
Crystallization-induced room-temperature phosphorescence in fumaramides†
Andrea
Nitti
 a,
Chiara
Botta
*b,
Alessandra
Forni
*c,
Elena
Cariati
d,
Elena
Lucenti
c and
Dario
Pasini
*a
a,
Chiara
Botta
*b,
Alessandra
Forni
*c,
Elena
Cariati
d,
Elena
Lucenti
c and
Dario
Pasini
*a
aDepartment of Chemistry and, INSTM Research Unit, University of Pavia, Via Taramelli 12, 27100 Pavia, Italy. E-mail: dario.pasini@unipv.it
bSCITEC-CNR, Consiglio Nazionale delle Ricerche and, INSTM Research Unit, Istituto di Scienze e Tecnologie Chimiche ‘G. Natta’, Via Corti 12, 20133 Milano, Italy
cSCITEC-CNR, Consiglio Nazionale delle Ricerche and, INSTM Research Unit, Istituto di Scienze e Tecnologie Chimiche ‘G. Natta’, Via Golgi 19, 20133 Milano, Italy
dDepartment of Chemistry, INSTM Research Unit, University of Milan, Via Golgi 19, 20133 Milano, Italy
First published on 14th October 2020
Abstract
Novel fumaramides exhibit room-temperature phosphorescence in the solid state once molecular design and positioning of the carbonyl and bromine atoms allow for the formation of strong intermolecular halogen bonding interactions. The extremely long phosphorescence lifetimes at room temperature are promising for solid state applications as organic phosphors.
Materials with stable and persistent room-temperature phosphorescence (RTP) in the solid state are attractive because of their applications spanning from optoelectronics to photomedicine and sensing.1 RTP phosphors have traditionally been developed from inorganic compounds and organometallic complexes;2 considered to be practically non-phosphorescent, pure (metal-free) organic compounds were very rarely explored before 2010.3 Organic chromophores with solid-state phosphorescence are in fact exciting materials,4 but they need to possess suitable molecular properties able to overcome the obstacles imposed by the aggregation-caused quenching (ACQ), which is usually observed in conventional chromophores in the condensed phase, and results in the switching off of all emissive properties.
Crystallization-induced phosphorescence (CIP), first reported by B. Z. Tang and co-workers,5 is an effective approach to achieve efficient pure organic material RTP, developing crystal engineering paradigms. After gaining a comprehensive understanding of the phosphorescence mechanisms in the solid state, different methodologies, including polymer aggregation, directional halogen and hydrogen bonding mediated aggregation, and self-assembly, have been used to achieve CIP.6–10 These strategies control the aggregation behavior promoting the stabilization of triplet excited states by suppressing their non-radiative-decay pathways; as such, they can be considered as part of the broader family of aggregation-induced emissive (AIE) phenomena.11
A precise control of the molecular design is fundamental to activate the intersystem crossing (ISC) process and achieve persistent RTP emission.12 Most of the structures are made up of rather complex organic compounds, whose RTP properties have been serendipitously discovered. All the reported organic structures for RTP possess π-extended, aromatic units as the key chromophores in the molecule. No structural unit based on a carbon–carbon double bond chromophore has been reported so far,13 yet suitably tetrasubstituted ethylene chromophores are amongst the prototypical and most efficient moieties showing AIE properties. We have been previously involved in the realization of small organic chromophores, constructed from tri- or tetrasubstituted ethylene cores, with peculiar AIE and solid state thermo- and mechanochromic properties.14
Herein, we report a new, simple organic amide, compound 1, which is constructed from a 2,3-dibromofumaryl central unit flanked with two N-methylaniline units and we show its unique fluorescence–phosphorescence dual-emission behavior and CIP properties with ultralong lifetimes in the solid state. To better understand its structure–property relationship, we report and characterize structurally related compounds 2 and 3, in which the key functional groups have been systematically varied (Fig. 1).
