
Highly sensitive detection of DNA damage in living cells by SERS and electrochemical measurements using a flexible gold nanoelectrode†
Jing
Zhou
a,
Dan
Yang
 a,
Guohui
Liu
a,
Siying
Li
a,
Wennan
Feng
a,
Guocheng
Yang
a,
Jin
He
*b and
Yuping
Shan
*a
a,
Guohui
Liu
a,
Siying
Li
a,
Wennan
Feng
a,
Guocheng
Yang
a,
Jin
He
*b and
Yuping
Shan
*a
aSchool of Chemistry and Life Science, Advanced Institute of Materials Science, Changchun University of Technology, Changchun 130012, China. E-mail: shanyp@ciac.ac.cn
bPhysics Department, Florida International University, Miami, 33199, USA. E-mail: jinhe@fiu.edu
First published on 11th February 2021
Abstract
Guanine (G) oxidation products, such as 8-hydroxy-2′-deoxyguanosine (8-OHdG) and 8-oxo-guanine (8-OXOG), have been widely studied as promising biomarkers for DNA oxidative damage. In this work, we develop a new method to detect G oxidative products released from live cells after chromium (VI) ion or hydrogen peroxide treatments by using a glass nanopipette-based flexible gold nanoelectrode (fGNE). Specific response to G oxidative products with high sensitivity can be detected from the fGNE tip through integrated electrochemical measurements and surface-enhanced Raman spectroscopy. The fGNE apex can be positioned very close to the cell membrane noninvasively because of its high flexibility and nanoscale tip size. With the assistance of the electrophoretic force, the fGNEs can effectively collect and detect the G-derived DNA damage products released from individual cells in the cell culture medium with high sensitivity.
Introduction
Reactive oxygen species (ROSs), formed during endogenous metabolic processes and exposure to environmental factors, can induce various lesions in nuclear and mitochondrial DNA.1,2 Among more than 100 types of DNA damage, guanine (G) oxidative derivatives are the most abundant forms because G is the one most susceptible to oxidation among the four DNA bases.3,4 Fortunately, life has developed various systems to quickly detect and repair DNA damage.2,5 The oxidized G base is mainly repaired through base excision repair (BER) and the base oxidation products, 8-oxo-guanine (8-OXOG) or its tautomer 8-hydroxyguanine (8-OHGua), are efficiently removed from the DNA strand. Meanwhile, their nucleotide forms, 8-oxo-2′-deoxyguanosine (8-OXOdG) and its tautomer 8-hydroxy-2′-deoxyguanosine (8-OHdG), can also be generated inside the cell through different repair pathways such as nucleotide excision repair (NER) or directly from the free nucleotide pool.6,7 Both 8-OXOG and 8-OHdG have become important biomarkers of oxidative stress and reflect the dynamic balance between cellular DNA damage and repair.8–10 It should be noted that the tautomer structures are often not clearly differentiated in the previous literature. Between the base and nucleoside forms, more analytical methods have been developed to detect and quantify 8-OHdG outside cells, which may be related to its better cross-membrane capability.7 8-OHdG is usually detected at an elevated level in the urine or serum of patients with increased oxidative stress or risk of diseases, including neurodegenerative diseases, cancers, cardiovascular diseases and diabetes.11–13Classical analytical methods have been developed for 8-OHdG and 8-OXOG detection mainly in urine and blood samples, including high-performance liquid chromatography (HPLC),10,14,15 gas chromatography (GC)–mass spectrometry (MS), liquid chromatography (LC)–MS,14,16,17 and enzyme-linked immunosorbent assay (ELISA).18,19 These methods have extremely high sensitivity (nanomolar or better) and specificity, but extensive sample preparations are needed, and only an ensemble average can be obtained. At the cellular level, 8-OXOG within the cells can often be detected by fluorescence-based methods.8,20 However, the research is still limited to directly detect the trace amount of 8-OXOG or 8-OHdG released from individual living cells,21 which are also critical for fundamental studies of oxidative DNA damage and early diagnosis of diseases.22
In recent years, various new methods have been developed for living cell sensing and analysis, especially down to the single-cell level. These new technical developments can provide a new solution for the detection of G-derived DNA damage products. Because 8-OXOG/8-OHGua and their nucleotide forms can be further oxidized and thus be detected electrochemically, well-developed micro/nanoelectrode-based electrochemical (EC) techniques can be utilized.23–25 For example, carbon fiber microelectrodes have been utilized for real-time detection of 8-OHdG released by individual lung epithelial cells after exposure to nicotine.26 In recent years, nanoelectrode (NE) and nanopipette based electrochemical methods have made great progress for single-cell studies since they can ensure the integrity and physiological activity of the living cells during detection.27–29 In addition to micro/nanoelectrode-based electrochemical techniques, surface-enhanced Raman spectroscopy (SERS) has been used together with nanoelectrodes and nanopipettes in qualitative single-cell analysis because of its high sensitivity and selectivity at the molecular level.30–32 Furthermore, SERS has been successfully used to differentiate four DNA bases and has been combined with EC techniques for biosensing.29–31 Therefore, an integrated EC and SERS approach may provide a promising new approach to probe the trace amount of oxidized G derivatives released from live cells.
In this report, we have explored an integrated EC and SERS approach for extracellular detection of oxidized G derivatives in the cell culture medium at the single-cell level. The measurements were achieved by using a glass nanopipette-based flexible gold nanoelectrode (fGNE). In the live cell experiment, the nanoscale and flexible tip of the fGNE can be positioned very close to the cell membrane with minimal damage to the cell. Without further chemical modification, the oxidized G base and nucleotide can attach to the exposed gold surface of the fGNE mainly through the strong Au–N interaction.33,34 After treatments with the often-used DNA damage agents chromium(VI) ion35,36 or hydrogen peroxide (H2O2),20,37,38 increased amounts of oxidized G derivatives are released from individual cells and can be effectively collected by the fGNE positioned very close to the cells. The fGNE shows good responses to G oxidative products, including 8-OHdG and 8-OXOG, in differential pulse voltammetry (DPV) and SERS measurements. By comparing with spiked 8-OHdG in a controlled environment, we conclude that the fGNE-based method can detect about 0.1 nM concentration of G oxidative products in the cell culture medium.
