
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolybdenum carbide and oxycarbide from carbon-supported MoO3 nanosheets: phase evolution and DRM catalytic activity assessed by TEM and in situ XANES/XRD methods†
Alexey
Kurlov
 a,
Xing
Huang
*b,
Evgeniya B.
Deeva
a,
Paula M.
Abdala
a,
Alexey
Fedorov
*a and
Christoph R.
Müller
*a
a,
Xing
Huang
*b,
Evgeniya B.
Deeva
a,
Paula M.
Abdala
a,
Alexey
Fedorov
*a and
Christoph R.
Müller
*a
aETH Zürich, Department of Mechanical and Process Engineering, Leonhardstrasse 21, CH 8092 Zürich, Switzerland. E-mail: fedoroal@ethz.ch; muelchri@ethz.ch
bETH Zürich, The Scientific Center for Optical and Electron Microscopy (ScopeM), Otto-Stern-Weg 3, CH 8093, Zürich, Switzerland. E-mail: xing.huang@scopem.ethz.ch
First published on 9th June 2020
Abstract
Molybdenum carbide (β-Mo2C) supported on carbon spheres was prepared via a carbothermal hydrogen reduction (CHR) method from delaminated nanosheets of molybdenum(VI) oxide (d-MoO3/C). The carburization process was followed by combined in situ XANES/XRD analysis revealing the formation of molybdenum oxycarbide Mo2CxOy as an intermediate phase during the transformation of d-MoO3/C to β-Mo2C/C. It was found that Mo2CxOy could not be completely carburized to β-Mo2C under a He atmosphere at 750 °C, instead a reduction in H2 is required. The β-Mo2C/C obtained showed activity and stability for the dry reforming of methane at 800 °C and 8 bar. In situ XANES/XRD evaluation of the catalyst under DRM reaction conditions combined with high resolution TEM analysis revealed the evolution of β-Mo2C/C to Mo2CxOy/C. Notably, the gradual oxidation of β-Mo2C/C to Mo2CxOy/C correlates directly with the increased activity of the competing reverse water gas shift reaction.
Introduction
Since the 1970s reports on the noble-metal-like catalytic activity of Mo2C1 and WC,2 transition metal carbides have remained in the focus of the catalysis community.3–11 For instance, molybdenum carbide has been studied as a catalyst for various heterogeneous reactions including methane reforming,12,13 methane dehydroaromatization,14,15 water gas shift,16–18 and deoxygenation reactions.19–22 The dry reforming of methane (DRM, eqn (1)) is a particularly relevant reaction because it allows producing synthesis gas from the two main greenhouse gases, CH4 and CO2, although it requires high temperatures for reaching high conversions (typically, 800 °C and above).23 However, Mo2C catalyzes also the reverse water gas shift reaction (RWGS, eqn (2)),24–26 which can occur in parallel with the DRM reaction consuming the hydrogen produced, resulting in a decreased H2/CO ratio of the produced syngas and the formation of steam.23,27,28 Currently, it remains unclear if the active sites for DRM and RWGS in Mo2C catalyst are identical.| CH4 + CO2 ↔ 2CO + 2H2 ΔH298 K = +247 kJ mol−1 | (1) |
| CO2 + H2 ↔ CO + H2O ΔH298 K = +41 kJ mol−1 | (2) |
The oxophilicity of Mo2C and, ultimately, its instability against oxidation to MoO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 12,29,30 in CO2-rich feeds at high operating temperatures make the DRM challenging for Mo2C-based catalysts.12,13,30–33 To attenuate this oxidative deactivation, DRM is often conducted at elevated pressure (2–10 bar) and/or with CH4-rich feeds.12,29,31,34 Those reports have indicated that a pristine Mo2C surface evolves to a molybdenum oxycarbide surface layer or a phase (Mo2CxOy), which plays a key role in both the DRM and RWGS reactions. It is also known that the oxygen coverage of the molybdenum carbide surface influences substantially its activity and selectivity, for instance in CO2-assisted propane dehydrogenation.35
12,29,30 in CO2-rich feeds at high operating temperatures make the DRM challenging for Mo2C-based catalysts.12,13,30–33 To attenuate this oxidative deactivation, DRM is often conducted at elevated pressure (2–10 bar) and/or with CH4-rich feeds.12,29,31,34 Those reports have indicated that a pristine Mo2C surface evolves to a molybdenum oxycarbide surface layer or a phase (Mo2CxOy), which plays a key role in both the DRM and RWGS reactions. It is also known that the oxygen coverage of the molybdenum carbide surface influences substantially its activity and selectivity, for instance in CO2-assisted propane dehydrogenation.35
Supported Mo2C-based catalysts are usually prepared by a so-called temperature programed reduction (TPR) method, i.e. a gas–solid reduction of molybdenum oxide (or ammonia molybdate precursor) by a mixture of H2 and a carbonaceous gas such as CH4 or C2H6.12,20,21,24,36 However, this procedure may also deposit passivating carbon on the catalytically active carbide surface.36 Alternatively, a carbothermal hydrogen reduction (CHR) method involving a reaction between solid carbon (or a carbon containing material) and molybdenum oxide under reducing (H2) or inert atmosphere can be exploited.37–39 The relatively low carburization temperatures (typically, <800 °C) and the absence of carbonaceous gases favour the production of high surface area nanostructured Mo2C, which is beneficial for catalytic applications.
Here, we studied the CHR reaction of two dimensional delaminated MoO3 nanosheets (d-MoO3) supported on carbon spheres, which served as carbon source for carburization and also provided a suitable morphological and phase contrast for transmission electron microscopy. Combination of in situ XANES/XRD analysis18,40,41 and high resolution TEM revealed that Mo2CxOy is an intermediate phase during the carbothermal CHR reaction of d-MoO3/C to β-Mo2C/C. Additionally, we demonstrate that under DRM conditions, β-Mo2C/C evolves into Mo2CxOy/C, which is found in active catalyst. Detection of the H2O produced during the in situ XANES/XRD (DRM conditions) experiments allowed us to link the oxidation of β-Mo2C/C to the emergence of the competing RWGS reaction. High resolution TEM of the active catalyst demonstrates two different morphologies, i.e. aggregates of nanoplatelets and nanorods, corresponding to β-Mo2C and Mo2CxOy structures, respectively.
