
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTubularenes†
Saber
Mirzaei‡
 ,
Edison
Castro‡
and
Raúl Hernández
Sánchez
*
,
Edison
Castro‡
and
Raúl Hernández
Sánchez
*
Department of Chemistry, University of Pittsburgh, 219 Parkman Ave., Pittsburgh, Pennsylvania 15260, USA. E-mail: raulhs@pitt.edu
First published on 14th July 2020
Abstract
We report the synthesis and characterization of conjugated, conformationally rigid, and electroactive carbon-based nanotubes that we term tubularenes. These structures are constructed from a resorcin[nb]arene base. Cyclization of the conjugated aromatic nanotube is achieved in one-pot eight-fold C–C bond formation via Suzuki–Miyaura cross-coupling. DFT calculations indicate a buildup of strain energy in excess of 90 kcal mol−1. The resulting architectures contain large internal void spaces >260 Å3, are fluorescent, and able to accept up to 4 electrons. This represents the first scaffolding approach that provides conjugated nanotube architectures.
Introduction
Macromolecules in the nanosize regime with host–guest capabilities offer opportunities to develop novel applications in electronic devices,1,2 purification systems,3–6 and sensing.7–10 Frequently, their preparation involves the bending of aromatic systems, further increasing their strain and complexity.11–26 As a result, overcoming the build-up of strain energy has to be carefully considered during their design process. The syntheses reported for [n]cyclo-para-phenylenes (CPPs) – the smallest circular segment forming an armchair carbon nanotube (CNT) – elegantly showcases the importance of this design process.27–29 These [n]CPPs or nanorings, with sizable strain energies (<120 kcal mol−1), were first27 synthesized by exploiting non-aromatic intermediates with sp3 carbon atoms to facilitate establishing the overall connectivity before re-aromatizing the nanoring in the last step. This synthetic breakthrough gave access to CPPs of varying sizes and concomitantly uncovered unexpected structure–function relationships,30 while also developing novel applications.31 Despite these advances, streamlined synthesis to extend the nanoring along its main axis to yield tubular systems comprised of contorted aromatic species are exceedingly rare.21,32–36To expand current approaches, our group started exploring the possibility of using macrocycles to template the synthesis of strained conjugated aromatics, with the ultimate goal of extending this methodology to longer and more complex nanotube structures. Herein we describe the first members of this class of highly-strained aromatic architectures having a tubular shape – that we have termed tubularenes.