Experimental
Reagents and materials
NaCl, KCl, Na2HPO4·2H2O, KH2PO4, trypan blue and H2SO4 of ACS grade or better were purchased from Shanghai Aladdin without further purification. 1×PBS was prepared from deionized water (∼18.2 MΩ cm, Millipore). AgNO3, Ru(NH3)6Cl3, DNA bases (A, T, G, C), 8-hydroxy-2′-deoxyguanosine (8-OHdG), and K2CrO4 were purchased from Sigma-Aldrich. HAuCl4·4H2O and H2O2 were purchased from Shanghai Macklin and Beijing Chemical Works, respectively. Silver nanoparticles (AgNPs, ∼59 nm) were synthesized as previously described.39 The characterization of AgNPs is described in the ESI.†Fabrication of the fGNE
The details of fGNE fabrication can be found in the ESI.† In brief, a clean quartz capillary (QF100-70-7.5) was pulled by using a laser pipette puller (P-2000, Sutter Instrument) with the following parameters: HEAT = 650, FIL = 4, VEL = 60, DEL = 170, and PUL = 180. The nanopore size was determined by ionic current measurements. Then CNEs were formed inside the nanopipette barrel by the pyrolysis of butane gas, which flows through the nanopipette barrel with controlled pressure.40 The fabricated CNEs were then placed in 1×PBS solution with 10 mM HAuCl4. Gold was electrochemically deposited on the CNE apex as well as the nearby glass surface at a constant −70 mV bias for 8 min (see Fig. S3†).41 The electrode surface area was determined by the diffusion-limited current of cyclic voltammograms (CVs). The nanopipette, CNE and fGNE were also imaged by SEM (JEOL JSM-7610F).Characterization of nanopipettes, CNEs, and fGNEs by electrochemical measurements
The electrochemical measurements of G and 8-OHdG were conducted using a potentiostat (CHI 852D). An Ag/AgCl wire electrode was used as the quasi-reference electrode (0.2 mm in diameter) and a Pt wire (0.5 mm in diameter) was used as the counter electrode. For the measurements in the controlled environment, the DPV measurements were performed in 10 mM PBS (pH 7.0–7.5) with various G or 8-OHdG concentrations. The DPV measurement of 8-OHdG was also conducted in the cell culture medium. To study G oxidation, CVs were obtained continuously with two different potential sweep rates (20 V s−1 and 0.05 V s−1) for 50 cycles in 10 mM PBS solution with 10 μM G to obtain the oxidation products of G on the fGNE surface. Subsequently, DPV measurements were performed using fGNEs with the adsorbed oxidation products of G in 10 mM PBS solution.SERS measurements for products of DNA oxidative damage on the fGNE
Before the SERS measurements, the fGNE tethered with products of DNA oxidative damage was placed in 10 mM PBS with about 80 pM of 59 nm size AgNPs for 5 min to enhance the SERS signal. SERS measurements were performed using a confocal Raman microscope system (Horiba LabRAM HR evolution). The samples were excited with a 532 nm laser with 20 mW power, and a ×50 objective was used in the measurements to focus the laser beam spot (about 2 μm size) on the fGNE tip. The typical exposure time was 10 s. The Raman spectral resolution was 0.3 cm−1. The acquired Raman spectra were smoothened by a moving average method (neighboring 5 points) using Origin software (OriginLab).Cell culture, DNA damage treatment, and DNA oxidative damage product collection
Three types of cells, human lung cancer cells (A549), human cervical cancer cells (HeLa), and African green monkey kidney cells (Vero), were purchased from the Institutes of Biological Sciences (Shanghai, China). A549 and HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM), and Vero cells were cultured in minimum Eagle's medium (MEM). Each type of medium was supplemented with 10% fetal bovine serum, penicillin (100 μg mL−1), and streptomycin (100 μg mL−1). The cells were cultured at 37 °C in a humidified incubator containing 5% CO2. The cells were subcultured until 80% of the Petri dish was covered by cells. For DNA damage treatment, concentrated K2CrO4 or H2O2 solution was added into the Petri dish to reach a final concentration of 20 μM, and the treatment time was always 2 h. Before measurements, cells attached to the glass slides were washed with 1×PBS and rinsed with the medium. A micromanipulator (TransferMan 4r, Eppendorf) was used to precisely control the distance between the fGNE and the cell membrane surface. The fGNEs collected the DNA oxidative damage products at an applied bias of −70 mV for different times. In the experiments of collecting DNA oxidative damage products, a temperature control system (Warner Instruments, TC-344C) maintained the temperature at 37 °C.DFT calculation
Gaussian 09 software was used for geometric optimization and Raman spectra calculation of all four DNA bases and 8-OHdG. The Raman spectra calculations were conducted by using the functional B3LYP and the basis set 6-311+G**. A scaling factor of 0.963 was used for all the vibrational bands.Results and discussion
Experimental setup for detecting the DNA oxidative damage products
