaller amino acid (i.e. glycine or alanine). The resultant unique hole accommodated halogens, and alkyl or aromatic groups, on modified (bumped) nicotinamide-inspired inhibitors that don't inhibit wild-type PARPs. Intriguingly, structure-based sequence alignments revealed that PARP4 is unique among the PARP family members in that it contains a threonine (Thr484, human PARP4 numbering) in the ceiling position whereas all other H–Y–E PARPs contain a lysine and HYΦ-PARPs contain a tyrosine or branched hydrophobic amino acid in this position (Fig. 1A). While there is no crystal structure of PARP4, a homology model of PARP4 shows that Thr484 is in the ceiling position adjacent to the nicotinamide sub-site and partially overlaps with Lys903 of PARP1 (Fig. 1B). We reasoned that the unique ceiling position threonine in PARP4 compared to other PARP family members may be exploited for the development of selective inhibitors of PARP4.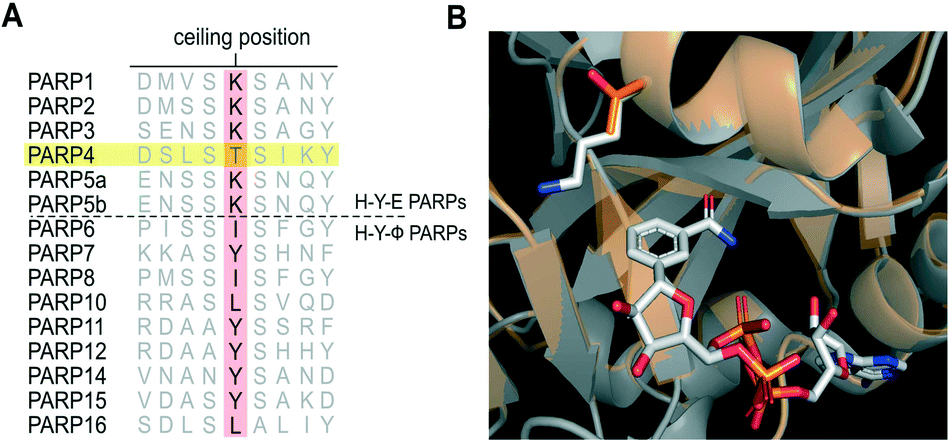 | ||
| Fig. 1 PARP4 has a unique threonine in the ceiling position. A) Structure-based sequence alignment of PARP family members (Clustal Omega). B) Overlay of the crystal structure of PARP1 (gray, PDB: 6BHV) bound to BAD (non-hydrolyzable NAD+ analog) with the PARP4 homology model (orange) generated using PARP2 bound to olaparib (4TVJ) as a template (SWISS-MODEL). | ||
To develop a selective PARP4 inhibitor we envisioned converting a pan-PARP inhibitor into a selective PARP4 inhibitor by adding an appropriately positioned substituent that would be guided by structural analysis. We based our PARP4 inhibitors on STO1102, a quinazolin-4(3H)-one (QDR)-based pan-PARP inhibitor.29 A crystal structure of STO1102 in complex with human PARP15 (PDB: 4F0E) shows the C-8 position of STO1102 closely abuts the ceiling position (Tyr598) in PARP15 (Fig. 2A).30 We hypothesized that substituents at the C-8 position would clash with Tyr598 in PARP15 and large ceiling position amino acids in other PARP family members, but not with the smaller Thr484 of PARP4 (Fig. 2B). We initially synthesized a small series of modified analogs of STO1102 that contain small groups such as a methyl (AEP01) and bromo (AEP02), and larger groups such as an iodo (AEP03), propynyl (AEP04) and a phenyl-propynyl (AEP05) (Scheme 1).