Experimental section
Materials
The orthorhombic α-MoO3 nanobelts were synthesized by a reported hydrothermal method using ammonium heptamolybdate tetrahydrate (AHM, Sigma-Aldrich, 99.98% trace metals basis) and nitric acid (70%, Sigma-Aldrich, ACS reagent grade).42 In a standard experiment, the pH of AHM (1 g) solution in deionized (DI) water (20 mL) was adjusted to 1 by the dropwise addition of HNO3 (ca. 5 mL). The reaction mixture was then kept at 180 °C for 24 h in a Teflon-lined autoclave (45 mL). The obtained material was washed with DI water until a pH of ca. 7 was reached and subsequently dried at 100 °C.d-MoO3 nanosheets were synthesized according to a reported method.43 The orthorhombic α-MoO3 (1 g) was ground in an agate mortar with acetonitrile (0.2 mL, Sigma-Aldrich, ACS reagent, ≥99.5% purity) and the resulting material was dispersed by sonication in 50% aqueous ethanol (15 mL) for 2 h. After sonication, the suspension was centrifuged (8000 rpm, 30 min) and the supernatant containing the dissolved d-MoO3 nanosheets was collected and used for impregnation onto carbon spheres.
Carbon spheres were synthesized via a hydrothermal method from xylose (Sigma-Aldrich, ≥99% purity) in water.44,45 Xylose (6 g) was dissolved in DI water (15 mL) and the mixture was kept at 180 °C for 20 h in a Teflon-lined autoclave (45 mL).
d-MoO3/C material was obtained via wet impregnation of the supernatant solution of d-MoO3 (ca. 1.5 mg mL−1 by the thermogravimetric analysis) onto carbon spheres. The following treatment was applied: annealing of d-MoO3/C in N2 (800 °C, 1.5 h, 5 °C min−1) followed by annealing in 10 vol% H2 in N2 (30 min, 800 °C) giving ultimately β-Mo2C/C.
Characterization
Ex situ X-ray powder diffraction (XRD) data were collected on a PANalytical Empyrean X-ray diffractometer equipped with a Bragg–Brentano HD mirror and operated at 45 kV and 40 mA using CuKα radiation (λ = 1.5418 nm). The materials were examined within the 2θ range of 5–90° using a step size of 0.0167°. The scan time per step was 3 s. Thermogravimetric analysis (TGA) experiments were performed in a Mettler Toledo TGA/DSC 3 instrument. Typically, 750 μL of a colloidal solution of d-MoO3 was placed in a sapphire crucible (900 μL) that was heated to 80 °C (5 °C min−1) and kept for 1 h. Inductively coupled plasma atomic emission spectroscopy (ICP-AES) analysis was performed by the Mikroanalytisches Labor Pascher (Remagen, Germany). Scanning electron microscopy (SEM) was performed on a Zeiss LEO Gemini 1530 microscope. All electron microscopy images were taken at an acceleration voltage of 5 kV. Prior to imaging the materials were coated with a ca. 2 nm conductive layer of platinum.Transmission electron microscopy (TEM) samples were prepared by dry-deposition of the sample powders onto Cu TEM grids covered with a thin holey carbon layer. TEM, high resolution TEM (HR-TEM) and high angle annular dark field scanning TEM (HAADF-STEM) images of the samples were taken by an aberration-corrected JEOL JEM-ARM 300F GrandARM transmission electron microscope operated at 300 kV. The energy-dispersive X-ray spectroscopy (EDX) elemental mapping was performed using dual silicon drift EDX detectors with a total detection area of 200 mm2. The electron energy loss (EELS) spectra were recorded in dual EELS mode by a GIF Quantum ER System (Model 965) that is attached to the TEM.
Combined X-ray absorption spectroscopy (XAS) and powder diffraction (XRD) experiments were performed at the Swiss-Norwegian Beamlines (SNBL, BM31) at the European Synchrotron Radiation Facility (ESRF, Grenoble, France). XAS spectra were collected at the Mo K-edge using a double-crystal Si (111) monochromator with continuous scanning in transmission mode. XRD data were collected using a DEXELA-PerkinElmer 2923 CMOS pixel detector46 and a Si (111) channel-cut monochromator set at a wavelength of λ = 0.5 Å. The in situ dry reforming of methane (DRM) experiment was performed in a quartz capillary reactor.47 Calibration of the XAS data was based on Mo foil set at 20000.0 eV. In a typical XAS experiment ca. 2 mg of d-MoO3/C was placed between two quartz wool plugs in a capillary reactor (outer diameter 1.5 mm, wall thickness 0.1 mm). The carburization step was performed in pure He in the temperature range from 50 to 750 °C (5 mL min−1, ca. 4.5 °C min−1); a final annealing was performed in 50 vol% H2 in He (750 °C, 10 mL min−1, ca. 20 min). DRM tests were performed at 8 bar (CH4:CO2:He = 4:3:3) with the total flow rate varying in the range 1.75–3.5 mL min−1 (space velocity (SV) of ca. 3500–7000 L gMo−1 h−1; catalyst weight/volume flow rate (W/F) ratio of ca. 0.5–1 ms gMo mL−1) at 730 °C. The composition of the outlet gases was followed online by a mass spectrometer (MS). Ex situ XAS data were collected from pellets of reference materials with an optimized amount of sample mixed with cellulose. Activated materials were handled in a N2-filled glove box to prepare specimen for XAS analysis in air-tight sealed Al bags. XAS data were processed using the Athena software (Demeter 0.9.25 software package).48In situ time resolved XANES data were analyzed using a Multivariate Curve Resolution-Alternating Least Squares (MCR-ALS) method.40,49 MCR-ALS analysis was performed with a MATLAB software package using the multivariate curve resolution toolbox.50 The experimental spectra were analyzed in the 19950–20100 eV energy range. The number of components characterizing the whole XANES spectra dataset was determined using principal component analysis (PCA). Non-negative constraints for both the phase concentration and spectra profiles as well as cumulative concentration profile closure to 1 were applied in the analysis.49 The MCR-ALS routine was regarded successful when the convergence criterion fell below 0.1%.
Catalytic testing
The laboratory DRM tests were carried out in a metal fixed-bed reactor (Hastelloy X, 8 mm inner diameter) at 8 bar. In a typical experiment, 75 mg of d-MoO3/C was placed in between two quartz wool plugs. Prior to the catalytic tests, d-MoO3/C was transformed in situ into β-Mo2C/C by thermal treatment in (1) N2 (800 °C, 1.5 h, 20 mL min−1, 5 °C min−1) followed by (2) 10 vol% H2 in N2 (800 °C, 30 min, 20 mL min−1) at atmospheric pressure. After this pretreatment, the pressure was increased to 8 bar (N2) and the DRM feed was introduced (CH4:CO2:N2 = 4:3:3, a total flow rate was 10 mL min−1, SV ca. 550 L gMo−1 h−1, W/F ca. 6.5 ms gMo mL−1, 800 °C, 8 bar). The composition of the off-gas was analyzed via a gas chromatograph (GC, PerkinElmer Clarus 580) equipped with a thermal conductivity detector (TCD).