The fabrication of fGNEs has been reported previously.42 The details are also shown in Fig. S1.† In brief, the empty barrel of the single-barrel nanopipette was filled with a carbon nanoelectrode (CNE) after the pyrolysis of butane gas. Then, gold was electro-deposited on the exposed CNE at the orifice. Fig. 1A shows the optical microscopy images of the side view of the nanopipette (i) and fabricated fGNE tip (ii) and SEM images of the apex of the nanopipette (iii) and fGNE (iv). The mean diameter of the nanopore is about 102 ± 16 nm as determined by ionic current measurements (Fig. S2†). The cross section of the fGNE shows gold plating at the apex of the CNE. After gold deposition on the CNE, the diffusion-limited current in the CV increased by about 6-fold (see Fig. S3A†). Accordingly, the mean effective surface area increased from 0.78 ± 0.41 μm2 for the CNEs to 5.06 ± 0.89 μm2 for the fGNEs (Fig. S3B and C†). A stable and pronounced gold reduction peak appeared in the CV during the repeated potential scans in 0.5 M H2SO4 (Fig. S4†), confirming the successful fabrication of stable fGNEs.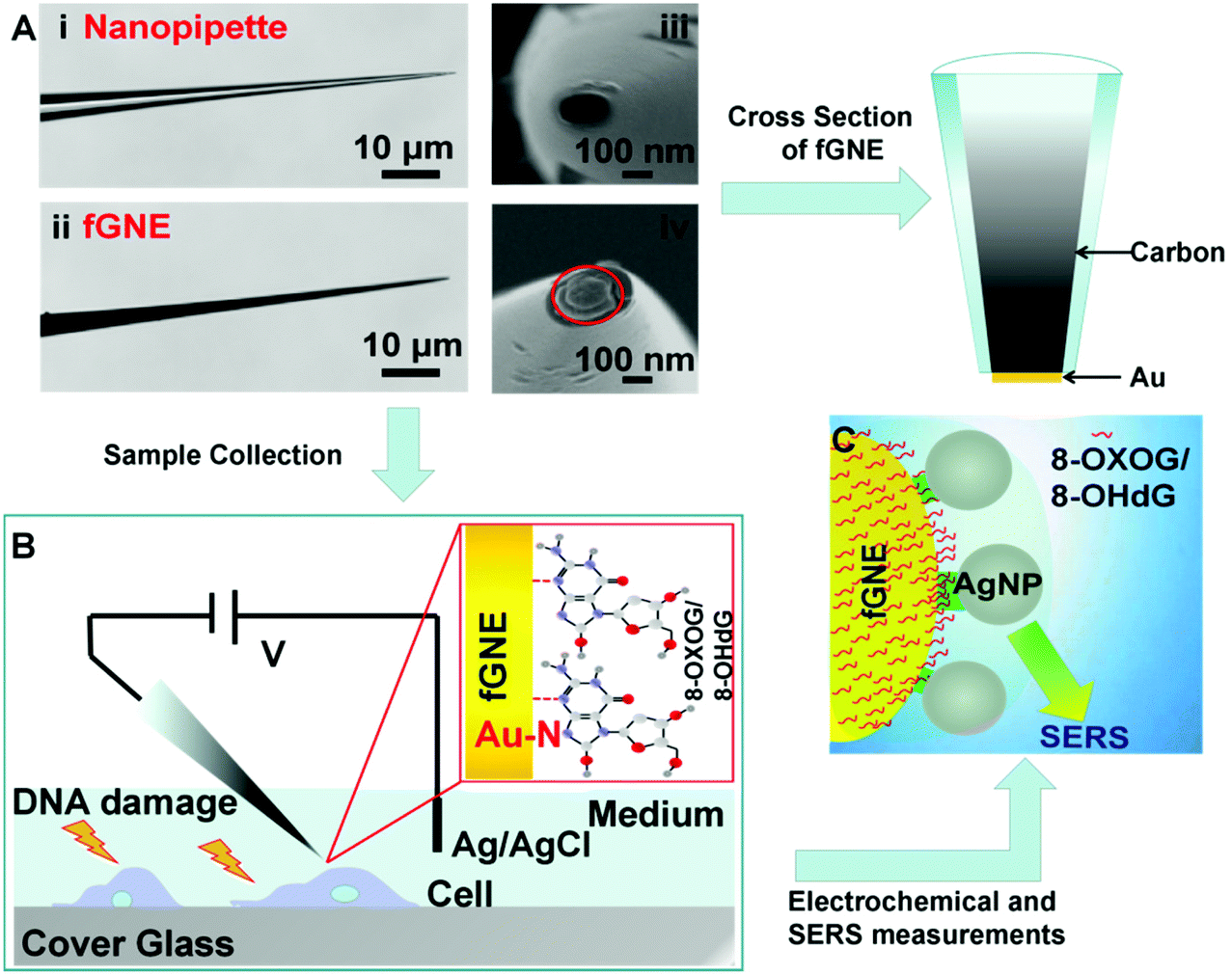 | ||
| Fig. 1 Schematic diagram of the experimental setup. (A) Optical (left) and SEM images (right) of the nanopipette (i and iii) and fGNE (ii and iv). The orifice pointed by the red circle indicates the successful deposition of gold on the carbon nanoelectrode. (B) Diagram of the fGNE collecting DNA damage products (only the 8-OHdG structure is shown here) from individual cells. (C) Illustration of SERS detection of cellular DNA damage products on the fGNE with AgNPs. | ||
In single-cell analysis, we used a micromanipulator to position the fGNE apex near the membrane surface of individual live cells that have been treated with the DNA damage agents K2CrO4 or H2O2 for 2 h (see Fig. 1B). Both reagents have often been used to induce cellular DNA damage. Fast release and increased levels of an oxidized G base and a nucleotide, including 8-OHdG and 8-OXOG, were reported.35–38 A negative bias (−70 mV) was applied on the fGNE to help attract the negatively charged DNA damage oxidative products on to the fGNE through the relatively strong Au–N interaction.33,43 The strong interaction between 8-OHdG and gold was also confirmed by the AuNP experiment using UV–vis spectroscopy (Fig. S5†).
The collected G and its derivatives on the fGNE apex can be detected and identified by differential pulse voltammetry (DPV) measurements based on the oxidation peak. We further used SERS to detect them. Before SERS measurements, AgNPs with a mean size of about 59 nm (Fig. S6†) were first deposited on the fGNE surface to enhance the SERS signal, as shown in Fig. 1C. Fig. S6C† shows an 8-OHdG-modified fGNE apex covered with AgNPs.
Electrochemical detection and oxidation of G and 8-OHdG
To evaluate the sensitivity and selectivity of fGNEs for detecting G and 8-OHdG, we first use the DPV method, which has a current noise level of 0.1 pA. Fig. 2A and C show the DPVs with a pronounced oxidation peak for G and 8-OHdG, respectively. The oxidation potentials are about 0.72 V for G and 0.43 V for 8-OHdG.44,45 We also conducted the DPV measurement in the solution with both 10 μM G and 8-OHdG. Both peaks of G and 8-OHdG can be observed (Fig. S7A†). In addition, the peaks of G or 8-OHdG are also lower than the oxidation peaks of other bases and thus can be clearly differentiated in the mixture (Fig. S7B†).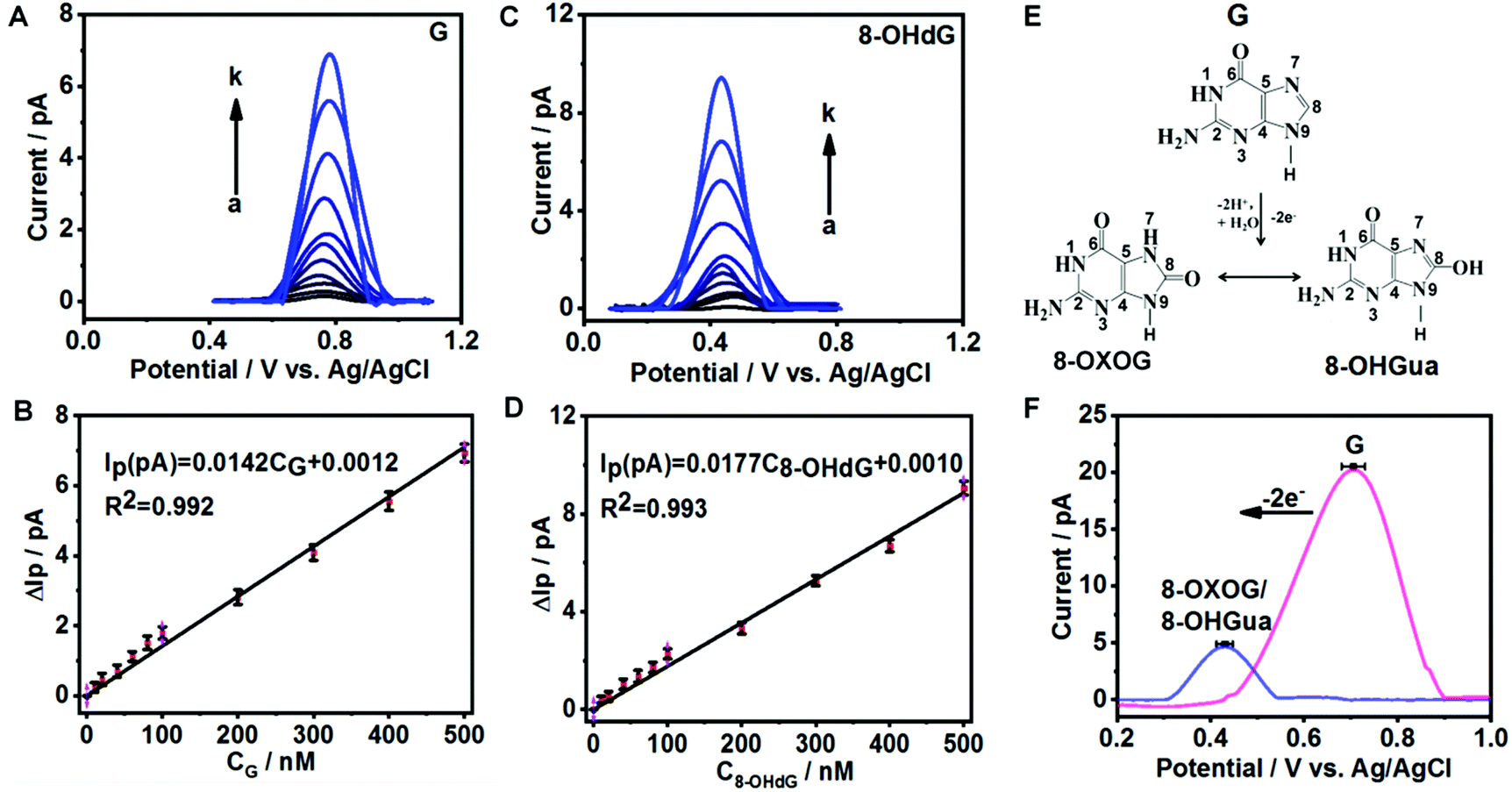 | ||
| Fig. 2 Electrochemical detection of G and 8-OHdG with the fGNE. (A, C) DPVs of G and 8-OHdG at different concentrations (from a to k, 0, 10, 20, 40, 60, 80, 100, 200, 300, 400, and 500 nM, respectively) in 10 mM PBS. The scan rate is 50 mV s−1. (B, D) The peak current (ΔIP) at different concentrations of G and 8-OHdG. The error bars represent standard deviations. (E) Diagram of the two-electron electrochemical oxidation of G on the surface of fGNEs. (F) The oxidative peaks before (pink) and after (purple) the completion of the two-electron transfer process of G. The error bar represents the standard deviation. | ||
The DPV peak height (ΔIP) increases proportionally with the concentration of G or 8-OHdG in the range from 0 to 500 nM (Fig. 2B and D). The stepwise increase of ΔIP with increasing concentrations of G and 8-OHdG shows the fast response and high sensitivity of the fGNE to G and 8-OHdG. The slopes of the ΔIPvs. concentration plots for G and 8-OHdG in the range of 0–500 nM are 0.014 pA nM−1 and 0.018 pA nM−1, respectively. With a S/N ratio of 3, the limits of detection (LoDs) for G and 8-OHdG are 5 nM and 10 nM, respectively, under the conditions of a collection time of a few minutes and in 10 mM PBS. In the control experiment, the ΔIP values generated by CNEs at the same concentration of G and 8-OHdG are lower than the corresponding values acquired from the fGNEs, indicating the higher sensitivity of the fGNE after gold deposition of the CNE (Fig. S8A and C†).