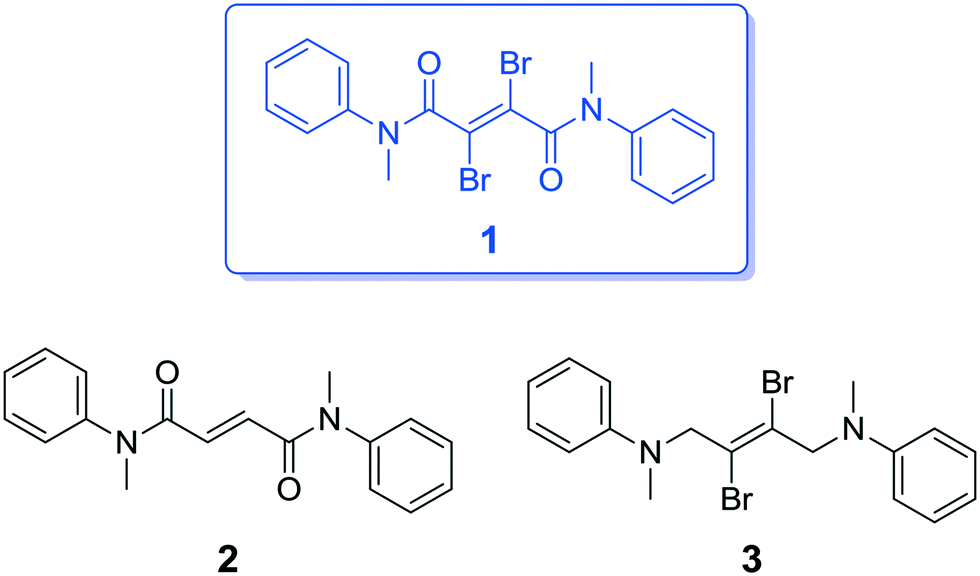 | ||
| Fig. 1 Chemical structures of the compounds studied in this work. | ||
Compound 1 was synthesized starting from commercially available acetylenedicarboxylic acid by means of the initial stereospecific bromination of the triple bond, followed by the formation of the corresponding acid dichloride and coupling with N-methylaniline through the Schotten–Baumann protocol. Compound 2 was synthesized in a similar manner starting from fumaric acid. With respect to 1, it lacks the bromine atoms, which are substituted by hydrogen atoms. Compound 3 was synthesized by double nucleophilic substitution of N-methylaniline on (E)-1,2,3,4-tetrabromobut-2-ene; with respect to 1, it lacks the carbonyl groups, which are substituted by CH2 groups. The synthetic procedures for the compounds, along with their full chemical characterization and temperature-dependent NMR spectra are reported in the ESI.†
In the case of compound 1, the 1H NMR spectra showed the presence of different conformers in slow equilibrium on the NMR timescale at room temperature (Fig. S1†), associated with rotation about the (Br)C–C(O) and/or the formally single (O)C–N bonds. On the other hand, such conformational exchange was already fast at room temperature on the NMR timescale in both compounds 2 and 3. The higher rigidity of 1 is likely the consequence of the presence, in 1, of the bulky bromine atoms (if compared with 2) and the carbonyl groups (if compared with 3). From DFT geometry optimization of 1, 2 and 3 (Fig. S2†), the (Br)C–C(O) and (O)C–N bond lengths measure 1.513, 1.497, and 1.522 Å and 1.360, 1.372, and 1.440 Å, respectively. Meanwhile, free rotation around (O)C–N is clearly hindered in 1 and 2 by its partial double bond character, and the steric effect associated with Br atoms in 1 and 3 manifests in their strongly twisted conformation, different from the rather planar one of 2. The C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C–C–N torsion angles measure in fact 106, 170 and 139° in 1, 2 and 3, respectively.
C–C–N torsion angles measure in fact 106, 170 and 139° in 1, 2 and 3, respectively.