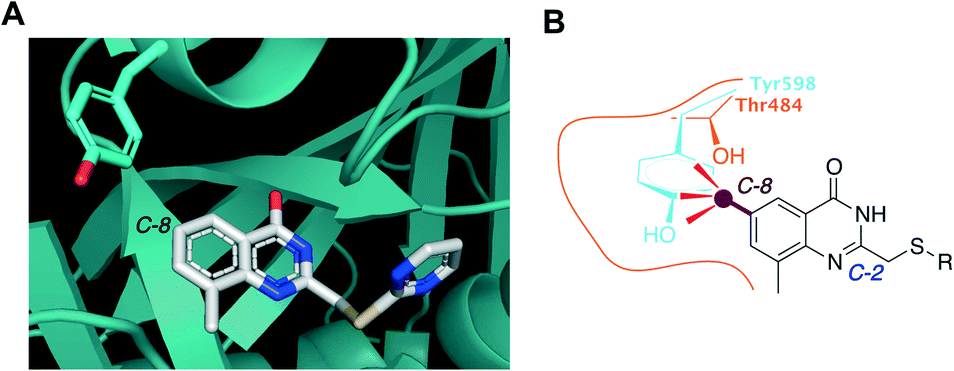 | ||
| Fig. 2 Rational design of PARP4 inhibitors exploiting a threonine at the ceiling position. A) Crystal structure of PARP15 bound to STO1102 (PDB: 4F0E). B) Graphic model showing how C-8 substituted QDR analogs clash with Tyr598 (or similarly large ceiling position amino acids) of PARP15 but not with Thr484 of PARP4. | ||
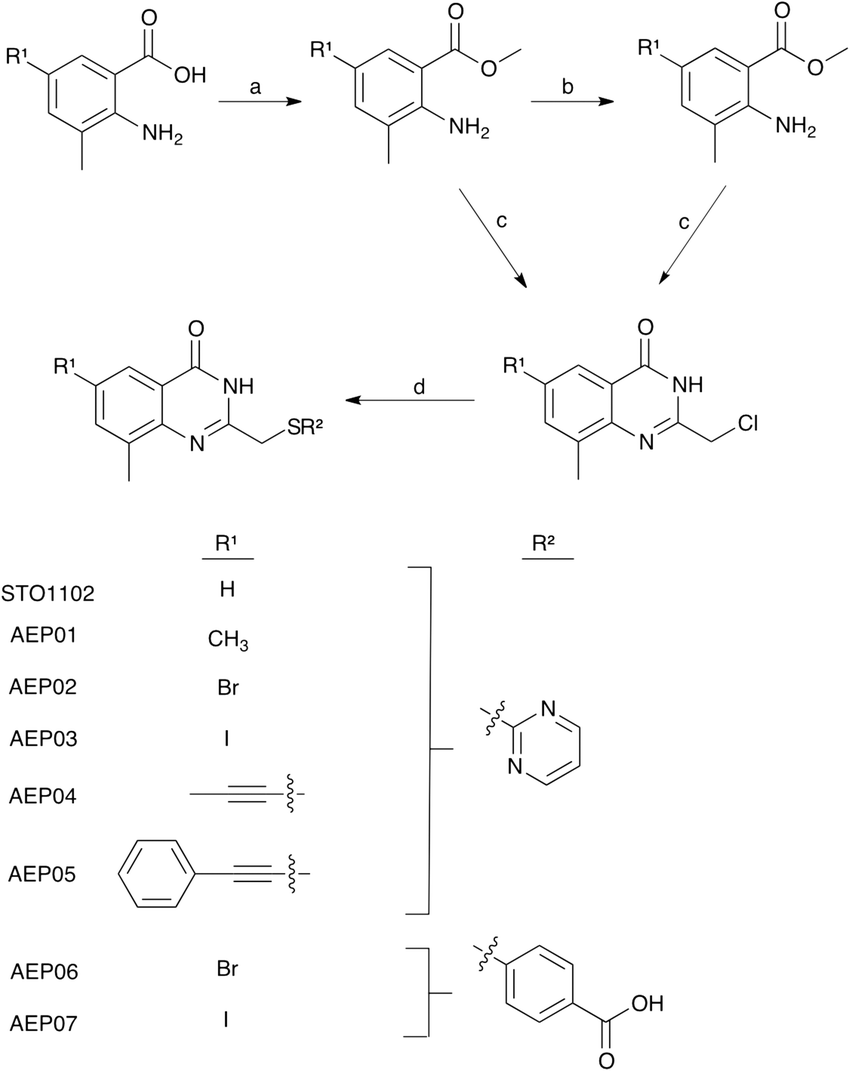 | ||
| Scheme 1 Synthesis of STO1102 and QDR analogs with substitutions at C-8 (R1) and C-2 (R2). Reagents and conditions: a. H2SO4, MeOH, 90 °C, 18 h; (AEP04-05) b. bromo benzoate analog, Pd(PPh3)4, R1–SnBu3, toluene, 115 °C, 3 h; c. 4 M HCl, chloroacetonitrile, 110 °C, 8 h; d. R2-SH, NaH, DMF, RT, 1 h. | ||
First, we screened STO1102 and AEP01-05 against several members PARP family using our PARP activity screening and inhibitor testing assay (PASTA)16,24,31 (Fig. 3, and Table S1†). We found that the unsubstituted STO1102 most potently inhibited PARP2 and PARP11 (IC50 = 0.8 μM and 0.2 μM respectively). STO1102 did not inhibit PARP4 up to 10 μM. By contrast, AEP01, which contains a simple methyl group at the C-8 position of the QDR scaffold, potently inhibited PARP4 (IC50 = 0.5 μM); however, AEP01 was more potent against PARP1 (IC50 = 0.1 μM). AEP02, which contains a bromo group at C-8 of the QDR scaffold, had similar potency to AEP01 against PARP4 (IC50 = 0.8 μM), but was 25-fold less potent than AEP01 against PARP1 (IC50 = 2.5 μM). AEP02 also inhibited PARP2 with moderate potency (IC50 = 2.0 μM). Beyond PARP1, PARP2, and PARP4, AEP02 did not appreciably inhibit other PARP family members tested. AEP03 was less potent than AEP02 against PARP4 (IC50 = 2.7 μM), suggesting that. While AEP03 was less potent than AEP02 against PARP4, it did exhibit better family-wide selectivity (Fig. 3 and Table S1†). Neither the 1-propyne- (AEP04) nor the 1-phenyl-propyne (AEP05) derivatives appreciably inhibited PARP4 or other PARP family members. The one exception is moderate inhibition of PARP15 by AEP04 (IC50 = 3.3 μM). Taken together, these results show that QDR-based inhibitors with small substituents at the C-8 position are promising selective inhibitors of PARP4.
