
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePeptide late-stage C(sp3)–H arylation by native asparagine assistance without exogenous directing groups†
Yiyi
Weng
 *ab,
Xingxing
Ding
a,
João C. A.
Oliveira
b,
Xiaobin
Xu
a,
Nikolaos
Kaplaneris
b,
Meijie
Zhu
a,
Hantao
Chen
a,
Zhuo
Chen
a and
Lutz
Ackermann
*b
*ab,
Xingxing
Ding
a,
João C. A.
Oliveira
b,
Xiaobin
Xu
a,
Nikolaos
Kaplaneris
b,
Meijie
Zhu
a,
Hantao
Chen
a,
Zhuo
Chen
a and
Lutz
Ackermann
*b
aCollege of Pharmaceutical Sciences, Zhejiang University of Technology, Hangzhou 310014, P. R. China. E-mail: wengyoyo@163.com
bInstitut fuer Organische und Biomolekulare Chemie, Georg-August-Universitaet Goettingen, Tammannstrasse 2, Goettingen 37077, Germany. E-mail: Lutz.Ackermann@chemie.uni-goettingen.de
First published on 12th August 2020
Abstract
There is a strong demand for novel native peptide motifs for post-synthetic modifications of peptides without pre-installation and subsequent removal of directing groups. Herein, we report an efficient method for peptide late-stage C(sp3)–H arylations assisted by the unmodified side chain of asparagine (Asn) without any exogenous directing group. Thereby, site-selective arylations of C(sp3)–H bonds at the N-terminus of di-, tri-, and tetrapeptides have been achieved. Likewise, we have constructed a key building block for accessing agouti-related protein (AGRP) active loop analogues in a concise manner.
Introduction
Peptides are increasingly important drug candidates, which are largely employed to treat metabolic disorders, cancer, allergy, and immune and cardiovascular diseases.1 They also represent key tools that modulate biological events mediated by protein–protein interactions (PPIs).2 Native peptides usually suffer from poor pharmacological features due to lack of structural diversity or enzymatic degradation,3 but chemically modified non-natural peptides could feature higher binding affinities to the target, as well as improved pharmacokinetics, stability, and cell permeability.4The late-stage modification represents an effective strategy to obtain structurally diverse peptides and peptidomimetics. Thus, late-stage modification methods of peptides have been achieved in terms of arylations,5 alkylations,6 and cycloadditions.7 Over the past few years, C–H activation has been recognized as an atom- and step-economical pathway towards molecular syntheses,8 with remarkable applications in materials science,9 the agrochemical industry,10 and drug discovery,11 among others.12 To the best of our knowledge, studies on late-stage functionalizations of peptides via C(sp3)–H activation have been scarcely reported. In this context, Yu13 successfully implemented C(sp3)–H activation of peptides using native N,O- or N,N-bidentate coordination without external auxiliary (Fig. 1a). On a different note, Noisier/Albericio14 reported the synthesis of a novel class of stapled peptides. Likewise, research studies of post-synthetic modification of peptides through C(sp3)–H activation by installing exogenous auxiliary assistance have been pursued. In 2017, Ackermann15 developed a strategy of triazole (Tzl)-assisted C(sp3)–H arylations of peptides. In the same year, Chen16 described 8-aminoquinoline (AQ)-directed C(sp3)–H arylation to generate cyclophane-braced peptide macrocycles. Recently, Shi17 established a palladium-catalyzed site selective γ-C(sp3)–H silylation and δ-C(sp3)–H alkylation of amino acids and peptides utilizing picolinamide (PA) auxiliary (Fig. 1b). The installation and subsequent removal of DGs often implies additional and non-trivial steps. Considering the atom- and step-economy of late-stage modification of peptides, we intended to utilize the natural amino acid embedded in the peptide backbone for chelation assistance. To our knowledge, C(sp3)–H functionalizations of peptides assisted by the unmodified side chain of a natural amino acid has not been accomplished thus far.