The absorption spectra of 1–3 in ACN (10−5 mol L−1) show UV bands with the maxima at 231 nm for 1, 285 nm for 2, and 300 and 250 nm for 3 (Fig. S4†). In the case of 2, the low energy absorption band shows a long tail extending above 300 nm, suggesting the presence of low intensity transitions at lower energy. On the basis of DFT/TDDFT calculations, the first singlet excited state (S1) of 1, 2 and 3 is computed at 252, 269 and 254 nm with an oscillator strength (f) equal to 0, 0.007 and 0.079, respectively. The low f values of S1 for 2 are in agreement with the expected weak transitions at 300–330 nm in their observed UV spectra. The overall shapes of the simulated spectra closely resemble the observed ones (Fig. S3†), although a hypsochromic shift of about 50 nm is observed. Compounds 1 and 2 are non-emissive in solution, whereas 3 displays a very weak, sharp fluorescence emission (PLQY < 0.001%) at 330 nm at 298 K (ACN 10−5 M) that shifts to 387 nm at 77 K (Fig. S5†). The red-shifted emission observed at 77 K might be associated with molecular aggregation in solid ACN. In fact, by adding a non-solvent (H2O) to the ACN solution, an increase in the emission intensity, with a concomitant red-shift (from 330 to 380 nm), is observed (Fig. S6†), demonstrating the AIE behavior of the compound.
Powders of compound 1 show dual-mode (fluorescence and phosphorescence) emission. In fact, the solid state emission spectra (Fig. 2, top) are composed of structured fluorescence with peaks at 460, 490 and 522 nm having a lifetime of 3.82 ns (Fig. S7†) and long-lived structured phosphorescence, redshifted by 0.37 eV from fluorescence, with peaks at 534, 577 and 622 nm and a very long lifetime of 55.6 ms (Fig. S8†). Compound 2 as a powder exhibits weak fluorescence peaked at 480 nm lacking phosphorescence emission (Fig. S9†). Powders of 3 (Fig. 2, bottom) show an emission spectrum composed of a very broad fluorescence emission (covering the 400–700 nm spectral region) centered at 503 nm (τav = 1.53 ns, Fig. S10†) and two phosphorescence components (Fig. S11†) at about 480 nm (τav = 1.03 ms) and 600 nm (τav = 5.92 ms). For all the compounds, the fluorescence excitation spectra are centered at the edge of the absorption spectra (Fig. S12†), while the excitations of the lower energy phosphorescence emission of compounds 1 and 3 display bands well below the absorption edge (Fig. S13 and S14†), confirming the triplet nature of the electronic states responsible for these emissions. Powder X-ray diffraction (PXRD) analysis of 1–3 demonstrated their crystalline nature (Fig. S15–S17†).
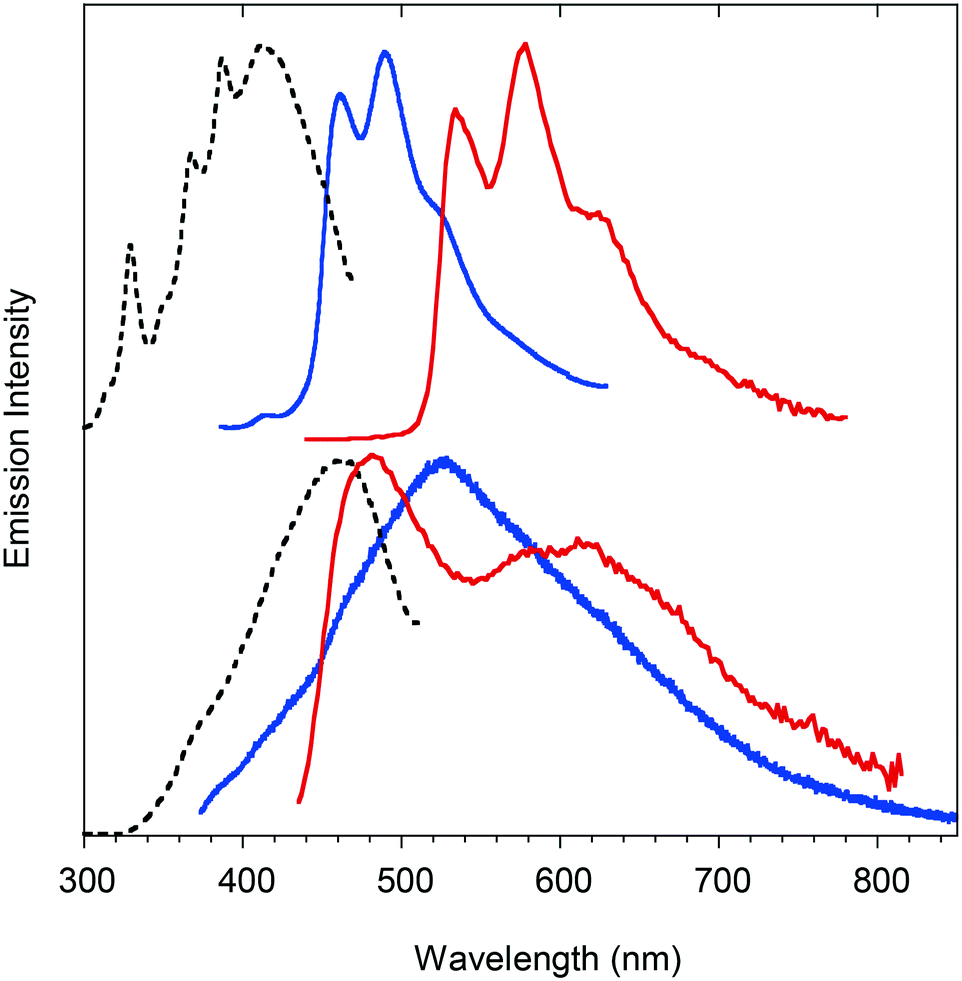 | ||
| Fig. 2 Emissive behavior of compounds 1 (top) and 3 (bottom) as crystalline powders. PL spectra (blue solid lines; 1λex = 330 nm, 3λex = 335 nm), PLE spectra (black dashed lines; 1λem = 490 nm, 3λem = 525 nm) and phosphorescence spectra (red solid lines; 1λex = 410 nm, time delay 1 ms, window 10 ms; 3λex = 420 nm, time delay 5 ms, window 15 ms). | ||
