
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDurability of anion exchange membrane water electrolyzers
Dongguo
Li†
a,
Andrew R.
Motz†
b,
Chulsung
Bae
c,
Cy
Fujimoto
d,
Gaoqiang
Yang
e,
Feng-Yuan
Zhang
e,
Katherine E.
Ayers
*b and
Yu Seung
Kim
 *a
*a
aMPA-11: Materials Synthesis & Integrated Devices, Los Alamos National Laboratory, Los Alamos, NM 87545, USA. E-mail: yskim@lanl.gov
bNel Hydrogen, Wallingford, CT 06492, USA. E-mail: kayers@nelhydrogen.com
cDepartment of Chemistry and Chemical Biology, Rensselaer Polytechnic Institute, Troy, New York 12180, USA
dNanoscale Science Department, Sandia National Laboratories, Albuquerque, New Mexico 87185, USA
eDepartment of Mechanical, Aerospace & Biomedical Engineering, UT Space Institute, University of Tennessee, Knoxville, TN 37388, USA
First published on 27th April 2021
Abstract
Interest in the low-cost production of clean hydrogen is growing. Anion exchange membrane water electrolyzers (AEMWEs) are considered one of the most promising sustainable hydrogen production technologies because of their ability to split water using platinum group metal-free catalysts, less expensive anode flow fields, and bipolar plates. Critical to the realization of AEMWEs is understanding the durability-limiting factors that restrict the long-term use of these devices. This article presents both durability-limiting factors and mitigation strategies for AEMWEs under three operation modes, i.e., pure water-fed (no liquid electrolyte), concentrated KOH-fed, and 1 wt% K2CO3-fed operating at a differential pressure of 100 psi. We examine extended-term behaviors of AEMWEs at the single-cell level and connect their behavior with the electrochemical, chemical, and mechanical instability of single-cell components. Finally, we discuss the pros and cons of AEMWEs under these operation modes and provide direction for long-lasting AEMWEs with highly efficient hydrogen production capabilities.
 Dongguo Li | Dongguo Li received his bachelor's degree from University of Science and Technology of China in 2008. He finished his PhD study in chemistry at Brown University in 2014. He then became a postdoc at Argonne National Laboratory focusing on fuel cell catalysts and electrochemistry. After that, he joined the fuel cell research team at Los Alamos National Laboratory as a postdoc. His current research topic covers AEM electrolyzers and PEM fuel cells. |
 Andrew R. Motz | Andrew Motz is Project Manager of Cell Stack Materials at Nel Hydrogen. He received his BS in chemical engineering from the Ohio State University and went on to pursue a PhD in chemical engineering from Colorado School of Mines. Upon graduation in 2018, Andrew was recognized by the faculty for having the top thesis in the department. Next, he joined the research and development group at Nel Hydrogen and has worked on a broad range of electrolysis research projects covering both PEMWE and AEMWE. Andrew has been an active member of the Electrochemical Society since 2014. |
 Chulsung Bae | Chulsung Bae is Ford Foundation Professor at Department of Chemistry & Chemical Biology, Rensselaer Polytechnic Institute. He received BS in Polymer Science & Engineering at Inha University, MS in Materials Science at POSTECH, MS in Chemistry at University of Massachusetts at Lowell, and PhD in Chemistry at University of Southern California under the guidance of Surya Prakash and Nobel Laureate George Olah. He conducted postdoctoral research with John F. Hartwig at Yale University before starting independent academic career in 2004. Bae's research areas focus on the development of ionic functional polymers for applications in clean energy technologies. |
 Cy Fujimoto | Cy Fujimoto is a Principle member of the Technical Staff at Sandia National Laboratories (SNL). He obtained his BS degree in Chemistry from the University of Hawaii, Manoa (1996) and his PhD degree from the University of California, Irvine in Organometallic chemistry in 2001. He then worked as a post-doc at SNL from 2001–2004 in developing ion conducting Diels Alder poly(phenylenes) after which he was promoted to the Technical Staff. He received a joint (with Yu Seung Kim at LANL) award in Outstanding Technical Contribution in the area from the US DOE Hydrogen and Fuel Cell Program in 2016. |
 Katherine E. Ayers | Katherine Ayers is Vice President of R&D for Nel Hydrogen US, with responsibility for developing and executing Nel's technology strategy in proton exchange membrane electrolysis. Her team is primarily responsible for the development of advanced cell stack materials, designs and manufacturing processes, including external collaborations. She is also the lead for the Hydrogen and Fuel Cell Technologies Office funded project on benchmarking and protocol development in water splitting. She received her BS degree in Chemistry/Chemical physics at UC San Diego, and her PhD in chemistry from Caltech. |
 Yu Seung Kim | Yu Seung Kim is a senior staff scientist at Los Alamos National Laboratory (LANL), USA. He currently leads the Membrane Working Group of US DOE Hydrogen and Fuel Cell Technologies Office. He received his BS degree from Korea University (1994) and his PhD from Korea Advanced Institute of Science and Technology (1999) in the Department of Chemical Engineering. He spent three years as a postdoc at Virginia Tech before joining the fuel cell group at Los Alamos (2003). He received Outstanding Innovation Tech Transfer Awards three times from LANL. His research interest is material design for fuel cells and electrolyzers. |
Broader contextHydrogen is an attractive energy carrier that can be stored, re-electrified on demand, and used to produce ammonia and other industrially important chemicals and materials. Currently, a majority (>95%) of hydrogen is produced from fossil fuels by steam reforming which releases massive amounts of carbon dioxide and atmospheric pollutants. Consequently, interest in hydrogen production from renewable sources such as biomass, geothermal, solar, or wind through water-splitting technology is growing. The anion exchange membrane water electrolyzer (AEMWE) is an alternative water-splitting technology to the well-established alkaline or proton exchange membrane water electrolyzers. AEMWEs use less expensive platinum group metal-free electrocatalysts like alkaline water electrolyzers, and have the capability to produce pressurized hydrogen at a high hydrogen production rate. However, the performance and durability of AEMWEs need to be significantly improved for practical use. In this article, we present the performance and durability of state-of-the-art AEMWEs. Then we discuss the durability-limiting factors of AEMWEs based on our own and other's key publications. Comprehensive degradation mechanisms and in-depth discussions on the mitigation strategies will provide future directions to develop commercially viable AEMWE systems. |
1. Introduction
As the U.S. Department of Energy pushes the H2@Scale initiative, research for a more efficient and cost-effective water electrolyzer has received substantial attention due to large-scale hydrogen production and utilization requirements for the resiliency of the power generation and transmission sectors.1,2 The most common hydrogen generation method is the steam reforming of methane or other hydrocarbons which results in high emissions of carbon dioxide. Consequently, water electrolyzers that electrochemically split water into hydrogen and oxygen have garnered great interest.3,4 For high-temperature operation (700–950 °C), solid-oxide steam electrolyzer cells (SOECs) have been developed and demonstrated at the laboratory and experimental plant scale (Fig. 1a). The high operating temperatures of SOECs provide benefits when operating at a relatively low cell voltage with negligible kinetic limitations (high heating value (HHV) electrolysis efficiency is close to 100% at current densities of ∼1 A cm−2). However, problems associated with high-temperature operations such as: long start-stop time, rapid degradation due to high-temperature interdiffusion of the cell components, and poisoning by the corrosion products, currently make SOECs challenging to deploy in the market.3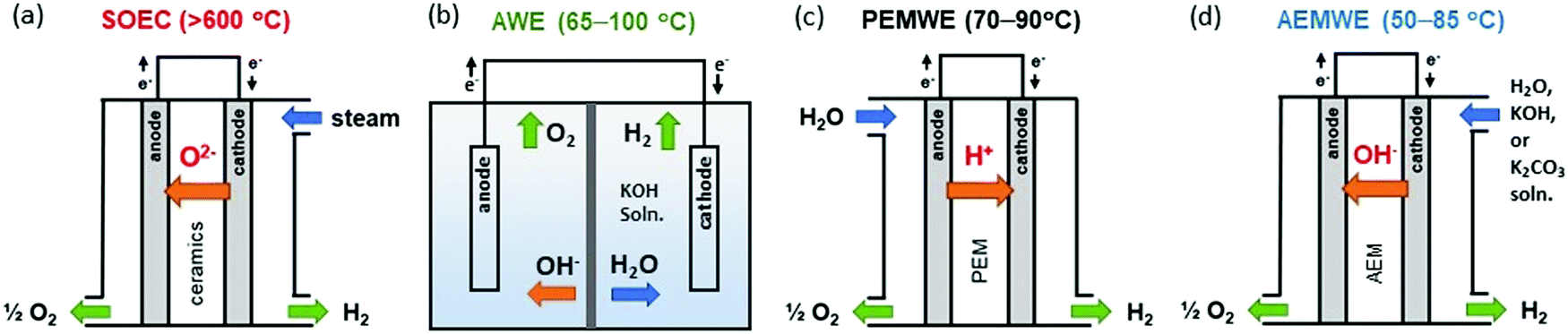 | ||
| Fig. 1 Schematic single cell configurations of different water electrolyzers. (a) SOEC, (b) AWE, (c) PEMWE, and (d) AEMWE. | ||
For low-temperature operation (<100 °C), alkaline water electrolyzers (AWEs) are the well-established technology. AWEs use an aqueous KOH solution as a liquid electrolyte and a porous diaphragm separator (Fig. 1b). The research activity to develop platinum group metal (PGM)-free electrocatalysts for hydrogen and oxygen evolution reactions (HER and OER, respectively) is well documented.5–7 Current research is moving towards leveraging chloralkali cell configurations, e.g., the zero-gap design, to increase current density or add pressure. The hydrogen production rate of AWEs is low, typically 200 mA cm−2 at a cell voltage of 1.8 V with an energy efficiency of 75%HHV.8,9
Proton exchange membrane water electrolyzers (PEMWEs) use a proton exchange membrane (PEM) and ionomer in the electrode which allows cell operation without circulating liquid electrolytes. In this configuration, both electrodes (anode and cathode) are in physical contact with a non-porous PEM resulting in compact cell arrangements (zero-gap configuration) (Fig. 1c). This zero-gap design allows the operation of PEMWEs at ∼2 A cm−2 with an efficiency of 74%HHV.9 Besides, the non-porous membrane of PEMWEs allows for differential pressure operation which produces high-pressure hydrogen at the cathode and atmospheric pressure oxygen at the anode. Such differential pressure operations can minimize the need for second stage mechanical compression to pressurize for hydrogen storage. Despite these advantages, the high costs of the electrocatalysts, such as iridium oxide and platinum, and the corrosion-resistant current collectors and separator plates in acidic environments may become a limitation for very large systems as the cell stack becomes a larger contributor to total system cost.10 Both AWEs and PEMWEs are considered matured technologies and have been deployed at a commercial scale depending on the specific needs of the application.
Anion exchange membrane water electrolyzers (AEMWEs) operate under an alkaline environment in which PGM-free catalysts could be used. Anion exchange membranes (AEMs) are non-porous hydroxide-conducting polymers that contain immobilized, positively charged functional groups on their backbone or pendant side chains, enabling a zero-gap configuration and differential pressure operation (Fig. 1d). The overall reaction of AEMWEs consists of HER and OER. Water or alkaline liquid electrolyte is circulated through the cathode where water is reduced to hydrogen and hydroxide ions by adding two electrons from the anode (H2O + 2e− → H2 + OH−). The hydroxide ions diffuse through the AEM to the anode while the electrons are transported to the cathode through the external circuit. In the anode, the hydroxide ions recombine as oxygen and water and produce two electrons (2OH− → ½O2 + H2O + 2e−). The hydrogen and oxygen form as bubbles at the surface of the HER and OER catalysts, respectively. Similar to PEMWEs, the zero-gap configuration of AEMWEs employing a non-porous membrane can produce hydrogen at a high rate and minimize the need for mechanical hydrogen compression for storage.11 It is worth noting that AEMWEs take advantage of both AWEs (PGM-free catalyst) and PEMWEs (zero-gap configuration and non-porous membrane). Interestingly, in stark contrast to PEMWEs that use only polymer electrolytes, many AEMWEs often use liquid electrolytes (e.g., KOH or K2CO3 solutions) in addition to polymer electrolytes. A recent modeling study suggested that the additional liquid electrolyte not only reduces the ohmic resistance of the membrane and catalyst layer, but also improves the reaction kinetics.12 By adding a liquid electrolyte to the cell, the local pH increases at the catalyst–electrolyte interface and an additional electrochemical interface is generated. Industrial AEMWEs comprised of Ni-based catalysts produced hydrogen with ∼1.8 A cm−2 at 2 V in 1 M KOH which approaches the performance of conventional PEMWEs under ambient pressure.13 Due to the low-cost of catalysts and hardware, applicable zero-gap configurations, and differential pressure operation, interest in hydrogen production via AEMWEs is growing. A bibliometric analysis of the publications of AEMWEs by Journal Citation Reports (JCR) indicated that the articles’ publication number of AEMWE-related research rapidly increased over the last three years (Fig. 2), reaching 7.2% of the publication number of water electrolyzer research in 2020.
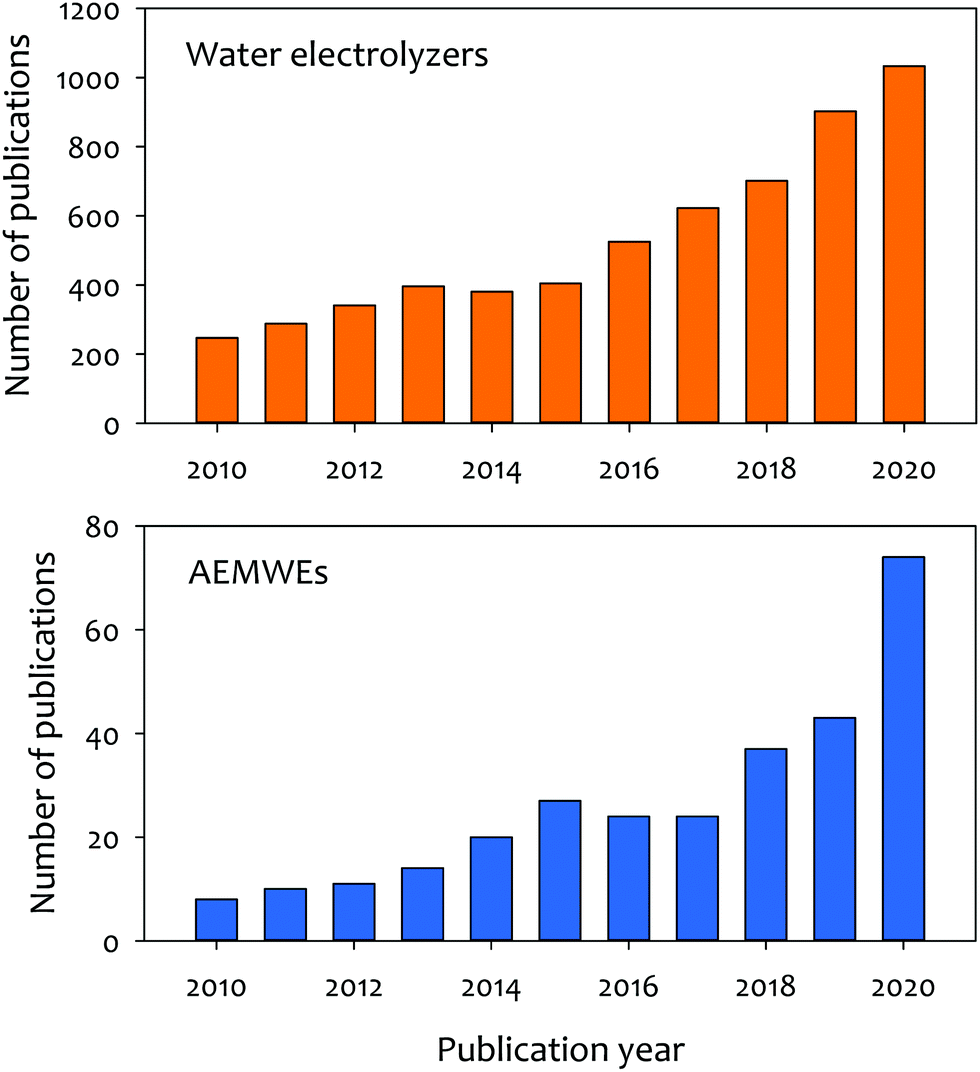 | ||
| Fig. 2 Bibliometric analysis of the publications for water electrolyzers and AEMWEs. “Water” and “electrolyzer” were used to search words for water electrolyzers. “Anion exchange membrane” and “electrolyzer” were used to search words for AEMWEs. | ||
The most critical technical challenge for AEMWEs in commercially viable systems is their durability. The durability of AEMWEs normally means the longevity or lifetime of the devices. In the early stage of AEMWE development, the longevity measurement of AEMWEs is relatively easy because the lifetime of the cell is short (<500 hours). However, as more durable AEMWEs are developed, measuring the longevity of AEMWEs becomes cumbersome. Note that running a cell over 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 hours takes more than a year. Therefore, the durability of AEMWEs has often been evaluated by the voltage change rate during an extended-term test (100–1000 hours) or by an accelerated stress test (AST) using degradation accelerating parameters such as higher operating temperatures, high current density, etc. However, one should note that extended-term tests using the voltage change rate and longevity under AST conditions cannot accurately predict the lifetime of AEMWEs because the cell lifetime is affected by the combination of several degradation modes and often limited by catastrophic failure. Therefore, it is still important to obtain the lifetime of the cell by continuously running the cell under normal operating conditions.
000 hours takes more than a year. Therefore, the durability of AEMWEs has often been evaluated by the voltage change rate during an extended-term test (100–1000 hours) or by an accelerated stress test (AST) using degradation accelerating parameters such as higher operating temperatures, high current density, etc. However, one should note that extended-term tests using the voltage change rate and longevity under AST conditions cannot accurately predict the lifetime of AEMWEs because the cell lifetime is affected by the combination of several degradation modes and often limited by catastrophic failure. Therefore, it is still important to obtain the lifetime of the cell by continuously running the cell under normal operating conditions.
While the stack lifetime of commercial PEMWEs is close to 20000 to 60000 hours, the reported longevity of most AEMWEs is <3000 hours. Moreover, most AEMWEs have been tested under ambient pressure conditions. In the early stage of research, the chemical stability of AEMs under high pH conditions was regarded as the most critical durability-limiting factor of AEM-based electrochemical devices and consequently, extensive research was accordingly devoted. To date, several hydroxide-conducting polymers comprising of an alkaline-stable cationic functional group and polymer backbone, are available for AEM-based electrochemical devices.14–20 Those alkaline-stable polymers showed less than 5% loss in ion exchange capacity (IEC) even after several thousand hours in 1–4 M KOH at 80–95 °C.21,22 However, many AEM fuel cells (AEMFCs) and AEMWEs have shown a substantial reduction in performance over the first 100–200 hours of operation.23–27 These results suggest that there are other durability-limiting factors besides the alkaline instability of AEMs.
Here, we present the durability-liming factors of AEMWEs. We have structured our discussion based on the following considerations. First, when comparing the durability of AEMFCs and AEMWEs, the durability of AEMWEs seems to be better. When the same quaternized Diels–Alder poly(phenylene) AEM and ionomer were used, the lifetime of the AEMFC using the polymer was only 300 hours.28 In contrast, the lifetime of the AEMWE was more than 2000 hours.29 The higher longevity of AEMWEs is also evidenced by the fact that approximately 2000 hours of durability of AEMFCs was demonstrated recently after extensive research,30 whereas the reported lifetime of a 1 M KOH circulated AEMWE was 12000 hours even though AEMWE research was still in infancy.31 This result suggests that the durability-limiting factors for the two AEM-based devices may be different and previous articles on AEMFC durability32–34 may not cover all aspects of the AEMWE-specific degradation pathways. Second, when determining durability-limiting factors, the performance of AEMWEs needs to be considered because there are often trade-offs between performance and durability. This aspect is critical since a commercially viable AEMWE system requires both good performance as well as durability. The performance of AEMWEs using PGM-free catalysts is of particular interest as the primary benefit of AEMWEs is their ability to use PGM-free catalysts. Third, the durability of AEMWEs strongly depends on the operation mode, i.e., the type of liquid electrolyte and applied pressure level. Counterintuitively, the corrosive concentrated KOH-circulating AEMWEs showed more stable performance than pure water-fed AEMWEs which suggests that the primary durability-limiting factor may change depending on operating environments. Therefore, it is critical to address the durability-limiting factors of AEMWEs in terms of operation modes. Fourth, transient operations including start-up/stop, may affect the lifetime of AEMWEs. However, transient operations for AEMWEs are much less extensive than automotive fuel cells for which large voltage swings and frequent start-up/stops are expected.35 Since there are limited durability studies for the transient operation of AEMWEs, we herein provide the durability perspective of AEMWEs under steady-state operating conditions.
2. State-of-the-art performance and durability of AEMWEs
The performance and durability of AEMWEs are strongly dependent on circulating alkaline electrolytes. In this section, we begin with a discussion of the effect of liquid electrolytes on AEMWE performance. Then we present the progress of AEMWE's performance and the state-of-the-art (SOA) durability under three electrolyte circulation modes, i.e., pure water-fed (no liquid electrolyte), concentrated KOH-fed, and 1 wt% K2CO3-fed AEMWEs.2.1. Effect of liquid electrolyte on AEMWE performance
Circulating liquid electrolytes can dramatically improve AEMWE performance. A possible reason for the notable performance increase with the additional liquid electrolyte circulation is attributed to the fact that the liquid electrolyte provides higher hydroxide ion transport and extended catalyst–electrolyte interfacial area. For PEMWEs, a perfluorosulfonic acid (PFSA) ionomer can provide a highly acidic environment without liquid electrolytes as the sulfonic acid functional group of the ionomer is more acidic than the liquid electrolyte, i.e., sulfuric acid. The quaternized ionomers have limited ability to provide a highly basic environment as the quaternary ammonium groups in the ionomers are less basic than alkali metals (pKa of conjugated acid of quaternary ammonium and KOH = ∼10 and 15.7, respectively).36Fig. 3 explains the impact of circulating liquid electrolytes on AEMWE performance with a schematic of half-cells using hexamethyltrimethylammonium functionalized Diels–Alder poly(phenylene) (HTMA-DAPP) under pure water-fed, 1 wt% K2CO3-fed, and 1 M KOH-fed conditions. Under the pure water-fed conditions, only the polymeric material provides hydroxide conduction pathways as in the case of PEMWEs. The hydroxide conductivity (σbulk) of the AEM is 18.4 mS cm−1 at 50 °C. At the electrode, the effective conductivity (σeffective) of the ionomeric binder is calculated to 1.2 mS cm−1 from eqn (1).