These macromolecules bottom-up approach starts from resorcin[nb]arenes – macrocycles resulting from the acid-catalyzed condensation of resorcinol and an aldehyde – as templates.37,38 Given the resulting shape and template origin of the molecules reported herein, we decided to term these tubular[nb,n,m,r]arenes (tubularenes for short), where nb (b stands for basal) comes from the resorcin[nb]arene parent, n and m are the CNT chiral indices, and r corresponds to the number of aromatic rings making up the armchair nanoring in the upper termini (disregarding the pyrazine ring). In this report we describe the synthesis and characterization of tubular[4,8,8,8]arene (1) and tubular[4,8,8,12]arene (2), as shown in Scheme 1.
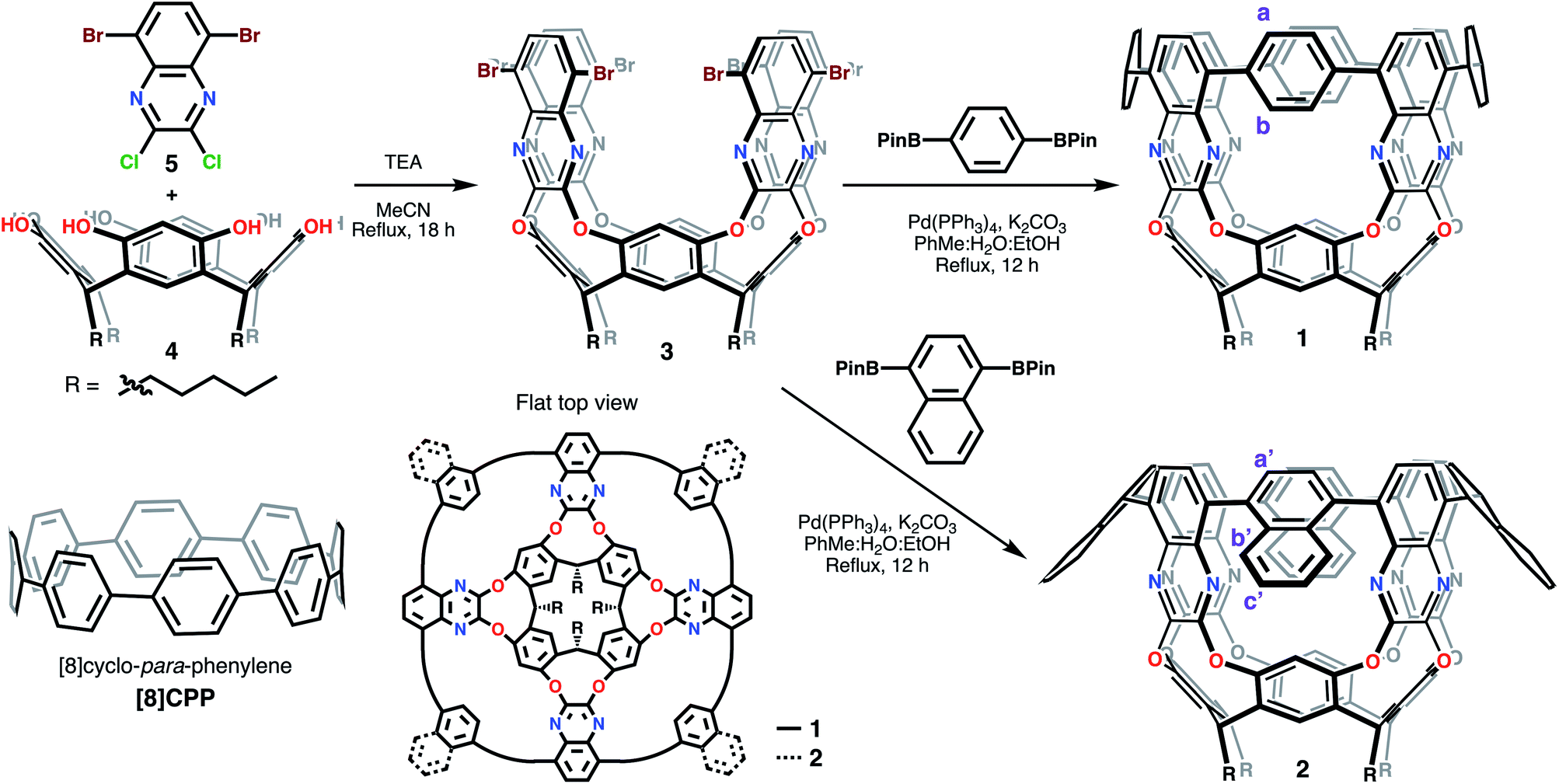 | ||
| Scheme 1 Synthesis of tubular[nb,n,m,r]arenes 1 and 2. | ||
Results and discussion
Tubularene construction
Starting materials 4 (ref. 37) and 2,3-dichloro-5,8-dibromoquinoxaline (5) can be synthesized on a multigram scale. Reaction between 4 and 5 under basic conditions using triethylamine (TEA) in acetonitrile under reflux produces octabromo-derivative 3 in 57% yield. Under similar reaction conditions, Suzuki–Miyaura cross coupling of 3 with either 1,4-benzenediboronic or 1,4-naphthalenediboronic acid bis(pinacol) ester affords 1 and 2 (Scheme 1), respectively, after an eight-fold C–C bond formation. The isolated yield after the cross-coupling step to give 1 is ∼1.3% (0.8% for 2). Similarly, other contorted aromatics initially reporting low yields have been optimized up to seven-fold.20,39 The cross-coupling cyclization step provides rigidity and aromatic conjugation to 1 and 2, much like the walls of CNTs. In 1, this newly formed upper nanoring resembles [8]CPP,29 although with fewer degrees of freedom since only four phenyl rings may rotate as opposed to eight in [8]CPP. We hypothesize this rigid conformation is vital to maintaining electron or hole delocalization across the conjugated framework, especially since it is known that departure from the radially oriented π system breaks the spin density distribution in radical monocationic CPPs.40Tubular structure
MALDI-TOF MS of tubularenes 1 and 2 display matching simulated values and isotopic distributions as shown in Fig. 1a. In solution, 1H NMR of 1 and 2 is readily assigned indicating C4v symmetry (Fig. 1b). To our delight, slow evaporation of a dichloromethane/acetonitrile solution of 1 at room temperature affords high quality yellow-green crystals. In contrast, crystals of 2 are needle-like and weakly diffracting. The rigid framework expected for 1 is observed in its molecular crystal structure shown in Fig. 1c. In the same figure, its internal cavity is readily apparent from the top view of the sphere packing model. To determine the size of this cavity, we obtain a volume of ∼360 Å3, if we consider the average diameter of 1 as ∼10 Å, and having a height of ∼4.6 Å (as described in Fig. S11†). Alternatively, by employing the solvent accessible calculator in Olex2 (ref. 41) we find a volume of ∼266 Å3 (Fig. S12†), as described in the ESI.†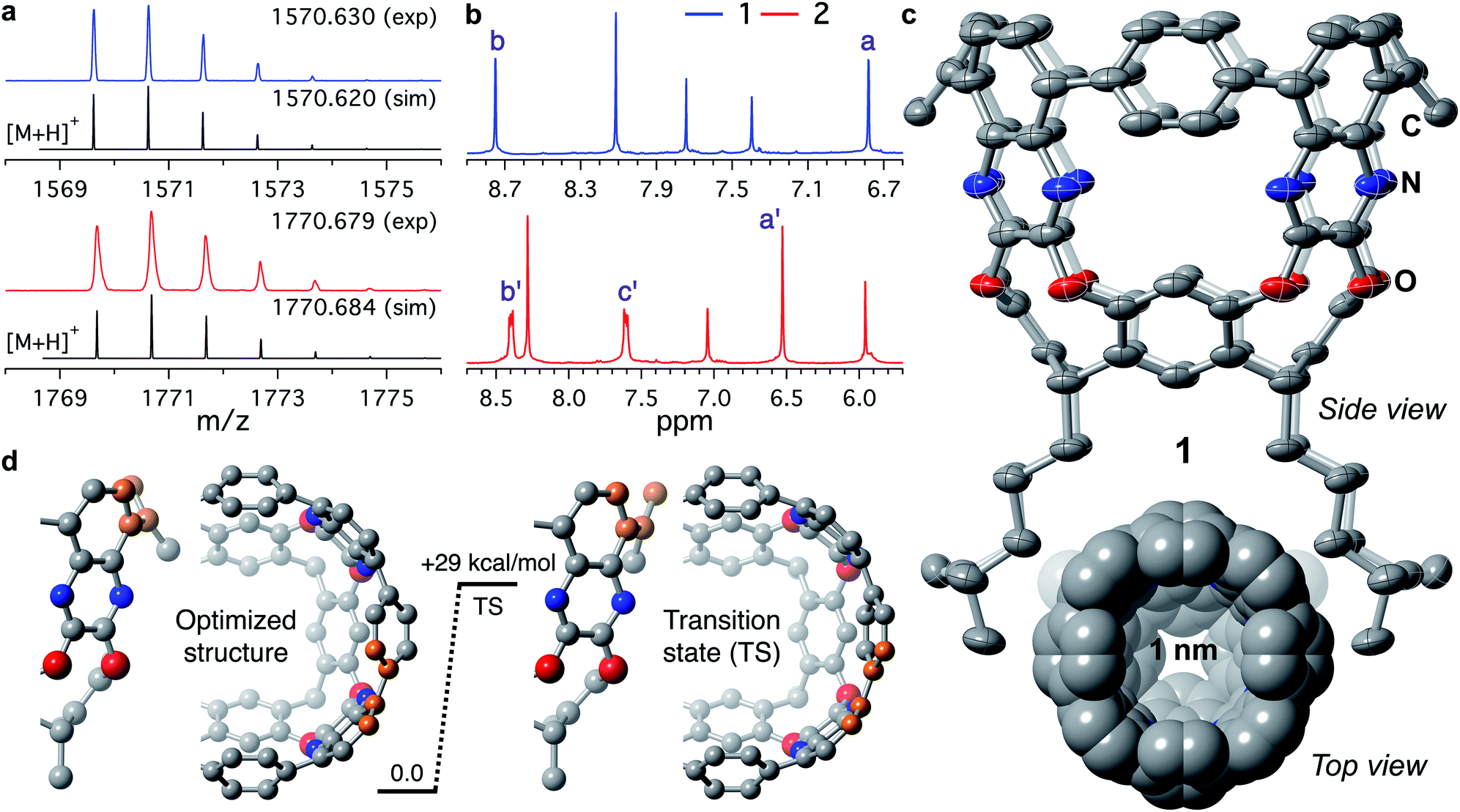 | ||