Results and discussion
Orthorhombic α-MoO3 nanobelts were synthesized via a reported route.42 The nanobelt morphology of α-MoO3 was confirmed by scanning electron microscopy (SEM, Fig. S1†) and phase purity by X-ray powder diffraction (XRD) analysis (space group Pbnm, Fig. S2†). Note that increasing the Mo concentration in the hydrothermal synthesis from 0.56 to 2 mol L−1 yields a hexagonal polymorph h-MoO3 (space group P63/m, Fig. S3†). Sonication of grinded α-MoO3 and its dispersion in 50% aqueous ethanol yielded, after centrifugation, a transparent colloidal solution of delaminated MoO3 films (d-MoO3). Transmission electron microscopy (TEM) imaging of a dried aliquot of this solution revealed a few-layer-thin nanosheets of d-MoO3 (Fig. S4†). High resolution TEM (HR-TEM) images show lattice fringes separated by 3.72 Å, which is characteristic for the distance of the (1 1 0) planes in α-MoO3 (Fig. S4†). The wet impregnation of carbon spheres (Fig. S5†) with the colloidal solution of d-MoO3 nanosheets (ca. 1.5 mg mL−1 by thermogravimetric analysis, Fig. S6†) yielded, after drying at 100 °C, d-MoO3/C (1.49 wt% Mo by elemental analysis). High angle annular dark field scanning TEM (HAADF-STEM) imaging of d-MoO3/C shows carbon spheres (light contrast) covered by a Mo-rich phase (heavier contrast) that segregates as poorly crystalline halo-like patterns around the carbon spheres (Fig. 1b–d and S7†). We also observed small poorly ordered crystalline clusters imbedded in the amorphous matrix of carbon spheres, as shown in Fig. 1d and S7.† Energy-dispersive X-ray spectroscopy (EDX) mapping indicates that the Mo-rich phase contains both Mo and O, consistent with d-MoO3, as well as C from, possibly, bound acetonitrile/ethanol solvent molecules (Fig. S8†). In agreement with EDX, electron energy loss spectroscopy (EELS) analysis indicates that the material contains C, O, and Mo (Fig. S9†). According to the shape and energy position of the O K-edge peak (ca. 530 eV), Mo atoms have an oxidation state between +4 and +6 (Fig. S10†).51,52 The partial reduction of Mo +6 states is likely due to the electron beam reduction during the acquisition of EELS data, consistent with the previous data.51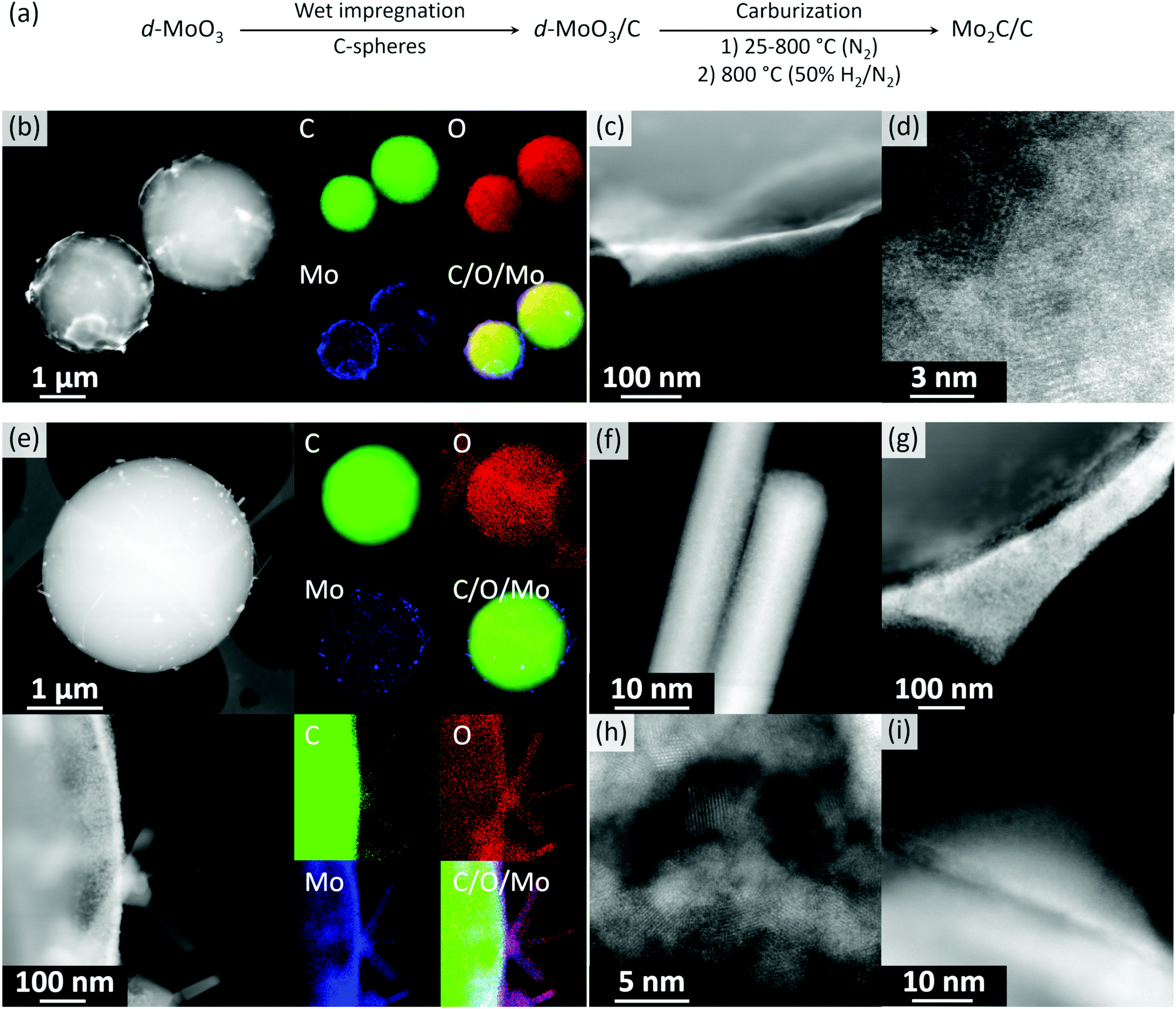 | ||
| Fig. 1 (a) Synthesis of β-Mo2C/C from delaminated MoO3 films; (b) HAADF-STEM images and EDX maps of d-MoO3/C; (c) and (d) HR-STEM images of d-MoO3/C; (e) HAADF-STEM images and EDX maps of β-Mo2C/C; and (f)–(i) HR-STEM images of different morphologies decorating the surface of carbon spheres in β-Mo2C/C. | ||
Annealing of d-MoO3/C at 800 °C in N2 (1.5 h) followed by an additional annealing step in 10 vol% H2 (800 °C, 30 min) gave Mo2C/C. STEM imaging of this material, after exposure of the specimen to air during sample transfer, reveals crystalline nanorods of ca. 100–500 nm in length decorating the surface of carbon spheres (Fig. 1f). Additionally, crystalline agglomerates (Fig. 1g and h) and core/shell nanoparticles (Fig. 1i) were found on the surface of the carbon spheres. HAADF-STEM images uncover that the core/shell structures have a crystalline core and an amorphous shell (Fig. 1i). EELS analysis performed on all observed morphologies demonstrates a shift of the Mo M2,3 edge to a lower energy when compared to d-MoO3/C, which indicates a more reduced Mo. However, the oxidization state of Mo is between +2 to +4 (Fig. S11†). Furthermore, EDX mapping confirms the oxidized state of Mo, revealing that all structures contain Mo, C and O (Fig. 1e). These results indicate that an oxidation of molybdenum carbide took place when exposing the activated reduced material to ambient air during sample transfer, consistent with the high reactivity of the activated, highly oxophilic Mo2C surface.53