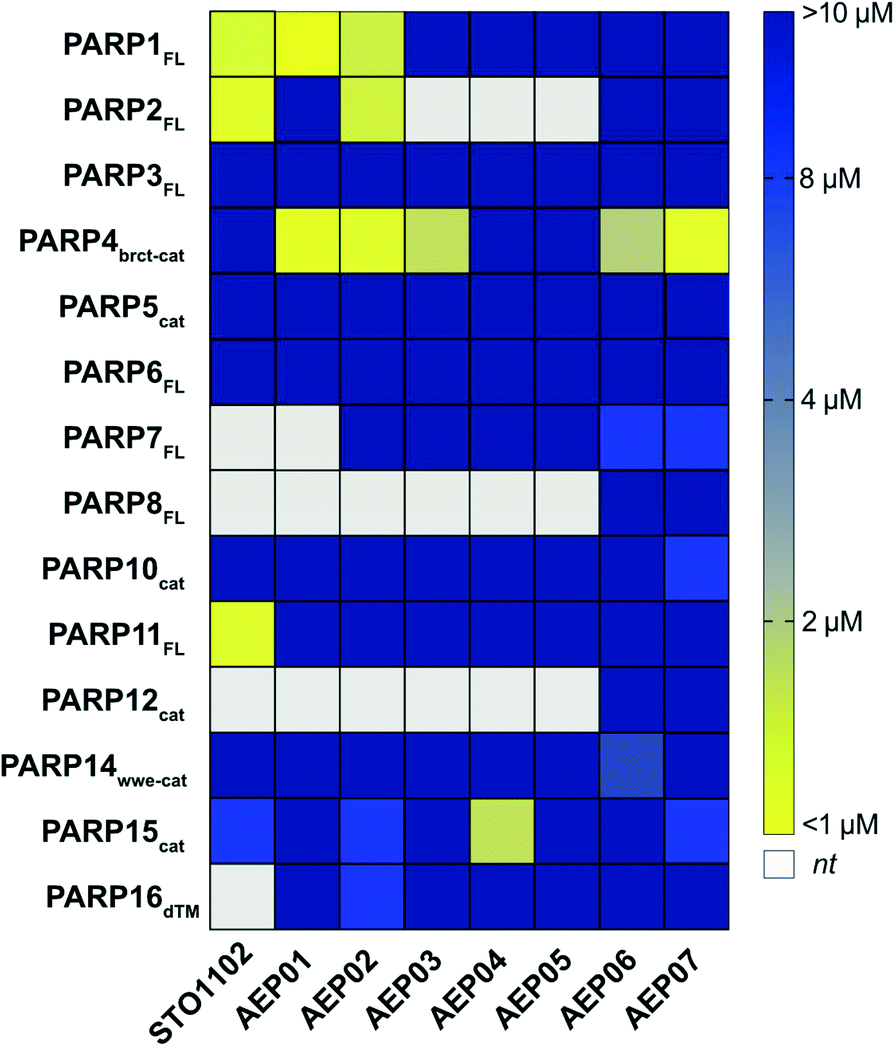 | ||
| Fig. 3 PASTA data for AEP series of compounds. Heat map representation of IC50 data [yellow = more potent, blue = less potent; white: not tested (nt)]. | ||
We sought to further improve the PARP4 selectivity of C-8 substituted QDR compounds, with a particular focus on eliminating PARP1/2 inhibition. The C-2 position on the QDR scaffold of STO1102 (and related compounds) interacts with the ADP-ribose sub-site of the NAD+ pocket.30 Unlike the nicotinamide sub-site, the amino acid composition of the ADP-ribose sub-site—particularly in a region referred to as the donor loop (D-loop)—is divergent across PARP family members. Indeed, previous studies from our lab and others found that the nature of the C-2 substituent of QDR-based inhibitors can impact family-wide selectivity and inhibitor potency.24,25 For example, we found that changing the pyrimidine to a para-benzoic acid at the C-2 position of C-7 modified QDR-based inhibitors decreased potency against PARP1 and PARP2. We therefore synthesized the C-2 para-benzoic acid analogs of AEP02 and AEP03: AEP06 (C-2 bromo) and AEP07 (C-2 iodo) (Scheme 1). We screened AEP06 and AEP07 across the PARP family using PASTA (Fig. 3 and Table S1†). AEP06 showed improved selectivity over AEP02 against PARP1 and PARP2, but was 2.5-fold less potent than AEP02 against PARP4 (IC50 = 1.9 μM). AEP07 was 4-fold more potent than AEP03 against PARP4 (IC50 = 0.8 μM), and exhibited excellent family-wide selectivity. It is interesting, with respect to PARP4 inhibition, that AEP07 trends in the opposite direction of AEP06 when comparing their C-2 pyrimidine counterparts (AEP03 and AEP02, respectively). One possibility is that the C-2 substituent influences the interactions at C-8 by subtly changing the orientation of the QDR scaffold in the NAD+ binding site of PARP4. Obtaining crystal structures of these compounds bound to PARP4 will provide insight into this hypothesis. Taken together, these results show that the C-2 position can impact the selectivity and potency of C-8 halogen substituted QDR-based PARP4 inhibitors.
Conclusions
Using rational design based on structural bioinformatics, we found that substituting the C-8 position of QDR-based inhibitors with halogens can lead to selective and reasonably potent inhibitors of PARP4. AEP07, which contains an iodo at C-8 and a para-benzoic acid at C-2, is the most potent and selective PARP4 inhibitor described to date. Further studies are required to evaluate the efficacy of AEP07 in inhibiting PARP4 in cells. AEP07 contains a benzoic acid which may impact its membrane permeability. It should be noted, however that a related QDR analog (ITK6) with a propynyl at the C-7 position and a para-benzoic acid at C-2 inhibited PARP10 in cells.24 Even if AEP07 is not membrane permeable it will still serve as a good lead compound for further optimization. Ultimately, membrane permeable PARP4 inhibitors will be a useful pharmacological tool for elucidating the function of PARP4-mediated MARylation in cells and for exploring the therapeutic potential of inhibiting the catalytic activity of PARP4.How AEP07 impart PARP4-selective inhibition is not clear, but could involves a combination of steric and stereoelectronic effects. Obtaining a crystal structure of AEP07 bound to PARP4 could reveal how the iodo at C-8 interacts with the nicotinamide sub-site and how the para-benzoic acid at C-2 interacts with the ADP-ribose sub-site. This information could guide the design of next generation PARP4 inhibitors with improved potency and selectivity.
Experimental section
Cloning
cDNA encoding full length human PARP1, full length human PARP2, full length human PARP3, human PARP4 brct-catalytic domain, human PARP5 catalytic domain, human PARP10 catalytic domain, full length human PARP11, human PARP12 catalytic domain, human PARP14 wwe-catalytic domain, human PARP15 catalytic domain, PARP16 (Δ transmembrane, TM), and SRPK2 were obtained as previously described.24Individual cDNA constructs encoding full length human PARP6, full length human PARP7, and full length human PARP8 were obtained using gBlock gene fragments containing the PARP11 gene as template (IDT) and the following primers (IDT) for subsequent Gibson assembly cloning:
PARP6FL: forward: TACTTCCAATCCAATGCAGACATCAAAGGCC; AGTTCTGGA reverse: TTATCCACTTCCAATGTTATTAGTTTGTGTAAACCTGAGTTCCGATCA.