It has been demonstrated that the electrochemical oxidation of G can be controlled by the electrode potential sweep rate.23 As shown in Fig. 2E, when the sweep rate is high (20 V s−1), G undergoes the two-electron oxidation process, forming 8-OXOG and its minor tautomer 8-OHGua. In contrast, when the electrode potential sweep rate is low (V = 0.05 V s−1), G loses four electrons to form 8-OXOG (OX) (Fig. S9A†). As shown in Fig. 2F, after we sweep the electrode potential on the fGNE at 20 V s−1 for 50 cycles, a clear oxidation peak appears at +0.42 V in the DPV, which is down-shifted from +0.71 V. This oxidation peak becomes stable in the DPV at 50 mV s−1 and the peak position is consistent with the peak of 8-OXOG reported before.46–49 Therefore, the down-shifted oxidation peak confirms the two-electron oxidation transformation from G to 8-OXOG/8-OHGua on the fGNE apex surface. In contrast, after continuously sweeping the G-modified fGNE for 50 cycles with a low sweep rate of 50 mV s−1, the oxidation peak is up-shifted from +0.71 V to +0.78 V in the DPV (Fig. S9B†), suggesting that the four-electron oxidation occurs and the oxidation product is not 8-OXOG or 8-OHGua. We also noticed that the 8-OXOG/8-OHGua oxidation peak is very close to the oxidation peak of 8-OHdG in Fig. 2C. Therefore, we cannot further differentiate the base or nucleotide forms of oxidative G from the oxidation peak in the DPV measurements.
SERS detection of G and its oxidative derivatives
G or its derivatives attached to fGNEs were also measured by SERS. The tip of the fGNE was first immersed in an aqueous solution (DI water) with 10 nM G or 8-OHdG for a few minutes. AgNPs were then introduced to the fGNE apex to enhance the intensity of SERS spectra, as described in the experimental methods and shown in Fig. 1C.The typical Raman results are shown in Fig. 3. The G peaks can be identified in the normal Raman spectrum of G powder, as shown in Fig. 3A. The peaks in the SERS spectrum of G are generally broader and slightly shifted. The most noticeable peaks in the SERS spectrum of G are at 643, 934, 1342, 1381 and 1529 cm−1. The assignments of these peaks are shown in Table S1.†
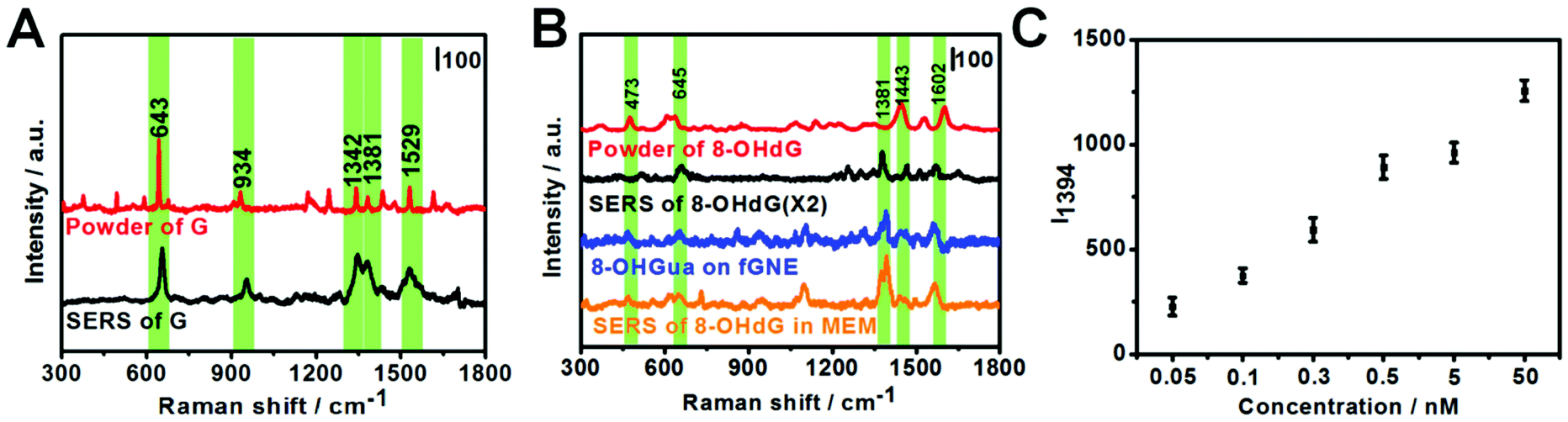 | ||
| Fig. 3 Raman spectra of G derivatives. (A) Raman spectrum of the G powder and SERS spectrum of G. (B) Raman spectrum of the 8-OHdG powder (red), SERS spectrum of the fGNE after immersing in DI water with 10 nM 8-OHdG (black), SERS spectrum of 8-OHGua oxidized from G on the fGNE (blue), and SERS spectrum of the fGNE after immersing in MEM with 0.1 nM 8-OHdG (orange). The spectra are offsetted for clarity. (C) The SERS peak intensity of I1394 for fGNEs that were immersed in MEM solutions with different concentrations of 8-OHdG. The error bar represents the standard deviations. | ||
The Raman spectrum of the 8-OHdG powder and the SERS spectrum of adsorbed 8-OHdG on the fGNE tip are shown in Fig. 3B. For the 8-OHdG powder, four characteristic peaks appeared at 473, 637, 1443 and 1602 cm−1. Compared with the normal Raman spectra, more spectral fluctuations are observed in the SERS spectra, as shown in three SERS spectra of 8-OHdG collected from different fGNEs under the same conditions in Fig. S10.† New peaks are observed near 1305 and 1381 cm−1. The peaks at 660, 1381 and 1580 cm−1 almost always appeared. In particular, the 1580 cm−1 peak is downshifted from 1602 cm−1 in the normal Raman spectrum. These changes are likely induced by the metal–molecule interactions on the gold surface.