To obtain information about the state of the annealed material at the specific temperatures and gas atmospheres, we turned to in situ experimentation and followed the CHR process (d-MoO3/C → β-Mo2C/C) by a combined in situ XANES/XRD experiment.18,40,41 The XANES spectra were collected at the Mo K-edge during annealing of d-MoO3/C from 50 to 750 °C (highest attainable temperature of the set up) under He and 50% H2/He at 750 °C (total flow rates of 5 and 10 mL min−1, respectively), while XRD patterns were collected at 50, 400 and 750 °C during the same experiment. For the latter temperature, data were collected under a flow of pure helium or 50% H2/He. The XRD pattern collected at 50 °C contains no Bragg peaks and shows a dominant amorphous halo due to the C phase, confirming the low crystallinity of the d-MoO3/C material as observed by TEM (Fig. 2a and 1d). At 400 °C, Bragg peaks corresponding to the MoO2 phase (distorted rutile type structure, space group P21/c) appear, indicating a reduction of d-MoO3 to MoO2 by the carbon spheres (reducing agent), accompanied by the crystallization of MoO2 (Fig. 2a). Interestingly, while at 750 °C under a He atmosphere only reflections of MoO2 are present, flowing H2 through the specimen immediately reduced it to β-Mo2C (Fig. 2a). Our in situ XRD results agree well with previous studies that have identified MoO2 as an intermediate phase during the carburization of MoO3 to Mo2C, both via TPR25,54 and CHR39,55,56 routes. In addition, it was also reported that annealing of carbon-supported AHM in pure He at 800 °C for 2 h does not lead to its complete carburization, giving a mixture of MoO2 and β-Mo2C.56
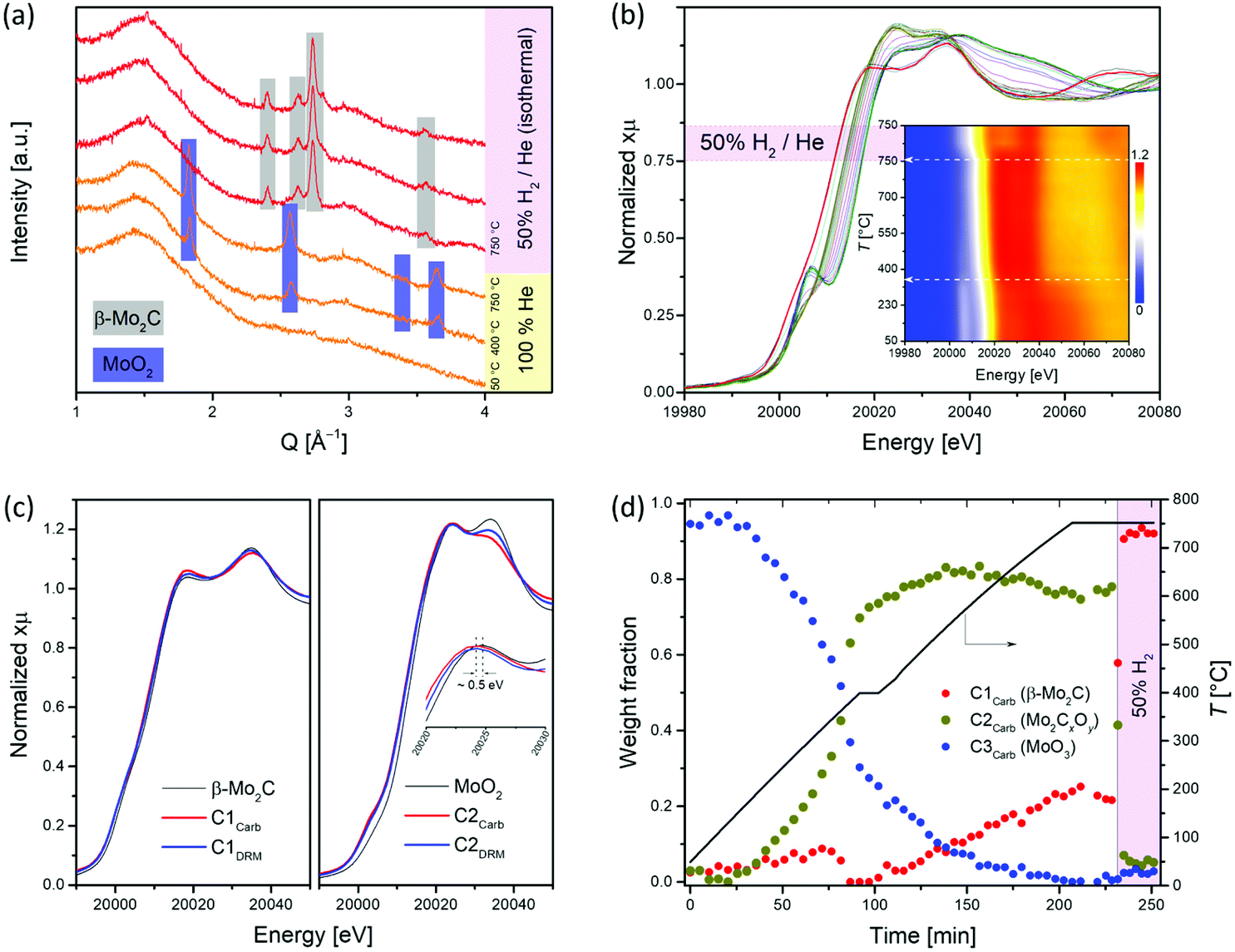 | ||
| Fig. 2
In situ carburization of d-MoO3/C under He and H2 as followed by (a) XRD (Q = 4πsin(θ)/λ) and (b) Mo K-edge XANES; the inset shows the evolution of the pre-edge and edge features with increasing temperature. (c) Comparison of the extracted C1 and C2 components spectra from the MCR-ALS analysis performed during the carburization of d-MoO3/C (labelled Carb) to β-Mo2C/C and during the dry reforming of methane (labelled DRM, vide infra) along with the references β-Mo2C and MoO2. (d) Phase evolution during the carburization process according to MCR-ALS of the XANES data. | ||
The in situ XANES data collected during annealing of d-MoO3/C indicates clearly a gradual reduction of Mo6+ to Mo4+ when heating under He to 750 °C and the formation of β-Mo2C, upon switching to 50 vol% H2/He, as confirmed by a shift of the Mo K edge energies from 20016.3 (at room temperature) to, respectively 20011.6 and 20000.6 eV (without and with co-feed H2 at 750 °C; Fig. 2b).18 Two notable changes are observed in the in situ XANES spectra. The first change occurs at ca. 360 °C and is associated with the disappearance of the characteristic pre-edge feature, typical for molybdenum in a non-centrosymmetric environment (Fig. 2b, inset),57,58 that can be explained by the reduction/transformation of d-MoO3 to MoO2. From 360 °C to 750 °C (heating up in He), a slight shift of the Mo K-edge position towards lower energies is observed, consistent with a partial reduction. The second change occurs immediately after introducing H2 at 750 °C and is associated with a prominent shift of the Mo K-edge position from 20011.6 to 20000.6 eV, highlighting a further reduction of the Mo phase. In agreement with the in situ XRD data, the XANES spectrum of the final phase corresponds to the reference spectrum of β-Mo2C.