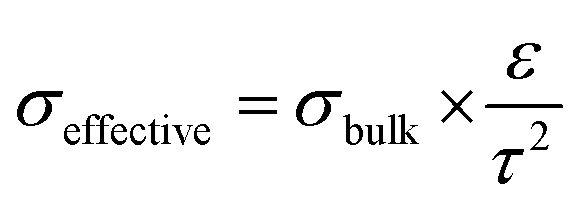 | (1) |
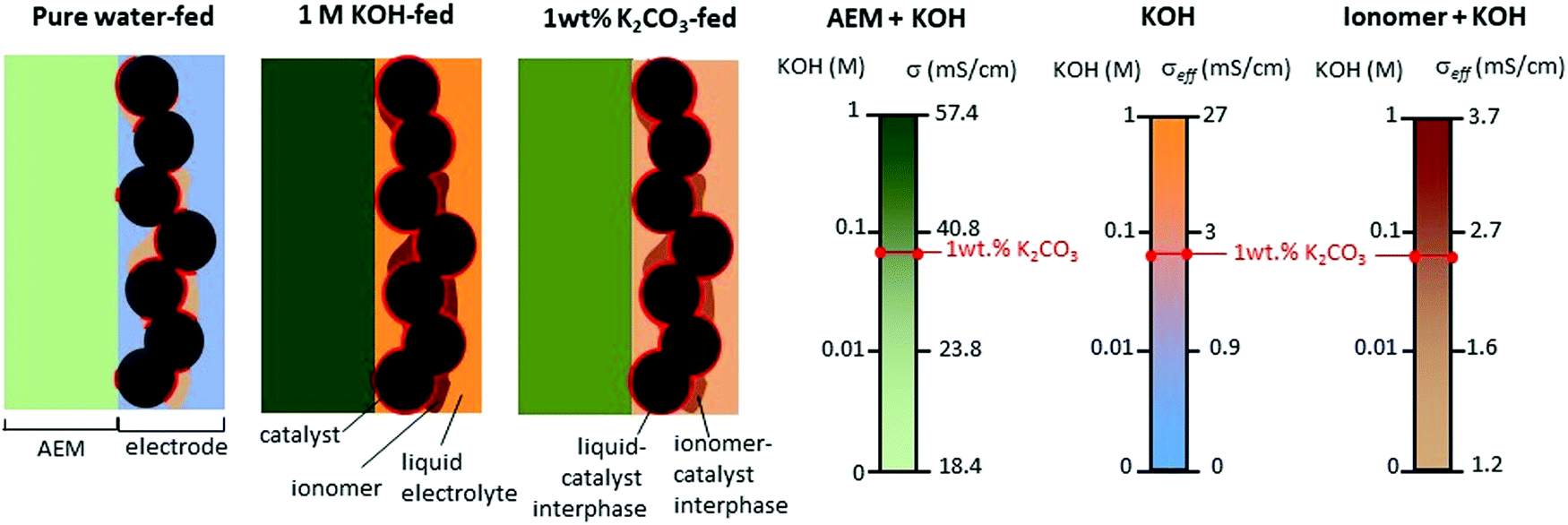 | ||
| Fig. 3 Schematic illustration of a half cell with supplying pure water, 1 M KOH, or 1 wt% K2CO3 solution.12 We estimated the conductivity of AEM from the high-frequency resistance of the cell using an HTMA-DAPP AEM as a function of KOH concentration at 50 °C. | ||
Under the 1 M KOH-fed conditions, the conductivity of the AEM increases to 57.4 mS cm−1. At the electrode, the σeffective of the ionomeric binder and the liquid electrolyte increases to 27 mS cm−1, respectively. Note that the conductivity increase in the liquid electrolyte phase is much more significant. Adding the highly concentrated KOH solution improves the catalyst utilization five times. The improvement of the catalyst utilization is due to the expanded catalyst–electrolyte interfacial area and the improved intrinsic kinetics of the catalysts. Under the 1 wt% K2CO3-fed conditions (equivalent to 0.07 M KOH), the conductivities of the AEM and the ionomeric binder are 37 and 2.4 mS cm−1, respectively. Like the 1 M KOH-fed conditions, the added K2CO3 electrolyte (σeffective = 2.46 mS cm−1) provides an additional pathway of hydroxide conduction that contributes to increased catalyst utilization 1.5 times that of the pure water-fed conditions. As a result of the low ohmic resistance and high catalyst utilization, the performance of the liquid electrolyte-fed AEMWEs is much better than that of pure water-AEMWE. The current density of the pure water-, 1 wt% K2CO3- and 1 M KOH-fed AEMWEs at 60 °C were 450, 1200, and 1700 mA cm−2 at 2 V, respectively.
2.2. Performance progression of AEMWEs
When comparing AEMFCs,32,34,37 the performance improvement of AEMWEs is more impressive because it has been achieved with PGM-free catalysts. Various PGM-free HER catalysts including: transition metals, and the transition metal's alloys, and compounds with oxides, nitrides, chalcogenides, phosphides, carbides, borides, etc., have been made to exhibit a certain degree of activities. Ni-Based catalysts showed relatively high HER activity among PGM-free catalysts.38 Still, as far as we know, the best alkaline HER catalysts are platinum-based alloys or defects-doped platinum.39–43 As for OER catalysts for AEMWEs, a large variety of metals show high OER activity and corrosion stability in an alkaline environment. The most promising transition metals are found in the fourth row of the periodic table (3d elements).44–48 The order of OER catalytic activity of metal catalysts from highest to lowest are reported in the order of Ni > Co > Fe > Mn.49 Various metal oxides, halide, nitride and phosphide with different structures including perovskites, spinels, and rutiles that also have high OER activity.50–54 The earth-abundant transition metal and metal oxide OER catalysts have shown extended-term stability (ca. 100 hours) and potential cycling stability (including ≥5000 cycles) under half-cell experiments.55 In this section, we present the current status of AEMWE performance using PGM-free catalysts at different cell operation modes. It is worth noting that a large number of papers reported pure water-fed AEMWE performance using PGM catalysts because many PGM-free catalysts were developed for concentrated KOH solution circulating AWEs and studies on ionomer-catalyst interactions for HER and OER are still scarce.Fig. 4a and b shows the chronological progress in cell performance of pure water-fed AEMWEs employing PGM-free catalysts. The AEMWE performance of MEAs employing a PGM-free anode and PGM cathode increased from 0.2 to 0.85 A cm−2 at 2.0 V in publications from the year 2011 to 2015. Then, a substantially higher performance (>0.5 A cm−2 at 1.7 V) was obtained by 2020 and 2021 cells (Fig. 4a). The striking performance improvement is primarily due to the high IEC (∼3.3 mequiv. g−1) of the ionomer that provides a high pH environment.56 Additionally, the AEMWEs used thin membranes (thickness: 20–35 μm) and operated at high temperatures (85 and 90 °C). The AEMWE performance of MEAs employing PGM-free catalysts for both electrodes also increased from 0.24 to 1.0 A cm−2 at 1.8 V from the year 2012 to 2020 (Fig. 4b). When compared to PGM catalyzed AEMWEs (black dash line), the PGM-free AEMWEs exhibited lower performance due to the limited activity of the PGM-free cathode. The AEMWE using a PGM cathode and PGM-free anode (blue line in Fig. 4a) outperformed the PGM-catalyzed AEMWE (black dash line Fig. 4b).
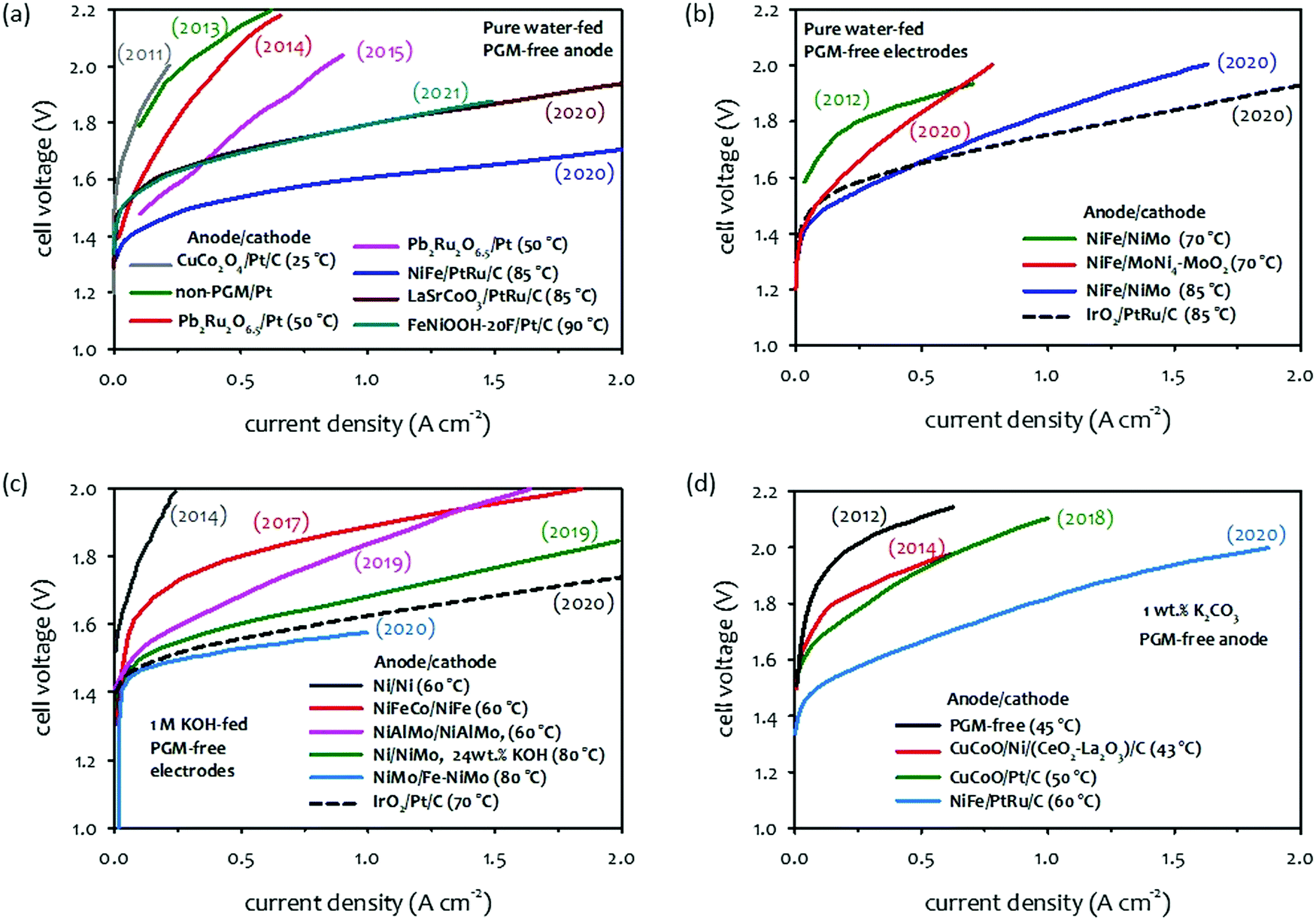 | ||
| Fig. 4 Selected performance of AEMWEs in the literature. (a) Pure water-fed AEMWEs with PGM-free anode/PGM cathode.57–63 Adapted with permission.58,63 Copyright 2013, IOP publishing, Copyright 2021, American Chemical Society. (b) Pure water-fed AEMWEs with PGM-free anode/PGM-free cathode.62,64–66 (c) 1 M KOH-fed AEMWEs.66–71 Adapted with permission.67–71 Copyright 2014, Elsevier, Copyright 2017, IOP Publishing, Copyright 2019, American Chemical Society, Copyright, 2020, John Wiley and Sons. (d) 1 wt% K2CO3-fed AEMWEs.72–74 Adapted with permission.72–74 Copyright 2012, 2018, Elsevier, Copyright 2014, John Wiley and Sons. The 2020 cell data was produced at Los Alamos National Laboratory. AEM: alkyl ammonium functionalized poly(styrene-b-ethylene-b-styrene) triblock copolymer (SES-TMA, 35 μm thick); anode: NiFe (4 mg cm−2); cathode: PtRu/C (50 wt% Pt, 25 wt% Ru, 2 mgPt cm−2). | ||
Like pure water-fed AEMWEs, the concentrated KOH-fed AEMWE performance of MEAs significantly improved over the last years (Fig. 4c). The current density of 0.1 A cm−2 at 1.8 V of the 2014 cell (dark gray line) increased to 1.0 A cm−2 for the 2019 cell (pink line). The performance of 24 wt% KOH-fed AEMWE reported in 2019 showed higher performance (1.7 A cm−2 at 2.0 V, green line), presumably due to the highly concentrated (4.4 M) KOH electrolyte. The 1.0 M KOH-fed 2020 AEMWE cell employing a commercially reinforced membrane (X37-50-T, thickness: 50 μm, Dioxide Materials) exhibited substantially higher performance (1 A cm−2 at 1.57 V, light blue line) at 80 °C with an efficiency of 75.1%HHV that was even higher than the PGM-catalyzed AEMWE (black dash line). Compared to the performance of pure water-fed AEMWEs, the performance of KOH-fed AEMWEs was notably better (0.3 A cm−2 at 1.57 V for water-fed vs. 1 A cm−2 for 1 M KOH-fed). Although no longer-term stability was reported for the 2020 cell, notable performance loss was observed over the 25 hour test at 50 °C and a constant current density of 500 mA cm−2.50
Fig. 4d shows the chronological progress of the single-cell performance of 1 wt% K2CO3-fed AEMWEs from 2014 to 2020. The performance of 1 wt% K2CO3-fed AEMWEs is relatively low. The current density of the concentrated KOH-fed AEMWEs (PGM-free electrodes) is >0.5 A cm−2 at 1.6 V (green and light blue lines in Fig. 4c), while the current density of the 1 wt% K2CO3-fed 2020 AEMWE cell (PGM-free anode) is 0.34 A cm−2 at 1.6 V (light blue line). One of the reasons for this low performance is the high cell resistance of 1 wt% K2CO3-fed AEMWEs because of their lower mobility, approximately 3–4 times in the presence of carbonate and bicarbonate ions, compared to a carbonate ion-free system.75,76
The literature survey indicates that the performance of current PGM-free catalyzed AEMWEs increases in the order: concentrated KOH-fed (Fig. 4a, light blue) ≫ pure water-fed (Fig. 4a, blue) > 1 wt% K2CO3-fed (Fig. 4b, light blue). The high performance of the concentrated KOH-fed AEMWEs is mainly due to the high pH environment. The higher performance of the SOA pure water-fed AEMWEs compared to 1 wt% K2CO3-fed AEMWEs contradicts the results shown in the electrolyte impact (Section 2.1). However, one should note that the pure water-fed AEMWE performance in literature was obtained at relatively high operating temperatures and more advanced ionomeric binders that provide a high local pH at the catalyst–electrolyte interface. Also, one should consider that other cell issues, such as soft shorts or gas mixing, may cause the very low voltages of highly-performing AEMWE cells.
2.3. Durability of AEMWEs
The papers that reported high performance of AEMWEs typically discussed cell durability after relatively short-term (<100 hours) operations and often under less rigorous conditions (low current density, low operating temperature, and ambient pressure). This is because the literature focused more on the material development viewpoint at the cost of cell durability. Consequently, the best durability of AEMWEs was reported in separate papers under less rigorous conditions. Also, it was noted that in most cases, PGM catalyst-containing cathodes were used to evaluate AEMWE's durability because PGM catalysts have better HER activity and stability. In this section, we survey the longest lifetime of AEMWEs under the three operating modes. Additionally, we present the reported lowest voltage degradation rate of AEMWEs to provide the current status of AEMWE durability.The long-term performance of pure water-fed AEMWEs was reported in 2012–2014 (Fig. 5a).29,77 All three pure water-fed AEMWEs have more than 500 hours of lifetime at 50 °C and a constant current density of 200 mA cm−2. The degradation rate of the cells was relatively high because less stable AEMs and ionomers were used for the tests. The quaternized Radel® poly(sulfone) AEM used in one of the cells had an aryl ether group in the polymer backbone that is susceptible to the nucleophilic substitution, SN2, under high pH conditions. Ex situ alkaline stability of the benzyltrimethyl ammonium functionalized Diels–Alder poly(phenylene) (BTMA-DAPP) polymer indicated that the hydroxide conductivity of the polymer decreased from 13 to 1 mS cm−1 after 550 hours exposure in 0.5 M NaOH at 80 °C and 95% RH due to the SN2 of the BTMA group.15 The reported longest lifetime of the pure water-fed AEMWE under steady-state conditions is the BTMA-DAPP based AEMWE that operated 2200 hours before the test was halted due to other testing commitments. However, the voltage degradation rate of the BTMA-DAPP-based AEMWE was high, ca. 0.2 mV h−1 at the low current density. Recently, Xu et al. reported a more stable PGM catalyzed pure water-fed AEMWE at 80 °C and a constant current density of 500 mA cm−2. The cell voltage increased from 1.7 to 1.75 V after 500 hours (voltage degradation rate: 0.1 mV h−1).78 Challenges in the durability of water fed-AEMWEs include demonstrating >10000 hours of operation with higher hydrogen production rates and lower degradation rates. Demonstration of durability under differential pressure conditions remains a future task as well.
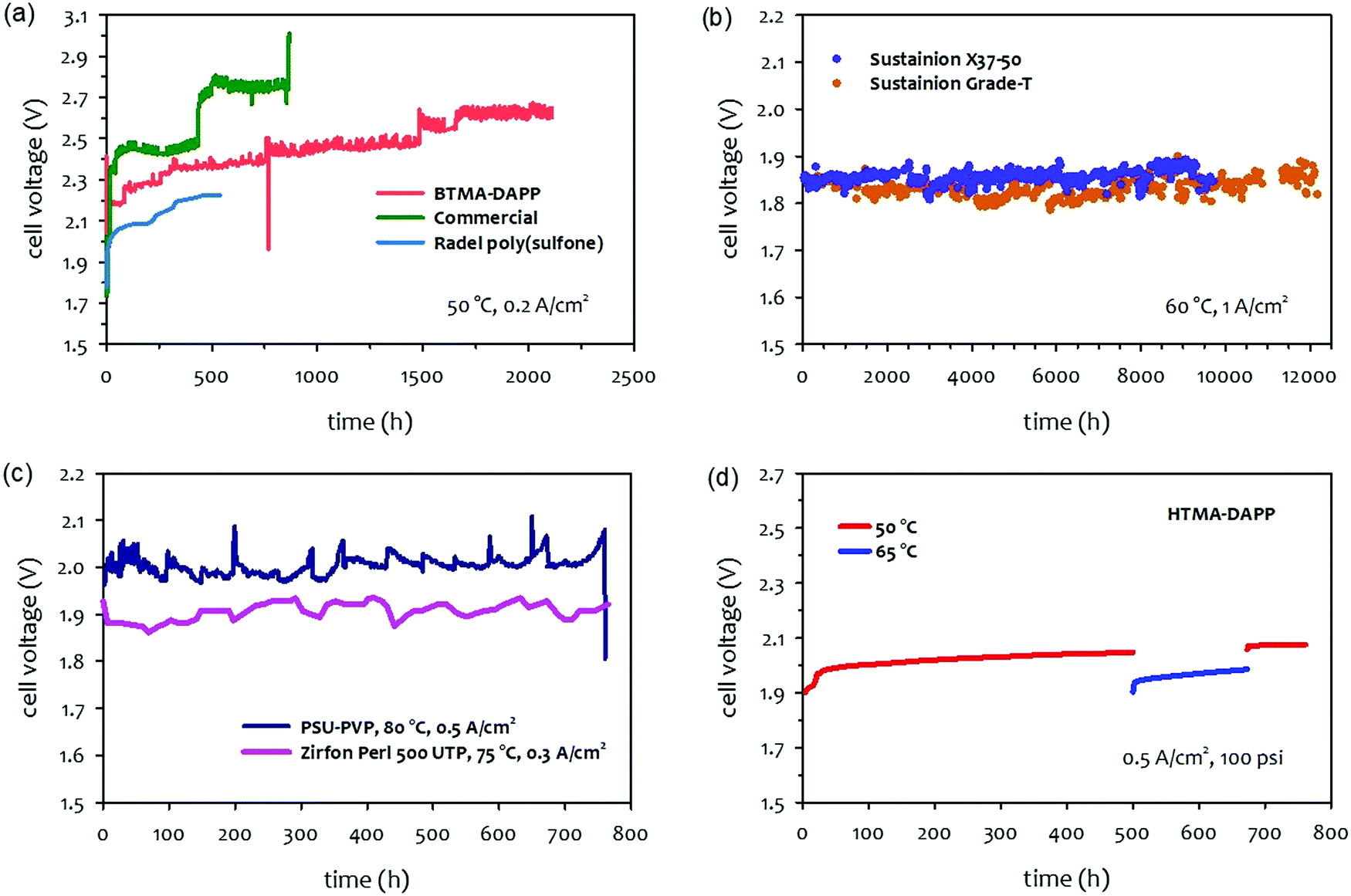 | ||
| Fig. 5 Durability of AEMWEs. (a) Pure water-fed AEMWEs under ambient pressure.29 (b) 1 M KOH-fed AEMWEs at a current density of 1 A cm−2. AEM: Sustainion® Grade T or Sustainion® X37-50 (50 μm thick), anode: NiFe2O4 (1.8 mg cm−2), cathode: rainy nickel on nickel fiber paper (14.5 mg cm−2).31 Reproduced with permission.31 Copyright 2021, Elsevier. (c) Concentrated KOH-fed AEMWEs.79 Cell 1: the cell voltage was measured at 500 mA cm−2 and 80 °C with 20 wt% (3.6 M) KOH solution. AEM: PSU-PVP (120 μm thick), ionomer: Nafion, anode: Ni form; cathode: proprietary. Reproduced with permission.79 Copyright 2020, Elsevier. Cell 2: the cell voltage was measured at 300 mA cm−2 and 75 °C with 6 M KOH solution. AEM: Zirfon Perl (500 UPT AGFA) sandwiched by two pieces of polypropylene, anode: stainless steel 316L, cathode: Ra-Ni.80 (d) 1 wt% K2CO3-fed AEMWE at a current density of 500 mA cm−2 and under 100 psi differential pressure. The AEMWE cell data was produced at Nel Hydrogen. AEM: HTMA-DAPP (78 μm thick). Ionomer: Aemion, anode: Co3O4 (3 mg cm−2), cathode: Pt black (3 mg cm−2). | ||
Fig. 5b shows the long-term test of a 1 M KOH-fed AEMWE at the current density of 1 A cm−2 using commercial AEMs (Sustainion®) and PGM-free catalysts. The cell using Sustainion® Grade T AEM was able to operate >12000 hours with a voltage degradation rate of 0.7 μV h−1.31 The concentrated KOH-fed AEMWE cells using a non-quaternized membrane also showed stable performance (voltage degradation rate = ∼30 μV h−1) over 700 hours at 75–80 °C (Fig. 5c).79,80 Note that the AEMWE using the non-quaternized membrane requires a higher concentrated KOH solution (3.6–6 M) for a high hydrogen production rate. In the AEMWE cell with 20 wt% KOH, relatively large voltage fluctuations were observed due to the KOH concentration change suggesting that it may be critical to control the KOH concentration for highly concentrated KOH-fed systems.