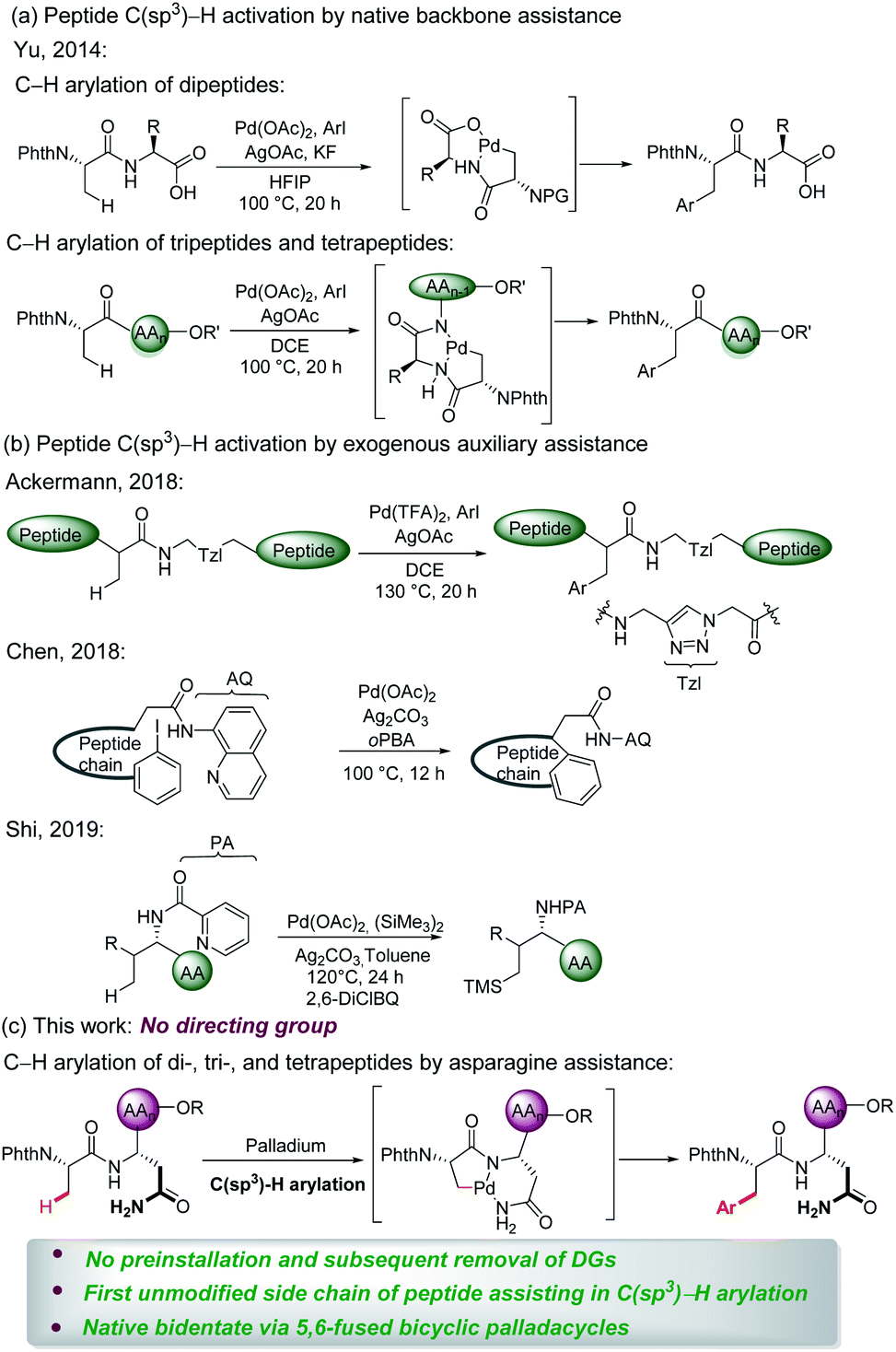 | ||
| Fig. 1 Palladium-catalyzed C(sp3)–H functionalization of peptides. | ||
Asn is a natural amino acid with a side chain bearing a primary amide and could potentially be exploited as a directing group. This prompts us to survey whether the side chain and backbone of Asn could coordinate with palladium, leading to a bidentate coordinated palladium complex. Therefore, we introduce Asn as an internal bidentate DG to accomplish C(sp3)–H activation of peptides. Simultaneously, Asn is a common residue contained in many bioactive peptides, which display a range of biological activities, such as antioxidant activity,18 blocking the neprilysin activity,19 and inhibiting ACE activity.20 Remarkably, Phe–Asn is an essential sequence that exists in some bioactive peptides, for example novel ACE inhibitory peptides,21 anticancer peptides,22 and AGRP.23 Inspired by the significant work by Ackermann et al.,15,24 we provide a useful strategy employing Asn as an internal directing group for C(sp3)–H functionalization of peptides. The unmodified side chain of Asn combined with the backbone was utilized as the N,N- bidentate coordination via 5,6-fused bicyclic palladacycles (Fig. 1c) to perform the late-stage peptide C(sp3)–H arylation.