Further analysis of the in situ XANES data via principal component analysis (PCA) indicates the presence of three components during the annealing of d-MoO3/C (Fig. S12†). Using the information of the presence of three phases, MCR-ALS analysis was performed to extract the three components spectra that reproduce the full data set (Fig. S13†). Comparison of the obtained spectra with the available references revealed that the spectrum of the component C1Carb is identical to that of β-Mo2C (Fig. 2c). The spectrum of component C2Carb resembles that of MoO2 except that the edge and the white line feature are shifted to a lower energy (and the shape of the white line is slightly different), indicating overall a reduced Mo oxidation state compared to Mo4+, i.e. an oxidation state between MoO2 and Mo2C (Fig. 2c). This finding suggests assigning the C2Carb component to molybdenum oxycarbide (Mo2CxOy). Combining this new insight with XRD analysis, we speculate that the Mo2CxOy phase may be formed due to the incorporation of C into the MoO2 lattice or surface layers during annealing, thus retaining the bulk structure of MoO2 (distorted rutile type structure, space group P21/c). The formation of such a MoO2/Mo2CxOy phase is in a good agreement with a previous report indicating that MoO2 can coexist with oxycarbide phases.59 For brevity, we will refer to the MoO2/Mo2CxOy phase below as Mo2CxOy. Notably, the shape of the obtained XANES spectra of the C2Carb component resembles a recently reported molybdenum oxycarbide Mo2CxOy, although here, the Mo K-edge is shifted to lower energies indicating a more reduced Mo state, probably due to the higher C content in the oxycarbide.60 Lastly, the edge position and the oscillation profile of component C3Carb is similar to that of MoO3, which allows ascribing C3Carb to MoO3. The changes of weight fractions of the C1Carb–C3Carb components with annealing is presented in Fig. 2d. Mo2CxOy that is formed via the reduction of MoO3 by the carbon of the spheres, starts to appear at ca. 200 °C and reaches an asymptotic level of ca. 75–80% at 400 °C (Fig. 2d). Surprisingly, MCR indicates that the molybdenum carbide phase appears already at ca. 400 °C. This has not been observed in previous carburization studies of supported Mo oxides and is explained by the use of a carbon support as the carburizing agent in place of the traditionally used CH4/H2 carburizing gas mixture and thin sheets of d-MoO3. The fraction of MoO3 reduces continuously from ca. 170 °C until it disappears at 750 °C. At this stage Mo2CxOy and Mo2C coexist in fraction of 75% and 25%, respectively (Fig. 2d). However, as discussed above, a complete carburization by the internal carbon source could not be accomplished at 750 °C in He. To attain the complete conversion of the remaining Mo2CxOy to β-Mo2C, a gas containing 50 vol% H2 in He was passed through the reactor at 750 °C. Under such conditions the remaining Mo2CxOy was immediately carburized to β-Mo2C according to MCR analysis (Fig. 2d).
To summarize, although MCR-ALS analysis reveals a coexistance of Mo2CxOy and β-Mo2C at 750 °C in He, in situ XRD data showed only peaks corresponding to distorted rutile type MoO2. This observation can be explained by (i) the amorphous nature of the molybdenum oxycarbide phase and/or (ii) the incorporation of variable amounts of carbon into the MoO2 phase, forming effectively the Mo2CxOy phase with a reduced Mo oxidation state (and the corresponding Mo K-edge position), yet maintaining the same crystal structure as MoO2.
Next, we optimized the DRM conditions to obtain a stable catalytic performance of β-Mo2C/C. The condition identified is a pressure of 8 bar at 800 °C and a CH4-rich feed, i.e. CH4:CO2:N2 = 4:3:3 (Fig. 3a). In particular, under this condition no considerable deactivation was observed over 8 h. The space time yield (STY) of CO was 0.22 mol(CO) mol(Mo)−1 s−1 while the methane conversion rate was 0.11 mol(CH4) mol(Mo)−1 s−1 (X(CH4) was ca. 45%, Fig. 3b). However, the obtained H2/CO ratio was ca. 0.7, which indicates that the RWGS and DRM reactions compete under these conditions.