PARP7FL: forward: TACTTCCAATCCAATGCAGAAACCACCGCCGAACCTGAG; reverse: TTATCCACTTCCAATGTTATTAAATGGAAACAGTGTTACTGACTTC.
PARP8FL: forward: TACTTCCAATCCAATGCAGGGATGTGTTCAAGGCAAGAGAG; reverse: TTATCCACTTCCAATGTTATTAACCAGTCGCAGTCTGATTACCAATC.
The amplified fragments were gel purified and cloned into a pET-His-SUMO-TEV LIC cloning vector (1B), a gift from Scott Gradia (Addgene plasmid #29653) by an isothermal assembly protocol using the Gibson assembly mix (NEB). The human PARP3 plasmid was a gift from Herwig Schuler (Lund University).
PARP4 homology model
A PARP4 homology model was generated using SWISS-MODEL (https://swissmodel.expasy.org/).32 In brief, the catalytic domain of human PARP4 (aa 369–573) was used to search for a template. PARP2 bound to the PARP1/2 inhibitor olaparib (4TVJ) was identified as the best template and was used to generate a model of the PARP4 catalytic domain.Expression and purification of proteins
The following protein were expressed and purified as previously described: N-terminal hexahistidine (His6) human PARP1, PARP2, PARP3, PARP5bcat, PARP10cat, PARP14wwe-cat, PARP15cat, SRPK2; pET-His-SUMO-TEV human PARP4, PARP6, PARP11, and PARP16.24pET-His6-SUMO-TEV-PARP6FL, pET-His6-SUMO-TEV-PARP7Fl, or pET-His6-SUMO-TEV-PARP8FL plasmids was transformed into Escherichia coli BL21 (DE3) competent cells (Millipore) and grown on an LB agar plate with kanamycin (50 mg mL−1) and chloramphenicol (34 mg mL−1) overnight at 37 °C. A swath of cells was inoculated into a 50 mL starter culture of LB media with kanamycin (50 mg mL−1) and chloramphenicol (34 mg mL−1) at 225 rpm, 37 °C overnight. For each protein of interest 1–2 liters of terrific broth (TB) media (12 g bacto tryptone, 24 g yeast extract, 0.4% glycerol, 17 mM KH2PO4, 72 mM K2HPO4, 1% glucose, 50 μg mL−1 kanamycin, 34 μg mL−1 chloramphenicol) was inoculated with the starter culture and grown to an OD = 0.8–1.0 at 37 °C, 225 rpm. Isopropyl-β-thiogalactoside, IPTG (Sigma-Aldrich) was added to 0.4 mM to induce protein expression for 18–24 h at 16 °C, 225 rpm. Cells were harvested by centrifuging, resuspended in lysis buffer [20 mM HEPES, pH 7.5, 1 mM β-mercaptoethanol, 1 mM benzamidine, 0.2% NP-40, 0.2% TWEEN-20, 500 mM NaCl, 1 mM phenylmethylsulfonyl fluoride (PMSF), 8.3 mg L−1 DNAse I (Roche)] and lysed by sonication at 0 °C (Branson sonifier 450). Lysates were incubated with pre-washed Ni-NTA agarose resin (50% slurry, Qiagen) with end-over-end rotation at 4 °C for 1 h. Following extensive washing with buffer B1+20 (20 mM HEPES, pH 7.5, 1 mM β-Me, 1 mM PMSF, 1 mM benzamidine, 500 mM NaCl, 20 mM imidazole). Protein was eluted in four fractions of B1 containing 100–400 mM imidazole. Fractions were analyzed by SDS page gel stained with coomassie blue, or western blot visualized with ponceau stain. Fractions found to contain the desired protein as judged by molecular weight were dialyzed to 50 mM Tris–HCl, pH 7.5, 0.1 mM EDTA, 1 mM β-Me, 0.4 M NaCl at 4 °C. Protein concentration was determined from a standard curve of His6-PARP1FL of a known concentration on western blot visualized with a monoclonal anti-His antibody (Proteintech). Proteins were obtained in >90% purity.
PASTA (PARP inhibitor screening assay)
96-Well nickel coated plate (Pierce) was incubated with 350 ng His6-tagged SRPK2 in hB (50 mM HEPES pH 7.5, 100 mM NaCl, 4 mM MgCl2, 0.2 mM TCEP) for 60 min at RT. After extensively washing the plate, 4–400 nM (2×) of recombinant PARP enzyme in hB were added to individual wells of the 96-well plate. PARP3FL was activated by Dnick 5’P as described previously [26]. PARPs were initially screened at 10 μM (and up to 30 μM for PARP1, which is more tolerant of DMSO, for AEP07 and AEP08). For compounds that exhibited inhibition below 10 μM, half-maximal inhibitory concentration (IC50) values were obtained by varying concentrations of each inhibitor (0–200 μM). Inhibitors were pre-incubated with 200 μM 6-a-NAD+ (2×) in hB at room temperature for 5–10 min, then added to the plate. This reaction proceeded for 60 min at 30 °C, the plate was then washed three times with 1X PBST (1X PBS, 0.01% Tween-20), once with 1X PBS, then click conjugation was performed in CB (100 μM biotin-PEG3-azide, 100 μM Tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA, Sigma), 1 mM CuSO4, 1 mM TCEP, 1X PBS) for 30 min at RT. The plate was then washed three times with 1X PBST, once with 1X PBS, and then blocked with 1% milk (Carnation) in 1X PBST for 30 min at RT. The plate was then washed three times with 1X PBST, once with 1X PBS, and then incubated with Strep-HRP (300 ng μL−1 BSA, 0.05 ng μL−1 Strep-HRP, 1X PBS) for 30 min at RT. The plate was then washed three times with 1X PBST, once with 1X PBS, and then developed with QuantaRed™ enhanced chemifluorescent HRP substrate (Thermo) for 30–45 s before quenching with Quanta Red Stop Solution. Fluorescence for each sample and control was read at excitation 570 nM and emission 600 nM with a Spectra Max i3 (molecular devices) within five min of development. Inhibitor dose response curves were fit using linear regression in Prism 7 (GraphPad™ Software). The mean IC50 for each compound was calculated from at least three independent assays.Chemistry
Methy 2-amino-3-methylbenzoate. From 2-amino-3-methylbenzoic acid (500 mg, 3.3 mmol); yield 348 mg (64%). 1H NMR (400 MHz, chloroform-d) δ 7.77 (ddd, J = 8.1, 1.6, 0.6 Hz, 1H), 7.19 (ddq, J = 7.2, 1.7, 0.8 Hz, 1H), 6.59 (dd, J = 8.1, 7.2 Hz, 1H), 5.83 (s, 2H), 3.87 (s, 3H), 2.17 (d, J = 0.7 Hz, 3H).