The durability of 1 wt% K2CO3-fed AEMWEs was evaluated typically over 500–700 hours under 100 psi differential pressure conditions. Fig. 5d shows an 1 wt% K2CO3-fed AEMWE employing the HTMA-DAPP AEM at a constant current density of 0.5 A cm−2. The voltage decay rate at the operating temperature of 50 °C decreased over time and started to stabilize around ∼50 μV h−1 at the end of the 500 hour test. After 500 hours, the operating temperature of the cell was raised to 65 °C. The voltage of the cell dropped by ca. 50 mV, but the degradation rate increased to ∼200 μV h−1. After 200 hours of operation at 65 °C, the cell's operating temperature was returned to 50 °C to see whether the good durability at 50 °C was recoverable. The cell's voltage was higher than that of the initial 500 hour test. However, the cell was stabilized with a voltage decay rate of ∼1 μV h−1. At the end of the 750 hour test, the AEMWE did not show signs of cross cell leak or electronic-short failures, and the cell was able to run with a current of up to 2 A cm−2. Throughout the durability test, the AEMWE cell was held at 100 psi differential pressure.
A literature survey indicates that the highest longevity of AEMWEs demonstrated was obtained with 1 M KOH-fed AEMWE for 12000 hours with a degradation rate of 0.7 μV h−1. The longevity of pure water-fed and 1 wt% K2CO3-fed AEMWEs were only demonstrated for <3000 hours. The voltage degradation rate of SOA pure water-fed and 1 wt% K2CO3-fed AEMWEs are higher than that of the SOA 1 M KOH-fed AEMWE (50–200 μV h−1 over 500 to 700 hours at a constant current density of 500 mA cm−2). It is noted that the good durability of the pure water-fed AEMWE cell was obtained at a relatively high temperature (80 °C) and low operating voltage (1.7–1.75 V). In contrast, the good durability of the 1 wt% K2CO3-fed AEMWE was obtained at a lower temperature (50–65 °C) and higher operating voltage (1.9–2.0 V). The different optimum operating conditions in terms of operating temperature and cell voltage suggest that the primary durability-limiting factors for these AEMWEs with these two operation modes may be different. In the next section, firstly, we provide background information on AEMWE durability that includes cell voltage behaviors during an extended-term test of AEMWEs and the effects of bipolar plates and gas diffusion layers (GDLs) on AEMWE durability. Then we discuss the durability-limiting factors of AEMWEs with these three operation modes: pure water-fed, concentrated KOH-fed, and 1 wt% K2CO3-fed operating with differential pressure.
3. Durability evaluation of AEMWE single cell
3.1. Cell voltage behaviors during an extended-term test
The durability of AEMWEs is typically evaluated by measuring the cell voltage over time at a constant current density. Fig. 6 shows several cell voltage behaviors at a constant current density that were commonly observed during an extended-term operation (ca. 100 hours). Fig. 6a shows the stable performance of AEMWEs in which the cell voltage is constant. A slight decrease in cell voltage may occur during the first ten hours most likely due to the cell break-in during which the optimal interface between the electrocatalyst and electrolyte is formed. Fig. 6b shows the gradual cell voltage increase over time (Type 1). This is the most common degradation behavior and it often appears concurrently with other voltage behaviors. The primary cause of this behavior is the loss of a cationic functional group of the AEM that increases the cell's resistance. Catalyst dissolution/aggregation or other slow degradation of MEA components can also be the cause of this voltage behavior. In this mode of degradation, the cell degradation rate (V h−1) is well defined. Fig. 6c shows a stepwise increase in voltage (Type 2). This type of voltage behavior originates from rapid performance decay due to catalyst activity loss or local pH change by ionomer oxidation. Fig. 6d shows a spike type of voltage behavior (Type 3). Cell degradation occurs within a short period, but this degradation is recoverable. Change in the liquid electrolyte concentration or water transport issues due to evolving gas81 may cause such voltage behavior. Fig. 6e shows a gradual voltage decrease (Type 4), which is the opposite behavior of Fig. 6b. A gradual increase of hydrogen crossover by AEM thinning may cause this behavior. This voltage behavior makes the durability evaluation by cell degradation rate difficult as the cell degradation rate does not accurately reflect the degradation process. If AEM thinning occurs with cationic group degradation, then the cell voltage may look constant. Therefore, the cell showing a stable cell voltage may need to be placed in a longer-term test or have the faradaic efficiency measured. Fig. 6f shows catastrophic failure. The mechanical failure or pinhole formation of AEM can result in such behavior (Type 5).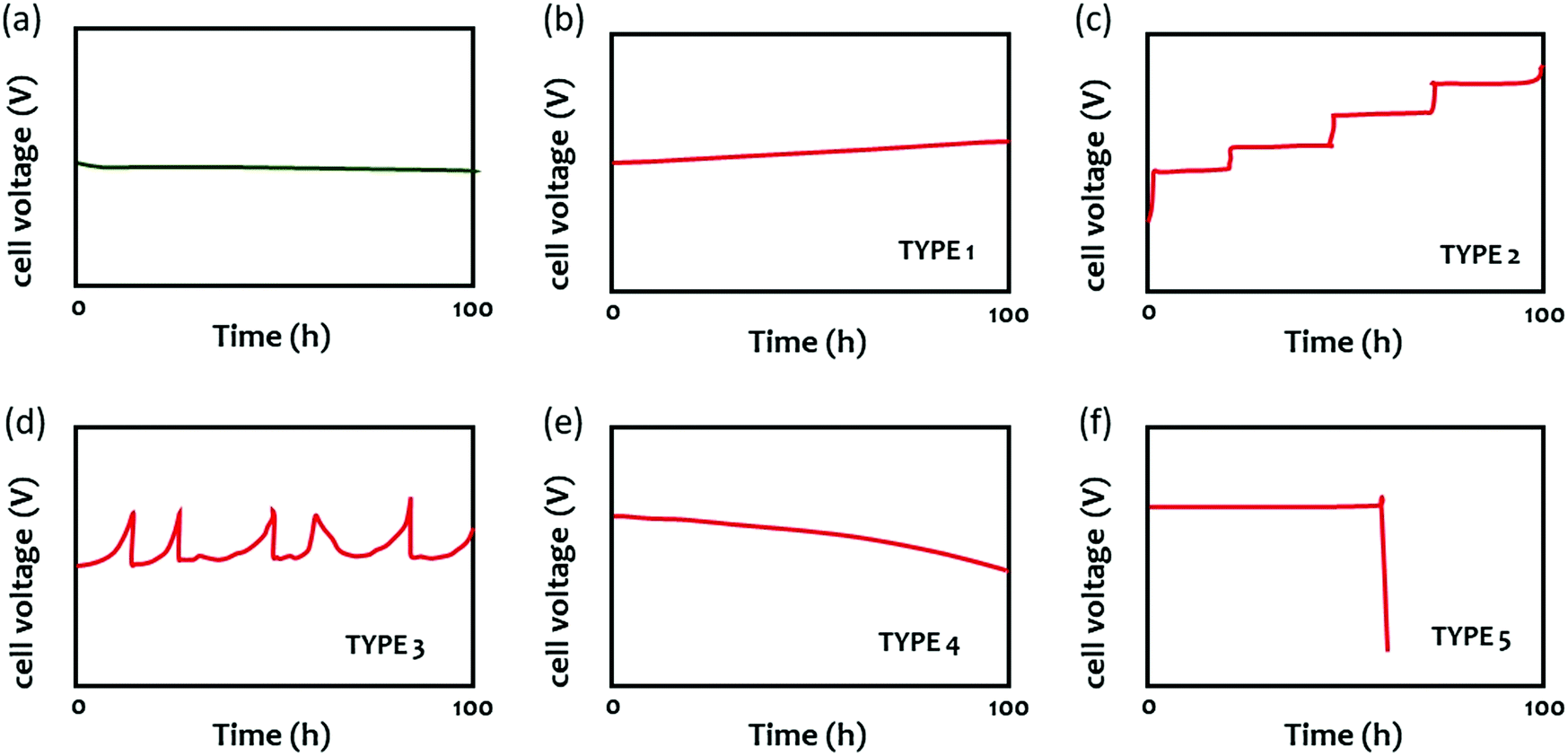 | ||
| Fig. 6 Various voltage behaviors of AEMWEs during 100 hour extended-term test at a constant current density. | ||
3.2. Effect of bipolar plates and gas diffusion layers on AEMWE's durability
The choice of proper bipolar plates (BPs) with flow fields and gas diffusion layers (GDLs) is crucial to AEMWE performance and durability. Fig. 7a shows the schematic of BP and GDLs in a single cell. In AEMWEs, BPs are used to conduct current and heat, distribute reactants (H2O) into and products (H2 and O2) out of the active area, and provide mechanical support as well as separation for the stack. Due to the less corrosive alkaline environment in AEMWEs compared to PEMWEs, cheap materials such as stainless steel (SS) and graphite can be used for BPs in the research which would greatly reduce the stack cost.69,82,83 However, the degradation and corrosion of the BPs in AEMWEs may still occur with an improper selection of the chemical composition of BP materials and surface property as well as AEMWE operating conditions. Although SS materials can provide good corrosion resistance in alkaline solutions in AEMWEs, SS alloys with increasing Ni and Cr contents will provide severe passivation and form metal hydroxides (FeOOH, CrOOH) on the anode of the BP's surfaces leading to high interfacial contact resistances (ICRs) and high overpotentials in the stacks.84,85 Graphite BPs are not suitable for long-term use with high potentials in the anode since carbon materials are susceptible to corrosion under OER conditions in the presence of OH− ions, which are excellent nucleophiles.11 Compared to PEMWEs, very few publications discuss the durability of BPs in AEMWEs. Therefore, more studies that explore the corrosion resistance, flow field patterns, and surface treatments for durable BPs in AEMWEs are needed.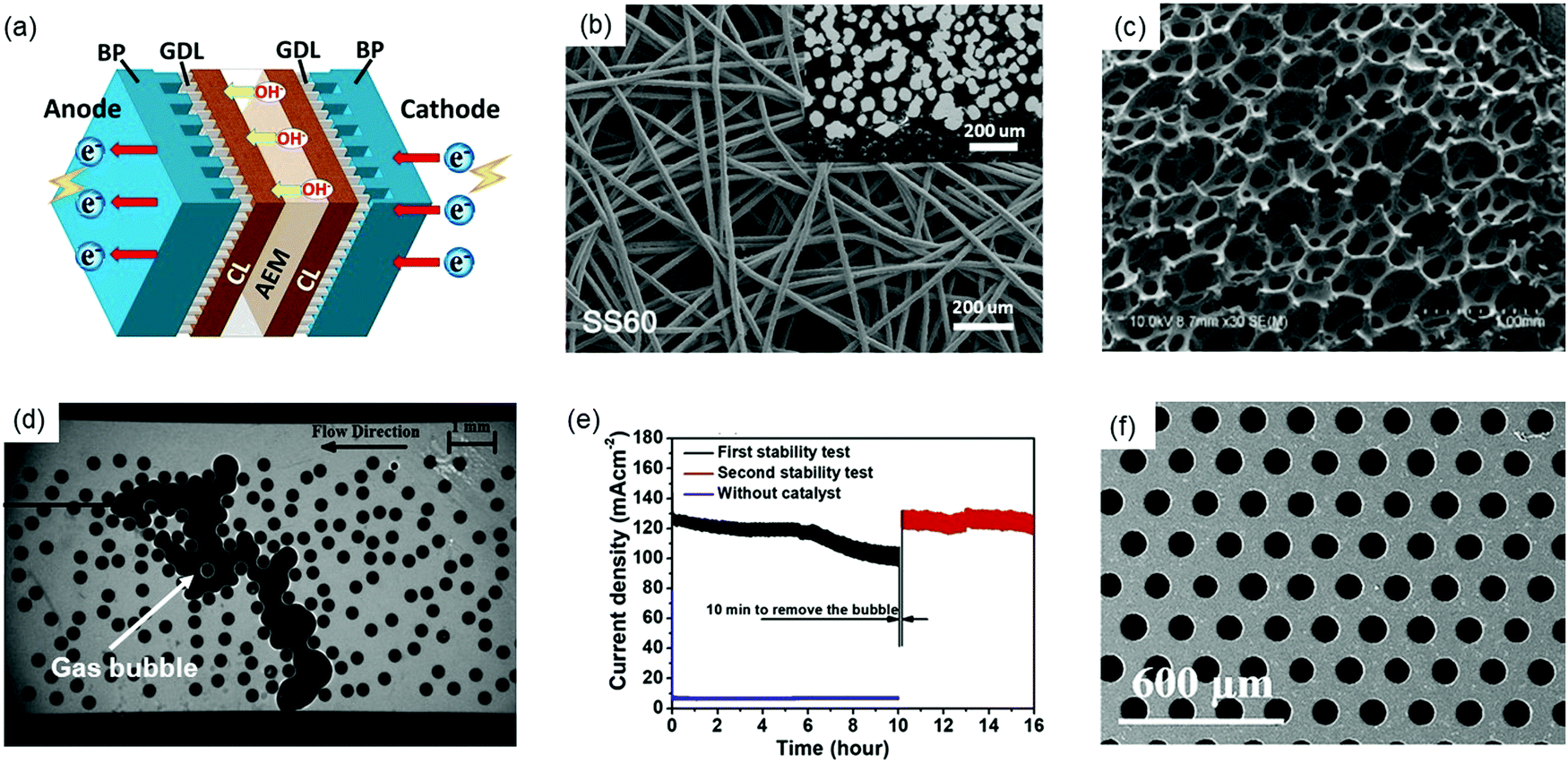 | ||
| Fig. 7 (a) Typical components in AEMWEs with AEM, bipolar plates (BPs) and gas diffusion layers (GDLs). (b) Felt-type GDL.86 Reproduced with permission.86 Copyright 2021, American Chemical Society. (c) Foam-type GDL.87 Copyright 2013, Elsevier. (d) Visualization image of trapped gas bubbles in the microfluidic chip representing a foam GDL.88 Reproduced with permission.88 Copyright 2014, Elsevier. (e) Stability test of the water electrolyzer, showing the first 10 hour stability test, removing bubbles, and then the second 6 hour stability test.89 Reproduced with permission.89 Copyright 2019, Elsevier. (f) Thin well tunable GDLs with straight pores and gradient porosity.90 Copyright 2017, Royal Society of Chemistry. | ||
The GDL is a porous transport layer located between the catalyst layer and the BP for transferring electron, mass and heat between the two components. Coating a GDL with catalysts results in a gas diffusion electrode (GDE) or a catalyst-coated substrate (CCS). Commonly used GDLs in AEMWEs include carbon paper or cloth, SS felt or mesh, Ti felt or foam, Ni foam, etc. (Fig. 7b and c).4 Mechanical/physical problems (e.g., over-compression, low permeability and wettability) and chemical/electrochemical failures (e.g. corrosion, erosion, and oxidation) can contribute to GDL degradation, further reducing the durability of AEMWEs. As aforementioned, since the OER condition and OH− ions lead to the corrosion of carbon materials during long-term operation, carbon GDLs can be more easily corroded than graphite BPs due to the higher concentration of OH− near the AEM. Similar to BPs, the passivation layer on SS and Ti GDLs can ensure AEMWE durability, which also leads to a high ICR. Additionally, the corrosion of metallic GDLs occurs when the voltage is above 2 V.91 Therefore, the corrosion of SS GDLs leads to the degradation of contact points between GDLs and catalyst layers causing increased MEA resistance and weak durability of AEMWEs. Although a lower operation voltage (<1.9 V) may reduce the corrosion of GDLs and improve the durability, the current density will be sacrificed, resulting in a lower hydrogen production rate.
More importantly, GDLs play an essential role in the mass transport of reactants and products. Recent studies have found that the mass transport aspect of GDLs also impacts the stability and durability of AEMWEs. During the electrolysis operation, oxygen bubbles on the anode must be removed from the GDL as soon as possible to prevent them from blocking catalyst active sites, especially at high current densities. Bazylak et al. visualized the gas bubble transport in GDLs to study how GDL geometry affects the multiphase flow in electrolyzers.88 Microscopy images showed that gas bubbles were trapped in the microfluidic chip representing a porous foam-based GDL in Fig. 7d. This can cause a greater mass transport problem in foam-based GDLs compared to felt-based GDLs. In addition, the researchers also found that the porosity, thickness, and pore diameter of GDLs greatly affect bubble accumulation in GDLs. Another study confirmed that the trapped gas or gas accumulation in GDLs greatly impacted the stability of AEMWEs, as shown in Fig. 7e.89 In the stability test of AEMWEs, catalyst-coated carbon clothes were used as GDEs, and 1 M KOH was pumped into AEMWEs with a flow rate of 60 mL min−1. In the first 10 hours (black line in Fig. 7e), slow degradation of 3 mA cm−2 h−1 was observed. Then, the power supply for the AEMWE was turned off, but the water pump kept working to pump out H2/O2 bubbles from the cell for 10 min. Interestingly, the performance of the AEMWE almost fully recovered after resuming the test (red line in Fig. 7e), and the current density returned to its initial value, indicating that the decay in the first 10 hours was due to the bubble accumulation and the blockage of water transportation in GDLs. Bubble accumulation and the resulting dehydration of catalyst layers and AEMs caused the blockage of reaction sites, starvation, and reduction on OH− conductance. This process may lead to not only short-term stability problems but also severe degradation over a longer period of time.
To the best of our knowledge, few studies have been reported on the BPs and GDLs for durable AEMWEs. Thus, more investigation is needed. Pt coated Ti BP/Pt coated Ti GDL and graphite BP/carbon paper could be used at the anode and cathode, respectively, as baseline materials to evaluate other components of AEMWEs for benchmark comparisons.62 In this way, the influence of BPs and GDLs on MEA durability could be minimized. Following the previous point, it is worth studying the contribution of GDLs and BPs to the degradation of AEMWEs to provide insights into the material selection and associated treatments of GDLs and BPs. Moreover, surface modification and structural optimization should be conducted for enhancing the mass transport and degradation resistance of GDLs. The modification and optimization processes include the followings: increasing the hydrophilicity of GDLs using chemical etching or surface coating; optimizing the GDL's structural and physical parameters by reducing pore size, producing graded pore dimensions, increasing porosity, and providing smooth surfaces; surface coating with TiN, Pt, Nb, or CrN for good corrosion resistance and low ICRs; using thin and well tunable GDLs with straight pores and gradient porosity to promote mass transport and reduce ICRs (Fig. 7f).90,92,93
4. Durability-limiting factors
4.1. Durability-limiting factor of pure water-fed AEMWEs
The durability of pure water-fed AEMWEs is relatively low. Pandiarajan et al. reported that the cell voltage of spinel ferrite catalyzed AEMWEs at 200 mA cm−2 and room temperature increased from 1.6 to 1.75 V within 3 hours despite the stable OER activity of the catalyst at >100 hours in the rotating disk electrode (RDE) experiment.94 Chu et al. reported that the iridium oxide catalyzed AEMWEs at 200 mA cm−2 and 50 °C exhibited a voltage increase from 1.75 to 2.3 V in 35 hours, although they did not observe any notable degradation of the piperidinium-functionalized AEM from the post-mortem test.27 Since operating AEMWEs with only flowing water and without additional liquid electrolytes is less corrosive, the durability-limiting factors of pure water-fed AEMWEs are not related to the alkaline stability of MEA components, but associated with a high operating cell voltage and current density. In this section, we discuss two durability-limiting factors, i.e., ionomer detachment and poisoning under high cell voltage and current density conditions. Those ionomer-related degradations are accelerated under high cell voltage and current density conditions.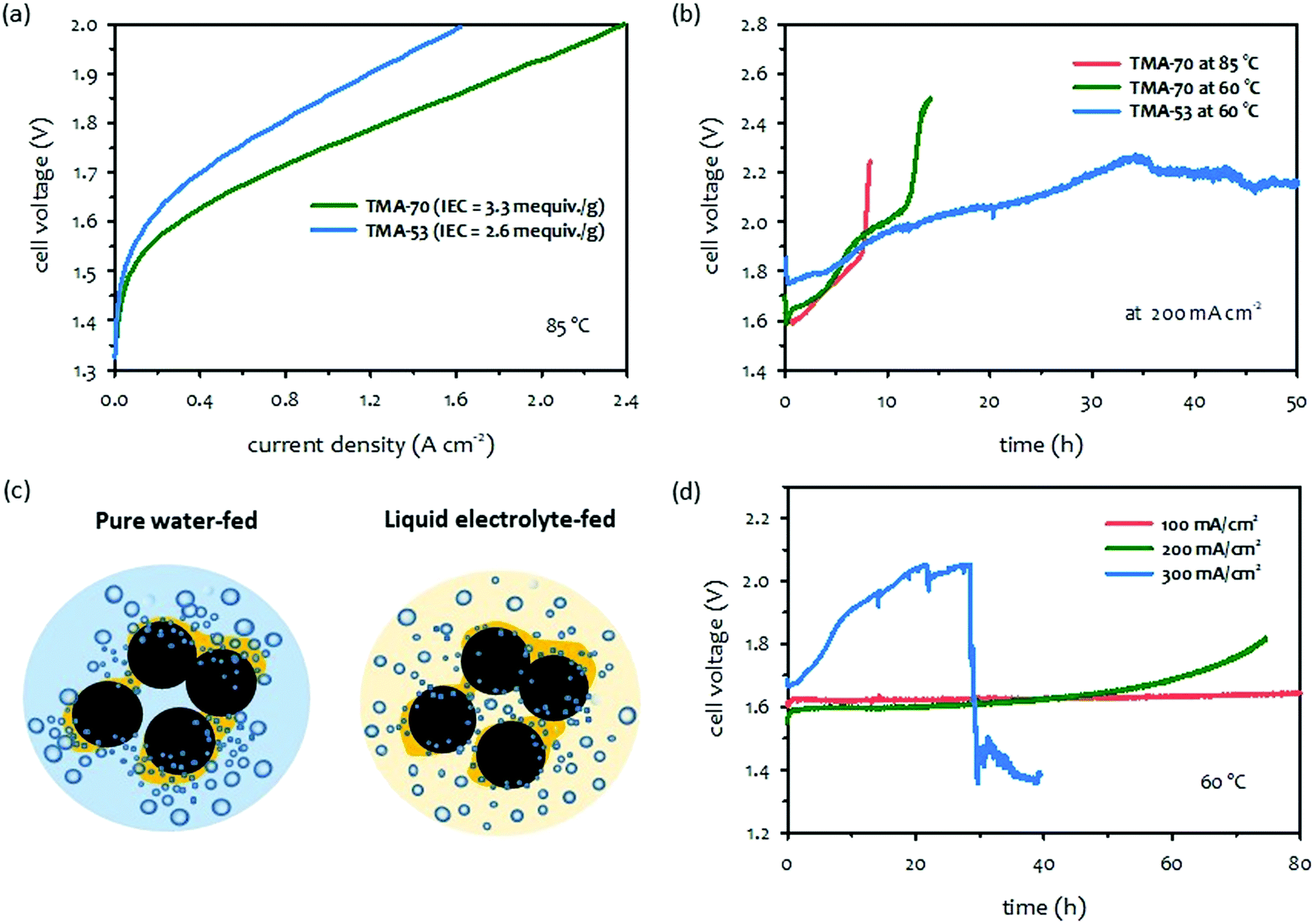 | ||
| Fig. 8 Performance-durability trade-off of pure water-fed AEMWEs.62 (a) Effect of IEC of ionomer on performance and (b) Effect of IEC of ionomers on extended-term performance. AEM: HTMA-DAPP (26 μm thick); anode: IrO2 (2.5 mg cm−2); cathode: NiFe nanofoam (3 mg cm−2). (c) Schematic of gas evolution at a high current density for pure water-fed and liquid electrolyte-fed AEMWEs. (d) Effect of current density on extended-term performance. AEM: SES-TMA (35 μm thick); ionomer: FLN-55; anode: IrO2 (2.5 mg cm−2); cathode: PtRu/C (50 wt% Pt, 25 wt% Ru, 2 mgPt cm−2). The AEMWE data was produced at Los Alamos National Laboratory. | ||
The detachment of the ionomeric binder is problematic for ionomers with a high IEC and high water uptake. These ionomers have greater dimensional change under fully hydrated conditions which weakens the adhesion of the ionomer on the catalyst's surface. The ionomer detachment by gas evolution occurs more severely in pure water-fed AEMWEs because the catalyst–electrolyte interfacial area of pure water-fed AEMWEs is relatively small. Thus, gas evolution occurs more inhomogeneously at a given current density (Fig. 8c). Since the gas permeability of polymeric materials is much lower than that of the KOH solution,95 it is difficult to remove the evolved gas fast enough from the catalyst-ionomer interface under high current operating conditions. Compared to PEMWEs, the gas bubble-induced ionomer detachment occurs more easily in AEMWEs because hydrocarbon-based quaternized ionomers have lower gas permeability,96,97 and a much lower adhesion due to their excessive swelling with water.98Fig. 8d shows the durability of pure water-fed AEMWEs as a function of the current density. The cell voltage of the AEMWE operated at 100 mA cm−2 was stable over the 100 hours while cell failure occurred within 40 hours when operating the cell at 300 mA cm−2 indicating that higher gas generation conditions are detrimental to pure water-fed AEMWEs. A similar effect was found with a nickel-iron oxyhydroxide catalyzed pure water-fed AEMWEs.63
The mitigation strategy of degradation caused by ionomer detachment is to use ionomers with a low IEC and to operate at a low operating temperature although substantially lower performance is anticipated. Alternatively, it may be plausible to develop high IEC ionomers with low to moderate water uptake. Polymer synthetic strategies to achieve low water-swollen hydrophobic ionomers include introducing multi-cations,99–102 polar interactions,103 and cross-linking.104–106 Several technical challenges in the design of high IEC ionomers with low water uptake need to be addressed. First, the conductivity of low water uptake of quaternized ionomers is low which reduces the hydrogen generation rate. Second, introducing multi-cations and polar group interactions into quaternized polymers often decreases the chemical stability of the ionomers. Third, the synthetic process of high IEC ionomers with low water uptake is likely more complex and expensive. Enhancing the robustness of the ionomeric film by a dispersing agent is another plausible approach. In general, non-aqueous dispersing agents increase chain entanglement of ionic group functionalized polymers,107,108 which provides better adhesion and mechanical robustness for the ionomer thin-film. Implementing a better dispersing agent may also produce a more uniform distribution of ionomers in the electrode making gas evolution more homogenous and improving cell stability under high current conditions. Reducing the size of catalyst nanoparticles may also help to a more uniform distribution of gas evolution reactions.
