
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAn overview of methods for the structural and functional mapping of epitopes recognized by anti-SARS-CoV-2 antibodies
Irene M.
Francino-Urdaniz
 and
Timothy A.
Whitehead
*
and
Timothy A.
Whitehead
*
Department of Chemical and Biological Engineering, University of Colorado, JSC Biotechnology Building, 3415 Colorado Avenue, Boulder, CO 80305, USA. E-mail: timothy.whitehead@colorado.edu; Tel: +1 303-735-2145
First published on 29th September 2021
Abstract
This mini-review presents a critical survey of techniques used for epitope mapping on the SARS-CoV-2 Spike protein. The sequence and structures for common neutralizing and non-neutralizing epitopes on the Spike protein are described as determined by X-ray crystallography, electron microscopy and linear peptide epitope mapping, among other methods. An additional focus of this mini-review is an analytical appraisal of different deep mutational scanning workflows for conformational epitope mapping and identification of mutants on the Spike protein which escape antibody neutralization. Such a focus is necessary as a critical review of deep mutational scanning for conformational epitope mapping has not been published. A perspective is presented on the use of different epitope determination methods for development of broadly potent antibody therapies and vaccines against SARS-CoV-2.
Irene M. Francino-Urdaniz obtained her BS in Chemical Engineering on 2019 at Universitat de Barcelona, Spain, and her MSc on 2021 in Chemical Engineering at the University of Colorado Boulder. She is currently a third year PhD student at the University of Colorado Boulder. Her research focuses on identifying antibody escape mutants and developing broadly neutralizing antibodies using yeast surface display. |
Tim Whitehead trained in Chemical Engineering at Vanderbilt University (BE) and the University of California-Berkeley (PhD). After a short postdoctoral position at the University of Washington under David Baker, he started his independent group in 2011 at Michigan State University. Since 2019 he has been at University of Colorado Boulder, where his lab focuses on solving data-driven protein engineering and design challenges in the fields of antibody engineering, enzyme engineering, and biosensor development. |
Introduction
The interaction of proteins with other proteins is foundational to cellular life.1 Understanding the structural, functional, and mechanistic basis of such noncovalent protein–protein interactions can help rationalize emergent cellular behavior,1 can be exploited for design of biologics like antibodies2 and can also be used to map and predict the next moves in the trench warfare between humoral immunity and pathogen evasion and evolution.3An important class of protein–protein interactions are antibody interactions with antigens. Here, the epitope is defined as the antigenic surface recognized by a given antibody. Identifying the structures, sequences, and sequence constraints on such antigen epitopes is essential for solving difficult problems in basic and applied immunology. For example, a key idea in modern vaccine design has been that antigen structures can be modified rationally to present critical epitopes that elicit antibodies that neutralize infection (neutralizing antibodies or nAbs) that, in turn, confer long-lasting protection. The first proof of concept demonstration of such structure-based vaccine design in Phase I clinical trials was published4 for an immunogen mimicking a key conformational epitope of a viral protein in respiratory syncytial virus. Similarly, the search for a universal influenza A vaccine was jump-started by the structural and sequence identification of a conserved epitope on the influenza surface protein haemagglutinin.5–7 Antibodies targeting this haemagglutinin epitope are able to neutralize broadly across different influenza A subtypes. This structural definition of an epitope led to immunogen designs that elicit high levels of broadly neutralizing antibody titers in a recently completed phase I clinical trial.8 Thus, therapeutic and prophylactic strategies are informed by, and often start with, a sequence and structural definition of an antigenic epitope.
There exist several relatively mature technologies available to delineate the sequences, structures, or sequence constraints of epitopes. In fact, several comprehensive reviews of individual methods have been published in this century.9–16Table 1 lists common experimental methods for epitope mapping. There are two major classifications of epitopes primarily based on the experimental method used for their identification. Linear epitopes are those that involve sequential residues in the primary amino acid sequence and can be identified using techniques like peptide microarrays, phage, or bacterial display. By contrast, conformational epitopes involve surfaces recognized by antibodies only when a protein is folded in its tertiary or quaternary state. Such conformationally sensitive epitopes are typically resolved by structural determination using X-ray crystallography or electron microscopy (EM). Less commonly, hydrogen–deuterium exchange coupled to mass spectrometry (HDX-MS)16 or deep mutational scanning17 can be employed. All methods have their relative strengths and drawbacks, but generally it has been difficult to compare directly between methods as not all are typically performed on the same set of proteins.
| Category | Technique | Information Obtained | Comparative Advantage | Comprehensive review |
|---|---|---|---|---|
| Linear epitopes | Peptide arrays | Linear peptide sequence recognized by antibody | Massive parallelization allows proteome-size scalability | Katz et al.12 |
| Phage and bacterial display | Can use linear and constrained peptides in a high throughput format | Pande et al.14 | ||
| Conformational epitopes | Electron microscopy (cryo-/negative stain) | Atomic structure of an antigen-antibody complex | Structural determination of large, complex complexes with only small amounts of material needed | Renaud et al.11 |
| X-Ray crystallography | Highest quality atomic structural determination | Malito et al.10 | ||
| HDX-MS | Antigenic surfaces shielded from solvent in presence of antibody | Description of dynamic conformations | Sun et al.16 | |
| Deep mutational scanning | Comprehensive antigenic sequence determinants to binding/competitive inhibition | High resolution sequence constraints on antigenic epitopes and evaluation of point mutants |
The emergence of SARS-CoV-218 has led to intense research on its virology, epidemiology, and therapeutic and prophylactic interventions.19 During this time, dozens of research groups around the world identified antibodies raised against natural SARS-CoV-2 infection.20–24 This outpouring of research represents a natural experiment for the relative strengths, weaknesses, and types of information inherent in different epitope mapping methods. Thus, in this review we critically survey techniques used for epitope mapping on SARS-CoV-2. However, we do not intend an in-depth explanation of all the methods since exhaustive modern reviews already exist and are cited. Nonetheless, an additional focus on this mini-review is given on epitope mapping and identification of mutants which escape antibody neutralization using deep mutational scanning,17 as to our knowledge no comprehensive review exists. Thus, the second half of this review is given to the critical appraisal of different deep mutational scanning strategies because since the effect of individual mutations on binding can be studied, deep mutational scanning is especially relevant when developing antibodies against evolving viruses.
Given that well over a hundred thousand papers have been published on SARS-CoV-2,19 a comprehensive review is impractical for this short mini-review format. We apologize to colleagues whose work we have failed to cite.