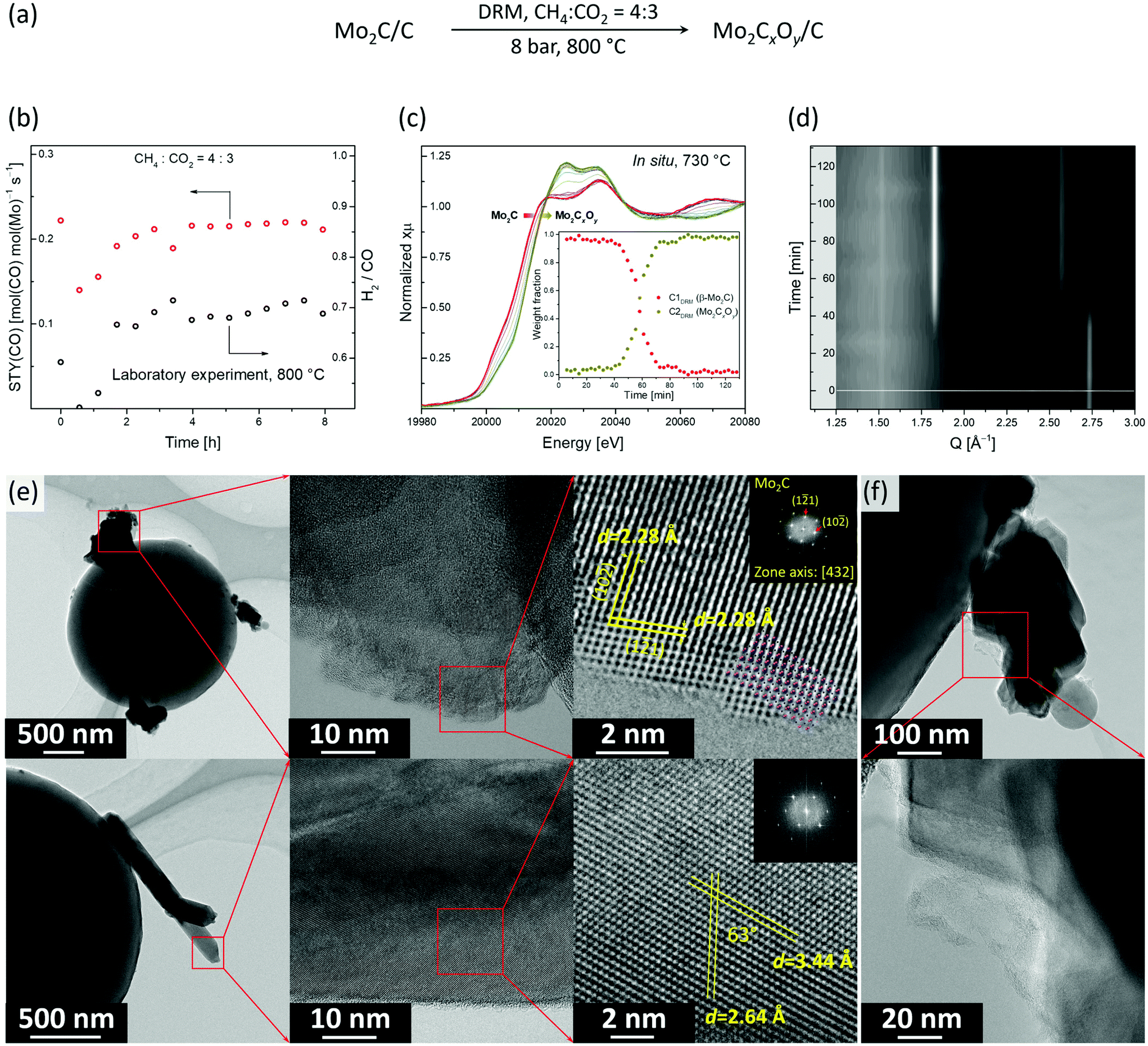 | ||
| Fig. 3 (a) Schematic of the experimental conditions during the DRM experiment; (b) catalytic performance of β-Mo2C/C in DRM (laboratory scale reactor, 800 °C, 8 bar, SV = 550 L gMo−1 h−1); DRM experiment followed by (c) Mo K-edge XANES, where the inset shows the phase evolution according to MCR-ALS, and (d) XRD (Q = 4πsin(θ)/λ) (capillary reactor, 730 °C, 8 bar, SV = 3500 L gMo−1 h−1); (e) TEM and HR-TEM images of the spent Mo2CxOy/C catalyst revealing two different morphologies, i.e. aggregates of nanoplates (top) and nanorods (bottom); (f) TEM images of amorphous carbon deposited on the active Mo2CxOy/C catalyst. | ||
To identify the active state and to follow the structural evolution of the β-Mo2C/C catalyst under DRM conditions, an in situ XANES/XRD experiment was carried out in a capillary reactor. After the carburization pre-treatment, the feed was switched to DRM conditions (CH4:CO2 = 4:3 at 8 bar and 730 °C) while analyzing the composition of the outlet gas by MS. Note that due to the heater limitation, the actual temperature in our catalytic in situ XANES/XRD experiment was 730 °C, instead of 800 °C as in the laboratory experiment, yet qualitatively similar results can be expected at those temperatures (Fig. 3c and d). Laboratory and capillary synchrotron DRM experiments differ also in catalyst weight/volume flow rate (W/F) ratio, which were W/F = 6.5 and 1 ms gMo mL−1, respectively. PCA analysis indicates that two components are necessary to account for changes of XANES spectra with time on stream (TOS, Fig. S12†). MCR-ALS analysis of the in situ XANES data yielded the XANES spectra for these two components, i.e. C1DRM and C2DRM (Fig. S13†). Comparison of the calculated spectra with references and the components obtained in the carburization MCR-ALS analysis, identified them as β-Mo2C and Mo2CxOy, respectively (Fig. 2c and S13†). Notably, the spectrum of the component C1DRM, identified as β-Mo2C, is identical to the spectrum of the component C1Carb obtained for the carburization process (Fig. 2c). Interestingly, while the shape and edge position of the spectrum of C2DRM (Mo2CxOy) is very similar to C2Carb, it bears a notable difference in the intensities of the white line features (Fig. 2c). This can probably be explained by the different C content and/or C distribution within the oxycarbide, formed by incorporation of oxygen atoms into β-Mo2C (DRM conditions) or by incorporation of carbon atoms into MoO2 (CHR conditions). No changes in XANES spectra and XRD data of β-Mo2C occur during the first 30–40 min TOS (Fig. 3c and d). Note that the products CO and H2 of the DRM reaction are detected by MS during the entire experiment (Fig. S14†). After this initial period, a shift of the Mo K-edge position to higher energies sets in, indicating the gradual oxidation of β-Mo2C. After ca. 1 h of TOS, β-Mo2C was transformed to Mo2CxOy according to the concentration profiles obtained by MCR analysis (Fig. 3c). Consistent with XANES data, the XRD peaks corresponding to MoO2 (or Mo2CxOy) appear after ca. 40 min of TOS with a simultaneous reduction of the intensities of the peaks due to β-Mo2C (Fig. 3d). After 50 min TOS, the MoO2 peaks reach their maximal intensities and only a low intensity (1 2 1) peak of β-Mo2C (Q = 2.74 Å−1) could be detected at this time. Interestingly, the MS data reveals an increase of the H2O signal starting from TOS = 45 min, arising from the competing RWGS reaction. The increase in the MS H2O signal coincides with the beginning of the oxidation of β-Mo2C observed by XANES and XRD (Fig. S14†). This result suggests that the RWGS reaction is favoured on a more oxygen-rich Mo2CxOy phase.
Flowing pure CH4 for 1 h through the reacted catalyst after 130 min TOS does not lead to a re-carburization, leaving the catalyst composition unchanged according to the XANES data (Fig. S15†).