The complex has facilitated the inert C(sp3)–H bond arylation in peptides. Thereby, arylated di-, tri-, and tetrapeptides containing Asn have been assembled. The salient features of our approach comprise (a) C(sp3)–H activation of peptides assisted by a natural amino acid which circumvent the preinstallation and removal of DGs; (b) the first unmodified side chain of the natural amino acid as the endogenous auxiliary assistance applied in C(sp3)–H activation; and (c) discovery of native bidentate assistance through less-strained 5,6-fused bicyclic palladacycles.25
Results and discussion
Optimization of reaction conditions
To validate our hypothesis, we initiated our studies by exploring reaction conditions for the palladium(II)-catalyzed primary C(sp3)–H arylation of N-phthaloyl protected dipeptide 1 with 3-iodotoluene (Tables 1 and S1 in the ESI†). Initial optimization revealed DCE to be the best solvent of choice (Table S1,† entries 1–5), with KF being identified as the optimal additive (entries 1–3). By replacing Pd(OAc)2 by PdCl2 as the catalyst the yield of product 2a was excitingly increased to 67% when the amount of AgOAc and KF was increased to 2.5 equivalent (entry 4). Notably, the reaction failed to proceed using AgOTf as the additive (entry 5), while Cu(OAc)2 gave a dramatically decreased yield (entry 6). Encouraged by the good efficiency of PdCl2, other palladium catalysts were further investigated. Gratifyingly, Pd(MeCN)2Cl2 was found to slightly improve the yield of peptide 2a to 72% (entries 7–9). It is noteworthy that other metal catalysts, based on ruthenium, rhodium or cobalt, were ineffective (entries 10–12). The control experiment verified the essential role of the palladium catalyst (Table S1,† entry 20).|
|
||||
|---|---|---|---|---|
| Entry | [TM] | Oxidant | Additives | Yield/% |
| a Reaction conditions: 1 (0.20 mmol), 3-Me–C6H4I (0.40 mmol), oxidant (0.40 mmol), [TM] (10 mol%), additive (0.40 mmol), DCE (2.0 mL), 130 °C, 12 h, yields of isolated products. b Oxidant (0.50 mmol), additive (0.50 mmol). | ||||
| 1 | Pd(OAc)2 | AgOAc | NaOAc | 26 |
| 2 | Pd(OAc)2 | AgOAc | Cs2CO3 | Trace |
| 3 | Pd(OAc)2 | AgOAc | KF | 41 |
| 4 | PdCl2 | AgOAc | KF | 67b |
| 5 | PdCl2 | AgOTf | KF | —b |
| 6 | PdCl2 | Cu(OAc)2 | KF | 10b |
| 7 | Pd(MeCN)2Cl2 | AgOAc | KF | 72b |
| 8 | Pd2(dba)3 | AgOAc | KF | 36b |
| 9 | Pd(PPh3)2Cl2 | AgOAc | KF | 35b |
| 10 | [RuCl2(p-cymene)]2 | AgOAc | KF | —b |
| 11 | [Cp*RhCl2]2 | AgOAc | KF | —b |
| 12 | Co(OAc)2·4H2O | AgOAc | KF | —b |
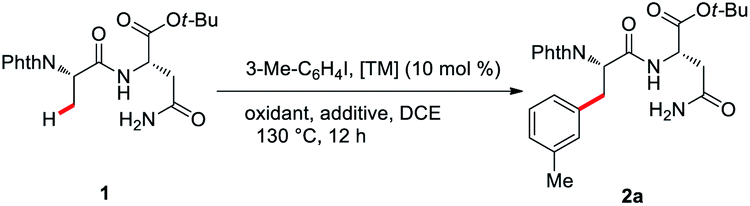
Substrate scope
With the optimal reaction conditions in hand, the substrate scope of a range of aryl iodides was investigated, and the results are summarized in Scheme 1. Both substrates with electron-donating (Me–, MeO–, and t-Bu–) and electron-withdrawing (F–, Cl–, Br–, CF3–, and CO2Me–) groups reacted smoothly and afforded the desired products in good yields. Pleasingly, biphenyl and naphthyl moieties were also tolerated, leading to the corresponding products (2m and 2n) in 63% and 64% yields. The reaction performed with good chemo-selectivity. | ||
| Scheme 1 Scope of ArI C(sp3)–H arylation of dipeptides. Reaction conditions: 1 (0.2 mmol), ArI (2.0 equiv.), Pd(MeCN)2Cl2 (10 mol%), AgOAc (2.5 equiv.), KF (2.5 equiv.), DCE (2.0 mL), 130 °C in air, 12 h, yields of isolated products. | ||
Encouraged by the success of the arylation of dipeptides, we next investigated the feasibility of applying this approach to the arylation of tripeptides and tetrapeptides (Scheme 2). Using tripeptide 3a as the substrate, through minor adjustment of the reaction conditions (Table S2 in the ESI†), we were pleased to find that the arylation of 3a with 1-iodo-4-methoxybenzene 4a could deliver the expected product 5aa in 61% isolated yield. Then, the scope of substrates was evaluated under the optimized reaction conditions. Satisfyingly, a wide range of aliphatic amino acids, including Leu, Ala, Val, and Lys, at the C-terminus of the tripeptides are compatible with these conditions. In addition, aryl iodides bearing electron-donating as well as electron-withdrawing substituents were tolerated, affording products 5aa–5ej. Given the feasibility of the tripeptide arylation, we expanded the peptide substrates to structurally complex tetrapeptides. The arylation products of tetrapeptides 6fa–6gh could be obtained in moderate yields (50–58%). Phe-containing tetrapeptide 3h could also be arylated albeit with lower yields (6hg–6hk, 25–36%). While considerable progress has been made in C(sp3)–H activation,26 our strategy enabled position-selective arylation of Ala assisted by N,N-bidentate coordination of the Asn in tri- and tetra-peptides.