Comparing the photophysical properties of the powders of the three compounds, the role of the bromine atom in activating phosphorescence through spin–orbit coupling (SOC) effects at the molecular and/or intermolecular level is evident. Moreover, it is observed that compound 1 possesses RTP with lifetimes longer than those of 3, suggesting the better capability of its aggregates to suppress non-radiative relaxations and, at the same time, stabilize the lowest triplet excited states. Since 1 and 3 share the same type and number of heavy atoms, it is expected that the intrinsic SOC constants are similar for the two compounds. The observed longer RTP lifetimes in 1 could be therefore explained on the basis of intermolecular effects, in particular to the presence of strong Br⋯O halogen bonds governing the crystal packing of 1 (see below). This interaction, on the one hand, is able to impose a more effective rigidification of the molecular structure of 1 in the crystal and, on the other hand, allows for a more efficient SOC through extrinsic heavy atom effects.15 To support this hypothesis, we compared the optical properties of 1 in the crystalline and amorphous states. The amorphous state, obtained by melting and quickly cooling the crystalline powders (see PXRD patterns in Fig. S18†), displayed a more structured and blue shifted fluorescence emission accompanied by weaker and shorter lived (τav = 4.03 ms) phosphorescence (see Fig. S19 and S20†). Another indirect proof of these deductions came from the comparison of the dual-mode (fluorescence and phosphorescence) emission properties of geminate crystals (Fig. S21†) and crystalline powders of 3 that showed different lifetimes and spectral shapes of the phosphorescence components. The dependence of the phosphorescence properties of compound 3 on the nature of aggregation is a consequence of the absence of strong intermolecular interactions able to lock the structure in a rigid and well-defined lattice.
Single crystals of 1 and 2 suitable for X-ray analysis have been grown successfully as thin laminae from ACN solutions. Both compounds crystallize in the P21/c space group with half a molecule (1) or three half molecules (2) (labelled A, B and C) in the asymmetric unit (see Table S1†). In all cases, the molecules lie about an inversion center. Their conformation closely resembles the gas-phase computed one, with a highly distorted molecular skeleton in 1 and an almost coplanar one in 2, as denoted by the C1′C1–C2–N1 torsion angles (see Fig. S22 and S23† for the atom labelling). In fact, they measure 106.0(1)° in 1 and 172.4(7), −179.0(6) and 172.1(6)° in molecules A, B and C, respectively, of 2. The phenyl rings disrupt the planarity of 2, being rotated by 57.8(3) (A), 73.9(2) (B) and 63.7(3)° (C) with respect to the plane through atoms C1, C2, N1 and C3. The crystal structure of 1 (Fig. 3) is governed by the formation of infinite chains along the a crystallographic axis where centrosymmetry-related molecules are interconnected on both sides by two strong centrosymmetry-related Br⋯O halogen bonds (rBr⋯O = 3.114(2) Å, shorter by 7.6% than the sum of Br and O van der Waals radii; the CO⋯Br angle is equal to 111.2(1)°). This relative disposition allows for the formation of an additional Br⋯Br interaction (rBr⋯Br = 3.707(1) Å, comparable with two times the bromine van der Waals radius). Moreover, strong π–π interactions between aromatic moieties (the centroid–centroid distance is 3.906 Å) stabilize the overall structure.