SARS-CoV-2 spike protein as a model system
Comparisons between epitope mapping methods can be accomplished on antibodies targeting the same protein. In this mini-review we will focus on the SARS-CoV-2 Spike protein (S) as it is a highly glycosylated surface exposed protein on the virus and the focus of the overwhelming majority of SARS-CoV-2 epitope studies published to date (Fig. 1a).21,25,26 The S protein is a homotrimer in which each protomer is arranged as two subunits, S1 and S2. A furin cleavage site separates each S subunit and after cleavage the subunits are noncovalently associated in the prefusion metastable structure.27 The S1 subunit binds to angiotensin-converting enzyme 2 (ACE2) via its receptor binding domain (RBD)28 and contains an N-terminal domain (NTD), while the S2 subunit containing the C-terminal domain (CTD) is critical for the fusion of the viral and host cell membranes. The S2 subunit is more conserved than S1, perhaps because most of the surface exposed portion of the virus is on S1.29 Similar to other coronaviruses, the prefusion metastable structure of S undergoes two major conformations: a conformation where the RBD is in the “up” state and a conformation with RBD “down”.20,30 The biological relevance for these conformations is that the ACE2 receptor binding motif (RBM) is exposed to solvent only when the RBD is in the “up” state. Thus, at least one RBD must be in the “up” state for cell entry via ACE2 recognition.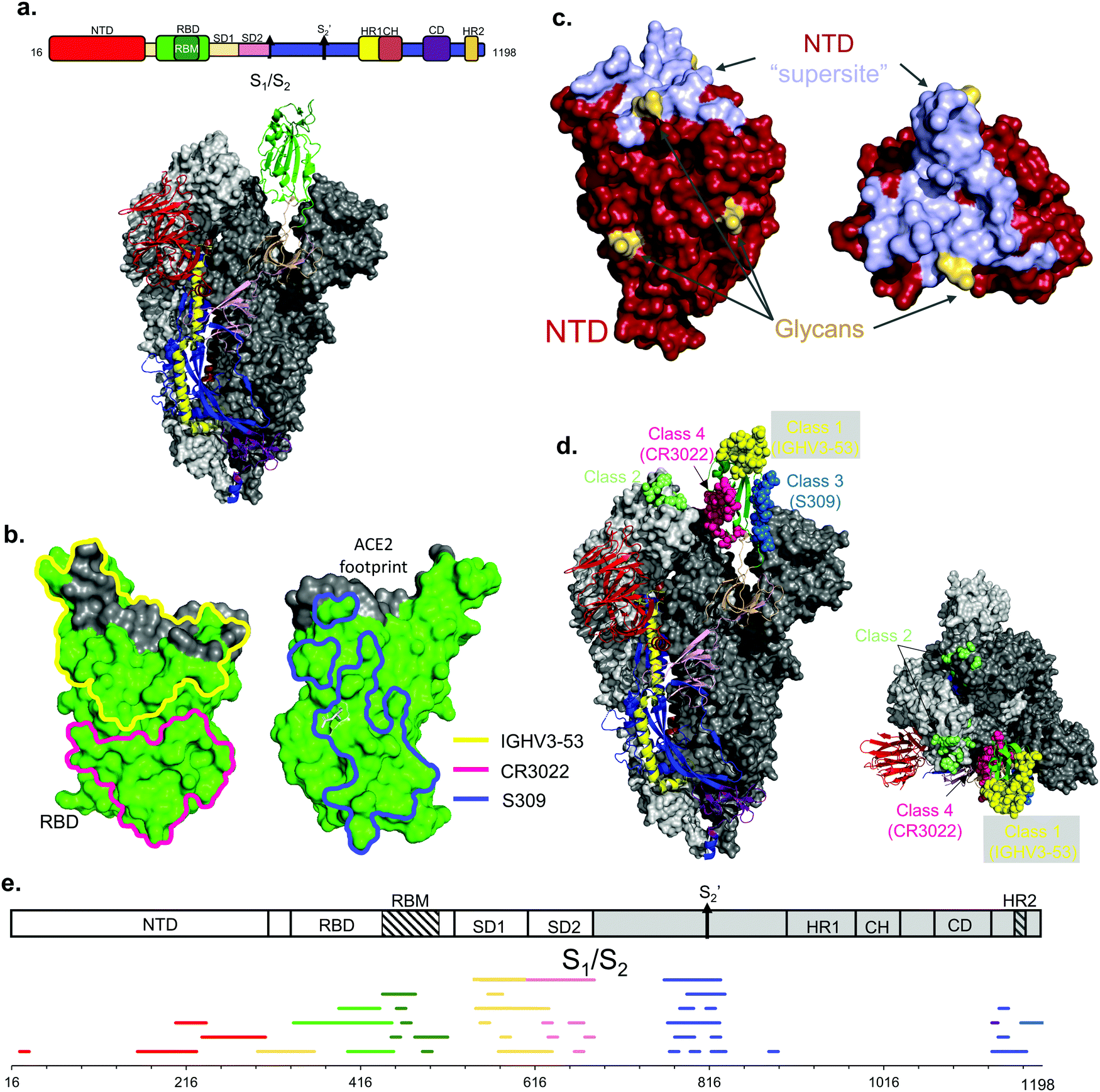 | ||
Fig. 1 Epitope mapping techniques in the context of SARS-CoV-2. a. SARS-CoV-2 Spike ectodomain schematic with labelled regions. NTD: N-terminal domain, RBD: receptor-binding domain, RBM: receptor-binding motif, SD1: subdomain 1, SD2: subdomain 2, S1/S2: furin cleavage site, 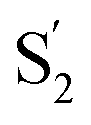 : : 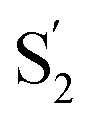 cleavage site, HR1: heptad repeat 1, CH: central helix, CD: connector domain, HR2: heptad repeat 2. Below is the structure of the Spike glycoprotein color coded with one protomer shown with RBD in the up conformation. The other two protomers are shown in different shades of grey and have the RBD in the down position. b. RBD structure (in green) showing epitopes identified by X-ray crystallography for anti-RBD IGHV3-53 (yellow), CR3022 (pink), and S309 (blue) antibodies. c. NTD structure (in chocolate brown) shown with the “supersite” epitope (pale blue). Glycans are shown in yellow. d. Common epitopes represented as spheres for the identified antibody classes on S. Class 1 binds on the RBM only available on the ‘up’ conformation. Class 2 can recognize the RBD on the ‘up’ and ‘down’ position. Class 3 binds in the same region as the previously identified S309 nAb. Class 4 in a non-neutralizing group of antibodies that bind a conserved epitope only available on the ‘up’ conformation, previously identified with CR3022. e. Linear epitope mapping along the Spike protein. Epitopes are color coded by domain as in a. Note the diversity of epitopes, including at SD1 and SD2 domains that are underrepresented in X-ray crystallography and EM structural studies. cleavage site, HR1: heptad repeat 1, CH: central helix, CD: connector domain, HR2: heptad repeat 2. Below is the structure of the Spike glycoprotein color coded with one protomer shown with RBD in the up conformation. The other two protomers are shown in different shades of grey and have the RBD in the down position. b. RBD structure (in green) showing epitopes identified by X-ray crystallography for anti-RBD IGHV3-53 (yellow), CR3022 (pink), and S309 (blue) antibodies. c. NTD structure (in chocolate brown) shown with the “supersite” epitope (pale blue). Glycans are shown in yellow. d. Common epitopes represented as spheres for the identified antibody classes on S. Class 1 binds on the RBM only available on the ‘up’ conformation. Class 2 can recognize the RBD on the ‘up’ and ‘down’ position. Class 3 binds in the same region as the previously identified S309 nAb. Class 4 in a non-neutralizing group of antibodies that bind a conserved epitope only available on the ‘up’ conformation, previously identified with CR3022. e. Linear epitope mapping along the Spike protein. Epitopes are color coded by domain as in a. Note the diversity of epitopes, including at SD1 and SD2 domains that are underrepresented in X-ray crystallography and EM structural studies. | ||
X-Ray crystallography
X-Ray crystallography allows atomic resolution of the antigen–antibody interaction and is the acknowledged gold standard for epitope determination. Epitopes can be determined amazingly fast: the first structure of an anti-SARS-CoV-2 antibody in complex with S RBD20 was reported on a preprint server only 9 weeks after the genetic sequences of SARS-CoV-2 were made public. Closely following this initial study, other reports described neutralizing and non-neutralizing epitopes for antibody complexes with individual S domains like the RBD (Fig. 1b) or NTD (Fig. 1c).30–36 These early studies helped define the structural basis of neutralizing and non-neutralizing epitopes on these individual domains.The RBD is a major target for neutralizing antibodies since it is responsible for binding ACE2.27 In the early days of the COVID-19 pandemic, antibodies from SARS-CoV convalescent patients were screened against SARS-CoV-2 S RBD. An early cross-reactive antibody is CR3022,20 and this antibody defines one non-neutralizing