The SERS spectrum of 8-OXOG/8-OHGua is also shown in Fig. 3B, which is collected after the two-electron oxidation of G on the fGNE. The major peaks are similar to the ones of adsorbed 8-OHdG on the fGNE. The 1381 cm−1 peak shifted slightly to around 1390 cm−1. Other than that, it is generally difficult to differentiate 8-OXOG/8-OHGua from 8-OHdG in the SERS spectra. Between the SERS spectra of G and its two-electron oxidation derivatives 8-OXOG/8-OHGua or 8-OHdG, the characteristic peak of G at 643 cm−1 is much stronger than the corresponding peak of 8-OHdG/8-OHGua/8-OXOG near 660 cm−1. Therefore, this peak can be used to differentiate G from its two-electron oxidative derivatives. We also measured the SERS spectrum of 8-OXOG (OX) after the four-electron oxidation on the fGNE (see Fig. S9C†). The SERS spectra of 8-OXOG (OX) are noticeably different from those of G or 8-OXOG/8-OHGua/8-OHdG. The SERS spectra of the other three DNA bases, A, T and C, were also measured on the fGNE surface. The characteristic peaks of G and its two-electron oxidation derivatives are quite different from those of the other three DNA bases and thus can be identified using the SERS spectra (Fig. S11†).50
To mimic the cell detection experiment (see the next paragraph), the fGNE was also immersed in MEM with various 8-OHdG concentrations, and a −70 mV bias voltage was applied for 40 min before the SERS measurements. Fig. 3B shows the typical SERS spectrum (orange color) for 0.1 nM 8-OHdG. With the applied bias and 40 min collection time, the intensity of the SERS peaks is obviously higher. Compared with the SERS spectrum of 8-OHdG adsorbed in DI water, the peak near 473 cm−1 appeared more often. The 1381 cm−1 peak often splitted into two peaks at 1371 and 1394 cm−1. The peak at 1580 cm−1 is further downshifted to 1560 cm−1. These spectral changes are likely due to the co-adsorbed interfering molecules, which are from the MEM medium, on the fGNE. The 1394 cm−1 peak is always the highest peak in the spectra. Therefore, we used the 1394 peak as the signature peak for 8-OHdG detection in MEM. As shown in Fig. 3C, the intensity of the 1394 peak (I1394) increased linearly with an increase of the 8-OHdG concentration in MEM. The corresponding spectra of these data points are shown in Fig. S12.† Considering a S/N ratio of 3, the LOD is about 0.1 nM for the SERS detection of 8-OHdG by the fGNE using −70 mV bias and 40 min collection time in MEM. Regarding the LOD, the SERS method is better than the DPV method.
In Table 1, we have presented the characteristic Raman peaks of 8-OHdG generated from the DFT calculation and the 8-OHdG powder and the SERS peaks from 8-OHdG-modified fGNEs with their adsorption in DI water or MEM.
| Calculated | Powder | SERS (adsorbed in DI water) | SERS (adsorbed in MEM) | Assignments |
|---|---|---|---|---|
| R5, five-membered ring; R6, six-membered ring; bend, bending; def, deformation; rock, rocking; sciss, scissoring; sqz, squeezing; str, stretching; wag, wagging. | ||||
| 453 | 473 | 431 | 473 | Wag N7–H, N9–H |
| 646 | 637 | 660 | 650 | Whole-molecule ring breathing |
| 1300 | 1305 | 1295 | Rock N1–H, str C5–C6 | |
| 1372 | 1382 | 1371/1394 | Rock N1–H, bend N1–C2–N3, N9–C4, def R5, R6 | |
| 1455 | 1443 | 1468 | Str N1–C2, sqz C4–C5, rock NH2 | |
| 1568 | 1602 | 1580 | 1560 | Str N7–C8O–N9, C6O–C5–C6 |
Probing the oxidative DNA damage products in living cells by the fGNE
After demonstrating 8-OXOG/8-OHGua and 8-OHdG detection by the fGNE using electrochemical and SERS techniques in a controlled environment, we applied the fGNE for detecting oxidation damage products of DNA in live cells. The kidney epithelial Vero cells were exposed to 20 μM K2CrO4 or H2O2 for 2 h. It has been found that the cells can complete the DNA damage repair within two hours and maintain the normal physiological state of the cells.51 The cell viability after the DNA damage treatment was also tested using the trypan blue exclusion test, and almost no dead cell was found at the used dosage (Fig. S13†). Then, the treated cells were placed in a fresh serum-free medium. Controlled by a micromanipulator, the fGNE slowly approached toward the cell until a small movement deformation of the cell membrane was observed in the optical image under a ×20 objective lens, suggesting that the apex already touched the cell surface (Fig. S14†). Then the tip apex was withdrawn about 2 μm, at which distance the cellular deformation disappeared. We defined this point as d0, and other fGNE–cell distances were determined by withdrawing a defined distance from d0 (see Fig. 4A). Facilitated by an applied bias of −70 mV to the fGNE, negatively charged DNA oxidation damage products were collected and attached to the surface of the fGNE. During the entire collection process of 40 min, the cells appear in a good condition in the culture medium MEM, without changes in their shape, volume, morphology, and substrate attachment.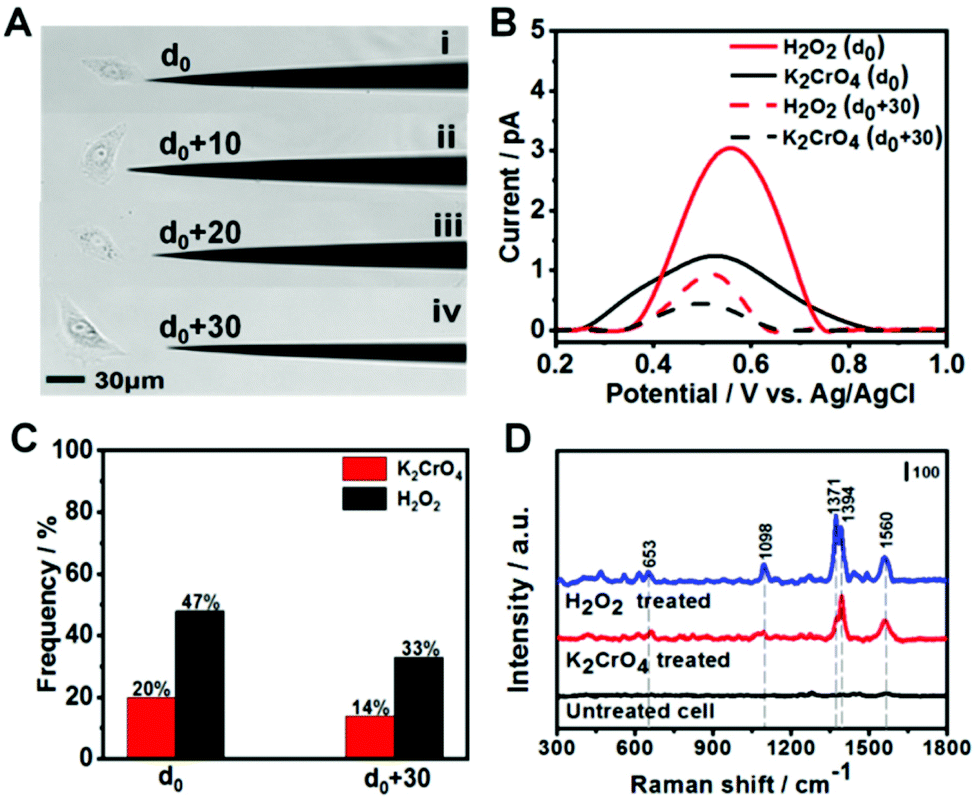 | ||
| Fig. 4 Detecting the DNA damage products in living cells. (A) Optical image of the different distances between the fGNE and cell membrane controlled by a micromanipulator. (B) The DPV peaks of damage products collected on the fGNE with different distances (d0, solid line; d0 + 30 μm, dotted line) after treating cells with a final concentration of 20 μM K2CrO4 (black) and H2O2 (red). (C) The probability of observing DPV peaks of 8-OHdG on fGNEs after collecting the damage products at distances d0 and d0 + 30 μm (N = 15). (D) SERS of the DNA damage products on the fGNEs measured at distance d0. The collection time is always 40 min. | ||