Methyl 2-amino-3,5-dimethylbenzoate. From 2-amino-3,5-dimethylbenzoic acid (250 mg, 1.52 mmol); yield 200 mg (74%). 1H NMR (400 MHz, DMSO-d6) δ 7.43 (d, J = 2.2 Hz, 1H), 7.04 (d, J = 2.2 Hz, 1H), 6.31 (s, 2H), 3.77 (s, 3H), 2.11 (dt, J = 20.3, 0.7 Hz, 6H).
Methyl 2-amino-5-bromo-3-methylbenzoate. From 2-amino-5-bromo-3-methylbenzoic acid (50 mg, 0.22 mmol); yield 42.8 mg (90%). 1H NMR (400 MHz, DMSO-d6) δ 7.87 (d, J = 2.2 Hz, 1H), 7.48 (dd, J = 2.1, 1.0 Hz, 1H), 6.70–6.62 (m, 2H), 3.79 (s, 3H), 2.09 (s, 3H).
Methyl 2-amino-3-methyl-5-(prop-1-yn-1-yl)benzoate (novel compound). Methyl 2-amino-5-bromo-3-methylbenzoate (200 mg, 0.8 mmol) was dissolved in anhydrous toluene and concentrated (3 × 4 mL) then dried under vacuum for 1 h. The flask was evacuated and refilled with argon gas (3×), then anhydrous toluene (8 mL) was added via syringe and the solution degassed with argon 10 min. Tributyl(1-propynyl)tin (0.3 mL, 0.88 mmol) was added via syringe and the solution degassed 5 min. Pd(PPh3)4 (95 mg, 0.08 mmol) was added quickly and the solution degassed with argon for 3 min. The reaction mixture was refluxed at 115 °C for 2.5 h. TLC analysis (10% EtOAc in hexanes) showed consumption of starting benzoate. The reaction was cooled to RT and concentrated in vacuo. The crude residue was purified via a Combiflash Companion system (4 g Redisep silica column; 0–20% EtOAc in hexanes). Product was isolated as a yellow oil: 75 mg (46%). 1H NMR (400 MHz, chloroform-d) δ 7.86 (s, 1H), 7.22 (s, 1H), 5.93 (s, 2H), 3.86 (d, J = 0.9 Hz, 3H), 2.13 (t, J = 0.8 Hz, 3H), 2.02 (d, J = 0.9 Hz, 3H).
Methyl 2-amino-3-methyl-5-(phenylethynyl)benzoate (novel compound). Methyl 2-amino-5-bromo-3-methylbenzoate (150 mg, 0.6 mmol) was dissolved in anhydrous toluene and concentrated (3 × 4 mL) then dried under vacuum for 1 h. The flask was evacuated and refilled with argon gas (3×), then anhydrous toluene (8 mL) was added via syringe and the solution degassed with argon 10 min. Tributyl(phenylethynyl)stannane (0.23 mL, 0.66 mmol) was added via syringe and the solution degassed 5 min. Pd(PPh3)4 (70 mg, 0.06 mmol) was added quickly and the solution degassed with argon for 3 min. The reaction mixture was refluxed at 115 °C for 3 h. TLC analysis (10% EtOAc in hexanes) showed consumption of starting benzoate. The reaction was cooled to RT and concentrated in vacuo. The crude residue was purified via a Combiflash Companion system (4 g Redisep silica column; 0–20% EtOAc in hexanes). Product was isolated as an orange oil: 100 mg (65%). 1H NMR (400 MHz, chloroform-d) δ 8.02 (d, J = 2.0 Hz, 1H), 7.49 (dd, J = 8.0, 1.7 Hz, 2H), 7.37 (s, 1H), 7.34–7.31 (m, 3H), 6.05 (s, 2H), 3.89 (s, 3H), 2.18 (d, J = 0.7 Hz, 3H).
2-(Chloromethyl)-8-methylquinazolin-4(3H)-one. From methyl 2-amino-3-methylbenzoate (250 mg, 1,52 mmol); yield 210 mg (48%); 1H NMR (400 MHz, DMSO-d6) δ 12.57 (s, 1H), 7.97 (d, J = 7.9 Hz, 1H), 7.70 (d, J = 7.5 Hz, 1H), 7.44 (t, J = 7.6 Hz, 1H), 4.56 (d, J = 1.3 Hz, 2H), 2.53 (d, J = 9.2 Hz, 3H).
2-(Chloromethyl)-6,8-dimethylquinazolin-4(3H)-one. From methyl 2-amino-3,5-dimethylbenzoate (100 mg, 0.6 mmol); yield 125 mg (94%). 1H NMR (400 MHz, DMSO-d6) δ 12.49 (s, 1H), 7.76 (d, J = 2.1 Hz, 1H), 7.54 (d, J = 2.1 Hz, 1H), 4.55 (s, 2H), 2.40 (s, 3H).
6-Bromo-2-(chloromethyl)-8-methylquinazolin-4(3H)-one. From methyl 2-amino-5-bromo-3-methylbenzoate (50 mg, 0.22 mmol); yield 50 mg (79%); 1H NMR (400 MHz, chloroform-d) δ 9.80 (s, 1H), 8.26 (d, J = 2.3 Hz, 1H), 7.75–7.72 (m, 1H), 4.58 (s, 2H), 2.57 (s, 3H).