 | ||
| Scheme 2 Scope of C(sp3)–H arylation of tripeptides and tetrapeptides. aReaction conditions: 3 (0.2 mmol), ArI (2.5 equiv.), PdCl2 (15 mol%), AgOAc (3.0 equiv.), KF (2.0 equiv.), DCE (3.0 mL), 110 °C in air, 12 h, yields of isolated products. bAgOAc (2.5 equiv.). cKF (1.0 equiv.) d120 °C, no base. e130 °C. | ||
To further demonstrate that the reaction coordination site is the primary amide of Asn, the control reaction and competition reaction were investigated under the standard conditions (Scheme 3). First, we removed the Asn side chain of dipeptide 1 and replaced it with a methyl group, while retaining the tert-butyl ester of the dipeptide. Therefore, N-phthaloyl protected dipeptide 7 was independently prepared, and subjected to the optimized reaction conditions. It failed to afford arylated products of arylation of C(sp3)–H bonds at the N-terminus (Scheme 3a). Since tripeptides or tetrapeptides both contain Asn bidentate and backbone amide bidentate, it is important to analyze the key role of Asn bidentate in promoting C(sp3)–H functionalization. For the competition experiment between tripeptides 3c and 8a, product ratio of approximately (5cg![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9a = 6:1) (Scheme 3b) was obtained.
:9a = 6:1) (Scheme 3b) was obtained.
 | ||
| Scheme 3 (a) Control experiment under the standard conditions. (b) Competition experiments under optimized conditions. | ||
Mechanistic investigation
Additionally, we probed the catalyst mode of action by means of computational studies at the PW6B95-D4/def2-TZVP+SMD (DCE)//PBE0-D3BJ/def2-SVP level of theory (Fig. 2).27 A detailed analysis between the C–H activation and reductive elimination elementary steps provided support for the C–H activation to be the rate-determining step with an activation energy of 19.6 kcal mol−1, with oxidative addition being energetically more favorable by only 1 kcal mol−1. An alternative pathway where the NH2 of the terminal amide is deprotonated was also taken into consideration (Fig. S1, see the ESI†). The latter was shown to be overall energetically disfavored, with reductive elimination as the rate-determining step with a high energy barrier of 30.9 kcal mol−1. These studies provide strong support for the palladium-catalyzed C(sp3)–H arylation to occur through a Pd(II/IV) pathway where the NH of the internal, instead of the terminal amide is deprotonated.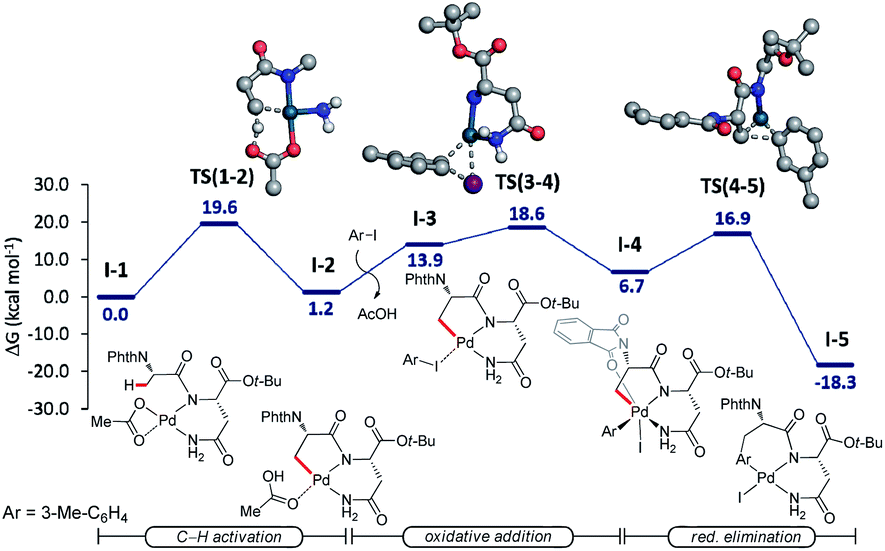 | ||
| Fig. 2 Computed relative Gibbs free energies (ΔG403.15) in kcal mol−1 for palladium(II)-catalyzed C(sp3)–H arylation at the PW6B95-D4/def-TZVP+SMD(DCE)//PBE0-D3BJ/def2-SVP level of theory. Non-relevant hydrogen atoms on the transition state structures were omitted for clarity. | ||
Based on previous reports on palladium-catalyzed amide-directed C–H bond activation and computational studies, we propose a plausible catalytic cycle to be initiated by a facile organometallic C–H activation (Scheme 4). Initially, the palladium catalyst coordinates covalently with the deprotonated NH of the internal amide generating a bidentate coordinated palladium(II) complex A. Subsequently, complex A undergoes slow C(sp3)–H bond cleavage to form the 5,6-fused bicyclic palladium complex B. The oxidative addition of the aryl iodide to B affords palladium(IV) intermediate C, which then undergoes reductive elimination followed by protonation leading to the formation of the corresponding arylated product. The silver salt is proposed to accelerate the rate of the oxidative addition or the reductive elimination, while likewise acting as a halide scavenger.8i,24a,28