After collecting the damage products, the fGNE was placed in 10 mM PBS for DPV measurements. A clear peak around a potential of 0.5 V was observed (Fig. 4B), which is slightly higher than the oxidative peak of 8-OHdG or oxidized G base products 8-OXOG/8-OHGua near 0.43 V in the previous characterization (Fig. 2C). Considering that the DNA damage products were collected in the cell culture medium, we speculated that other substances adsorbed on the electrode surface hinder the electron transfer of oxidized G bases and nucleotides and increase their oxidation potential. The electrochemical current is also dependent on the collecting distance between the fGNE tip and cell membrane. The DPV peak current was higher at a distance d0 than that at d0 + 30 μm. This is attributed to the adsorption of more competitive substances (such as proteins) to the electrode surface. These competitive substances likely suppressed or blocked the electron transfer of the newly attached oxidized G bases and nucleotides, which is called electrode fouling. Therefore, chemical modification to the gold surface will be needed to improve the sensitivity of the DPV method in live cell measurements.
Although the DPV cannot accurately measure the released amount of DNA damage products, the DPV results still suggest that both K2CrO4 and H2O2 treatments can cause damage to the DNA of Vero cells and lead to the release of oxidized G bases and nucleotides. As we have shown in the previous section, the fGNE also effectively collects them. However, due to electrode fouling, only a fraction of fGNEs can observe the DPV peak near 0.5 V. This percentage also depends on the type of DNA damage agent and the distance between the fGNE tip and the cell membrane. As shown in Fig. 4C, when the fGNEs are at d0, the probability to detect the DPV peak near 0.5 V is 20% for K2CrO4 and 47% for H2O2, respectively. When the fGNEs are at d0 + 30 μm, the appearance probability is 14% for K2CrO4 and 33% for H2O2, respectively. Between K2CrO4 and H2O2 treatments, the latter always induces a higher DPV peak at a potential of 0.5 V. This is reasonable considering H2O2 is a stronger oxidant; it causes greater oxidative damage to DNA, leading to a higher level of released oxidized G bases and nucleotides after self-repairing.
To further analyze the DNA damage products, SERS measurements were conducted to detect the attached DNA damage products on the apex surface of fGNEs. Fig. 4D shows the typical SERS spectra from fGNEs after collecting DNA damage products from Vero cells treated with 20 μM K2CrO4 or H2O2 for 2 h. No clear SERS peaks can be identified for untreated Vero cells. In contrast, clear and almost identical peaks can be observed after K2CrO4 or H2O2 treatment. In both spectra, pronounced peaks appeared at 653, 1394, and 1560 cm−1. These peaks are consistent with the SERS peaks of 8-OHdG collected in MEM. Therefore, the SERS measurement further confirmed the successful collection of 8-OHdG or 8-OXOG on the fGNE apex. The signature peak of G near 643 cm−1 cannot be clearly identified in the spectra. Therefore, the amount of free G or dG and GTP in the extracellular medium is very low compared with the oxidized G products, which were released by the cell after various DNA repair mechanisms. By using Fig. 3C with spiked 8-OHdG as the calibration curve, the amount of oxidized G products released from a single cell can be roughly estimated by comparing the intensity of I1394. Although there are cell-to-cell fluctuations in I1394, the concentration of the released oxidized G products near the cell surface is generally less than 0.1 nM after K2CrO4 treatment. It is also found that the SERS intensity of the peaks of the oxidized G products from H2O2-treated cells is always higher than that of K2CrO4-treated cells, suggesting that more oxidized G products were released after H2O2 treatment. This is consistent with the DPV measurement.
To optimize the collection of oxidized G products, we first varied the distance between the fGNE apex and cell surface. As shown in Fig. 5A, I1394 increases when the fGNE apex is moved away from the cell membrane surface. Interestingly, this is opposite to the DPV results. When the fGNE is away from the cell membrane surface, it has the chance to collect more oxidized G products released from nearby cells. However, more competitive substances are also likely attached to the fGNE. These interferences made a bigger impact on the electrochemical current but contribute much less to the SERS signal. This result also underscores the importance of combined electrochemical and SERS measurements. Subsequently, we varied the collection time while fixing the distance between the fGNE and cell membrane at d0. As shown in Fig. 5B, I1394 increases with the increase of the collection time and reaches a maximum at 60 min. Interestingly, beyond 60 min, I1394 gradually decreases. One plausible explanation is that the cells actively repair DNA damage induced by the treatment in the first 60 min. After 60 min, the level of released oxidized G products gradually decreases. However, the decrease of I1394 may also be due to the saturation of the electrode. To test this possibility, we performed control experiments in the cell culture medium MEM (but without cells) with various concentrations of 8-OHdG (Fig. S15†). We found that I1394 continues to increase beyond 60 min as the collection time increases. Therefore, the decrease of the I1394 peak after 60 min in cellular measurements should be related to the recovery of cells after self-repair.