2-(Chloromethyl)-6-iodo-8-methylquinazolin-4(3H)-one (novel compound). From methyl 2-amino-5-iodo-3-methylbenzoate (250 mg, 0.85 mmol); yield 275 mg (97%). 1H NMR (400 MHz, DMSO-d6) δ 12.75 (s, 1H), 8.23 (d, J = 2.1 Hz, 1H), 8.05 (d, J = 2.1 Hz, 1H), 4.55 (s, 2H), 2.46 (s, 3H).
2-(Chloromethyl)-8-methyl-6-(prop-1-yn-1-yl)quinazolin-4(3H)-one (novel compound). From methyl 2-amino-3-methyl-5-(prop-1-yn-1-yl)benzoate (30 mg, 0.15 mmol); yield 20 mg (54%). Taken on crude.
2-(Chloromethyl)-8-methyl-6-(phenylethynyl)quinazolin-4(3H)-one (novel compound). from methyl 2-amino-3-methyl-5-(phenylethynyl)benzoate (100 mg, 0.4 mmol); yield 80 mg (67%). Taken on crude.
8-Methyl-2-((pyrimidin-2-ylthio)methyl)quinazolin-4(3H)-one, STO1102. From 2-(chloromethyl)-8-methylquinazolin-4(3H)-one (112 mg, 0.54 mmol); reaction was concentrated in vacuo and product recovered as a white solid; yield 55.3 mg (36%); 1H NMR (400 MHz, DMSO-d6) δ 12.46 (s, 1H), 8.70 (d, J = 4.9 Hz, 2H), 7.98–7.94 (m, 1H), 7.70–7.64 (m, 1H), 7.40 (t, J = 7.6 Hz, 1H), 7.29 (t, J = 4.9 Hz, 1H), 4.48 (s, 2H), 2.48 (s, 3H); Rt: 5.81 min. HRMS m/z [M + H]+ for C14H13N4OS: 285.08046, found 285.08128.
6,8-Dimethyl-2-((pyrimidin-2-ylthio)methyl)quinazolin-4(3H)-one (novel compound), AEP01. From 2-(chloromethyl)-6,8-dimethylquinazolin-4(3H)-one (25 mg, 0.11 mmol); product precipitated as fine white solids in water; yield 10 mg (30%). 1H NMR (400 MHz, DMSO-d6) δ 12.33 (s, 1H), 8.74–8.60 (m, 2H), 7.72 (s, 1H), 7.47 (s, 1H), 7.26 (d, J = 5.1 Hz, 1H), 4.42 (s, 2H), 2.39 (d, J = 14.1 Hz, 6H). MS m/z [M + H]+ for C14H13N4OS: 299.0, found 299.0. Rt = 4.94 min. HRMS m/z [M + H]+ for C15H14N4OS: 299.09611, found 299.09897.
6-Bromo-8-methyl-2-((pyrimidin-2-ylthio)methyl)quinazolin-4(3H)-one (novel compound), AEP02. From 6-bromo-2-(chloromethyl)-8-methylquinazolin-4(3H)-one (25 mg, 0.09 mmol); product precipitated as fine white solids in water; yield 12 mg (36%). 1H NMR (400 MHz, DMSO-d6) δ 12.61 (s, 1H), 8.66 (d, J = 4.8 Hz, 2H), 8.19 (d, J = 2.1 Hz, 1H), 7.98 (s, 1H), 7.25 (t, J = 4.9 Hz, 1H), 4.42 (s, 2H), 2.40 (s, 3H). MS m/z [M + H]+ for C14H11BrN4OS: 362.9, 364.9, found 362.8, 364.8. Rt = 5.37 min. HRMS m/z [M + H]+ for C14H11BrN4OS: 364.98895, found 364.9917.
6-Iodo-8-methyl-2-((pyrimidin-2-ylthio)methyl)quinazolin-4(3H)-one (novel compound), AEP03. From 2-(chloromethyl)-6-iodo-8-methylquinazolin-4(3H)-one (25 mg, 0.07 mmol) product precipitated as fine white solids in water; yield 15 mg (54%). 1H NMR (400 MHz, DMSO-d6) δ 12.60 (s, 1H), 8.65 (d, J = 4.9 Hz, 2H), 8.19 (d, J = 2.1 Hz, 1H), 7.98 (s, 1H), 7.25 (t, J = 4.9 Hz, 1H), 4.42 (s, 2H), 2.39 (s, 3H). MS m/z [M − H]− for C14H11IN4OS: 408.9, found 408.5. Rt = 5.57 min. HRMS m/z [M + H]+ for C14H11IN4OS: 410.97710, found 410.97996.
8-Methyl-6-(prop-1-yn-1-yl)-2-((pyrimidin-2-ylthio)methyl)quinazolin-4(3H)-one (novel compound), AEP04. From 2-(chloromethyl)-8-methyl-6-(prop-1-yn-1-yl)quinazolin-4(3H)-one (30 mg, 0.08 mmol)); product precipitated as fine white solids in water; yield 23 mg (92%). 1H NMR (400 MHz, DMSO-d6) δ 8.70–8.63 (m, 2H), 7.84 (s, 1H), 7.61 (s, 1H), 7.25 (t, J = 4.8 Hz, 1H), 4.43 (d, J = 5.4 Hz, 2H), 2.40 (s, 3H), 2.07 (s, 3H). MS m/z [M − H]− for C17H14N4OS: 322.09, found 320.6. Rt = 5.17 min. HRMS m/z [M + H]+ for C17H14N4OS: 323.09611, found 323.09896.