 | ||
| Scheme 4 Proposed mechanism. | ||
Agouti-related protein (AGRP) is a potent orexigenic peptide that antagonizes the melanocortin-3 and melanocortin-4 receptors (MC3R and MC4R).29 This protein has been physiologically implicated in regulating food uptake, body weight control, and energy homeostasis.30 In attempts to improve the antagonist activity and selectivity of AGRP active loop, previous studies have applied a substitution strategy to prepare AGRP active loop analogues.31 The results have indicated that some substitutions of amino acid could increase potency of AGRP. However, the synthesis of AGRP loop analogues requires the introduction of modified unnatural amino acids. Some unnatural amino acids are expensive and difficult to synthesize, such as L-4,4′-biphenylalanine (Bip) and 3-(2-naphthyl)-L-alanine (Nal(2′)). Through C–H activation, the functional group could be installed directly into native peptides, such an approach is highly efficient, step- and atom-economical. Thus, we attempted to apply our strategy to synthesize new AGRP loop analogues. The arylation products 6 through deprotection of phthaloyl (Phth) gave NH2-free tetrapeptides 10 (details see the ESI†). Tetrapeptides 10a and 10b subsequently were coupled with Cbz–DPro–Pro–Arg(Pbf)–Phe–OH to obtain linear octapeptides, which were cyclized to access AGRP loop analogues. AGRP loop analogues 11a and 11b were obtained through this strategy (see the ESI† synthesis of AGRP loop analogues); the introduction of a bromide atom in 11b potentially enables further late-stage derivatization of this peptide (Scheme 5).
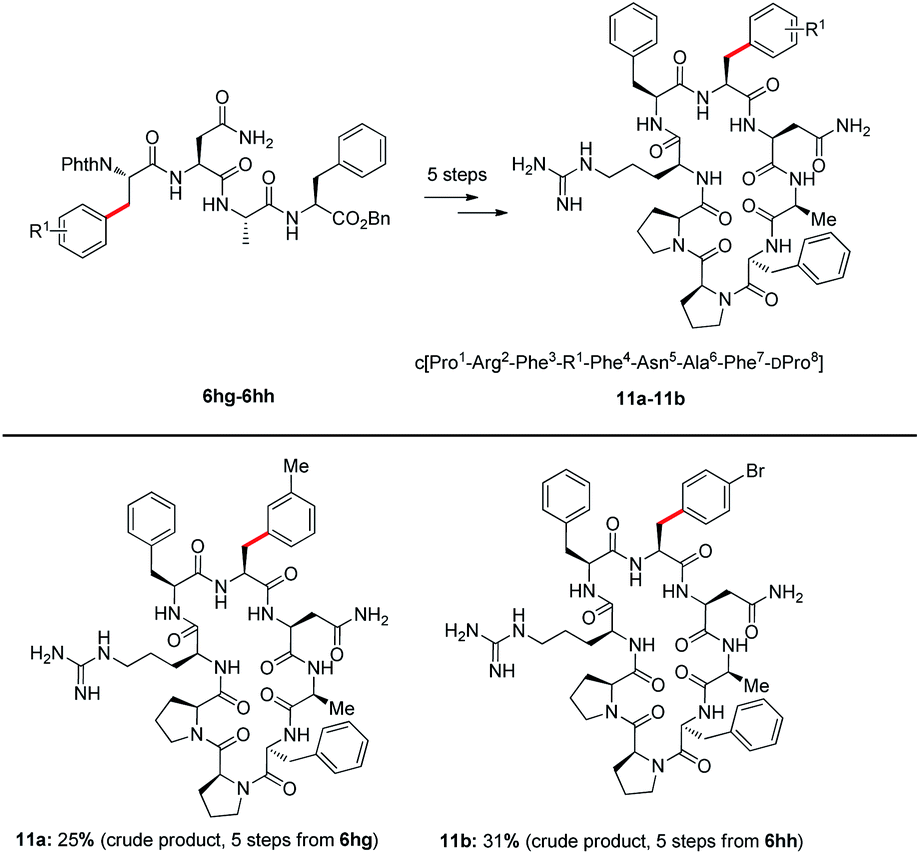 | ||
| Scheme 5 Synthesis of AGRP loop analogues, details see the ESI.† | ||
Conclusion
In conclusion, we have developed an efficient strategy for palladium(II/IV)-catalyzed late-stage C(sp3)–H arylations of peptides using unprecedented internal Asn. The protocol avoids the additional requirement for installation and removal of exogenous directing groups. Importantly, our approach has provided a novel synthetic route to access the key building block for the synthesis of AGRP loop analogues.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Generous support by the NSFC (Grant No. 21978273 and 21506190), the CSC (Scholarship to Y. Weng), the DFG (Gottfried-Wilhelm-Leibniz award to L. A.), the University of Goettingen and the Onassis Foundation (fellowship to N. K.) is gratefully acknowledged.Notes and references
- (a) A. A. Kaspar and J. M. Reichert, Drug Discovery Today, 2013, 18, 807–817 CrossRef CAS PubMed; (b) F. Albericio and H. G. Kruger, Future Med. Chem., 2012, 4, 1527–1531 CrossRef CAS PubMed; (c) S. R. Gracia, K. Gaus and N. Sewald, Future Med. Chem., 2009, 1, 1289–1310 CrossRef CAS PubMed.