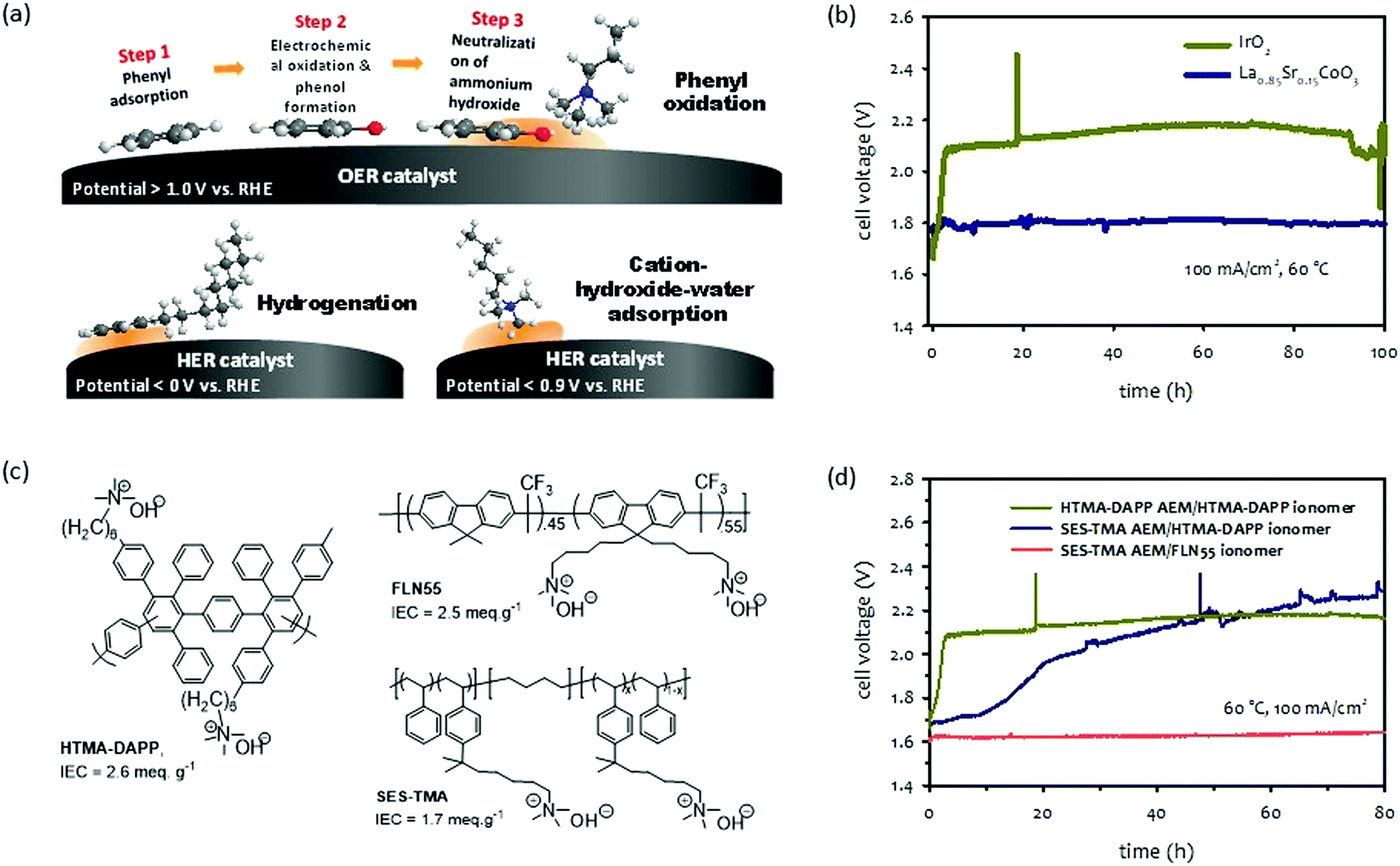 | ||
| Fig. 9 (a) Schematic diagram of degradation mechanisms by electrochemical phenyl oxidation,109 hydrogenation and cation-hydroxide-water co-adsorption. (b) Short-term durability of pure water-fed La0.85Sr0.15CoO3 or IrO2 catalyzed AEMWEs. AEM: HTMA-DAPP (35 μm thick); ionomer: HTMA-DAPP; anode: La0.85Sr0.15CoO3 (2 mg cm−2) or IrO2 (1 mg cm−2); cathode: Pt/C (0.6 mgPt cm−2). The durability was measured under ambient pressure.114 (c) Chemical structure of polymer electrolytes used for phenyl oxidation study. (d) Extended-term AEMWE durability employing different AEM and ionomer combinations. AEM: HTMA-DAPP (35 μm thick) or SES-TMA (35 μm thick), ionomer: HTMA-DAPP, SES-TMA, or FLN55, anode: IrO2 (1 mg cm−2); cathode: PtRu/C (50 wt% Pt, 25 wt% Ru, 0.5 mgPt cm−2). | ||
Because electrochemical oxidation occurs with adsorbed phenyl groups on OER catalyst surfaces, the adsorption energy of phenyl groups on the surface of OER catalysts plays a critical role in the degradation process. The density functional theory (DFT) found that the adsorption energies of the phenyl group in BTMAOH that are parallelly oriented to the surface of iridium oxide(110) is higher (−1.2 to −2.2 eV at 1.6 V) than that of the La0.85Sr0.15CoO3 perovskite catalyst (−0.18 to −0.42 eV at 1.6 V).109 The RDE data is consistent with the calculation data that the phenyl oxidation rate on the surface of the iridium oxide is approximately three times higher than that on the surface of the perovskite catalyst. Fig. 9b shows the durability of the iridium oxide and perovskite catalyzed AEMWEs employing the HTMA-DAPP ionomer.114 The cell voltage of the iridium oxide catalyzed AEMWE rapidly increased from 1.7 to 2.1 V during the first 5 hours of operation. In contrast, the perovskite catalyzed AEMWE was stable at ∼1.8 V over 100 hours.
The mitigation strategy for the degradation caused by the electrochemical oxidation of the phenyl group is to use OER catalysts with low phenyl group adsorption energy. While the phenyl group adsorption energy on transition metal surfaces such as Pt, Pd, or Ir is relatively high,115,116 alloy catalysts can dramatically reduce the adsorption energy by changing the electronic structure in the d-band center. For example, the phenyl adsorption energy of the BTMA group parallel to the Pt(111) surface is −2.30 eV, whereas the phenyl adsorption energy of the BTMA parallel to the surface of Pt1Ru1(111) is −1.30 eV.117 As demonstrated in Fig. 8b, perovskite catalysts have minimal phenyl adsorbing surface properties which is beneficial for long-term operation without much performance loss. Furthermore, the lower pH dependency of perovskite catalysts may be beneficial to the performance of pure water-fed AEMWEs.118–120 Another approach is to use polymer electrolytes with less adsorption energy of phenyl groups. Quaternized polyolefins have lower adsorption energy than quaternized polyaromatics.121 Non-rotatable phenyl groups, such as fluorene or carbazole, have lower adsorption energy than the rotatable phenyl groups, such as biphenyl.66,122,123
The effects of electrochemical oxidation of phenyl groups on AEMWEs’ durability can be seen in Fig. 9c which compares the short-term voltage behaviors of three MEAs prepared by AEMs and ionomers with different phenyl adsorption characteristics. The first MEA was fabricated with HTMA-DAPP for both the AEM and ionomer. HTMA-DAPP has biphenyl and terphenyl units in its backbone so a high level of electrochemical oxidation of phenyl groups was expected.112,121 The second MEA was fabricated with a trimethylalkylammonium functionalized poly(styrene-b-ethylene-b-styrene) triblock copolymer (SES-TMA) AEM and HTMA-DAPP ionomer. The SES-TMA AEM has no phenyl group in its polymer backbone, and thus, a lower degree of phenyl oxidation is expected. The third MEA was fabricated with SES-TMA AEM and a quaternized poly(fluorene) ionomer (FLN55). The poly(fluorene) ionomer has a non-rotatable phenyl group that can minimize phenyl oxidation. Fig. 9d compares the durability of the MEAs employing those combinations of AEMs and ionomers. The performance of the MEA employing HTMA-DAPP for the AEM and ionomer decreased quickly during the first five hours of operation due to the phenyl oxidation of the ionomer and AEM. The MEA having SES-TMA AEM and HTMA-DAPP ionomer showed a gradual decrease over 80 hours. The slower degradation rate of the MEA is because the phenyl oxidation rate of the interdiffused SES-TMA and HTMA-DAPP phase in the electrode is lower than the HTMA-DAPP MEA. In contrast, the MEA employing SES-TMA AEM and FLN55 ionomer showed relatively stable performance over 80 hours under the same operating conditions.
Deactivation of HER electrocatalysts by hydrogenation of ionomer fragments and cumulative cation-hydroxide-water co-adsorption can impact the device's performance and durability (Fig. 9a). Although hydrogenation of the ionomeric binder at the AEMWE's cathode has not been systematically investigated at a single cell level, hydrogenation of phenyl, ketone, and alkene compounds on precious metal group catalysts is well documented.124–127 Strong adsorption of the hydroxide ion on the metal surface by cation-hydroxide-water coadsorption may lower the device's durability as well. Cumulative hydroxide adsorption can reduce water access to the catalyst's surface due to lower water solubility at the high hydroxide concentrated ionomeric layer.49,128 However, the cumulative hydroxide adsorption occurs mostly in HOR potentials, ca. 0.1 V vs. RHE129,130 and thus, the impact may be less than other ionomer poisonings.
Both durability-limiting factors of pure water-fed AEMWEs are related to the ionomeric binder. Because ionomer detachment from the surface of electrocatalysts occurs when there is a lack of adhesion and ionomer poisoning occurs with the adsorption of ionomer fragments on the surface of OER catalysts, degradation of pure water-fed AEMWEs proceeds on either degradation pathway. High durability may be possible under a low hydrogen-generating rate due to the performance-durability trade-off, suggesting that achieving high performance and durability of pure water-fed AEMWEs simultaneously may be a great technical challenge for the development of commercially viable systems.
4.2. Durability-limiting factor of concentrated KOH-fed AEMWEs
The primary durability-limiting factors of the concentrated KOH-fed AEMWEs are different from those of pure water-fed AEMWEs due to the different operating environments created by the additional electrolyte. The circulated liquid electrolyte provides a catalyst–electrolyte interface by allowing water-splitting reactions which maintains the catalyst utilization of the HER and OER. As a result, there may be no need for high IEC ionomers. Therefore, the performance loss by the ionomer detachment could be avoided. Moreover, the electrochemical oxidation of phenyl groups in the AEM and ionomer is less critical because the liquid electrolyte can neutralize the phenols without significantly changing the local pH at the vicinity of the catalyst–electrolyte interface. The performance tolerance of ionomer degradation on the concentrated alkaline solution-fed AEMWEs is shown in Fig. 10a. The cell voltage increased from 1.7 to 1.95 V during a short-term test for NiFe catalyzed pure water-fed AEMWE suggesting that the electrochemical oxidation of phenyl groups occurred. When compared to the AEMWE performance under pure water-fed conditions, a notable voltage loss (∼80 mV loss at 50 mA cm−2) confirmed AEMWE degradation (Fig. 10b). However, when the performance before and after the short-term test was compared to the 0.1 M NaOH flowing, negligible performance loss was detected because the local pH at the catalyst–electrolyte interface was not varied by the concentrated alkali hydroxide solution with the electrochemical phenyl oxidation. Although circulating a concentrated alkali hydroxide solution improves AEMWE performance and performance tolerance to ionomer degradation, the corrosive liquid electrolyte accelerates the degradation of AEMs. Therefore, chemical and electrochemical stability of AEMs becomes major concerns of concentrated KOH-fed AEMWEs. In the previous perspective article on AEM stability for AEMFC applications, several key degradation pathways of AEMs such as SN2 benzyl substitution, SNAR aryl ether cleavage and heterocycle deprotonation were mentioned.33 In this section, we focus more on other degradation pathways of polymer electrolytes and non-quaternized polymers that have been known as “alkaline-stable”. Also, we provide information on radical-induced degradation and crosslinking reaction which can be accelerated with corrosive electrolyte circulation. Possible degradation mechanisms of electrocatalysts under high pH conditions is also discussed in this section.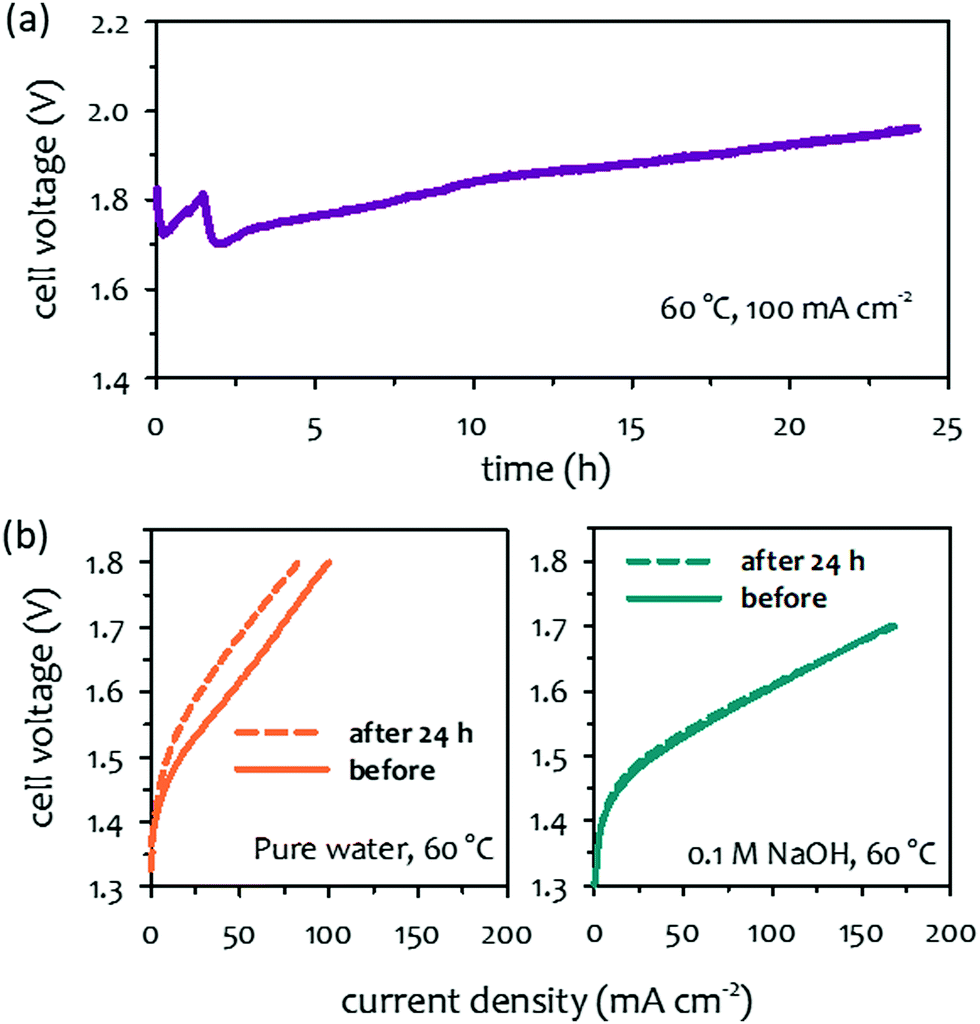 | ||
| Fig. 10 Impact of concentrated alkaline solution circulation on AEMWE performance after durability test. (a) Cell voltage change during 24 hour operation of a pure water-fed AEMWE. (b) Performance comparison after 24 hour operation for pure water-fed and 0.1 M NaOH-fed conditions. AEM and ionomer: HTMA-DAPP (35 μm thick), anode: NiFe nanoform (2 mg cm−2); cathode: PtRu/C (50 wt% Pt, 25 wt% Ru, 0.5 mgPt cm−2). The durability was measured at 60 °C under ambient pressure. The AEMWE data was produced at Los Alamos National Laboratory. | ||
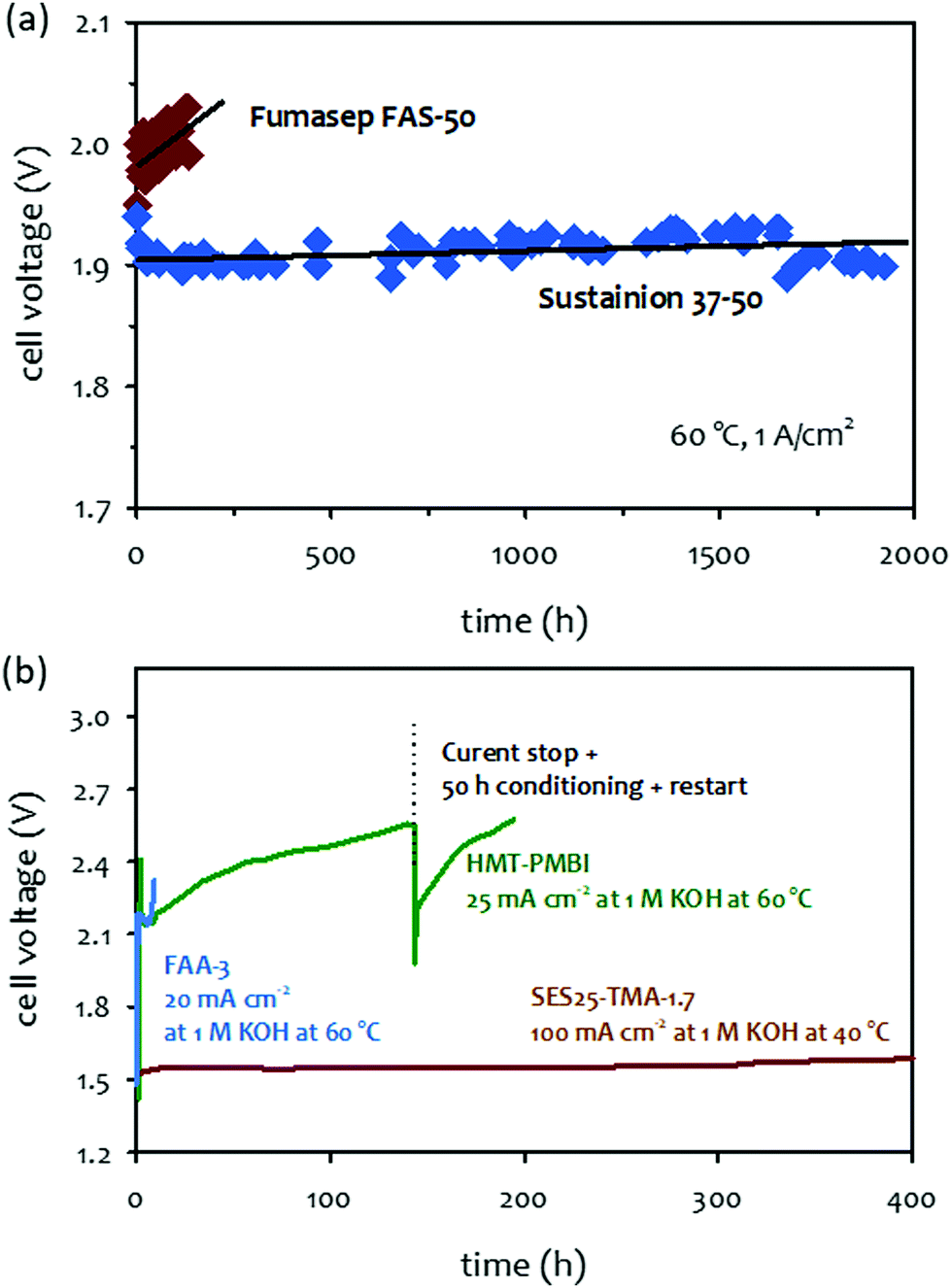 | ||
| Fig. 11 (a) 1 M KOH-fed AEMWEs at 60 °C at a current density of 1 A cm−2.131 AEM: Fumasep® FAS-50 (50 μm thick) or Sustainion® 37–50 (50 μm thick), ionomer: Nafion, anode: NiFe2O4 (2 mg cm−2); cathode: NiFeCo (2 mg cm−2). The durability was measured at 60 °C under ambient pressure. Reproduced with permission.131 Copyright 2017, Elsevier. (b) Short-term test of 1 M KOH-fed AEMWEs employing alkaline-stable AEMs. AEM/ionomer: HMT-PBI (43 μm thick)/HMT-PBI, Fumatech FAA-3 (50 μm thick)/FAA-3, anode and catalysts: Pt/C (0.5 mgPt cm−2), AEM/ionomer: SES25-TMA-1.7 (53 μm thick)/FLN-55, anode: IrO2 (1 mg cm−2); cathode: PtRu/C (50 wt% Pt, 25 wt% Ru, 0.5 mgPt cm−2). Reproduced with permission.18 Copyright 2016, Royal Society of Chemistry. | ||
Fig. 11b compares the 1 M KOH-fed AEMWE durability employing three different AEMs: FAA-3, hexamethyl-p-terphenyl poly(benzimidazolium) (HMT-PBI), and SES-TMA. While the AEMWE using a commercial FAA-3 AEM quickly degraded over 10 hours of operation at 60 °C and a constant current density of 20 mA cm−2, the AEMWE using an aryl ether-free HMT-PBI AEM showed a gradual performance decay at a constant current density of 25 mA cm−2 over 200 hours.146 The 1 M KOH-fed AEMWE cell employing SES-TMA AEM also showed stable performance over 400 hours. However, it is not certain that the AEMWEs using the alkaline-stable AEMs can operate for >20000 hours, as in the case of the SOA PEMWEs because some degradation of the polymer was observed under higher stress accelerated conditions. In this following section, we discuss possible degradation mechanisms of AEMs that are known as “alkaline-stable”.