We have also investigated by TEM the active state of the Mo2C/C catalyst, i.e. the material recovered after cooling down the working catalyst to room temperature in N2 after 480 min TOS (Mo2C/CTOS480) at the stable performance stage (Fig. 3d). The specimen was transferred in ambient air before the TEM analysis. Mo2C/CTOS480 features aggregates of nanoplates and nanorods as the two main morphologies (Fig. 3d and S16†). We observe that the crystallinity of the material surface layer (for both morphologies) is lower compared to the crystallinity of the bulk structure (Fig. S17†). Based on the EELS data, the Mo M2,3 edges position shows that the molybdenum oxidation state is below +4. Interestingly, the maximum of the Mo M3 edge of Mo2C/CTOS480 is found at a lower energy compared to that of Mo2C/C (395.5 eV and 397.5 eV, respectively, Fig. S11†), although both materials were exposed to ambient air during the transfer. This difference might be owing to the oxidation of β-Mo2C during the DRM reaction, which enriches the catalyst surface layers with O, thereby preventing a further oxidation in air at room temperature. For both morphologies, the EDX mapping shows the presence of Mo, C and O elements, however, a higher oxygen to molybdenum ratio is observed in the nanorods compared to the nanoparticles (Fig. S18†). Additionally, a higher oxygen content is observed in the surface layer compared to the bulk structure for both morphologies (Fig. S19†), in line with the presence of an oxycarbidic layer. The structural analysis of a plate-like nanoparticle by HR-TEM reveals that its crystal structure matches that of the orthorhombic Mo2C (Fig. 3e, top). In particular, the observed structure fits perfectly the [4 3 2] zone axis of β-Mo2C (Pbcn). In contrast, the nanorod structure does not correspond to β-Mo2C and it fits neither to MoO3 or MoO2 with a distorted rutile structure (Fig. S20†). This observation combined with a high O content, in addition to the simultaneous presence of Mo and C elements in the EDX maps, allows us to attribute the nanorods to a crystalline oxycarbidic phase Mo2CxOy. Additionally, amorphous carbon was found attached to the surface of Mo2C/CTOS480 (Fig. 3e). The origin of the carbon observed can be either the partial degradation of the carbon spheres support or catalyst coking under reaction conditions. However the latter is unlikely due to the low coking rate of carbide based catalysts.12 Noteworthy, the TEM analysis of Mo2C/CTOS480 reveals no presence of MoO2 proving the high stability of Mo2CxOy towards oxidation under operating conditions.
Conclusions
We have shown that delaminated MoO3 nanosheets dispersed on carbon spheres, are carburized only partially by annealing in an inert gas and require H2 to form β-Mo2C/C quantitatively. The carburized material shows a stable performance in the DRM reaction at an elevated pressure of 8 bar. The combination of advanced in situ XANES/XRD analysis and TEM characterization allowed us to attribute a combination of β-Mo2C and Mo2CxOy to the active state of the catalyst. In addition, the higher oxidation states of β-Mo2C/Mo2CxOy catalyst correlate with higher rates of the competing, undesired RWGS reaction.Conflicts of interest
The authors declare that there is no conflict of interest regarding the publication of this article.Acknowledgements
The authors thank ScopeM (ETH Zürich) for the use of their electron microscopy facilities. The Swiss-Norwegian Beamline (SNBL) at European Synchrotron Radiation Facility (ESRF, Grenoble) is acknowledged for provision of beamtime through proposal CH5410. SNBL staff members, Dr Wouter van Beek and Dr Dragos Stoian, are thanked for support during the experiment. Dr Felix Donat is acknowledged for assistance with the MCR analysis. This project has received funding from the Bundesamt für Energie (SI/500881-01) and ETH Zurich (ETH-40 17-2).Notes and references
- J. H. Sinfelt and D. J. Yates, Nat. Phys. Sci., 1971, 229, 27–28 CrossRef CAS.
- R. B. Levy and M. Boudart, Science, 1973, 181, 547–549 CrossRef CAS PubMed.
- S. T. Oyama, The Chemistry of Transition Metal Carbides and Nitrides, Springer, 1996 Search PubMed.
- H. H. Hwu and J. G. Chen, Chem. Rev., 2005, 105, 185–212 CrossRef CAS PubMed.
- A. M. Alexander and J. S. Hargreaves, Chem. Soc. Rev., 2010, 39, 4388–4401 RSC.
- A. L. Stottlemyer, T. G. Kelly, Q. Meng and J. G. Chen, Surf. Sci. Rep., 2012, 67, 201–232 CrossRef CAS.
- W. F. Chen, J. T. Muckerman and E. Fujita, Chem. Commun., 2013, 49, 8896–8909 RSC.
- Y. Ma, G. Guan, X. Hao, J. Cao and A. Abudula, Renewable Sustainable Energy Rev., 2017, 75, 1101–1129 CrossRef CAS.
- W. Wan, B. M. Tackett and J. G. Chen, Chem. Soc. Rev., 2017, 46, 1807–1823 RSC.
- M. M. Moyer, C. Karakaya, R. J. Kee and B. G. Trewyn, ChemCatChem, 2017, 9, 3090–3101 CrossRef CAS.
- R. Dronskowski, S. Kikkawa and A. Stein, Handbook of Solid State Chemistry, Wiley, 2017 Search PubMed.
- J. B. Claridge, A. P. E. York, A. J. Brungs, C. Marquez-Alvarez, J. Sloan, S. C. Tsang and M. L. H. Green, J. Catal., 1998, 180, 85–100 CrossRef CAS.
- A. J. Brungs, A. P. E. York, J. B. Claridge, C. Marquez-Alvarez and M. L. H. Green, Catal. Lett., 2000, 70, 117–122 CrossRef CAS.
- I. Lezcano-Gonzalez, R. Oord, M. Rovezzi, P. Glatzel, S. W. Botchway, B. M. Weckhuysen and A. M. Beale, Angew. Chem., Int. Ed., 2016, 55, 5215–5219 CrossRef CAS PubMed.
- N. Kosinov, F. J. A. G. Coumans, E. A. Uslamin, A. S. G. Wijpkema, B. Mezari and E. J. M. Hensen, ACS Catal., 2016, 7, 520–529 CrossRef.
- J. Patt, D. J. Moon, C. Phillips and L. Thompson, Catal. Lett., 2000, 65, 193–195 CrossRef CAS.
- P. Liu and J. A. Rodriguez, J. Phys. Chem. B, 2006, 110, 19418–19425 CrossRef CAS PubMed.
- E. B. Deeva, A. Kurlov, P. M. Abdala, D. Lebedev, S. M. Kim, C. P. Gordon, A. Tsoukalou, A. Fedorov and C. R. Müller, Chem. Mater., 2019, 31, 4505–4513 CrossRef CAS.
- W.-S. Lee, A. Kumar, Z. Wang and A. Bhan, ACS Catal., 2015, 5, 4104–4114 CrossRef CAS.
- J. A. Schaidle, J. Blackburn, C. A. Farberow, C. Nash, K. X. Steirer, J. Clark, D. J. Robichaud and D. A. Ruddy, ACS Catal., 2016, 6, 1181–1197 CrossRef CAS.
- M. M. Sullivan, C.-J. Chen and A. Bhan, Catal. Sci. Technol., 2016, 6, 602–616 RSC.
- H. Wang, S. Liu and K. J. Smith, Energy Fuels, 2016, 30, 6039–6049 CrossRef CAS.