and broadly conserved epitope on RBD distal to its RBM (Fig. 1b). Another early described broadly conserved epitope is the one recognized by mAb S30931 (Fig. 1b), which recognizes an epitope defined by a conserved N-linked glycan at Asn343. In contrast to the CR3022 epitope, antibodies at this S309 epitope neutralize both SARS-CoV and SARS-CoV-2. Further into the pandemic, SARS-CoV-2 specific nAbs were identified from convalescent patients and, for some, their epitopes were structurally determined by X-ray crystallography. Some examples are P2B-2F6,37 P4A1,38 CB6;39 some other antibodies such as PR107740 were isolated from immunized mice. A large fraction of these nAbs bind at or adjacent to the ACE2 binding site. In particular, P4A138 covers the majority of the ACE2 footprint. As one example, nAbs from the IGHV3-53 germline class represent the most common antibodies elicited from natural infection from the original Wuhan-Hu-1 strain.32 Structures of IGHV3-53 nAbs CC12.1, CC12.3, and B38 define the basis of neutralization by competitive inhibition of ACE2 recognition32,33 (Fig. 1b).
Antibodies can also neutralize SARS-CoV-2 by binding at the NTD, with several crystallographic studies pinpointing the key epitopes.34,35 There are conserved epitopes between SARS-CoV and SARS-CoV-2 NTD but all are non-neutralizing; conversely, the key non-conserved epitope is neutralizing and has been named ‘supersite’34,36 (Fig. 1c). Most of the NTD surface is covered by a glycan shield, and the supersite is one of the only exposed proteinaceous surfaces on NTD. Structural studies show that antibodies from different germline classes bind this key aglycosylated epitope.35 Unfortunately, this supersite undergoes extensive antigenic variation, and many variants of concern (VoC) are no longer neutralized using supersite nAbs elicited from the original Wuhan-Hu-1 strain.34
Overall, X-ray crystallography has been a key technique in the study of SARS-CoV-2 epitopes as it was used to define individual conserved and non-conserved epitopes on the RBD and NTD of the SARS-CoV-2 S. Key limitations of this technique include the difficulty of the preparation of high diffraction quality crystal of full-length S ectodomain, limiting determination of epitopes to those entirely contained within individual RBD and NTD domains.
Electron microscopy
The 2017 Nobel Prize in Chemistry was awarded “for developing cryo-electron microscopy [cryo-EM] for the high-resolution structure determination of biomolecules in solution”. The use of electron microscopy, and cryo-EM in particular, has exploded in popularity over the past decade for the method's ability to determine structures of large protein complexes like antibodies in complex with S. In fact, cryo-EM was used to determine the atomic structure of S ectodomain less than two months after the publication of the S sequence.27,30Dozens of cryo-EM and, less commonly, negative stain-EM structures41 of potent neutralizing antibodies in complex with S have been reported. We list here a few of the antibodies that can be grouped in two representative examples of the types of epitopes that can be analyzed using electron microscopy. In the first example, a study led by Adimab scientists used cryo-EM to determine the epitope of a broadly neutralizing antibody developed by Adimab that binds to S RBD.42 Regeneron too has developed an antibody cocktail binding to S RBD whose epitope has been mapped using this same technique.43 Likewise, these specific complexes could have been determined by X-ray crystallography since the epitope is entirely contained within an S RBD monomer. Novel epitopes such as the one of H014 on RBD44 and the anti-S NTD antibody 4A836 can also be determined using EM. In another example, a different group used cryo-EM to characterize the epitope for a nAb that binds simultaneously to two of the three RBDs contained in the S trimeric complex.45 This specific complex would be difficult to determine by X-ray crystallography. Thus, cryo-EM can be used for complexes both amenable and refractory to solution by X-ray crystallography.
Cryo-EM and X-ray crystallography can be combined to define the structural epitopes recognized by antibodies elicited from natural infection. An excellent example of a joint study was reported by Barnes et al., who defined the four major classes of antibodies binding to RBD epitopes46 (see definitions in Fig. 1d).
Linear epitopes from synthetic peptide arrays
Linear epitopes are commonly identified using synthetic peptide arrays12,13 or peptide libraries coupled with phage or bacterial display.14,15 Most of the published linear epitopes for SARS-CoV-2 have been from synthetic peptide arrays; to our knowledge there have been no published reports on the use of phage display to determine epitopes on SARS-CoV-2. A unique strength about determining epitopes using synthetic peptide arrays, compared with other techniques covered in this review, is that individual antibodies as well as a bulk serological response can be studied.Synthetic peptide arrays have been used to study epitopes of monoclonal antibodies and convalescent patient serum on the whole S protein by several groups.47–52 Even though this review focuses on the S protein epitope mapping, one group has used synthetic peptide arrays to identify proteome-wide epitopes for SARS-CoV-2 and other coronaviruses,47 highlighting the advantage of scale for synthetic peptide arrays.