Park et al. investigated the degradation pathways of the HTMA-DAPP AEM that has an aryl ether-free wholly aromatic polymer backbone and pendant alkyltrimethyl ammoniums.15 They found that a common nucleophilic methyl substitution (SN2) reaction occurred only under relatively high temperatures (>80 °C). Instead, the β-elimination (E2) reaction (also known as Hofmann elimination) was observed to occur over a prolonged period (Fig. 12a). Under 80 °C and 0.5 M NaOH conditions, degradation by β-elimination in the alkyl chain extender started to occur after 3500 hours and continued until the end of the experiment (after 11000 hours, 50% loss in hydroxide conductivity was observed). Under 80 °C and 4 M NaOH conditions, the degradation due to β-elimination started to occur at 1800 hours, and ∼40% loss in hydroxide conductivity was observed after 3500 hours. The delayed degradation via the β-elimination degradation process is consistent with other polymers with a long alkyl chain quaternary ammonium (more than four carbons in the alkyl chain linker).147,148 Fan et al. investigated the degradation behaviors of poly(arylimidazolium)s (Fig. 12b).149 The polymers with ethyl-, propyl-, and butyl-substituted side chains showed excellent alkaline stability. The chemical stability of these polymers after immersion of the AEM in 10 M KOH at 80 °C indicated that 94 to 98% of imidazolium remained intact after 240 hours in highly caustic solutions. The possible degradation mechanisms of the poly(arylimidazolium)s were evaluated with the less stable methyl-substituted poly(arylimidazolium). The 1H NMR analysis after the alkaline stability test suggested that the degradation of the AEMs occurs via both dealkylation (major) and ring-opening (minor) pathways.
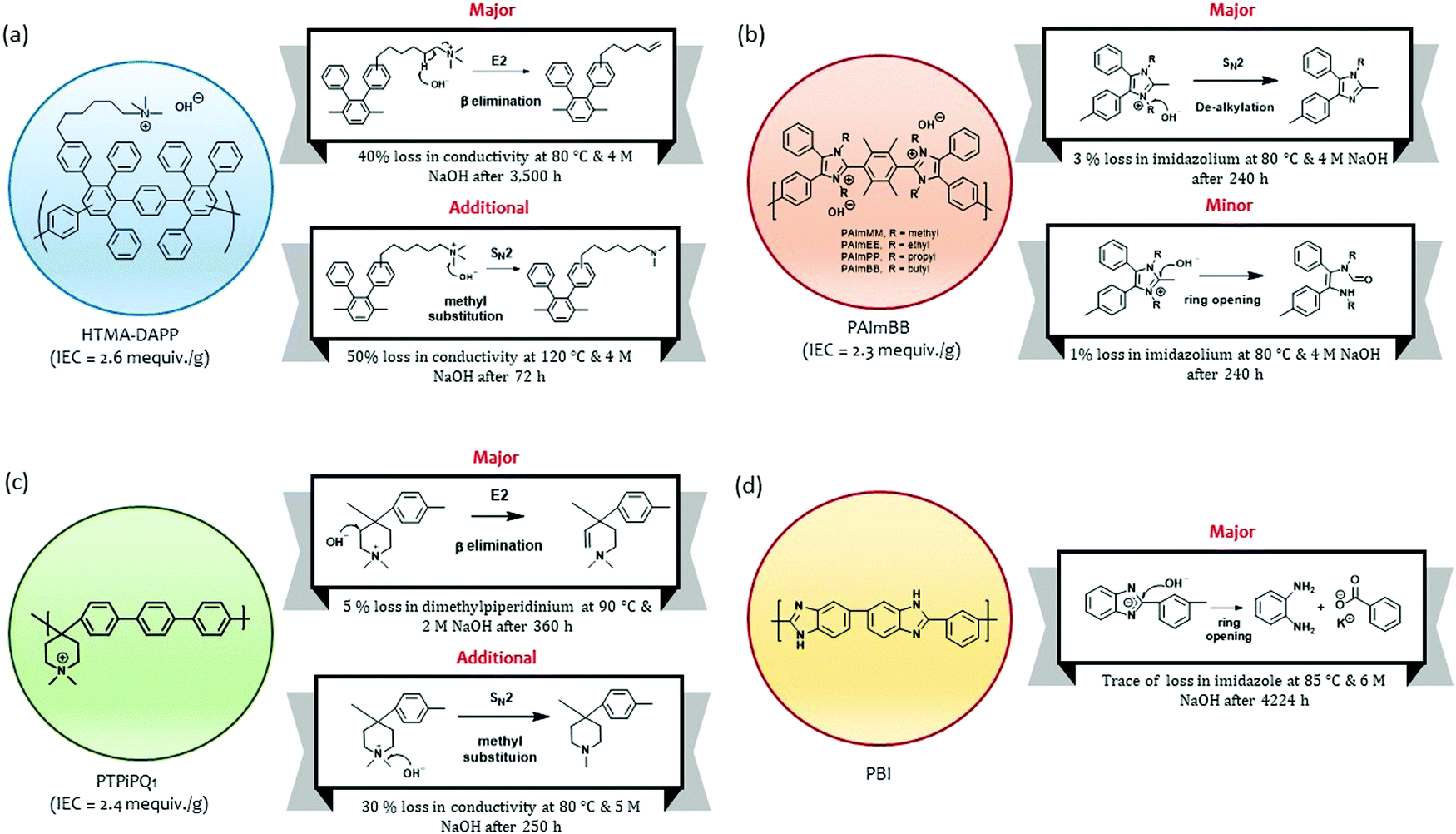 | ||
| Fig. 12 Major degradation mechanisms of alkaline-stable polymers under high pH conditions (a) hexamethyl trimethyl ammonium functionalized Diels–Alder poly(phenylene) (HTMA-DAPP). (b) Butyl substituted poly(arylimidazolium) (PAImBB). (c) Poly(arylene piperidinium)s (PTPiPQ1). (d) Polybenzimidazole. | ||
Olsson et al. investigated the degradation mechanisms of poly(arylene piperidinium)s with methyl, butyl, hexyl, and octyl substitutes (Fig. 12c).18 The polymers showed no degradation under 2 M NaOH at 60 °C for 15 days. The degradation mechanisms of the polymers were investigated under harsher conditions (2 M NaOH at 90 °C for 360 hours). The study indicated that the major degradation pathway is the β-elimination in the piperidinium ring. A nucleophilic α-methyl substitution reaction was also identified for alkyl piperidinium, particularly with a higher KOH concentration.150,151 Aili et al. examined the chemical stability of polybenzimidazole (PBI) under 6 M NaOH at 85 °C for 176 days (Fig. 12d).152 The chemical structural change of the PBI was not detected after 116 days of aging, but a trace of structural change was detected after 176 days of aging. The 1H NMR analysis indicates that the base-catalyzed hydrolysis of PBI primarily occurs through a nucleophilic attack by hydroxide ions on the benzimidazole C2 position.153,154
The mitigation strategies of AEM degradation have been investigated. Pham suggested that higher alkaline stability of cyclic cation functionalized polymers may be achieved by introducing a pendant group that may reduce the ring strain and restrict conformation.16 Alternatively, alkaline-stable non-quaternized polymers can be used instead of quaternized polymers. PBIs have been extensively studied for this purpose. Kraglund et al. demonstrated the performance of 20 wt% (3.6 M) KOH-fed AEMWE cell employing poly(2,2′-(m-phenylene)-5,5-bibenzimidazole) (m-PBI).155 The cell's performance gradually decreased due to the base-catalyzed hydrolysis of amino groups in the polymer backbone. A more alkaline-stable poly(2,2′-(m-mesitylene)-5,5′-bibenzimidazole) (mes-PBI) was developed from the same research group.156 The mes-PBI membrane was stable in 0–10 wt% KOH at 88 °C for 207 days. In a 50 wt% aqueous KOH solution, the molecular weight of the mes-PBI membrane gradually decreased, suggesting that the polymer backbone degradation was initiated from the nucleophilic addition at the benzimidazolide C2 position. Although the alkaline stability of mes-PBI is superior to most quaternized polymers, one should note that the use of non-quaternized membranes requires a higher concentration of KOH solution to meet the conductivity requirement. Reasonably high performance and durability are possible with this approach when using non-quaternized polymer electrolytes.
Ayers et al. showed the mechanical property deterioration of quaternized poly(arylene ether) AEMs after being subjected to the Fenton's test for up to 5 hours.58 Visual inspection of the AEMs after the Fenton's test by optical microscopy showed surface cracking and possible AEM dissolution (Fig. 13a). The dominating reaction in the degradation processes of polyaromatics is the loss of OCH3 from the methoxy substituted compound. This mechanism is relevant to aryl ether-containing polymers such as polysulfones and polyether ketones as it can lead to bond breaking within the C–O–C connections (Fig. 13b).167 Owing to its electrophilic nature, the HO˙ radical preferentially attacks the aromatic ring near the aryl ether bond-forming phenols under high pH conditions (Fig. 13c).167 The attack of free radicals on the vulnerable carbon (carbon in para position for the trimethyl ammonium hydroxide group of vinyl benzyl chloride grafts) degrades the polymer backbone of the quaternized polystyrenes (Fig. 13d).165,168–170 Cation degradation by a radical attack on the benzyl triethyl ammonium is also depicted in Fig. 13e.171 In this mechanism, hydroxide ions attack the quaternary ammonium groups of the AEMs to produce ylides and water molecules by the abstraction of a proton from the α-carbon. Then, the oxygen molecules in the alkaline solution capture the electron of the ylides to produce superoxide anion radicals and quaternary ammonium radicals, respectively. The quaternary ammonium radicals subsequently degrade into ethylenes and tertiary amines. Mitigation strategies for electro-oxidative degradation by radical species include using an aryl ether-free polymer backbone172,173 and introducing cross-linking reactions.174,175 Radical inhibitors such as p-ethyl phenol that could protect the AEMs against the attack may be used.171
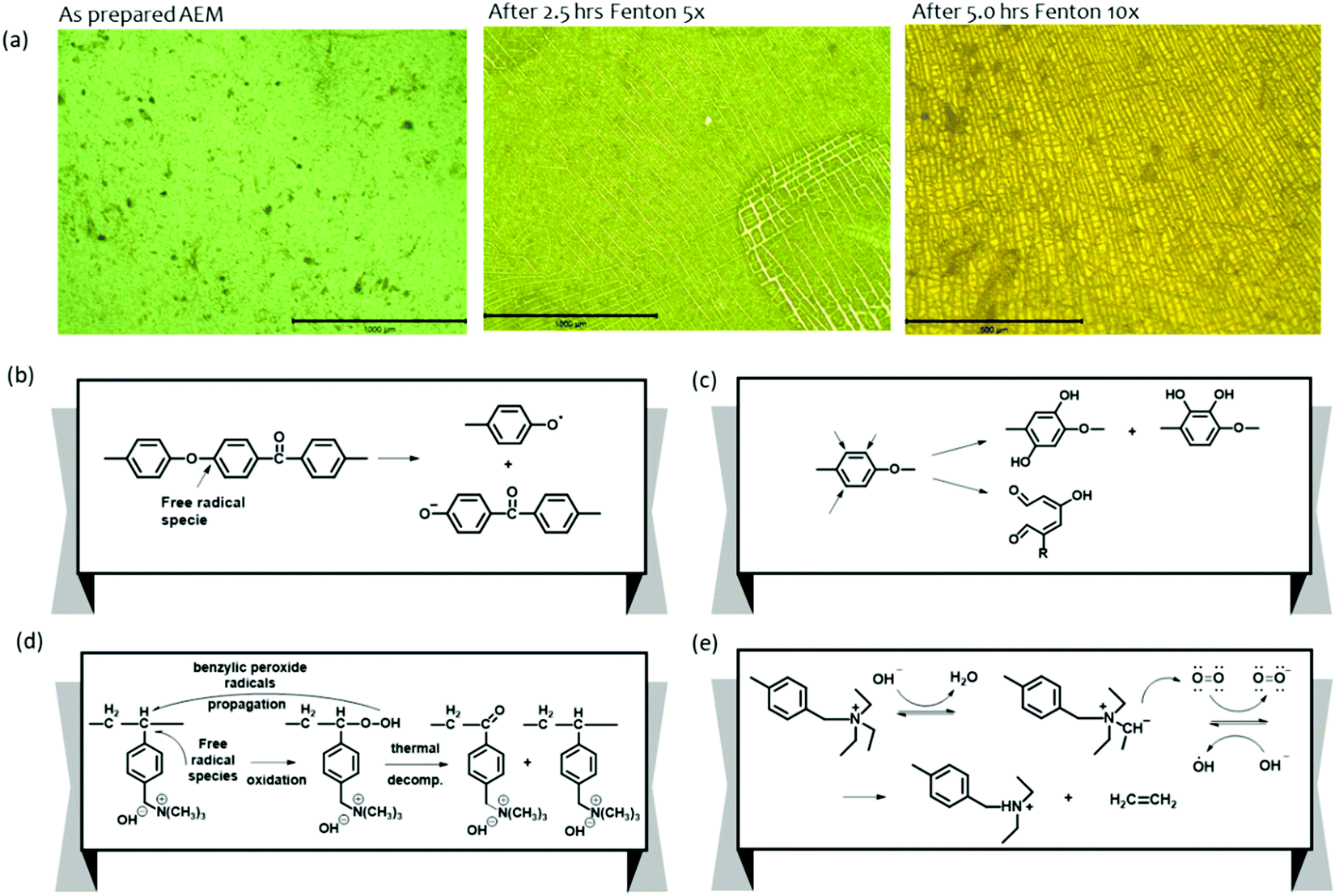 | ||
| Fig. 13 (a) Optical microscopy analysis of AEM subjected to Fenton's test as a function of exposure time.58 Major polymer degradation by oxygen radical species. Adapted with permission.58 Copyright 2013, IOP publishing. (b) Aryl ether polymer backbone degradation. (c) Phenyl group degradation by formation of phenolates. (d) Polymer backbone degradation of quaternized polystyrene (e) Cationic group degradation. | ||
 | ||
| Fig. 14 Several cross-linking pathways of AEMs under high pH conditions. | ||
Identification of the major degradation mechanism of AEMs in a working cell can be tricky because AEM degradations can simultaneously occur in concentrated KOH-fed AEMWEs unlike ionomer poisoning and ionomer detachment found in pure water-fed AEMWEs does not occur simultaneously. However, operating parameter changes may help to identify the main degradation mechanism of AEMs. Radical-induced hydrolysis is proportional to the operating voltage, temperature, and catalysts. Alkaline instability and cross-linking are more dependent on the concentration of KOH. The degradation by cross-linking reaction occurs mostly during the first 100 hours of the test, but the degradation rate by hydroxide attack of AEMs is rather constant. Another difference in degradation behavior between pure water-fed and concentrated KOH-fed AEMWEs is the performance-durability trade-off. Because using a highly concentrated KOH electrolyte allows lower voltage operation which can reduce radical-induced AEM hydrolysis, a less clear performance-durability trade-off is observed in concentrated KOH-fed AEMWEs.
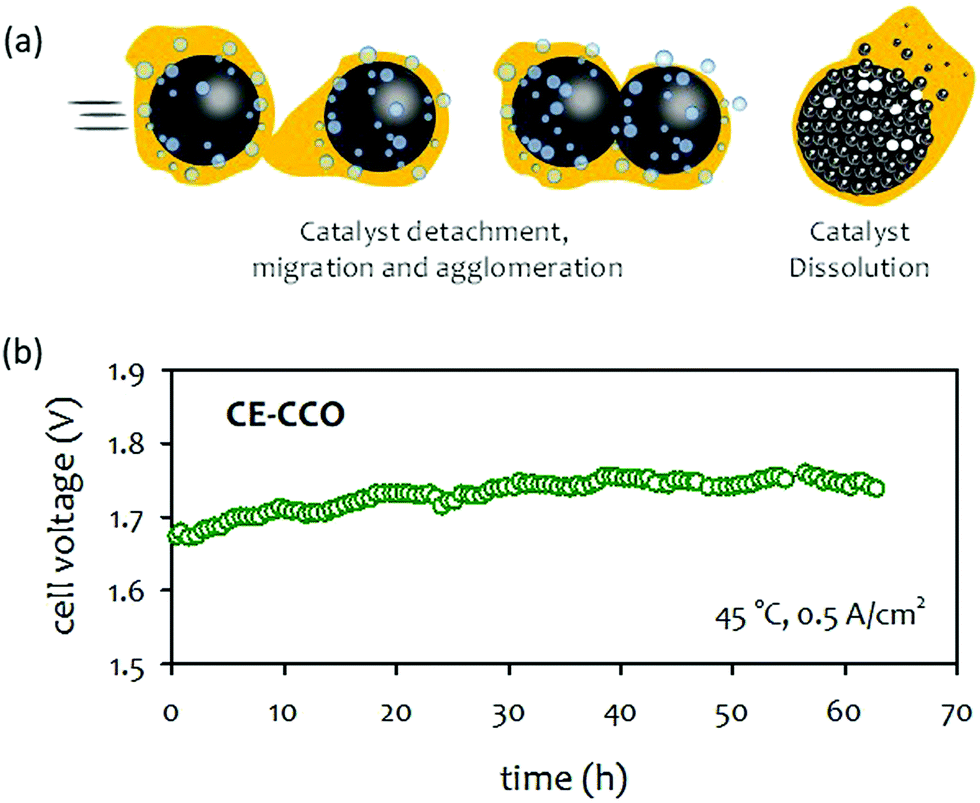 | ||
| Fig. 15 (a) Catalyst degradation pathways under AEMWE operating conditions. (b) Durability of 1 M KOH-fed AEMWE at 500 mA cm−2 at 45 °C. AEM: Sustainion® X37-50, anode: CuCo-oxide on nickel form (CE-CCO), cathode: Pt/C (1 mgPt cm−2).92 Adapted with permission.92 Copyright 2020, Elsevier. | ||
To overcome the issue of catalyst degradation, various efforts have been made to develop effective material design strategies such as doping,192–196 surface-structure modification,197–199 and formation of protective layers on the catalyst surface.200–203 Such approaches are effective to improve catalytic activity and stability. However, one should note that the long-term stability of electrocatalysts has been evaluated mostly at a relatively small current density (10 mA cm−2).192,204–207 For concentrated KOH-fed conditions, there has been plenty of literature reviews covering the OER catalyst durability in which the actual test was often carried out in a half-cell immersed in concentrated alkaline solution. For pure water-fed or 1 wt% K2CO3-fed conditions, half-cell tests may not be a viable option, thus the durability of the OER catalysts needs to be evaluated in a AEMWE cell. In this case, device performance loss often originates from AEM and ionomer failure. Therefore, it may be difficult to dissect the cell and determine the degree of liability stemming from the catalyst degradation. In addition, the catalyst degradation may be accelerated by poisoning due to the ionomer as discussed in Section 4.1.2. For these reasons, the catalyst stability should ultimately be evaluated with an AEMWE cell under conditions such as (i) high current density, (ii) sufficiently long time (>1000 hours) and (iii) target electrolyte circulation.
Limited studies on the impact of catalysts on AEMWE durability was performed. Moderate device degradation for pure water-fed and carbonated solution-fed AEMWEs was demonstrated at a higher cell voltage (1.8–2.0 V) over several hundreds hours.208,209 The cell degradation rates of the AEMWEs were ∼0.2 mV h−1. For concentrated KOH-fed AEMWEs, the duration of most durability tests was ≤100 hours and degradation rates were higher.210–212 For example, Park et al. reported on the durability of surface-etched CuCo-oxide (CE-CCO) catalysts for OER.92 A half-cell test indicated that the CE-CCO catalyst was durable, exhibiting stable OER activity for 3600 hours at a constant current density of 20 mA cm−2. The performance of 1 M KOH-fed AEMWE employing the CE-CCO OER catalysts was reasonably good (1390 mA cm−2 at 1.8 V) at 45 °C. However, a notable voltage degradation, ca. 1 mV h−1, was observed during the 65 hours of extended-term test at a constant current density of 500 mA cm−2 (Fig. 15b). Although it was not clear that the performance decay of the AEMWEs originated only from the OER catalysts, the result suggests that a deeper analysis is needed to establish whether the high activity and stability of electrocatalysts are transferred to the device's performance and durability.