| Fig. 1 (a) Experimental MALDI-TOF MS molecular ion peaks of 1 (blue trace) and 2 (red trace). Black traces represent the simulated [M + H]+ isotopic distributions. (b) 1H NMR of (top) 1 and (bottom) 2 in CD2Cl2 at 20 °C. Proton labels according to Scheme 1. (c) Molecular crystal structure of 1 obtained at 150 K. Thermal ellipsoids are set at 50% probability level. The C, N, and O atoms are coloured grey, blue, and red, respectively. The H atoms are omitted for clarity. Bottom: top view of the sphere packing model (van der Waals radii) of 1. (d) DFT calculated barrier for ring flipping. Rotation of the phenyl moiety was followed by tracking the dihedral angle between the highlighted carbon atoms in orange. | ||
The upper nanoring of 1 resembles [8]CPP, albeit with fewer degrees of freedom. The strain energy reported by Itami et al. for [8]CPP is 72.2 kcal mol−1, obtained by DFT at the B3LYP/6-31G(d) level of theory, considering a homodesmotic reaction (Fig. S13†).42 Following a similar approach at the same level of theory, we determine the strain energy of 1 to be 92.4 kcal mol−1. DFT calculations at different levels of theory produced consistent values around 89 ± 3 kcal mol−1 (Table S2†). This larger value for tubularene 1 relative to [8]CPP is expected, especially since 1 has only four out of eight phenyl rings that can freely rotate. In fact, free rotation is hampered at room temperature as observed in the vastly different chemical environments of a versus b protons at 6.8 and 8.7 ppm (Fig. 1b), respectively. DFT-calculated nucleus-independent chemical shifts (NICS) establish a shielding effect at the interior of the tubularene (Fig. S14†).43 We find by DFT analysis a rotational barrier of ∼29 ± 1 kcal mol−1 for this phenyl group (Fig. 1d, S15, and Table S3†). Comparing to other highly strained molecules, this barrier is similar in magnitude to several others where temperatures well above 150 °C are required for rotation of the aromatic group or inversion of stereochemistry.44–51 Note that in the latter examples DFT calculations reproduce closely the experimental rotational barriers. In contrast, similar phenyl rotation in [8]CPP is lower than 4 kcal mol−1 (Table S4†). We hypothesize the locked conformation in 1 to be particularly relevant to maintaining conjugation across the tubularene. Indeed, DFT calculations show the HOMO density evenly distributed across the upper rim of 1, whereas the LUMO has significant orbital density at the pyrazine fragments (Fig. 2a).