The identified linear epitopes on S are clustered in defined regions (Fig. 1e) mainly at cleavage sites or sites necessary for conformational changes for viral entry, like the S1/S2 cleavage site,32 the S2′ cleavage site, and the CTD.48,49 While the majority of the linear epitopes are found outside of the RBD, several have also been identified on the RBD.50 These combined studies highlight the diversity of the antibody response on the entire S protein and pinpoint immunodominant epitopes as well as epitopes that are relatively occluded from antibody recognition. However, there is a lack of information on the correlates of protection for these identified epitopes, and the structural basis for recognition must be inferred by structural information given by cryo-EM and X-ray crystallography.
Hydrogen deuterium exchange
Epitope determination using hydrogen–deuterium exchange coupled to mass spectrometry (HDX-MS) is based on the biophysical principle that amide hydrogens can exchange with deuterium in deuterated solvent faster when unbound than bound with antibody. Epitopes are determined by identifying locations on protein surfaces with lower exchange rates. The S RBD-ACE2 interface as well as the soluble ACE2 have been mapped by HDX-MS, which contributed to our understanding of the conformational changes on the S protein upon binding to ACE2.53 HDX-MS has also been used to determine and explain antibody epitopes.54,55 Regeneron in particular used HDX to understand the mechanism by which non-competitive antibodies bind simultaneously to the RBD.55 These results can help create a cocktail of neutralizing antibodies that would not overlap or block each other while simultaneously binding the RBD on the ACE2 footprint.While HDX-MS can facilitate the understanding of the conformational dynamics of binding, it may give recurrent false positives and the experimental proposal must fulfill an exacting list of requirements to obtain good results.16 Thus, HDX-MS is usually coupled to methods like cryo-EM to marry conformational dynamics with structural insight.
Deep mutational scanning
Deep mutational scanning, independently developed by the Fields17 and the Bolon56 groups, leverages next generation sequencing to observe the functional effect of individual mutants in a large population.17 The power of this method is scale, as tens of thousands of mutants can be assessed in a mixed pool. In 2015, conformational epitope mapping of protein–protein interactions using deep mutational scanning was independently developed by different labs.57–59 In the last year, three independent groups have used similar epitope mapping approaches to understand and engineer interactions between S RBD and antibodies or the ACE2 receptor (Fig. 2). The Procko group identified mutations on human ACE2 that increase binding affinity to S RBD.60 The Bloom group identified the sequence determinants of S RBD for ACE2 recognition and mapped anti-RBD antibody epitopes.26,61,62 Finally, the Whitehead group has developed a method to determine the set of mutations on S RBD which can escape monoclonal antibody neutralization.3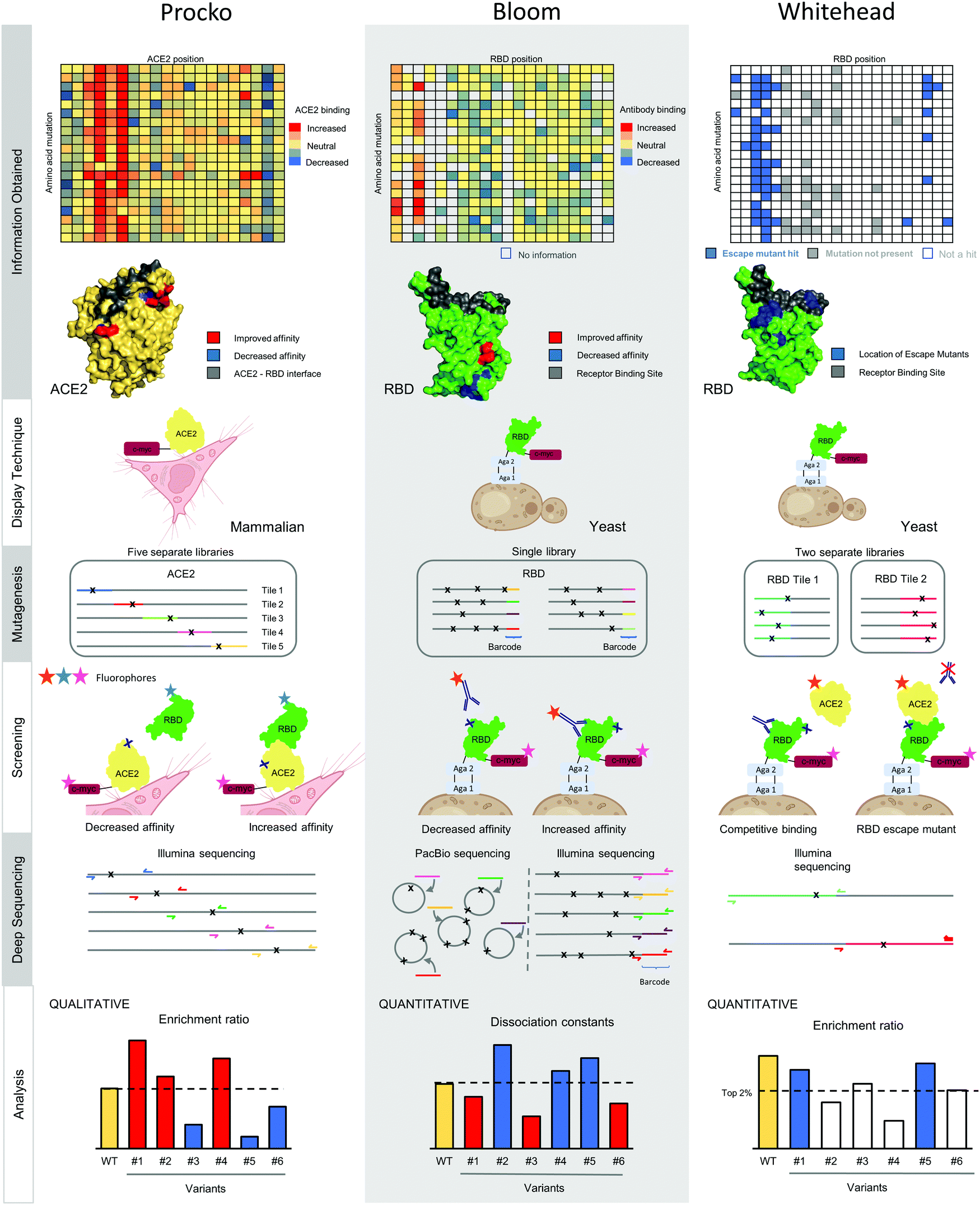 | ||
| Fig. 2 Overview of independent deep mutational scanning workflows for conformational epitope mapping. | ||
In deep mutational scanning, the antigenic sequence dependence on binding can be assessed for nearly every single point mutant in the protein sequence. This information is used to identify conformational epitopes under the assumption that epitope positions are less tolerant of mutations than non-epitope positions. Deep mutational scanning workflows for conformational epitope mapping are similar at a superficial level. The antigen of choice is displayed on the surface of a eukaryotic cell. Next, binding to an antibody or receptor is monitored using a flow cytometer after cell labeling with appropriate fluorophores. Comprehensive mutagenesis of the antigen gene is performed thus generating a library of antigen mutants that can be transformed into the relevant cell type. A population of cells, where each cell displays a distinct antigen mutant, is split and incubated in several different binding conditions. For example, each reaction could contain a different amount (or none) of antibody. After fluorophore labeling, the cells are screened using a cell sorter. Different populations are distinguished using gates on different light scattering or fluorescent values. For example, a gate is typically set to identify cells maintaining high antibody binding as inferred by a high fluorescence signal in the appropriate channel. Populations of cells are sorted according to these gates by fluorescence activated cell sorting (FACS), regrown, plasmids harvested and prepared for deep sequencing, and then sequenced. For each sorted population the frequency of each variant is enumerated; along with other information about sorting conditions, this information is processed either qualitatively or quantitatively to identify the effect of each introduced mutation on the binding considered in the assay.