8-Methyl-6-(phenylethynyl)-2-((pyrimidin-2-ylthio)methyl)quinazolin-4(3H)-one (novel compound), AEP05. From 2-(chloromethyl)-8-methyl-6-(phenylethynyl)quinazolin-4(3H)-one (20 mg, 0.07 mmol); product precipitated as fine white solids in water; yield 18 mg (62%). 1H NMR (400 MHz, DMSO-d6) δ 12.61 (s, 1H), 8.67 (d, J = 4.9 Hz, 2H), 8.05 (s, 1H), 7.80 (s, 1H), 7.62–7.58 (m, 2H), 7.48–7.43 (m, 3H), 7.26 (t, J = 4.9 Hz, 1H), 4.46 (s, 2H), 2.45 (s, 3H). MS m/z [M + H]+ for C22H16N4OS: 384.1, found 384.8. Rt = 7.41 min. HRMS m/z [M + H]+ for C22H16N4OS: 385.11176, found 385.11469.
4-(((6-Bromo-8-methyl-4-oxo-3,4-dihydroquinazolin-2-yl)methyl)thio)benzoic acid (novel compound), AEP06. From 6-bromo-2-(chloromethyl)-8-methylquinazolin-4(3H)-one (20 mg, 0.07 mmol); product precipitated as tan solids in water; yield 5 mg (14%). 1H NMR (400 MHz, DMSO-d6) δ 12.92 (s, 1H), 12.61 (s, 1H), 7.99 (s, 1H), 7.84 (d, J = 7.1 Hz, 3H), 7.61 (d, J = 8.2 Hz, 2H), 4.25 (s, 2H), 2.43 (s, 3H). MS m/z [M − H]− for C17H13BrN2O3S: 402.9, 404.9, found 402.5, 404.5. Rt = 5.02 min. HRMS m/z [M + H]+ for C17H13BrN2O3S: 40698895, found 406.99078.
4-(((6-Iodo-8-methyl-4-oxo-3,4-dihydroquinazolin-2-yl)methyl)thio)benzoic acid (novel compound), AEP07. From 2-(chloromethyl)-6-iodo-8-methylquinazolin-4(3H)-one (25 mg, 0.08 mmol) product precipitated as fine white solids in water; yield 15 mg (58%). 1H NMR (400 MHz, DMSO-d6) δ 12.93 (s, 1H), 12.60 (s, 1H), 8.18 (d, J = 2.1 Hz, 1H), 7.98 (t, J = 1.4 Hz, 1H), 7.87–7.82 (m, 2H), 7.62 (d, J = 8.5 Hz, 2H), 4.24 (s, 2H), 2.40 (s, 3H). MS m/z [M − H]− for C17H13IN2O3S: 450.9, found 450.5. Rt = 4.79 min. HRMn/z [M + H]+ for C17H13IN2O3S: 452.97643, found 452.97873.
Funding
This work was supported by grants from the Pew Biomedical Scholars program (M. S. C.), OHSU Biomedical Innovation Program (M. S. C.), and NIH NINDS (NIH 2R01NS088629 to M. S. C.).Author contributions
I. T. K. and M. S. C. conceived of this study. I. T. K. and A. E. P. synthesized the compounds and executed all experiments. I. T. K., A. E. P., and M. S. C. wrote the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thanks members of the Cohen laboratory for thoughtful reading of the manuscript.References
- L. Weixler, et al. ADP-ribosylation of RNA and DNA: from in vitro characterization to in vivo function, Nucleic Acids Res., 2021, 49, gkab136 CrossRef PubMed.
- M. S. Cohen and P. Chang, Insights into the biogenesis, function, and regulation of ADP-ribosylation, Nat. Chem. Biol., 2018, 14, 236–243 CrossRef CAS PubMed.
- S. Vyas, et al. Family-wide analysis of poly(ADP-ribose) polymerase activity, Nat. Commun., 2014, 5, 4426 CrossRef CAS PubMed.
- G. T. Marsischky, B. A. Wilson and R. J. Collier, Role of Glutamic Acid 988 of Human Poly-ADP-ribose Polymerase in Polymer Formation EVIDENCE FOR ACTIVE SITE SIMILARITIES TO THE ADP-RIBOSYLATING TOXINS (*), J. Biol. Chem., 1995, 270, 3247–3254 CrossRef CAS PubMed.
- V. Rolli, M. O'Farrell, J. M. Murcia and G. Murcia, Random Mutagenesis of the Poly(ADP-ribose) Polymerase Catalytic Domain Reveals Amino Acids Involved in Polymer Branching †, Biochemistry, 1997, 36, 12147–12154 CrossRef CAS PubMed.
- M. O. Hottiger, P. O. Hassa, B. Lüscher, H. Schüler and F. Koch-Nolte, Toward a unified nomenclature for mammalian ADP-ribosyltransferases, Trends Biochem. Sci., 2010, 35, 208–219 CrossRef CAS PubMed.
- N. L. Kedersha and L. H. Rome, Isolation and characterization of a novel ribonucleoprotein particle: large structures contain a single species of small RNA, J. Cell Biol., 1986, 103, 699–709 CrossRef CAS PubMed.
- V. A. Kickhoefer, et al. The 193-Kd Vault Protein, Vparp, Is a Novel Poly(Adp-Ribose) Polymerase, J. Cell Biol., 1999, 146, 917–928 CrossRef CAS PubMed.
- N. L. Kedersha and L. H. Rome, Vaults: Large cytoplasmic RNP's that associate with cytoskeletal elements, Mol. Biol. Rep., 1990, 14, 121–122 CrossRef CAS PubMed.
- C.-L. Zheng, et al. Characterization of MVP and VPARP assembly into vault ribonucleoprotein complexes, Biochem. Biophys. Res. Commun., 2004, 326, 100–107 CrossRef PubMed.
- A. Zon, M. H. Mossink, R. J. Scheper, P. Sonneveld and E. A. C. Wiemer, The vault complex, Cell. Mol. Life Sci., 2003, 60, 1828–1837 CrossRef PubMed.
- S. Raval-Fernandes, V. A. Kickhoefer, C. Kitchen and L. H. Rome, Increased Susceptibility of Vault Poly(ADP-Ribose) Polymerase–Deficient Mice to Carcinogen-Induced Tumorigenesis, Cancer Res., 2005, 65, 8846–8852 CrossRef CAS PubMed.