- D. Pakhare and J. Spivey, Chem. Soc. Rev., 2014, 43, 7813–7837 RSC.
- M. D. Porosoff, X. Yang, J. A. Boscoboinik and J. G. Chen, Angew. Chem., Int. Ed., 2014, 53, 6705–6709 CrossRef CAS PubMed.
- W. Xu, P. J. Ramirez, D. Stacchiola and J. A. Rodriguez, Catal. Lett., 2014, 144, 1418–1424 CrossRef CAS.
- S. Posada-Pérez, P. J. Ramírez, R. A. Gutiérrez, D. J. Stacchiola, F. Viñes, P. Liu, F. Illas and J. A. Rodriguez, Catal. Sci. Technol., 2016, 6, 6766–6777 RSC.
- M. C. J. Bradford and M. A. Vannice, J. Catal., 1999, 183, 69–75 CrossRef CAS.
- J. Kehres, J. G. Jakobsen, J. W. Andreasen, J. B. Wagner, H. Liu, A. Molenbroek, J. Sehested, I. Chorkendorff and T. Vegge, J. Phys. Chem. C, 2012, 116, 21407–21415 CrossRef CAS.
- D. C. LaMont, A. J. Gilligan, A. R. S. Darujati, A. S. Chellappa and W. J. Thomson, Appl. Catal., A, 2003, 255, 239–253 CrossRef CAS.
- T.-c. Xiao, A. Hanif, A. P. E. York, Y. Nishizaka and M. L. H. Green, Phys. Chem. Chem. Phys., 2002, 4, 4549–4554 RSC.
- A. P. E. York, J. B. Claridge, A. J. Brungs, S. C. Tsang and M. L. H. Green, Chem. Commun., 1997, 1, 39–40 RSC.
- D. C. LaMont and W. J. Thomson, Chem. Eng. Sci., 2005, 60, 3553–3559 CrossRef CAS.
- P. Liang, H. Gao, Z. Yao, R. Jia, Y. Shi, Y. Sun, Q. Fan and H. Wang, Catal. Sci. Technol., 2017, 7, 3312–3324 RSC.
- T. Xiao, H. Wang, J. Da, K. S. Coleman and M. L. H. Green, J. Catal., 2002, 211, 183–191 CAS.
- M. M. Sullivan and A. Bhan, J. Catal., 2018, 357, 195–205 CrossRef.
- J. S. Lee, S. T. Oyama and M. Boudart, J. Catal., 1987, 106, 125–133 CrossRef CAS.
- S. Chaudhury, S. K. Mukerjee, V. N. Vaidya and V. Venugopal, J. Alloys Compd., 1997, 261, 105–113 CrossRef CAS.
- H. M. Wang, X. H. Wang, M. H. Zhang, X. Y. Du, W. Li and K. Y. Tao, Chem. Mater., 2007, 19, 1801–1807 CrossRef CAS.
- E. Ochoa, D. Torres, R. Moreira, J. L. Pinilla and I. Suelves, Appl. Catal., B, 2018, 239, 463–474 CrossRef CAS.
- A. Tsoukalou, P. M. Abdala, D. Stoian, X. Huang, M. G. Willinger, A. Fedorov and C. R. Muller, J. Am. Chem. Soc., 2019, 141, 13497–13505 CrossRef CAS PubMed.
- M. A. Naeem, P. M. Abdala, A. Armutlulu, S. M. Kim, A. Fedorov and C. R. Müller, ACS Catal., 2020, 10, 1923–1937 CrossRef CAS.
- A. Chithambararaj, N. Rajeswari Yogamalar and A. C. Bose, Cryst. Growth Des., 2016, 16, 1984–1995 CrossRef CAS.
- F. Ji, X. Ren, X. Zheng, Y. Liu, L. Pang, J. Jiang and S. Liu, Nanoscale, 2016, 8, 8696–8703 RSC.
- M.-M. Titirici, M. Antonietti and N. Baccile, Green Chem., 2008, 10, 1204–1212 RSC.
- M. A. Naeem, A. Armutlulu, Q. Imtiaz, F. Donat, R. Schaublin, A. Kierzkowska and C. R. Muller, Nat. Commun., 2018, 9, 2408 CrossRef PubMed.
- P. M. Abdala, H. Mauroy and W. van Beek, J. Appl. Crystallogr., 2014, 47, 449–457 CrossRef CAS.
- W. van Beek, O. V. Safonova, G. Wiker and H. Emerich, Phase Transitions, 2011, 84, 726–732 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- A. Martini, E. Borfecchia, K. A. Lomachenko, I. A. Pankin, C. Negri, G. Berlier, P. Beato, H. Falsig, S. Bordiga and C. Lamberti, Chem. Sci., 2017, 8, 6836–6851 RSC.
- J. Jaumot, A. de Juan and R. Tauler, Chemom. Intell. Lab. Syst., 2015, 140, 1–12 CrossRef CAS.
- D. Wang, D. S. Su and R. Schlögl, Z. Anorg. Allg. Chem., 2004, 630, 1007–1014 CrossRef CAS.
- L. Lajaunie, F. Boucher, R. Dessapt and P. Moreau, Ultramicroscopy, 2015, 149, 1–8 CrossRef CAS PubMed.
- W. Wu, Z. Wu, C. Liang, P. Ying, Z. Feng and C. Li, Phys. Chem. Chem. Phys., 2004, 6, 5603–5608 RSC.
- H. J. Guzmán, W. Xu, D. Stacchiola, G. Vitale, C. E. Scott, J. A. Rodríguez and P. Pereira-Almao, Can. J. Chem., 2013, 91, 573–582 CrossRef.
- G. Vitale, H. Guzmán, M. L. Frauwallner, C. E. Scott and P. Pereira-Almao, Catal. Today, 2015, 250, 123–133 CrossRef CAS.
- W. F. Chen, C. H. Wang, K. Sasaki, N. Marinkovic, W. Xu, J. T. Muckerman, Y. Zhu and R. R. Adzic, Energy Environ. Sci., 2013, 6, 943–951 RSC.
- S. Takenaka, T. Tanaka, T. Funabiki and S. Yoshida, J. Phys. Chem. B, 1998, 102, 2960–2969 CrossRef CAS.
- A. M. Beale and G. Sankar, Chem. Mater., 2003, 15, 146–153 CrossRef CAS.
- S. R. J. Likith, C. A. Farberow, S. Manna, A. Abdulslam, V. Stevanović, D. A. Ruddy, J. A. Schaidle, D. J. Robichaud and C. V. Ciobanu, J. Phys. Chem. C, 2018, 122, 1223–1233 CrossRef CAS.
- Y. Zheng, Y. Tang, J. R. Gallagher, J. Gao, J. T. Miller, I. E. Wachs and S. G. Podkolzin, J. Phys. Chem. C, 2019, 123, 22281–22292 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0nr02908d |
| This journal is © The Royal Society of Chemistry 2020 |