The Procko group used deep mutational scanning to identify ACE2 mutations that increase binding to SARS-CoV-2 S RBD60 in order to develop a receptor trap prophylactic and therapeutic against SARS-CoV-2. Key mutations found to increase ACE2 binding to S were those removing N-linked glycans that partially shield the ACE2 surface recognized by the S RBD. The best engineered soluble ACE2 (sACE2) variant can outcompete natural ACE2 for binding to S RBD. Further, the authors showed that sACE2 can neutralize different coronaviruses, including SARS-CoV and SARS-CoV-2.63 To engineer this receptor, ACE2 was displayed on the surface of mammalian cells and incubated with soluble S RBD. The variants that bind tighter to the S RBD than native ACE2 were collected and identified by an increase in frequency in the binding population relative to a control.56
The Bloom research group used deep mutational scanning for the quantitative assessment of the sequence dependence of S RBD on ACE2 binding affinity.61 This same platform was also used to map epitopes and escape mutants for several monoclonal antibodies,26,62 predicting in advance the N501Y mutation observed in several VoC. S RBD is displayed on the surface of yeast and labeled either with soluble ACE2 or mAb at multiple different concentrations. Cell populations collected depend on whether epitopes or escape mutants are identified, and sequence data is processed using a quantitative maximum likelihood estimation method.64
The Whitehead group has developed a method that identifies the near-comprehensive set of escape mutants on S RBD for neutralizing antibodies that directly compete with ACE2 for binding.3 Several antibodies can be tested in parallel. Most escape mutations identified in the study are located adjacent to but not directly on the ACE2 binding footprint. Most intriguing, many escape mutants map to K417, including K417N which is present in the circulating Delta plus VoC (B.1.617.2 + K417N) and in the Beta VoC and K417T present in the Gamma VoC.65,66 To identify escape mutants, an aglycosylated S RBD construct is displayed on the surface of yeast and a competitive binding experiment is performed between a given antibody and soluble ACE2. Cells harboring RBD variants able to maintain ACE2 binding in the presence of a nAb are collected, and a novel algorithm is used to identify escape mutants.
The above studies all performed different strategies, shown in Fig. 2, with these differences instructive for those setting up a deep mutational scanning experiment. One major difference between groups is the display technique. One group displayed bona fide ACE2, including its membrane-spanning pass, on mammalian cells, while the other groups used an artificial genetic fusion of S RBD to a yeast cell surface protein. The yeast display set-up maintains several advantages for deep mutational scanning: relatively fast growth rates, excellent genetics and high transformation efficiency, robust cells, and validated protocols.67 In our hands 11 of 12 tested antibodies targeting S RBD maintained binding to the engineered construct on yeast,3 attesting to the fidelity of the platform. Still, it remains difficult to display complicated glycoproteins in the active form,3 and yeast has different N-linked glycosylation patterns involving heavy mannosylation relative to mammalian cells.68 Therefore, antibodies that target across S protomers, that involve glycan recognition, or that bind on the S2 protein cannot be considered using yeast display. While mammalian cell display has several disadvantages relative to yeast display, the key advantage is displaying a membrane protein in its native context. In the Procko case, using the native ACE2 conformation was essential to identify that the removal of the glycans increases the binding affinity to RBD.
The two next steps in deep mutational scanning are (i.) performing comprehensive mutagenesis of the gene to be scanned; and (ii.) transforming the resulting DNA libraries into cells. Comprehensive mutagenesis on plasmid DNA can be performed using several methods like PFunkel,69 nicking,70 or overlap extension PCR mutagenesis.60 Illumina sequencing platforms typically utilize 250 base pair DNA sequencing, which limits the linear stretch of the gene which contains mutations to typical 250–350 bp. Covering an entire gene like ACE2 or S RBD, which are both larger than 350 bp, requires multiple libraries for coverage. These libraries are colloquially referred to as ‘tiles’. Both the Procko and Whitehead groups used this tiling strategy (Fig. 2). The main disadvantage of tiling is handling each library independently – separate labeling, sorting, and DNA prep steps must be performed for each tile. In contrast, the Bloom group encoded all mutations on S RBD in a single library. Then, they utilized PacBio long read sequencing to haplotype each set of mutants on S RBD to a unique barcode (Fig. 2). Illumina short read sequencing of the short barcode could then be used to identify frequencies of each mutant. This approach has a higher upfront cost of library haplotyping (the PacBio step) but has more streamlined downstream steps with less expensive sequencing on the backend.
All groups used FACS to screen cell populations. Both Procko and Bloom groups used direct labeling either with antigen or antibody. In contrast, the Whitehead group developed a competitive ACE2 binding screening assay for a neutralizing antibody to infer the set of escape mutants. All groups also used Illumina for next generation sequencing of library DNA. The Procko and Whitehead groups screened and sequenced each tile separately, while the Bloom group sequenced library barcodes only. Best practices for these screening steps involve making true biological replicates (DNA libraries prepared and transformed independently) and sorting replicate libraries on different days.
In the final step, sequencing results are analyzed with a method appropriate for each approach. The analysis results are qualitative or quantitative and depend on factors in the experimental approach like the choice of display format, the type of mutagenesis performed, and screening strategy. In deep mutational scanning workflows the first step is to enumerate the frequency of each variant for each sequenced population. The simplest qualitative analysis is to compare the frequency of a selected population with a reference population that has passed through the cell sorter but is otherwise not screened for binding. The log transform of this frequency change between populations is called an 'enrichment ratio'. The Procko group used this qualitative analysis to determine the relative binding for their ACE2 variants. Such qualitative analyses are simple to perform and suitable for engineering goals like developing superior ACE2 receptor traps. However, this enrichment ratio analysis is subject to consider noise resulting from complexity bottlenecks in the FACS screening, DNA preparation, and sequencing steps. Thus, one drawback from a qualitative analysis is hit identification – how does one determine high enrichment ratios that result from binding events rather than ones that occur by chance? The Whitehead group solved this problem by independently sorting a control population subject to the same screening criteria as their competitively inhibited yeast cells. This control population was then used to set an empirical False Discovery Rate at which an enrichment ratio is not expected to occur by chance in a population of a given size.