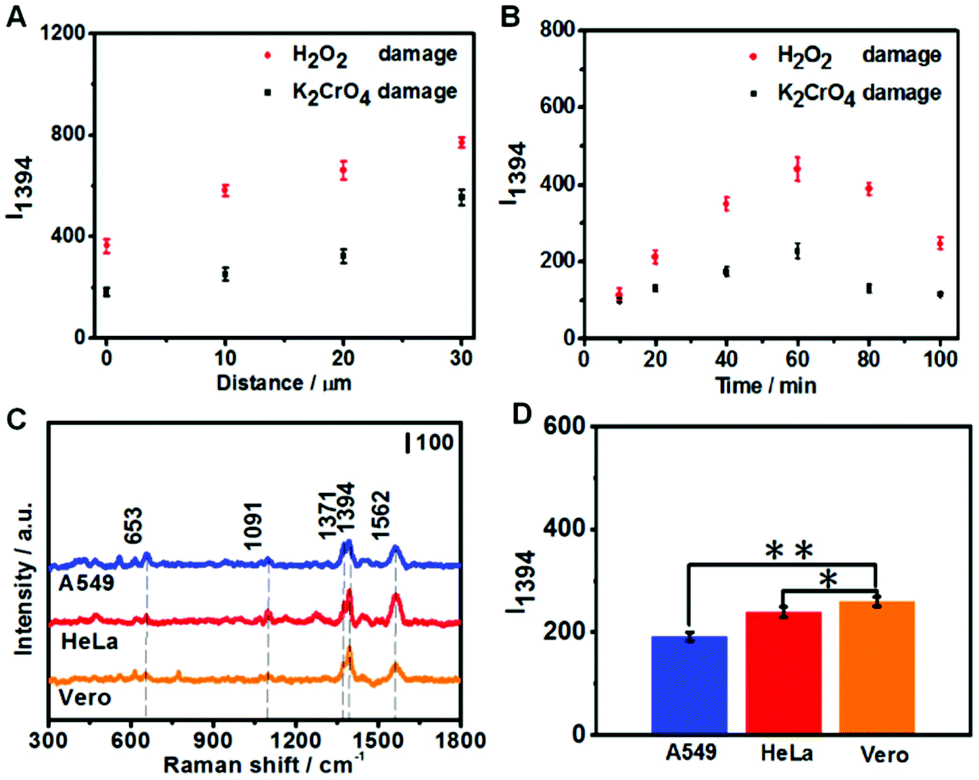 | ||
| Fig. 5 The SERS peak intensity I1394 of fGNEs under different conditions. (A) The I1394 value that collects the damage products for 40 min at different distances from the cell after K2CrO4 or H2O2 treatment. (B) The I1394 value that collects the damage products of cells for different times with distance d0 after K2CrO4 or H2O2 treatments. (C) The SERS spectra of fGNEs that collect the damage products resulting from three different cell lines at distance d0 after treatment with K2CrO4. (D) The intensity histograms of I1394 for fGNEs that collect the damage products resulting from three different cell lines at distance d0 after treatment with K2CrO4. Two-tailed Student's t-test: *P < 0.05, **P < 0.01. | ||
We further performed the measurements on two cancer cell lines (A549 and HeLa) under the same conditions. Three cell lines were both treated with 20 μM K2CrO4 for 2 h; then the fGNE was used to collect DNA damage products from individual cells at d0 for 40 min. It can be seen from Fig. 5C that similar SERS spectra were acquired from all three cell lines. However, the I1394 value of Vero cells is slightly higher than those of the other two cell lines (Fig. 5D), which is attributed to the higher resistance of cancer cells to environmental damage.
Conclusions
In summary, we have successfully prepared a fGNE for collecting and detecting DNA damage products released by individual live cells. To evaluate its capability to detect DNA damage products, we first tested the performance of the fGNE by using the most common DNA damage biomarkers 8-OHdG and 8-OXOG. The base form of G oxidative products can be acquired by electrochemical oxidation of the tethered G on the electrode. When the electrode potential scanning speed is high, G is mainly oxidized to 8-OXOG. fGNEs show high sensitivity and selectivity toward oxidative G products in the integrated electrochemistry and SERS measurements. By analyzing the oxidation potential of the DPV peak and the molecular fingerprints in the SERS spectra, we can confirm that the attached molecules are mainly oxidative G products instead of G or 8-OXOG (OX) or other non-G interferences. However, we cannot further determine the detailed structure of oxidized G products, including whether they are in base or nucleotide forms and with which tautomer structures.In the cellular measurements, the cells were treated with K2CrO4 or H2O2. By controlling the distance between the fGNE tip and the cell membrane, the collection time and the applied bias, we can optimize the collecting conditions and enhance the detection of released oxidative DNA damage products. Based on the DPV and SERS results, we can determine that mainly G oxidative products were released from the cell after both treatments. We found that at the same treatment dosage, more oxidative G products were generated by H2O2 treatment than K2CrO4 treatment. Similar detection results have been acquired in three different cell lines, HeLa, A549, and Vero. However, the cancer cells A549 and HeLa normally generated less DNA damage products than the normal cells Vero. The SERS measurement can overcome electrode fouling and show much better LOD (∼0.1 nM) for oxidative G and dG products than the DPV measurement. The detection limit by SERS is comparable to or better than a lot of methods for 8-OHdG detection in biofluids.25,52,53 Here, the electrode is not functionalized, and we relied on the strong Au–N interaction between gold and oxidative G. In recently reported works, aptamer-involved EC and spectroscopy methods can further improve the detection limit of 8-OHdG to the picomolar concentration.52,53 Similar chemical modification strategies, as well as other nanostructured materials,25 should also be adopted by the fGNE method to further improve its specificity and sensitivity and enable real-time detection by the electrochemical method. We also envision that the fGNE-based integrated analytical approaches can be applied for both intracellular detection and extracellular detection of other important cellular biomarkers at the single-cell level.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the National Natural Science Foundation of China (21673023 and 21773017). We acknowledge Jianghao Zhou for help with DFT calculations.Notes and references
- M. S. Cooke, M. D. Evans, M. Dizdaroglu and J. Lunec, FASEB J., 2003, 17, 1195–1214 CrossRef CAS.
- S. P. Jackson and J. Bartek, Nature, 2009, 461, 1071–1078 CrossRef CAS.
- J. Cadet, J. R. Wagner, V. Shafirovich and N. E. Geacintov, Int. J. Radiat. Biol., 2014, 90, 423–432 CrossRef CAS.
- S. Steenken and S. V. Jovanovic, J. Am. Chem. Soc., 1997, 119, 617–618 CrossRef CAS.
- E. C. Friedberg, Nature, 2003, 421, 436–440 CrossRef.
- M. S. Cooke, M. D. Evans, K. E. Herbert and J. Lunec, Free Radical Res., 2000, 32, 381–397 CrossRef CAS.
- C.-Y. Ock, E.-H. Kim, D. J. Choi, H. J. Lee, K.-B. Hahm and M. H. Chung, World J. Gastroenterol., 2012, 18, 302–308 CrossRef CAS.
- S. F. Lee and S. Pervaiz, in Methods in Cell Biology, ed. Z. Darzynkiewicz, E. Holden, A. Orfao, W. Telford and D. Wlodkowic, Academic Press, 2011, vol. 103, pp. 99–113 Search PubMed.
- B. Halliwell and M. Whiteman, Br. J. Pharmacol., 2004, 142, 231–255 CrossRef CAS.
- H. J. Helbock, K. B. Beckman and B. N. Ames, in Methods in Enzymology, Academic Press, 1999, vol. 300, pp. 156–166 Search PubMed.
- L. L. Wu, C.-C. Chiou, P.-Y. Chang and J. T. Wu, Clin. Chim. Acta, 2004, 339, 1–9 CrossRef CAS.
- M. A. Lovell and W. R. Markesbery, Nucleic Acids Res., 2007, 35, 7497–7504 CrossRef CAS.
- I. Dalle-Donne, R. Rossi, R. Colombo, D. Giustarini and A. Milzani, Clin. Chem., 2006, 52, 601–623 CrossRef CAS.
- M. Dizdaroglu, P. Jaruga and H. Rodriguez, Nucleic Acids Res., 2001, 29, e12–e12 CrossRef CAS.
- S. Koide, Y. Kinoshita, N. Ito, J. Kimura, K. Yokoyama and I. Karube, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2010, 878, 2163–2167 CrossRef CAS.