- (a) A. Russo, C. Aiello, P. Grieco and D. Marasco, Curr. Med. Chem., 2016, 23, 748–762 CrossRef CAS PubMed; (b) A. Sandomenico, S. M. Monti, M. Sabatella, A. De Capua, L. Tornatore, N. Doti, F. Viparelli, N. A. Dathan, C. Pedone, M. Ruvo and D. Marasco, Chem. Biol. Drug Des., 2009, 73, 483–493 CrossRef CAS PubMed; (c) P. Chène, ChemMedChem, 2006, 1, 400–411 CrossRef PubMed.
- (a) D. J. Craik, D. P. Fairlie, S. Liras and D. Price, Chem. Biol. Drug Des., 2013, 81, 136–147 CrossRef CAS PubMed; (b) K. Fosgerau and T. Hoffmann, Drug Discovery Today, 2015, 20, 122–128 CrossRef CAS PubMed.
- (a) C. M. Grison, G. M. Burslem, J. A. Miles, L. K. A. Pilsl, D. J. Yeo, Z. Imani, S. L. Warriner, M. E. Webb and A. J. Wilson, Chem. Sci., 2017, 8, 5166–5171 RSC; (b) Y. S. Chang, B. Graves, V. Guerlavais, C. Tovar, K. Packman, K.-H. To, K. A. Olson, K. Kesavan, P. Gangurde, A. Mukherjee, T. Baker, K. Darlak, C. Elkin, Z. Filipovic, F. Z. Qureshi, H. Cai, P. Berry, E. Feyfant, X. E. Shi, J. Horstick, D. A. Annis, A. M. Manning, N. Fotouhi, H. Nash, L. T. Vassilev and T. K. Sawyer, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, E3445–E3454 CrossRef CAS PubMed; (c) G. H. Bird, N. Madani, A. F. Perry, A. M. Princiotto, J. G. Supko, X. He, E. Gavathiotis, J. G. Sodroski and L. D. Walensky, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14093–14098 CrossRef CAS PubMed; (d) M. Werle and A. Bernkop-Schnürch, Amino Acids, 2006, 30, 351–367 CrossRef CAS PubMed.
- H. G. Lee, G. Lautrette, B. L. Pentelute and S. L. Buchwald, Angew. Chem., Int. Ed., 2017, 56, 3177–3181 CrossRef CAS PubMed.
- E. L. Tyson, Z. L. Niemeyer and T. P. Yoon, J. Org. Chem., 2014, 79, 1427–1436 CrossRef CAS PubMed.
- V. D. Bock, R. Perciaccante, T. P. Jansen, H. Hiemstra and J. H. van Maarseveen, Org. Lett., 2006, 8, 919–922 CrossRef CAS PubMed.
- (a) P. Gandeepan and L. Ackermann, Chem, 2018, 4, 199–222 CrossRef CAS; (b) J. C. K. Chu and T. Rovis, Angew. Chem., Int. Ed., 2018, 57, 62–101 CrossRef CAS PubMed; (c) W. Ma, P. Gandeepan, J. Li and L. Ackermann, Org. Chem. Front., 2017, 4, 1435–1467 RSC; (d) J. He, M. Wasa, K. S. L. Chan, Q. Shao and J.-Q. Yu, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed; (e) J. A. Leitch and C. G. Frost, Chem. Soc. Rev., 2017, 46, 7145–7153 RSC; (f) T. Gensch, M. N. Hopkinson, F. Glorius and J. Wencel-Delord, Chem. Soc. Rev., 2016, 45, 2900–2936 RSC; (g) B. Su, A. Bunescu, Y. Qiu, S. J. Zuend, M. Ernst and J. F. Hartwig, J. Am. Chem. Soc., 2020, 142, 7912–7919 CrossRef CAS PubMed; (h) S. Rej, Y. Ano and N. Chatani, Chem. Rev., 2020, 120, 1788–1887 CrossRef CAS PubMed; (i) S. Guin, P. Dolui, X. Zhang, S. Paul, V. K. Singh, S. Pradhan, H. B. Chandrashekar, S. S. Anjana, R. S. Paton and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 5633–5638 CrossRef CAS PubMed; (j) A. Dey, S. Pimparkar, A. Deb, S. Guin and D. Maiti, Adv. Synth. Catal., 2017, 359, 1301–1307 CrossRef CAS; (k) R. Srinivasan, A. Dey, N. S. Nagarajan, R. S. Kumaran, T. Gandhi and D. Maiti, Chem. Commun., 2017, 53, 11709–11712 RSC; (l) N. Thrimurtulu, A. Dey, A. Singh, K. Pal, D. Maiti and C. M. R. Volla, Adv. Synth. Catal., 2019, 361, 1441–1446 CrossRef CAS; (m) L. Liu, Y.-H. Liu and B.-F. Shi, Chem. Sci., 2020, 11, 290–294 RSC.