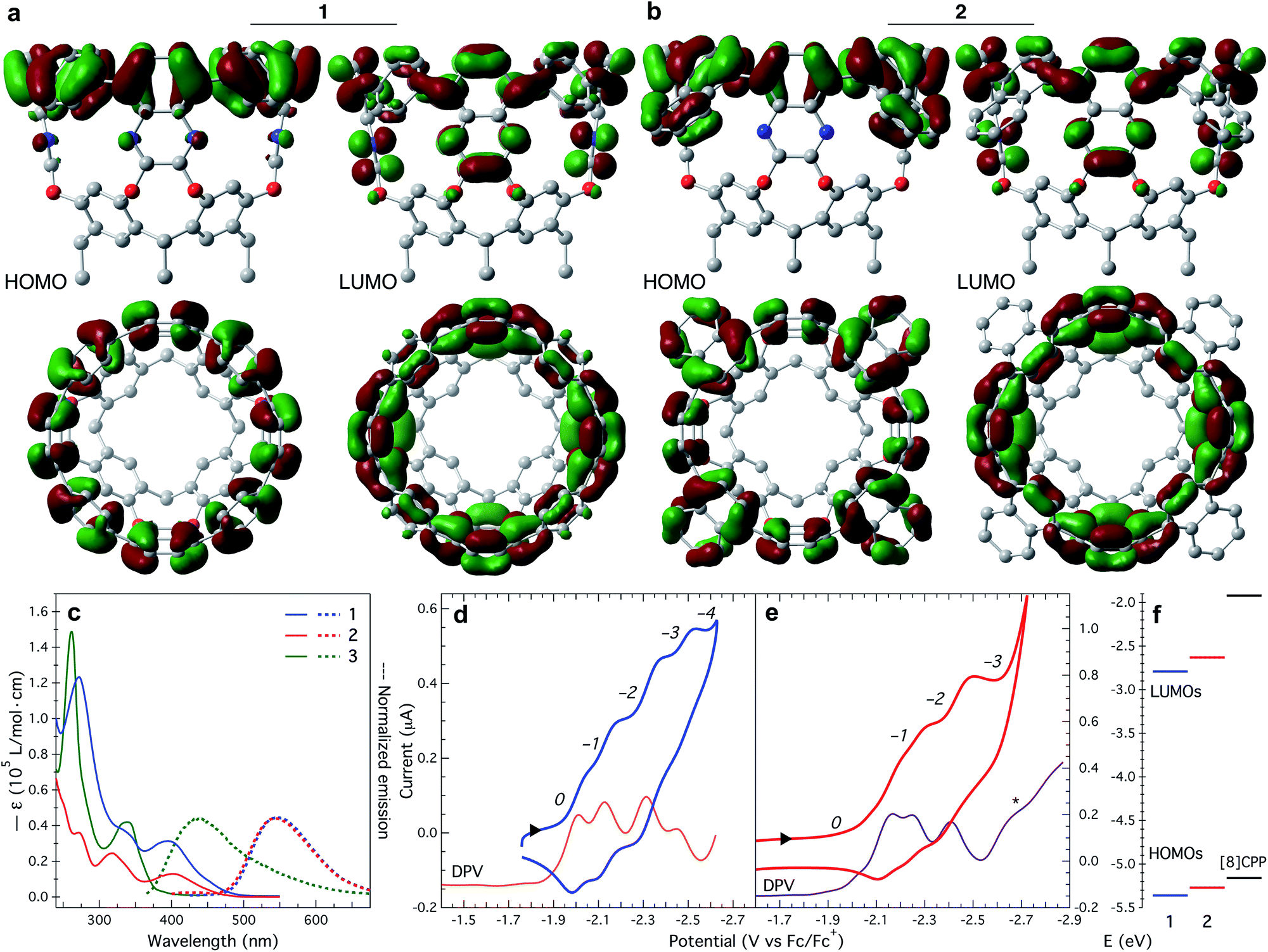 | ||
| Fig. 2 HOMO and LUMO density plots (±0.02 au) of (a) 1, and (b) 2. (c) UV-Vis absorption (solid trace) and emission spectra (dotted trace) of 1, 2, and 3, collected in CH2Cl2 at room temperature. Cyclic (CV) and differential pulse voltammetry (DPV) for (d) 1, and (e) 2 in ortho-dichlorobenzene at room temperature. CV scan rate: (d) 100 mV s−1, and (e) 50 mV s−1. A 0.1 M [n-Bu4N][PF6] solution was used as supporting electrolyte. The labels on italics correspond to the oxidation level q represented as: (d) [1]q, and (e) [2]q. The E1/2 potentials were obtained from the DPV data. DPV traces are shifted down for better visualization of the data. (f) Experimentally determined HOMO–LUMO energy levels of 1, 2, and [8]CPP.31 | ||
The 1,4-naphthylene fragments in tubularene 2 are significantly more restricted to rotation. In fact, due to steric clashing of the naphthylene moiety with the resorcin[4]arene base we were not able to calculate the rotational barrier via DFT. At room temperature, there is a single resonance for protons a′, b′, and c′ (Fig. 1b). Furthermore, we determine by DFT the strain energy in 2 to be ∼81 ± 5 kcal mol−1. The magnitude of this strain energy is substantially higher than that of the similar nanoring [8]cyclo-1,4-naphthylene ([8]CN) at 50.6 kcal mol−1.52 Looking at the DFT optimized structure of 2, we note it converges to a conformation where all naphthylene units are almost horizontally aligned pointing away from the tubularene (Fig. S17†). This naphthylene bending results in large dihedral angles (∼55°) and is likely the reason of the increased strain.42
Optoelectronic properties
Species 1 and 2 display lowest energy absorption bands at λmax of 394 (ε = 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 380 L mol−1 cm−1) and 402 (ε = 12970 L mol−1 cm−1) nm, respectively. These bands are red-shifted in comparison to the lowest energy transition of precursor 3 at λmax = 338 (ε = 42030 L mol−1 cm−1) nm (Fig. 2c). Regarding emission, the fluorescence envelope of 1 and 2 is also red-shifted in comparison to precursor 3 (λem = 435 nm). Emission bands of 1 and 2 are almost identical to each other only slightly shifted in λem from 546 to 542 nm, respectively. Using these data we extracted the optical band gap (Egap)53 for both tubularenes. Egap for 1 and 2 is 2.57 and 2.64 eV, respectively.
380 L mol−1 cm−1) and 402 (ε = 12970 L mol−1 cm−1) nm, respectively. These bands are red-shifted in comparison to the lowest energy transition of precursor 3 at λmax = 338 (ε = 42030 L mol−1 cm−1) nm (Fig. 2c). Regarding emission, the fluorescence envelope of 1 and 2 is also red-shifted in comparison to precursor 3 (λem = 435 nm). Emission bands of 1 and 2 are almost identical to each other only slightly shifted in λem from 546 to 542 nm, respectively. Using these data we extracted the optical band gap (Egap)53 for both tubularenes. Egap for 1 and 2 is 2.57 and 2.64 eV, respectively.