- B. Halliwell and M. Dizdaroglu, Free Radical Res. Commun., 1992, 16, 75–87 CrossRef CAS.
- S. Mahajan, T.-C. Lee, F. Biedermann, J. T. Hugall, J. J. Baumberg and O. A. Scherman, Phys. Chem. Chem. Phys., 2010, 12, 10429 RSC.
- B. Yin, R. M. Whyatt, F. P. Perera, M. C. Randall, T. B. Cooper and R. M. Santella, Free Radicals Biol. Med., 1995, 18, 1023–1032 CrossRef CAS.
- K. Shimoi, H. Kasai, N. Yokota, S. Toyokuni and N. Kinae, Cancer Epidemiol., Biomarkers Prev., 2002, 11, 767–770 CAS.
- R. P. Soultanakis, R. J. Melamede, I. A. Bespalov, S. S. Wallace, K. B. Beckman, B. N. Ames, D. J. Taatjes and Y. M. W. Janssen-Heininger, Free Radicals Biol. Med., 2000, 28, 987–998 CrossRef CAS.
- A. R. Collins, J. Cadet, L. Mőller, H. E. Poulsen and J. Viña, Arch. Biochem. Biophys., 2004, 423, 57–65 CrossRef CAS.
- K. S. Korkmaz, B. Debelec Butuner and D. Roggenbuck, J. Lab. Precis. Med., 2018, 3, 95–95 CrossRef.
- E. E. Ferapontova, Electrochim. Acta, 2004, 49, 1751–1759 CrossRef CAS.
- J. Langmaier, Z. Samec and E. Samcová, Electroanalysis, 2003, 15, 1555–1560 CrossRef CAS.
- A. M. Chiorcea-Paquim and A. M. Oliveira-Brett, Mikrochim. Acta, 2021, 188, 58 CrossRef CAS.
- S. Prabhulkar and C.-Z. Li, Biosens. Bioelectron., 2010, 26, 1743–1749 CrossRef CAS.
- P. Actis, S. Tokar, J. Clausmeyer, B. Babakinejad, S. Mikhaleva, R. Cornut, Y. Takahashi, A. López Córdoba, P. Novak, A. I. Shevchuck, J. A. Dougan, S. G. Kazarian, P. V. Gorelkin, A. S. Erofeev, I. V. Yaminsky, P. R. Unwin, W. Schuhmann, D. Klenerman, D. A. Rusakov, E. V. Sviderskaya and Y. E. Korchev, ACS Nano, 2014, 8, 875–884 CrossRef CAS.
- J. Zhang, J. Zhou, R. Pan, D. Jiang, J. D. Burgess and H.-Y. Chen, ACS Sens., 2018, 3, 242–250 CrossRef CAS.
- S.-M. Lu and Y.-T. Long, TrAC, Trends Anal. Chem., 2019, 117, 39–46 CrossRef CAS.
- J. Guo, A. Sesena Rubfiaro, Y. Lai, J. Moscoso, F. Chen, Y. Liu, X. Wang and J. He, Analyst, 2020, 145, 4852–4859 RSC.
- S. Hanif, H.-L. Liu, S. A. Ahmed, J.-M. Yang, Y. Zhou, J. Pang, L.-N. Ji, X.-H. Xia and K. Wang, Anal. Chem., 2017, 89, 9911–9917 CrossRef CAS.
- E. A. Vitol, Z. Orynbayeva, M. J. Bouchard, J. Azizkhan-Clifford, G. Friedman and Y. Gogotsi, ACS Nano, 2009, 3, 3529–3536 CrossRef CAS.
- P. H. Acioli and S. Srinivas, J. Mol. Model., 2014, 20, 2391 CrossRef.
- X. Zhang, C. Q. Sun and H. Hirao, Phys. Chem. Chem. Phys., 2013, 15, 19284–19292 RSC.
- K. D. Sugden, C. K. Campo and B. D. Martin, Chem. Res. Toxicol., 2001, 14, 1315–1322 Search PubMed.
- H. W. Kuo, Occup. Environ. Med., 2003, 60, 590–594 CrossRef CAS.
- Y. Fujii, K. Tomita, H. Sano, A. Yamasaki, Y. Hitsuda, I. M. Adcock and E. Shimizu, Cancer Cell Int., 2002, 2, 16 CrossRef.
- H. Kasai, P. F. Crain, Y. Kuchino, S. Nishimura, A. Ootsuyama and H. Tanooka, Carcinogenesis, 1986, 7, 1849–1851 CrossRef CAS.
- S. Agnihotri, S. Mukherji and S. Mukherji, RSC Adv., 2014, 4, 3974–3983 RSC.
- N. Panday, G. Qian, X. Wang, S. Chang, P. Pandey and J. He, ACS Nano, 2016, 10, 11237–11248 CrossRef CAS.
- D. Yang, G. Liu, H. Li, A. Liu and J. He, Analyst, 2020, 145(3), 1047–1055 RSC.
- F. Lussier, T. Brule, M. J. Bourque, C. Ducrot, L. É. Trudeau and J. F. Masson, Faraday Discuss., 2017, 205, 387–407 RSC.
- H. Schmidbaur, H. G. Raubenheimer and L. Dobrzańska, Chem. Soc. Rev., 2014, 43, 345–380 RSC.
- J. Zhang, D. Han, S. Wang, X. Zhang, R. Yang, Y. Ji and X. Yu, Electrochem. Commun., 2019, 99, 75–80 CrossRef CAS.
- I. Pandey and J. D. Tiwari, Sens. Actuators, B, 2019, 285, 470–478 CrossRef CAS.
- G. V. Martins, A. P. M. Tavares, E. Fortunato and M. G. F. Sales, Sci. Rep., 2017, 7, 14558 CrossRef.
- Z. Guo, X. Liu, Y. Liu, G. Wu and X. Lu, Biosens. Bioelectron., 2016, 86, 671–676 CrossRef CAS.
- F. D'Amico, F. Cammisuli, R. Addobbati, C. Rizzardi, A. Gessini, C. Masciovecchio, B. Rossi and L. Pascolo, Analyst, 2015, 140, 1477–1485 RSC.
- N. Jayanth, S. Ramachandran and M. Puranik, J. Phys. Chem. A, 2009, 113, 1459 CrossRef CAS.
- J.-M. Yang, L. Jin, Z.-Q. Pan, Y. Zhou, H.-L. Liu, L.-N. Ji, X.-H. Xia and K. Wang, Anal. Chem., 2019, 91, 6275–6280 CrossRef CAS.
- A. S. Rubfiaro, P. S. Tsegay, Y. Lai, E. Cabello, M. Shaver, J. Hutcheson, Y. Liu and J. He, ACS Appl. Bio Mater., 2021, 4(2), 1632–1639 CrossRef CAS.
- Y. Liu, M. Wei, L. Zhang, Y. Zhang, W. Wei, L. Yin, Y. Pu and S. Liu, Anal. Chem., 2016, 88, 6509–6514 CrossRef CAS.
- J. Fan, Y. Liu, E. Xu, Y. Zhang, W. Wei, L. Yin, Y. Pu and S. Liu, Anal. Chim. Acta, 2016, 946, 48–55 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1an00220a |
| This journal is © The Royal Society of Chemistry 2021 |