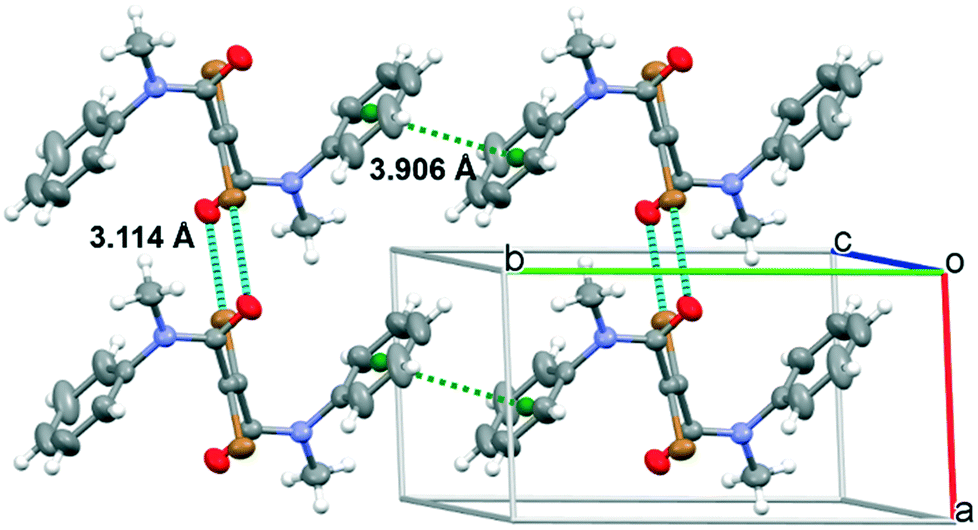 | ||
| Fig. 3 Crystal packing of 1 showing the Br⋯O halogen bonds and distances between rings' centroids (cyan and green dashed lines, respectively). Ellipsoids drawn at 50% probability. | ||
The crystal packing of compound 2 (Fig. S24†) consists of layers of A molecules, placed in the crystallographic bc plane, alternating with layers of B and C molecules. Only weak C–H⋯π hydrogen bonds (HBs, rH8A⋯C2A = 2.87 Å) keep the A molecules together, while the B and C molecules are interconnected by relatively strong π⋯π interactions between CC double bonds (rC1B⋯C1C = 3.309(9) Å) and C–H⋯O HBs (rH5B⋯O1C = 2.48 Å) along b and by C–H⋯O HBs (rH3B2⋯O1C = 2.58 Å) along c. The layers are connected to each other by weaker C–H⋯O HBs (rH8C⋯O1A = 2.67 Å). The comparison between the crystal packing of 1 and 2 clearly suggests the higher rigidity of the former structure due to the presence of the strong Br⋯O halogen bonds.
The comparison between the emissive properties of the three compounds demonstrates the importance of the bromine atoms and halogen bonds in the activation of long-lived RTP. In fact, the bromine atoms, besides activating the ISC process through spin–orbit coupling, are strongly involved in the rigidification of the crystal structure by the halogen bonds, thus increasing the phosphorescence lifetime at room temperature.13,14h
In conclusion, we have introduced a completely new class of simple fumaramide derivatives showing RTP in the solid state with very long lifetimes, the result of crystallization-induced activation of the emission. The introduction of crystal engineering paradigms, and, in particular, the use of directional and specific halogen bonds allow for the molecules to efficiently rigidify in the solid state in the case of 1, and to activate a dual mode fluorescence–phosphorescence radiative mechanism. The pronounced Stokes shift can be promising for spectral conversion applications.16 We are currently pursuing the introduction of suitable functionalities on the aryl fragments flanking the central chromophore unit in 1, as a way to reinforce and modulate the solid-state interactions and the excitation wavelength for the RTP response, which can be highly relevant for applications in bioimaging.