To gain further insight of the photophysical properties of 1 and 2 we performed time-dependent (TD) DFT calculations at various levels of theory (Table S5†). For both tubularenes, we found that the HOMO-to-LUMO transition is forbidden, as in [n]CPPs.54 Du et al. established a structure–function relationship between octamethoxy-[8]CPP (λem = 458 nm) and [8]CPP (λem = 535 nm) indicating that a bathochromic shift in emission corresponds to increased radial π-conjugation.55 Extrapolating this correlation to the emissive properties of 1 and 2 supports our claim that tubularene's rigidity assists to maintain a large π-conjugated surface, although not as much as [8]CN (λem = 570 nm).52
Electrochemically, tubularenes 1 and 2 display a rich series of reductive events in ortho-dichlorobenzene. Cyclic voltammograms (CVs) exhibit onset of reduction at around −1.95 and −2.05 V vs. Fc/Fc+ for 1 and 2 (Fig. 2d and e), respectively. Initially, these values are counterintuitive since we expect the LUMO of 2 to be more accessible and lower in energy than 1 due to the larger π surface of 2. Inspection of the DFT optimized structure of 2 (Fig. S17†), and its LUMO density plot shown in Fig. 2b, provides an explanation for this unexpected behaviour. As a result of the naphthylene fragments bending outwards these are devoid of LUMO density, hence effectively reducing the extent of conjugation of the π surface in 2.
The CV of 1 displays four reduction events on the cathodic scan, but since these are clustered together, it is not possible to extract the half-wave potentials (E1/2) from the CV alone. Fortunately, by differential pulse voltammetry (DPV, orange trace in Fig. 2d) we observe four peak maximums at −2.01, −2.13, −2.31, and −2.45 V vs. Fc/Fc+, each corresponding to a reduction event. In contrast to [8]CPP,56,57 oxidation events for 1 or 2 are not observed up to ca. +0.8 V vs. Fc/Fc+. Similar to 1, tubularene 2 displays an equally crowded CV (Fig. 2e). As in tubularene 1, DPV of 2 is well-resolved and shows three reduction peaks at −2.17, −2.25, and −2.41 V vs. Fc/Fc+ (purple trace in Fig. 2e). The fourth reduction of 2 is presumed to occur concomitant with solvent reduction based on the shoulder observed at around −2.65 V vs. Fc/Fc+ (marked with an asterisk in Fig. 2e). Note however how for the same number of electrons added (q), the E1/2 for 2 is 100 to 150 mV more cathodic relative to 1, indicating that 2 is harder to reduce than 1.