- P. C. Lara, M. Pruschy, M. Zimmermann and L. A. Henríquez-Hernández, MVP and vaults: a role in the radiation response, Radiat. Oncol., 2011, 6, 148 CrossRef CAS PubMed.
- W. Berger, E. Steiner, M. Grusch, L. Elbling and M. Micksche, Vaults and the major vault protein: Novel roles in signal pathway regulation and immunity, Cell. Mol. Life Sci., 2008, 66, 43 CrossRef PubMed.
- T. J. Wigle, et al. Forced Self-Modification Assays as a Strategy to Screen MonoPARP Enzymes, SLAS Discovery, 2019, 25, 241–252 Search PubMed.
- I. T. Kirby, R. K. Morgan and M. S. Cohen, A Simple, Sensitive, and Generalizable Plate Assay for Screening PARP Inhibitors, Methods Mol. Biol., 2018, 1813, 245–252 CrossRef CAS PubMed.
- A.-G. Thorsell, et al. Structural Basis for Potency and Promiscuity in Poly(ADP-ribose) Polymerase (PARP) and Tankyrase Inhibitors, J. Med. Chem., 2017, 60, 1262–1271 CrossRef CAS PubMed.
- I. T. Kirby and M. S. Cohen, Small-Molecule Inhibitors of PARPs: From Tools for Investigating ADP-Ribosylation to Therapeutics, Curr. Top. Microbiol. Immunol., 2018, 55, 7706–7721 Search PubMed.
- J. M. Gozgit, M. M. Vasbinder, R. P. Abo, K. Kunii, K. G. Kuplast-Barr, B. Gui, A. Z. Lu, J. R. Molina, E. Minissale, K. K. Swinger, T. J. Wigle, D. J. Blackwell, C. R. Majer, Y. Ren, M. Niepel, Z. A. Varsamis, S. P. Nayak, E. Bamberg, J.-R. Mo, W. D. Church, A. S. A. Mady, J. Song, L. Utley, P. E. Rao, T. J. Mitchison, K. W. Kuntz, V. M. Richon and H. Keilhack, PARP7 negatively regulates the type I interferon response in cancer cells and its inhibition triggers antitumor immunity, Cancer Cell, 2021, 39(9), 1214–1226.e10 CrossRef CAS PubMed.
- R. K. Morgan, I. T. Kirby, A. Vermehren-Schmaedick, K. Rodriguez and M. S. Cohen, Rational Design of Cell-Active Inhibitors of PARP10, ACS Med. Chem. Lett., 2019, 10, 74–79 CrossRef CAS PubMed.
- S. Murthy, et al. 4-(Phenoxy) and 4-(benzyloxy)benzamides as potent and selective inhibitors of mono-ADP-ribosyltransferase PARP10/ARTD10, Eur. J. Med. Chem., 2018, 156, 93–102 CrossRef CAS PubMed.
- J. Holechek, et al. Design and synthesis and evaluation of potent and selective inhibitors of mono-(ADP-ribosyl)transferases PARP10 and PARP14, Bioorg. Med. Chem. Lett., 2018, 28, 2050–2054 CrossRef CAS PubMed.
- H. Venkannagari, et al. Small-Molecule Chemical Probe Rescues Cells from Mono-ADP-Ribosyltransferase ARTD10/PARP10-Induced Apoptosis and Sensitizes Cancer Cells to DNA Damage, Cell Chem. Biol., 2016, 23, 1251–1260 CrossRef CAS PubMed.
- I. T. Kirby, et al. A Potent and Selective PARP11 Inhibitor Suggests Coupling between Cellular Localization and Catalytic Activity, Cell Chem. Biol., 2018, 25, 1547–1553 CrossRef CAS PubMed.
- L. B. Schenkel, J. R. Molina, K. K. Swinger, R. Abo, D. J. Blackwell, A. Z. Lu, A. E. Cheung, W. D. Church, K. Kunii, K. G. Kuplast-Barr, C. R. Majer, E. Minissale, J.-R. Mo, M. Niepel, C. Reik, Y. Ren, M. M. Vasbinder, T. J. Wigle, V. M. Richon, H. Keilhack and K. W. Kuntz, A potent and selective PARP14 inhibitor decreases protumor macrophage gene expression and elicits inflammatory responses in tumor explants, Cell Chem. Biol., 2021, 28(8), 1158–1168.e13 CrossRef CAS PubMed.
- A. C. Bishop, et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase, Nature, 2000, 407, 395–401 CrossRef CAS PubMed.
- R. K. Morgan, I. Carter-O'Connell and M. S. Cohen, Selective inhibition of PARP10 using a chemical genetics strategy, Bioorg. Med. Chem. Lett., 2015, 25, 4770–4773 CrossRef CAS PubMed.
- I. Carter-O'Connell, H. Jin, R. K. Morgan, L. L. David and M. S. Cohen, Engineering the Substrate Specificity of ADP-Ribosyltransferases for Identifying Direct Protein Targets, J. Am. Chem. Soc., 2014, 136, 5201–5204 CrossRef PubMed.
- E. Wahlberg, et al. Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors, Nat. Biotechnol., 2012, 30, 283–288 CrossRef CAS PubMed.
- C. D. Andersson, et al. Discovery of Ligands for ADP-Ribosyltransferases via Docking-Based Virtual Screening, J. Med. Chem., 2012, 55, 7706–7718 CrossRef CAS PubMed.
- I. T. Kirby, D. J. Sanderson and M. S. Cohen, PASTA: PARP activity screening and inhibitor testing assay, STAR protocols, 2021, 2, 100344 CrossRef PubMed.
- A. Waterhouse, et al. SWISS-MODEL: homology modelling of protein structures and complexes, Nucleic Acids Res., 2018, 46, gky427 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1md00195g |
| This journal is © The Royal Society of Chemistry 2021 |