The most sophisticated approach for analysis came from the Bloom group, who sought to quantitatively estimate binding dissociation constants for S RBD mutants. Their approach involved sorting using many different labeling concentrations of ACE2 or antibody and using a maximum likelihood estimation approach to infer dissociation constants.64 This protocol is very exhaustive, with precision coming at the expense of throughput. Thus, this is a suitable protocol to analyze a few antibodies in depth.
In summary, these three groups’ contributions show how different experimental observables result from different experimental strategies.
Conclusions and perspective
Hundreds of antibodies and nanobodies have had their epitopes mapped on SARS-CoV-2 S with a handful also having their escape mutants determined. This accumulated knowledge has contributed to the mitigation of COVID-19 through the development of monoclonal antibody therapies and novel vaccines. All techniques surveyed were quite useful for different facets of epitope mapping. Electron microscopy and X-ray crystallography were essential in the early days by defining the structural basis of many common neutralizing and non-neutralizing epitopes on S. Linear peptide arrays showed the diversity of the antibody response against S and identified several common immunodominant epitopes. Deep mutational scanning was essential for understanding the impact of individual mutations on both S RBD/ACE2 and anti-S antibody/S recognition. These mutational constraints on binding led to predictive understanding of the recognition landscape for currently circulating VoCs.Our mini-review described at length different conformational epitope mapping methods by deep mutational scanning as no in-depth review for this methodology exists. We are especially excited about the ability to delineate the sequence constraints on binding by both ACE2 and nAbs, as these constraints dictate the boundaries of the emerging arms race between future mutations on SARS-CoV-2 VoC and the ability of the humoral response in the vaccinated and naturally infected population to respond. It remains to be seen whether deep mutational scanning can inform the next generation of design of monoclonal antibody therapies and vaccine candidates.
Author contributions
Wrote paper: IMFU, TAW.Conflicts of interest
The authors declare no competing interests.Acknowledgements
We thank members of the Whitehead group (M. B. K., Z. T. B, M. N. K.) for critical reading of the manuscript and helpful suggestions. Funding: Research reported in this publication was supported by the National Institute Of Allergy And Infectious Diseases of the National Institutes of Health under Award Number R01AI141452 to T. A. W. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.References
- X. Peng, J. Wang, W. Peng, F. Wu and Y. Pan, Protein – protein interactions: detection, reliability assessment and applications, Briefings Bioinf., 2017, 18(2016), 798–819 CAS.
- D. W. Kulp and W. R. Schief, Advances in structure-based vaccine design, Curr. Opin. Virol., 2013, 3(3), 322–331 CrossRef CAS PubMed.
- I. M. Francino-Urdaniz, P. J. Steiner, M. B. Kirby, F. Zhao, C. M. Haas and S. Barman, et al., One-shot identification of SARS-CoV-2 S RBD escape mutants using yeast screening, Cell Rep., 2021, 109627 CrossRef CAS PubMed.
- M. C. Crank, T. J. Ruckwardt, M. Chen, K. M. Morabito, E. Phung and P. J. Costner, et al., A proof of concept for structure-based vaccine design targeting RSV in humans, Science, 2019, 509, 505–509 CrossRef PubMed.
- D. C. Ekiert, G. Bhabha, M. Elsliger, R. H. E. Friesen, M. Jongeneelen and M. Throsby, et al., Antibody Recognition of a Highly Conserved Influenza Virus Epitope, Exposure, 2009, 1857, 246–251 CrossRef PubMed.
- M. Throsby, E. Brink, M. van den, Jongeneelen, L. L. M. Poon, P. Alard and L. Cornelissen, et al., Heterosubtypic neutralizing monoclonal antibodies cross-protective against H5N1 and H1N1 recovered from human IgM + memory B cells, PLoS One, 2008, 3(12), e3942 CrossRef PubMed.
- J. Sui, W. C. Hwang, S. Perez, G. Wei, D. Aird and L. Chen, et al., Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses, Nat. Struct. Mol. Biol., 2009, 16(3), 265–273 CrossRef CAS PubMed.
- R. Nachbagauer, J. Feser, A. Naficy, D. I. Bernstein, J. Guptill and E. B. Walter, et al., A chimeric hemagglutinin-based universal influenza virus vaccine approach induces broad and long-lasting immunity in a randomized, placebo-controlled phase I trial, Nat. Med., 2021, 27(1), 106–114 CrossRef CAS PubMed.
- G. E. Morris, Epitope mapping, Methods Mol. Biol., 2005, 295, 255–268 CAS.
- E. Malito, A. Carfi and M. J. Bottomley, Protein Crystallography in Vaccine Research and Development, Int. J. Mol. Sci., 2015, 13106–13140 CrossRef CAS PubMed.
- J.-P. Renaud, A. Chari, C. Ciferri, W. Liu, H.-W. Rémigy and H. Stark, et al., Cryo-EM in drug discovery: achievements, limitations and prospects, Nat Rev. Drug Discovery, 2018, 17(7), 471–492 CrossRef CAS PubMed.
- C. Katz, L. Levy-beladev, S. Rotem-bamberger, T. Rito, S. G. D. Ru and C. Katz, Studying protein – protein interactions using peptide arrays, Chem. Soc. Rev., 2011, 2131–2145 RSC.
- R. Liu, A. M. Enstrom and K. S. Lam, Combinatorial peptide library methods for immunobiology research, Exp. Hematol., 2003, 31, 11–30 CrossRef CAS PubMed.
- J. Pande, M. M. Szewczyk and A. K. Grover, Phage display: Concept, innovations, applications and future, Biotechnol. Adv., 2010, 28(6), 849–858 CrossRef CAS PubMed.
- M. B. Irving, O. Pan and J. K. Scott, Random-peptide libraries and antigen-fragment libraries for epitope mapping and the development of vaccines and diagnostics, Curr. Opin. Chem. Biol., 2001, 314–324 CrossRef CAS.
- H. Sun, L. Ma, L. Wang, P. Xiao, H. Li and M. Zhou, et al., Research advances in hydrogen – deuterium exchange mass spectrometry for protein epitope mapping, Anal. Bioanal. Chem., 2021, 2345–2359 CrossRef CAS PubMed.
- D. M. Fowler and S. Fields, Deep mutational scanning: a new style of protein science, Nat. Methods, 2014, 11(8), 801–807 CrossRef CAS PubMed.
- K. G. Andersen, A. Rambaut, W. I. Lipkin, E. C. Holmes and R. F. Garry, The proximal origin of SARS-CoV-2, Nat. Med., 2020, 26, 450–452 CrossRef CAS PubMed.
- H. Else, How a torrent of COVID science changed research publishing – in seven charts, Nature, 2020, 588(7839), 553 CrossRef CAS PubMed.