We calculated the electrochemical LUMO levels (ELUMO) for 1 and 2 by employing E1/2 of the first reduction event according to ELUMO = −[E1/2 + 4.80] eV.58 Additionally, the HOMO energy level is obtained by subtracting Egap from ELUMO. HOMO and LUMO levels for 1 and 2 are plotted in Fig. 2f (see Table S6†). For comparison, the computed HOMO and LUMO positions for [8]CPP are −5.16 and −1.92 eV, respectively.31 Therefore, by extending the conjugation to the quinoxaline walls, while maintaining a radial orientation of the π surface, tubularene 1 and 2 are able to bring down the LUMO energy level. In contrast, HOMO–LUMO levels in [n]CPPs are determined by the number of 1,4-phenylene units making up the nanoring.59
Last, to gain further insight into the rigidity effect of tubularene 1 into electron delocalization across the aromatic framework, we performed DFT calculations on the radicals formed in 1 after removing an electron from the HOMO (1+˙) and adding an electron to the LUMO (1−˙). Results show the spin density of the radical completely delocalized across the conjugated backbone of 1 (Fig. S20†), in stark contrast to [8]CPP40 and other similar contorted aromatics,60 where the radical is mostly localized into a portion of the molecule.
Conclusions
Herein we describe the synthesis of a new family of compounds that we have termed tubularenes. These offer a wide range of potential applications given their permanent internal void space, tubular rigidity, conjugation, and electrochemical activity. Tubularene's overall architecture is highly modular and tunable. For example, modifications to the “wall” (compound 5), diboronate coupling partner, or the basal resorcin[nb]arene are envisioned as the tuning knobs of tubularene solubility profile (hydrophobic vs. hydrophilic), diameter, and overall electronic properties. Expansion of the tubularene family will open up a new landscape for discovery of novel properties and applications of contorted aromatic systems.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank startup funding from the University of Pittsburgh and a seed grant from the Central Research Development Fund (CRDF) at the University of Pittsburgh for their support. This research was supported in part by resources allocated by the University of Pittsburgh Center for Research Computing. S. M. acknowledges the support from a Dietrich School of Arts & Sciences Graduate Fellowship.Notes and references
- B. Zhang, R. Hernández Sánchez, Y. Zhong, M. Ball, M. W. Terban, D. Paley, S. J. L. Billinge, F. Ng, M. L. Steigerwald and C. Nuckolls, Nat. Commun., 2018, 9, 1957 CrossRef PubMed.
- N. Song, T. Kakuta, T.-a. Yamagishi, Y.-W. Yang and T. Ogoshi, Chem, 2018, 4, 2029–2053 CAS.
- A. Alsbaiee, B. J. Smith, L. L. Xiao, Y. H. Ling, D. E. Helbling and W. R. Dichtel, Nature, 2016, 529, 190–194 CrossRef CAS PubMed.
- T. Ogoshi, K. Saito, R. Sueto, R. Kojima, Y. Hamada, S. Akine, A. M. P. Moeljadi, H. Hirao, T. Kakuta and T.-a. Yamagishi, Angew. Chem., Int. Ed., 2018, 57, 1592–1595 CrossRef CAS PubMed.
- K. Jie, M. Liu, Y. Zhou, M. A. Little, A. Pulido, S. Y. Chong, A. Stephenson, A. R. Hughes, F. Sakakibara, T. Ogoshi, F. Blanc, G. M. Day, F. Huang and A. I. Cooper, J. Am. Chem. Soc., 2018, 140, 6921–6930 CrossRef CAS PubMed.
- Y. Liu, W. Zhao, C.-H. Chen and A. H. Flood, Science, 2019, 365, 159 CAS.
- J. W. Trzciński, R. Pinalli, N. Riboni, A. Pedrini, F. Bianchi, S. Zampolli, I. Elmi, C. Massera, F. Ugozzoli and E. Dalcanale, ACS Sens., 2017, 2, 590–598 CrossRef PubMed.
- R. Kumar, A. Sharma, H. Singh, P. Suating, H. S. Kim, K. Sunwoo, I. Shim, B. C. Gibb and J. S. Kim, Chem. Rev., 2019, 119, 9657–9721 CrossRef CAS PubMed.
- M. Rickhaus, M. Jirasek, L. Tejerina, H. Gotfredsen, M. D. Peeks, R. Haver, H.-W. Jiang, T. D. W. Claridge and H. L. Anderson, Nat. Chem., 2020, 12, 236–241 CrossRef CAS PubMed.