- (a) J. Zhang, L. J. Kang, T. C. Parker, S. B. Blakey, C. K. Luscombe and S. R. Marder, Molecules, 2018, 23, 922 CrossRef PubMed; (b) K. Shin, H. Kim and S. Chang, Acc. Chem. Res., 2015, 48, 1040–1052 CrossRef CAS PubMed; (c) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369–375 CrossRef CAS PubMed; (d) A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177–185 CrossRef CAS PubMed; (e) L. Ackermann, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed.
- (a) T. Bura, J. T. Blaskovits and M. Leclerc, J. Am. Chem. Soc., 2016, 138, 10056–10071 CrossRef CAS PubMed; (b) L. G. Mercier and M. Leclerc, Acc. Chem. Res., 2013, 46, 1597–1605 CrossRef CAS PubMed.
- J. Hubrich, T. Himmler, L. Rodefeld and L. Ackermann, ACS Catal., 2015, 5, 4089–4093 CrossRef CAS.
- (a) S. K. Sinha, G. Zanoni and D. Maiti, Asian J. Org. Chem., 2018, 7, 1178–1192 CrossRef CAS; (b) M. Seki, Org. Process Res. Dev., 2016, 20, 867–877 CrossRef CAS; (c) L. Ackermann, Org. Process Res. Dev., 2015, 19, 260–269 CrossRef CAS.
- W. Gong, G. Zhang, T. Liu, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 16940–16946 CrossRef CAS PubMed.
- A. F. Noisier, J. García, I. A. Ionut and F. Albericio, Angew. Chem., Int. Ed., 2017, 56, 314–318 CrossRef CAS PubMed.
- (a) M. Bauer, W. Wang, M. M. Lorion, C. Dong and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 203–207 CrossRef CAS PubMed; (b) W. Wang, M. M. Lorion, O. Martinazzoli and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 10554–10558 CrossRef CAS PubMed.
- X. Zhang, G. Lu, M. Sun, M. Mahankali, Y. Ma, M. Zhang, W. Hua, Y. Hu, Q. Wang, J. Chen, G. He, X. Qi, W. Shen, P. Liu and G. Chen, Nat. Chem., 2018, 10, 540–548 CrossRef CAS PubMed.
- (a) B.-B. Zhan, J. Fan, L. Jin and B.-F. Shi, ACS Catal., 2019, 9, 3298–3303 CrossRef CAS; (b) B.-B. Zhan, Y. Li, J.-W. Xu, X.-L. Nie, J. Fan, L. Jin and B.-F. Shi, Angew. Chem., Int. Ed., 2018, 57, 5858–5862 CrossRef CAS PubMed.
- J. Tao, Y.-Q. Zhao, C.-F. Chi and B. Wang, Mar. Drugs, 2018, 16, 100 CrossRef PubMed.
- M. Sobocińska, A. Giełdoń, J. Fichna and E. Kamysz, Amino Acids, 2019, 51, 1201–1207 CrossRef PubMed.
- W. Fu, C. Chen, H. Zeng, J. Lin, Y. Zhang, J. Hu and B. Zheng, LWT--Food Sci. Technol., 2019, 110, 54–63 CrossRef CAS.
- (a) T. Zhang, M. Su, X. Jiang, Y. Xue, J. Zhang, X. Zeng, Z. Wu, Y. Guo and D. Pan, J. Agric. Food Chem., 2019, 67, 5544–5551 CrossRef CAS PubMed; (b) Y. Zheng, Y. Li, Y. Zhang, X. Ruan and R. Zhang, J. Funct. Foods, 2017, 28, 48–58 CrossRef CAS.
- S. H. Cheong, E.-K. Kim, J.-W. Hwang, Y.-S. Kim, J.-S. Lee, S.-H. Moon, B.-T. Jeon and P.-J. Park, J. Agric. Food Chem., 2013, 61, 11442–11446 CrossRef CAS PubMed.