4.3. Durability-limiting factor of 1 wt% K2CO3-fed AEMWEs
As circulating a corrosive concentrated KOH solution into AEMWEs becomes a concern in MEA component degradation, several research groups have reported the possible use of an aqueous potassium (or sodium) carbonate solution.72,73 Although the cell performance of K2CO3-fed AEMWEs is inferior to that of the KOH-fed AEMWEs at the same molar concentration,132 the performance of K2CO3-fed AEMWE is superior to that of the KOH-fed AEMWE at the same pH level.74 This suggests that the K2CO3-fed AEMWE is an attractive approach to hydrogen production with improved durability and limited performance trade-off. Circulating a less corrosive K2CO3 solution creates less concern on MEA component degradation which allows operation with differential pressure across the membrane. Economic production of pressurized hydrogen offers additional benefits. In typical commercial PEMWEs, hydrogen is pressurized to 15–30 bars inside the cell/stack via simultaneous water splitting and electrochemical compression.213 The influence of differential pressure on 10 wt% K2CO3-fed AEMWE performance is small, (∼20 mV loss at 1 A cm−2 when the pressure increased from 1.0 to 8.5 bar), suggesting an advantage of hydrogen production with minimal energy penalty.214 Besides, for differential pressure operation, oxygen crossover is less critical for AEMWEs since the oxygen permeation rate at ambient pressure is much lower than that of hydrogen. The differential pressure operation enables a high purity hydrogen production without the additional burden of oxygen removal downstream. However, high differential pressure conditions place a high amount of stress on the membrane that may cause premature mechanical failure of AEM. In this section, we investigate the impact of AEM mechanical properties on 1 wt% K2CO3-fed AEMWEs operating with a differential pressure of 100 psi.Mechanical toughness or the tensile energy of AEMs can be obtained from the area under the stress–strain curve. Herein, we consider two types of hydrocarbon AEMs that have reasonably high toughness. One is an elastomeric material like acidic PFSA membranes that have low modulus and strength but high elongation. The other is a polyaromatic membrane that has high modulus and strength but low elongation. The stress–strain curves of two representative polymers (HTMA-DAPP and SES-TMA) are compared in Fig. 16a. The tensile energy for the SES-TMA AEM was ∼30% higher than that of the HTMA-DAPP. The durability of 1 wt% K2CO3-fed AEMWEs employing the HTMA-DAPP and SES-TMA AEMs was compared at 100 psi differential pressure. The cell employing HTMA-DAPP AEM showed stable voltage during the 500 hours at a constant current density of 0.5 A cm−2 and 50 °C. In contrast, the cell employing SES-TMA did not hold the differential pressure for 500 hours (Fig. 16b). Initially, the cell held the differential pressure of ∼100 psi, but after about 200 hours, the cell could not hold the full differential pressure. The SES-TMA cell also showed more voltage fluctuation with a higher decay rate (310 μV h−1) although it still produced hydrogen until the end of the 500 hour-test. This result suggests that the AEMs having low stiffness and tensile strength would not be suitable for long-term operation under differential pressure conditions. For a membrane to withstand the challenging conditions of differential pressure operation, a stiffer (but not brittle) membrane is desirable. The quaternized SES-TMA polymer has large elongation and a relatively low yield strength. It is conceivable that such a low-stress membrane is slowly yielded under differential pressure conditions eventually leading to a membrane rupture. When a less corrosive K2CO3 liquid electrolyte is used, the main durability-limiting factor of K2CO3-fed AEMWE is associated with the differential pressure. The differential pressure holding capability may require an increase in the thickness of the AEM. However, because hydrocarbon AEMs generally have lower hydrogen gas permeability, thinner membranes and lower cell resistance, the best mitigation strategy for differential pressure operation is to use a rigid polyaromatic-based polymer instead of elastomeric polyolefin-based polymers. Other popular strategies include reinforcements,218 composites,219,220 and cross-linkings.221,222
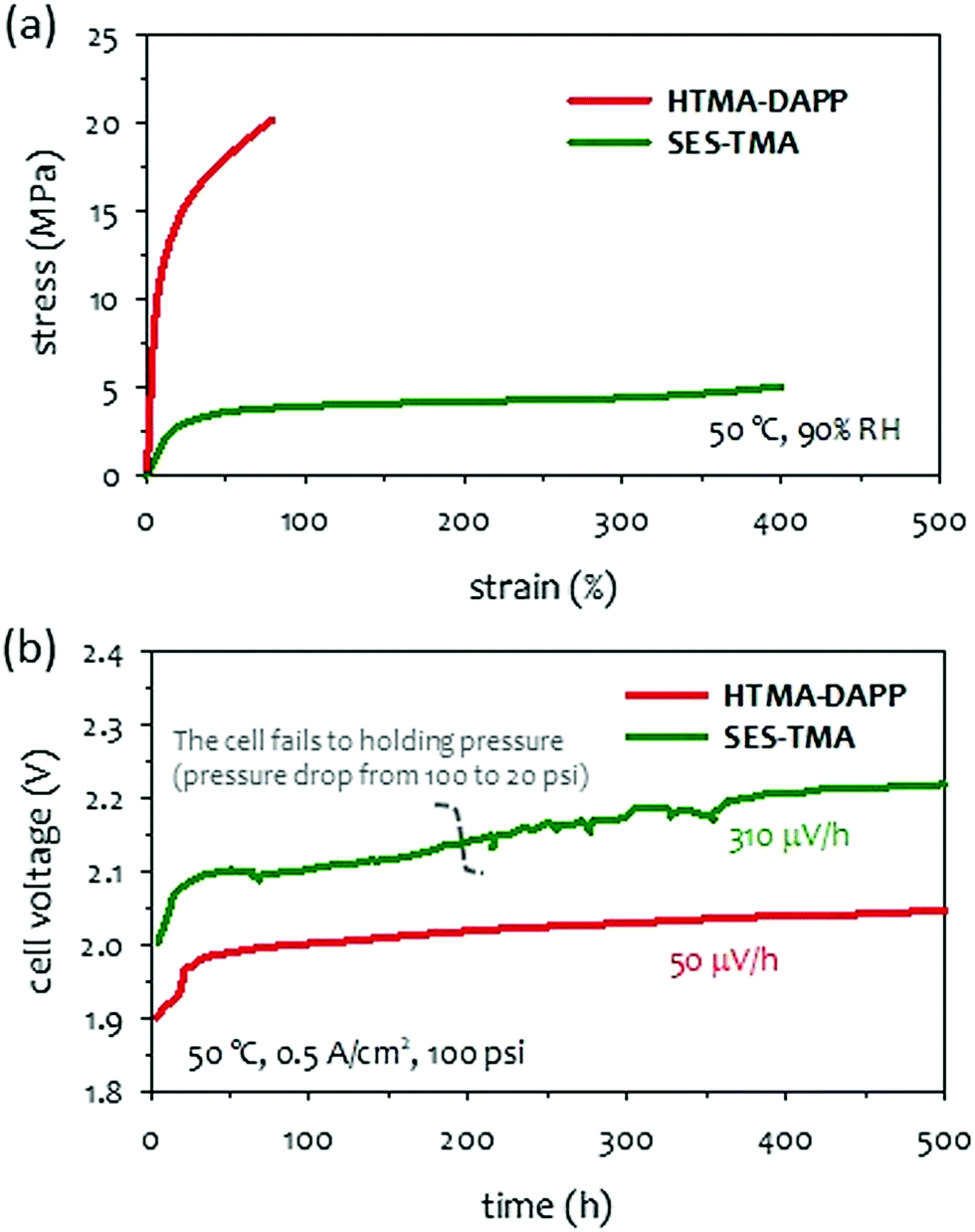 | ||
| Fig. 16 (a) Stress–strain curves of HTMA-DAPP and SES-TMA AEMs. (b) Comparison of long-term performance of AEMWEs using HTMA-DAPP (78 μm thick) and SES-TMA (53 μm thick) AEMs. Ionomer: Aemeon, Co3O4 (3 mg cm−2), cathode: Pt black (3 mgPt cm−2). The AEMWE cell data was produced at Nel Hydrogen. | ||
5. Summary and outlook
This paper discusses the durability-limiting factors of AEMWEs that are closely related to electrolyte-fed conditions. Table 1 summarizes the durability of SOA AEMWEs, durability-limiting factors and degradation behaviors of AEMWEs under these three operation modes.| Pure water-fed | Concentrated KOH-fed | 1 wt% K2CO3-fed, 100 psi differential pressure | |
|---|---|---|---|
| a PGM anode and cathode. | |||
| Durability of SOA AEMWE | |||
| Voltage degradation rate (μV h−1) | 100 at 0.5 A cm−2 (80 °C) | 0.7–30 at 0.3–1 A cm−2 (60–80 °C) | 1–50 at 0.5 A cm−2 (50 °C) |
| Longevity demonstrated (hours) | >2000a | >12000 |
>700 |
| Ref. | 29 | 31, 79 and 80 | |
| Durability-limiting factor (degradation behavior) | |||
| Ionomer detachment (Type 1 & 4) | ++ | − | − |
| Ionomer poisoning (Type 1 & 2) | ++ | + | + |
| Alkaline instability (Type 1, 3, 4 & 5) | − | ++ | − |
| Radical induced hydrolysis (Type 1) | + | ++ | + |
| Cross-linking reaction (Type 1 & 5) | + | ++ | + |
| Catalyst & degradation (Type 1) | + | + | + |
| Mechanical failure (Type 1 & 5) | − | − | ++ |
The major degradation mechanisms of pure water-fed AEMWEs is related to the ionomeric binder at the electrodes. The first ionomer-related degradation is the ionomer detachment from electrocatalysts. This degradation path can be observed with ionomeric binders with a higher IEC which provide high pH environment without circulating an additional liquid electrolyte. Because ionomeric binders with high IEC generally have high water affinity, lack of adhesion of the ionomeric binders often causes ionomer detachment from the electrode that limits the durability of the cell. Ionomer detachment causes Type 1 behavior (cell voltage increase), as well as Type 4 behavior (cell voltage decrease) at a later stage. Electrochemical phenyl oxidation is the other durability-limiting factor that is related to the ionomeric binder. Electrochemical oxidation of the phenyl groups in the ionomeric binder causes a rapid voltage jump, (Type 2) as the local pH change in the electrode occurs in a relatively short time. The voltage degradation rate of SOA pure water-fed AEMWE was reported to be 100 mV h−1 which is still higher than the liquid electrolyte-fed AEMWEs suggesting that the commercial viable pure water-fed AEMWEs may not be realized in the near future. This is also partly because the complete overcoming of ionomer-related degradation pathways is technically challenging. An ionomer synthetic strategy towards high IEC with low water uptake may help to find a sweet spot for a highly performance with good durability.56,223 Completely removing phenyl groups in the anode ionomers224 can resolve the issues associated with the electrochemical oxidation of ionomer.
The concept of the concentrated KOH (pH ≥ 14)-fed AEMWEs was developed by combining traditional AWEs with the zero-gap configuration of PEMWEs. The performance stability of the concentrated KOH-fed AEMWEs was evaluated at a higher current density compared to pure water-fed AEMWEs. During a short-term test, the cell voltage behavior of extended-term tests shows a mixed pattern of Type 1 (cationic group degradation), Type 3 (electrolyte concentration change), and Type 4 (polymer backbone degradation), which makes it hard to interpret whether the cell is stable or unstable from an extended-term test. Occasionally, the cell also stops working with catastrophic failure behavior (Type 5). Since additional high pH electrolytes are available in the catalyst layer, the concentrated KOH-fed AEMWEs can largely remove the ionomer detachment and phenyl oxidation issues found in pure water-fed AEMWEs. Instead, the membrane and ionomer degradation under high pH conditions can cause performance loss over time. Compared to systems employing quaternized or non-quaternized membranes, there are trade-offs between performance and durability. When using a quaternized AEM, circulating relatively low concentrated KOH (1 M) is enough to produce high performance. When using a non-quaternized polymer, a higher concentration of KOH (3–5 M) is required for high performance. Quaternized polymers may be less stable than non-quaternized polymers under high pH conditions because the degradation of the polymer primarily starts with the cationic functional group, or the cationic functional group often triggers polymer backbone degradation. This means that alkaline stable non-quaternized polymers may survive with highly concentrated KOH solutions for only a prolonged period. The concentration of KOH also affects the selection of electrode binders. For the AEMWEs using a relatively low concentration of KOH (1 M) and a quaternized ionomeric binder may be beneficial for AEMWE performance. However, for AEMWEs using highly concentrated KOH (≥3 M), a more alkaline-stable non-quaternized ionomer such as Nafion might be used. Significant advancements of alkaline stability of polymeric materials over the past decade have allowed for the operation of concentrated KOH-fed AEMWEs using a quaternized AEM > 12000 hours of durability with a relatively low voltage degradation rate (0.7 μV h−1). For the AEMWEs using non-quaternized AEMs, more than 700 hours of durability with a low voltage degradation rate (30 μV h−1) was demonstrated. For a commercially viable system, additional performance and durability validation at a stack level is required. Moreover, the higher durability of such AEMWEs under differential pressure conditions needs to be demonstrated for an economically competitive system.
The 1 wt% K2CO3-fed AEMWEs are unique configurations for AEMWEs that increase the durability of the device at the cost of performance. Degradation behaviors of the 1 wt% K2CO3-fed AEMWEs when operating under differential pressure during an extended-term test may be more complex as different degradation limiting factors can play a role in addition to the behaviors associated with gas crossover increase (Type 1) and mechanical failure (Type 5). Although long-term performance data is limited, the durability of 1 wt% K2CO3-fed AEMWEs with differential pressure is reasonably good. The improved material stability at lower pH environments enables the accommodation of differential pressure operation. Operation of AEMWE cells under differential pressure conditions is important, especially in smaller scales, because mechanical compression is not economical for ambient pressure operation. However, operation under differential pressure conditions may narrow the AEM material choice to endure the highly pressurized conditions. Particularly, the AEMs having low stiffness and mechanical strength have limitations to hold differential pressure. The SOA 1 wt% K2CO3-fed AEMWEs exhibited reasonably good device durability (>700 hours) with a low voltage degradation rate (1–50 μV h−1). One remaining technical challenge for 1 wt% K2CO3-fed AEMWEs is performance improvement. The SOA 1 wt% K2CO3-fed AEMWE system showed relatively low performance compared to other electrolyte-fed AEMWE systems. Because of carbonation issues, a more conductive AEM and ionomer may be essential for 1 wt% K2CO3-fed AEMWE systems. This approach may be plausible to keep high durability with improved performance, as 1 wt% K2CO3 circulation provides less corrosive environments than concentrated KOH circulation. In addition, more active catalysts under the lower pH conditions are critical.
This article indicates that polymer electrolytes play a critical role in the durability AEMWEs. Over the past decade, various anion exchange polymer electrolytes have been developed for electrochemical applications and have mainly been used in AEMFCs. As AEMFCs have the major benefit of utilizing PGM-free oxygen reduction catalysts, the development of AEMFCs has been focused on automotive applications. In automotive AEMFC applications, the temperature difference between the fuel cell and the ambient is relatively small creating a significant challenge for designing a lightweight, small-size cooling system.225 Therefore, much effort has been devoted to designing highly stable polymer electrolytes at high operating temperatures (80–95 °C). In addition, the relatively high resistance of AEMs under desirable low RH conditions drives the reduction in thickness of AEMs. For AEMWE applications, the heat rejection constraint does not exist and the device operates under nearly liquid water saturated conditions. Instead, substantial benefits of differential pressure operation of AEMWEs drive the increase of AEM thickness or stiffness to withstand differential pressure. Increased resistance of thick AEMs can be compensated by using liquid electrolytes and a liquid water environment. Due to the less stringent requirements of AEMs in AEMWE applications, more AEM materials are available for AEMWEs applications. Several commercial AEMs were tested in the AEMWE mode.226 Fumatech's FAA3 AEMs are a poly(arylene ether) based material. Although an AEMWE using an unsupported FAA3 AEM showed reasonably high durability of up to 1000 hours of cell voltage cycling (1–1.8 V) at 50 °C,227 the alkaline stability of the aryl ether bond of AEMs is a concern for longer term and harsher conditions. Ionomr's AEMION AEMs, a methylated polybenzimidazole-based polymer (HMT-PBI), showed stable AEMWE performance at 1 A cm−2 at 60 °C for 150 hours.69 The AEM stability evaluation after longer-term operation of the AEMWE is still needed for the practical use of this AEM. Dioxide Materials’ Sustainion is based on poly(4-vinyl benzyl chloride-co-styrene) chemistry. Long-term stability (∼12000 hours) of AEMWEs using a PTFE-reinforced Sustainion AEM was demonstrated at 1 A cm−2 at 60 °C (see Fig. 5b).31 The high durability of the AEMWE cell may be obtained by maintaining the hydroxide conductivity by circulating KOH solution as the alkaline stability of the benzyl tetramethyl imidazolium cationic group of the polymer is low.144 Orion Polymer's m-TPN AEMs are based on aryl ether-free terphenylene-based materials.26 For this polymer, unsupported and reinforced composite membrane (Durion, Xergy) are available. The m-TPN-based AEMs are known for their excellent alkaline stability but the mechanical properties for differential pressure operation needs to be validated. No long-term performance data of AEMWEs using these AEMs are reported. Diaphragm membranes such as AGFA's Zirfon80,95 or ion-solvating membranes such as PBI79 showed good durability (see Fig. 5c) but those non-functionalized membranes can only be used with a highly concentrated KOH solution. Polyolefinic AEMs developed originally for AEMFCs228–230 need to verify their mechanical robustness, particularly under partial differential pressure conditions. Other aryl ether-free polyaromatics231–233 also have good potentials for AEMWE applications, but more characterization in a working electrolyzer cell is required.
From an industrialization perspective, it is clear that the AEMWE community has made great strides in recent years towards improved cell performance and has demonstrated the ability for performance to reach levels that would be viable compared to current PEM technology. Despite these developments, the technology still poses a much larger technical risk than the incumbent technologies (PEMWE and AWE) and the academic community will need to continue research to further derive the technology. Primary topics that, if researched, could enable commercialization are as follows: (i) a fundamental understanding of in situ failure mechanisms (ii) more studies showing durability (>100 hours) at practical current densities of >0.75 A cm−2 (ideally >1.0 A cm−2) and (iii) durability testing with earth abundant bipolar plates and porous transport layers. While degradation of individual cell components have been studied, there is a need to relate these to failure modes experienced within the cell during operation. One example of this could be proving an ex situ degradation protocol that relates to in situ failure mechanisms. Another example would be developing an in situ AST. To become cost competitive, cost models show that AEMWEs will need to operate at current densities >0.5 A cm−2 and potentially >1.0 A cm−2 even when assuming large reductions in MEA cost. At these current densities, it is common to see accelerated degradation rates. A key potential benefit of AEMWE is enabling the use of more earth abundant materials during construction. While some testing has used materials such as stainless steel and nickel, there should be more investigation into the impacts on cell durability. Furthermore, greater access to these materials would be beneficial to the community. In conclusion, a large amount of progress has been made in the field of AEMWE and the remaining challenge for the research community is improving and gaining a better understanding of AEMWE durability.
6. Concluding remarks
Significant advancement in the performance of AEMWE technology, especially with PGM-free anode catalysts, shows that AEMWE technology is moving towards a commercially viable option. From the durability perspective, it seems that achieving robust durability of AEMWEs is less problematic than AEMFCs, although there are several durability-limiting factors depending on the cell operating modes. However, it is important to know that if AEMWEs were to replace PEMWE and AWE technologies, that AEMWE MEAs would have to meet all the same performance and durability requirements. Currently, AEMWEs do not meet all requirements, indicating much needed improvement in specific areas before being placed in products. Although it is hard to say which operating modes for AEMWEs is more beneficial to performance, durability, cost, and system complexity, increasing research efforts in the academic community targeting the improved performance and durability of the AEMWEs retains hope that this promising and economically viable technology will eventually find its place in a rapidly emerging hydrogen economy.7. Experimental
7.1. Materials
For the AEM, we chose two quaternized polymers. The first is an alkyltrimethyl ammonium functionalized poly(styrene-b-ethylene-b-styrene) triblock copolymer (SES-TMA). The SES-TMA polymer was synthesized by Friedel–Crafts bromoalkylation of polystyrene block copolymers.181 Due to the low glass transition temperatures of the polyethylene segment and the polystyrene hard segment, this block copolymer has a rubbery nature (i.e., low strength/modulus and high elongation). The second polymer is a hexamethyltrimethyl ammonium functionalized Diels–Alder poly(phenylene) (HTMA-DAPP).139 This polymer has a wholly aromatic backbone which possesses a stiff backbone nature (high strength/modulus and low elongation). All items have good chemical stability under high pH conditions. For the ionomeric binder, we used three ionomers: a commercial ionomer (Aemion AP1-HNN8),234 HTMA-DAPP, and quaternized poly(fluorene) (FLN-55).1227.2. MEA fabrication and extended-term test
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We gratefully acknowledge research support from the HydroGEN Advanced Water Splitting Materials Consortium (Award Number: 2.2.0.401), established as part of the Energy Materials Network under the US Department of Energy, Office of Energy Efficiency and Renewable Energy (EERE), Hydrogen and Fuel Cell Technologies Office (HFTO). F. Y. Z. greatly appreciate the support from U.S. Department of Energy, EERE, HFTO under the Award Number DE-EE0008426. We thank Professor Yuehe Lin at Washington State University for providing NiFe catalysts. Additionally, we would like to thank Alex Keane from the Nel team for fabricating electrodes, building cell stacks, and running the durability test. Los Alamos National Laboratory is operated by Triad National Security, LLC under US Department of Energy Contract Number 89233218CNA000001.References
- B. Pivovar, N. Rustagi and S. Satyapal, Electrochem. Soc. Interface, 2018, 27, 47–52 CrossRef CAS.
- E. L. Miller, S. T. Thompson, K. Randolph, Z. Hulvey, N. Rustagi and S. Satyapal, MRS Bull., 2020, 45, 57–64 CrossRef.
- S. A. Grigoriev, V. N. Fateev, D. G. Bessarabov and P. Millet, Int. J. Hydrogen Energy, 2020, 45, 26036–26058 CrossRef CAS.