- A. F. Sierra, D. Hernández-Alonso, M. A. Romero, J. A. González-Delgado, U. Pischel and P. Ballester, J. Am. Chem. Soc., 2020, 142, 4276–4284 CrossRef CAS PubMed.
- F. Zhang, G. Götz, H. D. F. Winkler, C. A. Schalley and P. Bäuerle, Angew. Chem., Int. Ed., 2009, 48, 6632–6635 CrossRef CAS PubMed.
- J. K. Sprafke, D. V. Kondratuk, M. Wykes, A. L. Thompson, M. Hoffmann, R. Drevinskas, W.-H. Chen, C. K. Yong, J. Kärnbratt, J. E. Bullock, M. Malfois, M. R. Wasielewski, B. Albinsson, L. M. Herz, D. Zigmantas, D. Beljonne and H. L. Anderson, J. Am. Chem. Soc., 2011, 133, 17262–17273 CrossRef CAS PubMed.
- L. T. Scott, E. A. Jackson, Q. Zhang, B. D. Steinberg, M. Bancu and B. Li, J. Am. Chem. Soc., 2012, 134, 107–110 CrossRef CAS PubMed.
- E. Kayahara, T. Iwamoto, H. Takaya, T. Suzuki, M. Fujitsuka, T. Majima, N. Yasuda, N. Matsuyama, S. Seki and S. Yamago, Nat. Commun., 2013, 4, 2694 CrossRef PubMed.
- P. J. Evans, E. R. Darzi and R. Jasti, Nat. Chem., 2014, 6, 404–408 CrossRef CAS PubMed.
- E. Kayahara, V. K. Patel and S. Yamago, J. Am. Chem. Soc., 2014, 136, 2284–2287 CrossRef CAS PubMed.
- K. Asai, A. Fukazawa and S. Yamaguchi, Chem. Commun., 2015, 51, 6096–6099 RSC.
- M. R. Golder and R. Jasti, Acc. Chem. Res., 2015, 48, 557–566 CrossRef CAS PubMed.
- Y.-Y. Liu, J.-Y. Lin, Y.-F. Bo, L.-H. Xie, M.-D. Yi, X.-W. Zhang, H.-M. Zhang, T.-P. Loh and W. Huang, Org. Lett., 2016, 18, 172–175 CrossRef CAS PubMed.
- G. Povie, Y. Segawa, T. Nishihara, Y. Miyauchi and K. Itami, Science, 2017, 356, 172–175 CrossRef CAS PubMed.
- Z. Sun, K. Ikemoto, T. M. Fukunaga, T. Koretsune, R. Arita, S. Sato and H. Isobe, Science, 2019, 363, 151–155 CrossRef CAS PubMed.
- K. Y. Cheung, S. J. Gui, C. F. Deng, H. F. Liang, Z. M. Xia, Z. Liu, L. F. Chi and Q. Miao, Chem, 2019, 5, 838–847 CAS.
- F. S. Conrad-Burton, T. Liu, F. Geyer, R. Costantini, A. P. Schlaus, M. S. Spencer, J. Wang, R. Hernández Sánchez, B. Zhang, Q. Xu, M. L. Steigerwald, S. Xiao, H. Li, C. P. Nuckolls and X. Zhu, J. Am. Chem. Soc., 2019, 141, 13143–13147 CrossRef CAS PubMed.
- Y. Ni, T. Y. Gopalakrishna, H. Phan, T. Kim, T. S. Herng, Y. Han, T. Tao, J. Ding, D. Kim and J. Wu, Nat. Chem., 2020, 12, 242–248 CrossRef CAS PubMed.
- Q. Zhang, Y.-E. Zhang, S. Tong and M.-X. Wang, J. Am. Chem. Soc., 2020, 142, 1196–1199 CrossRef CAS PubMed.
- T.-H. Shi, Q.-H. Guo, S. Tong and M.-X. Wang, J. Am. Chem. Soc., 2020, 142, 4576–4580 CrossRef CAS PubMed.
- R. Jasti, J. Bhattacharjee, J. B. Neaton and C. R. Bertozzi, J. Am. Chem. Soc., 2008, 130, 17646–17647 CrossRef PubMed.
- H. Takaba, H. Omachi, Y. Yamamoto, J. Bouffard and K. Itami, Angew. Chem., Int. Ed., 2009, 48, 6112–6116 CrossRef CAS PubMed.
- S. Yamago, Y. Watanabe and T. Iwamoto, Angew. Chem., Int. Ed., 2010, 49, 757–759 CrossRef CAS PubMed.
- M. Fujitsuka, D. W. Cho, T. Iwamoto, S. Yamago and T. Majima, Phys. Chem. Chem. Phys., 2012, 14, 14585–14588 RSC.
- E. J. Leonhardt and R. Jasti, Nat. Rev. Chem., 2019, 3, 672–686 CrossRef CAS.