- (a) C. Haskell-Luevano and E. K. Monck, Regul. Pept., 2001, 99, 1–7 CrossRef CAS PubMed; (b) M. R. Tota, T. S. Smith, C. Mao, T. MacNeil, R. T. Mosley, L. H. T. Van der Ploeg and T. M. Fong, Biochemistry, 1999, 38, 897–904 CrossRef CAS PubMed.
- (a) J. Wu, N. Kaplaneris, S. Ni, F. Kaltenhäuser and L. Ackermann, Chem. Sci., 2020, 11, 6521–6526 RSC; (b) W. Wang, P. Subramanian, O. Martinazzoli, J. Wu and L. Ackermann, Chem.–Eur. J., 2019, 25, 10585–10589 CrossRef CAS PubMed; (c) N. Kaplaneris, T. Rogge, R. Yin, H. Wang, G. Sirvinskaite and L. Ackermann, Angew. Chem., Int. Ed., 2019, 58, 3476–3480 CrossRef CAS PubMed; (d) W. Wang, M. M. Lorion, J. Shah, A. R. Kapdi and L. Ackermann, Angew. Chem., Int. Ed., 2018, 57, 14700–14717 CrossRef CAS PubMed; (e) Y. Zhu, M. Bauer and L. Ackermann, Chem.–Eur. J., 2015, 21, 9980–9983 CrossRef CAS PubMed; (f) Y. Zhu, M. Bauer, J. Ploog and L. Ackermann, Chem.–Eur. J., 2014, 20, 13099–13102 CrossRef CAS PubMed.
- G. Xia, J. Weng, L. Y. Liu, P. Verma, Z. Q. Li and J.-Q. Yu, Nat. Chem., 2019, 11, 571–577 CrossRef CAS PubMed.
- (a) P. Dolui, J. Das, H. B. Chandrashekar, S. S. Anjana and D. Maiti, Angew. Chem., Int. Ed., 2019, 58, 13773–13777 CrossRef CAS PubMed; (b) S. Guin, A. Deb, P. Dolui, S. Chakraborty, V. K. Singh and D. Maiti, ACS Catal., 2018, 8, 2664–2669 CrossRef CAS; (c) A. Deb, S. Singh, K. Seth, S. Pimparkar, B. Bhaskararao, S. Guin, R. B. Sunoj and D. Maiti, ACS Catal., 2017, 7, 8171–8175 CrossRef CAS; (d) N. Thrimurtulu, S. Khan, S. Maity, C. M. R. Volla and D. Maiti, Chem. Commun., 2017, 53, 12457–12460 RSC; (e) T. Bhattacharya, S. Pimparkar and D. Maiti, RSC Adv., 2018, 8, 19456–19464 RSC.
- For detailed information, see the ESI.†.
- (a) Y.-F. Yang, G. Chen, X. Hong, J.-Q. Yu and K. N. Houk, J. Am. Chem. Soc., 2017, 139, 8514–8521 CrossRef CAS PubMed; (b) Y.-F. Yang, G.-J. Cheng, P. Liu, D. Leow, T.-Y. Sun, P. Chen, X. Zhang, J.-Q. Yu, Y.-D. Wu and K. N. Houk, J. Am. Chem. Soc., 2014, 136, 344–355 CrossRef CAS PubMed; (c) C. Colletto, A. Panigrahi, J. Fernandez-Casado and I. Larrosa, J. Am. Chem. Soc., 2018, 140, 9638–9643 CrossRef CAS PubMed; (d) B. Bhaskararao, S. Singh, M. Anand, P. Verma, P. Prakash, C. Athira, S. Malakar, H. F. Schaefer and R. B. Sunoj, Chem. Sci., 2020, 11, 208–216 RSC.
- Y.-K. Yang, D. A. Thompson, C. J Dickinson, J. Wilken, G. S. Barsh and S. B. H. Kent, Mol. Endocrinol., 1999, 13, 148–155 CrossRef CAS PubMed.
- (a) M. M. Ollmann, B. D. Wilson, Y. K. Yang, J. A. Kerns, Y. Chen, I. Gantz and G. S. Barsh, Science, 1997, 278, 135–138 CrossRef CAS PubMed; (b) M. Graham, J. R. Shutter, U. Sarmiento, I. Sarosi and K. L. Stark, Nat. Genet., 1997, 17, 273–274 CrossRef CAS PubMed.
- (a) K. A. Fleming, K. T. Freeman, M. D. Ericson and C. Haskell-Luevano, J. Med. Chem., 2018, 61, 7729–7740 CrossRef CAS PubMed; (b) M. D. Ericson, K. T. Freeman, S. M. Schnell, K. A. Fleming and C. Haskell-Luevano, J. Med. Chem., 2017, 60, 8103–8114 CrossRef CAS PubMed; (c) M. D. Ericson, A. Wilczynski, N. B. Sorensen, Z. Xiang and C. Haskell-Lueyano, J. Med. Chem., 2015, 58, 4638–4647 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0sc03830j |
| This journal is © The Royal Society of Chemistry 2020 |