- I. Vincent and D. Bessarabov, Renewable Sustainable Energy Rev., 2018, 81, 1690–1704 CrossRef CAS.
- A. S. Aliyev, R. G. Guseynova, U. M. Gurbanova, D. M. Babanly, V. N. Fateev, I. V. Pushkareva and D. B. Tagiyev, Chem. Probl., 2018, 3, 283–306 CrossRef.
- C. Liang, P. Zou, A. nairan, Y. Zhang, J. Liu, K. Liu, S. Hu, F. Kang, H. J. Fan and C. Yang, Energy Environ. Sci., 2020, 13, 86–95 RSC.
- H. Zhou, F. Yu, Q. Zhu, J. Sun, F. Qin, L. Yu, J. Bao, Y. Yu, S. Chen and Z. Ren, Energy Environ. Sci., 2018, 11, 2858–2864 RSC.
- O. Schmidt, A. Gambhir, I. Staffell, A. Hawkes, J. Nelson and S. Few, Int. J. Hydrogen Energy, 2017, 42, 30470–30492 CrossRef CAS.
- K. E. Ayers, E. B. Anderson, C. B. Capuano, B. D. Carter, L. T. Dalton, G. Hanlon, J. Manco and M. Niedzwiecki, ECS Trans., 2010, 33, 3–15 CrossRef CAS.
- S. M. Saba, M. Muller, M. Robinius and D. Stolten, Int. J. Hydrogen Energy, 2018, 43, 1209–1223 CrossRef CAS.
- H. A. Miller, K. Bouzek, J. Hnat, S. Loos, C. I. Bernacker, T. Weibgarber, L. Rontzsch and J. Maeier-Haack, Sustainable Energy Fuels, 2020, 4, 2114–2133 RSC.
- J. Liu, M. R. Gerhardt, D. Li, M. Pak, Z. Kang, S. M. Alia, G. Bender, Y. S. Kim and A. Z. Weber, ECS Meet. Abstr., 2020, MA2020-02, 2443 CrossRef.
- J. J. Kaczur, H. Yang, Z. Liu, S. D. Sajjad and R. I. Masel, Front. Chem., 2018, 6, 263 CrossRef PubMed.
- J. T. Fan, A. G. Wright, B. Britton, T. Weissbach, T. J. G. Skalski, J. Ward, T. J. Peckham and S. Holdcroft, ACS Macro Lett., 2017, 6, 1089–1093 CrossRef CAS.
- E. J. Park, S. Maurya, M. R. Hibbs, C. H. Fujimoto, K. D. Kreuer and Y. S. Kim, Macromolecules, 2019, 52, 5419–5428 CrossRef CAS.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Am. Chem. Soc., 2017, 139, 2888–2891 CrossRef CAS PubMed.
- A. M. A. Mahmoud, A. M. M. Elsaghier, K. Otsuji and K. Miyatake, Macromolecules, 2017, 50, 4256–4266 CrossRef CAS.
- J. S. Olsson, T. H. Pham and P. Jannasch, Adv. Funct. Mater., 2018, 28, 1702758 CrossRef.
- W. H. Lee, E. J. Park, J. Han, D. W. Shin, Y. S. Kim and C. Bae, ACS Macro Lett., 2017, 6, 566–570 CrossRef CAS.
- N. Chen, C. Lu, Y. Li, C. Long and H. Zhu, J. Membr. Sci., 2019, 572, 246–254 CrossRef CAS.
- E. J. Park and Y. S. Kim, J. Mater. Chem. A, 2018, 6, 5456–15477 Search PubMed.
- J. Wang, Y. Zhao, B. P. Setzler, S. Rojas-Carbonell, C. B. Yehuda, A. Amel, M. Page, L. Wang, K. Hu, L. Shi, S. Gottesfeld, B. Xu and Y. Yan, Nat. Energy, 2019, 4, 392–398 CrossRef CAS.
- L. Liu, X. M. Chu, J. Y. Liao, Y. D. Huang, Y. Li, Z. Y. Ge, M. A. Hickner and N. W. Li, Energy Environ. Sci., 2018, 11, 435–446 RSC.
- A. M. A. Mahmoud and K. Miyatake, J. Mater. Chem. A, 2018, 6, 14400–14409 RSC.
- G. Gupta, K. Scott and M. Mamlouk, Fuel Cells, 2018, 18, 137–147 CrossRef CAS.
- E. J. Park, C. B. Capuano, K. E. Ayers and C. Bae, J. Power Sources, 2018, 375, 367–372 CrossRef CAS.
- X. Chu, Y. Shi, L. Liu, Y. Huang and N. Li, J. Mater. Chem. A, 2019, 7, 7717–7727 RSC.
- C. Fujimoto, D. S. Kim, M. Hibbs, D. Wrobleski and Y. S. Kim, J. Membr. Sci., 2012, 423-424, 438–449 CrossRef CAS.
- Y. K. Choe, C. Fujimoto, K. S. Lee, L. T. Dalton, K. Ayers, N. J. Henson and Y. S. Kim, Chem. Mater., 2014, 26, 5675–5682 CrossRef CAS.
- N. U. Hassan, M. Mandal, G. Huang, H. A. Firouzjaie, P. A. Kohl and W. E. Mustain, Adv. Energy Mater., 2020, 2001986 CrossRef.
- B. Motealleh, Z. Liu, R. I. Masel, J. P. Sculley, Z. R. Ni and L. Meroueh, Int. J. Hydrogen Energy, 2021, 46, 3379–3386 CrossRef CAS.
- S. Gottesfeld, D. R. Dekel, M. Page, C. Bae, Y. Yan, P. Zelenay and Y. S. Kim, J. Power Sources, 2018, 375, 170–184 CrossRef CAS.
- W. Mustain, M. Chatenet, M. Page and Y. S. Kim, Energy Environ. Sci., 2020, 13, 2805 RSC.
- D. R. Dekel, J. Power Sources, 2018, 375, 158–169 CrossRef CAS.
- E. S. Dvydova, F. D. Speck, M. T. Y. Paul, D. R. Dekel and S. Cherevko, ACS Catal., 2019, 9, 6837–6845 CrossRef.
- H. K. Hall, J. Am. Chem. Soc., 1957, 79, 5441–5444 CrossRef CAS.
- M. Mandal, ChemElectroChem, 2020, 7, 1–11 CrossRef.
- K. Zeng and D. K. Zhang, Prog. Energy Combust. Sci., 2010, 36, 307–326 CrossRef CAS.
- R. Subbaraman, D. Tripkovic, D. Strmcnik, K. C. Chang, M. Uchimura, A. P. Paulikas, V. Stamenkovic and N. M. Markovic, Science, 2011, 334, 1256–1260 CrossRef CAS PubMed.
- N. Danilovic, R. Subbaraman, D. Strmcnik, K. C. Chang, A. P. Paulikas, V. R. Stamenkovic and N. M. Markovic, Angew. Chem., Int. Ed., 2012, 51, 12495–12498 CrossRef CAS PubMed.
- D. Strmcnik, P. P. Lopes, B. Genorio, V. R. Stamenkovic and N. M. Markovic, Nano Energy, 2016, 29, 29–36 CrossRef CAS.
- J. Nash, J. Zheng, Y. Wang, B. J. Xu and Y. S. Yan, J. Electrochem. Soc., 2018, 165, J3378–J3383 CrossRef CAS.
- C. Q. Li and J. B. Baek, ACS Omega, 2020, 5, 31–40 CrossRef CAS PubMed.
- L. Trotochaud, S. L. Young, J. K. Ranney and S. W. Boettcher, J. Am. Chem. Soc., 2014, 136, 6744–6753 CrossRef CAS PubMed.
- M. B. Stevens, C. D. M. Trang, L. J. Enman, J. Deng and S. W. Boettcher, J. Am. Chem. Soc., 2017, 139, 11361–11364 CrossRef CAS PubMed.
- J. S. Kim, B. Kim, H. Kim and K. Kang, Adv. Energy Mater., 2018, 8, 1702774 CrossRef.
- B. R. Wygant, K. Kawashima and C. B. Mullins, ACS Energy Lett., 2018, 3, 2956–2966 CrossRef CAS.
- D. Y. Chung, P. P. Lopes, P. F. B. D. Martins, H. Y. He, T. Kawaguchi, P. Zapol, H. D. You, D. Tripkovic, D. Strmcnik, Y. S. Zhu, S. Seifert, S. S. Lee, V. R. Stamenkovic and N. M. Markovic, Nat. Energy, 2020, 5, 222–230 CrossRef.
- R. Subbaraman, D. Tripkovic, K. C. Chang, D. Strmcnik, A. P. Paulikas, P. Hirunsit, M. Chan, J. Greeley, V. Stamenkovic and N. M. Markovic, Nat. Mater., 2012, 11, 550–557 CrossRef CAS PubMed.
- E. Fabbri, A. Habereder, K. Waltar, R. Kotz and T. J. Schmidt, Catal. Sci. Technol., 2014, 4, 3800–3821 RSC.
- W. T. Hong, M. Risch, K. A. Stoerzinger, A. Grimaud, J. Suntivich and Y. Shao-Horn, Energy Environ. Sci., 2015, 8, 1404–1427 RSC.
- A. Grimaud, K. J. May, C. E. Carlton, Y. L. Lee, M. Risch, W. T. Hong, J. G. Zhou and Y. Shao-Horn, Nat. Commun., 2013, 4, 2439 CrossRef PubMed.
- E. Fabbri, M. Nachtegaal, T. Binninger, X. Cheng, B. J. Kim, J. Durst, F. Bozza, T. Graule, R. Schaublin, L. Wiles, M. Pertoso, N. Danilovic, K. E. Ayers and T. J. Schmidt, Nat. Mater., 2017, 16, 925–931 CrossRef CAS PubMed.
- W. J. Yin, B. C. Weng, J. Ge, Q. D. Sun, Z. Z. Li and Y. F. Yan, Energy Environ. Sci., 2019, 12, 442–462 RSC.
- M. I. Jamesh and X. M. Sun, J. Power Sources, 2018, 400, 31–68 CrossRef CAS.
- Y. S. Kim, ACS Appl. Polym. Mater., 2021, 3(3), 1250–1270 CrossRef CAS.
- X. Xu and K. Scott, J. Mater. Chem., 2011, 21, 12344–12351 RSC.
- K. E. Ayers, E. B. Anderson, C. B. Capuano, M. Niedzweicki, M. Hickner, C.-Y. Wang, Y. Len and W. Zhou, ECS Trans., 2013, 45, 121–130 CrossRef.
- J. Parrondo, C. G. Arges, M. Niedzwiecki, E. B. Anderson, K. Ayers and V. Ramani, RSC Adv., 2014, 4, 9875–9879 RSC.
- J. Parrondo, M. George, C. Capuano, K. E. Ayers and V. Ramani, J. Mater. Chem. A, 2015, 3, 10819–10828 RSC.
- H. T. Chung, presented in part at the 2020 Annual Merit Review Proceedings, Washington, DC, 2020.
- D. Li, E. J. Park, W. Zhu, Q. Shi, Y. Zhou, H. Tian, Y. Lin, A. Serov, B. Zulevi, E. D. Baca, C. Fujimoto, H. T. Chung and Y. S. Kim, Nat. Energy, 2020, 5, 378–385 CrossRef CAS.
- J. Xiao, A. M. Oliveira, L. Wang, Y. Zhao, T. Wang, J. Wang, B. P. Setzler and Y. Yan, ACS Catal., 2021, 11, 264–270 CrossRef CAS.
- L. Xiao, S. Zhang, J. Pan, C. X. Yang, M. L. He, L. Zhuang and J. T. Lu, Energy Environ. Sci., 2012, 5, 7869–7871 RSC.
- P. Thangavel, M. Ha, S. Kumaraguru, A. Meena, A. N. Singh, A. M. Harzandi and K. S. Kim, Energy Environ. Sci., 2020, 13, 3447–3458 RSC.
- M. S. Cha, J. E. Park, S. Kim, S.-H. Han, S.-H. Shin, S. H. Yang, T.-H. Kim, D. M. Yu, S. So, Y.-T. Hong, S. J. Yoon, S.-G. Oh, Y. K. Sun, O.-H. Kim, H. S. Park, B. Bae, Y.-E. Sung, Y.-H. Cho and J. Y. Lee, Energy Environ. Sci., 2020, 13, 3633–3645 RSC.
- S. H. Ahn, B.-S. Lee, I. Choi, S. J. Yoo, H.-J. Kim, E. Cho, D. Henkensmeier, S. W. Nam, S.-K. Kim and J. H. Jang, Appl. Catal., B, 2014, 154-155, 197–205 CrossRef CAS.
- Z. Liu, S. D. Sajjad, Y. Gao, J. J. Kaczur and R. I. Masel, ECS Trans., 2017, 77, 71–73 CrossRef CAS.
- L. Wang, T. Weissbach, R. Reissner, A. Ansar, A. S. Gago, S. Holdcroft and K. A. Friedrich, ACS Appl. Energy Mater., 2019, 2, 7903–7912 CrossRef CAS.
- M. R. Kraglund, M. Carmo, G. Schiller, S. A. Ansar, D. Aili, E. Christensen and J. O. Jensen, Energy Environ. Sci., 2019, 12, 3313–3318 RSC.
- P. Chen and X. Hu, Adv. Energy Mater., 2020, 10, 202285 Search PubMed.
- M. Faraj, M. Boccia, H. Miller, F. Martini, S. Borsacchi, M. Geppi and A. Pucci, Int. J. Hydrogen Energy, 2012, 37, 14992–15002 CrossRef CAS.
- C. C. Pavel, F. Cecconi, C. Emiliani, S. Santiccioli, A. Scaffidi, S. Catanorchi and M. Comotti, Angew. Chem., Int. Ed., 2014, 53, 1378–1381 CrossRef CAS.
- H. Ito, N. Kawaguchi, S. Someya, T. Munakata, N. Miyazaki, M. Ishiba and A. Nakano, Int. J. Hydrogen Energy, 2018, 43, 17030–17039 CrossRef CAS.
- N. Ziv, A. N. Mondal, T. Weissbach, S. Holdcroft and D. R. Dekel, J. Membr. Sci., 2019, 586, 140–150 CrossRef CAS.
- A. G. Divekar, A. C. Yang-Neyerlin, C. M. Antunes, D. J. Strasser, A. R. Motz, S. S. Seifert, X. B. Zuo, B. S. Pivovar and A. M. Herring, Sustainable Energy Fuels, 2020, 4, 1801–1811 RSC.
- Y. Leng, G. chen, A. J. Mendoza, T. B. Tighe, M. A. Hickner and C.-Y. Wang, J. Am. Chem. Soc., 2012, 134, 9054–9057 CrossRef CAS PubMed.
- H. Xu, 2020 ARPA-E REFUEL Virtual Annual Program Review Meeting, 2020.
- D. Aili, M. R. Kraglund, J. Tavacoli, C. Chatzichristodoulou and J. O. Jensen, J. Membr. Sci., 2020, 598, 117674 CrossRef CAS.
- A. N. Colli, H. H. Girault and A. Battistel, Materials, 2019, 12, 1336 CrossRef CAS.
- A. K. Niaz, A. Akhtar, J.-Y. Park and H.-T. Lim, J. Power Sources, 2021, 481, 229093 CrossRef CAS.
- D. Y. Xu, M. B. Stevens, M. R. Cosby, S. Z. Oener, A. M. Smith, L. J. Enman, K. E. Ayers, C. B. Capuano, J. N. Renner, N. Danilovic, Y. G. Li, H. Z. Wang, Q. H. Zhang and S. W. Boettcher, ACS Catal., 2019, 9, 7–15 CrossRef CAS.
- J. E. Park, S. Y. Kang, S. H. Oh, J. K. Kim, M. S. Lim, C. Y. Ahn, Y. H. Cho and Y. E. Sung, Electrochim. Acta, 2019, 295, 99–106 CrossRef CAS.
- T. Nakagawa, H. Matsushima, M. Ueda and H. Ito, ECS Meeting Abstract, 2020, MA2020-01, 984 CrossRef.
- D. P. Davies, P. L. Adcock, M. Turpin and S. J. Rowen, J. Power Sources, 2000, 86, 237–242 CrossRef CAS.
- Q. Xu, S. Z. Oener, G. Lindquist, H. Jiang, C. Li and S. W. Boettcher, ACS Energy Lett., 2021, 6, 305–312 CrossRef CAS.
- S. Seetharaman, R. Balaji, K. Ramya, K. S. Dhathathreyan and M. Velan, Int. J. Hydrogen Energy, 2013, 38, 14934–14942 CrossRef CAS.
- F. Arbabi, A. Kalantarian, R. Abouatallah, R. Wang, J. S. Wallace and A. Bazylak, J. Power Sources, 2014, 258, 142–149 CrossRef CAS.
- G. C. Chen, T. H. Wondimu, H. C. Huang, K. C. Wang and C. H. Wang, Int. J. Hydrogen Energy, 2019, 44, 10174–10181 CrossRef CAS.
- Z. Y. Kang, J. K. Mo, G. Q. Yang, S. T. Retterer, D. A. Cullen, T. J. Toops, J. B. Green, M. M. Mench and F. Y. Zhang, Energy Environ. Sci., 2017, 10, 166–175 RSC.
- X. Wu and K. Scott, Int. J. Hydrogen Energy, 2013, 38, 3123–3129 CrossRef CAS.
- Y. S. Park, J. Yang, J. Lee, M. J. Jang, J. Jeong, W. S. Choi, Y. Kim, Y. D. Yin, M. H. Seo, Z. W. Chen and S. M. Choi, Appl. Catal., B, 2020, 278, 119276 CrossRef CAS.
- Z. Y. Kang, G. Q. Yang, J. K. Mo, Y. F. Li, S. L. Yu, D. A. Cullen, S. T. Retterer, T. J. Toops, G. Bender, B. S. Pivovar, J. B. Green and F. Y. Zhang, Nano Energy, 2018, 47, 434–441 CrossRef CAS.
- T. Pandiarajan, L. John Berchmans and S. Ravichandran, RSC Adv., 2015, 5, 34100–34108 RSC.
- M. Schalenbach, W. Lueke and D. Stolten, J. Electrochem. Soc., 2016, 163, F1480–F1488 CrossRef CAS.
- D. S. Kim, C. Welch, R. P. Hjelm, Y. S. Kim and M. D. Guiver, in Polymer Science: A Comprehensive Reference, ed. M. Moeller and K. Matyjaszewski, Elsevier, 2012, vol. 10, pp. 691–720 Search PubMed.
- L. Zhang, C. S. Ma and S. Mukerjee, Electrochem. Acta, 2003, 48, 1845–1859 CrossRef CAS.
- Y. S. Kim and B. S. Pivovar, J. Electrochem. Soc., 2010, 157, B1616 CrossRef CAS.
- L. Zhu, J. Pan, Y. Wang, J. Han, L. Zhuang and M. A. Hickner, Macromolecules, 2016, 2016, 815–824 CrossRef.
- L. Li, C. X. Lin, X. Q. Wang, Q. Yang, Q. G. Zhang, A. M. Zhu and Q. L. Liu, J. Membr. Sci., 2018, 553, 209–217 CrossRef CAS.
- N. Chen, C. Lu, Y. Li, C. Long, Z. Li and H. Zhu, J. Membr. Sci., 2019, 588, 17120 Search PubMed.
- J. Pan, C. Chen, Y. Li, L. Wang, L. Tan, G. Li, X. Tang, L. Xiao, J. Lu and L. Zhuang, Energy Environ. Sci., 2013, 7, 354–360 RSC.
- E. J. Park, S. Maurya, U. Martinez, Y. S. Kim and R. Mukundan, J. Membr. Sci., 2021, 617, 118565 CrossRef CAS.
- L. Zhu, T. J. Zimudzi, Y. Wang, X. Yu, J. Pan, J. Han, D. I. Kushner, L. Zhuang and M. A. Hickner, Macromolecules, 2017, 50, 2329–2337 CrossRef CAS.
- X. L. Gao, L. X. Sun, H. Y. Wu, Z. Y. Zhu, N. Xiao, J. H. Chen, Q. Yang, Q. G. Zhang, A. M. Zhu and Q. L. Liu, J. Mater. Chem. A, 2020, 8, 13065–13076 RSC.
- S.-B. Lee, C.-M. Min, J. Jang and J.-S. Lee, Polymer, 2020, 192, 122331 CrossRef.
- Y. S. Kim, C. F. Welch, R. P. Hjelm, N. H. Mack, A. Labouriau and E. B. Orler, Macromolecules, 2015, 48, 2161–2172 CrossRef CAS.
- Y. S. Kim, C. F. Welch, N. H. Mack, R. P. Hjelm, E. B. Orler, M. E. Hawley, K. S. Lee, S.-D. Yim and C. M. Johnston, Phys. Chem. Chem. Phys., 2014, 16, 5927–5932 RSC.
- D. Li, I. Matanovic, A. S. Lee, E. J. Park, C. Fujimoto, H. T. Chung and Y. S. Kim, ACS Appl. Mater. Interfaces, 2019, 11, 9696–9701 CrossRef CAS PubMed.
- S. Maurya, A. S. Lee, D. Li, E. J. Park, D. P. Leonard, S. Noh, C. Bae and Y. S. Kim, J. Power Sources, 2019, 436, 226866 CrossRef CAS.
- D. Li, H. T. Chung, S. Maurya, I. Matanovic and Y. S. Kim, Curr. Opin. Electrochem., 2018, 12, 189–195 CrossRef CAS.
- I. Matanovic, S. Maurya, E. J. Park, J. Y. Jeon, C. Bae and Y. S. Kim, Chem. Mater., 2019, 31, 4195–4204 CrossRef CAS.
- M. Jonsson, J. Lind and G. Merenyi, J. Phys. Chem. A, 2002, 106, 4758–4762 CrossRef CAS.
- H. T. Chung, presented in part at the 2019 Annual Merit Review Proceedings, Washington, DC, 2019.
- W. Liu, J. Carrasco, B. Santra, A. Michaelides, M. Scheffler and A. Tkatchenko, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 245405 CrossRef.
- S. Lehwald, H. Ibach and J. E. Demuth, Surf. Sci., 1978, 78, 577–590 CrossRef CAS.
- I. Matanovic, H. T. Chung and Y. S. Kim, J. Phys. Chem. Lett., 2017, 8, 4918–4924 CrossRef CAS PubMed.
- B. H. Han, A. Grimaud, L. Giordano, W. T. Hong, O. Diaz-Morales, L. Yueh-Lin, J. Hwang, N. Charles, K. A. Stoerzinger, W. Yang, M. T. M. Koper and Y. Shao-Horn, J. Phys. Chem. C, 2018, 122, 8445–8454 CrossRef CAS.
- B. H. Han, M. Risch, Y. L. Lee, C. Ling, H. F. Jia and Y. Shao-Horn, Phys. Chem. Chem. Phys., 2015, 17, 22576–22580 RSC.