- M. Yuan, N. C. Wu, X. Zhu, C. C. D. Lee, R. T. Y. So and H. Lv, et al., A highly conserved cryptic epitope in the receptor binding domains of SARS-CoV-2 and SARS-CoV, Science, 2020, 368(6491), 630–633 CrossRef CAS PubMed.
- T. F. Rogers, F. Zhao, D. Huang, N. Beutler, A. Burns and W. He, et al., Isolation of potent SARS-CoV-2 neutralizing antibodies and protection from disease in a small animal model, Science, 2020, 369(6506), 956 LP–963 CrossRef PubMed.
- B. B. Banach, G. Cerutti, A. S. Fahad, C.-H. Shen, M. Olivera de Souza and P. S. Katsamba, et al., Paired heavy and light chain signatures contribute to potent SARS-CoV-2 neutralization in IGHV3-53/3-66 public antibody responses, Cell Rep., 2021 DOI:10.1101/2020.12.31.424987.
- T. Noy-porat, E. Makdasi, R. Alcalay, A. Mechaly, Y. Levy and A. Bercovich-kinori, et al., A panel of human neutralizing mAbs targeting SARS-CoV-2 spike at multiple epitopes Tal, Nat. Commun., 2020, 1–7 Search PubMed.
- X. Zeng, L. Li, J. Lin, X. Li, B. Liu and Y. Kong, et al., Isolation of a human monoclonal antibody specific for the receptor binding domain of SARS-CoV-2 using a competitive phage biopanning strategy, Antibody Ther., 2020, 3(2), 95–100 CrossRef CAS PubMed.
- A. Baum, B. O. Fulton, E. Wloga, R. Copin, K. E. Pascal and V. Russo, et al., Antibody cocktail to SARS-CoV-2 spike protein prevents rapid mutational escape seen with individual antibodies, Science, 2020, 1018, 1014–1018 CrossRef PubMed.
- A. J. Greaney, T. N. Starr, P. Gilchuk, S. J. Zost, E. Binshtein and A. N. Loes, et al., Complete Mapping of Mutations to the SARS-CoV-2 Spike Receptor-Binding Domain that Escape Antibody Recognition, Cell Host Microbe, 2020, 29(1), 44–57 CrossRef PubMed.
- A. C. Walls, Y.-J. Park, M. A. Tortorici, A. Wall, A. T. McGuire and D. Veesler, Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein, Cell, 2020, 180, 1–12 CrossRef PubMed.
- R. Yan, Y. Zhang, Y. Li, L. Xia, Y. Guo and Q. Zhou, Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2, Science, 2020, 367(6485), 1444 LP–1448 CrossRef PubMed.
- M. A. Tortorici and D. Veesler, Structural insights into coronavirus entry, Advances in Virus Research, Elsevier Inc., 1st edn, 2019 Search PubMed.
- D. Wrapp, N. Wang, K. S. Corbett, J. A. Goldsmith, C. Hsieh and O. Abiona, et al., Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation, Science, 2020, 1263, 1260–1263 CrossRef PubMed.
- D. Pinto, Y.-J. Park, M. Beltramello, A. C. Walls, M. A. Tortorici and S. Bianchi, et al., Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody, Nature, 2020, 583(7815), 290–295 CrossRef CAS PubMed.
- M. Yuan, H. Liu, N. C. Wu, C.-C. D. Lee, X. Zhu and F. Zhao, et al., Structural basis of a shared antibody response to SARS-CoV-2, Science, 2020, 369(6507), 1119 LP–1123 CrossRef PubMed.
- Y. Wu, F. Wang, C. Shen, W. Peng, D. Li and C. Zhao, et al., A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2, Science, 2020, 368(6496), 1274 LP–1278 CrossRef PubMed.
- M. McCallum, A. De Marco, F. A. Lempp, D. Corti, M. S. Pizzuto and D. Veesler, et al., N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2, Cell, 2021, 184(9), 2332–2347 CrossRef CAS PubMed.
- G. Cerutti, Y. Guo, T. Zhou, Z. Sheng, P. D. Kwong and L. Shapiro, et al., Potent SARS-CoV-2 neutralizing antibodies directed against spike N-terminal domain target a single supersite, Cell Host Microbe, 2021, 29(5), 819–833 CrossRef CAS PubMed.
- X. Chi, R. Yan, J. Zhang, G. Zhang, Y. Zhang and M. Hao, et al., A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2, Science, 2020, 369(6504), 650 LP–655 CrossRef PubMed.
- B. Ju, Q. Zhang, J. Ge, R. Wang, J. Sun and X. Ge, et al., Human neutralizing antibodies elicited by SARS-CoV-2 infection, Nature, 2020, 584, 115–119 CrossRef CAS PubMed.
- Y. Guo, L. Huang, G. Zhang, Y. Yao, H. Zhou and S. Shen, et al., A SARS-CoV-2 neutralizing antibody with extensive Spike binding coverage and modified for optimal therapeutic outcomes, Nat. Commun., 2021, 12, 2623 CrossRef CAS PubMed.
- R. Shi, C. Shan, X. Duan, Z. Chen, P. Liu and J. Song, et al., A human neutralizing antibody targets the receptor-binding site of SARS-CoV-2, Nature, 2020, 584, 120–124 CrossRef CAS PubMed.
- D. Fu, G. Zhang, Y. Wang, Z. Zhang, H. Hu and S. Shen, et al., Structural basis for SARS-CoV-2 neutralizing antibodies with novel binding epitopes, PLoS Biol., 2021, 2, 1–22 Search PubMed.
- C. O. Barnes, A. P. West, K. E. Huey-Tubman, M. A. G. Hoffmann, N. G. Sharaf and P. R. Hoffman, et al., Structures of Human Antibodies Bound to SARS-CoV-2 Spike Reveal Common Epitopes and Recurrent Features of Antibodies, Cell, 2020, 182(4), 828–842 CrossRef CAS PubMed.
- C. G. Rappazzo, L. V. Tse, C. I. Kaku, D. Wrapp, M. Sakharkar and D. Huang, et al., Broad and potent activity against SARS-like viruses by an engineered human monoclonal antibody, Science, 2021, 371, 823–829 CrossRef CAS PubMed.
- R. Copin, A. Baum, E. Wloga, A. J. Murphy, G. D. Yancopoulos and C. A. Kyratsous, et al., The monoclonal antibody combination REGEN-COV protects against SARS-CoV-2 mutational escape in preclinical and human studies, Cell, 2021, 184(15), 3949–3961 CrossRef CAS PubMed.
- Z. Lv, Y. Deng, Q. Ye, L. Cao, C. Sun and C. Fan, et al., Structural basis for neutralization of SARS-CoV-2 and SARS-CoV by a potent therapeutic antibody, Science, 2020, 1509, 1505–1509 CrossRef PubMed.