- F. E. Golling, M. Quernheim, M. Wagner, T. Nishiuchi and K. Müllen, Angew. Chem., Int. Ed., 2014, 53, 1525–1528 CrossRef CAS PubMed.
- D. Myśliwiec, M. Kondratowicz, T. Lis, P. J. Chmielewski and M. Stępień, J. Am. Chem. Soc., 2015, 137, 1643–1649 CrossRef PubMed.
- P. Neuhaus, A. Cnossen, J. Q. Gong, L. M. Herz and H. L. Anderson, Angew. Chem., Int. Ed., 2015, 54, 7344–7348 CrossRef CAS PubMed.
- D. Lu, G. Zhuang, H. Wu, S. Wang, S. Yang and P. Du, Angew. Chem., Int. Ed., 2017, 56, 158–162 CrossRef CAS PubMed.
- Z. Sun, T. Mio, K. Ikemoto, S. Sato and H. Isobe, J. Org. Chem., 2019, 84, 3500–3507 CrossRef CAS PubMed.
- L. M. Tunstad, J. A. Tucker, E. Dalcanale, J. Weiser, J. A. Bryant, J. C. Sherman, R. C. Helgeson, C. B. Knobler and D. J. Cram, J. Org. Chem., 1989, 54, 1305–1312 CrossRef CAS.
- P. Timmerman, W. Verboom and D. N. Reinhoudt, Tetrahedron, 1996, 52, 2663–2704 CrossRef CAS.
- G. Povie, Y. Segawa, T. Nishihara, Y. Miyauchi and K. Itami, J. Am. Chem. Soc., 2018, 140, 10054–10059 CrossRef CAS PubMed.
- M. R. Talipov, R. Jasti and R. Rathore, J. Am. Chem. Soc., 2015, 137, 14999–15006 CrossRef CAS PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- Y. Segawa, H. Omachi and K. Itami, Org. Lett., 2010, 12, 2262–2265 CrossRef CAS PubMed.
- P. v. R. Schleyer, C. Maerker, A. Dransfeld, H. Jiao and N. J. R. van Eikema Hommes, J. Am. Chem. Soc., 1996, 118, 6317–6318 CrossRef CAS PubMed.
- R. H. Martin and M.-J. Marchant, Tetrahedron Lett., 1972, 13, 3707–3708 CrossRef.
- H. Omachi, Y. Segawa and K. Itami, Org. Lett., 2011, 13, 2480–2483 CrossRef CAS PubMed.
- S. Hitosugi, W. Nakanishi and H. Isobe, Chem.–Asian J., 2012, 7, 1550–1552 CrossRef CAS PubMed.
- A. Yagi, Y. Segawa and K. Itami, J. Am. Chem. Soc., 2012, 134, 2962–2965 CrossRef CAS PubMed.
- M. Gingras, G. Félix and R. Peresutti, Chem. Soc. Rev., 2013, 42, 1007–1050 RSC.
- Z. Sun, P. Sarkar, T. Suenaga, S. Sato and H. Isobe, Angew. Chem., Int. Ed., 2015, 54, 12800–12804 CrossRef CAS PubMed.
- Z. Sun, T. Suenaga, P. Sarkar, S. Sato, M. Kotani and H. Isobe, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8109 CrossRef CAS PubMed.
- J. Barroso, J. L. Cabellos, S. Pan, F. Murillo, X. Zarate, M. A. Fernandez-Herrera and G. Merino, Chem. Commun., 2018, 54, 188–191 RSC.
- K. Okada, A. Yagi, Y. Segawa and K. Itami, Chem. Sci., 2017, 8, 661–667 RSC.
- J. C. S. Costa, R. J. S. Taveira, C. F. R. A. C. Lima, A. Mendes and L. M. N. B. F. Santos, Opt. Mater., 2016, 58, 51–60 CrossRef CAS.
- S. E. Lewis, Chem. Soc. Rev., 2015, 44, 2221–2304 RSC.
- S. Cui, G. Zhuang, J. Wang, Q. Huang, S. Wang and P. Du, Org. Chem. Front., 2019, 6, 1885–1890 RSC.
- E. Kayahara, T. Kouyama, T. Kato, H. Takaya, N. Yasuda and S. Yamago, Angew. Chem., Int. Ed., 2013, 52, 13722–13726 CrossRef CAS PubMed.
- A. V. Zabula, A. S. Filatov, J. Xia, R. Jasti and M. A. Petrukhina, Angew. Chem., Int. Ed., 2013, 52, 5033–5036 CrossRef CAS PubMed.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367–2371 CrossRef CAS PubMed.
- E. R. Darzi and R. Jasti, Chem. Soc. Rev., 2015, 44, 6401–6410 RSC.
- J. Niklas, K. L. Mardis and O. G. Poluektov, J. Phys. Chem. Lett., 2018, 9, 3915–3921 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental methods, 1H, 13C, COSY, and DOSY NMR spectra, crystallographic details, and DFT calculations. CCDC 1994725. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0sc03384g |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2020 |