This work was supported by the University of Pavia (postdoctoral fellowship to AN), and MIUR (PRIN 2017 BOOSTER Prot. 2017YXX8AZ to DP). A. F. gratefully thanks Pietro Colombo for support in the single crystal X-ray data collection. A. N. and D. P. thank Davide Callegari for support with the PXRD experiments.
Conflicts of interest
There are no conflicts to declare.Notes and references
-
(a) A. Kishimura, T. Yamashita, K. Yamaguchi and T. Aida, Nat. Mater., 2005, 4, 546–549 CrossRef CAS
; (b) J. P. Celli, B. Q. Spring, I. Rizvi, C. L. Evans, K. S. Samkoe, S. Verma, B. W. Pogue and T. Hasan, Chem. Rev., 2010, 110, 2795–2838 CrossRef CAS
-
(a) Y. Li, B. Ma, Y. Long, Z. Song, J. Su, Y. Wang, R. Liu, G. Song and H. Zhu, J. Mater. Chem. C, 2020, 8, 2467–2474 RSC
-
(a) V. Ramamurthy, D. F. Eaton and J. V. Caspar, Acc. Chem. Res., 1992, 25, 292–299 CrossRef
- Selected examples:
(a) N. Zhang, D. Wang, C. Qian, Z. An, W. Huang and Y. Zhao, Nat. Commun., 2020, 11, 944 CrossRef
- Y. Gong, G. Chen, Q. Peng, W. Z. Yuan, Y. Xie, S. Li, Y. Zhang and B. Z. Tang, Adv. Mater., 2015, 27, 6195–6201 CrossRef CAS
-
(a) Kenry, C. Chen and B. Liu, Nat. Commun., 2019, 10, 2111 CrossRef CAS
- G. Zhang, J. Chen, S. J. Payne, S. E. Kooi, J. N. Demas and C. L. Fraser, J. Am. Chem. Soc., 2007, 129, 8942–8943 CrossRef CAS
- O. Bolton, D. Lee, J. Jung and J. Kim, Chem. Mater., 2014, 26, 6644–6649 CrossRef CAS
- E. Lucenti, A. Forni, C. Botta, L. Carlucci, C. Giannini, D. Marinotto, A. Previtali, S. Righetto and E. Cariati, J. Phys. Chem. Lett., 2017, 8, 1894–1898 CrossRef CAS
- A. Fermi, G. Bergamini, M. Roy, M. Gingras and P. Ceroni, J. Am. Chem. Soc., 2014, 136, 6395–6400 CrossRef CAS
-
(a) S. Xu, Y. Duan and B. Liu, Adv. Mater., 2020, 32, 1903530 CrossRef CAS
-
(a) W. Zhao, Z. He, J. W. Y. Lam, Q. Peng, H. Ma, Z. Shuai, G. Bai, J. Hao and B. Z. Tang, Chem, 2016, 1, 592 CrossRef CAS
- A. Forni, E. Lucenti, C. Botta and E. Cariati, J. Mater. Chem. C, 2018, 6, 4603–4626 RSC
-
(a) A. Nitti, F. Villafiorita-Monteleone, A. Pacini, C. Botta, T. Virgili, A. Forni, E. Cariati, M. Boiocchi and D. Pasini, Faraday Discuss., 2017, 196, 143–161 RSC
- E. Lucenti, A. Forni, C. Botta, C. Giannini, D. Malpicci, D. Marinotto, A. Previtali, S. Righetto and E. Cariati, Chem. – Eur. J., 2019, 25, 2452–2456 CrossRef CAS
- K. Kanosue and S. Ando, ACS Macro Lett., 2016, 5, 1301–1305 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details and characterization of the new compounds, additional spectroscopic experiments, and details for X-ray crystal structures and calculations. CCDC 2020250 and 2020251. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d0ce01253j |
| This journal is © The Royal Society of Chemistry 2020 |