- L. Liu, P. Wang, M. S. Nair, J. Yu, M. Rapp and Q. Wang, et al., Potent neutralizing antibodies against multiple epitopes on SARS-CoV-2 spike, Nature, 2020, 584, 450–456 CrossRef CAS PubMed.
- C. O. Barnes, C. A. Jette, M. E. Abernathy, K. A. Dam, S. R. Esswein and H. B. Gristick, et al., SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies, Nature, 2020, 588, 682–687 CrossRef CAS PubMed.
- N. Mishra, X. Huang, S. Joshi, C. Guo, J. Ng and R. Thakkar, et al., Immunoreactive peptide maps of SARS-CoV-2, Commun Biol., 2021, 1–7 Search PubMed.
- Y. Li, M. Ma, Q. Lei, Y. Li, M. Ma and Q. Lei, et al., Linear epitope landscape of the SARS-CoV-2 Spike protein constructed from 1051 COVID-19 patients, Cell Rep., 2021, 34(13), 108915 CrossRef CAS PubMed.
- L. Farrera-soler, J. Daguer, S. B. Id, O. V. Id, P. Cohen and S. Pagano, et al., Identification of immunodominant linear epitopes from SARS-CoV-2 patient plasma, PLoS One, 2020, 1–15 Search PubMed.
- S. Ravichandran, E. M. Coyle, L. Klenow, J. Tang, G. Grubbs and S. Liu, et al., Antibody signature induced by SARS-CoV-2 spike protein immunogens in rabbits, Sci. Transl. Med., 2020, 1–9 Search PubMed.
- Y. Li, D. Y. Lai, Q. Lei, Z. W. Xu, F. Wang and H. Hou, et al., Systematic evaluation of IgG responses to SARS-CoV-2 spike protein-derived peptides for monitoring COVID-19 patients, Cell Mol Immunol., 2021, 18(3), 621–631 CrossRef CAS PubMed.
- C. M. Poh, G. Carissimo, B. Wang, S. N. Amrun, C. Y. P. Lee and R. S. L. Chee, et al., Two linear epitopes on the SARS-CoV-2 spike protein that elicit neutralising antibodies in COVID-19 patients, Nat Commun., 2020, 11, 2806 CrossRef CAS PubMed.
- P. V. Raghuvamsi, N. K. Tulsian, F. Samsudin, X. Qian, K. Purushotorman and G. Yue, et al., SARS-CoV-2 S protein: ACE2 interaction reveals novel allosteric targets, eLife, 2021, 1–17 Search PubMed.
- B. E. Jones, P. L. Brown-augsburger, K. S. Corbett, K. Westendorf, J. Davies and T. P. Cujec, et al., The neutralizing antibody, LY-CoV555, protects against SARS-CoV-2 infection in nonhuman primates, Sci. Transl. Med., 2021, 1–18 Search PubMed.
- J. Hansen, A. Baum, K. E. Pascal, V. Russo, S. Giordano and E. Wloga, et al., Studies in humanized mice and convalescent humans yield a SARS-CoV-2 antibody cocktail, Science, 2020, 1014, 1010–1014 CrossRef PubMed.
- R. T. Hietpas, J. D. Jensen and D. N. A. Bolon, Experimental illumination of a fitness landscape, Proc. Natl. Acad. Sci. U. S. A., 2011, 108(19), 7896–7901 CrossRef CAS PubMed.
- C. A. Kowalsky, M. S. Faber, A. Nath, H. E. Dann, V. W. Kelly and L. Liu, et al., Rapid Fine Conformational Epitope Mapping Using Comprehensive Mutagenesis and Deep Sequencing, J. Biol. Chem., 2015, 290(44), 26457–26470 CrossRef CAS PubMed.
- T. Van Blarcom, A. Rossi, D. Foletti, P. Sundar, S. Pitts and C. Bee, et al., Precise and Efficient Antibody Epitope Determination through Library Design, Yeast Display and Next-Generation Sequencing, J. Mol. Biol., 2015, 427(6), 1513–1534 CrossRef CAS PubMed.
- K. M. Doolan and D. W. Colby, Conformation-dependent epitopes recognized by prion protein antibodies probed using mutational scanning and deep sequencing, J. Mol. Biol., 2015, 427(2), 328–340, DOI:10.1016/j.jmb.2014.10.024.
- K. K. Chan, D. Dorosky, P. Sharma, S. A. Abbasi, J. M. Dye and D. M. Kranz, et al., Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2, Science, 2020, 1265(369), 1261–1265 CrossRef PubMed.
- T. N. Starr, A. J. Greaney, S. K. Hilton, N. P. King, D. Veesler and J. D. Bloom, et al., Deep Mutational Scanning of SARS-CoV-2 Receptor Binding Domain Reveals Constraints on Folding and ACE2 Binding, Cell, 2020, 182(5), 1295–1310 CrossRef CAS PubMed.
- T. N. Starr, A. J. Greaney, A. S. Dingens and J. D. Bloom, Complete map of SARS-CoV-2 RBD mutations that escape the monoclonal antibody LY-CoV555 and its cocktail with LY-CoV016, Cell Rep. Med., 2021, 2(4), 100255 CrossRef PubMed.
- K. K. Chan, T. J. C. Tan, K. K. Narayanan and E. Procko, An engineered decoy receptor for SARS-CoV-2 broadly binds protein S sequence variants, Sci. Adv., 2021, 1–10 Search PubMed.
- R. M. Adams, T. Mora, A. M. Walczak and J. B. Kinney, Measuring the sequence-affinity landscape of antibodies with massively parallel titration curves, eLife, 2016, 1–27 CAS.
- C. D. C. SARS-CoV-2 Variant Classifications and Definitions [Internet]. 2021. Available from: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html#unweighted-proportions-substitutions-of-therepeutic-concern.
- J. Corum and C. Zimmer Coronavirus Variants and Mutations. The New York Times [Internet]. 2021. Available from: https://www.nytimes.com/interactive/2021/health/coronavirus-variant-tracker.html.
- G. Chao, W. L. Lau, B. J. Hackel, S. L. Sazinsky, S. M. Lippow and K. D. Wittrup, Isolating and engineering human antibodies using yeast surface display, Nat. Protoc., 2006, 1(2), 755–768 CrossRef CAS PubMed.
- Y. Jigami, Yeast Glycobiology and Its Application, Biosci. Biotechnol. Biochem, 2008, 72(3), 637–648 CrossRef CAS PubMed.
- E. Firnberg and M. Ostermeier, PFunkel: Efficient, Expansive, User-Defined Mutagenesis, PLoS One, 2012, 7(12), 1–10 CrossRef PubMed.
- P. J. Steiner, Z. T. Baumer and T. A. Whitehead, A Method for User-defined Mutagenesis by Integrating Oligo Pool Synthesis Technology with Nicking Mutagenesis, Bio-Protoc., 2020, 10(15), e3697 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2021 |