- H. Liu, J. Yu, J. Sunarso, C. Zhou, B. Liu, Y. Shen, W. Zhou and Z. Shao, Electrochem. Acta, 2018, 282, 324–330 CrossRef CAS.
- S. Maurya, C. H. Fujimoto, M. R. Hibbs, C. N. Vilarrubia and Y. S. Kim, Chem. Mater., 2018, 30, 2188–2192 CrossRef CAS.
- S. Maurya, S. Noh, I. Matanovic, C. H. Park, C. N. Vilarrubia, U. Martinez, J. Han, C. Bae and Y. S. Kim, Energy Environ. Sci., 2018, 11, 3283–3291 RSC.
- R. Soni, S. Miyanishi, H. Kuroki and T. Yamaguchi, ACS Appl. Energy Mater., 2020, 4, 1053–1058 CrossRef.
- M. S. Ide, B. Hao, M. Neurock and R. J. Davis, ACS Catal., 2012, 2, 671–683 CrossRef CAS.
- B. Yoon, H. B. Pan and C. M. Wai, J. Phys. Chem. C, 2009, 113, 1520–1525 CrossRef CAS.
- K. M. Bratlie, H. Lee, K. Komvopoulos, P. D. Yang and G. A. Somorjai, Nano Lett., 2007, 7, 3097–3101 CrossRef CAS PubMed.
- S. Maurya, J. H. Dumont, C. N. Villarrubia, I. Matanovic, D. G. Li, Y. S. Kim, S. Noh, J. Y. Han, C. Bae, H. A. Miller, C. H. Fujimoto and D. R. Dekel, ACS Catal., 2018, 8, 9429–9439 CrossRef CAS.
- T. J. Schmidt, P. N. Ross and N. M. Markovic, J. Electroanal. Chem., 2002, 524, 252–260 CrossRef.
- H. T. Chung, U. Martinez, I. Matanovic and Y. S. Kim, J. Phys. Chem. Lett., 2016, 7, 4464–4469 CrossRef CAS PubMed.
- J. H. Dumont, A. J. Spears, R. P. Hjelm, M. Hawley, S. Maurya, D. G. Li, G. C. Yuan, E. B. Watkins and Y. S. Kim, ACS Appl. Mater. Interfaces, 2020, 12, 1825–1831 CrossRef CAS PubMed.
- Z. Liu, S. D. Sajjad, Y. Gao, H. Yang, J. J. Kaczur and R. I. Masel, Int. J. Hydrogen Energy, 2017, 42, 29661–29665 CrossRef CAS.
- J. Hnat, M. Paidar, J. Schauer and K. Bouzek, Int. J. Hydrogen Energy, 2014, 39, 4779–4787 CrossRef CAS.
- C. G. Arges and V. Ramani, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 2490–2495 CrossRef CAS PubMed.
- A. D. Mohanty, S. E. Tignor, J. A. Krause, Y. K. Choe and C. Bae, Macromolecules, 2016, 49, 3361–3372 CrossRef CAS.
- T. H. Pham, A. Allushi, J. S. Olsson and P. Jannasch, Polym. Chem., 2020, 11, 6953–6963 RSC.
- J. H. Dong, N. Yu, X. F. Che, R. H. Liu, D. Aili and J. S. Yang, Polym. Chem., 2020, 11, 6037–6046 RSC.
- H. Kuroki, S. Miyanishi, A. Sakakibara, Y. Oshiba and T. Yamaguchi, J. Power Sources, 2019, 438, 226997 CrossRef CAS.
- C. Fujimoto, E. Sorte, N. Bell, C. Poirier, E. J. Park, S. Maurya, K. S. Lee and Y. S. Kim, Polymer, 2018, 158, 190–197 CrossRef CAS.
- M. R. Hibbs, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 1736–1742 CrossRef CAS.
- Z. Zhang, L. Wu, J. R. Varcoe, C. Li, A. L. Ong, S. Poynton and T. Xu, J. Mater. Chem. A, 2013, 1, 2595–2601 RSC.
- A. D. Mohanty and C. Bae, J. Mater. Chem. A, 2014, 2, 17314–17320 RSC.
- M. G. Marino and K. D. Kreuer, ChemSusChem, 2015, 8, 513–523 CrossRef CAS PubMed.
- A. G. Wright, T. Weissbach and S. Holdcroft, Angew. Chem., Int. Ed., 2016, 55, 4818–4821 CrossRef CAS PubMed.
- K. M. Hugar, H. A. Kostalik and G. W. Coates, J. Am. Chem. Soc., 2015, 137, 8730–8737 CrossRef CAS PubMed.
- Y. S. Park, J. H. Lee, M. J. Jang, J. Jeong, S. M. Park, W.-S. Choi, Y. Kim, J. Yang and S. M. Choi, Int. J. Hydrogen Energy, 2020, 45, 36–45 CrossRef CAS.
- A. G. Wright, J. T. Fan, B. Britton, T. Weissbach, H. F. Lee, E. A. Kitching, T. J. Peckham and S. Holdcroft, Energy Environ. Sci., 2016, 9, 2130–2142 RSC.
- H.-S. Dang and P. Jannasch, Macromolecules, 2015, 48, 5742–5751 CrossRef CAS.
- S. A. Nunez, C. Capparelli and M. A. Hickner, Chem. Mater., 2016, 28, 2589–2598 CrossRef CAS.
- J. Fan, S. Willdorf-Cohen, E. M. Schibli, Z. Paula, W. Li, T. J. G. Skalski, A. T. Sergeenko, A. Hohenadel, B. J. Frisken, E. Magliocca, W. E. Mustain, C. E. Diesendruck, D. R. Dekel and S. Holdcroft, Nat. Commun., 2019, 10, 2306 CrossRef PubMed.
- X. Chu, L. Liu, Y. Huang, M. D. Guiver and N. Li, J. Membr. Sci., 2019, 578, 239–250 CrossRef CAS.
- X. Hu, Y. Huang, L. Liu, Q. Ju, X. Zhou, X. Qiao, Z. Zheng and N. Li, J. Membr. Sci., 2021, 621, 118964 CrossRef CAS.
- D. Aili, M. K. Hansen, R. F. Renzaho, Q. Li, E. Christensen, J. O. Jensen and N. J. Bjerrum, J. Membr. Sci., 2013, 447, 424–432 CrossRef CAS.
- O. D. Thomas, K. J. W. Y. Soo, T. J. Peckham, M. P. Kulkarni and S. Holdcroft, J. Am. Chem. Soc., 2012, 134, 10753–10756 CrossRef CAS PubMed.
- D. Henkensmeier, H. R. Cho, H. J. Kim, C. N. Kirchner, J. Leppin, A. Dyck, J. H. Jang, E. Cho, S. W. Nam and T. H. Lim, Polym. Degrad. Stab., 2012, 97, 264–272 CrossRef CAS.
- M. R. Kraglund, D. Aili, K. Jankova, E. Christensen, Q. Li and J. O. Jensen, J. Electrochem. Soc., 2016, 163, F3125–F3131 CrossRef CAS.
- D. Aili, A. G. Wright, M. R. Kraglund, K. Jankova, S. Holdcroft and J. O. Jensen, J. Mater. Chem., 2017, 5, 5055–5066 RSC.
- A. B. LaConti, M. Hamdan and R. C. McDonald, in Handbook of Fuel Cells – Fundamentals, Technology and Applications, Volume 3: Fuel Cell Technology and Applications, ed. W. Vielstich, H. A. Gasteiger and A. Lamm, Wiley, Chichester, UK, 2003, vol. 3 Search PubMed.
- F. D. Speck, P. G. Santori, F. Jaouen and S. Cherevko, J. Phys. Chem. C, 2019, 123, 25267–25277 CrossRef CAS.
- G. Wu, N. Li, D.-R. Zhou, K. Mitsuo and B.-Q. Xu, J. Solid State Chem., 2004, 177, 3682–3692 CrossRef CAS.
- K. Juodkazis, J. Juodkazyte, R. Vilkauskaite and V. Jasulaitiene, J. Solid State Electrochem., 2008, 12, 1469–1479 CrossRef CAS.
- S. Loos, I. Zaharieva, P. Chernev, A. Lißner and H. Dau, ChemSusChem, 2019, 12, 1966–1976 CrossRef CAS PubMed.
- S. Wierzbicki, J. C. Douglin, A. Kostuch, D. R. Dekel and K. Kruczala, J. Phys. Chem. Lett., 2020, 11, 7630–7636 CrossRef PubMed.
- J. Parrondo, Z. Wang, M.-S. J. Jung and V. Ramani, Phys. Chem. Chem. Phys., 2016, 18, 19705–19712 RSC.
- H. Kaczmarek, L. A. Linden and J. F. Rabek, Polym. Degrad. Stab., 1995, 47, 175–188 CrossRef CAS.
- R. Espiritu, B. T. Golding, K. Scott and M. Mamlouk, J. Power Sources, 2018, 375, 373–386 CrossRef CAS.
- R. Borup, J. Meyers, B. S. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. L. More, K. Stroh, T. A. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K. I. Kimijima and N. Iwashita, Chem. Rev., 2007, 107, 3904–3951 CrossRef CAS PubMed.
- G. Hubner and E. Roduner, J. Mater. Chem., 1999, 9, 409–418 RSC.
- Y.-F. He, R.-M. Wang, Y.-Y. Liu, Y. Chang, Y.-P. Wang, C.-G. Xia and J.-S. Suo, J. Mol. Catal. A: Chem., 2000, 159, 109–113 CrossRef CAS.
- R. Espiritu, B. T. Golding, K. Scott and M. Mamlouk, J. Mater. Chem. A, 2017, 5, 1248–1267 RSC.
- I. Di Somma, R. Andreozzi, M. Canterino, V. Caprio and R. Sanchirico, AIChE J., 2008, 54, 1579–1584 CrossRef CAS.
- N. Ye, Y. Xu, D. Zhang, J. Yang and R. He, Polym. Degrad. Stab., 2018, 153, 298–306 CrossRef CAS.
- M. Adamski, T. J. G. Skalski, B. Britton, T. J. Peckham, L. Metzler and S. Holdcroft, Angew. Chem., Int. Ed., 2017, 56, 9058–9061 CrossRef CAS PubMed.
- S. Zhang, X. L. Zhu and C. H. Jin, J. Mater. Chem. A, 2019, 7, 6883–6893 RSC.
- B. Hu, L. Y. Miao, Y. X. Zhao and C. L. Lu, J. Membr. Sci., 2017, 530, 84–94 CrossRef CAS.
- J. K. Hao, X. Q. Gao, Y. Y. Jiang, H. J. Zhang, J. S. Luo, Z. G. Shao and B. L. Yi, J. Membr. Sci., 2018, 551, 66–75 CrossRef CAS.
- Q. Q. Ge, J. Ran, J. B. Miao, Z. J. Yang and T. W. Xu, ACS Appl. Mater. Interfaces, 2015, 7, 28545–58553 CrossRef CAS PubMed.
- N. W. Li, M. D. Guiver and W. H. Binder, ChemSusChem, 2013, 6, 1376–1383 CrossRef CAS PubMed.
- L. Zhu, T. J. Zimudzi, N. Li, J. Pan, B. Lin and M. A. Hickner, Polym. Chem., 2016, 7, 2464–2475 RSC.
- Y. B. He, L. Wu, J. F. Pan, Y. Zhu, X. L. Ge, Z. J. Yang, J. Ran and T. W. Xu, J. Membr. Sci., 2016, 504, 47–54 CrossRef CAS.
- C. X. Lin, Y. Z. Zhuo, E. N. Hu, Q. G. Zhang, A. M. Zhu and Q. L. Liu, J. Membr. Sci., 2017, 539, 23–33 Search PubMed.
- J. Y. Jeon, S. Park, J. Han, S. Maurya, A. D. Mohanty, D. Tian, N. Saikia, M. Hickner, C. Y. Ryu, M. E. Tuckerman, S. J. Paddison, Y. S. Kim and C. Bae, Macromolecules, 2019, 52, 2139–2147 CrossRef CAS.
- M. M. Hossain, L. Wu, X. Liang, Z. J. Yang, J. Hou and T. Xu, J. Power Sources, 2018, 390, 234–241 CrossRef CAS.
- S. Miyanishi and T. Yamaguchi, Polym. Chem., 2020, 11, 3812–3820 RSC.
- Q. Y. Wang, Y. Gao, Z. Y. Ma, Y. Zhang, W. P. Ni, H. A. Younus, C. Zhang, Z. J. Chen and S. G. Zhang, J. Energy Chem., 2021, 54, 342–351 CrossRef.
- P. Paciok, M. Schalenbach, M. Carmo and D. Stolten, J. Power Sources, 2017, 365, 53–60 CrossRef CAS.
- H. Jin, B. Ruqia, Y. S. Park, H. J. Kim, H. S. Oh, S. I. Choi and K. Lee, Adv. Energy Mater., 2021, 11, 2003188 CrossRef CAS.
- J. Staszak-Jirkovsky, C. D. Malliakas, P. P. Lopes, N. Danilovic, S. S. Kota, K. C. Chang, B. Genorio, D. Strmcnik, V. R. Stamenkovic, M. G. Kanatzidis and N. M. Markovic, Nat. Mater., 2016, 15, 197–203 CrossRef CAS PubMed.
- Y. Zhang, L. Gao, E. J. M. Hensen and J. P. Hofmann, ACS Energy Lett., 2018, 3, 1360–1365 CrossRef CAS PubMed.
- S. Cherevko, A. R. Zeradjanin, G. P. Keeley and K. J. J. Mayrhofer, J. Electrochem. Soc., 2014, 161(12), H822–H830 CrossRef.
- M. Schalenbach, O. Kasian, M. Ledendecker, F. Speck, A. Mingers, K. Mayrhofer and S. Cherevko, Electrocatalysis, 2018, 9, 153–161 CrossRef CAS.
- S. Cherevko, S. Geiger, O. Kasian, N. Kulyk, J. P. Grote, A. Savan, B. R. Shrestha, S. Merzlikin, B. Breitbach, A. Ludwig and K. J. J. Mayrhofer, Catal. Today, 2016, 262, 170–180 CrossRef CAS.
- C. Tang, R. Zhang, W. B. Lu, L. B. He, X. Jiang, A. M. Asiri and X. P. Sun, Adv. Mater., 2017, 29, 1602441 CrossRef PubMed.
- H. Osgood, S. V. Devaguptapu, H. Xu, J. Cho and G. Wu, Nano Today, 2016, 11, 601–625 CrossRef CAS.
- J. W. D. Ng, M. Garcia-Melchor, M. Bajdich, P. Chakthranont, C. Kirk, A. Vojvodic and T. F. Jaramillo, Nat. Energy, 2016, 1, 16053 CrossRef CAS.
- T. T. Liu, A. M. Asiri and X. P. Sun, Nanoscale, 2016, 8, 3911–3915 RSC.
- C. Guan, W. Xiao, H. J. Wu, X. M. Liu, W. J. Zang, H. Zhang, J. Ding, Y. P. Feng, S. J. Pennycook and J. Wang, Nano Energy, 2018, 48, 73–80 CrossRef.
- F. Yu, H. Q. Zhou, Z. Zhu, J. Y. Sun, R. He, J. M. Bao, S. Chen and Z. F. Ren, ACS Catal., 2017, 7, 2052–2057 CrossRef CAS.
- K. W. Liu, C. L. Zhang, Y. D. Sun, G. H. Zhang, X. C. Shen, F. Zou, H. C. Zhang, Z. W. Wu, E. C. Wegener, C. J. Taubert, J. T. Miller, Z. M. Peng and Y. Zhu, ACS Nano, 2018, 12, 158–167 CrossRef CAS PubMed.
- P. Z. Chen, K. Xu, Z. W. Fang, Y. Tong, J. C. Wu, X. L. Lu, X. Peng, H. Ding, C. Z. Wu and Y. Xie, Angew. Chem., Int. Ed., 2015, 54, 14710–14714 CrossRef CAS PubMed.
- J. Mahmood, M. A. R. Anjum, S. H. Shin, I. Ahmad, H. J. Noh, S. J. Kim, H. Y. Jeong, J. S. Lee and J. B. Baek, Adv. Mater., 2018, 30, 1805606 CrossRef PubMed.
- L. H. Ai, T. Tian and J. Jiang, ACS Sustainable Chem. Eng., 2017, 5, 4771–4777 CrossRef CAS.
- Y. Y. Feng, H. J. Zhang, L. Fang, Y. P. Mu and Y. Wang, ACS Catal., 2016, 6, 4477–4485 CrossRef CAS.
- L. Du, L. L. Luo, Z. X. Feng, M. Engelhard, X. H. Xie, B. H. Han, J. M. Sun, J. H. Zhang, G. P. Yin, C. M. Wang, Y. Wang and Y. Y. Shao, Nano Energy, 2017, 39, 245–252 CrossRef CAS.
- O. Mabayoje, A. Shoola, B. R. Wygant and C. B. Mullins, ACS Energy Lett., 2016, 1, 195–201 CrossRef CAS.
- W. Chen, H. T. Wang, Y. Z. Li, Y. Y. Liu, J. Sun, S. H. Lee, J. S. Lee and Y. Cui, ACS Central Sci., 2015, 1, 244–251 CrossRef CAS PubMed.
- X. Xu, F. Song and X. L. Hu, Nat. Commun., 2016, 7, 12324 CrossRef CAS PubMed.
- L. Guo, F. Luo, F. Guo, Q. Zhang, K. G. Qu, Z. H. Yang and W. W. Cai, Chem. Commun., 2019, 55, 7623–7626 RSC.
- J. Chi, H. M. Yu, G. F. Li, L. Fu, J. Jia, X. Q. Gao, B. L. Yi and Z. G. Shao, RSC Adv., 2016, 6, 90397–90400 RSC.
- D. Pletcher, X. H. Li and S. P. Wang, Int. J. Hydrogen Energy, 2012, 37, 7429–7435 CrossRef CAS.
- Y. K. Li, G. Zhang, H. Huang, W. T. Lu, F. F. Cao and Z. G. Shao, Small, 2020, 16, 2005184 CrossRef CAS PubMed.
- A. Loh, X. H. Li, O. O. Taiwo, F. Tariq, N. P. Brandon, P. C. Wang, K. Xu and B. G. Wang, Int. J. Hydrogen Energy, 2020, 45, 24232–24247 CrossRef CAS.
- I. Vincent, E. C. Lee and H. M. Kim, RSC Adv., 2020, 10, 37429–37438 RSC.
- J. Renner, K. Ayers and E. Anderson, in PEM Electrolysis for Hydrogen Production, ed. D. Bessarabov, H. Wang, H. Li and M. Zhao, CRC Press, New York, 2015, ch. 9 Search PubMed.
- H. Ito, N. Kawaguchi, S. Someya and T. Munakata, Electrochim. Acta, 2019, 297, 188–196 CrossRef CAS.
- U. Babic, M. Suermann, F. N. Buchi, L. Gubler and T. J. Schmidt, J. Electrochem. Soc., 2017, 164, F387–F399 CrossRef CAS.
- A. Albert, A. O. Barnett, M. S. Thomassen, T. J. Schmidt and L. Gubler, ACS Appl. Mater. Interfaces, 2015, 7, 22203–22212 CrossRef CAS PubMed.
- D. S. Kim, C. Fujimoto, M. R. Hibbs, A. Labouriau, Y.-K. Choe and Y. S. Kim, Macromolecules, 2013, 46, 7826–7833 CrossRef CAS.
- G. Huang, M. Mandal, X. Peng, A. C. Yang-Neyerlin, B. S. Pivovar, W. E. Mustain and P. A. Kohl, J. Electrochem. Soc., 2019, 166, F637–F644 CrossRef CAS.
- M. Y. Niu, C. M. Zhang, G. H. He, F. X. Zhang and X. M. Wu, Int. J. Hydrogen Energy, 2019, 44, 15482–15493 CrossRef CAS.
- A. M. Park, R. J. Wycisk, X. M. Ren, F. E. Turley and P. N. Pintaouro, J. Mater. Chem. A, 2016, 4, 132–141 RSC.
- X. M. Du, Z. Wang, H. Y. Zhang, W. C. Liu, Z. Y. Chen and J. M. Xu, J. Membr. Sci., 2020, 587, 117178 CrossRef.
- Z. H. Mo, R. Yang, S. Hong and Y. X. Wu, Int. J. Hydrogen Energy, 2018, 43, 1790–1804 CrossRef CAS.
- X. Luo, D. I. Kushner, J. Li, E. J. Park, Y. S. Kim and A. Kusoglu, Adv. Funct. Mater., 2021, 2008778 CrossRef CAS.
- X. D. Liu, X. H. Luo, X. H. Chen, S. B. Zou, X. Liu, J. J. Li, H. Li and D. H. Dong, J. Electroanal. Chem., 2020, 871, 114283 CrossRef CAS.
- R. K. Ahluwalia, X. Wang and A. J. Steinbach, J. Power Sources, 2016, 309, 178–191 CrossRef CAS.
- D. Henkensmeier, M. Najibah, C. Harms, J. Žitka, J. Hnát and K. Bouzek, J. Electrochem. Energy Convers. Storage, 2020, 18, 024001 CrossRef.
- A. Carbone, S. Campagna Zignani, I. Gatto, S. Trocino and A. S. Aricò, Int. J. Hydrogen Energy, 2020, 45, 9285–9292 CrossRef CAS.
- W. You, E. Padgett, S. N. MacMillan, D. A. Muller and G. W. Coates, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 9726–9734 Search PubMed.
- M. Mandal, G. Huang, N. Ul Hassan, W. E. Mustain and P. A. Kohl, J. Mater. Chem. A, 2020, 8, 17568–17578 RSC.
- K. M. Meek, C. M. Reed, B. Pivovar, K. D. Kreuer, J. R. Varcoe and R. Bance-Soualhi, RSC Adv., 2020, 10, 36476–36477 RSC.
- X. M. Chu, Y. Shi, L. Liu, Y. D. Huang and N. W. Li, J. Mater. Chem. A, 2019, 7, 7717–7727 RSC.
- T. H. Pham, J. S. Olsson and P. Jannasch, J. Mater. Chem. A, 2018, 6, 16537–16547 RSC.
- N. Chen, C. Hu, H. H. Wang, S. P. Kim, H. M. Kim, W. H. Lee, J. Y. Bae, J. H. Park and Y. M. Lee, Angew. Chem., Int. Ed., 2021, 60, 7710–7718 CrossRef CAS PubMed.
- P. Fortin, T. Khoza, X. Z. Cao, S. Y. Martinsen, A. O. Barnett and S. Holdcroft, J. Power Sources, 2020, 451, 227814 CrossRef CAS.
Footnote |
| † Equal contributor. |
| This journal is © The Royal Society of Chemistry 2021 |